

Abstract
The novel corona virus disease or COVID-19 caused by a positive strand RNA virus (PRV) called SARS-CoV-2 is plaguing the entire planet as we conduct this study. In this study a multifaceted analysis was carried out employing dinucleotide signature, codon usage and codon context to compare and unravel the genomic as well as genic characteristics of the SARS-CoV-2 isolates and how they compare to other PRVs which represents some of the most pathogenic human viruses. The main emphasis of this study was to comprehend the codon biology of the SARS-CoV-2 in the backdrop of the other PRVs like Poliovirus, Japanese encephalitis virus, Hepatitis C virus, Norovirus, Rubella virus, Semliki Forest virus, Zika virus, Dengue virus, Human rhinoviruses and the Betacoronaviruses since codon usage pattern along with the nucleotide composition prevalent within the viral genome helps to understand the biology and evolution of viruses. Our results suggest discrete genomic dinucleotide signature within the PRVs. Some of the genes from the different SARS-CoV-2 isolates were also found to demonstrate heterogeneity in terms of their dinucleotide signature. The SARS-CoV-2 isolates also demonstrated a codon context trend characteristically dissimilar to the other PRVs. The findings of this study are expected to contribute to the developing global knowledge base in countering COVID-19.
Abbreviations: PRV, Positive strand RNA Virus; SARS-CoV-2, Severe Acute Respiratory Syndrome Coronavirus 2; SARS, Severe Acute Respiratory Syndrome; MERS, Middle East Respiratory Syndrome; HCV, Hepatitis C Virus; CRS, Congenital Rubella Syndrome; CNS, Central Nervous System; PCA, Principal Component Analysis; Nc, Effective Number of Codons; GC1, Guanine and Cytosine content on the first position of the codon; GC2, Guanine and Cytosine content on the second position of the codon; GC3, Guanine and Cytosine content on the third position of the codon; RSCU, Relative Synonymous Codon Usage; CAI, Codon Adaptation Index; SCUO, Synonymous Codon Usage Order; Fop, Frequency of optimal codons; CUB, Codon Usage Bias; RCDI, Relative Codon De-Optimization Index; SiD, Similarity Index; MFE, Minimum Free Energy
Keywords: SARS-CoV-2, Codon usage bias, Codon context, Positive strand RNA virus, Coronaviruses, COVID-19
1. Introduction
The single strand RNA viruses with positive polarity (PRVs) are a group of viruses which are particularly infamous for their high degree of infectivity and incorporates more than one-third of all virus genera (Ahlquist et al., 2003). The viruses within these group include the Poliovirus, Dengue virus, Zika virus, Japanese encephalitis virus, Hepatitis C virus, Norovirus, Rubella virus, Coronavirus, and others. The most recent addition to this list is the novel Severe acute respiratory syndrome related coronavirus 2 (SARS-CoV-2).
This study was designed to make a comparative analysis of the dinucleotide signature and codon biology of the PRVs with special emphasis on the most recently discovered novel SARS-CoV-2 strain, since from the genomic view point, codon usage pattern plays a vital role in deciphering the basic biological and evolutionary processes (Wang and Chen, 2013). The study of codon usage bias in viruses have been carried out to understand host adaptation of the virus, host-pathogen interaction, host immune system evasion (Khandia et al., 2019; Butt et al., 2016; Castells et al., 2017; Pinto et al., 2018; Mortazavi et al., 2016; Karumathil et al., 2018) and translational kinetics (Karumathil et al., 2018; Deka et al., 2019). Codon usage bias study also provides a firm understanding of the evolutionary history of virus, their phylogeny, selection pressure (Hanson et al., 2018; Yao et al., 2019; Gu et al., 2019a), and provides vital insight for developing medications, and new treatment regimes (Castells et al., 2017; Mortazavi et al., 2016).
The PRVs include some of the most pathogenic viruses, and PRVs like Dengue virus, Zika virus and Middle East respiratory syndrome-related coronavirus pose a serious threat to public health (Brechot et al., 2019; Nelemans and Kikkert, 2019). PRVs either rapidly infect new host or develop mechanisms to tackle host defence machineries (Hilleman, 2004; Beachboard and Horner, 2016; García-Sastre, 2017). These viruses are also thought to hide viral replication intermediates in intercellular replication factories to escape from host defence pathway (Harak and Lohmann, 2015; Overby et al., 2010). Extensive studies have unravelled the fact that complete ability to escape from these defence mechanisms may be one of the potent reasons that helps PRVs like Dengue, Hepatitis C, Zika, and some coronaviruses to cause disease outbreak in humans (Chen et al., 2017; Uno and Ross, 2018; Gokhale et al., 2014; Kindler et al., 2016; Pardy et al., 2019). The single strand RNA with positive polarity in both Middle East respiratory syndrome-related coronavirus and Severe acute respiratory syndrome-related coronaviruses encode structural proteins like membrane protein (M), envelope (E), nucleocapsid (N), non-structural polyproteins and spike (S) protein which along with glycoprotein and enzymes controls severity in disease exaggeration (Subbaram et al., 2017; Phan, 2020; Ortega et al., 2020).
The PRVs included in this study along with SARS-CoV-2 are very potent human pathogens. These include the Poliovirus, Japanese encephalitis virus, Hepatitis C virus, Norovirus, Rubella virus, Semliki Forest virus, Zika virus, Dengue virus, Human rhinoviruses and the Betacoronaviruses from sub genera Embecovirus, Hibecovirus, Merbecovirus, Nobecovirus and Sarbecovirus. Of all the known RNA viruses, coronaviruses possess the largest genomes (Woo et al., 2009; Almazán et al., 2014; Ruan et al., 2003), and its members are spread across the phylum Riboviria within the different genera of the family Coronaviridae under the order Nidovirales (Almazán et al., 2014; Corman et al., 2018; Gorbalenya et al., 2020; Cui et al., 2019). Most of the Betacoronaviruses included in this study are zoonotic in origin out of which three have at some point of time jumped into humans (Rehman et al., 2020; Menachery et al., 2020; Woo et al., 2006). The Betacoronaviruses that have jumped into humans cause severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS) and COVID-19. These viruses are members of the sub-genera Sarbecovirus and Merbecovirus respectively which causes illness linked to the respiratory tract and in extreme cases death due to pneumonia and even multi organ failures (Zaki et al., 2012; Assiri et al., 2013; Farcas et al., 2005; Huang et al., 2005; Yin et al., 2004; Napoli, n.d.).
Among the other PRVs included in this study, Poliovirus, the causative agent of the communicable disease poliomyelitis (Kitamura et al., 1981; Xu et al., 2019) is a prominent member of Picornaviridae family with a RNA genome of approximately 7500 nucleotides, covalently linked to virus-coded VPg at 5′ end and a poly(A) tail at the 3′ end (Yogo and Wimmer, 1972; Flanegan et al., 1977; Lee et al., 1977; Racaniello and Baltimore, 1981). Another PRV of approximately 7200 bp, included in this study are the human rhinoviruses of the Picornaviridae family. Some of the most significant health concerns arise due to PRVs from the family Flaviviridae and we have included these in our study. The flavivirus Japanese encephalitis virus is responsible for the mosquito borne zoonotic viral disease Japanese encephalitis and it is the most important causative agent behind epidemic viral encephalitis in the Southeast Asian and Western Pacific regions, China, and the Indian subcontinent (Li et al., 2019; Cherian and Walimbe, 2015). Zika virus is an arbovirus from the family Flaviviridae primarily transmitted by Aedes mosquito ( Musso and Gubler, 2016 ; Plourde and Bloch, 2016 ) causing disease with commonly reported symptoms including rash, fever, arthralgia, myalgia, fatigue, headache, and conjunctivitis. Infection by Zika virus in human cortical neural progenitor cells results into stunted cell growth and transcriptional dysregulation (Tang et al., 2016), neonatal microcephaly (Oliveira Melo et al., 2016). Dengue virus is another mosquito borne Flavivirus transmitted by Aedes aegypti and Aedes albopictus. Although primary infection with Dengue virus may include rash and fever, but many infections are asymptomatic, while secondary infection causes severe complications with mortality rate up to 20% (Uno and Ross, 2018; Guzman and Harris, 2015). Another PRV of the Flaviviridae family, the Hepatitis C virus targets human hepatocytes (Zeisel et al., 2013; Chan and Ou, 2017), and leads to severe liver diseases including cirrhosis and hepatocellular carcinoma. The Hepatitis A virus is another important liver affecting virus which is highly contagious and belongs to the genus Hepatovirus of the family Picornaviridae ( McKnight and Lemon, 2018 ). The Caliciviridae is represented by Norovirus, infamous for causing acute infectious gastroenteritis with symptoms including rapid onset, abundant vomiting or diarrhoeal disease (Furuta et al., 2003; Maunula et al., 2012; Anttila et al., 2010). The human pathogen Rubella virus of the genus Rubivirus of family Togaviridae ( Kanbayashi et al., 2018 ; Mangala Prasad and Klose, 2017 ) which is responsible for foetal death or congenital rubella syndrome (CRS) in pregnant women has also been included in this study. Semliki Forest virus, a PRV of the genus Alphavirus from Togaviridae family is reported to cause lethal encephalitis by infection of the central nervous system (CNS) (Atkins et al., 1999).
Now-a-days in the era of next generation sequencing, the rapid progress and robustness in sequence analysis techniques have provided an opportunity to compare genomic sequences on a large scale to unravel many hidden facets of the genome and its evolution. Simultaneously, this enriches the field of comparative genomics-based study to comprehend the pathogenic viruses to a large extent. A slight change in the codon and codon pair usage pattern may influence viral pathogenicity on a massive scale and hence these studies may provide critical pointer in the development of proper vaccine or therapy against the virus (Alexaki et al., 2019; Baker et al., 2015). Moreover, synonymous codon usage study may also unveil information regarding genic evolution and assist to detect the horizontal gene transfer events (RoyChoudhury and Mukherjee, 2010). Furthermore, translational kinetics can be better understood, and protein structure may be predicted through such codon usage data analysis (Athey et al., 2017; Cheng et al., 2017). Although codon usage analysis of SARS-CoV-2 have been conducted in some isolates (Kandeel et al., 2020; Tort et al., 2020), but till now no study is available where the SARS-CoV-2 and other coronaviruses have been compared with other PRVs to find out how they relate to each other in terms of their genomic codon usage affinities. We have included the whole genomes of many SARS-CoV-2 isolates from different regions of the world available in the publicly available sequence databases to find out if there exist any dissimilarities within the codon and codon pair usage pattern among the different SARS-CoV-2 isolates both on the genomic scale as well as gene wise, and how these isolates relate in terms of dinucleotide signature, codon usage and codon pair usage with respect to the other PRVs. We have emphasised on the genomic signatures of all the viral genomes with reference to their dinucleotide composition, codon usage bias, amino acid usage trend and codon context pattern. We have also tried to comprehend the codon usage pattern of all the viruses included in this study by comparing them alongside with highly expressed human genes such as those encoding the human ribosomal proteins. We anticipate the findings of this study will be useful in understanding the biology of SARS-CoV-2 virus to a certain extent and contribute towards the knowledge base that is being developed to counter SARS-CoV-2.
