

Abstract
Nonacholic fatty liver disease, or hepatic steatosis, is the most common liver disorder affecting the western world and currently has no pharmacologic cure. Thus, many investigations have focused on alternative strategies to treat or prevent hepatic steatosis. Our laboratory has shown that chronic heat treatment (HT) mitigates glucose intolerance, insulin resistance, and hepatic steatosis in rodent models of obesity. Here, we investigate the direct bioenergetic mechanism(s) surrounding the metabolic effects of HT on hepatic mitochondria. Utilizing mitochondrial proteomics and respiratory function assays, we show that one bout of acute HT (42°C for 20 min) in male C57Bl/6J mice (n = 6/group) triggers a hepatic mitochondrial heat shock response resulting in acute reductions in respiratory capacity, degradation of key mitochondrial enzymes, and induction of mitophagy via mitochondrial ubiquitination. We also show that chronic bouts of HT and recurrent activation of the heat shock response enhances mitochondrial quality and respiratory function via compensatory adaptations in mitochondrial organization, gene expression, and transport even during 4 weeks of high-fat feeding (n = 6/group). Finally, utilizing a liver-specific heat shock protein 72 (HSP72) knockout model, we are the first to show that HSP72, a protein putatively driving the HT metabolic response, does not play a significant role in the hepatic mitochondrial adaptation to acute or chronic HT. However, HSP72 is required for the reductions in blood glucose observed with chronic HT. Our data are the first to suggest that chronic HT (1) improves hepatic mitochondrial respiratory efficiency via mitochondrial remodeling and (2) reduces blood glucose in a hepatic HSP72-dependent manner.
Keywords: mitophagy, HSP72, autophagy, liver, redox, ubiquitin, mitochondrial organization, chaperone-mediated autophagy
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disorder in the Western world and affects over one-third of the general population.1–3 Importantly, NAFLD contributes to the etiology of Type 2 diabetes, cardiovascular disease, and gross metabolic dysfunction.4 One primary factor contributing to the development of NAFLD is impaired hepatic mitochondrial function.5 To this end, our group and others have shown that defects in mitochondrial quality control pathways, such as mitophagy (the targeted degradation of damaged mitochondria), increase the susceptibility to hepatic steatosis in preclinical rodent models.6–9 While we have shown that treatments such as exercise can alter mitophagy and mitochondrial respiratory function during high-fat feeding,9 alternative strategies that improve hepatic mitochondrial quality control and function and ameliorate steatosis are needed as contraindications can exist for exercise in certain patient populations.10 One potential strategy that our group has explored to great effect in animals and primary hepatocytes is heat treatment (HT).11,12
First established by Philip Hooper in 1999,13 we and others have consistently shown that chronic whole-body HT positively impacts glucose homeostasis in both obese humans and animals.11,12,14–17 Importantly, the current hypothesis surrounding the positive metabolic effects of HT is tied to increased Heat Shock Protein 72 (HSP72) levels.18 This hypothesis has been propagated by findings that HSP72 content is reduced in both liver and muscle tissue during obese conditions15,19 and that restoration of HSP72 levels via HT is associated with positive metabolic benefits including lowered hepatic lipid storage.11,12,14,15,20 Moreover, increased HSP72 content via genetic overexpression or pharmacologic activation in animals fed a high-fat diet (HFD) is shown to benefit insulin sensitivity and skeletal muscle mitochondrial content.15,21–24 We have also recently shown that acute knockdown of HSP72 in primary hepatocytes causes increased lipid uptake, reduced fatty acid oxidation (FAO), and depolarization of mitochondria.11 However, difficulties in generating a tissue-specific knockout model (floxed alleles) have kept the field from definitively showing a role for HSP72 in muscle and/or liver metabolic adaptations with HT.
While chronic HT and/or increased HSP72 chaperone content lead to positive metabolic benefits during high-fat feeding, very little is known about the underlying molecular mechanisms by which HT-induced activation of the acute heat shock response leads to long-term metabolic adaptations. Previous literature shows that HT acutely induces mitochondrial control mechanisms, such as mitophagy, to protect the cell during HT-induced stress.25–27 Increasing evidence suggests that these acute mitochondrial/metabolic control mechanisms may be carried out in a chaperone-dependent manner. For instance, chaperone-mediated autophagy (CMA), the targeted degradation of proteins via lysosomal translocation, is key in degrading glycolytic enzymes,28 but it has not been shown if HT induces CMA or if the induction of mitophagy is HSP72-dependent. Although it has not been examined, functional and proteomic changes at the mitochondrial level may be potential mechanism underlying the metabolic benefits of HT. Specifically, the recurrent activation of heat stress pathways may lead to adaptive changes in the mitochondrial proteome, enhancing mitochondrial quality and efficiency. It is possible that these adaptive changes ultimately enable the net metabolic benefit of HT and could point to new therapeutic targets to treat metabolic dysfunction.
We conducted detailed functional and quantitative studies on isolated hepatic mitochondria from mice subjected to acute (single bout) and chronic (multiple bouts) of HT. We are the first group to generate a liver-specific HSP72 knockout mouse (L-HSP72KO) to determine whether hepatic HSP72 was required for the acute (single bout) or chronic (multiple bouts) metabolic response to HT. Using both low-fat and high-fat diets (LFD and HFD, respectively), we confirmed that HT causes an acute bioenergetic stress (defined here as the mitochondrial heat shock response)—ultimately triggering mitochondrial quality control pathways such as mitophagy independent of other metabolic challenges such as diet. Importantly, we are the first to show that recurrent activation of the mitochondrial heat shock response via chronic bouts of HT leads to enhanced mitochondrial quality and respiratory function during high-fat feeding. Using a novel liver-specific L-HSP72KO model, we also show that hepatic HSP72 does not play a significant role in mitochondrial adaptations to HT but is required for HT-induced reductions in blood glucose over time.
Materials and Methods
Ethical Approval
The animal protocol used for this study was approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH guide, 8th edn, 2011), as well as the ARRIVE guidelines for reporting animal experiments. Mice were anesthetized with pentobarbital sodium (100 mg kg−1) before the terminal procedure.
Animals
For the initial, acute postheat time course studies, 12-week-old male wildtype (WT) C57Bl/6J mice were used for all experiments, n = 6/group (sham [SHM] or heat-treated [HT]), per time point (0, 0.5, 1, and 2 h post-HT). Follow-up mitophagy flux studies using in vivo injections of the autophagy inhibitor leupeptin (LEU) also used 12-week-old male WT C57Bl/6J mice, n = 8/group (SHM or 24 h post-HT). To determine the role of hepatic HSP72 in the response to HT, we created a novel liver‐specific, conditional HSP72 null (L-HSP72KO) mouse maintained on the C57Bl/6J background. Briefly, we inserted synthetic introns (via guide RNA) into mouse embryonic stem cells, where loxP sites were then inserted flanking the functional domain of Hspa1a to create a premature stop codon (Figure S1). C57Bl/6J mice with the floxed Hspa1a gene were bred together and subsequently, 10-week-old male floxed mice were exposed to AAV8-Alb-Cre (107787-AAV8) or AAV8-Alb-GFP (105535-AAV8) obtained from Vector Biolabs (Malvern, PA, USA) via tail vein injection (1 × 1011 CG/mL diluted in 200 µL of saline) to generate L-HSP72KO and WT littermates, respectively (Figure S1). All WT and L-HSP72KO mice were left for 14 days postviral injection to ensure viral delivery and thus they were 12 weeks of age at the beginning of experimentation (n = 6/group for acute and chronic studies; SHM vs HT, WT vs l-HSP72KO). All mice were housed at thermoneutrality (∼30°C) on a reverse light cycle (dark 10.00–22.00 h) with ad libitum access to water and a matched low‐fat diet (LFD; D12110704: 10% kcal fat, 3.5% kcal sucrose, and 3.85 kcal g−1 energy density, Research Diets, New Brunswick, NJ, USA) or HFD (D12492: 60% kcal fat, 7% kcal sucrose, and 5.21 kcal g−1) as indicated. Animal weights and food intake were measured weekly and every 72 h, respectively, throughout the 4‐week study. All mice were euthanized, and tissue was collected at the end of their light cycle (10.00). Mice were not fasted prior to HT or sacrifice.
