

Abstract
Purpose
Most patients with pancreatic ductal adenocarcinoma (PDAC) present with surgically unresectable cancer. As a result, endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) is the most common biospecimen source available for diagnosis in treatment-naïve patients. Unfortunately, these limited samples are often not considered adequate for genomic analysis, precluding the opportunity for enrollment on precision medicine trials.
Experimental Design
Applying an Epithelial cell adhesion molecule (EpCAM)-enrichment strategy, we show the feasibility of using real-world EUS-FNAs for in depth, molecular-barcoded, whole-exome sequencing (WES) and somatic copy number alteration (SCNA) analysis in 23 PDAC patients.
Results
Potentially actionable mutations were identified in >20% of patients. Further, an increased mutational burden and higher aneuploidy in WES data were associated with an adverse prognosis. To identify predictive biomarkers for first line chemotherapy, we developed an SCNA based complexity score (CS) that was associated with response to platinum-based regimens in this cohort.
Conclusions
Collectively, these results emphasize the feasibility of real-world cytology samples for in depth genomic characterization of PDAC and show the prognostic potential of SCNA for PDAC diagnosis.
Keywords: fine needle aspiration, core biopsy, whole exome sequencing, molecular barcoding, pancreatic ductal adenocarcinoma
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a disease with a dire prognosis and one of the few cancers with a rising incidence, leading to estimates of it becoming the second leading cause of cancer related death in the United States within the next decade (1). A series of studies on large-scale molecular characterization of PDAC have elucidated the comprehensive genomic landscape of this neoplasm, including the so-called “long tail” of potentially actionable mutations (many at as low prevalence rates as ~1%), which may form the basis for precision oncology and clinical trial inclusion (2)(3)(4)(5)(6). Most of these prior “discovery” studies have used archival surgically resected specimens with optimal tumor cellularity (e.g. >60% for the TCGA cohort), limiting much of our current knowledge to localized, and therefore resectable tumors (3)(4). Nonetheless, the majority of PDAC patients (~80–85%) are diagnosed with locally advanced or metastatic disease, precluding surgical options, which, in turn, restricts many patients from the opportunity for in-depth genomic analyses from resected specimens (1)(7).
All PDAC patients, irrespective of disease stage, are required to have histological or cytological confirmation of their underlying diagnosis prior to onset of therapy (7). The two most common avenues for obtaining diagnostic biospecimens in advanced disease include either endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) of the primary tumor, performed by a gastroenterologist, or a percutaneous core biopsy of a biopsy-amenable metastatic lesion, when possible, the latter performed by an interventional radiologist. Overall, with the gastroenterology clinic being the typical “portal of entry” for symptomatic individuals, an EUS-FNA represents one of the most common, if not the most common, sample type obtained in PDAC patients at the time of de novo presentation. Although both sources of biopsies are comprised of minimal tissue material, the challenges of using EUS-FNA from PDAC for next generation sequencing (NGS) studies are unique. In contrast to the generally high cellularity of metastatic samples obtained by core biopsy, EUS-FNA samples of desmoplastic primary PDAC tend to be hypocellular, and potentially contaminated with non-neoplastic gastric and duodenal mucosa, replete with mucosal inflammatory cells, which can “drown” cancer-associated alterations (8). Further, the limited neoplastic DNA yield from EUS-FNA samples often precludes the use of even the currently available targeted NGS panels, let alone more comprehensive WES. Consequently, most published studies have been limited to assessing a few common “hotspot” mutations in PDAC, such as KRAS or BRAF (9). At the same time, it is worth noting that the currently available targeted NGS panels are geared towards common actionable genes, and not included are the aforementioned low frequency alterations (so-called “long tail”). Third, the most commonly used first-line, and by some measures, most efficacious therapy in advanced PDAC is FOLFIRINOX, a platinum-containing multi-drug regimen (10). While preclinical data supports that patients with PDAC with homologous recombination repair defects (HRD) are likely to bear greatest susceptibility to platinum agents like oxaliplatin (an integral component of FOLFIRINOX), the targeted NGS panels only interrogate a fraction of HRD, such as the ~5% of patients with deleterious BRCA1/2 mutations. Yet, prior studies have suggested that the full compendium of HRD in PDAC, as reflected in the “unstable genome” phenotype comprised of multiple structural abnormalities, might be several fold higher in prevalence (3). As a result, most first-line therapy decisions in the advanced disease setting are made empirically, warranting strategies that can provide improved treatment prediction information from the PDAC genome.
In this study, we use real-world limited biospecimens obtained via EUS-FNA, almost all in the diagnostic “first encounter” that a PDAC patient has with their gastroenterologist, for in-depth genomic analysis of PDAC. Applying a relatively facile enrichment strategy, we were able to significantly increase neoplastic cell fraction, which allowed us to detect actionable mutations using molecularly barcoded WES for clinical decision making. Further, we demonstrate the ability to use the WES information paired with bioinformatics pipelines to determine potential HRD status in the cases, which can help guide the choice of first line regimen in a clinically feasible timeline. Our data establishes the feasibility for WES in the most commonly obtained diagnostic biospecimen in PDAC, EUS-FNA samples, with consistent results compared to previous studies.
Materials and Methods
Patient cohort
Investigators obtained written informed consent under MD Anderson protocol Lab00–396 from each patient prior to tissue and blood sampling. The study was performed in accordance with standard ethical guidelines approved by the institutional review board (IRB) and in accordance with the Declaration of Helsinki. All patients had clinically and histologically confirmed localized or metastatic PDAC. In the pilot phase, resected tissue specimens were only included if tumor cellularity was below 10%. (Supplementary Figure 1b). All biopsies (CT guided core or EUS-FNA) were taken within routine clinical procedures. No EUS-fine needle biopsy (FNB) samples have been included. All research passes harvested were sampled after routine diagnostic EUS-FNA passes and ROSE-rapid on-site cytology assessment which showed adequate cellularity in diagnostic specimens.
No complications have been reported in any tissue sampling.
Tissue digestion and EpCAM pulldown
Biopsy samples were mechanically and enzymatically digested into single cell suspension as described before (11). The cell suspension was then processed using the EasySepTM Human EpCAM Positive Selection Kit (Stemcell, Vancouver, Canada, cat#18356) following the manufacturer’s protocol to enrich for EpCAM positive epithelial cells. In brief, cell suspensions were incubated with EpCAM antibodies and magnetic beads, followed by subsequently clean-up steps using magnetic separation (Supplementary Figure 1c).
DNA isolation, QC, and Digital Droplet PCR Analysis
Bulk genomic DNA was extracted from enriched and non-enriched cell suspensions using the QIamp DNA Micro Kit (Qiagen, Hilden, cat#56304) and DNeasy Blood & Tissue Kit for PBMCs (Qiagen, Hilden, cat#69506) following the manufacturer’s protocol. At least two different quantification methods for DNA quality and quantity were performed in parallel using a QubitTM dsDNA BR Assay Kit (Thermofisher, cat#Q32853), a NanoDropTM 2000/2000c spectrophotometers (Thermo Fisher, Waltham, cat#ND2000) and/or high sensitivity D1000 screentape (HSD1000) with a Tapestation 2200 system (Agilent, Santa Clara, cat#5067–5584). Droplet digital polymerase chain reaction was performed for validation of KRAS mutations and MYC amplification as described in the supplementary materials.
Library construction and sequencing
A median of 160ng of enriched tumor DNA or 200ng of matched PBMC DNA was diluted in a total volume of 52ul low TE buffer and fragmented to 150–200bq using a Covaris LE 220 ultrasonicators system (Covaris, Woburn USA). The following optimized settings were used: Duty Factor 30%, Peak Incident Power PIP W 450, cycles per burst 200, time 300s, temperature 7°C and water level 6. Adequate fragmentation was documented using HSD1000. Molecular-barcoded libraries were constructed following the SureSelect XT HT targeted enrichment protocol (Version A1, July 2017). In brief, end repair and dA-tailing were followed by ligating individual molecular-barcoding to each strand, PCR- amplification and bead-based cleaning. Libraries are then hybridized and incubated with a whole-exome capture library (SureSelect Clinical Research Exome V2, cat#5190–9492, Agilent, Santa Clara), captured to streptavidin-coated beats, washed and amplified. Libraries were multiplexed, denatured and diluted to a final concentration of 1.8 pM for sequencing and cluster generation as per manufacturer’s recommendation. Clustered flow-cells were sequenced on the Illumina NextSeq 500 instrument (Illumina, San Diego, USA) using standard Illumina paired-end primers and chemistry. A median of 74,991,096 reads/PBMC (range: 25,067,034 – 251,278,724 reads) and 144,308,769 reads/enriched tumor (range: 25,451,842 – 236,573,016 reads) was detected. Median on-target coverage reached 56x (range: 23–157x) for matched PBMC and 108x (range:14–177x) for enriched tumor samples for family size of 1 library.
Sequencing Data Analysis:
Sequencing data was processed as detailed in the supplementary materials. Three algorithms were used for SNV calling and these were filtered as detailed in the supplementary materials. Of note, the median mutational burden of non-PBMC paired samples (n=5) was higher than in paired samples but did not reach statistical significance (4.89 mut/Mb, range: 0.72 – 23.91 vs. 1.63 mut/Mb, range: 0.29 – 21.46, p=0.06) and seemed to had no impact our ability to identify patients with increased TMB (Supplementary Figure 3c).
When available, clinical CLIA reports were used for SNV validation. These reports were generated from resected or core biopsied tissue samples during the course of treatment. Reports were available for one core biopsy patient (pilot phase) and for four FNA patients (second phase). Four of the CLIA reports were generated in-house at MDACC with a PCR-based sequencing platform and assembled using GRCh37/hg19 builds. MDACC CLIA reports included a panel of 134 individual genes (Solid Tumor Genomic Assay V1 report). The fifth patient report was performed by an outside CLIA-certified laboratory using a company specific gene panel (Perthera).