2. Materials and methods
2.1. Retrieval of whole genome sequence data
The completed whole genome sequences of SARS-CoV-2 and 49 other PRV genomes (primarily reference genomes) were downloaded from GenBank (Benson et al., 2013) and NCBI Virus, a virus variation resource (Hatcher et al., 2017). Only complete genomes were included in the study, and those with incomplete sequencing status along with improper annotation, incomplete data regarding location and date of collection were discarded from the study. The different ORFs and coding sequences comprising the genome were also obtained from GenBank and NCBI Virus and the annotation data related to these were thoroughly scrutinised. The presence of additional meta data available with the whole genome data were also utilized for sorting the different ORFs and coding sequences of the different PRVs included in this study. Detailed information regarding the accession number and other features of the viral genomes is given in Supplementary Table 1.
2.2. Dinucleotide frequency abundance and representation
The dinucleotide frequency or dinucleotide abundance in every genome bears a great significance in determining genome pattern and evolution which is specific for each organism. This can be regarded as a genomic signature (Prabha and Singh, 2014) since it varies little despite the diversity between species (Jernigan and Baran, 2002). Dinucleotide frequencies of all the possible 16 dinucleotide combinations were computed for all the genomes included in the study to determine whether there is a preference for specific dinucleotide pairs (Pandit et al., 2013). The statistical over- and underrepresentation of all the 16 dinucleotide combinations was computed for all the PRV genomes as well as the individual coding sequences of the SARS-CoV-2 isolates and compared utilizing a z-score statistic with a base model for the whole genomes and coding sequences, and a codon model and syncodon model for all the individual coding sequences (Palmeira et al., 2006; Gautier et al., 1985; Karlin and Cardon, 1994). The R package ‘seqinr’ was used to perform this analysis. The statistical over- and underrepresentation of dinucleotides in a sequence was further subjected to a multivariate data analysis technique called principal component analysis (PCA) to differentiate the PRV genomes based on their dinucleotide representation and detect a genomic signature both within the genomes and the coding sequences of SARS-CoV-2. The multivariate data analysis was carried out using the R package ‘factoextra’ and ‘FactoMineR’. PCA plots were constructed using the R package ‘ggplot2’.
2.3. Codon usage pattern analysis
To comprehend the codon usage pattern prevailing within the virus genomes, the parameters like effective number of codons (Nc) (Wright, 1990), guanine and cytosine content on the first, second and third position of the codon (GC1, GC2 and GC3 respectively) (Wright, 1990), relative synonymous codon usage (RSCU), gene length, hydrophobicity (Kyte and Doolittle, 1982), codon adaptation index (CAI) (Sharp and Li, 1987), synonymous codon usage order (SCUO) (Wan et al., 2004), and frequency of optimal codons (Fop) (Ikemura, 1985; Xu et al., 2013) were calculated using INCA 2.1 (Supek and Vlahovicek, 2004), CodonW (Peden, 1999), in house developed Perl scripts and the R packages- ‘coRdon’ and ‘seqinr’.
The Nc is considered as one of the best quantitative measure to evaluate the degree of bias for the usage of codons. The Nc value ranges from 20 to 61 where lower value indicates higher codon usage bias while higher value denotes reduced codon usage bias (Wright, 1990). In the course of genome evolution, GC content on the third base of codon along with GC1 and GC2 has been reported as a key component in regulating gene expression (Genereux, 2002), while hydrophobicity actually predicts the nature of cellular protein encoded by the gene present within the genome (Saha et al., 2019). RSCU measures the non-uniform usage of synonymous codons in a coding sequence and represents the number of times a codon is used in comparison to the number of times that codon would be observed in case of uniform usage. CAI can also be envisaged as another effective measure for calculating the expression levels of gene sequences (Sharp and Li, 1987; Prabha et al., 2017). This is an important index for CUB analysis, with values ranging from 0 to 1. This is a well-accepted parameter for estimating the relative adaptation of codon usage of a gene towards the codon usage of highly expressed genes (Ayon et al., 2014). CAI was calculated following an improved implementation (Xia, 2007). The human ribosomal protein coding genes were used as reference for measuring CAI. CAI is of great importance for estimating translational efficiency and predicting cellular protein levels (Baha et al., 2019). SCUO ranges from 0 to 1, and represents the synonymous codon usage bias (Wan et al., 2004) which is mainly based on Shannon Information theory explaining entropy of codon sequences of genes (Behura and Severson, 2012). Larger value denotes a higher codon usage bias with less entropy. For comparative CUB analysis, this parameter is being widely applied (Prabha et al., 2017). Fop encodes the proportion of optimal codons accounting for all synonymous codons. The value varies from 0 to 1 (Ikemura, 1985). A gene with no optimal codon possess a Fop value of ‘0’ whereas the gene comprising of a significant number of optimal codons possess the value ‘1’ (Nakamura and Ikemura, 1995).
To further explore the codon usage bias in each of the strain and to depict the correlation between Nc and GC3, Nc plot was constructed for each of the gene present within the viral genome. Such a plot is generally used to elucidate the mechanistic forces influencing CUB (Pal et al., 2019). The neutrality plot explaining the interrelationship between different GC attributes like GC1, GC2, GC3 content of the prevailing genes within the genome was also utilized (Xiang et al., 2015). To decipher the genic organization within the viral genome and to investigate the mutation-selection equilibrium in shaping codon usage bias, this serves as an important tool. A plot regression with a slope of 1 indicates complete neutrality while a slope of 0 indicates no effect of directional mutation pressure (Kumar et al., 2016). The GC content in the first two codon position of each genic sequence was plotted against the respective GC3 content and regression values were estimated (Franzo et al., 2018).
2.4. Relative codon de-optimization index (RCDI)
To understand host-virus relationship this index has been proved to be useful as virus evolution occurs along with its host (Puigbò et al., 2010). This can be defined as a measure of codon usage deoptimization by comparing the codon usage of a gene with that of the reference genome. This value indicates the cumulative effects of codon biases on the expression of gene (D'Andrea et al., 2019). The concept of RCDI (Mueller et al., 2006), also explains the rate of viral gene translation where RCDI value closest to 1 indicates higher adaptation of a virus with its host. Similarly, it also predicts higher translation rate within the host genome (Khandia et al., 2019; Mueller et al., 2006).
2.5. Similarity index (SiD)
The similarity index value (Zhou et al., 2013) is a codon usage parameter which evaluates the host potentiality and is often used to decipher the influence of codon usage bias on host genome (Zhou et al., 2019; Cao et al., 2018). SiD is also suggested to be helpful in obtaining a more accurate assessment of adaptation between the virus and its host (Silverj and Rota-Stabelli, 2020). The value ranges from 0 to 1 representing the effect of codon usage on host, and a higher value is suggestive of low codon usage similarity (Silverj and Rota-Stabelli, 2020).
2.6. Minimum free energy (MFE) calculation of mRNA structure
Calculating the minimum free energy released by the mRNA secondary structure during the transcription process, has a great significance in assessing the mRNA stability. The value may range from negative to positive (Ringnér and Krogh, 2005). Higher value indicates comparatively greater loss of energy by the mRNA molecule sustaining a stable mRNA conformation (Deb and Uddin, 2020). The MFE was calculated using Quikfold hosted at DINAMelt Web Server (http://unafold.rna.albany.edu/?q=DINAMelt/Quickfold) (Markham and Zuker, 2005).
2.7. Codon pair analysis and codon context study
This represents the basic preliminary feature of a gene which influence the mRNA decoding fidelity (Moura et al., 2007). Codon pair utilization bias present in each of the different organisms can be better explored through this parameter to infer on phylogeny (Moura et al., 2011). Codon context analysis was carried out using the software ANACONDA 2 to generate the frequency table of codon pair context (Baha et al., 2019), and followed by subsequent statistical analysis, to identify preferred set of codon pairs in a coding sequence (Pal et al., 2015; Nasrullah et al., 2015). Along with codon context analysis, the codon pair ratio or CPR was also calculated for each viral genome to find out how many codon pairs have been utilized in coding the different coding sequences relative to the number of codon pairs combinations that are feasible (Pal et al., 2015).
3. Results and discussion
In RNA viruses, mono and dinucleotide frequencies in genomes or mRNAs may significantly fluctuate (Belalov and Lukashev, 2013). To capture the variation in AU/GC content of all the PRV genomes included in this study (data given in Supplementary Table 2), individual Mann-Whitney Rank Sum Tests utilizing the respective AU and GC content of the viruses was carried out after normality check failed using Shapiro-Wilk test (p < 0.05). The results demonstrated that the difference in the median values between the AU and GC is greater than would be expected by chance, suggesting a significant difference in AU as well as GC content between SARS-CoV-2 and the other PRVs (U = 1024.000; p < 0.01). The SARS-CoV-1 virus demonstrated an average 2.78% less AU content than the SARS-CoV-2 isolates. The highest AU content (67.94%) among all the PRVs considered in this study was observed in Human coronavirus HKU1 (NC_006577), whereas the highest GC content (69.60%) was observed in Rubella virus (NC_001545.2). The average AU and GC content of the SARS-CoV-2 isolates was found to be about 62% and 38% respectively. A graph depicting the relative fluctuations in AU and GC content among the PRVs is given in Fig. 1, and it suggests that in these viruses there is a strict positive correlation between AU and genome size (r = 0.72, p < 0.01) where larger genomes are dominated by AU nucleotides.
Fig. 1.