In Vivo SHM or HT
Mice in both SHM and HT groups were brought to the laboratory at the end of their light cycle (10.00). All mice were then anesthetized with 75 mg/kg Ketamine + 7.5 mg/kg Xylazine in two doses (a 50 mg/kg Ketamine + 5 mg/kg Xylazine dose followed by a 25 mg/kg Ketamine + 2.5 mg/kg Xylazine dose 10 min later). After mice were fully anesthetized, a rectal probe (RET-3; Braintree Scientific; Braintree, MA, USA) was inserted and taped to the tail to monitor core temperature via waterproof thermometers (THS-232-101; ThermoWorks, American Fork, UT, USA). The probed animal was placed into metal beads (A1254301; Thermo Fisher Scientific; Waltham, MA, USA) warmed in a water bath (15-462-5Q; Thermo Fisher Scientific; Waltham, MA, USA) set to either 40°C or 45°C for SHM and HT, respectively. Animals started their SHM or HT when their core temperatures reached 37°C or 41°C, respectively. Animals were maintained at core temperatures 37°C–38°C or 41°C–42°C for the respective SHM and HT groups for 20 min. After 20 min of SHM or HT, animals were removed from the beads, the rectal probe was removed, and mice were placed back into their cages for recovery. All animals received a dose of saline commensurate (v/v) to their dose of anesthetic solution. For acute HT experiments, animals were subjected to a single bout of SHM or HT and were sacrificed at the times indicated post-HT (Figure S2A). For chronic HT experiments, animals were subjected to a total of nine SHM or HT bouts spread across the 4-week period (every 72 h) and all animals were sacrificed 72 h after their last bout of SHM or HT to observe basal adaptations (Figure S2B). We elected to place 72 h recovery periods between treatments during the entirety of the experiment, as well as before the final sacrifice, because previous literature suggests that there is a 72 h refractory period between consecutive HTs in terms of heat preconditioning.29
Tissue Collection and Mitochondrial Isolation
After anesthetizing mice, livers were quickly removed to be either flash-frozen in liquid nitrogen or placed into 8 mL ice-cold mitochondrial isolation buffer (220 mM mannitol, 70 mM sucrose, 10 mM Tris, 1 mM EDTA, pH adjusted to 7.4 with KOH) and minced. Using the minced liver, hepatic mitochondria were prepared as previously described.30 Briefly, minced livers were homogenized on ice in 8 mL of mitochondrial isolation buffer placed in a 15 mL glass tube with a Teflon pestle. Liver homogenates were placed into 50 mL conical tubes and centrifuged (4°C, 10 min, 1500 g). After centrifugation, the supernatant was filtered through sterile gauzed (to remove any debris) and transferred into a 30 mL round‐bottom tube and centrifuged (4°C, 10 min, 8000 g). Postcentrifugation, the supernatant was discarded, and the pellet was resuspended in 6 mL isolation buffer using a glass‐on‐glass Dounce homogenizer for five passes. The resuspension was then transferred back into the 30 mL round‐bottom tube and centrifuged (4°C, 10 min, 6000 g). Again, the supernatant was discarded, and the pellet was resuspended in 4 mL isolation buffer containing 0.1% fatty acid‐free BSA using Dounce homogenization, transferred back into the 30 mL round‐bottom tube, and centrifuged (4°C, 10 min, 4000 g). After this final centrifugation step, the isolated mitochondrial pellet was resuspended in 350 μL modified MiR05 mitochondrial respiration buffer (0.5 mM EGTA, 3 mM MgCl2, 60 mM KMES, 20 mM glucose, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 0.1% BSA, adjust pH to 7.1 with KOH). Protein concentrations were determined by bicinchoninic acid assay (BCA) per the manufacturer’s instructions (23225; Thermo Fisher Scientific; Waltham, MA, USA).
Mitochondrial Respiration and H2O2 Emission
Mitochondrial respiratory capacity (pmol s−1 mL−1) and H2O2 emission (pmol s−1 ml−1) were measured simultaneously via the Oroboros O2k fluorometer (Oroboros Instruments, Innsbruck, Austria) as described previously.31 All O2k data were analyzed using DatLab 7 (Oroboros Instruments). All chambers were calibrated for atmospheric oxygen and H2O2 emission prior to each run. Mitochondrial isolates were added to the chamber containing the modified MiR05 mitochondrial respiration buffer described above with the addition of malate (2 mM), free CoA (63.5 μM), and l‐carnitine (2.5 mM) to total volume of 2 mL (temperature of 30°C). This modified MiR05 mitochondrial respiration buffer did not contain lactobionic acid or taurine as these antioxidants may have interfered with our H2O2 emission measurements. Coupled maximal respiration rate (State 3) was determined after the addition of adenosine 5′‐disphosphate (ADP; 2.5 mM). Maximal respiration rates for ADP-dependent respiration (State 3) were measured in two independent protocols utilizing either palmitoyl-CoA (10 μM) or pyruvate (5 mM) in mitochondrial isolates. Maximal respiratory rates for ADP+succinate-dependent respiration (State 3S) were determined with the addition of succinate (10 mM) for both substrates. Finally, maximal uncoupled respiration was determined with titrations of carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (1 μM) for each substrate. All data were normalized to mitochondrial protein content within each chamber as determined by the BCA mentioned above. H2O2:O2 ratios were calculated as State 3S H2O2 emission/3S respiration to infer the quantity of H2O2 emission at a given respiratory rate.
Serum Metabolic Measures
Blood was collected immediately upon sacrifice via cardiac puncture, placed in a 1.5 mL microtube at room temperature for 30 min, and placed on ice thereafter. All samples were then centrifuged (4°C, 10 min, 1500 g) and serum was collected and placed into a fresh screw-top tube, then frozen and stored at −80°C prior to analysis. Serum nonesterified fatty acids (NEFAs; 999-34691, 991-34891, 993-35191; Wako Chemicals; Richmond, VA, USA), glucose (CBA086; Sigma-Aldrich; St. Louis, MO, USA), insulin (90080; Crystal Chem; Elk Grove Village, IL, USA), lactate (MAK064; Sigma-Aldrich; St. Louis, MO, USA), and β-hydroxybutyrate (2440-058; Stanbio Laboratory; Boerne, TX, USA) were measured using the manufacturer’s protocols.
Hepatic Triacylglyceride Analysis
A commercially available kit (TR0100; Sigma‐Aldrich; St. Louis, MO, USA) was used to determine hepatic triacylglycerol concentration as previously described.32
Western Blotting and Immunoprecipitation Assays
Mitochondrial isolations were completed as described above in mitochondrial isolation buffer; however, all samples were diluted 1:1 with a 2× NP-40 solution (300 mM NaCl, 2% NP-40, 100 mM Tris, 10 mM EDTA, and 2× protease/phosphatase inhibitor cocktail [78429 and 78420, respectively; Thermo Fisher Scientific; Waltham, MA, USA]) to create samples for western blotting and immunoprecipitation assays. For whole-cell lysate samples, frozen livers were smashed and homogenized in a 1× NP-40 solution as mentioned above with a bead TissueLyser II (Qiagen; Hilden, DE). The whole-cell lysate was used for immunoprecipitation assays, which were conducted using a commercially manufactured kit per the manufacturer’s instructions (17–500; Millipore Sigma; Burlington, MA, USA). Western‐ready Laemmli samples were produced from liver tissue homogenate, immunoprecipitated protein isolates, and isolated mitochondria. SDS‐PAGE was used to separate samples, which were then transferred to polyvinylidene difluoride membrane and probed with primary antibodies at a concentration of 1:2000 (specific antibody information listed below). Densitometry was used to quantify individual protein bands with Image Lab software (Bio‐Rad Laboratories, Hercules, CA, USA). All values were normalized to total protein or mitochondrial protein using 0.1% amido‐black (Sigma‐Aldrich; St. Louis, MO, USA) staining as previously described.30 Immunoprecipitation assays were only visualized and not quantified.
Quantitative Determination of Mitochondrial Fission and Mitophagy
For quantitative assessment of mitochondrial fission/fusion, we used western blotting in our mitochondrial isolates to determine the content of putative mitochondrial fission and fusion proteins: mitofusin 2 (MFN2; 9482S), OPA1 Mitochondrial Dynamin Like GTPase (OPA1; 80471S), and Dynamin-Related Protein 1 (DRP1; 8570S). For quantitative assessment of mitophagy, we used western blotting in our mitochondrial isolates to determine the content of canonical and putative mitophagy proteins: total ubiquitin (Ubiq; sc-8017), Sequestosome 1 (p62; 5114S), Microtubule-Associated Protein 1 Light Chain 3A/B (LC3A/B; 12741S), BCL2 Interacting Protein 3 (BNIP3; 3769S), HSP72 (ADI-SPA-810), Parkin (4211S), Carboxy Terminus of Hsp70-Interacting Protein (CHIP; ab2917), and Heat Shock Cognate 71 KDa Protein (HSC70; 10654-1-AP). Moreover, we measured electron transport proteins (Total OXPHOS; ab110413) to determine the specificity of degradation with regard to mitochondrial catalytic function. Finally, we utilized immunoprecipitation assays to determine binding between Ubiq, HSC70, CHIP, p62, and total OXPHOS. MFN2, OPA1, DRP1, p62, LC3A/B, BNIP3, and Parkin antibodies were purchased from Cell Signaling Technologies (Danvers, MA, USA). CHIP and Total OXPHOS antibodies were purchased from Abcam (Cambridge, UK). Ubiq, HSP72, and HSC70 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), Enzo Life Sciences (Farmingdale, NY, USA), and Thermo Fisher Scientific (Waltham, MA, USA), respectively.
Transmission Electron Microscopy and Confocal Microscopy for Mitochondrial Fission and Mitophagy
To obtain a qualitative visual representation of mitochondrial fission/fusion, we used transmission electron microscopy to visualize hepatic mitochondria 0 h post-HT or SHM condition (n = 3/group). Briefly, the liver tissue was excised and immediately fixed using a 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) solution (16536-15; Electron Microscopy Sciences; Hatfield, PA, USA), postfixed in 1% osmium tetroxide, rinsed and dehydrated in a series of ethanol followed by two changes of propylene oxide. Samples were then polymerized in Embed 812 resin. Blocks were trimmed and sectioned into thin 70 nm section. Sections were placed on grids and contrasted with 3% uranyl acetate followed by Reynold’s lead citrate.
