

Abstract
Low-temperature soft chemical synthesis routes to transition-metal nitrides are of interest as an alternative to conventional high-temperature ammonolysis reactions involving large volumes of chemotoxic NH3 gas. One such method is the reaction between metal oxides and NaNH2 at ca. 200 °C to yield the counterpart nitrides; however, there remains uncertainty regarding the reaction mechanism and product phase assemblage (in particular, noncrystalline components). Here, we extend the chemical tool box and mechanistic understanding of such reactions, demonstrating the nitridation of Fe3O4 by reaction with NaNH2 at 170–190 °C, via a pseudomorphic reaction. The more reduced Fe3O4 precursor enabled nitride formation at lower temperatures than the previously reported equivalent reaction with Fe2O3. The product phase assemblage, characterized by X-ray diffraction, thermogravimetric analysis, and 57Fe Mössbauer spectroscopy, comprised 49–59 mol % ε-Fe2+xN, accompanied by 29–39 mol % FeO1–xNx and 8–14 mol % γ″-FeN. The oxynitride phase was apparently noncrystalline in the recovered product but could be crystallized by heating at 180 °C. Although synthesis of transition-metal nitrides is achieved by reaction of the counterpart oxide with NaNH2, it is evident from this investigation that the product phase assemblage may be complex, which could prove a limitation if the objective is to produce a single-phase product with well-defined electrical, magnetic, or other physical properties for applications. However, the significant yield of the FeO1–xNx oxynitride phase identified in this study opens the possibility for the synthesis of metastable oxynitride phases in high yield, by reaction of a metal oxide substrate with NaNH2, with either careful control of H2O concentration in the system or postsynthetic hydrolysis and crystallization.
Short abstract
Fe3O4 is reacted with NaNH2 with the aim of low-temperature iron nitride synthesis. The reaction is mediated by a NaNH2−NaOH molten salt. Investigation of the phase assemblage by XRD, Mössbauer spectroscopy, and TGA-MS reveals a mixture of crystalline nitride and amorphous oxynitride phases.
Introduction
Transition-metal nitrides have considerable technological potential, with broad applications as functional materials with exploitable physical and chemical properties,1,2 and advancements in their synthesis have been the focus of considerable interest. Synthesis of a nitride from a readily available oxide precursor is preferential as similarities in ionic radius and electronegativity (3.50 and 3.07 for oxygen and nitrogen, respectively3) allow for substitution of nitrogen for oxygen, providing the host compound is capable of charge compensation. However, doing so with gaseous nitrogen is not possible due to unfavorable thermodynamics. The triple N≡N bond has an enthalpy of 941 kJ/mol, whereas the double O=O bond has an enthalpy of only 500 kJ/mol, and thus, nitride formation cannot be achieved.4 This can be overcome by use of high pressure as well as high temperature to manipulate the thermodynamics to promote nitride stabilization. However, the energy and instrumentation requirements for this can be prohibitive. A detailed discussion and visualization of nitride synthesis methodologies was recently presented by Miura et al.4
Typical gaseous nitridation is achieved by using high flow rates of NH3 gas at high temperatures, in the region of 600–1200 °C3 acting upon oxide or halide precursors. Although the use of NH3 has been successfully applied for the synthesis of most known nitride compounds, a large number of diffusion steps and high temperature are essential to activate the diffusion. The high processing temperature may induce the decomposition of NH3 into N2 and H2, which is an undesirable side reaction. In addition, gas exchange between the solid and the ammonia is a limiting factor, sometimes resulting in samples that have reacted at the surface only. Thus, several ammonolysis steps with intermediate regrinding are often required.3 As such, these reactions demand large volumes of NH3 gas, which comes with safety and material implications due to the chemotoxic and corrosive nature of NH3.
Several alternate solid-state nitrogen sources have been developed for nitride synthesis. Urea, CO(NH2)2, has been used to synthesize a number of nanoparticulate binary nitrides such as TiN, VN, NbN, GaN,5 and AlN.6 Ethanol-based solutions of a metal precursor and urea form a gel-like intermediate product, which is then heated to 800 °C or greater under N2, to yield the nitride. Melamine, C3H6N6, decomposes to C3N4, which can then be used as a carbothermal reducing and nitriding agent.7,8 Similar to the urea route, the mixed metal precursor and nitriding agent are then heat-treated in the region of 650–800 °C. While these methods offer a more easily implemented nitriding source compared to NH3 gas, they still require high-temperature processing and are subject to residual carbon impurities in the final product.
A solid-state nitrogen source that does not require high synthesis temperatures is sodium amide, NaNH2. Nitrides synthesized by this method include TiN,9−11 VN,12 Cu3N,13 InN,14,15 Si3N4,1616 BN,17 Fe3N1+x,18 and a variety of manganese nitrides.18−20 These reactions predominantly used oxide precursors, excluding examples of TiN,9,10 VN,12 and MnN,20 which were formed from their respective chlorides, and BN synthesized from liquid BBr3.17 All reactions utilized a similar methodology performing reactions in stainless steel autoclaves and the end product being washed with ethanol to destroy and remove excess NaNH2. Typical reaction temperatures range from 170 to 300 °C. Synthesis via NaNH2 therefore offers a route to nitride formation at much lower temperatures than those required for ammonolysis or reactions using other solid-state nitrogen sources and avoids the use of toxic NH3 gas.
Of interest to this study is the reported formation of Fe2+xN (or, equivalently, Fe3N1+x),18 which has broad applications and research potential in corrosion-resistant coatings, magnetic data recording, catalysis, and biomedical uses.1 Synthesis of Fe2+xN was previously achieved from reaction of Fe2O3 with an excess of NaNH2 at 240 °C for 36 h.18 Previous work indicates that the reactions are highly reducing in nature, with Cu metal being formed from reaction of CuO with NaNH2 at 190 °C for 60 h, compared to Cu3N being formed at 170 °C for 60 h.13 As reduction is key to the reaction mechanism, we opted to use Fe3O4 as a comparatively reduced counterpart to Fe2O3 to explore the potential for synthesis of Fe2+xN under more mild reaction conditions. In this work, we extend the NaNH2 synthetic approach to consider reactions using Fe3O4 with lower reaction temperatures of 170–190 °C and variable duration. In particular, we extend the suite of characterization applied to these phases to develop a more complete understanding of the product phase assemblage and reaction mechanism. In all reaction products, we determine a mixture of ε-Fe2+xN and γ″-FeN nitride compositions along with an oxynitride phase FeO1–xNx, demonstrating complexity of the phase assemblage not previously considered.