DFCI cohort data were processed using the same bioinformatics pipeline used in our study with the exception of unique molecular identifiers specific processing.
Actionable mutation
We evaluated each sample for presence of 24 potentially targetable genes/alterations and SCNAs: BRCA1, BRCA2, PALP2, FGFR1, FGFR2, FGFR3, FGFR4, PDGFR, c- Kit/CD117, ROS1 (fusion), MET, NOTCH1, JAK1, JAK2, JAK3, mTOR, BRAF, RNF43, PI3K /PIK3CA, AKT, NTRK, MYC, KRAS wild type (wt), HER2 amplification.
Tumor Mutational Burden (TMB)
Tumor mutational burden was defined by the sum of all synonymous SNV, non- synonymous SNV, stopgain, stoploss, frameshifts or indels per sample that passed filter criteria as described above. For PBMC matched samples germline mutations called by HaplotypeCaller were excluded but for mutations in important PDAC genes like e.g. BRCA1/2, PALB2 or ATM (12). Non-PBMC matched samples are indicated in the analysis workflow and throughout results. TMB was then calculated by dividing the total number of mutations/patient by the coding region target of the SureSelect Clinical Research Exome V2 panel used (67.3 Mb).
Mutational signatures
Mutational signature analysis of FNAs was performed using MutationalPatterns following the steps outlined for cancer/COSMIC signature analysis (13). Only FNA samples with a matched germline control (PBMC sample) and those from treatment naive tumors were included in the mutational analysis.
Detection of chromosomal alterations
Two algorithms were used for detection and classification of somatic copy number alterations SCNAs (amplifications, deletions, and cnLOH). HapLOHseq, was used for detection of genomic regions exhibiting allelic imbalance (AI). Results from this algorithm were combined with output from standard log2 copy ratio segmentation data as described in the supplementary materials.
This approach detects B-allele frequency (BAF) shifts at germline heterozygous sites, indicative of AI, allowing for detection of chromosomal alterations in low mutant cell fraction settings (14,15). Germline heterozygous sites with a depth greater than or equal to 10 were included as input for AI analysis. The hidden Markov model of hapLOHseq was used to compute the probability that a set of adjacent markers span a region of AI. An AI event was defined as a continuous set of markers with posterior probabilities exceeding the threshold of 0.85.
GATK (16) was used for segmentation of log2 copy ratio data. SCNA’s were called by overlaying HapLOHseq AI and GATK segmentation calls (Supplementary Figure 5a). For SCNA calls exclusive to GATK only those with a log2 copy ratio below −1 and above 0.58 for deletion/amplifications were included in the final call set. This approach allows for the inclusion of balanced amplifications/losses, which do not result in AI, as well as focal SCNAs spanning a modest number of germline heterozygous sites. Genomic regions with posterior probability of AI > 0.85 were included in the final SCNA call set and were classified using GATK segmentation values, log2 copy ratio below −0.41 and above 0.32 were called as deleted and amplified, respectively. Copy neutral loss of heterozygosity (cnLOH) was defined as genomic regions with a log2 copy ratio between ±0.15 and a BAF deviation >0.1. Events with a posterior probability of > 0.85 and log2 copy ratio between ±0.15 and −0.41 or +0.32 were defined as undetermined (Supplementary Fig. S4b). An aneuploidy score was calculated for each sample. It was defined as the number of chromosome arms (out of 39) with arm-level aneuploidy, following principles previously used in the analysis of genomic instability (17). A chromosome arm (except for the short arm of acrocentric chromosomes 13, 14, 15, 21 and 22) was categorized as aneuploid when an amplification, deletion, cnLOH, or undeterminable SCNA(s) spanned more than 75% of the chromosome arm. To accommodate for high-confidence AI events, with subtle log2 copy ratio deviations, an arm-level SCNA was also called in cases where one type of SCNA (amplification, deletion, or cnLOH) plus undeterminable SCNA(s) spanned more than 75% of the arm, but a single type of SCNA did not reach the 75% threshold.
For identification of SCNA patterns suggestive of DNA damage repair deficiencies, we calculated a new and easy to compute score. This complexity score was calculated by summing the number of chromosome arms with two SCNAs with opposing classification (ie gain and loss, or gain cn-LOH). Undetermined events and those smaller than 1Mb were excluded. This chromosome-arm level signature is inferred to have arisen from double strand breaks, thus higher scores are present in patients with deficiencies in DNA- repair.
Statistical analysis:
PFS was defined as time period from the start of any treatment (chemo, radiation or surgery) to date of disease progression (defined by RECIST 1.1 guidelines (18)) or death. OS was defined as time from tissue sampling (surgery, core biopsy or EUS-FNA) to death for any reason. Survival curves were estimated using the Kaplan–Meier method. Statistical analyses were performed with SPSS statistical software, version 24 (IBM, Armonk, NY) or Prism 8 (GraphPad Software, Inc, San Diego, USA). All tests were two- sided. Statistical significance was defined as a P value of <0.05. We used bootstrapping to estimate the sampling distribution of the CS score in the DFCI cohort.
Results
Patient cohort and workflow
To analyze the feasibility and impact of epithelial cell enrichment, we utilized samples from either surgically resected primary PDAC (N = 5) or CT guided metastatic core biopsies (N = 4, 3 patients) in a pilot “first phase” of this study. The nine pilot phase samples were obtained from eight independent patients. Subsequently, for the main (“second phase”) study, we assessed 23 cytology samples from 23 PDAC patients undergoing endoscopic FNAs between April 2017 and November 2018 at MD Anderson Cancer Center (MDACC). Patient details and processing workflow for both phases are summarized in Supplementary Table 1 and Supplementary Figure 1a. Matched blood samples were obtained in 24/31 patients (77.4%).
Pilot phase: Significance and feasibility of EpCAM enrichment
A major obstacle for genomic characterization of small biopsies, especially using whole exome sequencing, is the limited amount of starting material and the generally low tumor cellularity of pancreatic cancer. To overcome this known hurdle, we used a magnetic EpCAM-pulldown approach to enrich for epithelial tumor cells. In the pilot phase, we used five fresh resected tissues samples with a low cellularity (<10%) and four CT or ultrasound guided core biopsies (liver and lung metastases, N=2 each, Supplementary Figure 1a and b). All samples were mechanically and enzymatically dissociated into a single cell suspension before further processing. EpCAM-enrichment was performed using an EpCAM-based magnetic pulldown followed by DNA isolation (Supplementary Figure 1c). Digital droplet PCR (ddPCR) showed a significant increase in KRAS MAF (median 4.3% vs. 21.7%, P=0.049) (Figure 1a and b). In four patients with a non-enriched KRAS MAF falling below the cut-off for inclusion in CLIA reports (<5%), enrichment increased the MAF above this threshold (Figure 1a and b). Additionally, we compared performance for WES SNV and somatic copy number alterations (SCNA) calls between EpCAM- enriched samples from resected PDAC and matched archived formalin-fixed paraffin- embedded (FFPE) slides of the matched tumor at resection (n=2). While this comparison showed only a few alterations in FFPE tissues, a greater number of genomic alterations were observed in the enriched samples, underscoring the value of EpCAM-enrichment (Supplementary Table 2 and Supplementary Figure 2a and b). For example, in patient 13, sequencing of the FFPE block did not identify any mutations in PDAC driver genes, whereas they were detected in the EpCAM-enriched sample (KRAS, SMAD4 and RNF43) (Supplementary Table 2). Additionally, no major allelic imbalance events were found in the matched FFPE-derived samples compared to the enriched counterpart in both patients (Supplementary Figure 2a and b).
Figure 1.