Sections viewed on a JEM-1400 electron microscope (JEOL; Akishima, Tokyo, JP).
To obtain a visual representation of mitophagy activation, we used an Adenoviral (Ad) Cox8-GFP-mCherry mitophagy reporter assay as we described previously.33 Male WT mice (n = 3/group) were injected with (Ad)-Cox8-GFP-mCherry (5 × 108 PFU/mouse diluted in 200 µL of saline) via tail vein injection. All animals were given 14 days to recover prior to experimentation to ensure viral uptake. Animals then underwent either the SHM or HT protocol described above and were sacrificed immediately (0 h) posttreatment. Liver tissue was removed and fixed using ice-cold 4% paraformaldehyde overnight. Tissues were then washed 3× with ice-cold PBS and transferred to chilled 20% sucrose overnight. Tissues were then embedded in OCT, frozen on dry ice, and sectioned with a cryostat (CM1860; Leica Biosystems; Buffalo Grove, IL, USA). Fluorescence images were then acquired analyzed under the confocal microscope (Leica TCS SPE Confocal Microscope; Leica Microsystems; Buffalo Grove, IL, USA).
Confirmation of Mitophagy Flux via LEU
An additional cohort of WT mice (n = 8/group) was used to determine if mitophagy flux was impacted by HT. First, WT mice subjected to SHM or HT were given intraperitoneal injections of either 40 mg kg − 1 LEU immediately post-HT. An additional dose of 20 mg kg − 1 LEU was given 4 h prior to sacrifice to ensure continued autophagy inhibition. All mice were sacrificed 24 h following SHM or HT. LEU was chosen because we, and others, have shown that LEU is a potent and specific inhibitor of hepatic autophagy/mitophagy in vivo.8,34,35 Mitophagy flux was inferred by the accumulation of mitophagy-related proteins including LC3A/B, p62, CHIP, and ubiq in isolated mitochondrial samples (“Methods and Materials” section) via western blotting.
Mitochondrial Proteomics
A subset (n = 3/group, WT SHM vs HT) of isolated mitochondrial fractions obtained for western blotting were used for proteomics. Proteomics was completed separately for acute and chronic HT samples, the parameters for each are listed below (first = acute and second = chronic). Proteins were reduced, alkylated, and purified by chloroform/methanol extraction prior to digestion with sequencing grade modified porcine trypsin (Promega; Madison, WI, USA). Tryptic peptides were labeled using a tandem mass tag 6-plex isobaric label reagent set (Thermo Fisher Scientific; Waltham, MA, USA). The labeled peptide multiplex was separated into 36 (acute) or 46 (chronic) fractions on a 100 × 1.0 mm Acquity BEH C18 column (Waters; Milford, MA, USA) using an UltiMate 3000 UHPLC system (Thermo Fisher Scientific; Waltham, MA, USA) with a 40 (acute) or 50 (chronic) min gradient from 99:1 to 60:40 buffer A:B ratio (Buffer A = 0.1% formic acid, 0.5% acetonitrile; Buffer B = 0.1% formic acid, 99.9% acetonitrile) under basic pH conditions (pH 10), and then consolidated into 18 super-fractions. Each super-fraction was then further separated by reverse-phase XSelect CSH C18 2.5 µm resin (Waters; Milford, MA, USA) on an in-line 150 × 0.075 mm column using an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific; Waltham, MA, USA). Peptides were eluted using a 60 min gradient from 98:2 to 60:40 buffer A:B ratio. Eluted peptides were ionized by electrospray (2.2 kV) followed by mass spectrometric analysis on an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific; Waltham, MA, USA) using multinotch MS3 parameters with real-time search enabled. MS data were acquired using the FTMS analyzer in top-speed profile mode at a resolution of 120000 over a range of 375–1500 m/z (acute) or 400–1600 m/z (chronic). Following CID activation with normalized collision energy of 35.0, MS/MS data were acquired using the ion trap analyzer in centroid mode and normal mass range. Using synchronous precursor selection, up to 10 MS/MS precursors were selected for HCD activation with normalized collision energy of 65.0 (acute) or 50.0 (chronic), followed by acquisition of MS3 reporter ion data using the FTMS analyzer at a resolution of 50000 (acute) or 15000 (chronic) over a range of 100–500 m/z.
Mitochondrial Proteomic Data Analysis
Proteins were identified and reporter ions quantified using MaxQuant (Max Planck Institute, version 1.6.12.0) against the UniprotKB Mus musculus (April 2020) with a parent ion tolerance of 3 ppm, a fragment ion tolerance of 0.5 Da, and a reporter ion tolerance of 0.003 Da. Scaffold Q + S (Proteome Software) was used to verify MS/MS-based peptide and protein identifications (protein identifications were accepted if they could be established with <1.0% false discovery and contained at least two identified peptides; protein probabilities were assigned by the Protein Prophet algorithm36) and to perform reporter ion-based statistical analysis.
Protein TMT MS3 reporter ion intensity values were assessed for quality using our in-house ProteiNorm app, a user-friendly tool for a systematic evaluation of normalization methods, imputation of missing values, and comparisons of different differential abundance methods.37 Popular normalization methods are evaluated including log2 normalization (Log2), median normalization (Median), mean normalization (Mean), variance stabilizing normalization (VSN),38 quantile normalization (Quantile),39 cyclic loess normalization (Cyclic Loess),40 global robust linear regression normalization (RLR),41 and global intensity normalization (Global Intensity).41 The individual performance of each method can be evaluated by comparing the following metrics: total intensity, Pooled intragroup Coefficient of Variation (PCV), Pooled intragroup Median Absolute Deviation (PMAD), Pooled intragroup estimate of variance (PEV), intragroup correlation, sample correlation heatmap (Pearson), and log2-ratio distributions. The data were normalized using cyclic loess as this method had the lowest sample variance and highest sample correlation metrics. The normalized data were analyzed for differential abundance using Linear Models for Microarray Data (limma) with empirical Bayes (eBayes) smoothing to the standard errors.40 Proteins with an FDR adjusted P < 0.05 and a fold change > 2 were considered significant (fold change was determined as change in HT group vs SHM control). All proteomic data sets are publicly available at https://doi.org/10.6084/m9.figshare.13360475. Genome set enrichment analysis (GSEA) was performed on all proteins deemed to have significant changes with HT using WebGestalt 2019.42 Using the Mus musculus database, gene ontology for nonredundant biological processes was determined with an FDR adjusted P < 0.05. All data are presented as normalized enrichment value for significant biological processes.
mRNA Expression
RNA was extracted using an RNeasy mini‐kit following the manufacturer’s instructions (74104; Qiagen; Hilden, DE) and cDNA was prepared as previously described.43 A QuantStudio 3 Real‐Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) and SYBR green mouse primers for PGC1α (PPARGC1A; fwd.: TCACCATATTCCAGGTCAAG and rev: TCATAGGCTTCATAGCTGTC; Sigma Aldrich; St. Louis, MO, USA) were used for real‐time quantitative PCR analysis. All mRNA values were normalized to the housekeeping gene, cyclophilin B (PPIB).
RNA-Seq
Using subsets of mRNA isolated per the method above (n = 3/group, WT SHM vs HT at various time points as indicated), the Stranded mRNA-Seq was performed using the Illumina NovaSeq 6000 Sequencing System at the University of Kansas Medical Center – Genomics Core (Kansas City, KS). Quality control on RNA submissions was completed using the Agilent TapeStation 4200 using the RNA ScreenTape Assay kit (5067–5576; Agilent Technologies; Santa Clara, CA, USA) with an Agilent RIN value threshold of ≥8.0. Total RNA (1 ug) was used to initiate the library preparation protocol. The total RNA fraction was processed by oligo dT bead capture of mRNA, fragmentation, reverse transcription into cDNA, end repair of cDNA, ligation with the appropriate Unique Dual Index (UDI) adaptors, strand selection, and library amplification by PCR using the Universal Plus mRNA-seq with NuQuant library preparation kit (0520-A01; Tecan Genomics; Männedorf, CH).
Library validation was performed using the D1000 ScreenTape Assay kit (5067–5582; Agilent Technologies; Santa Clara, CA, USA) on the Agilent TapeStation 4200. Concentration of each library was determined with the NuQuant module of the library prep kit using a Qubit 4 Fluorometer (Thermo Fisher Scientific; Waltham, MA, USA). Libraries were pooled based on equal molar amounts and the multiplexed pool was quantitated, in triplicate, using the Roche Lightcycler96 with FastStart Essential DNA Green Master (06402712001; Roche; Basel, CH) and KAPA Library Quant (Illumina; San Diego, CA, USA) DNA Standards 1–6 (KK4903; KAPA Biosystems; Wilmington, MA, USA). Using the qPCR results, the RNA-Seq library pool was adjusted to 2.125 nM for multiplexed sequencing.
Pooled libraries were denatured with 0.2 N NaOH (0.04 N final concentration) and neutralized with 400 mM Tris-HCl pH 8.0. A dilution of the pooled libraries to 425 pM is performed in the sample tube, on instrument, followed by onboard clonal clustering of the patterned flow cell using the NovaSeq 6000 S1 Reagent Kit (200 cycles; 20012864; Illumina; San Diego, CA, USA). A 2 × 101 cycle sequencing profile with dual index reads is completed using the following sequence profile: Read 1—101 cycles × Index Read 1—8 cycles × Index Read 2—8 cycles × Read 2–101 cycles . Following collection, sequence data is converted from .bcl file format to fastq file format using bcl2fastq software and demultiplexed into individual sequences for data distribution using a secure FTP site or Illumina BaseSpace for further downstream analysis.