A parallel plot depicting the relation between genome size and AU/GC content of the positive strand RNA viruses (PRVs) included in this study.
3.1. Genomic and genic dinucleotide signature analysis
A genome wide comparison of the relative under and overrepresentation of the different dinucleotide frequencies of the SARS-CoV-2 isolates and other PRVs was performed (Supplementary Table 3). The variation in dinucleotide composition in all the viruses was studied to find out if there is any bias towards specific dinucleotides which may have arisen due to host specific mutational pressure or inherent features of the virus. It was observed that in all the SARS-CoV-2 isolates there is a significant underrepresentation of CpG dinucleotides which is similar to other RNA viruses like the Influenza A virus (Gu et al., 2019b). This trend was also evident among the other PRVs included in this study except for Rubella virus which did not demonstrate CpG underrepresentation. Among the SARS-CoV-2 isolates we observed heterogeneity within the distribution of the CpG dinucleotides within the different genomes. The SARS-CoV-2 strains MT246488, MT159713, MT159717, MT246449 and MT066156 demonstrated substantial underrepresentation of CpG dinucleotides in their genome, whereas the isolates from MT020881, MT152824, MT246458, MT246472, MT246478, MT118835, and the isolates from China demonstrated a relatively lesser underrepresentation of the CpG dinucleotides. The AU dinucleotide was also found to be underrepresented along with UA, UC, GG and GA. The dinucleotides AU, UA and UC were also found to be underrepresented in all the other RNA viruses included in this study.
To determine the dinucleotide genomic signature of the viral genomes we designed and executed many PCAs utilizing the relative over and underrepresentation of the 16 different dinucleotide combinations. A genome wise PCA utilizing the dinucleotide representation of all the PRV genomes considered in this study demonstrated the first two dimensions to contribute towards 34% and 20% of the total variance which was the highest among all the dimensions. Analysis of the contributions of the different dinucleotide combinations to the first and second dimensions clearly demonstrated that most of the G containing dinucleotide combinations like GU, GG, GA and GC play a major role in differentiating the virus genomes (Supplementary Fig. 1). A bi-plot depicting the individual virus genomes and the different dinucleotide combinations (Fig. 2) shows that nearly all the SARS-CoV-2 genomes cluster together on the bi-plot except for a few one and segregate themselves quite prolifically from the rest of the other PRVs included in this study. The other PRVs represented by the genomes of Hepatitis A virus (NC_001489), Zika virus (NC_035889), Dengue virus (NC_001474), Semliki forest virus (NC_003215), Hepatitis C virus (NC_004102), Human rhinovirus (NC_038311), and Rubella virus (NC_001545) were found to occupy discrete locations on the left-hand side of the bi-plot, which is opposite to that of the SARS-CoV-2 genomes suggesting difference in genomic dinucleotide signature. To understand the uniqueness within the genomic dinucleotide signatures of the SARS-CoV-2 genomes, a PCA was carried out utilizing the 16 dinucleotide combinations and grouping the virus genomes based on their geographical location. The PCA bi-plot depicted in Fig. 3 shows that the first two dimensions contributes 16% and 11% of the total variance which is highest among all the dimensions. The dinucleotide combinations UA and UC was found to contribute the greatest in both the dimensions. To further comprehend the trend and uniqueness in the dinucleotide signatures within the genome of the SARS-CoV-2 isolates, a gene wise PCA analysis was carried out, since the sum of genes represents the genome in its near entirety in case of the PRVs. The statistical dinucleotide over- and underrepresentation of all the individual genes or coding sequences constituting the genome of SARS-CoV-2 were obtained based on the base model, codon model and syncodon model (Palmeira et al., 2006; Gautier et al., 1985; Karlin and Cardon, 1994). The data obtained (Supplementary Table 4) based on these models were analysed utilizing three separate PCAs. The PCA performed on the base model data (Supplementary Fig. 2) suggests that most of the SARS-CoV-2 genes has a characteristic dinucleotide signature. In this case, the first two dimensions accounts for 56.15% and 16.57% of the total variance with the dinucleotide combinations of UA, UC, AU and AG contributing the maximum to both the dimensions. These include the genes S, N, orf6, E, orf1ab and orf7b. The orf7a, orf3a and M clusters tightly together at the centre of the bi-plot which suggests that they share similar dinucleotide signature. The orf8 and orf10 also demonstrated some amount of overlap in terms of their genic dinucleotide signature. The PCA on codon model demonstrated a similar trend, except for orf1ab which was found to be segregated into two different clusters, one being shaped by GU and AC whereas the other is influenced by UG. In depth analysis of the two clusters clearly demonstrated geographical separation where the isolates from South and South-East Asia such as Japan, India, South Korea, Taiwan and Vietnam were found to share the same cluster along with that of Sweden, Australia, Italy and Brazil (Supplementary Fig. 3). The orf1ab from the isolates of Spain were found to be present in a separate cluster along with many of the orf1ab coding sequences isolated from USA. The orf1ab coding sequences from different provinces of China along with USA was found to intermingle in both the clusters, but those isolated from Wuhan province of China was found to cluster along with those from Asia suggesting similar dinucleotide signature. PCA analysis based on the syncodon model data that allows for random sequence generation through synonymous codon shuffling (Supplementary Fig. 4) also demonstrated similar trend for orf1ab. Based on the syncodon model data the dinucleotide signature of orf8, orf10, orf7a, orf7b and orf6 was found to be mostly influenced by CG, UA and UC.
Supplementary Fig. 1.

A bar plot depicting the contributions (in percentage) of the different dinucleotide combinations in the first and second dimension of the PCA showing the segregation of the positive strand RNA viruses (PRVs) based on the over and underrepresentation of the 16 dinucleotide combinations.
Fig. 2.

A PCA bi-plot showing the segregation of the positive strand RNA viruses (PRVs) based on the over and underrepresentation of the 16 dinucleotide combinations. The x-axis represents the 1st dimension which accounts for 33.9% of the total variance and the y-axis represents the 2nd dimension accounting for 20.2% of the total variance.
Fig. 3.

A PCA bi-plot showing the segregation of the SARS-CoV-2 isolates based on the over and underrepresentation of the 16 dinucleotide combinations. The x-axis represents the 1st dimension which accounts for 16% of the total variance and the y-axis represents the 2nd dimension accounting for 11.1% of the total variance.
Supplementary Fig. 2.

A PCA bi-plot showing the segregation of the different genes and ORFs of the SARS-CoV-2 isolates based on the base model data of over and underrepresentation of the different dinucleotide combinations. The different dinucleotide combinations contributing majorly to the variation in the first two dimensions of the plot is shown in the plot background.
Supplementary Fig. 3.

A PCA plot of the different genes and ORFs of the SARS-CoV-2 isolates based on the codon model data of over and underrepresentation of the different dinucleotide combinations demonstrating segregation of the ORFs based on geographical location.
Supplementary Fig. 4.