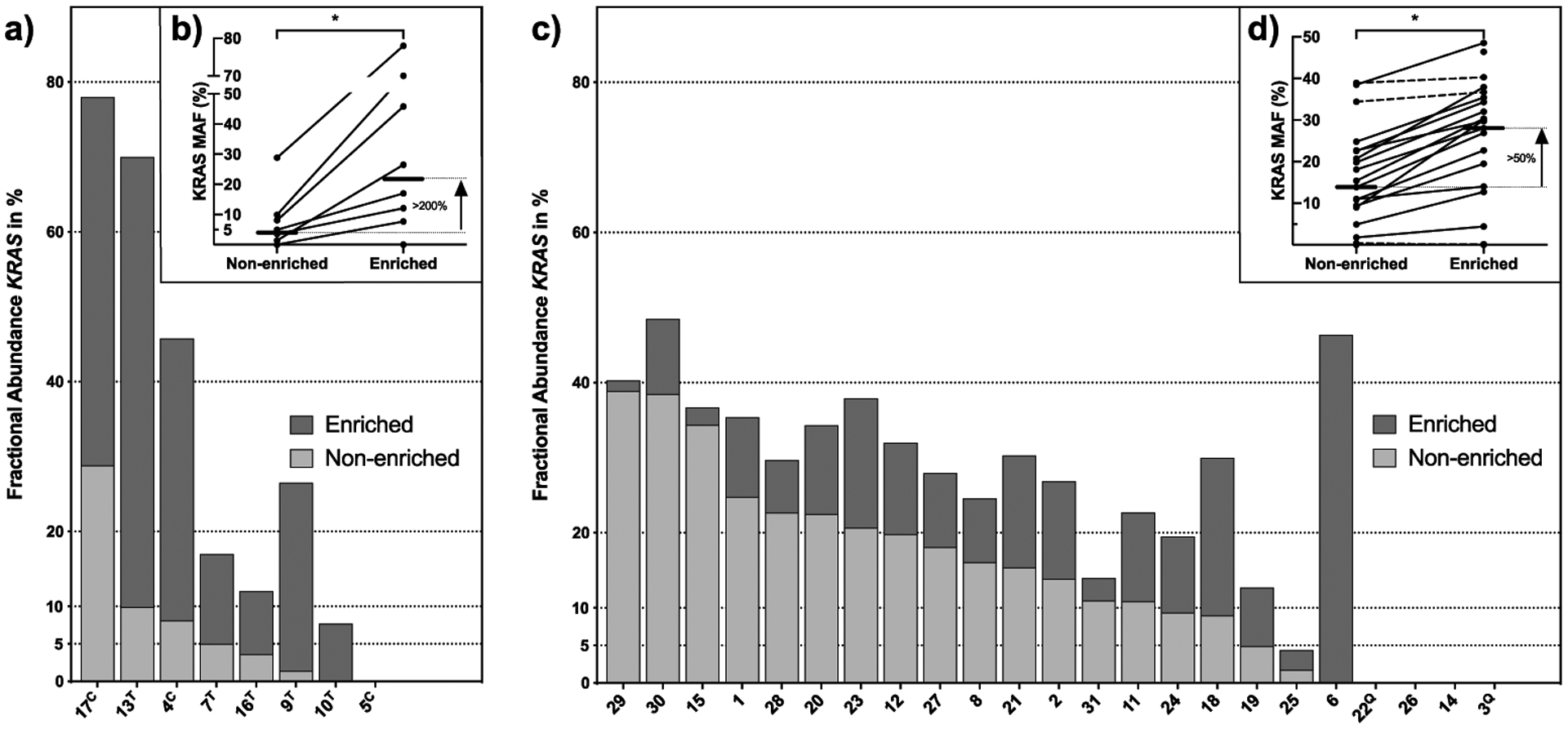
Detection of mutant KRAS in FNAs and core biopsies. a) and c) Fractional abundance of mutant KRAS in enriched (dark gray) and non-enriched (light gray) samples measured by ddPCR. Samples of the pilot phase derived from resected tumor specimens (n=5, marked with T) or core biopsies (n=3, marked with c) are shown in a) and b), one sequential core biopsy was not processed with ddPCR. Samples derived from FNAs in the second study phase (n=23) are summarized in c) and d). b) and d) gray lines indicate changes in KRAS MAF between non-enriched and enriched samples with a median increase of 95%. Straight lines indicate a percentage increase of >10%, connected lines show a difference <10%. Samples marked with Q represent KRAS mutations in Q61.
All but one sample (1/9) of this pilot phase (patient 16, resected tumor) passed sequencing quality control (QC), which was therefore excluded from further analysis, with the exception of ddPCR profiling. The genomic landscape of mutations in known PDAC- associated genes in this pilot phase generally match those previously reported (3,4)(19)(20) (Figure 2).
Figure 2.
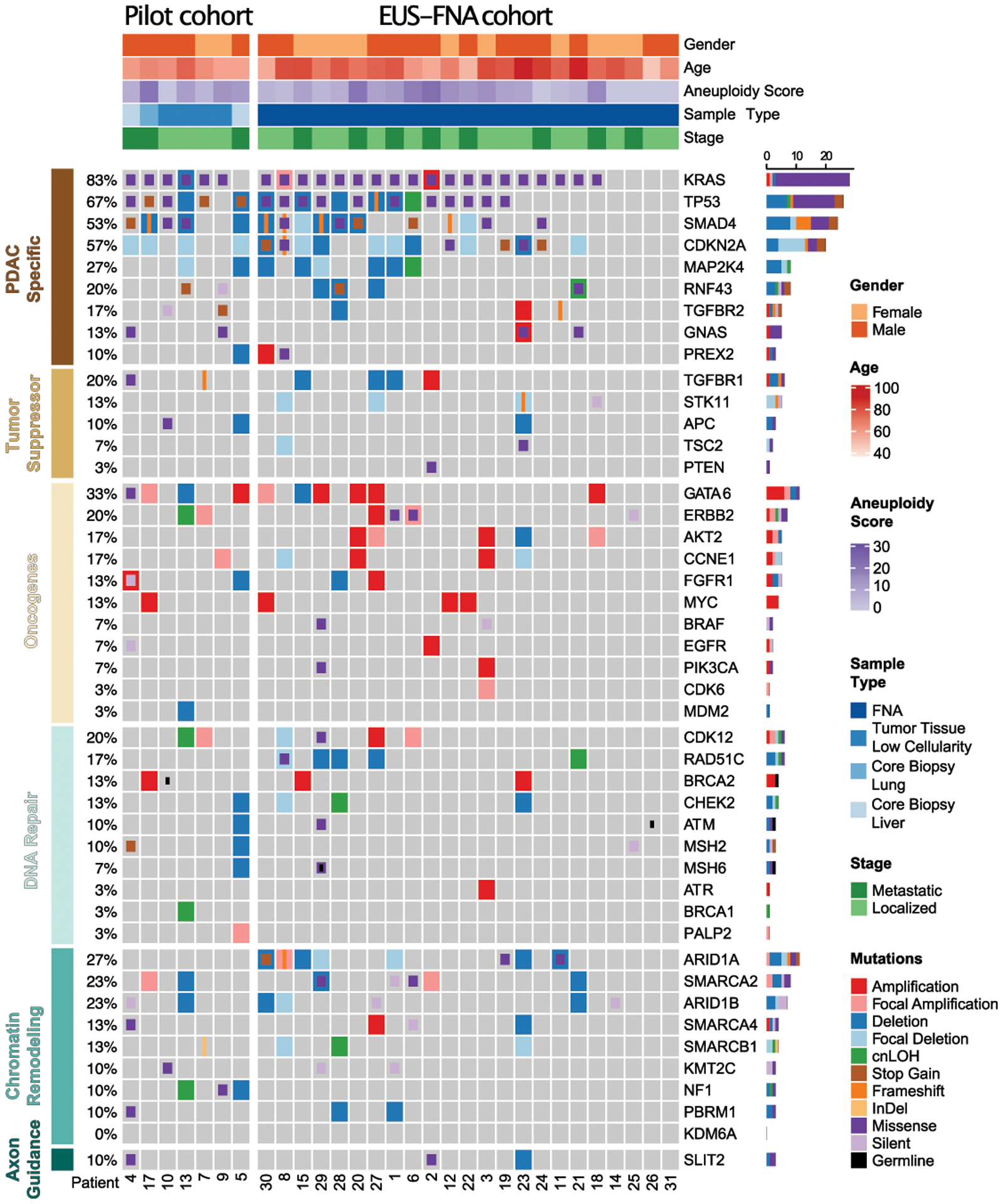
Genomic alterations identified during the pilot and subsequent study. Heatmap of genomic data for 30 patients (columns) for pilot phase including resected specimens and core biopsies (left side) and for EUS-FNA derived samples (right side). Heatmap includes SNV (classified as missense, silent, InDel, frameshift, stopgain, stoploss, and germline) as well as SCNA (classified as amplifications, deletions, and cnLOH) in selected PDAC driver genes organized by their functional classes. Germline mutations are only shown if identified in a CLIA certified clinical test (Supplementary Table 3) in addition to our calling algorithm. Patients’ clinical pathological data are shown as tracks at the top. The percentage of PDAC samples with an alteration of any type is noted at the left and the proportions of alterations per genes at the right.
Second phase: EUS-FNA biopsies resemble commonly reported PDAC genomic landscapes from high quality tissue sources
In the second phase of this study, we applied our EpCAM-enrichment strategy to 23 independent real world EUS-FNA samples collected during routine diagnostic procedures (18/23 with paired PBMC, median of one FNA pass, range 1–2). Comparable to the results of the pilot phase, EpCAM-enrichment followed by ddPCR showed a significant increase in KRAS MAF (median 13.9% vs. 28.0%, P=0.03) (Figure 1c and d). These results were also seen by sequencing as we split up one pooled FNA with two research passes (patient 18) to conduct WES on matched enriched and non-enriched DNA. The median MAF of all overlapping mutations and SCNAs, in the enriched versus the non-enriched samples, increased significantly (p<0.001, Supplementary Table 2 and Supplementary Figure 2c). Only three cases showed either no demonstrable KRAS or a MAF below 5% after EpCAM-enrichment (range KRAS MAF: 0–4.39%). Non-diagnostic EUS-FNA passes cannot be excluded in these cases as our single research passes were acquired after routine clinical passes and may therefore harbor lower levels of cancer cells (21).
All 23 enriched FNA samples had enough DNA to proceed with library preparation and passed sequencing quality control (QC). The genomic landscape of mutations in known PDAC-associated genes generally matched those from previous reports (3,4,10,15). In our cohort, significantly recurrent mutations (>10%) were identified in KRAS, TP53, CDKN2A, SMAD4, GNAS, RNF43 and ARID1A (Figure 2). Prevalence of KRAS mutations using sequencing alone was 82.6% (19/23 patients) whereas combination of both sequencing and ddPCR increased the KRAS mutation detection rate to 91.3% (21/23 patients). In order to confirm our methods for calling SNVs and CNVs in patients that underwent a subsequent resection or core biopsy, we used pre-generated targeted sequencing CLIA laboratory reports as the gold standard for validation. All but one SNV (patient 6: TP53) showed a concordance (92.3%) between the CLIA report and our sequencing results (Supplementary Table 3). Additionally, comparison of sequencing with ddPCR results for KRAS MAF and GNAS p.R201C showed significant correlation (R2=0.93, p<0.0001, Supplementary Figure 3a, b and Supplementary Table 4).