Gene Expression Analysis
Following sequencing and demultiplexing, all reads were trimmed for adapters, filtered based on Phred quality score, and aligned to the mouse genome using the STAR aligner. Resulting .bam files were imported in Seqmonk for gene-level quantification. RNA-seq quality metrics including the proportion of reads aligning to genic regions were calculated. Differential expression analyses (pairwise comparisons) were performed using packages in base R and the limma-voom pipeline.40 Genes with a P < 0.05 and a fold change > 2 were considered significant (fold change was determined as change in HT group vs SHM control). All RNA-seq datasets are publicly available at https://doi.org/10.6084/m9.figshare.13360475. GSEA was performed on all genes deemed to have significant changes with HT using WebGestalt 2019.42 Using the Mus musculus database, gene ontology for nonredundant biological processes was determined with an FDR adjusted P < 0.05. All data are presented as normalized enrichment value for significant biological processes.
Citrate Synthase Assay
The Citrate synthase activity was used to infer mitochondrial content and was completed with hepatic whole cell lysate as previously described.44 Citrate synthase has recently been validated as having significant correlation with mitochondrial content.45
Statistics
The main effects of condition (C; SHM vs HT), time (T; 0, 0.5, 1, and 2 h), and genotype (G; WT vs L-HSP72KO) were examined using separate Two‐way ANOVAs with GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). Specifically, acute time course studies analyzed the variance within and between C and T, whereas studies looking at the varying genotypes analyzed the variance within and between C and G. Upon discovering significant main effects, post hoc analyses were performed using the Sidak method to test for any specific pairwise differences. Statistical significance was set at P < 0.05. Significant outliers for all data sets were removed using the ROUT method. Finally, all data are presented as the mean with data point range.
Results
Acute HT Drives Down Hepatic Mitochondrial Respiration and Alters Metabolism
We sought to establish an acute time course analysis (0–2 h) for in vivo HT on tissue-specific mitochondrial function. HT results in acutely reduced hepatic mitochondrial State 3S respiration and mitochondrial efficiency (increased State 3S H2O2:O2 ratio) within 0.5 h post-HT compared to SHM treatment in isolated mitochondria (Figure 1A–F). Importantly, this decreases in respiratory capacity occurred with both palmitoyl-CoA (PCoA) and pyruvate (PYR) as metabolic substrates. A similar pattern of O2 and H2O2 flux post-HT for each respiratory state can be found in Figure S3A–L. Hepatic mitochondrial respiration was restored 24 h post-HT (Figure 1G–J).
Figure 1.

Acute HT Drives Down Hepatic Mitochondrial Respiration and Alters Metabolism. Liver State3S mitochondrial respiratory capacity was determined in isolated mitochondrial fractions from WT mice subjected to SHM or HT at post-HT 0, 0.5, 1, and 2 h using the Oroboros O2k fluorometer with PCoA (A) and PYR (B) as the metabolic substrates. H2O2 emission and State3S H2O2:O2 ratios for PCoA (C and E) and PYR (D and F) were also measured. Liver mitochondrial respiratory capacity and H2O2 emission were also determined in isolated mitochondrial fractions from WT mice subjected to SHM or HT at 24 h post-HT for all respiratory states using both PCoA (G and I) and PYR (H and J). Maximal respiratory capacity and H2O2 emission values were normalized to the mitochondrial protein content added to each chamber as measured via the BCA assay. Liver mRNA was used for RNA-Seq at 0 h and 24 h post-HT to determine GO biological processes impacted (FDR, P < 0.05) by HT (K). Serum NEFA (L), lactate (M), β-hydroxybutyrate (N), and glucose (O) for WT mice subjected to SHM or HT at 0, 0.5, 1, and 2 h were determined using commercially available colorimetric assays. Liver TAGs (P) and hepatic PGC1α expression (Q) were also measured. Data are presented as mean with range (n = 4–6/group) or normalized enrichment value (n = 3/group). C, P < 0.05 main effect for condition (SHM vs HT); T, P < 0.05 main effect time (0 h vs 0.5, 1, or 2 h); C × T, P < 0.05 condition by time interaction; c, P < 0.05 within time condition effect; t, P < 0.05 within condition time effect (vs 0 h).
Transcriptional analyses revealed an inflammatory response immediately post-HT (0 h time point; GO:0007178: transmembrane receptor protein serine/threonine kinase signaling pathway; Figure 1K). Genes upregulated in hepatic tissue contributing to this event included Jun Proto-Oncogene, AP-1 Transcription Factor Subunit (JUN, score 3.15), Cellular Communication Network Factor 1 (CCN1, score 2.71), and Foes Proto-Oncogene, AP-1 Transcription Factor Subunit (FOS, score 1.63). Despite observing a restoration of mitochondrial respiratory capacity 24 h post-HT, GSEA analysis from this timepoint displayed marked reductions in a variety of metabolic processes, notably the lipid catabolic process (GO:0016042) and the fatty acid derivative metabolic process (GO:1901568).
The time course of HT effects on systemic metabolic outcomes has not been previously examined. In this study, we found that serum NEFAs (0–2 h), serum β-hydroxybutyrate (2 h), and liver triacylglycerides (TAGs, main effect only) were reduced with HT, while serum lactate was increased (0.5 h; Figure 1L–N and P). Importantly, blood glucose remained stable with HT (Figure 1O). Finally, we found that HT suppressed fasting-induced increases in the primary transcriptional coactivator for mitochondrial biogenesis Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha (PGC1α) necessary for gluconeogenesis46 within the 2 h following HT (Figure 1Q).
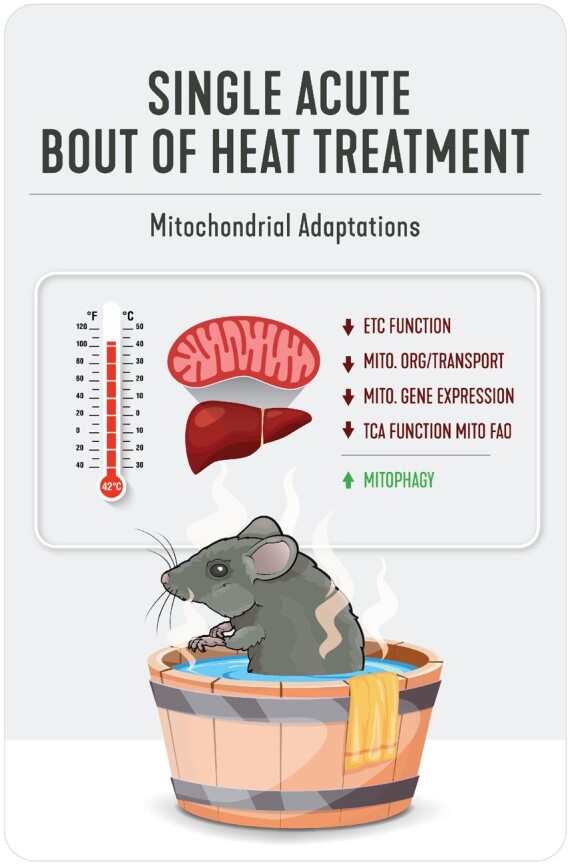
Acute HT significantly reduced respiratory chain Complexes I and II within liver mitochondrial isolates at 0 h and normalized thereafter, while Complexes III–V were unchanged by HT throughout the time course (Figure 2A–F). GSEA analysis on proteomic data from immediate post-HT (0 h) mitochondrial isolates suggests significant reductions in biological processes for mitochondrial organization (GO:0007005), mitochondrial gene expression (GO:0140053), NADH dehydrogenase complex assembly (GO:0010257), the tricarboxylic acid (TCA) metabolic process (GO:0072350), mitochondrial transport (GO:0006839) and protein localization (GO:0070585), and the pyruvate metabolic process (GO:0006090; Figure 2G). Conversely, we observed increases in biological processes such as vacuolar organization (GO:0007033) and vacuolar transport (GO: GO:0007034), as well as the proteasomal catabolic process (GO:0010498; Figure 2G). Taken together, our data suggest that acute HT reduces electron transport chain (ETC) function, ETC complex formation, TCA function, and mitochondrial organization (Figure 2H). Interestingly, we found that ETC complex content was not different in the whole-cell lysate (Figure S4A–F)—suggesting that ETCs are possibly transferred to lysosomes.
Figure 2.

Acute HT Reduces Mitochondrial Metabolic Complexes and Enzymes Immediately Post-HT. Protein content for ETC Complexes I (A), II (B), III (C), IV (D), and V (E) were measured using liver mitochondrial isolates from WT animals subjected to SHM or HT at post-HT 0, 0.5, 1, and 2 h via western blotting (F). All proteins quantified were normalized to total protein using amido black staining. Liver mitochondrial isolates were used for mitochondrial proteomics at 0 h post-HT to determine GO biological processes impacted (FDR, P < 0.05) by HT (G). Acute HT lowers hepatic mitochondrial function (H). Data are presented as mean with range (n = 4–6/group) or normalized enrichment value (n = 3/group). C, P < 0.05 main effect for condition (SHM vs HT); T, P < 0.05 main effect time (0 h vs 0.5, 1, or 2 h); C × T, P < 0.05 condition by time interaction; c, P < 0.05 within time condition effect; t, P < 0.05 within condition time effect (vs 0 h).