A PCA bi-plot showing the segregation of the different genes and ORFs of the SARS-CoV-2 isolates based on the syncodon model data of over and underrepresentation of the different dinucleotide combinations. The different dinucleotide combinations contributing majorly to the variation in the first two dimensions of the plot is shown in the plot background.
3.2. Analysis of codon usage pattern of the PRVs
The results of the genomic codon usage (NcGenome) pattern study (Supplementary Table 5) of the PRVs demonstrated that Rubella virus (NcGenome = 38.41) and Hepatitis A virus (NcGenome = 39.61) has the most codon biased genome, whereas Poliovirus (NcGenome = 53.60), Zika virus (NcGenome = 54.03), Norovirus (NcGenome = 54.67), Semliki forest virus (NcGenome = 55.66), Japanese encephalitis virus (NcGenome = 55.96) and Hepatitis C virus (NcGenome = 56.16) has the least biased genome in terms of codon usage. Among the coronaviruses, Rousettus bat coronavirus, a member of sub-genus Nobecovirus was found to demonstrate the least genomic codon bias (NcGenome = 55.14), whereas Human coronavirus HKU1 (NcGenome = 40.56), a member of sub-genus Embecovirus demonstrated the highest codon bias. The genomic codon bias of the SARS-CoV-2 isolates was found to range between 46.61 and 49.0 with the isolate MT049951 from China and MT233519 from Spain demonstrating the highest and lowest codon bias, respectively. This suggests that the SARS-CoV-2 along with the other coronaviruses has lower genomic codon bias, since an Nc value greater than 40 indicates low codon bias (Xu et al., 2013; Messier and Stewart, 1997). But when compared to the remaining PRVs except Rubella virus and Hepatitis A virus, the genomic codon bias is relatively higher.
The effect of GC3 on the codon usage pattern of the different viruses considered in this study was further explored with the help of a Nc plot. An Nc plot depicting the association between Nc and GC3 of different genes of the PRVs is shown in Fig. 4 which suggests that relative to the human ribosomal protein genes (which are generally highly expressed) (Lin et al., 2002; Luo et al., 2016) the positioning of the SARS-CoV-2 genes on the Nc plot is quite discrete. Nearly all the SARS-CoV-2 representatives on the Nc plot showed similar clustering pattern. Relative to SARS-CoV-1, the SARS-CoV-2 isolates demonstrated a different clustering pattern. A detailed study of Nc plot clearly shows that there is little overlap in the codon usage pattern among the coronaviruses, except for Middle East respiratory syndrome-related virus which resembles the aggregation pattern of SARS-CoV-2 on the Nc plot to a large extent. The PRVs (excluding all the different coronaviruses) included in this study demonstrated a Nc-GC3 relationship which was found to be significantly different from all the Betacoronaviruses but resembled that of the human ribosomal genes to a greater extent.
Fig. 4.

A Nc-plot depicting the relationship between GC content at the 3rd position of codon or GC3 (x-axis) with effective number of codons or Nc (y-axis) of all the genes present in the genomes of the positive strand RNA viruses (PRVs) included in this study. The dashed blue line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
3.2.1. Intra and inter genic codon usage analysis of different SARS-CoV-2 genomes
The codon usage pattern of all the genes constituting the SARS-CoV-2 genomes were analysed intricately (Supplementary Table 6). The codon bias of the genes coding for the structural components of the virus demonstrated that the structural component coding genes have low codon bias with gene E being the least biased one (Nc = 61) followed by gene M, N and S (Nc ~ 44.1). A similar trend was also observed for the other ORFs. A correlation of Nc with sequence length demonstrated a significant negative correlation in the case of orf1ab (ρ = −0.672, p < 0.01). This suggests that codon bias is positively correlated with length in these ORFs. The correlation between Nc and GC1 demonstrated a significant negative correlation in the case of orf1ab (ρ = −0.737, p < 0.01). Negative correlation was also observed between Nc and GC2 of orf1ab (ρ = −0.63, p < 0.01), S (ρ = −0.42, p < 0.01). A positive correlation was observed in case of orf8 (ρ = −0.869, p < 0.01) which is entirely contrary to the prevalent trend. In terms of association between Nc and GC3, significant positive correlation was observed in most of the cases for orf3a and orf8.
The relationship between Nc and GC3 was further explored to better understand the role of GC3 in determining the codon usage bias trend in all the 12 coding sequences constituting the genome of SARS-CoV-2. All the ORFs except orf6 and orf1ab were found to cluster together in proximity on the Nc plot shown in Fig. 5 . Apart from gene E and orf10, the other genes were found to lie below the null hypothesis curve suggesting the mechanistic effect of translational selection as a determinant of codon bias. The coding sequences of gene E was found to cluster tightly and lie above the null hypothesis curve along with orf10 indicating the presence of mutational pressure.
Fig. 5.

A Nc-plot depicting the relationship between GC content at the 3rd position of codon or GC3 (x-axis) with effective number of codons or Nc (y-axis) of all the genes present in the genomes of the SARS-CoV-2 isolates included in this study. The dashed blue line represents the null hypothesis curve which suggests that codon usage bias is solely due to mutation and not selection (Wright, 1990).
To have a better insight on the effect of GC3 on codon usage bias, the relation between GC3 and SCUO was analysed (Fig. 6 ), since GC3 has been found to depict a quantitative relationship with SCUO in microbial and archaeal genomes, and in some mammals (Wan et al., 2004; Pal et al., 2015; Zeeberg, 2002). Although a linear relationship between GC3 and SCUO was evident in case of orf1ab (R 2 = 1.0, F = 14,398,631.11) and orf7a (R 2 = 0.80, F = 482.56), such a trend was absent in most of the other coding sequences. In case of orf6 (R 2 = 0.99, F = 6232.84) and orf10 (R 2 = 1.0, F = 5.535E+14), a quadratic equation was found to best describe the association between GC3 and SCUO. A linear relationship was also found to exist in case of gene S (R 2 = 0.5, F = 124.85) and N (R 2 = 0.42, F = 88.02). No distinct association was found to exist between GC3 and SCUO of orf8, and M.
Fig. 6.

Plots depicting the relationship between GC content at the 3rd position of codon or GC3 (x-axis) and synonymous codon usage order or SCUO (y-axis) for the genes N, S and orf1ab, orf6, orf7 and orf10 of the SARS-CoV-2 isolates.
Analysis of neutrality plots depicting the relationship between GC1/GC2 with GC3 of the genes constituting the SARS-CoV-2 genome (Fig. 7) demonstrated a linear relationship in the case of orf1ab and orf1a. No such prominent relationship was found to exist in the case of the other genes (Supplementary Table 7). In a neutrality plot, a slope of 1 indicates complete neutrality while a slope of 0 indicates no effect of directional mutation pressure (Kumar et al., 2016).
Fig. 7.