Although not present in a majority of PDAC patients, low frequency, potentially actionable alterations were found in individual cases. Based on knowledge of recently published actionable mutations (2) and by including only highly deleterious SNVs from the Catalog of Somatic Mutations in Cancer (COSMIC) database and amplifications with more than 3 copies, a total of 5/23 EUS-FNA patients (21.7%) showed potentially actionable alterations such as MYC amplifications = 1 (SCNA with a level of 5.5), MTOR = 1 (nonsense mutation, p.A835S), BRAF = 1 (nonsense mutation, p.R509L), MSH6 = 1 (nonsense mutation, p.P1087R), POLE = 1 (frameshift insertion, p.F699Vfs*11)) (Figure 3a). These results match prevalent actionable mutations previously reported in 17%−48% of PDAC cases (19,20,22). Potential treatment options for these mutations include GSK525762 for MYC-dependent carcinomas, Everolimus, VS5584 or LY3023414 for MTOR-mutant tumors, Vemurafenib for BRAF-mutant PDACs and agents targeting tumors with mismatch repair defects caused by MSH6 and POLE such as Pembrolizumab and Nivolumab which have shown response rates of over 30% in non-colorectal cancer patients (2)(23). Nonetheless, it should be emphasized that estimates for the proportion of actionable mutations in PDAC vary widely depending on the inclusion criterion.
Figure 3. Clinicopathological significance of mutational burden (only EUS-FNA cohort, n=23).

a) Potential actionable alterations based on literature findings (2). Alterations included are deleterious SNVs found in the COSMIC database and gene amplifications with copy number of 3 or greater. b) Sum of all somatic mutations (missense, silent, frameshift, InDel, stopgain and stoploss) per patient (column) arranged in decreasing order. TMBhigh cases are defined as > 670 mutations/patient (>10 SNVs/Mb). Samples without paired PBMC for germline correction are marked with v, all other samples are FNAs with paired PBMC. c) TMB shown as tracks at the bottom with TMBhigh cases left of vertical dotted line and mutations in DDR genes highlighted (p<0.0001). d) Kaplan Meier curves showing progression free survival (PFS) comparing TMBhigh vs. TMBlow (p=0.09).
Taken together, we were able to show that EpCAM- enrichment of epithelial tumor cells significantly increases mutant allele frequency (MAF) and facilitates genomic profiling in PDAC, especially in challenging sample types like EUS-FNA and samples with low overall tumor cellularity.
Tumor mutational burden is correlated with mutations in DNA repair genes
PDAC is known for a relatively modest mutational burden, compared to melanoma or lung cancer (1–3)(4). Matching previous reports, our median mutational burden for (non-) synonymous SNVs (18/23 PBMC paired samples) was 1.63 mutations/Mb (range: 0.29 mut/Mb - 21.46 mut/Mb) (24). TMB was not affected by the presence of matching germline samples, stage, or tissue origin (Supplementary figure 3c and d, e).
Two of 23 individuals in our EUS-FNA cohort (8.7%) showed an increased mutational burden (TMBhigh), defined as carrying more than 10 mutations/Mb by recent Pan-cancer analysis (25) (Figure 3b). Both TMBhigh patients harbored SNVs in DNA-damage response (DDR) genes or mismatch repair defects genes, while the majority of TMBlow displayed no mutations in these genes (p<0.001) (Figure 3c), which is in line with previously reported results (26). Patient 29 harbored a MSH6 germline mutation (Lynch syndrome) which has been confirmed by clinical CLIA germline testing as a variant of unknown significance and an additional non-synonymous mutation (Supplementary Table 3). This patient had an extensive family history of multiple cancers including a mother with breast cancer, a sister with PDAC, a parental cousin with an aggressive melanoma, and two parental cousins with brain tumors. Patient 8 had a COSMIC annotated frameshift insertion in POLE (COSM2001733, p.F699Vfs*11, no clinical CLIA testing performed) and also reported a family history for cancer (gastrointestinal cancer: mother and grandfather). In our limited EUS-cohort, TMBhigh patients demonstrated a trend toward improved progression-free survival (PFS) (median PFS: 674 days vs 191 days, p=0.09) (Figure 3d) but not overall survival (OS) (417 days vs. undefined, p=0.19) compared to non-TMBhigh (Supplementary Figure 4a).
Prognostic impact of chromosomal alterations in WES
To ensure a rigorous standard for annotating somatic copy number alterations (SCNA), we used two independent algorithms: HapLOHseq (14), a powerful tool to analyze the presence of allelic imbalance (AI) in low purity samples and the genome analysis toolkit (GATK) standard segmentation pipeline (Supplementary Figure 4b). The results from these tools were merged to obtain a high confidence SNCA call set, which was validated by a significant correlation of the MYC amplification estimates between sequencing and ddPCR results (n=7, R2=0.91, p<0.0001, Supplementary Figure 5a–c).
Consistent with previous studies, deleterious SCNAs were mainly detected in known tumor suppressor genes such as CDKN2A (9/23, 39.1%), SMAD4 (7/23, 30.4%), TP53 (7/23, 21.7%) and ARID1A (6/23, 26.1%) (all p<0.05) (3,4,19,20). Interestingly, CDKN2A showed a large number of focal SCNA events (5/9, 55.5%), whereas most other loci showed a mixture of both focal and larger events. Not unexpectedly, amplifications were most commonly centered on established oncogenes such as GATA6 (5/23, 21.7%) or MYC (3/23, 13.0%) (Figure 2, Figure 4 and Supplementary Figure 6a). Mirroring results from previous PDAC publications, at least one arm-level SCNA occurred in 78.3% of patients. The majority of patients harbored multiple SCNAs, most frequently spanning chromosome arms 6q (9/23, 39.1%), 8p (8/23, 34.8%), 9p (7/23, 30.4%), 17p (11/23, 47.8%), 18p (7/23, 30.4%), 18q (11/23, 47.8%) and 19p (9/23, 39.1%) (4,17) (Figure 4 and Supplementary Figure 6).
Figure 4.
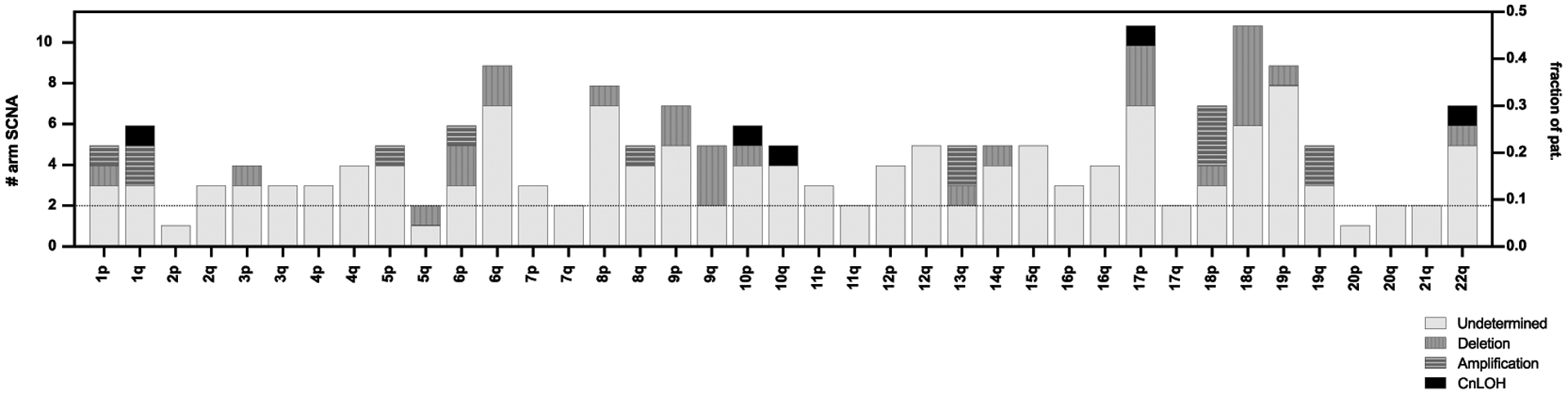
SCNA(s) across all patients. Arm-level SCNA(s) with classification information (deletion, amplification, cnLOH, or undetermined) per chromosomal arm (left y-axis) and fraction of patients with events (right y-axis)
Genomic instability from SCNA data across cancer types can be estimated using a number of different approaches. For PDAC, structural rearrangements contributing to an “unstable genome” (>200 rearrangements) have been used as a measure of genomic instability (3,27). We were unable to derive this previously described “unstable genome” phenotype from the exome capture platform (SureSelect, Agilent), as it only covers 71 possible breakpoints. Therefore other previously proposed quantitative metrics, such as HRD-LST (large-scale state transitions), HRD-LOH (loss of heterozygosity), the total SCNA burden, as well as aneuploidy were analyzed (17,28,29), and all of these quantitative assessments were readily feasible on EUS-FNA WES data. All scores were significantly correlated with each other, which is why we used aneuploidy in the subsequent analyses (all p<0.015, Supplementary Table 5). In our cohort, aneuploidy score was not affected by patient’s gender, primary tumor location, tissue source or age (Supplementary Figure 7a–d). Somatic TP53 mutation carriers showed a significantly higher aneuploidy score than patients without a TP53 mutation (median: 0, range 0–8 vs. 11, range 3–22, p=0.0002), as has been previously reported (Supplementary Figure 7e) (17). The magnitude of aneuploidy measured by the number of aneuploid chromosome arms on WES has previously been used as an indirect measure of genomic instability, the prognostic value of which is an area of active research (17). Correspondingly, studies in many solid tumors (30), showed a correlation of high aneuploidy with later stage disease and poorer prognosis (e.g. colorectal cancer (31) and esophageal cancer (32)). We therefore evaluated the prognostic potential of the aneuploidy score. Classification of patients based on the median aneuploidy level into a low-level aneuploidy (Aneuploidylow: bottom half=0–7 arm-level events), and high-level aneuploidy (Aneuploidyhigh: top half≥8 arm-level events) revealed a significant and deleterious impact on prognosis (Figure 5a– c). Aneuploidyhigh tumors showed a median PFS of 104.5 days, whereas tumors with low-level aneuploidy experienced a significantly longer PFS of 365 days (p=0.009) (Figure 5b). Additionally, Aneuploidyhigh showed a trend for worse OS (225 days vs. undefined, p=0.06) (Figure 5c). Importantly, the aneuploidy score maintained its prognostic trends also in localized patients (Figure 5d–e), which suggested that aneuploidy’s prognostic value is not based on advanced tumor stage. Correspondingly, after restricting the analysis to non-surgically treated patients (n=19) in this group, prognostic impact of aneuploidy remained significant (OS: 286.6±58.8 days vs. 466.9±69.5 days, p=0.039 and PFS: 130.0±51.4 days vs. 317.0±24.4 days, p=0.005). A potential confounder for our analysis may be the inclusion of TMBhigh samples as these samples demonstrated longer PFS and therefore results have to be interpreted with caution. To further validate the prognostic impact of the aneuploidy score, we used an independent cohort from the Dana-Farber Cancer Institute (DFCI) in Boston (MA) (20). There are several details that need to be highlighted when comparing the two datasets. In the DFCI cohort: 1) Most of the specimens (96%) were taken using core needles or intra-operative biopsies, primarily from metastatic lesions, while only 4% of cases were sampled by EUS-FNA with 9% coming from primary pancreatic lesions. 2) No enrichment of the biopsies was performed and the authors report a median cellularity of 40% (range 5–80%). 3) Most of the cohort (91%) and all of the patients with a cellularity >20% were diagnosed with metastatic disease. 4) No UMI-enhanced sequencing was used for WES.