Experimental Section
Caution
NaNH2 is a strong base that reacts violently with oxidizing agents and with water (producing ammonia); explosive peroxides may form on atmospheric exposure: use of this reagent must be rigorously risk-assessed; storage and handling under a dry inert atmosphere are advised.21
Reagents of Fe3O4 and NaNH2 (Sigma-Aldrich, 98% purity) were mixed in a 1:14 molar ratio and hand-ground under a dry nitrogen atmosphere for 5 min. Typically, 1 g of powder was placed in a lidded crucible (5 mL, fabricated from a type 316 stainless steel), which was sealed within a poly(tetrafluoroethylene) (PTFE)-lined stainless steel digestion vessel (45 mL, Parr model 4744); this was performed under a dry nitrogen atmosphere. The vessel was transferred to a temperature-controlled oven (170 or 190 °C, ±1 °C) for the reaction duration (24, 48, or 96 h). Vessels were removed from the oven, cooled at room temperature, and opened in a fume hood, due to the distinctive odor of ammonia evolved from the head space, confirmed by moist universal indicator paper. Inspection of the stainless steel crucible revealed a solid, fused, mass. The product was recovered by soaking in ethanol to dissolve residual NaNH2 forming (soluble) sodium ethoxide and ammonia gas (NaNH2 + C2H5OH → C2H5ONa + NH3). The product, a black powder, was washed twice in ethanol and recovered by filtration for further analysis.
Characterization of the nitrided powders was carried out by X-ray diffraction (XRD) performed on a Bruker D2 Phaser diffractometer in fluorescence mode with a Ni-filtered Cu Kα radiation of λ = 1.5418 Å and a Lynxeye position-sensitive detector; the energy discriminator window was adjusted to minimize the acceptance of Fe Kα fluorescence. Scanning electron microscopy coupled with energy-dispersive X-ray analysis (SEM-EDX) was performed first on a Hitachi TM3030 desktop unit with a Bruker Quantax EDX system and subsequently on an FEI Inspect F50 FEG SEM operating at 10 kV. 57Fe Mössbauer spectroscopy measurements were performed with a Wissel MRG-500 spectrometer in transmission mode using a 50 mCi 57Co source; calibration was performed with an α-Fe reference foil. Spectra were measured using a constant acceleration waveform with a velocity range of ±10 mm s–1. Data were analyzed using Recoil software,22 fitting Lorentzian line shapes. Thermogravimetric analysis mass spectrometry (TG-MS) measurements were made using a Netzsch STA 449 F3 Jupiter thermal analyzer coupled with a Netzsch QMS 403 Aelos Quadro quadrupole mass spectrometer and using an argon carrier gas.
Samples were prepared for transmission electron microscopy (TEM) using the crushed grain method. Samples were rapidly ground with isopropanol using an agate pestle and mortar and allowed to settle. A small amount of the resultant mixture was siphoned from the top of the solvent line and pipetted onto carbon-holey lined Cu grids (Agar Scientific). TEM was performed using a JEOL F200 operating at 200 keV. Images and electron diffraction patterns were recorded using a Gatan OneView camera under bright-field conditions. EDS was undertaken using dual silicon drift detectors. Electron energy loss spectroscopy (EELS) was recorded using a Gatan Image Filter Quantum ER, with a slit width of 200 eV, a dispersion of 25 eV/channel, and a 5 mm entrance aperture. The energy resolution was 0.8 eV. For each spectral acquisition, 40 frames with an exposure time of 0.5 s were recorded and summed. Conversion of two-dimensional (2D) polycrystalline electron diffraction patterns was achieved by taking several radial grayscale line scans of each pattern using the ImageJ software package,23 with the undiffracted (000) “reflection” taken as 1/d = 0. Sample specific line scans were normalized to the most intense reflection, summed to give an average for the whole pattern and inverted to produce a one-dimensional (1D) intensity against the d-spacing pattern.
Results and Discussion
Initial analysis of XRD data (Figure 1) determined all products to be composed of a mixture of crystalline ε-Fe2+xN (space group P6322) and a noncrystalline phase contributing to diffuse scattering most apparent in the regions 20° < 2θ < 50°. The relatively broad nature of the reflections associated with the Fe2+xN phase was interpreted as the characteristic of a nanocrystalline material, as later confirmed by TEM observation. Comparison of the XRD data of the ε-Fe2+xN products with reference patterns of ε-Fe2N (PDF 04–017–2325) and ε-Fe3N (PDF 01-083-0878) suggested a composition with an intermediate Fe/N ratio (0 < x < 1), as shown in Figure 1. The Fe–N phase diagram is known to be complex, but the ε-Fe2+xN solid solution, with 0 < x < 1, has been reasonably well characterized; the structure, in space group P6322, comprises a hexagonal close-packed array of Fe atoms with N occupying octahedral interstices in an ordered fashion.24−27 Reaction conditions were systematically varied in an attempt to optimize the crystallinity of the product and control the composition. The yield of Fe2+xN was not improved by increasing the reaction temperature from 170 to 190 °C for 24 h or increasing the reaction time from 24 to 96 h at 170 °C, as shown in Figure 1. However, the reflection full width at half-maximum was observed to increase with reaction temperature and time, indicative of a decrease in crystallite size. To estimate the average composition of the ε-Fe2+xN product phase, the Inorganic Crystal Structure Database28 was mined to establish a calibration of unit cell volume as a function of x in ε-Fe2+xN, assuming a linear dependence in accordance with Vegard’s law (see Table S1 and Figure S1 in the Supporting Information). The unit cell parameters of the Fe2+xN products and estimated compositions determined using this linear calibration are reported in Table 1 and establish x ≈ 0.12(2), corresponding to an average formal Fe oxidation state of Fe1.42+. The refined unit cell parameters show a trend of increasing unit cell volume with reaction time and temperature, as shown in Table 1, implying a small decrease in x, although this is within the estimated uncertainty of the linear calibration. It should be noted that the extracted value of unit cell volume relies upon XRD data with broad reflections and a subsequent calibration of these values to available data and thus represents an estimate of composition only. The trend of increasing cell volume, combined with the increase in full width at half maximum (FWHM) for the XRD data in Figure 1, implies decreased nitridation with increased reaction time and temperature. Miura et al.18 offer an explanation for this with consideration of the relative excess of NaNH2 present in the reactions. Where reduced concentrations of NaNH2 are present, the reaction is limited, with the formation of H2O and corresponding positive values for the Gibbs free energy of reaction. In this instance, prolonged reaction times and higher temperatures result in greater dissolution of NaNH2, thus reducing the available reagent at the latter stages of the reaction. Formation of H2O results in hydrolysis of the nitride content, adding to the amorphous phase and reducing the apparent nitridation of the end product.