Acute HT Initiates Mitochondrial Fission, Ubiquitination, and Degradation via Mitophagy
We sought to determine if HT-induced reductions in hepatic mitochondrial proteins were due to the targeted degradation of mitochondria via mitochondrial fission/fusion and mitophagy. Acute HT decreased Mitofusin 2 (MFN2), a key fusion protein, while increasing the mitochondrial fission protein DRP1 (Figure 3A–D). OPA1 Mitochondrial Dynamin Like GTPase (OPA1) was unchanged by acute HT. Electron microscopy revealed a robust increase in the amount of hyper-fissed and fissing mitochondria from livers immediately post-HT (Figure 3E, not quantified). Hyper-fissed mitochondria putatively can be subsequently degraded via mitophagy as indicated in Figure 3F.
Figure 3.

Acute HT Initiates Mitochondrial Fission. Protein content for mitochondrial fusion and fission proteins MFN2 (A), OPA1 (B), and DRP1 (C) were measured using liver mitochondrial isolates from WT animals subjected to SHM or HT at post-HT 0, 0.5, 1, and 2 h via western blotting (D). All proteins quantified were normalized to total protein using amido black staining. Qualitative visual confirmation of mitochondrial fission and fusion was determined via electron microscopy for WT animals subjected to SHM or HT at 0 h post-HT (E). Images were taken at 3000× and 12 000×, red arrows indicate fissing mitochondria and blue arrows indicate hyper-fissed mitochondria. Acute HT initiates mitochondrial fission, which likely leads to mitophagy (F). Data are presented as mean with range (n = 4–6/group). C, P < 0.05 main effect for condition (SHM vs HT); T, P < 0.05 main effect time (0 h vs 0.5, 1, or 2 h); C × T, P < 0.05 condition by time interaction; c, P < 0.05 within time condition effect; t, P < 0.05 within condition time effect (vs 0 h).
Acute HT dramatically increased ubiquitination of the hepatic mitochondrial fraction (Figure 4A and I). Moreover, we found that autophagy machinery such as Sequestosome 1 (p62) and Microtubule-Associated Proteins 1A/1B Light Chain 3A II (LC3II) was reduced with acute HT in the mitochondrial fraction, suggesting an increase in mitophagy flux (Figure 4B, C, and I). HT did not impact protein content of other purported modifiers of mitophagy (BCL2 Interacting Protein 3 (BNIP3), HSP72, and E3 ubiquitin ligases Parkin and Carboxy Terminus of Hsp70-Interacting Protein (CHIP), other than HSP72 increasing at the 2 h time point (Figure 4D–H and I). Acute HT did increase the primary effector of CMA Heat Shock Cognate 70 (HSC70; Figure 4H and I). Using immunoprecipitation (pulling down with both ubiquitin and HSC70), we found evidence of increased ubiquitin and HSC70 binding to Complexes II and IV of the mitochondrial respiratory chain acutely following HT (Figure 4J). However, the magnitude of ubiquitination binding was far higher than HSC70 binding (Figure 4J). HSC70 was constitutively bound to CHIP and p62, whereas ubiquitin binding to these proteins appears to be heat-dependent (increased p62 binding and reduced CHIP binding; Figure 4J).
Figure 4.

Acute HT Causes Mitochondrial Ubiquitination and Degradation via Mitophagy. Protein content for known and putative mitophagy/CMA-associated proteins Ubiquitin (A), p62 (B), LC3II (C), BNIP3 (D), HSP72 (E), Parkin (F), CHIP (G), and HSC70 (H) were measured using liver mitochondrial isolates from WT animals subjected to SHM or HT at post-HT 0, 0.5, 1, and 2 h via western blotting (I). All proteins quantified were normalized to total protein using amido black staining. Follow up immunoprecipitation experiments pulling down with Ubiquitin and HSC70 in whole liver lysates from the 0h post-SHM or -HT timepoints were used to determine complex-specific ETC protein binding (CI-V), CHIP, and p62 (J). Qualitative visual confirmation of mitophagy was obtained via confocal imaging of livers from WT animals exposed to the Ad-Cox8-GFP-mCherry fluorescent mitophagy reporter at 0h post-SHM or -HT timepoints (K). HSC70 and E3 ligase partners Parkin and CHIP putatively enable mitochondrial ubiquitination and subsequent recruitment to the auto phagolysosome via p62/LC3II (L). Data are presented as mean with range (n = 4–6/group) and representative images (immunoprecipitation and confocal imaging n = 3/group). C, P < 0.05 main effect for condition (SHM vs HT); T, P < 0.05 main effect for time (0h vs 0.5, 1, or 2 h); C × T, P < 0.05 condition by time interaction; c, P < 0.05 within time condition effect; t, P < 0.05 within condition time effect (vs 0 h).
To confirm that mitophagy flux was directly increased by HT, we employed an Ad fluorescent reporter for Cox8, whose GFP signal gets quenched with entry into the lysosome, combined with in vivo dosing of a hepatic autophagy inhibitor (LEU). As shown in Figure 4K, we confirmed that acute HT (0 h) increased hepatic mitophagy flux (color change from green to yellow and red). Moreover, we confirmed that mitophagy flux was increased 24 h post HT using LEU (Figure S5A–H). Thus, acute HT initiates hepatic mitophagy that appears to be linked to mitochondrial ubiquitination.
HSP72 Is Not Required for HT-induced Mitochondrial Ubiquitination, Mitophagy, and Respiratory Suppression
Our laboratory has hypothesized that HSP72 is the primary driver of the HT-induced improvements in hepatic mitochondrial function and lipid metabolism.11 To test this question, we developed a conditional liver-specific HSP72 knockout mouse (L-HSP72KO). In L-HSP72KO and WT mice fed a LFD, we found that mice lacking hepatic HSP72 also displayed reduced mitochondrial respiratory capacity and increased State 3S H2O2:O2 ratios following HT (with both PCoA and PYR substrates; Figure 5A–F). Similar to the WT animals, acute HT resulted in mitochondrial ubiquitination and mitophagy induction in the L-HSP72KO mice (Figure 5M–P). These trends in respiratory function and mitophagy induction remained largely similar in animals subjected to an acute bout of HT following 4 weeks of high-fat feeding (Figure 5G–T). However, L-HSP72KO fed a HFD and subjected to HT had reduced uncoupled respiratory capacity and increased H2O2 emission for the PYR condition compared to WT (Figure 5H, J, and L). Overall, these data suggest that loss of hepatic HSP72 mildly alters mitochondrial function but is not involved in the acute mitochondrial response to HT.
Figure 5.

HSP72 Is Not Required for HT-Induced Mitochondrial Ubiquitination, Mitophagy, and Respiratory Suppression. Liver mitochondrial respiratory capacity for all energetic states was determined in isolated mitochondrial fractions from L-HSP72KO and WT littermates subjected to acute SHM or HT on an LFD using the Oroboros O2k fluorometer with PCoA (A) and PYR (B) as the metabolic substrates 0 h post-HT. H2O2 emission for all states and State3S H2O2:O2 ratios for PCoA (C and E) and PYR (D and F) were also measured 0 h post-HT. The same functional mitochondrial measures for PCoA (G, I, and K) and PYR (H, J, and L) were completed in isolated mitochondrial fractions from L-HSP72KO and WT littermates subjected to acute SHM or HT on an HFD 1 h post-HT. Maximal respiratory capacity and H2O2 emission values were normalized to the mitochondrial protein content added to each chamber as measured via the BCA assay. Protein content for known and putative mitophagy-associated proteins Ubiquitin, HSP72, and LC3II was measured using liver mitochondrial isolates from L-HSP72KO and WT littermates subjected to acute SHM or HT on an LFD (M–P) or an HFD (Q–T) via western blotting (I). All proteins quantified were normalized to total protein using amido black staining. Data are presented as mean with range (n = 4–6/group). C, P < 0.05 main effect for condition (SHM vs HT); G, P < 0.05 main effect for genotype (L-HSP72KO vs WT littermate); C × G, P < 0.05 condition by genotype interaction; c, P < 0.05 within time condition effect; g, P < 0.05 within condition genotype effect (vs WT).
Similar to our initial time course data using an LFD, we observed that acute HT reduced serum NEFAs and β-hydroxybutyrate in both WT and L-HSP72KO mice on the HFD (Figure S6E and F). However, serum lactate and liver TAGs were unchanged in both WT and L-HSP72KO mice with HT despite being on the HFD (Figure S6C and G). Interestingly, we observed that serum glucose was reduced in the L-HSP72KO compared to WT in both SHM and HT conditions (Figure S6H). Of note, body mass was reduced by acute HT, but weekly energy intake was similar across all groups (Figure S6A and B).