Neutrality plot showing the relationship between GC1/GC2 with GC3 for the different genes of the SARS-CoV-2 genome.
The CAI which is a measure of the potential expression of genes was found to be relatively low for the genes N, M, E and orf10 whereas the gene coding for the surface glycoprotein and other non-structural genes had higher CAI values. To find out the effect of codon usage bias on gene expression, Spearman rank correlation was performed between CAI and the other codon usage bias determining parameters. Our results demonstrated significant negative correlation between Nc and CAI for most of the sequences such as orf7a, orf7b, orf1a, M and N. On the other hand, an inverse trend was observed in the case of orf1ab, orf6, orf8, orf3a and S. In terms of association, CAI was found to be significantly positively correlated for majority of the coding sequences like orf7a (ρ = 0.819; p ≤0.01), orf6 and E (ρ = 0.77; p ≤0.01), orf3a (ρ = 0.656; p ≤0.01) and orf7b (ρ = 0.61; p ≤0.01). Significant negative correlation was observed in case of orf1ab (ρ = −0.62; p ≤0.01) and M (ρ = −0.70; p < 0.01). In our study with the SARS-CoV-2 genomes, Fop was found to correlate the best with CAI. Apart from orf1ab and orf1a, Fop was found to correlate significantly in a positive manner with CAI for gene S, orf8, E, orf6, orf3a, etc. The only significant negative association between Fop and CAI was evident in case of orf1ab (ρ = −0.66; p < 0.01). A significant positive correlation was observed between CAI and hydrophobicity in orf7a, orf1ab, S, orf3a and N while the rest of the sequences were found to be negatively correlated. The orf10 and orf1a did not depict any statistically significant correlation. The GC content at different codon positions like GC1, GC2 and GC3 was found to correlate significantly with CAI in orf1ab whereas in rest of the coding sequences there was no substantial association between these parameters suggesting GC1, GC2 and GC3 do not play a significant role in determining the potential expression levels of the coding sequences.
3.2.2. Association between dinucleotide frequency and codon usage pattern
The correlation between CAI and dinucleotide abundance was performed to detect the effect of the latter on the potential expression level of a gene. The dinucleotide combinations UA (ρ = −0.266; p = 0.003), UC (ρ = −0.266; p = 0.003), UU (ρ = −0.240; p = 0.007), CC (ρ = −0.217; p = 0.016) and GC (ρ = −0.216; p = 0.017) was found to be negatively associated with CAI whereas CG and GC was found to positively influence the CAI levels. This suggests that the abundance of certain dinucleotide combinations within the coding sequences are responsible to a certain extent in determining the codon usage pattern that in turn influences gene expression.
3.2.3. Codon usage bias and mRNA minimum free energy (MFE)
The mRNA MFE is an indicator of stability of the mRNA molecule and plays an important role in translation and product formation. Our results suggest that on a gene-to-gene basis in the SARS-CoV-2 isolates, the genic GC content has a direct bearing on the stability of the mRNA positively in case of gene M (ρ = 0.81, p < 0.01) and orf8 CDS (ρ = 0.36, p < 0.01), whereas in orf3a (ρ = −0.90, p < 0.01) and gene E (ρ = −0.70, p < 0.01) an anti-correlation was observed. The other coding sequences did not show any relation with GC content in terms of mRNA stability. The mRNA MFE was also not found to correlate strongly with CAI in about all the coding sequences suggesting that the potential expression level of the viral mRNAs is not positively impacted by mRNA stability. In fact, in gene M, a feeble negative correlation (ρ = −0.20, p < 0.01) was even evident. When examined in conjunction with the Fop score, which is an indicator of the optimization level of synonymous codon choice of each gene to translation process, we find that the Fop values of most of the SARS-CoV-2 genes are low. Since the presence of optimal codons positively impact the translation elongation rate, which in turn increases mRNA stability (Hanson et al., 2018; Wu et al., 2019), the anti-correlation between CAI and MFE in the SARS-CoV-2 genes is quite expected. On the other hand, the RCDI was found to be the best descriptor of mRNA MFE in about all the coding sequences except orf6, orf7a and orf10. For orf3a (ρ = 0.34, p < 0.01), orf8 (ρ = 0.32, p < 0.01) and E (ρ = 0.58, p < 0.01) a significant positive correlation was detected whereas the structural genes S (ρ = −0.31, p < 0.01) and M (ρ = −0.81, p < 0.01) depicted a negative correlation. Except for orf8, the Nc (ρ = 0.44, p < 0.01) was not found to correlate significantly with MFE, but the SCUO was found to dictate codon usage bias to some extent in orf3a (ρ = 0.52, p < 0.01), orf8 (ρ = −0.42, p < 0.01), gene S (ρ = −0.30, p < 0.01) and M (ρ = −0.24, p < 0.01).
3.3. Analysis of RSCU
3.3.1. RSCU analysis of SARS-CoV-2 genes
The analysis of RSCU data (Supplementary Table 8) of the genes coding for the structural proteins of the SARS-CoV-2 isolates showed gene specific preference towards certain codons coding for the different amino acids. For glycine, the gene E and S demonstrated a strong bias towards GGU whereas GGC and GGA was preferred in gene N and M. In case of alanine, GCU was universally found to be the most and preferred codon in genes S, M, and N. Out of the six synonymous codons of leucine, CUU was found to be the most preferred by all the structural genes except N. In case of isoleucine, out of the 3 synonymous codons, preference towards the use of AUU in all the structural genes was visualized barring E where all the three synonymous codons were equally employed without any bias. Of the two synonymous codons of phenylalanine, UUC was the most preferred whereas UUU was the least preferred codon in E, M and N, while S exhibited an entirely opposite scenario. For tyrosine, UAC was found to preferred in E, M and N gene whereas this was UAU in S. In case of arginine, the different structural component coding genes depicted a heterogenous choice of codons. In case of the polar neutral amino acids like serine, threonine, asparagine, glutamine and cysteine, somewhat similar codon usage pattern has been observed in the structural genes.
3.3.2. Comparative RSCU analysis of SARS-CoV-2 with human ribosomal protein coding genes
A comparison of the highly expressed ribosomal protein coding genes of humans with that of SARS-CoV-2 demonstrated that for glycine, GGC is the preferred codon in most of the human ribosomal protein coding genes which was found to be in accordance with that of orf10, orf7a and N gene of SARS-CoV-2. Similarly, for phenylalanine and tyrosine, a bias towards the use of codons UUC and UAC respectively was observed in most of the human ribosomal protein coding genes and SARS-CoV-2 structural component coding genes. This trend was also evident in the usage of the codon CAC for histidine. For the amino acids proline, leucine, alanine, isoleucine, valine, and arginine, no significant similarity between the codon preference of human ribosomal protein coding genes and SARS-CoV-2 genes was found to exist. Furthermore, we observed that the most preferred codons for lysine, aspartate, glutamate, and cysteine in the different SARS-CoV-2 genes are least preferred in the different human ribosomal protein coding genes. These inferences were found to be in accordance with the different tRNA species predictions of the human genome (Chan and Lowe, 2008; Chan and Lowe, 2015). The preference of certain codons like GGC for glycine, UUC for phenylalanine, UAC for tyrosine and CAC for histidine was observed in both human and SARS-CoV-2 genes.
3.3.3. Comparative RSCU analysis of SARS-CoV-2 with other PRVs
In the rest of the PRVs considered in this study, except Hepatitis C virus, Norovirus, and Rubella virus the codon GGA was found to be preferred for glycine by all the seven PRVs (like Dengue virus, Zika virus, Hepatitis A virus, and Japanese encephalitis virus) which is in line with that of M gene of SARS-CoV-2. Similarly, the preferential utilization of the codon GGC for glycine in Hepatitis C virus, Norovirus, and Rubella virus was also found to be inline with that of orf10, orf7a and N gene of SARS-CoV-2. In case of alanine, the optimal usage of GCU in Hepatitis A virus (54%) and Human rhinovirus (45%) was noticed to be in accordance with that of S, M, and N gene of SARS-CoV-2. The preferential usage of AUU for isoleucine in Hepatitis A virus (66%) and Norovirus (53%) was also in accordance with the three structural genes of SARS-CoV-2. Besides, 80% of the studied PRVs were found to prefer UUC codon for phenylalanine and UAC for tyrosine, while 60% of the PRVs were observed to preferentially utilize CAC for histidine which is in line with our observation for SARS-CoV-2. Within the studied PRVs, only Hepatitis A virus was found to completely avoid GCG for alanine and CGG for arginine and in most situations, and its preferential codon usage pattern was found to differ somewhat with respect to the other PRVs.
3.4. Analysis of SiD values in the different PRVs
The analyses of SiD values of all the viruses in this study clearly indicates the presence of the SARS-CoV2 among with the other viruses which demonstrates optimisation of codon pattern to tune itself with the host human genome. The SiD value of all these PRVs is graphically depicted in Fig. 8 . The SiD value of SARS-CoV-2 was found to be harmonized with the rest of the other coronaviruses and slightly higher than that of SARS-CoV-1, Middle East respiratory syndrome-related virus and other highly infectious human viruses like Japanese encephalitis viruse, Dengue virus, Zika virus and Poliovirus. The SiD value of Rubella virus was the lowest among all the PRVs while Human rhinovirus demonstrated the highest SiD score.
Fig. 8.

A scatter plot showing the similarity index (SiD) values of the different positive strand RNA viruses (PRVs) included in this study.
3.5. Analysis of codon context pattern
The SARS-CoV-2 isolates were found to be biased towards the utilization of valine-initiated (GUU-UUA, GUU-GAA, GUU-UAU, GUU-GUA) and glycine-initiated codon pairs (GGU-GUU, GGU-GAU, GGU-AAA, GGU-GGU). Glutamate (GAA-GAA, GAA-GCU, GAA-ACU, GAA-GGU), alanine (GCU-GUU, GCU-UUA, GCU-UUU, GCU-UGU) and isoleucine (AUU-GUU, AUU-GCU, AUU-AAA, AUU-CUU, AUU-CAA) initiated codon pairs were also observed in higher frequency. In general, guanine initiated and uracil ending codon pairs dominated the entire genome sequence. A genome wise comparison of codon pair data of the other PRVs revealed near universal preference for ACU-GAU, AAG-AAA, AAU-GAU, AAG-AAG and UAC-AAG codon pairs. Specific lysine-initiated codon pairs like AAA-CAU, AAA-CAA, AAA-AUU, AAA-AUG; asparagine-initiated codon pairs like AAU-GGU, AAU-UUU; valine ending codon pairs like GGU-GUU, GAA-GUU, UCU-GUU, GAA-GUU; glutamate dominated codon pairs like GAA-GUU, GAA-GAU, GAA-GAA and GUU-UAU, UAU-AAU were found to be preferred in most of the coronaviruses. UCG-initiated codon pairs were least preferred among all these viral genomes. “Asparagine-pattern” (Pal et al., 2015) and valine initiated codon context pairs like GUU-UAU, GUU-GUU, GUU-GUG, GUU-GGU, GUU-GCU, GUU-GAU, GUU-AAC, GUU-AAU, GUU-AAA were manifested in most of the coronavirus species. Some aspartate-initiated codon pairs such as GAU-UUU was found be preferred by Bovine coronavirus, Human coronavirus OC43 and Human coronavirus HKU1. The GAU-GUU codon context pair was found to be extensively used by Bovine coronavirus, Human coronavirus HKU1, Rabbit coronavirus HKU14, Rat coronavirus Parker, Middle East respiratory syndrome-related coronavirus and Human rhinovirus. In SARS-CoV-1, the leucine-initiated codon pairs (CUC-AAA, CUC-AAC, CUC-AAG, CUC-ACU, CUC-AUG, CUC-AUU, CUU-AAU, CUU-AAA) were found to be preferred. Threonine and phenylalanine-initiating codon pairs were also found to be widely preferred by the coronaviruses.
The codon pair analysis data (Supplementary Table 9) of the SARS-CoV-2 isolates demonstrated the presence of five different types of CPR clusters based on a k-means clustering (Supplementary Fig. 5). This suggest that there is a certain amount of heterogeneity within the SARS-CoV-2 isolates in the utilization pattern of the different codon pairs. When compared to the other PRVs it was observed that the coronaviruses have a relatively higher CPR compared to all the other PRVs. Lower CPR values was evident in Hepatitis A virus (0.3199), Human rhinovirus (0.325) and Rubella virus (0.3632). This is in line with the overall codon usage nature of these viruses which is much more constricted or biased compared to SARS-CoV-2.
Supplementary Fig. 5.
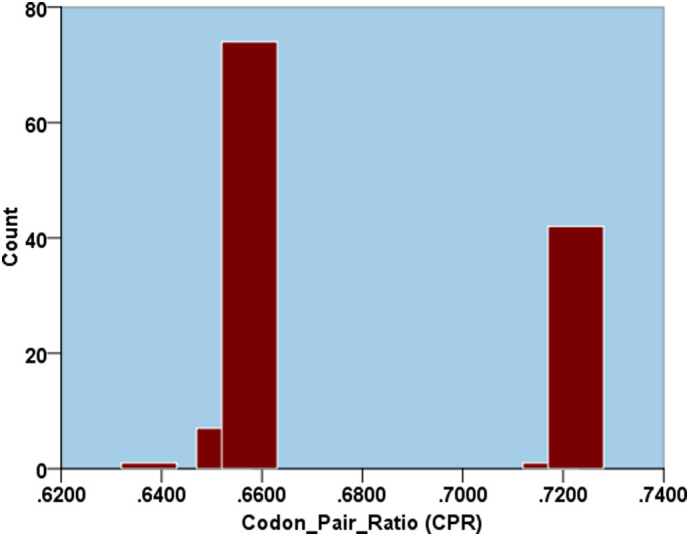
A histogram depicting the codon pair ratio (CPR) data of the SARS-CoV-2 isolates demonstrating the presence of five different types of CPR clusters utilizing k-means clustering.
To exploit the heterogeneity in codon pair usage of the SARS-CoV-2 isolates, multivariate data analysis in the form of PCA was performed to differentiate the genomes based on their genomic codon pair signature. The PCA analysis of the different SARS-CoV-2 isolates demonstrated that the first two dimensions account for 55.54% and 30.89% of the total variance. The PCA plot (Supplementary Fig. 6) clearly depicts five distinct codon pair signatures that are encircled by different colours. The isolates MT 240479 and MT 121215 were found to occupy characteristically isolated positions on the plot suggesting distinctively different codon pair signature. The isolates NC_045512 and MT 049951 from China was found to club together with the USA based isolates MT 163716 to MT 163719 on the plot, suggesting similarity in terms of their overall codon context. The remaining two clusters were found to be dense, populated by many of the remaining isolates. The magenta cluster was predominated by the isolates from USA (MT 246451 to MT 246490) and Spain (MT 233519, MT 233522 and MT 233523) suggesting they share a similar genomic codon context. Thus, these isolates apart from depicting a similar dinucleotide signature also shares similar codon context pattern. The blue cluster was found to be the most heterogenous assemblage with the maximum number of isolates from the other geographical regions clubbed together into it. Thus, we find that there is a differentiation in terms of codon context within the genome of SARS-CoV-2 and that is quite in in line with their dinucleotide signature.
Supplementary Fig. 6.