Figure 1.
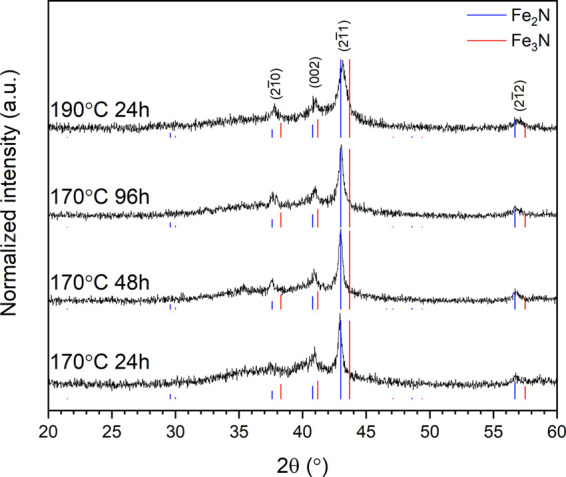
XRD data for the product of reaction of Fe3O4 and NaNH2 at 170 °C for (from bottom) 24, 48, and 96 h and at 190 °C for 24 h (top). XRD data are indexed in the space group P6322 structure of ε-Fe2+xN. Reference diffraction data for ε-Fe2N (PDF 04–017–2325) are shown in blue and ε-Fe3N in red (PDF 01-083-0878).
Table 1. Unit Cell Dimensions and Inferred Composition for ε-Fe2+xN Reaction Productsa.
| sample | a (Å) | c (Å) | volume (Å3) | x in Fe2+xN | estimated composition | avg. Fe oxidation state |
|---|---|---|---|---|---|---|
| 170 °C/24 h | 4.779(1) | 4.418(1) | 87.38(1) | 0.13(6) | Fe2.13N | 1.41 |
| 170 °C/48 h | 4.782(1) | 4.416(1) | 87.46(1) | 0.11(6) | Fe2.11N | 1.42 |
| 170 °C/96 h | 4.781(1) | 4.420(1) | 87.50(1) | 0.10(6) | Fe2.10N | 1.43 |
| 190 °C/24 h | 4.782(1) | 4.418(2) | 87.47(1) | 0.11(6) | Fe2.11N | 1.42 |
Unit cell parameters were calculated using Le Bail fits of XRD data. Estimation of the stoichiometry (x) was made from a calibration of unit cell volume from ICSD data28 (see Table S1 and Figure S1 in the Supporting Information).
Reducing the reaction temperature to 150 °C for 24 h afforded a negligible yield of the ε-Fe2+xN product. The reported melting point of NaNH2 is 200 °C; however, the presence of water (and the formation of NaOH) reduces the melting point to ca. 170 °C.29 This is consistent with our observation of the product forming a fused mass indicative of the reaction being mediated by a NaNH2–NaOH molten salt and negligible yield realized at 150 °C, below the eutectic melting temperature, where the kinetics of mass transport are limiting.29
In an effort to identify the noncrystalline phase contributing to the diffuse scattering apparent in the XRD data of Figure 1, we attempted to crystallize this phase by annealing at 180 °C for 24 h in air. The resulting XRD data exhibited relatively sharp Bragg reflections associated with ε-Fe2+xN, with no significant shift in 2θ position but evidently reduced full width at half-maximum (compare to Figure 2), indicative of larger crystallite size and/or narrower distribution of nonstoichiometry about the average of x ≈ 0.12(2). Moreover, very broad Bragg reflections were apparent at the location of previously observed diffuse intensity as shown in Figure 2 and a weak unindexed reflection at 2θ = 36° in Figure 1, which could be indexed on a face-centered cubic cell with a = 4.36(1) Å. Analysis of 57Fe Mössbauer data, discussed below, suggested that this phase is in fact an iron oxynitride, in the (wustite-Fe1–xO)1–y(γ‴–FeN)y solid solution, adopting a NaCl-type structure. Indeed, the determined unit cell parameter is intermediate between that of wustite Fe1–xO (a = 4.28–4.31 Å) and γ‴-FeN (a = 4.50 Å),30−32 consistent with such a solid solution. Hitherto, iron oxynitrides, of the type FeO1–xNx, have been reported only as thin films produced by magnetron sputtering or molecular beam epitaxy and exhibit unstrained unit cell parameters in the range 4.25 Å < a < 4.52 Å depending on composition (with (N + O)/Fe ≈ 1.0 and 0.13 < N/(N + O) < 0.65), consistent with our determined unit cell parameter.33−38 Analysis of 57Fe Mössbauer data also established the presence of minor γ″-FeN, adopting the zinc blende structure and a = 4.31 Å.39 This phase would also make a minor contribution to the broad Bragg reflections of the face-centered cubic phase indexed in Figure 2, based on the estimated phase fraction derived from 57Fe Mössbauer data.
Figure 2.