Chronic HT Improves Hepatic Mitochondrial Respiratory Function and Mitochondrial Quality Independent of HSP72
As previous results from our laboratory suggest that chronic HT ameliorates systemic weight gain and insulin resistance,14 we wanted to determine if these HT-induced improvements in glucose metabolism and liver fat storage tracked with enhanced hepatic mitochondrial function and if HSP72 was required for these adaptations. Chronic HT increased hepatic mitochondrial respiratory capacity, while reducing H2O2 emission, and thus State 3S H2O2:O2 ratios, for both PCoA and PYR conditions in L-HSP72KO and WT mice (Figure 6A–F). While mitochondrial content was generally higher in the L-HSP72KO mice (Figure 7H), chronic HT did not impact ETC protein expression (Figure 7A–G) and reduced mitochondrial content (inferred by the citrate synthase assay) in both L-HSP72KO and WT mice. It is possible that L-HSP72KO models have compensatory increases in mitochondrial content or reduced basal rates of mitophagy, however, more studies are needed to elucidate this difference.
Figure 6.

Chronic HT Improves Hepatic Mitochondrial Respiratory Function Independent of HSP72. Liver mitochondrial respiratory capacity for all energetic states was determined in isolated mitochondrial fractions from L-HSP72KO and WT littermates subjected to SHM or HT every 72 h for 4 weeks on an HFD using the Oroboros O2k fluorometer with PCoA (A) and PYR (B) as the metabolic substrates 72 h after their last SHM or HT. H2O2 emission for all states and State3S H2O2:O2 ratios for PCoA (C and E) and PYR (D and F) were also measured 72 h after their last SHM or HT. Maximal respiratory capacity and H2O2 emission values were normalized to the mitochondrial protein content added to each chamber as measured via the BCA assay. Data are presented as mean with range (n = 4–7/group). C, P < 0.05 main effect for condition (SHM vs HT); G, P < 0.05 main effect for genotype (L-HSP72KO vs WT littermate); C × G, P < 0.05 condition by genotype interaction; c, P < 0.05 within time condition effect; g, P < 0.05 within condition genotype effect (vs WT).
Figure 7.

Chronic HT Improves Mitochondrial Quality and Efficiency Independent of HSP72. Protein content for hepatic HSP72 (A) and ETC proteins (B–F) was determined in mitochondrial isolates via western blotting (G) from L-HSP72KO and WT littermates subjected to SHM or HT every 72 h for 4 weeks on an HFD. All proteins quantified were normalized to total protein using amido black staining. Hepatic mitochondrial content was inferred by measuring hepatic citrate synthase activity of whole-cell liver lysate (H). RNA-Seq (I) and proteomics (J) were determined using total hepatic mRNA and isolated mitochondrial samples, respectively, from WT littermates only (n = 3/group). GO biological processes impacted (FDR, P < 0.05) by HT for both RNA-Seq and proteomics are reported. Chronic HT improves mitochondrial function via increased mitochondrial quality control (Mito QC; K). The acute phase following HT suppresses mitochondrial function and increases mitophagy, while recurrent activation on these pathways chronically causes compensatory adaptations to improve mitochondrial function and quality—all of which are independent of HSP72 (L). Data are presented as mean with range (n = 4–7/group) or normalized enrichment value (n = 3/group, WT only). C, P < 0.05 main effect for condition (SHM vs HT); G, P < 0.05 main effect for genotype (L-HSP72KO vs WT littermate); C × G, P < 0.05 condition by genotype interaction; c, P < 0.05 within time condition effect; g, P < 0.05 within condition genotype effect (vs WT).
To determine the mechanism by which chronic HT improved hepatic mitochondrial respiration, we conducted RNA-Seq and proteomic analysis on whole liver samples and mitochondrial isolates. Chronic HT reduced the GO terms associated with fatty acid metabolism and gluconeogenesis (Figure 7I–J). In contrast, HT increased GO categories pertaining to metal ion homeostasis (RNA-Seq analysis) and increased mitochondrial organization (GO:0007005), mitochondrial gene expression (GO:0140053), mitochondrial transport (GO:0006839) and protein localization (GO:0070585), and protein folding (GO: 0006457; proteomic analysis; Figure 7I–J). Combined, these data suggest that HT causes adaptive changes in mitochondrial function via increased mitochondrial quality control rather than content.
Importantly, we observed these distinct HT-induced mitochondrial changes in association with reduced serum glucose in the WT but not the L-HSP72KO animals—suggesting that hepatic HSP72 is required for HT-induced reductions in serum glucose (Figure S7D). SHM and HT animals in both genotypes lost weight over the 4-week intervention, however, L-HSP72KO animals tended to lose less which may have contributed to their lack of change in blood glucose in response to HT (Figure S7A). Additionally, all HT animals tended to take in less energy—(Figure S7A and B). We saw no significant changes in circulating NEFAs or liver TAGs, but TAGs did display a similar trend to serum glucose albeit insignificant (Figure S7C, D, and F).
In summary, our results suggest that acute in vivo HT induces a hepatic mitochondrial heat shock response—acutely reducing mitochondrial respiratory capacity paired with induction of hepatic mitophagy. However, chronic bouts of HT result in a compensatory adaptation that improves mitochondrial respiratory function and mitochondrial quality via enhanced mitochondrial organization, transport, protein localization, and gene expression (GO terms determined via mitochondrial proteomics; Figure 7J). Importantly, neither the acute phase nor the chronic phase of mitochondrial adaptations to heat is dependent on hepatic HSP72 (Figure 7L). However, it does appear that hepatic HSP72 is required for HT-induced reductions in serum glucose. Thus, we believe that these time-dependent adaptations in mitochondrial function and quality control, as well as chronic induction of hepatic HSP72, contribute to the positive metabolic benefits of HT.
Discussion
Our laboratory and others have shown that repeated bouts of HT mitigate glucose intolerance, insulin resistance, and hepatic steatosis in preclinical rodent models of diet-induced obesity.11,12,14,15 Moreover, HT also improves glucose homeostasis in obese, insulin-resistant women.16 While HT is shown to increase muscle metabolism in humans,17 the direct bioenergetic mechanism(s) surrounding the positive metabolic effects of HT remain largely unknown, as well as whether similar findings occur in the liver. Here, we show that HT causes an acute mitochondrial heat shock response in the liver paired with a dramatic induction of mitophagy and short-term reductions in mitochondrial function. Importantly, we are the first to show that recurrent activation of these mitochondrial control pathways ultimately leads to enhanced mitochondrial quality and respiratory function when challenged with high-fat feeding. Finally, utilizing a novel liver-specific HSP72 KO model, we are the first to show that HSP72 does not play a significant role in the mitochondrial adaptation to HT but is required for HT-induced reductions in serum glucose.
One primary factor contributing to the development of hepatic steatosis is impaired hepatic mitochondrial function.5 Central to the findings in this study, others have determined that impairments to the degradation of damaged mitochondria via mitophagy increase the susceptibility to steatosis and impaired hepatic metabolism.6,7 Thus, strategies that restore hepatic mitophagy/mitochondrial quality control, such as exercise or fasting, are of increasing interest in the prevention of hepatic steatosis.8,9,47 Building on seminal work of others, we are the first to show that HT is an alternative strategy to exercise to restore hepatic mitophagy and quality control during high-fat feeding. This finding is particularly relevant for individuals in which exercise or fasting is contraindicated.
First established three decades ago, Gropper et al. found that subjecting murine mammary cells to heat stress causes increased proteolysis of long-lived proteins via ubiquitin-mediated pathways.48 Later it was determined that heat stress causes nuclear mutations via mitochondrially-derived reactive oxygen species (ROS) in Saccharomyces cerevisiae.49 These seminal studies lead to the discovery that HT induces the targeted degradation of damaged proteins (via autophagy; in HeLa cells) and ROS producing mitochondria (via mitophagy; in HeLa cells and rat liver) to prevent cellular apoptosis via the activation of NFκB and the reduction of cytoplasmic cytochrome c release, respectively.25–27 However, until now the metabolic bioenergetic consequences of acute and chronic, nonlethal HT remained unknown.
In agreement with the previous observations,50 we found that acute HT caused an acute inflammatory event (increased JUN and FOS expression) and increased mitochondrial ROS production (inferred by State 3S H2O2 expression). Moreover, we replicated previous findings in a variety of cell types and model organisms showing HT induces mitophagy to the liver in vivo and found that this process was associated with mass mitochondrial fission and ubiquitination. While, it has recently been shown that both outer- and inner-mitochondrial proteins can be ubiquitinated,51 the functional respiratory changes associated with these processes remained ill-defined. Here we show that HT indeed causes the ubiquitination of inner mitochondrial proteins (ie, Complex I) and subsequent degradation of key mitochondrial ETC proteins, enzymes involved in the TCA cycle, mitochondrial transport/organization proteins, and enzymes associated with mitochondrial gene expression in our mitochondrial isolates. These alterations resulted in dramatically reduced respiratory capacity in isolated hepatic mitochondria immediately post-HT. Interestingly, we found that respiratory capacity was largely restored to SHM levels within ∼1 h of HT despite ubiquitination remaining elevated—suggesting that ubiquitination of mitochondrial enzymes itself does not directly impact their function. After 24 h of recovery from HT, we no longer observed any differences in respiratory capacity, however, the expression of genes associated with lipid catabolism and fatty acid handling continued to remain reduced in HT vs SHM. It is possible that we missed measuring gene expression in a key timepoint and thus missed the peak transcription period post-HT in which key enzymes for FAO were replaced. It is also possible other metabolic enzymes compensated, or our ex vivo experimental procedures in isolated mitochondria failed to capture the loss of certain metabolic enzymes. While this acute bioenergetic stress appeared to be negative short term, recurrent activation of this response with chronic HT over a 4-week period resulted in net positive mitochondrial adaptations.