Figure 5.
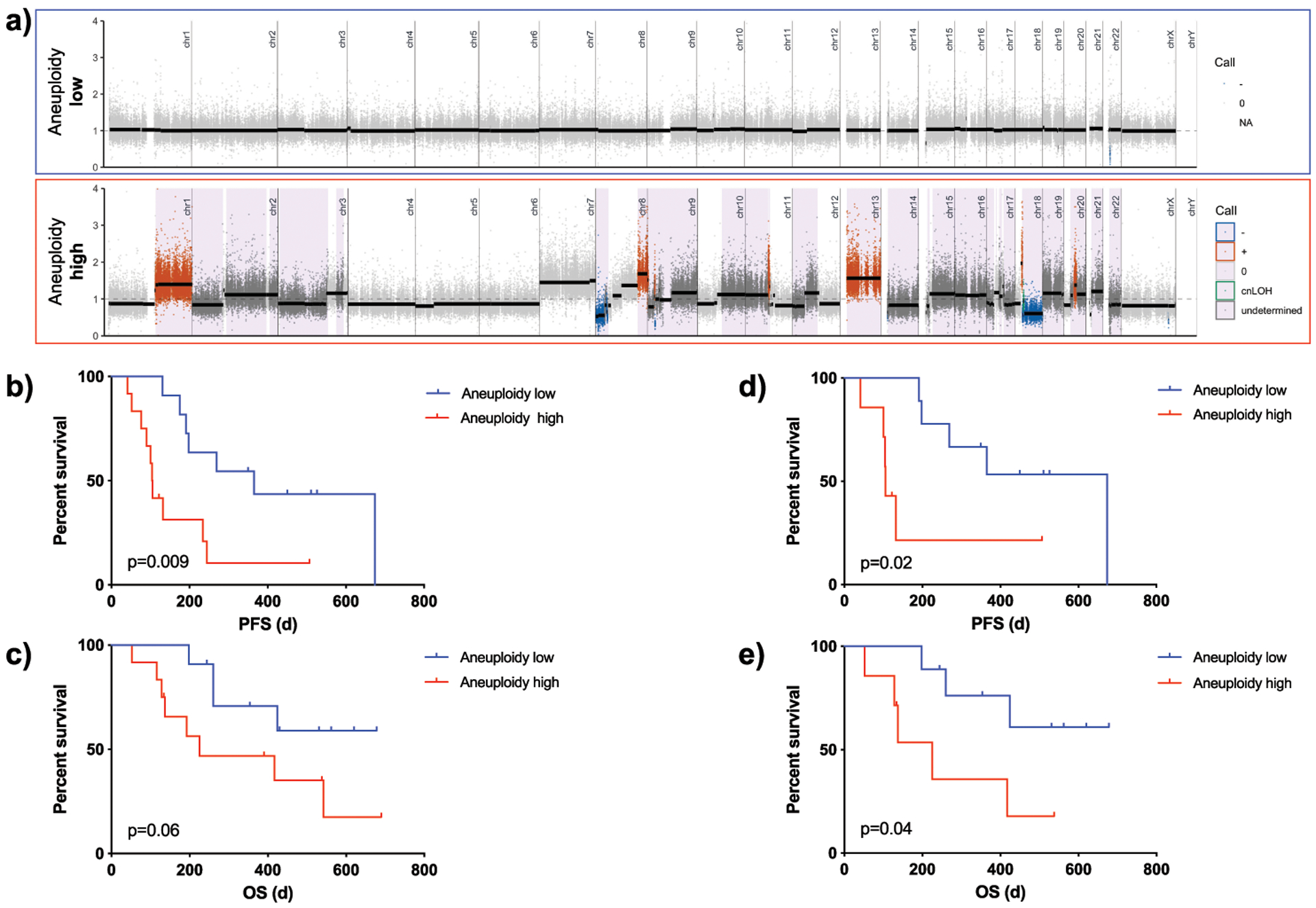
Prognostic significance of aneuploidy in patient samples. a) Exemplary segmentation plots intersected with HapLOHseq calls (lavender background) showing samples classifying into aneuploidy low (<=7 chromosomal arm events) and aneuploidy high (>=8). b) Kaplan Meier curves comparing progression free survival (PFS) of patients with low vs. high aneuploidy levels (p=0.009). c) Kaplan Meier curves comparing overall survival (OS) of patients with low vs. high aneuploidy levels (p=0.06). d) Kaplan Meier curves comparing progression free survival (PFS) of patients with low vs. high aneuploidy levels for patients with localized tumors only (p=0.02). e) Kaplan Meier curves comparing overall survival (OS) of patients with low vs. high aneuploidy levels in localized patients only (p=0.04). *For this and subsequent figures HapLOHseq calls shown pass a threshold of posterior probability of AI >0.85.
To minimize bias in the aneuploidy score related to low cellularity samples in the DFCI cohort, we restricted the analysis to specimens with a tumor cellularity of >20% (n=52/73). The median aneuploidy score of the DFCI cohort was 15 (range 2–33) and only 8/52 patients showed an aneuploidy score below 8. This may be due to the advanced stage in the DFCI cohort and mirrors our findings that metastatic patients show a trend for higher aneuploidy score compared to localized patients (median of 5.5 (range 0–18) vs. 11 (range 0–22), p=0.07). PFS for most patients in the DFCI cohort was unavailable; therefore we used OS data. Using our cutoffs for aneuploidyhigh (≥8) and aneuploidylow (≤7) there was no prognostic value for patients with aneuploidyhigh (median OS in days: 625±35.8 vs. 635±74.8, p=0.60). Because of the differences between the cohorts as detailed above, we then evaluated median aneuploidy score as a cohort specific cutoff value and found a significantly worse OS survival for aneuploidyhigh (≥15) patients (313.2±38.4 vs. 792.7±160.8, p=0.0013, Supplementary Figure 7f)).
Collectively, we speculate that increased aneuploidy in localized patients may be associated with more aggressive disease and earlier metastatic spread.
Prediction of platinum response using treatment naive EUS-FNA
Therapeutic decisions on first-line therapy in PDAC are currently mostly made on a clinical basis (e.g., performance status), without using biomarkers, unlike established markers for other cancer types, like colon cancer (33). There have been ongoing efforts using genomic or transcriptomic data to predict response to platinum-based therapy. In particular, cancers with BRCA1/2 mutations or HRD show superior response to platinum- based therapies in multiple cancers, including PDAC (22,34–36). We therefore evaluated the potential of WES data within our unique EUS-FNA set to predict response to platinum regimens.
Eight patients (four localized and four metastatic) whose EUS-FNA samples were sequenced, were treated with platinum-based therapies. Three of these showed a response to FOLF(IRIN)OX - defined as stable disease or partial response based on RECIST 1.1 criteria - while five patients progressed. None of the patients harbored deleterious SNVs in the following genes: BRCA1/2, PALB2, FANCM, XRCC4/6, CHEK2, BRIP1 or BARD1, except for two patients with ATM variants (patient 26 and 29). There was significance between patients with higher TMB and platinum response (8.86 mut/Mb, range 4.89–21.37 vs. 1.1 mut/Mb, range 0.79 – 3.36, p=0.04, Figure 6a), which is in line with previous reports (3). Previously proposed parameters including aneuploidy, HRD- LST and HRD-LOH (17,29) did not show a significant correlation (Figure 6b and Supplementary Table 5) with platinum response, while signature 3 showed a marginal positive association with response (p = 0.06), a finding that is in line with previous results (37).
Figure 6. Predictive value of SCNAs in platinum-based therapy response.