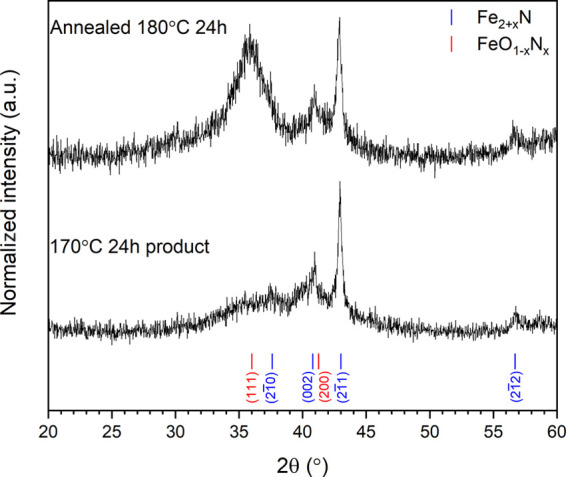
XRD patterns for product from reaction between Fe3O4 and NaNH2 at 170 °C/24 h (bottom) and post 180 °C/24 h anneal, showing the onset of broad Bragg reflections associated with a new face-centered cubic FeO1–xNx phase identified from Mössbauer analysis. Indexed reflections for ε-Fe2+xN (blue) and FeO1–xNx (red) are shown by tick marks; see the text for details.
SEM observation of the Fe3O4 reagent and ε-Fe2+xN product produced at 190 °C/24 h (Figure 3) revealed no significant change in the bimodal primary particle size. However, the ε-Fe2+xN product particles evidenced additional nanoscale texture compared to the reagent, consistent with the occurrence of an interfacial reaction occurring in a molten salt medium. EDX analysis (Figure S3) confirmed the presence of N in the product from the presence of N Kα emission, which was not observed in the reagent. Note that no Na was detected and hence residual NaNH2 is considered to have been removed by the washing procedure. Similar observations were made for the products obtained after reaction at 170 °C and are shown in Figure S2. The relative intensity of the N Kα emission was evidently reduced for ε-Fe2+xN (Figure S3) produced after reaction at 96 h consistent with an increase in the Fe/N ratio inferred from XRD analysis. From these data, we deduce that the synthesis of ε-Fe2+xN from Fe3O4 proceeds via a pseudomorphous reaction involving replacement of Fe3O4 by ε-Fe2+xN through reaction with NaNH2–NaOH molten salt. However, since the spinel structure of Fe3O4 and hexagonal ε-Fe2+xN do not have a three-dimensional correspondence, the reaction mechanism cannot readily be understood as being topotactic in nature.
Figure 3.

SEM images of untreated Fe3O4 (A) and product from reaction with NaNH2 at 190 °C for 24 h (B). Further SEM images of 170 °C reactions can be found in Figure S2 in the Supporting Information.
Further investigation of the product materials was made by transmission electron microscopy, which revealed the primary particles to be composed of nanoparticles of typically 5–20 nm dimension, shown in Figure 4. These nanoparticles exhibited both the presence and absence of fringes characteristic of both nanocrystalline and noncrystalline materials, consistent with XRD data. It is hypothesized that the distribution of amorphous material forms as a shell coating around intact cores of nitride material, as previously seen for amorphous MnOx layers around crystalline Mn4N2 particles.40 Selected area electron diffraction (SAED) patterns exhibited primarily rings consistent with sampling of many nanocrystalline domains, although the SAED data of the product arising from reaction at 190 °C/24 h also exhibited spots indicative of the presence of some single crystalline domains, consistent with the core–shell structures and inner crystalline SAED findings reported by Walter et al.40 Integration of the two-dimensional SAED data was performed, yielding one-dimensional diffraction patterns, to assist interpretation, as shown in Figure S4 in the Supporting Information. Consistent with XRD and 57Fe Mössbauer data shown in Figures 1 and 5, the SAED patterns can be interpreted as having contributions from ε-Fe2+xN (P6322) associated with diffraction maxima indexed as d(101) = 3.02 Å, d(110) = 2.39 Å, d(002) = 2.21 Å, d(111) = 2.10 Å, and d(112) = 1.62 Å, and an iron oxynitride, FeO1–xNx (Fm3̅m, a = 4.36 Å), associated with diffraction maxima indexed as d(111) = 2.52 Å, d(200) = 2.18 Å, d(220) = 1.52 Å, and d(311) = 1.34 Å.
Figure 4.

Electron diffraction and TEM images of product from reaction between Fe3O4 and NaNH2 reacted at 170 °C/24 h (top) and 190 °C/24 h (bottom).
Figure 5.
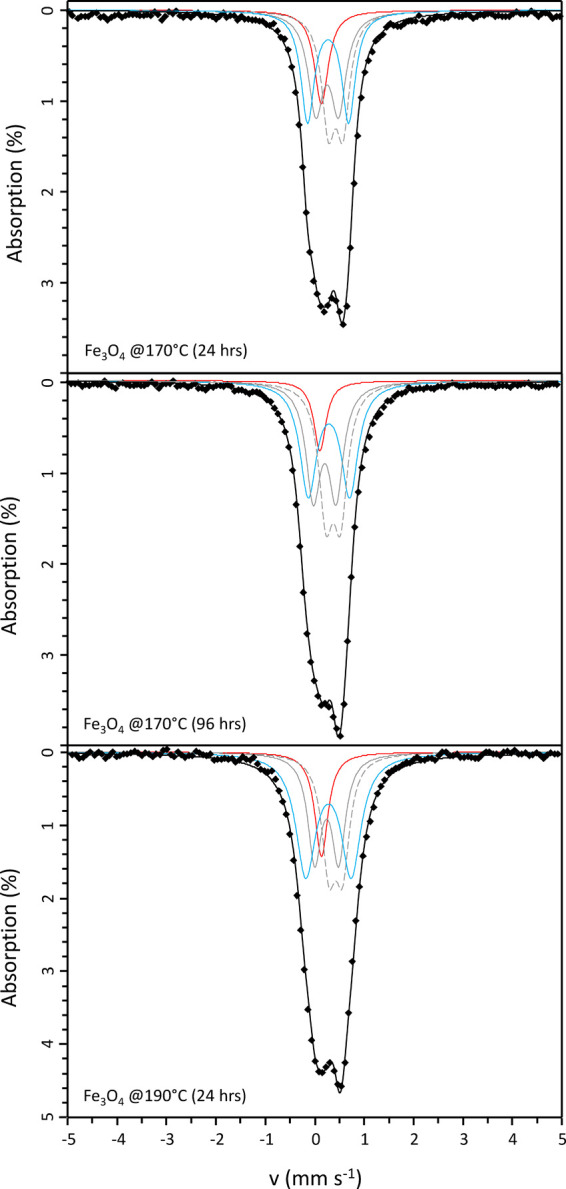
57Fe Mössbauer data from the product of reaction between Fe3O4 and NaNH2 at 170 °C/24 h and 96 h, and 190 °C for 24 h. Data are shown as solid points and the solid black line shows the overall model fit (components shown as blue, red, and gray lines; see the text for details).