In this study, we found that chronic activation of the mitochondrial heat shock response during high-fat feeding caused compensatory increases in hepatic proteins/genes associated with mitochondrial catalytic efficiency (metal ion homeostasis), organization, gene expression, transport, protein folding, and localization. These compensatory hepatic adaptations were associated with increased mitochondrial respiratory function and reduced mitochondrial ROS production. Importantly, these adaptations and functional changes occurred as a result of increased mitochondrial efficiency rather than elevated hepatic mitochondrial content as inferred by our citrate synthase data. Additionally, we are the first to show that both the acute and chronic mitochondrial effects of HT are not dependent on hepatic HSP72.
We and others have often attributed the positive metabolic effects of HT to HSP72.18 This idea originally stemmed from the fact that HSP72 is reduced in both liver and muscle tissue during obese conditions,15,19 whereas HT restores HSP72 levels in these tissues and was found to be associated with the metabolic benefits of HT.12,14,15,20 Moreover, increased HSP72 content via transgenic overexpression or pharmacologic activation in animals fed an HFD is shown to benefit whole-body metabolic measures and skeletal muscle mitochondrial content.15,21–24 However, as the gene encoding for HSP72 (HSPA1A) does not intrinsically contain introns, tissue-specific deletions of HSP72 in vivo was not possible to confirm its function.
Using the first available liver-specific HSP72 KO mouse model, we show that the acute and chronic effects of HT on hepatic mitochondria are not dependent on HSP72. Importantly, this may not be the case for all tissues. For instance, others have shown that HSP72 is involved in mitophagy in skeletal muscle.52 Thus, additional experiments using this novel model are needed to determine whether HSP72 is needed for mitochondrial adaptations in other tissues. However, we did find an association between another member of the HSP70 family, HSC70, and the acute activation of mitochondrial ubiquitination and mitophagy. HSC70 putatively coordinates a process known as CMA, which was recently shown to be activated by bioenergetic stress and impact hepatic metabolism.28,53–55 Moreover, the conical pentapeptide HSC70 binding motif (KFERQ) and KFERQ-like motifs are overrepresented in mitochondrial proteins associated with oxidative phosphorylation and fatty acid metabolism, suggesting they can be degraded via CMA.56 CMA is also putatively regulated by Carboxy Terminus of Hsp70-Interacting Protein (CHIP), an E3 ubiquitin ligase required for ubiquitination of HSC70-bound substrate proteins.57,58 We found that HSC70 is constitutively bound to CHIP and p62 (a protein required for autophagosome recruitment) and that acute HT increases the amount of HSC70 and p62 bound to both ubiquitin and ETC Complexes II and IV. These outcomes strongly suggest that HT initiates CMA of mitochondrial components, work that needs further delineation in the role of HT and mitochondrial quality control.
Another primary question concerns the ability of HT to contribute to mitochondrial remodeling through global metabolic changes. We found that acute HT caused a reduction in circulating NEFAs, β-hydroxybutyrate, and liver TAGs suggesting that HT induces increased mobilization and utilization of varying energy stores. Activation of beta-adrenergic receptors via stimuli like exercise causes increased serum NEFA concentration via enhanced lipolysis,59,60 and HT may increase parasympathetic tone. This makes sense when looking at recent data suggesting that HT causes increased flow-mediated dilation and reduces blood pressure.61 However, more research is needed to determine how substrate utilization and whole-body energy expenditure are impacted by HT and if changes in substrate flux and utilization play a critical role in transcriptional or epigenetic modifications.
We did see that chronic HT lowered circulating glucose and that this was dependent on hepatic HSP72 (Figure S7A)—a finding that supports previous data using HSP72 overexpression and HT.14,15 However, we saw no change in circulating NEFAs or liver TAGs, although liver TAGs displayed a similar trend to serum glucose (Figure S7A–F). We have previously reported that weekly HT in rats fed an HFD lowered TAGs,11 however, all animals (SHM and HT) lost weight during the chronic study (with the exception of 3 L-HSP72KO animals). It is possible that recurrent use of a rectal probe and/or anesthesia reduced food intake overall, causing the weight loss in all groups—something that should be considered for future studies. Additionally, we found that HT animals displayed reduced food intake when compared to SHM controls, a finding that has been previously attributed to increased leptin secretion/sensitivity with HT62 (Figure S7B). Thus, it is possible that we were unable to tease out gross metabolic differences as easily as previous studies.
Despite these methodological limitations, our data suggest that the mitochondrial adaptations mentioned above were not severely impacted by recurrent anesthesia or reductions in food intake in our model. However, due to the lack of additional global metabolic changes typically associated with HT and high-fat feeding (ie, reductions in liver TAGs, blood insulin, blood NEFAs, etc.), the direct contributions of hepatic HSP72 and mitochondrial adaptations to HT in metabolic health remain unclear. Despite this, our novel bioenergetic data combined with previous literature suggests that chronic HT may still be a viable strategy to reduce the burden of NAFLD and insulin resistance.
Conclusion
This study builds on previous work suggesting that HT causes an acute mitochondrial heat shock response that induces mitophagy. However, we are the first to show that recurrent activation of this mitochondrial heat shock response leads to enhanced hepatic mitochondrial quality, mitochondrial remodeling, and respiratory function during high-fat feeding. Finally, utilizing a novel liver-specific HSP72 KO model, we are the first to show that hepatic HSP72 does not play a significant role in the mitochondrial adaptation to HT but is required for HT-induced reductions in blood glucose. Thus, we believe both hepatic HSP72 and the independent mitochondrial adaptations to HT contribute to the metabolic benefits of heat. Future studies should aim to determine how HT initiates mitochondrial ubiquitination and degradation (ie, CMA). Insights into the acute activation of these mitochondrial control pathways could lead to the development of novel therapeutic targets to improve mitochondrial quality control and prevent or treat metabolic diseases such as NAFLD and insulin resistance.
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. Samuel G. Mackintosh and Dr. Stephanie Byrum at The University of Arkansas Medical Campus Proteomics Core (NIH R24GM137786 and P20GM121293) for their contribution to mitochondrial proteomics. We would also like to thank Dr. Randall Mynatt of Pennington Biomedical Research Center (COBRE [NIH 3P30-GM118430] and NIH-NORC [NIH 2P30-DK072476]), Dr. Jingying Zhang, and Dr. Jay Vivian for their help with generating the liver-specific HSP72 KO mouse. Additional thanks go to The University of Kansas Medical Center Genomics Core for their core support services for our RNA-Seq data acquisition.
Supplementary Material
Supplementary material is available at the APS Function online.
Funding
Funding for the Genomics core comes from Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation—COBRE (P30 GM122731-03), the NIH S10 High-End Instrumentation Grant (NIH S10OD021743), and the Frontiers CTSA grant (UL1TR002366) at the University of Kansas Medical Center, Kansas City, KS 66160. This work was also supported by a VA-Merit Grant 1I01BX002567 (J.P.T.), NIH R01 DK121497 (J.P.T.), and COBRE is P30 GM122731 (P.C.G.). This project was also supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103418. Finally, we would like to acknowledge the core staff and funding from the Madison and Lila Self Graduate Fellowship at The University of Kansas for salary support (A.T.V.S). All figures were created using BioRender.com and we would like to thank Gaige Larson for creating the graphical abstract.
Authors’ Contributions
A.T.V.S. contributed to the experimental conceptualization, data curation, formal data analysis, experimental investigation, experimental methodology, project administration, experimental validation, data visualization, and manuscript writing. F.D. contributed to experimental conceptualization, data curation, experimental investigation, experimental methodology, and manuscript revision. K.N.Z.F. contributed to data curation, experimental investigation, experimental methodology, and manuscript revision. E.F. contributed to data curation, experimental investigation, and manuscript revision. J.M. contributed to data curation, experimental investigation, and manuscript revision. J.A. contributed to data curation, experimental investigation, and manuscript revision. C.S.M. contributed to experimental conceptualization, experimental methodology, and manuscript revision. K.S. contributed to data curation, experimental methodology, formal data analysis, software implementation, and manuscript revision. W.-X.D. contributed to experimental conceptualization, experimental methodology, and manuscript revision. J.P.T. contributed to experimental conceptualization, funding acquisition, experimental methodology, supervision, and manuscript revision. P.C.G. contributed to experimental conceptualization, funding acquisition, experimental methodology, supervision, and manuscript revision.
Conflict of Interest Statement
The authors declare no competing interests.