A PCA plot showing the segregation of the different SARS-CoV-2 isolates included in this study based on the occurrence of the different codon pairs. The x-axis represents the 1st dimension which accounts for 55.8% of the total variance and the y-axis represents the 2nd dimension accounting for 33.9% of the total variance. The different coloured circles represent the five different clusters of the SARS-CoV-2 isolates based on their codon pair frequency.
To investigate the similarities and dissimilarities in the codon context pattern of the SARS-CoV-2 isolates with respect to the other PRVs included in this study, representatives from the five different clusters obtained previously was taken together with the 26 other PRVs included in this study and a PCA was performed. The PCA plot obtained (Fig. 9 ) demonstrated an interesting trend where it was observed that all the five different SARS-CoV-2 isolates demonstrated a codon context trend characteristically dissimilar to the other PRVs. While many of the other coronaviruses displayed a similar clustering on the plot, the SARS-CoV2 isolates did not overlap even with the other coronaviruses, neither with SARS-CoV-1 or the Middle East respiratory syndrome-related coronavirus. This is a very interesting finding since SARS-CoV-2 is a member of the coronavirus family but displays a codon context signature different from the other PRVs including coronaviruses.
Fig. 9.

A PCA plot showing the segregation of the different positive strand RNA viruses (PRVs) included in this study based on the occurrence of the different codon pairs. The x-axis represents the 1st dimension which accounts for 23.3% of the total variance and the y-axis represents the 2nd dimension accounting for 11.7% of the total variance. The red dashed square depicts the distribution of the SARS-CoV-2 isolates relative to the other coronaviruses (in the blue square) and the other PRVs (depicted by the green square).
4. Conclusion
The SARS-CoV-2 is a PRV and one among the three coronaviruses that have successfully jumped into human beings so far and have wreaked a havoc. This study was carried out to compare and comprehend the codon biology of the SARS-CoV-2 with respect to the other PRVs. The presence and continuous generation of a colossal amount of genome sequence data of SARS-CoV-2 is making it possible to intricately analyse the SARS-CoV-2 genome and gene features. But in comparison, the amount of genomic sequence data available for the other PRVs is quite low making it difficult to intricately analyse the same. Based on the limited amount of genome sequence data of the other PRVs in the public databases we observed that the codon usage bias analyses of many SARS-CoV-2 genomes and genes overall shows a great deal of heterogeneity in comparison to the other PRVs. In this comparative dinucleotide and codon biology-based analysis of the SARS-CoV-2 virus including other PRVs we found that among the SARS-CoV-2 isolates heterogeneity within the distribution of the CpG dinucleotides exist and SARS-CoV-2 genomes demonstrates a genomic dinucleotide signature different from the rest of the PRVs. In terms of codon usage, SARS-CoV-2 along with the other coronaviruses had relatively greater genomic codon bias compared to most of the other PRVs. Gene specific codon bias was observed in SARS-CoV-2, and the preference of certain codons were observed in both human ribosomal protein and SARS-CoV-2 coding sequences. The SARS-CoV-2 isolates were also found to demonstrate a codon context trend characteristically dissimilar to the other PRVs. RNA viruses are notorious in terms of mutation leading to continuous and ever-changing variability and hence a continuous vigil is required to monitor the genetic changes that occur in these. More studies concentrating on the other PRVs is of utmost necessity since if kept untracked, these viruses can resurface and catch us off-guard causing pandemics and wreaking havoc in the future.
The following are the supplementary data related to this article.
Details of the positive strand RNA viruses (PRVs) included in this study.
The mononucleotide and dinucleotide frequency of the positive strand RNA viruses (PRVs) included in this study.
Genomic over and underrepresentation of the 16 different dinucleotide combinations of the positive strand RNA viruses (PRVs) included in this study.
Gene wise over and underrepresentation data (base model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Gene wise over and underrepresentation data (codon model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Gene wise over and underrepresentation data (syncodon model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Codon usage (Nc) and GC (GC1, GC2, GC3) content data of the coding sequences of the positive strand RNA viruses (PRVs) included in this study.
Detailed codon usage data of the different SARS-CoV-2 isolates.
Linear regression representing the relationship between GC1/GC2 and GC3 for the different ORFs of the SARS-CoV-2 genome.
RSCU values of the different coding sequences of the SARS-CoV-2 genome.
Codon pair count data of all the positive strand RNA viruses (PRVs) included in this study.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
CRediT authorship contribution statement
Jayanti Saha: Conceptualization, Investigation, Methodology, Formal analysis, Writing – original draft, Writing – review & editing. Sukanya Bhattacharjee: Investigation, Methodology, Software, Formal analysis, Writing – original draft, Data curation, Writing – review & editing. Monalisha Pal Sarkar: Investigation, Methodology, Formal analysis, Writing – original draft, Writing – review & editing. Barnan Kumar Saha: Investigation, Formal analysis, Data curation, Writing – original draft. Hriday Kumar Basak: Investigation, Formal analysis, Software, Writing – original draft. Samarpita Adhikary: Investigation, Formal analysis, Data curation. Vivek Roy: Investigation, Formal analysis, Data curation. Parimal Mandal: Conceptualization, Investigation, Writing – original draft. Abhik Chatterjee: Supervision, Conceptualization, Methodology, Validation, Software, Writing – original draft. Ayon Pal: Supervision, Conceptualization, Methodology, Validation, Software, Formal analysis, Writing – original draft, Writing – review & editing.
Declaration of competing interest
All the authors declare that they have no conflict of interest.
References
- Ahlquist P. Host factors in positive-Strand RNA virus genome replication. J. Virol. 2003;77(15):8181. doi: 10.1128/JVI.77.15.8181-8186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexaki A. Codon and codon-pair usage tables (CoCoPUTs): facilitating genetic variation analyses and recombinant gene design. J. Mol. Biol. 2019;431(13):2434–2441. doi: 10.1016/j.jmb.2019.04.021. [DOI] [PubMed] [Google Scholar]
- Almazán F. Coronavirus reverse genetic systems: infectious clones and replicons. Virus Res. 2014;189:262–270. doi: 10.1016/j.virusres.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V.J., Nieminen T., Maunula L. The Noro story--viral gastoenteritis as a problem in inpatient facilities. Duodecim. 2010;126(13):1575–1581. [PubMed] [Google Scholar]
- Assiri A. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect. Dis. 2013;13(9):752–761. doi: 10.1016/S1473-3099(13)70204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athey J. A new and updated resource for codon usage tables. BMC Bioinformatics. 2017;18(1):391. doi: 10.1186/s12859-017-1793-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins G.J., Sheahan B.J., Liljeström P. The molecular pathogenesis of Semliki Forest virus: a model virus made useful? J Gen Virol. 1999;80(Pt 9):2287–2297. doi: 10.1099/0022-1317-80-9-2287. [DOI] [PubMed] [Google Scholar]
- Ayon P. The implication of codon usage design and expression level in determining the nature of selection and functionality amongst the amino acid biosynthetic pathway coding sequences of arthrobacter sp. FB24. Curr. Bioinforma. 2014;9(5):470–480. [Google Scholar]
- Baha S. Comprehensive analysis of genetic and evolutionary features of the hepatitis E virus. BMC Genomics. 2019;20(1):790. doi: 10.1186/s12864-019-6100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S.F., Nogales A., Martinez-Sobrido L. Downregulating viral gene expression: codon usage bias manipulation for the generation of novel influenza A virus vaccines. Future Virol. 2015;10(6):715–730. doi: 10.2217/fvl.15.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachboard D.C., Horner S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016;32:113–119. doi: 10.1016/j.mib.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura S.K., Severson D.W. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belalov I.S., Lukashev A.N. Causes and implications of codon usage bias in RNA viruses. PLoS One. 2013;8(2) doi: 10.1371/journal.pone.0056642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.A. GenBank. Nucleic Acids Res. 2013;41(Database issue):D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechot C. 2018 international meeting of the global virus network. Antivir. Res. 2019;163:140–148. doi: 10.1016/j.antiviral.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A.M. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerging microbes & infections. 2016;5(10):e107. doi: 10.1038/emi.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.-a. Analyses of nucleotide, synonymous codon and amino acid usages at gene levels of Brucella melitensis strain QY1. Infection, Genetics and Evolution. 2018;65:257–264. doi: 10.1016/j.meegid.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Castells M. Genome-wide analysis of codon usage bias in bovine coronavirus. Virol. J. 2017;14(1):115. doi: 10.1186/s12985-017-0780-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.P., Lowe T.M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Research. 2008;37(suppl_1):D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.P., Lowe T.M. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2015;44(D1):D184–D189. doi: 10.1093/nar/gkv1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, S.T. and J.J. Ou, Hepatitis C Virus-Induced Autophagy and Host Innate Immune Response. Viruses, 2017. 9(8). [DOI] [PMC free article] [PubMed]