57Fe Mössbauer spectroscopy analysis of the product materials revealed a paramagnetic doublet feature, with similar spectral envelope and the absence of any magnetic hyperfine splitting, as shown in Figure 5; fitting parameters are detailed in Table 2. Paramagnetic doublets indicate a composition ε-Fe2+xN with x < 0.4,41−45 consistent with XRD analyses. Hyperfine parameters have previously been reported in paramagnetic ε-Fe2+xN phases, characterized by several doublet subspectra contributing to the overall spectral envelope and associated with nitrogen ordering around the unique Fe(II) and Fe(III) sites44,45 (note that the nomenclature here is used to denote site designation, consistent with the literature, and does not relate to oxidation state). These are labeled D1 and D2 in our assignment in Table 2, represented by solid and dashed gray lines, respectively, in Figure 5. Refinement of the isomer shift and quadrupole shift of these contributions was followed by refinement of the site fractions, the refined parameters being consistent with previously reported values. Comparing the relative spectral area between the two components D1 and D2, the Fe(II) site, with two nearest-neighbor nitrogen atoms, contributes approximately 45% to the total signal for this phase. Based on the analysis of site fraction as a function of nitrogen content by Schaaf,45 the phase has a composition with 1.26 formula units of nitrogen ε-Fe3N1.26, otherwise expressed as ε-Fe2.38N. This is reasonably consistent with the interpretation of XRD data and the estimate of x = 0.12(2), described above. The inferred composition from XRD data relied upon the measurement of unit cell volume of comparatively broad reflections compared to the precision offered by Mössbauer spectroscopy, and, thus, the estimated compositions afforded from the Mössbauer spectroscopy data are preferred.
Table 2. Parameters Obtained from Fitting 57Fe Mössbauer Data of Products from Reaction between Fe3O4 and NaNH2a.
| sample | site | δ (mm s–1) | Δ (mm s–1) | Γ (mm s–1) | fraction (%) | assignment | approximate composition | average formal Fe oxidation state |
|---|---|---|---|---|---|---|---|---|
| 170 °C/24 h | S1 | 0.13 | 0.18 | 14 | γ″-FeN | FeNx | 3.00 | |
| D1 | 0.43 | 0.30 | 0.16 | 30 | ε-Fe2+xN Fe(III) | Fe2.38N | 1.26 | |
| D2 | 0.25 | 0.46 | 0.19 | 26 | ε-Fe2+xN Fe(II) | Fe2.38N | ||
| D3 | 0.27 | 0.83 | 0.17 | 29 | FeO1–xNx | FeO0.20N0.80 | 2.80 | |
| 170 °C/96 h | S1 | 0.16 | 0.14 | 8 | γ″-FeN | FeNx | 3.00 | |
| D1 | 0.44 | 0.29 | 0.18 | 31 | ε-Fe2+xN Fe(III) | Fe2.38N | 1.26 | |
| D2 | 0.25 | 0.46 | 0.18 | 28 | ε-Fe2+xN Fe(II) | Fe2.38N | ||
| D3 | 0.28 | 0.84 | 0.20 | 33 | FeO1–xNx | FeO0.25N0.75 | 2.75 | |
| 190 °C/24 h | S1 | 0.16 | 0.16 | 12 | γ″-FeN | FeNx | 3.00 | |
| D1 | 0.45 | 0.28 | 0.17 | 26 | ε-Fe2+xN Fe(III) | Fe2.38N | 1.26 | |
| D2 | 0.26 | 0.48 | 0.17 | 23 | ε-Fe2+xN Fe(II) | Fe2.38N | ||
| D3 | 0.30 | 0.92 | 0.24 | 39 | FeO1–xNx | FeO0.35N0.65 | 2.65 |
δ – Isomer Shift, Δ – Quadrupole Splitting, Γ – Half-Width Full Max. Δ is given relative to metallic Fe at room temperature. Estimated precision on δ, Δ, and Γ is 0.02 mm s–1 and 2% on the site fraction. Site assignment for each spectral component is indicated.
On using the analysis from Schaaf, we assume that the recoil-free fractions (f) of the Fe(II) and Fe(III) sites are equal and thus the spectral area is directly proportional to the number of absorbing nuclei. The recoil-free fraction (f) is a measure of the probability of recoil-free emission from the Fe atoms and is influenced by the strength of the interatomic forces between those atoms and the crystal lattice.46 The fraction therefore depends on the local structure of the surrounding lattice and according to the Debye model has contributions related to temperature, the energy of the γ ray, and the Debye temperature of the crystal.45 Limited information exists in the literature on the determination of recoil-free fractions in iron nitrides; however, similar Debye temperatures have been reported for ε-FexN (2 < x < 3),47,48 γ-FeNx,49 Fe4N,50 and FeOx51 phases. This suggests that all of the phases assigned in our model (and their individual subspectra) have similar recoil-free fractions and the assumption of f = 1, for all of the Fe sites, is reasonable.
The lower weighting of Fe(II) sites to Fe(III) sites is consistent with findings for the ε-FexN (2 < x < 3) system measured between 4.2 and 300 K.44 The compositions of ε-Fe2.47N and ε-Fe2.20N have Fe(II) site fractions of 58 and 27%, respectively, which places our intermediate ε-Fe2.38N product with 45% Fe(II) sites satisfactorily within this region.