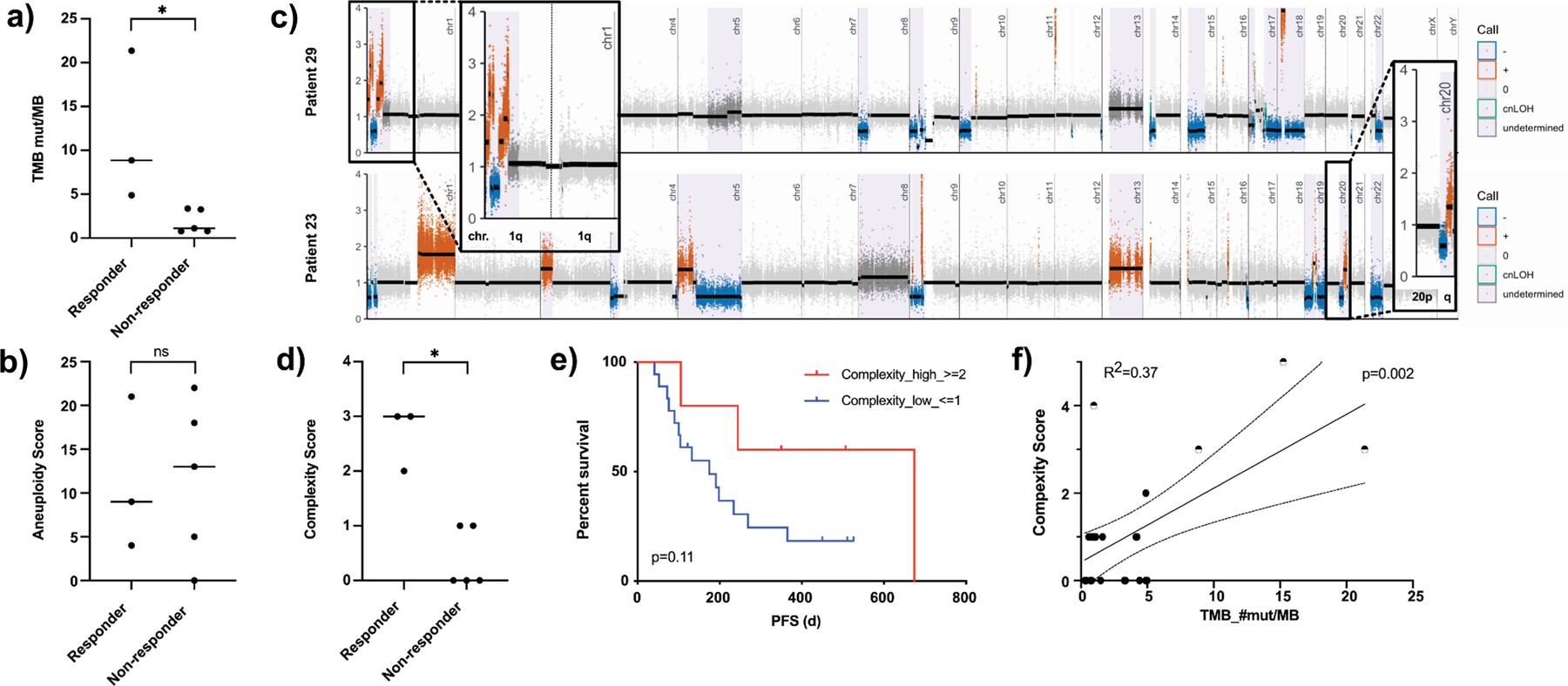
a) Scattered box plot showing the TMB level of responders vs. non-responders to platinum-based therapy (p=0.04). b) Scattered box plot showing the aneuploidy level of responders vs. non-responders to platinum-based therapy (p=0.73). c) Exemplary segmentation plots of two patients intersected with HapLOHseq calls (lavender background) showing samples with areas highlighted and zoomed in showing chromosomal arms rated positive for complexity score calculation. d) Scattered box plot showing the complexity score of responders vs. non-responders to platinum-based therapy (p=0.04). e) Kaplan Meier curves comparing progression free survival (PFS) of patients with low vs. high complexity score (p=0.11). f) Linear regression plotting complexity score vs. TMB (R2=0.37, p=0.002), half-filled dots mark samples with alterations in known DDR genes.
We therefore evaluated our SCNA data for new approaches to predict platinum response which might correlate with HRD or DDR deficiency. Based on the assumption that DDR deficiency and especially an impaired DNA double strand break repair contributes to HRD, we analyzed the number of chromosomal arms with opposing SCNA segments (e.g. gain and deletion present in the same chromosome arm, Figure 6c) and use the sum of arms exhibiting this pattern to calculate a “complexity score” (CS). Our assumption is that a higher CS is inversely correlated with DNA double strand repair capabilities. The CS is novel with regards to the integration of copy number (segmentation data) with allelic imbalance calls derived using a haplotype-based approach. However, this approach is similar in concept to using large-scale state transitions as a signature for homologous repair deficiency (HRD), which identifies adjacent chromosome segments with different copy number states (38). Patients with platinum therapy response showed a significantly higher CS than those with progressive disease (complexity score: 3, range 2–3 vs. 0, range 0–1, p=0.04, Figure 6d). Additionally, a high CS showed a trend for better PFS (674 days vs. 175 days, p=0.11, Figure 6e), OS (417 days vs. undefined, p=0.14, Supplementary Figure 7g) and a positive TMB correlation with tumors harboring a mutation in DDR genes with highest CS (R2=0.37, p=0.002, Figure 6f). In addition to the eight patients treated with a platinum-based therapy, eight patients of the FNA cohort received a Gemcitabine based regimen. Seven patients were excluded from this analysis due to trial treatment, upfront surgery, side effects, fragility or patient’s choice not to undergo any therapy. When examining CS for chemotherapy prediction, we observe a marginally significant association between the CS score and response: median1.5, range 0–3 for responders and median 1, range 0–1 for non-responders (p=0.07, Supplementary Figure 7h). We also attempted to test the predictive value in the DFCI cohort (see above). There were three responders in the data set (with 1 having an estimated tumor purity of only 4%), this severely restricted power to test for an association between the CS score and response to platinum therapy. However, we do note that patients with BRCA1/2 mutations (4 germline and 1 somatic) in the DFCI cohort have significantly higher CS scores than patients without these mutations (p = 0.008, mean = 3.6 CS for those with a mutation and mean = 0.7 CS for those without). In addition, we performed mutational signature analysis as described previously (13) on FNA samples from treatment naive tumors (Supplementary Figure 8). Herein, signature 3 which is associated with double strand repair deficiency showed a positive association with the complexity score (R2 = 0.49, P = 0.07).
Taken together CS shows potential as a predictive biomarker to estimate platinum responsiveness based on an indirect DDR evaluation even from limited FNA samples.
Longitudinal follow-up of an immune checkpoint treated patient gives insight into a possible mechanism of resistance
To demonstrate the clinical significance and feasibility of WES from limited clinical biopsies, we interrogated two consecutive endoscopic lung biopsies in a patient (patient 17) with recurrent metastatic PDAC to the lung (both biopsies were included in the pilot phase). The first biopsy (timepoint 1, T1) was taken prior to immunotherapy with a PD-L1 antibody (Durvalumab) and a STAT3-inhibitor (AZD9150) (39). The second endoscopic biopsy (timepoint 2, T2) was taken after progression on aforementioned therapy and prior to initiation of a hypoxia-activated prodrug regime with Evofosfamide (40). Before immune therapy initiation, this patient was diagnosed with a multifocal pulmonary recurrence two years after upfront distal pancreatectomy. The recurrent tumor was then treated with Xeloda and Oxaliplatin but progressed within a year (Figure 7a).
Figure 7.
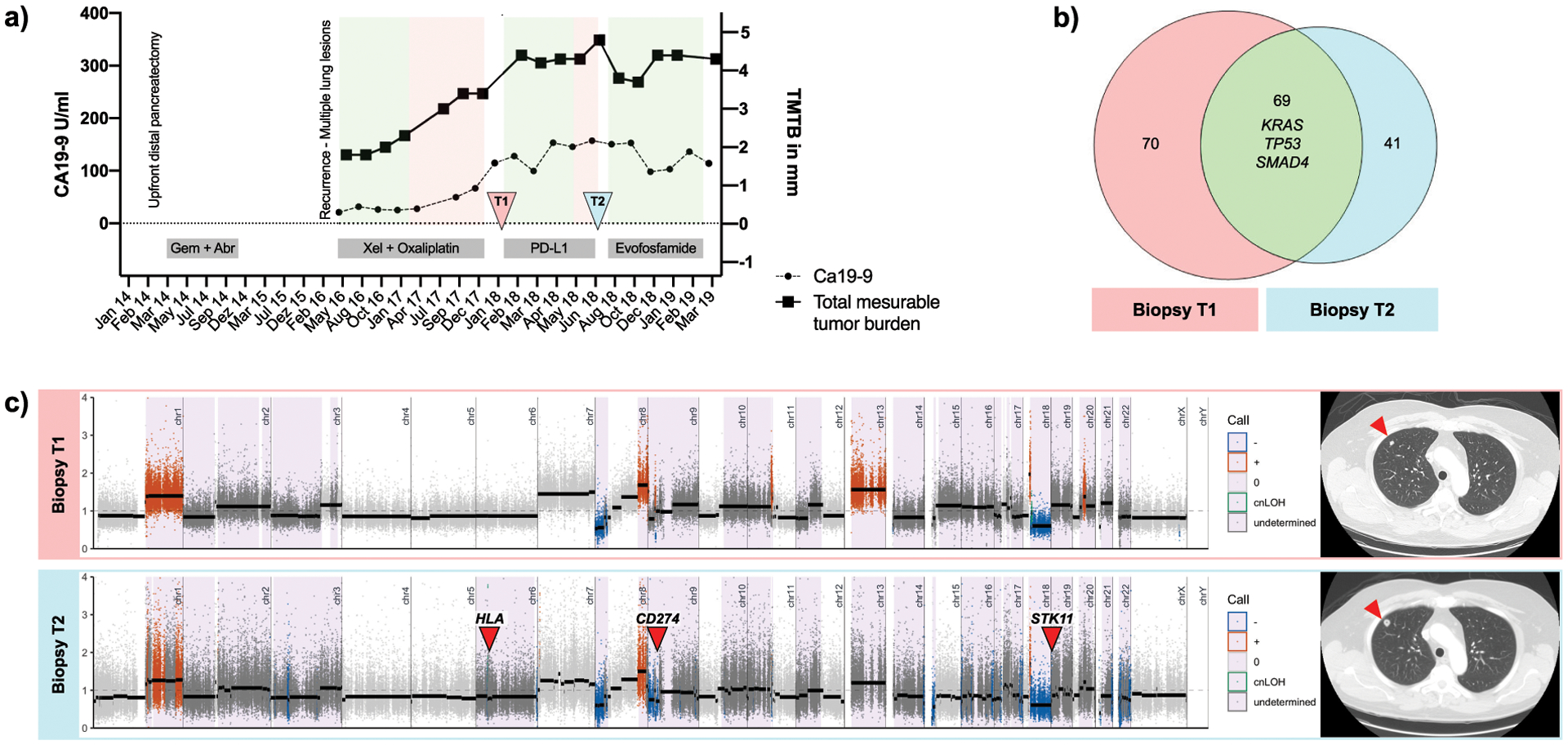
Longitudinal follow-up of patient 17. a) Time course of disease plotting the total measurable tumor burden (TMTB) (right y-axis) and Ca19–9 (left y-axis) with therapy regime administered at bottom. Green areas mark time periods with stable disease or partial response defined by RECIST 1.1, whereas red areas show progression. Time points of tissue sampling (T1 and T2) are indicated by arrows. b) Venn diagram plotting overlapping and distinct (non-) synonymous SNVs at T1 (n=139, left) and T2 (n=110, right). c) Left: Segmentation plots of T1 and T2 with intersected HapLOHseq calls (lavender background). Red arrows indicate loci that might be associated with acquired resistance to immune-therapy. Right: Exemplary CTC images showing an increase of pulmonal metastasis between T1 and T2 (red arrow).