- Chen S. Innate immune evasion mediated by flaviviridae non-structural proteins. 2017;9(10) doi: 10.3390/v9100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.Y.H. Development of live-attenuated arenavirus vaccines based on codon deoptimization of the viral glycoprotein. Virology. 2017;501:35–46. doi: 10.1016/j.virol.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian S.S., Walimbe A.M. Phylogeographic analysis of Japanese encephalitis virus in India (1956-2012) Arch. Virol. 2015;160(12):3097–3104. doi: 10.1007/s00705-015-2599-4. [DOI] [PubMed] [Google Scholar]
- Corman V.M. Chapter eight - hosts and sources of endemic human coronaviruses. In: Kielian M., Mettenleiter T.C., Roossinck M.J., editors. Advances in Virus Research. Academic Press; 2018. pp. 163–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea L. The critical role of codon composition on the translation efficiency robustness of the hepatitis a virus capsid. Genome Biology and Evolution. 2019;11(9):2439–2456. doi: 10.1093/gbe/evz146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb B., Uddin A. and S. Chakraborty, Codon usage pattern and its influencing factors in different genomes of hepadnaviruses. 2020;165(3):557–570. doi: 10.1007/s00705-020-04533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deka H. DNA compositional dynamics and codon usage patterns of M1 and M2 matrix protein genes in influenza a virus. Infect. Genet. Evol. 2019;67:7–16. doi: 10.1016/j.meegid.2018.10.015. [DOI] [PubMed] [Google Scholar]
- Farcas G.A. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J. Infect. Dis. 2005;191(2):193–197. doi: 10.1086/426870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanegan J.B. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. U. S. A. 1977;74(3):961–965. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzo G. The analysis of genome composition and codon bias reveals distinctive patterns between avian and mammalian circoviruses which suggest a potential recombinant origin for Porcine circovirus 3. PLoS One. 2018;13(6) doi: 10.1371/journal.pone.0199950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta I. Norwalk virus and Noro virus. Rinsho Biseibutshu Jinsoku Shindan Kenkyukai Shi. 2003;14(2):127–131. [PubMed] [Google Scholar]
- García-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22(2):176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier C., Gouy M., Louail S. Non-parametric statistics for nucleic acid sequence study. Biochimie. 1985;67(5):449–453. doi: 10.1016/s0300-9084(85)80263-7. [DOI] [PubMed] [Google Scholar]
- Genereux D.P. Evolution of genomic GC variation. Genome Biology. 2002;3(10) p. reports0058. [Google Scholar]
- Gokhale N.S., Vazquez C., Horner S.M. Hepatitis C virus. Strategies to evade antiviral responses. Future Virol. 2014;9(12):1061–1075. doi: 10.2217/fvl.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. p. 2020.02.07.937862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H. Dinucleotide evolutionary dynamics in influenza A virus. Virus Evol. 2019;5(2):vez038. doi: 10.1093/ve/vez038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H. Dinucleotide evolutionary dynamics in influenza A virus. Virus evolution. 2019;5(2):vez038. doi: 10.1093/ve/vez038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M.G., Harris E. Dengue. Lancet. 2015;385(9966):453–465. doi: 10.1016/S0140-6736(14)60572-9. [DOI] [PubMed] [Google Scholar]
- Hanson G. Translation elongation and mRNA stability are coupled through the ribosomal A-site. RNA. 2018;24(10):1377–1389. doi: 10.1261/rna.066787.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harak C., Lohmann V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology. 2015;479-480:418–433. doi: 10.1016/j.virol.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E.L. Virus variation resource - improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45(D1):D482–d490. doi: 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilleman M.R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc Natl Acad Sci U S A. 101 Suppl. 2004;2(Suppl. 2):14560–14566. doi: 10.1073/pnas.0404758101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.W. Acute renal failure in patients with severe acute respiratory syndrome. J. Formos. Med. Assoc. 2005;104(12):891–896. [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2(1):13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Jernigan R.W., Baran R.H. Pervasive properties of the genomic signature. BMC Genomics. 2002;3(1):23. doi: 10.1186/1471-2164-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbayashi D. Rubella virus genotype 1E in travelers returning to Japan from Indonesia, 2017. Emerg. Infect. Dis. 2018;24(9):1763–1765. doi: 10.3201/eid2409.180621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M. From SARS and MERS CoVs to SARS-CoV-2: moving toward more biased codon usage in viral structural and nonstructural genes. J. Med. Virol. 2020;92:660–666. doi: 10.1002/jmv.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S., Cardon L.R. Computational DNA sequence analysis. Annu. Rev. Microbiol. 1994;48:619–654. doi: 10.1146/annurev.mi.48.100194.003155. [DOI] [PubMed] [Google Scholar]
- Karumathil S. Evolution of synonymous codon usage bias in west African and central African strains of monkeypox virus. Evol. Bioinformatics Online. 2018;14 doi: 10.1177/1176934318761368. p. 1176934318761368-1176934318761368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandia R. Analysis of Nipah virus codon usage and adaptation to hosts. Front. Microbiol. 2019:10(886). doi: 10.3389/fmicb.2019.00886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler E., Thiel V., Weber F. Interaction of SARS and MERS coronaviruses with the antiviral interferon response. Adv. Virus Res. 2016;96:219–243. doi: 10.1016/bs.aivir.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 1981;291(5816):547–553. doi: 10.1038/291547a0. [DOI] [PubMed] [Google Scholar]
- Kumar N. Revelation of Influencing Factors in Overall Codon Usage Bias of Equine Influenza Viruses. 2016;11(4) doi: 10.1371/journal.pone.0154376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lee Y.F. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. U. S. A. 1977;74(1):59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Lethal encephalitis in seals with Japanese encephalitis virus infection, China, 2017. Emerg. Infect. Dis. 2019;25(8):1539–1542. doi: 10.3201/eid2508.181663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K. Conserved codon composition of ribosomal protein coding genes in Escherichia coli, Mycobacterium tuberculosis and Saccharomyces cerevisiae: lessons from supervised machine learning in functional genomics. Nucleic Acids Res. 2002;30(11):2599–2607. doi: 10.1093/nar/30.11.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S. Highly expressed ribosomal protein L34 indicates poor prognosis in osteosarcoma and its knockdown suppresses osteosarcoma proliferation probably through translational control. Sci. Rep. 2016;6(1):37690. doi: 10.1038/srep37690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangala Prasad V., Klose T. Assembly, maturation and three-dimensional helical structure of the teratogenic rubella virus. 2017;13(6):e1006377. doi: 10.1371/journal.ppat.1006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham N.R., Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Research. 2005;33(suppl_2):W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunula L. Presence of human noro- and adenoviruses in river and treated wastewater, a longitudinal study and method comparison. J. Water Health. 2012;10(1):87–99. doi: 10.2166/wh.2011.095. [DOI] [PubMed] [Google Scholar]
- McKnight K.L., Lemon S.M. Hepatitis A virus genome organization and replication strategy. Cold Spring Harb Perspect Med. 2018:8(12). doi: 10.1101/cshperspect.a033480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J. Virol. 2020;94(5) doi: 10.1128/JVI.01774-19. p. e01774-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier W., Stewart C.B. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385(6612):151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Mortazavi M. Retracted article: bioinformatic analysis of codon usage and phylogenetic relationships in different genotypes of the hepatitis C virus. Hepat. Mon. 2016;16(10) doi: 10.5812/hepatmon.39196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura G. Large scale comparative codon-pair context analysis unveils general rules that fine-tune evolution of mRNA primary structure. PLoS One. 2007;2(9) doi: 10.1371/journal.pone.0000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura G.R. Species-specific codon context rules unveil non-neutrality effects of synonymous mutations. PLoS One. 2011;6(10) doi: 10.1371/journal.pone.0026817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006;80(19):9687–9696. doi: 10.1128/JVI.00738-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D., Gubler D.J. Zika Virus. Clin. Microbiol. Rev. 2016;29(3):487–524. doi: 10.1128/CMR.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y., Ikemura T. Fop (frequency of optimal codon usage): WWW service with its distribution analysis. Genome Informatics. 1995;6:166–167. [Google Scholar]
- Napoli, M.C.M.R.A.C.S.C.D.R.D., Features, Evaluation and Treatment Coronavirus (COVID-19), in StatPearls [Internet]. 2020, StatPearls Publishing: Treasure Island (FL).