The remaining doublet D3 (blue line in Figure 5) is assigned to a paramagnetic iron oxynitride phase, which was identified in both XRD and SAED data, and implied by the relatively wide spectral envelope characteristic of oxynitrides with large quadrupole splitting. 57Fe Mössbauer spectra have previously been reported for iron oxynitrides with additional contributions assigned to ε-Fe2+xN (0 ≤ x ≤ 1) or γ″-FeN suggesting the simultaneous formation and coexistence of these phases.36,38,43 For iron oxynitride thin films, the relative contribution of the doublet to the total signal increased with increasing oxygen content, which was assigned to a composition FeyO1–xNx with y ≈ 1.36,38 Increased measured oxygen content in the films was correlated to the observed increase in isomer shift values. The isomer shift values for the doublets we observed (ca. 0.29 mm s–1) suggest an oxygen content of approximately 15 atom % equivalent to FeO0.32N0.38.
The singlet S1 (red line in Figure 5) was assigned to a binary nitride compound FeNx, for which face-centered cubic γ‴-FeN (NaCl structure) and γ″-FeN (ZnS structure) were considered as candidate species. Previous reports indicate that these can coexist and both present a singlet in Mössbauer measurements. However, the distinction can be made by the values of isomer shift; the isomer shift for γ″-FeN is approximately 0.1 mm s–1, while γ‴-FeN has an isomer shift of 0.6 mm s–1.52,53 This arises due to a different local geometry of the N environment. Our refined Mössbauer parameters are consistent with the γ″-FeN phase.42,54,55
Overall, the Mössbauer analysis confirms a phase assemblage for the reaction products of ε-Fe2+xN (x ≈ 0.38), FeO1–xNx (0.65 ≤ x ≤ 0.80), and γ″-FeN in a decreasing order of prevalence based upon refined site fractions. The identified phases are consistent with other characterization data presented here, similarly showing decreased total nitridation with increasing temperature and time. Estimated ε-Fe2+xN compositions of x ≈ 0.12 were made from XRD data (see Table 1), in comparison to x ≈ 0.38 implied from Mössbauer analysis, which are preferred as noted above. This corresponds to ε-Fe2+xN with estimated average formal Fe oxidation states in the range 1.43–1.26+.
We note that the analysis of the Mössbauer data may be complicated by the previously determined nanoparticle size. Nanoparticles can inhibit the presence of any ferromagnetic hyperfine splitting due to an absence of long-range magnetic order,56 as would impurities within those particles. Our analysis also extrapolates findings reported for thin films to the nanoparticulate system evident in the data. Thus, our determination of relative phase fractions and inferred stoichiometries should be interpreted with due caution.
Figure 6 shows the TGA-MS analysis of the product obtained from reaction at 170 °C for 24 h obtained under flowing Ar. These data show two distinctive regions of weight loss. The first region, from 25 to 270 °C, corresponds to a weight loss of 2.5 wt %, which was dominated by the loss of H2O, as evidenced by the MS signal corresponding to m/z = 18. The product therefore evidently retains both physisorbed water (lost below ca. 100 °C) and chemisorbed water (retained up to 270 °C). The second region, from 270 to 800 °C, corresponds to a further weight loss of 12.9 wt %, which was dominated by the loss of N2, as evidenced by the MS signal corresponding to m/z = 28. The material recovered after the TGA analysis was found to comprise Fe1–xO and Fe, indicating complete decomposition of the ε-Fe2+xN and FeO1–xNx phases, with evolution of N2 as evidenced by the MS data. Combined with the analysis of 57Fe Mössbauer data, assuming the site fractions are directly proportional to the spectral area in determining the initial phase assemblage, the weight loss above 270 °C is attributed to the following (approximate) decomposition reaction
| 1 |
Figure 6.
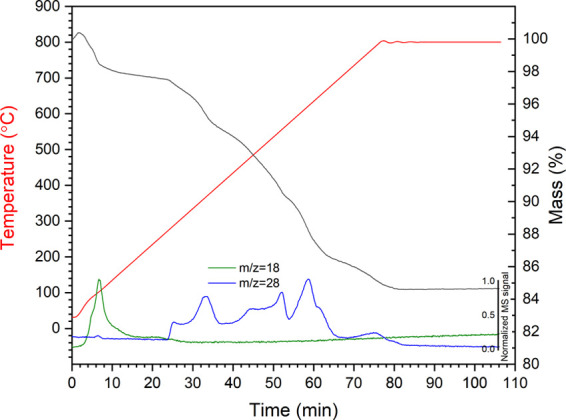
Thermogravimetry (TG, black) curve and mass spectrometry (MS) signals of 170 °C 24 h amide product (mass numbers 18 and 28 represent H2O and N2, respectively).
The expected weight loss for this reaction is between 11.8 wt % (x = 0.38, as implied from Mössbauer) and 12.6 wt % (x = 0.12, as implied from XRD). This is in reasonable agreement with the experimentally determined weight loss of 13.0 wt %, renormalized for loss of water below 270 °C, given the overall uncertainty in the precise stoichiometry of the component phases. TGA-MS data for the product obtained from reaction at 190 °C for 24 h, obtained under a flowing Ar, is shown in Figure S5 in the Supporting Information and exhibits two similar and distinctive weight loss events attributable to loss of water from 25 to 290 °C and loss of N2 from 290 to 800 °C. A similar calculation estimated the expected weight loss to be between 11.4 wt % (x = 0.38) and 12.2 wt % (x = 0.11), which was again in reasonable agreement with the experimentally determined weight loss of 10.7 wt %, renormalized for loss of water.