Comparative analysis of the biopsy taken before immunotherapy treatment (T1) and after progression (T2) revealed new-onset focal deletions of STK11, TSC2 and the CD274 locus (PD-L1) (Figure 7b). These loci have been previously reported to have a great impact on immunotherapy resistance. STK11 loss, for example, has been repeatedly linked to the development of immunotherapy resistance in KRAS-mutant lung cancer (41) and PD-L1 loss is associated with adaptive immune resistance in Hodgkin lymphoma and other neoplastic diseases (42). In addition to these focal deletions, chromosome 3q, 10q, 17q, 6p and 6q also developed new arm-wide SCNAs, indicating loss of heterozygosity at all major histocompatibility complex genes, as evident by high confidence AI calls. Excluding these focal changes, T1 and T2 had overall congruous allelic imbalance profiles. T1 and T2 biopsies shared focal deletions in major PDAC tumor suppressors such as SMAD4 and CDKN2A, as well as amplifications in GATA6 and MYC. T1 and T2 SNVs in established drivers, KRAS, SMAD4, and TP53 were shared (Figure 7c). Only two of the non-overlapping deleterious SNVs had a COSMIC annotation (CCDC42: p.R104W, COSM4443319 and RCBTB2: p.V347M, COSM6445879); upon inspection and literature review, these do not have a known mechanistic link that would explain the development of immunotherapy resistance. Overall, mutational burden remained roughly the same between T1 and T2 (2.1 mut/Mb vs. 1.6 mut/Mb). As illustrated by this example, paired longitudinal endoscopic biopsies obtained over the course of therapy in PDAC patients might elucidate genomic alterations that could explain the development of resistance to immunotherapy (or targeted therapies).
Discussion
Over the past decade, precision oncology has improved clinical outcomes for many patients with solid cancers. Melanoma and breast cancer patients are now routinely screened for their V600E proto-oncogene B-Raf (BRAF) (43) and human epidermal growth factor receptor type 2 (HER2) status, respectively (44). Targeting these aberrations has alleviated prognosis in both entities. Despite significant efforts, PDAC has proven less susceptible to targeted or immune therapies, mainly due its stroma- dense, immune “cold” tumors that additionally show a remarkable genomic heterogeneity (1–6).
Previous WES, whole genome sequencing (WGS), and whole transcriptomic approaches in molecular profiling of PDAC have used tissue from multiple imaging-guided core biopsies for DNA and RNA profiling. The amount of tissue from these multiple cores is substantially greater than our “one-pass” EUS-FNA starting material (20,22)(11,13). It is also important to mention that multiple EUS-FNA passes are not necessarily associated with an increased amount of neoplastic cells harvest (45,46) nor with an increased sensitivity of detection (21),and indeed, might increase the associated risk of complications (21,47). Other sampling techniques such as EUS-fine needle biopsies (FNB) have shown promising results for in-depth genetic analysis, but their applicability and wide-spread use can be constrained by their significant expense (48)(49). Additionally, a major advantage of EUS-FNA compared to EUS-FNB is that the former allows for ROSE-rapid on-site cytology assessment that ensures that there is adequate cellularity and a diagnostic specimen. Here, we report our efforts to characterize EpCAM-enriched real-world, limited EUS-FNA. Despite the fact that most PDAC patients are diagnosed using EUS-FNA, these samples have so far been deemed not suitable for in-depth molecular analysis such as WES (50,51). To increase the tumor to normal cell proportion, many studies have either enriched their specimens e.g. laser microdissection (time- and labor-intensive) or excluded low tumor cellularity samples from analysis altogether (2–6,20). In contrast, we used a simple and fast enrichment method to successfully isolate tumor cells resulting in an improved detection of deleterious mutations.
There is evidence that evaluation of aneuploidy as a marker for genomic instability may be a valid surrogate in metastatic PDAC (22,52). Aneuploidy demonstrated prognostic significance independent of disease stage within the current study and an independent dataset of pancreatic cancer patients. This finding is in line with results showing a worse prognosis and later disease stage correlated with increased aneuploidy (3,24,32). In this regard, tumor with high aneuploidy have shown increased tumor progression and chemotherapy resistance due to a higher intra-tumoral heterogeneity and ultimately an elevated capacity for adaptation (53). It is important to mention, that aneuploidy seems to be context-dependent and therefore might differently affects distinct tumor types (32).
One of the important clinical questions in PDAC is the susceptibility to platinum-based therapies as one of two first line regimens, which should ideally be discernible in WES data on limited samples material like EUS-FNA. In light of recent findings, up to 25% of PDAC patients harbor a genomic footprint of HRD (3) and many of these potential responders might be missed using only “classic” DDR mutations for identification, like BRCA1/2 or PALB2 that are present in 5–7% of PDAC patients (54). Other HRD causing alterations like BRCA1 promoter hypermethylation or biallelic loss of PALB2 are even more challenging, if not impossible, to detect by most commonly used targeted sequencing panels (2). In a cohort of breast cancer patients, over 30% of HRD positive tumors had no underlying genetic alteration associated with HRD and yet demonstrated platinum susceptibility (35). Within this aforementioned study, HRD has been estimated by an WGS algorithm called “HRDetect” but the algorithm performed suboptimally with WES data because essential predictor components such as rearrangement signatures and indels analysis are restricted (35,36).
We identified a relatively facile WES-based scoring system of the “complexity” of SCNA events within a chromosomal arm that predicts platinum response in this PDAC patient cohort. This “complexity score” (CS) analyzes the presence of alternating SCNA events (gains, losses and cnLOHs > 1M) within a single chromosomal arm boundary. This kind of event pattern can only occur upon a severe disruption of double strand DNA repair and results in mis- segregation of “shattered” arm fragments in mitosis. It is relatively easy to calculate using WES data and standard GATK segmentation pipeline which makes it an attractive possible biomarker. We therefore hypothesize that CS may be a marker for increased genomic instability indicating susceptibility to platinum-based treatment. Interestingly, CS positively correlated with increased TMB that by itself also showed a predictive value but did not show a correlation with other SCNA markers, like aneuploidy.
We acknowledge important drawbacks in our study. One downside of our approach is that it requires a freshly acquired aspiration biopsy with dissociated cells as starting material, as we did not attempt WES on archival cytology samples. On the other hand, dissociation of cells from FNAs is a relatively simple procedure when compared to the dissociation of core biopsies which is time consuming. We were also not able to conduct a transcriptional analysis to validate loss or gain of function of genes because EUS-FNA provided sparse material to conduct additional assays. Additionally, our WES library design refrained us from analyzing important RNA fusion patterns which are accessible primarily using WGS.
Although significant EpCAM expression is seen in epithelial derived carcinoma cells when compared to normal epithelial, up to one third of pancreatic cancers may have low to no EpCAM expression (55)(56). This underlines a potential limitation of the EpCAM enrichment approach of tumor cells, and may warrant alternative strategies such as negative selection by targeting cells expressing CD45, CD31 and FAP to clean the sample of immune/blood cells, endothelial cells and fibroblasts. Based on the minimal amount of cellular material acquired from a single EUS-FNA pass, we were not able to process each pass with multiple selection methods to compare intra-patient accuracy and performance. Additionally, with advances and improvements in sampling strategies (like EUS-FNB), enrichment in the context of adequate tissue acquisition may become redundant. Finally, due to the small number of overall patients, the non-CLIA setting of our assay and the inherent cohort heterogeneity, our conclusions need to be validated in a larger prospective EUS-FNA cohort.
In conclusion, in this proof-of-concept study we show the importance and ease of EpCAM enrichment for genomic analysis of real-world limited aspiration (EUS-FNA) samples. Using enriched EUS-FNA samples, we were able to perform an in-depth whole high- quality molecular barcoded WES analysis, reproducing genomic patterns consistent with previous results. We show the prognostic potential of SCNA (namely aneuploidy) for PDAC diagnosis, and also identify a novel predictive biomarker (complexity score, CS) for platinum responsiveness in our cohort. CS may be a relatively facile biomarker for predicting platinum responsiveness in newly diagnosed PDAC not harboring overt mutations in canonical DDR genes.