- Nasrullah I. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Marburg virus evolution. BMC Evol. Biol. 2015;15(1):174. doi: 10.1186/s12862-015-0456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelemans T., Kikkert M. Viral innate immune evasion and the pathogenesis of emerging RNA virus infections. 2019;11(10) doi: 10.3390/v11100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira Melo A.S. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet. Gynecol. 2016;47(1):6–7. doi: 10.1002/uog.15831. [DOI] [PubMed] [Google Scholar]
- Ortega J.T. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J. 2020;19:410–417. doi: 10.17179/excli2020-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overby A.K. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 2010;84(17):8470–8483. doi: 10.1128/JVI.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A. Deconstruction of archaeal genome depict strategic consensus in core pathways coding sequence assembly. PLoS One. 2015;10(2) doi: 10.1371/journal.pone.0118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A., Saha B.K., Saha J. Comparative in silico analysis of ftsZ gene from different bacteria reveals the preference for core set of codons in coding sequence structuring and secondary structural elements determination. PLoS One. 2019;14(12) doi: 10.1371/journal.pone.0219231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeira L., Guéguen L., Lobry J.R. UV-targeted dinucleotides are not depleted in light-exposed prokaryotic genomes. Mol. Biol. Evol. 2006;23(11):2214–2219. doi: 10.1093/molbev/msl096. [DOI] [PubMed] [Google Scholar]
- Pandit A., Vadlamudi J., Sinha S. Analysis of dinucleotide signatures in HIV-1 subtype B genomes. J. Genet. 2013;92(3):403–412. doi: 10.1007/s12041-013-0281-8. [DOI] [PubMed] [Google Scholar]
- Pardy R.D., Valbon S.F., Richer M.J. Running interference: interplay between Zika virus and the host interferon response. Cytokine. 2019;119:7–15. doi: 10.1016/j.cyto.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Peden J.F. Analysis of Codon Usage, in Department of Genetics. University of Nottingham. 1999 [Google Scholar]
- Phan T. Genetic diversity and evolution of SARS-CoV-2. Infection, Genetics and Evolution. 2020;81:104260. doi: 10.1016/j.meegid.2020.104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R.M. Hepatitis A virus codon usage: implications for translation kinetics and capsid folding. Cold Spring Harb Perspect Med. 2018:8(10). doi: 10.1101/cshperspect.a031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plourde A.R., Bloch E.M. A literature review of Zika virus. Emerg. Infect. Dis. 2016;22(7):1185–1192. doi: 10.3201/eid2207.151990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabha R., Singh D.P. Analysis of dinucleotide Bias and genomic signatures across cyanobacterial genomes. Journal of Advances in Biotechnology. 2014;3 [Google Scholar]
- Prabha R. Genome-wide comparative analysis of codon usage bias and codon context patterns among cyanobacterial genomes. Mar. Genomics. 2017;32:31–39. doi: 10.1016/j.margen.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Puigbò P., Aragonès L., Garcia-Vallvé S. RCDI/eRCDI: a web-server to estimate codon usage deoptimization. BMC Research Notes. 2010;3(1):87. doi: 10.1186/1756-0500-3-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V.R., Baltimore D. Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc. Natl. Acad. Sci. U. S. A. 1981;78(8):4887–4891. doi: 10.1073/pnas.78.8.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S.U. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens. 2020:9(3). doi: 10.3390/pathogens9030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringnér M., Krogh M. Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput. Biol. 2005;1(7) doi: 10.1371/journal.pcbi.0010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RoyChoudhury S., Mukherjee D. A detailed comparative analysis on the overall codon usage pattern in herpesviruses. Virus Res. 2010;148(1):31–43. doi: 10.1016/j.virusres.2009.11.018. [DOI] [PubMed] [Google Scholar]
- Ruan Y. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361(9371):1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha J. Comparative genomic analysis of soil dwelling bacteria utilizing a combinational codon usage and molecular phylogenetic approach accentuating on key housekeeping genes. Front. Microbiol. 2019:10(2896). doi: 10.3389/fmicb.2019.02896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.M., Li W.H. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverj A., Rota-Stabelli O. On the correct interpretation of similarity index in codon usage studies: comparison with four other metrics and implications for Zika and West Nile virus. Virus Res. 2020;286:198097. doi: 10.1016/j.virusres.2020.198097. [DOI] [PubMed] [Google Scholar]
- Subbaram K., Kannan H., Khalil Gatasheh M. Emerging developments on pathogenicity, molecular virulence, epidemiology and clinical symptoms of current Middle East respiratory syndrome coronavirus (MERS-CoV) Hayati. 2017;24(2):53–56. doi: 10.1016/j.hjb.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Vlahovicek K. INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics. 2004;20(14):2329–2330. doi: 10.1093/bioinformatics/bth238. [DOI] [PubMed] [Google Scholar]
- Tang H. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18(5):587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort F.L., Castells M., Cristina J. A comprehensive analysis of genome composition and codon usage patterns of emerging coronaviruses. Virus Res. 2020;283:197976. doi: 10.1016/j.virusres.2020.197976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno N., Ross T.M. Dengue virus and the host innate immune response. Emerg Microbes Infect. 2018;7(1):167. doi: 10.1038/s41426-018-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X.-F. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol. Biol. 2004;4(1):19. doi: 10.1186/1471-2148-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.-C., Chen F.-C. The evolutionary landscape of the Mycobacterium tuberculosis genome. Gene. 2013;518(1):187–193. doi: 10.1016/j.gene.2012.11.033. [DOI] [PubMed] [Google Scholar]
- Woo P.C., Lau S.K., Yuen K.-y. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr. Opin. Infect. Dis. 2006;19(5):401–407. doi: 10.1097/01.qco.0000244043.08264.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P.C.Y. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 2009;83(2):908. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Wu Q. Translation affects mRNA stability in a codon-dependent manner in human cells. eLife. 2019;8 doi: 10.7554/eLife.45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. An improved implementation of codon adaptation index. Evol. Bioinformatics Online. 2007;3:53–58. [PMC free article] [PubMed] [Google Scholar]
- Xiang H. Comparative analysis of codon usage bias patterns in microsporidian genomes. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0129223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C. Analysis of synonymous codon usage patterns in seven different citrus species. Evol. Bioinformatics Online. 2013;9:215–228. doi: 10.4137/EBO.S11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. Virus-like particle vaccines for poliovirus types 1, 2, and 3 with enhanced thermostability expressed in insect cells. Vaccine. 2019;37(17):2340–2347. doi: 10.1016/j.vaccine.2019.03.031. [DOI] [PubMed] [Google Scholar]
- Yao H., Chen M., Tang Z. Analysis of synonymous codon usage bias in flaviviridae virus. Biomed. Res. Int. 2019;2019:5857285. doi: 10.1155/2019/5857285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C.H. Clinical analysis of multiple organ dysfunction syndrome in patients suffering from SARS. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2004;16(11):646–650. [PubMed] [Google Scholar]
- Yogo Y., Wimmer E. Polyadenylic acid at the 3′-terminus of poliovirus RNA. Proc. Natl. Acad. Sci. U. S. A. 1972;69(7):1877–1882. doi: 10.1073/pnas.69.7.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zeeberg B. Shannon information theoretic computation of synonymous codon usage biases in coding regions of human and mouse genomes. Genome Res. 2002;12(6):944–955. doi: 10.1101/gr.213402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel M.B., Felmlee D.J., Baumert T.F. Hepatitis C virus entry. Curr. Top. Microbiol. Immunol. 2013;369:87–112. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- Zhou J.-h. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLOS ONE. 2013;8(10):e77239. doi: 10.1371/journal.pone.0077239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.-H. The genetic divergences of codon usage shed new lights on transmission of hepatitis E virus from swine to human. Infection, Genetics and Evolution. 2019;68:23–29. doi: 10.1016/j.meegid.2018.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of the positive strand RNA viruses (PRVs) included in this study.
The mononucleotide and dinucleotide frequency of the positive strand RNA viruses (PRVs) included in this study.
Genomic over and underrepresentation of the 16 different dinucleotide combinations of the positive strand RNA viruses (PRVs) included in this study.
Gene wise over and underrepresentation data (base model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Gene wise over and underrepresentation data (codon model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Gene wise over and underrepresentation data (syncodon model) of the 16 different dinucleotide combinations of the SARS-CoV-2 isolates included in this study.
Codon usage (Nc) and GC (GC1, GC2, GC3) content data of the coding sequences of the positive strand RNA viruses (PRVs) included in this study.
Detailed codon usage data of the different SARS-CoV-2 isolates.
Linear regression representing the relationship between GC1/GC2 and GC3 for the different ORFs of the SARS-CoV-2 genome.
RSCU values of the different coding sequences of the SARS-CoV-2 genome.
Codon pair count data of all the positive strand RNA viruses (PRVs) included in this study.