The TGA-MS data of the product from reaction at 170 °C for 24 h exhibits a staged weight loss in the second region between 270 and 800 °C, with onset at approximately (1) 270 °C, (2) 325 °C, (3) 425 °C, (3) 600 °C, and (4) 725 °C. Considering these events in order, we first attribute the onset of weight loss at 270 °C to the decomposition of the oxynitride FeO1–xNx phase to Fe1–xO (with the evolution of N2). This is consistent with the reported synthesis conditions required for FeO1–xNx oxynitrides, which were stabilized only in the thin film formed at low temperature, by magnetron sputtering with a substrate temperature estimated not to exceed 50 °C,38 or by molecular beam epitaxy with a substrate temperature of 250 °C.33,34 The weight loss at approximately 325 °C, associated with N2 loss, is consistent with the decomposition mechanism for γ″-FeN described by Suzuki et al.39 In the range of 320–340 °C, under vacuum, γ″-FeN was reported to decompose to ζ-Fe2N and ε-Fe2+xN with the release of N2. Decomposition of γ″-FeN was reported to be complete at 365 °C, yielding nearly single-phase ε-Fe2+xN. Widenmeyer et al.57 reported TGA analysis of the decomposition of a phase with composition ε-Fe2.17N under Ar. They established a two-stage mechanism of decomposition, which involves the onset of decomposition to γ′-Fe4+xN above 422 °C (with evolution of N2) and subsequent onset of decomposition to α-Fe above 622 °C (again, with evolution of N2), which is complete by 800 °C (these findings are also in general agreement with the findings of Suzuki et al.39). Neutron diffraction data has shown that this decomposition also involves the formation of austentite-type γ′-FeNz (<10 atom % N), which is fully converted to α-Fe at 679 °C. Thus, we attribute the weight losses observed in our data at 425, 600, and 725 °C, each accompanied by loss of N2 evidenced in the MS signal (Figure 6), to the sequential decomposition of ε-Fe2+xN → γ′-Fe4+xN → γ′-FeNz → α-Fe, broadly consistent with the reported literature data for the thermal stability of these phases.
Low-temperature nitridation of metal oxides by reaction with NaNH2 has proven an effective route to synthesis of InN (from LiInO215), ε-Fe2+xN (from Fe3O4 as shown here and Fe2O318), Mn6N5+x (from Mn2O318), and Cu3N (from CuO).13 Consideration of the thermodynamics of this reaction suggested that the driver is the high Gibbs free energy of NaOH combined with the extremely high local concentration of NH2–/NH3 achieved in the melt (with excess NaNH2).18 NaOH is formed in a metathesis reaction between the iron oxide and sodium amide by exchange of oxide and nitride anions. The formation of NaOH then results in further dissolution of NaNH2 and propagation of the metathesis and nitridation. The NaNH2 reaction is evidently reducing in nature, as exemplified by the reaction of CuO with NaNH2 to form Cu3N at 170 °C/60 h, but resulting in the formation of Cu at 190 °C/60 h.13 The more reduced nature of Fe3O4 therefore explains the exceptionally mild synthesis conditions required to yield ε-Fe2+xN by reaction with NaNH2 (170 °C/24 h), compared to that of Fe2O3 (240 °C/36 h). Hence, the redox character of the metal oxide substrate is recognized and demonstrated to be an important consideration in optimizing the conditions for NaNH2-mediated nitridation reactions. Furthermore, we achieved synthesis of ε-Fe2+xN by reaction between Fe3O4 and NaNH2, evidently mediated in a molten salt, although the reaction temperature is below the melting point of pure NaNH2 (210 °C). This suggests that trace water and NaOH byproduct play an important role in reducing the melting point in these reactions, consistent with the observations reported here, yielding a NaNH2–NaOH molten salt with a melting point between 210 °C and 170 °C.29 Our characterization data confirm the general understanding of a NaNH2 molten salt-mediated dissolution–precipitation reaction, which results in the pseudomorphous replacement of the metal oxide substrate with a nanocrystalline product in a metathesis reaction, consistent with reports of similar reactions.18 Similar solid-state metathesis reactions have yielded various binary, ternary, and quaternary nitrides, recent examples being Mn3N258 and Zn1–xMnxSiN2.59 The reaction between Fe3O4 and NaNH2 produced ε-Fe2+xN as the major product, but also a noncrystalline phase, as was also observed in the reaction with Fe2O3.18 We successfully crystallized this phase to yield a face-centered iron oxynitride, FeO1–xNx (and γ″-FeN), as verified using 57Fe Mössbauer spectroscopy. The presence of such an oxynitride phase in the product of reaction between Fe2O3 and NaNH2 was hypothesized by Miura et al., according to X-ray photoelectron spectroscopy data, which evidenced a material with an O-rich surface and N-rich interior. Miura et al. suggested that this phase was formed in the reaction workup by hydrolysis of the ε-Fe2+xN phase.18 This hypothesis is in agreement with a recently published analysis of solution thermochemical data, showing that Fe2N is stable in strongly alkaline and reducing conditions, otherwise decomposing to yield Fe2O3 or Fe3O4.60 However, we cannot exclude the possibility that the FeO1–xNx is formed, at least in part, by in situ reaction between ε-Fe2+xN and/or γ″-FeN with the NaOH byproduct. The presence of FeO1–xNx from this synthesis in substantial yield may also prove exploitable for metastable oxynitride synthesis. Substantial data harvesting, application of informatics tools, and subsequent density functional theory analyses have identified many metastable nitrides that are attainable experimentally,61−63 which the authors suggest is further applicable to oxynitrides.62 These rely upon “remnant metastability”, such that the nitride is synthesized in targeted thermodynamic conditions where it is stable and then kinetically retained when transitioned to metastable conditions,61 which the pseudomorphic reaction with NaNH2 in a sealed system and careful postreaction workup of the products may be able to achieve.