Supplementary Material
Statement of Translational Relevance.
Genomic characterization of PDAC is infeasible in most patients mainly due to limited tissue availability and quality. Using an enrichment protocol combined with molecular barcoded WES sequencing, we demonstrate feasibility, prognostic and predictive value of genomic characterization from limited FNA samples.
Acknowledgments:
We thank A. J. Aguirre, B. M. Wolpin, and the Hale Family Research Center at Dana-Farber Cancer Institute for providing an independent data set used for replication of our results. Most of all we thank the patients and their families. A.Maitra is supported by the MD Anderson Pancreatic Cancer Moon Shot Program, the Khalifa Bin Zayed Al-Nahyan Foundation, and the National Institutes of Health (NIH U01CA196403, U01CA200468, P50CA221707). A. Semaan is supported by the German Research Foundation (SE-2616/2-1). V. Bernard is supported by the NIH (U54CA096300, U54CA096297, T32CA217789). J.J. Lee is supported by the NIH (T32CA009599).
Disclosures: A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology, and this financial relationship is managed and monitored by the UTMDACC Conflict of Interest Committee. A.M. is also listed as an inventor on a patent that has been licensed by Johns Hopkins University to Thrive Earlier Detection. YAJ’s spouse is employed by Invitae and owns company stock.
References
- 1.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 2.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16:207–20. [DOI] [PubMed] [Google Scholar]
- 3.Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu, Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 7.Tempero MA. NCCN Guidelines Updates: Pancreatic Cancer. J Natl Compr Canc Netw. 2019;17:603–5. [DOI] [PubMed] [Google Scholar]
- 8.Roy-Chowdhuri S, Chen H, Singh RR, Krishnamurthy S, Patel KP, Routbort MJ, et al. Concurrent fine needle aspirations and core needle biopsies: a comparative study of substrates for next-generation sequencing in solid organ malignancies. Mod Pathol. 2017;30:499–508. [DOI] [PubMed] [Google Scholar]
- 9.Gleeson FC, Kerr SE, Kipp BR, Voss JS, Minot DM, Tu ZJ, et al. Targeted next generation sequencing of endoscopic ultrasound acquired cytology from ampullary and pancreatic adenocarcinoma has the potential to aid patient stratification for optimal therapy selection. Oncotarget. 2016;7:54526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 11.Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res. 2019;25:2194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.San Lucas FA, Sivakumar S, Vattathil S, Fowler J, Vilar E, Scheet P. Rapid and powerful detection of subtle allelic imbalance from exome sequencing data with hapLOHseq. Bioinformatics. 2016;32:3015–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakubek Y, Lang W, Vattathil S, Garcia M, Xu L, Huang L, et al. Genomic Landscape Established by Allelic Imbalance in the Cancerization Field of a Normal Appearing Airway. Cancer Res. 2016;76:3676–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 2018;33:676–89.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 19.Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology. 2019;156:2242–53.e4. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018;8:1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohamadnejad M, Mullady D, Early DS, Collins B, Marshall C, Sams S, et al. Increasing Number of Passes Beyond 4 Does Not Increase Sensitivity of Detection of Pancreatic Malignancy by Endoscopic Ultrasound–Guided Fine-Needle Aspiration [Internet]. Clinical Gastroenterology and Hepatology. 2017. page 1071–8.e2. Available from: 10.1016/j.cgh.2016.12.018 [DOI] [PubMed] [Google Scholar]
- 22.Aung KL, Fischer SE, Denroche RE, Jang G-H, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res. 2018;24:1344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord J-P, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020;38:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W-C, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell. 2017;171:1042–56.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphris JL, Patch A-M, Nones K, Bailey PJ, Johns AL, McKay S, et al. Hypermutation In Pancreatic Cancer. Gastroenterology. 2017;152:68–74.e2. [DOI] [PubMed] [Google Scholar]
- 27.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22:3764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–62. [DOI] [PubMed] [Google Scholar]
- 30.Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife [Internet]. 2018;7 Available from: 10.7554/eLife.37294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laubert T, Freitag-Wolf S, Linnebacher M, König A, Vollmar B, Habermann JK, et al. Stage-specific frequency and prognostic significance of aneuploidy in patients with sporadic colorectal cancer--a meta-analysis and current overview. Int J Colorectal Dis. 2015;30:1015–28. [DOI] [PubMed] [Google Scholar]
- 32.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2020;21:44–62. [DOI] [PubMed] [Google Scholar]
- 33.Sveen A, Kopetz S, Lothe RA. Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat Rev Clin Oncol. 2020;17:11–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldstein JB, Zhao L, Wang X, Ghelman Y, Overman MJ, Javle MM, et al. Germline DNA Sequencing Reveals Novel Mutations Predictive of Overall Survival in a Cohort of Patients with Pancreatic Cancer [Internet]. Clinical Cancer Research. 2020. page 1385–94. Available from: 10.1158/1078-0432.ccr-19-0224 [DOI] [PubMed] [Google Scholar]
- 35.Staaf J, Glodzik D, Bosch A, Vallon-Christersson J, Reuterswärd C, Häkkinen J, et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study [Internet]. Nature Medicine. 2019. page 1526–33. Available from: 10.1038/s41591-019-0582-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shahda S, Timms KM, Ibrahim AA, Reid JE, Cramer HM, Radovich M, et al. Homologous Recombination Deficiency in Patients With Pancreatic Ductal Adenocarcinoma and Response to Chemotherapy [Internet]. JCO Precision Oncology. 2018. page 1–11. Available from: 10.1200/po.17.00087 [DOI] [PubMed] [Google Scholar]
- 38.Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation [Internet]. Cancer Research. 2012. page 5454–62. Available from: 10.1158/0008-5472.can-12-1470 [DOI] [PubMed] [Google Scholar]
- 39.Shastri A, Choudhary G, Teixeira M, Gordon-Mitchell S, Ramachandra N, Bernard L, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest. 2018;128:5479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pourmorteza M, Rahman ZU, Young M. Evofosfamide, a new horizon in the treatment of pancreatic cancer. Anticancer Drugs. 2016;27:723–5. [DOI] [PubMed] [Google Scholar]
- 41.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. Mutations and PD-1 Inhibitor Resistance in -Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2 [Internet]. New England Journal of Medicine. 2001. page 783–92. Available from: 10.1056/nejm200103153441101 [DOI] [PubMed] [Google Scholar]
- 45.Cleveland P, Gill KRS, Coe SG, Woodward TA, Raimondo M, Jamil L, et al. An evaluation of risk factors for inadequate cytology in EUS-guided FNA of pancreatic tumors and lymph nodes. Gastrointest Endosc. 2010;71:1194–9. [DOI] [PubMed] [Google Scholar]
- 46.Savides TJ, Donohue M, Hunt G, Al-Haddad M, Aslanian H, Ben-Menachem T, et al. EUS-guided FNA diagnostic yield of malignancy in solid pancreatic masses: a benchmark for quality performance measurement. Gastrointest Endosc. 2007;66:277–82. [DOI] [PubMed] [Google Scholar]
- 47.Erickson RA, Sayage-Rabie L, Beissner RS. Factors predicting the number of EUS-guided fine-needle passes for diagnosis of pancreatic malignancies. Gastrointest Endosc. 2000;51:184–90. [DOI] [PubMed] [Google Scholar]
- 48.Dreyer SB, Jamieson NB, Evers L, Duthie F, Cooke S, Marshall J, et al. Feasibility and clinical utility of endoscopic ultrasound guided biopsy of pancreatic cancer for next-generation molecular profiling. Chin Clin Oncol. 2019;8:16. [DOI] [PubMed] [Google Scholar]
- 49.Vestrup Rift C, Melchior LC, Kovacevic B, Toxvaerd A, Klausen P, Karstensen JG, et al. Next-generation sequencing of endoscopic ultrasound guided microbiopsies from pancreatic cystic neoplasms. Histopathology. 2019;75:767–71. [DOI] [PubMed] [Google Scholar]
- 50.Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin Cancer Res. 2015;21:2029–37. [DOI] [PubMed] [Google Scholar]
- 51.Varadarajulu S, Tamhane A, Eloubeidi MA. Yield of EUS-guided FNA of pancreatic masses in the presence or the absence of chronic pancreatitis [Internet]. Gastrointestinal Endoscopy. 2005. page 728–36. Available from: 10.1016/j.gie.2005.06.051 [DOI] [PubMed] [Google Scholar]
- 52.Lohse I, Mason J, Cao PM, Pintilie M, Bray M, Hedley DW. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget. 2017;8:3064–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vitale I, Manic G, Senovilla L, Kroemer G, Galluzzi L. Karyotypic Aberrations in Oncogenesis and Cancer Therapy. Trends Cancer Res. 2015;1:124–35. [DOI] [PubMed] [Google Scholar]
- 54.Golan T, Kindler HL, Park JO, Reni M, Mercade TM, Hammel P, et al. Geographic and ethnic heterogeneity in the BRCA1/2 pre-screening population for the randomized phase III POLO study of olaparib maintenance in metastatic pancreatic cancer (mPC) [Internet]. Journal of Clinical Oncology. 2018. page 4115–4115. Available from: 10.1200/jco.2018.36.15_suppl.4115 [DOI] [Google Scholar]
- 55.Trzpis M, McLaughlin PMJ, de Leij LMFH, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007;171:386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spizzo G, Fong D, Wurm M, Ensinger C, Obrist P, Hofer C, et al. EpCAM expression in primary tumour tissues and metastases: an immunohistochemical analysis. J Clin Pathol. BMJ Publishing Group; 2011;64:415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.