References
- 1. Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology (Baltimore, MD) 2004;40(6):1387–1395. [DOI] [PubMed] [Google Scholar]
- 2. Smits MM, Ioannou GN, Boyko EJ, Utzschneider KM. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: results of a US national survey in three ethnic groups. Research Support, U.S. Gov't, Non-P.H.S. J Gastroenterol Hepatol 2013;28(4):664–670. [DOI] [PubMed] [Google Scholar]
- 3. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114(2):147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10(6):330. [DOI] [PubMed] [Google Scholar]
- 5. Dornas W, Schuppan D. Mitochondrial oxidative injury: a key player in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol 2020;319(3):G400–G411. [DOI] [PubMed] [Google Scholar]
- 6. Glick D, Zhang W, Beaton M, et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 2012;32(13):2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edmunds LR, Xie B, Mills AM, et al. Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Mol Metab 2020:41:101051. doi:10.1016/j.molmet.2020.101051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Von Schulze A, McCoin CS, Onyekere C, et al. Hepatic mitochondrial adaptations to physical activity: impact of sexual dimorphism, PGC1alpha and BNIP3-mediated mitophagy. J Physiol 2018;596(24):6157–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCoin CS, Von Schulze A, Allen J, et al. Sex modulates hepatic mitochondrial adaptations to high-fat diet and physical activity. Am J Physiol Endocrinol Metab 2019;317(2):E298–E311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elsawy B, Higgins KE. Physical activity guidelines for older adults. Am Fam Physician 2010;81(1):55–59. [PubMed] [Google Scholar]
- 11. Archer AE, Rogers RS, Von Schulze AT, et al. Heat shock protein 72 regulates hepatic lipid accumulation. Am J Physiol Regul Integr Comp Physiol 2018;315(4):R696–R707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rogers RS, Morris EM, Wheatley JL, et al. Deficiency in the heat stress response could underlie susceptibility to metabolic disease. Diabetes 2016;65(11):3341–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hooper PL. Hot-tub therapy for type 2 diabetes mellitus. New Engl J Med 1999;341(12):924–925. [DOI] [PubMed] [Google Scholar]
- 14. Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes 2009;58(3):567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chung J, Nguyen A-K, Henstridge DC, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci USA 2008;105(5):1739–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ely BR, Clayton ZS, McCurdy CE, et al. Heat therapy improves glucose tolerance and adipose tissue insulin signaling in polycystic ovary syndrome. Am J Physiol Endocrinol Metab 2019;317(1):E172–E182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hafen PS, Preece CN, Sorensen JR, Hancock CR, Hyldahl RD. Repeated exposure to heat stress induces mitochondrial adaptation in human skeletal muscle. J Appl Physiol (1985) 2018;125(5):1447–1455. [DOI] [PubMed] [Google Scholar]
- 18. Geiger PC, Gupte AA. Heat shock proteins are important mediators of skeletal muscle insulin sensitivity. Exerc Sport Sci Rev 2011;39(1):34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Naso FC, Porto RR, Fillmann HS, et al. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity (Silver Spring) 2015;23(1):120–129. [DOI] [PubMed] [Google Scholar]
- 20. Rogers RS, Beaudoin MS, Wheatley JL, Wright DC, Geiger PC. Heat shock proteins: in vivo heat treatments reveal adipose tissue depot-specific effects. J Appl Physiol (1985) 2015;118(1):98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupte AA, Bomhoff GL, Morris JK, Gorres BK, Geiger PC. Lipoic acid increases heat shock protein expression and inhibits stress kinase activation to improve insulin signaling in skeletal muscle from high-fat-fed rats. J Appl Physiol 2009:1425–1434. [DOI] [PubMed]
- 22. Henstridge DC, Bruce CR, Drew BG, et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014;63(6):1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kavanagh K, Flynn DM, Jenkins KA, Zhang L, Wagner JD. Restoring HSP70 deficiencies improves glucose tolerance in diabetic monkeys. Am J Physiol Endocrinol Metab 2011;300(5):E894–E901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zeng XY, Wang H, Bai F, et al. Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Research Support, Non-U.S. Gov't. Br J Pharmacol 2015;172(17):4303–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Y, Xing D, Zhou F, Chen Q. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem Biophys Res Commun 2010;395(2):190–195. [DOI] [PubMed] [Google Scholar]
- 26. Nivon M, Richet E, Codogno P, Arrigo A-P, Kretz-Remy C. Autophagy activation by NFκB is essential for cell survival after heat shock. Autophagy 2009;5(6):766–783. [DOI] [PubMed] [Google Scholar]
- 27. Oberley TD, Swanlund JM, Zhang HJ, Kregel KC. Aging results in increased autophagy of mitochondria and protein nitration in rat hepatocytes following heat stress. J Histochem Cytochem 2008;56(6):615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014;20(3):417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ilievska G, Dinevska-Kjovkarovska S, Miova B. Effect of single and repeated heat stress on chemical signals of heat shock response cascade in the rat's heart. Cell Stress Chaperones 2018;23(4):561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morris EM, Meers GM, Booth FW, et al. PGC-1alpha overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am J Physiol Gastrointest Liver Physiol 2012;303(8):G979–G992. doi:10.1152/ajpgi.00169.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krumschnabel G, Fontana-Ayoub M, Sumbalova Z, et al. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Mitochondrial Med 2015;1264:245–261. [DOI] [PubMed] [Google Scholar]
- 32. Rector RS, Thyfault JP, Morris RT, et al. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol 2008;294(3):G619–G626. [DOI] [PubMed] [Google Scholar]
- 33. Wang H, Ni H-M, Chao X, et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 2019;22:101148–101148. doi:10.1016/j.redox.2019.101148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haspel J, Shaik RS, Ifedigbo E, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011;7(6):629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 2003;75(17):4646–4658. doi:10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- 37. Graw S, Tang J, Zafar MK, et al. proteiNorm – a user-friendly tool for normalization and analysis of TMT and label-free protein quantification. ACS Omega 2020. doi: 10.1021/acsomega.0c02564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002;18(suppl_1):S96–S104. [DOI] [PubMed] [Google Scholar]
- 39. Bolstad B. preprocessCore: A collection of pre-processing functions 1.46. 0, R. package version. 2019. DOI: 10.18129/B9.bioc.preprocessCore.
- 40. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chawade A, Alexandersson E, Levander F. Normalyzer: a tool for rapid evaluation of normalization methods for omics data sets. J Proteome Res 2014;13(6):3114–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 2019;47(W1):W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morris EM, Jackman MR, Meers GM, et al. Reduced hepatic mitochondrial respiration following acute high-fat diet is prevented by PGC-1alpha overexpression. Am J Physiol Gastrointest Liver Physiol 2013;305(11):G868–G880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srere P. [1] Citrate synthase:[EC 4.1. 3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. Methods in Enzymology. Elsevier; 1969;13:3–11. [Google Scholar]
- 45. McLaughlin KL, Hagen JT, Coalson HS, et al. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci Rep 2020;10(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoon JC, Puigserver P, Chen G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001;413(6852):131–138. doi:10.1038/35093050 [DOI] [PubMed] [Google Scholar]
- 47. Kristensen CM, Olsen MA, Jessen H, Brandt N, Meldgaard JN, Pilegaard H. PGC-1α in exercise and fasting-induced regulation of hepatic UPR in mice. Pflugers Arch 2018;470(10):1431–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gropper R, Brandt R, Elias S, et al. The ubiquitin-activating enzyme, E1, is required for stress-induced lysosomal degradation of cellular proteins. J Biol Chem 1991;266(6):3602–3610. [PubMed] [Google Scholar]
- 49. Davidson JF, Schiestl RH. Mitochondrial respiratory electron carriers are involved in oxidative stress during heat stress in Saccharomyces cerevisiae. Mol Cell Biol 2001;21(24):8483–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gabai VL, Meriin AB, Mosser DD, et al. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem 1997;272(29):18033–18037. [DOI] [PubMed] [Google Scholar]
- 51. Sulkshane P, Duek I, Ram J, et al. Inhibition of proteasome reveals basal mitochondrial ubiquitination. J Proteom 2020;229:103949. doi: 10.1016/j.jprot.2020.103949. [DOI] [PubMed] [Google Scholar]
- 52. Drew BG, Ribas V, Le JA, et al. HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes 2014;63(5):1488–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cuervo AM, Palmer A, Rivett AJ, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem 1995;227(3):792–800. [DOI] [PubMed] [Google Scholar]
- 54. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018;19(6):365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodriguez-Navarro JA, Kaushik S, Koga H, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA 2012;109(12):E705–E714. doi:10.1073/pnas.1113036109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kirchner P, Bourdenx M, Madrigal-Matute J, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol 2019;17(5):e3000301. doi:10.1371/journal.pbio.3000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lizama BN, Palubinsky AM, Raveendran VA, et al. Neuronal preconditioning requires the mitophagic activity of C-terminus of HSC70-interacting protein. J Neurosci 2018;38(31):6825–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein. Autophagy 2013;9(12):1937–1954. [DOI] [PubMed] [Google Scholar]
- 59. Gagliardino J, Bellone C, Doria I, Sanchez J, Pereyra V. Adrenergic regulation of basal serum glucose, NEFA and insulin levels. Horm Metab Res 1970;2(06):318–322. [DOI] [PubMed] [Google Scholar]
- 60. Basu A, Passmore R, Strong J. The effect of exercise on the level of non‐esterified fatty acids in the blood. Q J Exp Physiol Cogn Med Sci 1960;45(3):312–317. [DOI] [PubMed] [Google Scholar]
- 61. Brunt VE, Howard MJ, Francisco MA, Ely BR, Minson CT. Passive heat therapy improves endothelial function, arterial stiffness and blood pressure in sedentary humans. J Physiol 2016;594(18):5329–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morera P, Basirico L, Hosoda K, Bernabucci U. Chronic heat stress up-regulates leptin and adiponectin secretion and expression and improves leptin, adiponectin and insulin sensitivity in mice. J Mol Endocrinol 2012;48(2):129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.