Conclusions
Reaction of Fe3O4 with excess NaNH2 produced ε-Fe2+xN under exceptionally mild conditions of 170 °C for 24 h. The reaction takes place by pseudomorphous replacement of the reagent by the product, mediated by a NaNH2–NaOH molten salt phase, involving dissolution of Fe3O4 and precipitation of nanocrystalline product. The recovered product contains an amorphous component, which was crystallized to yield an oxynitride, FeO1–xNx. 57Fe Mössbauer spectra could be fitted with contributions of ε-Fe2+xN, FeO1–xNx, and γ″-FeN in agreement with the observed phase assemblage. Interpretation of coupled thermogravimetric and mass spectroscopy data was consistent with this phase assemblage, with the observed mass loss attributed to N2 in reasonable agreement with that expected based on the relative proportions of phases estimated from 57Mössbauer spectroscopy and decomposition temperatures in reasonable agreement with available data for stepwise decomposition of ε-Fe2+xN to α-Fe. The yield of ε-Fe2+xN in the recovered product was 49–59 mol %, accompanied by 29–39 mol % FeO1–xNx, as determined from refined site fractions from 57Mössbauer spectroscopy. It is not yet clear whether FeO1–xNx is formed as a primary reaction product or by hydrolysis of ε-Fe2+xN in the reaction workup. Either way, this may pose a limitation to the synthesis of ε-Fe2+xN, and other metal nitrides, if the objective is to produce a single-phase product with well-defined electrical, magnetic, or other physical property for applications. On the other hand, the reported FeO1–xNx oxynitride phase identified in this study is an interesting outcome and opens the possibility for the synthesis of metastable oxynitride phases in high yield, by reaction of a metal oxide substrate with NaNH2, with either careful control of H2O concentration in the system or postsynthetic hydrolysis and crystallization.
Acknowledgments
This research utilized the HADES/MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/1.64 The authors are grateful for financial support from EPSRC under grant numbers EP/L015390/1 (S.E.O., N.C.H.), EP/S011935/1 (S.E.O., S.-K.S., N.C.H.), EP/S01019X/1 and EP/P013600/1 (S.-K.S., N.C.H.), and EP/N017374/1 (S.M.L., C.L.C.). The authors wish to acknowledge the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P02470X/1, and EP/P025285/1, for JEOL JEM-F200 access at Royce@Sheffield. Y.M. is also supported partly by JSPS Grant-in-Aid for Scientific Research on Innovative Areas “Mixed anion” (Grant Number JP16H06439).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c03452.
ICSD data used for stoichiometry estimation (Table S1); calibration graph of ICSD data for stoichiometry estimation (Figure S1); SEM of 170 °C for 24 and 96 h products (Figure S2); EDX spectra for starting reagent and reaction products (Figure S3); integrated 1d line scans from electron diffraction data (Figure S4); and TGA data for 190 °C/24 h sample (Figure S5) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bhattacharyya S. Iron Nitride Family at Reduced Dimensions: A Review of Their Synthesis Protocols and Structural and Magnetic Properties. J. Phys. Chem. A 2015, 119, 1601–1622. 10.1021/jp510606z. [DOI] [Google Scholar]
- Rasaki S. A.; Zhang B.; Anbalgam K.; Thomas T.; Yang M. Synthesis and application of nano-structured metal nitrides and carbides: A review. Prog. Solid State Chem. 2018, 50, 1–15. 10.1016/j.progsolidstchem.2018.05.001. [DOI] [Google Scholar]
- Ebbinghaus S. G.; Abicht H.-P.; Dronskowski R.; Müller T.; Reller A.; Weidenkaff A. Perovskite related oxynitrides recent developments in synthesis, characterisation and investigations of physical properties. Prog. Solid State Chem. 2009, 37, 173–205. 10.1016/j.progsolidstchem.2009.11.003. [DOI] [Google Scholar]
- Miura A. Low-temperature synthesis and rational design of nitrides and oxynitrides for novel functional material development. J. Ceram. Soc. Jpn. 2017, 125, 552–558. 10.2109/jcersj2.17055. [DOI] [Google Scholar]
- Giordano C.; Erpen C.; Yao W.; Milke B.; Antonietti M. Metal nitride and metal carbide nanoparticles by a soft urea pathway. Chem. Mater. 2009, 21, 5136–5144. 10.1021/cm9018953. [DOI] [Google Scholar]
- Gao Z.; Wan Y.; Xiong G.; Guo R.; Luo H. Synthesis of aluminum nitride nanoparticles by a facile urea glass route and influence of urea/metal molar ratio. Appl. Surf. Sci. 2013, 280, 42–49. 10.1016/j.apsusc.2013.04.075. [DOI] [Google Scholar]
- Masubuchi Y.; Tadaki M.; Kikkawa S. Synthesis of the perovskite SrTaO2N using C3N4 for both reduction and nitridation. Chem. Lett. 2018, 47, 31–33. 10.1246/cl.170953. [DOI] [Google Scholar]
- Zhao H.; Lei M.; Chen X.; Tang W. Facile route to metal nitrides through melamine and metal oxides. J. Mater. Chem. 2006, 16, 4407–4412. 10.1039/b611381h. [DOI] [Google Scholar]
- Feng X.; Bai Y.-J.; Lu B.; Wang C.-G.; Qi Y.-X.; Liu Y.-X.; Geng G.-L.; Li L. Low Temperature Induced Synthesis of TiN Nanocrystals. Inorg. Chem. 2004, 43, 3558–3560. 10.1021/ic049841n. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Xie Y.; Wang X.; Lv S.; Hou T.; Bai C. Synthesis of uniform titanium nitride nanocrystalline powders via a reduction–hydrogenation–dehydrogenation–nitridation route. J. Am. Ceram. Soc. 2005, 88, 249–251. 10.1111/j.1551-2916.2004.00050.x. [DOI] [Google Scholar]
- Huang Y.; Gu Y.; Zheng M.; Xu Z.; Zeng W.; Liu Y. Synthesis of nanocrystalline titanium nitride by reacting titanium dioxide with sodium amide. Mater. Lett. 2007, 61, 1056–1059. 10.1016/j.matlet.2006.06.048. [DOI] [Google Scholar]
- Chen L.; Gu Y.; Shi L.; Yang Z.; Ma J.; Qian Y. A room-temperature synthesis of nanocrystalline vanadium nitride. Solid State Commun. 2004, 132, 343–346. 10.1016/j.ssc.2004.07.041. [DOI] [Google Scholar]
- Miura A.; Takei T.; Kumada N. Synthesis of Cu3N from CuO and NaNH2. J. Asian Ceram. Soc. 2014, 2, 326–328. 10.1016/j.jascer.2014.08.007. [DOI] [Google Scholar]
- Barick B.; Dhar S. Surface and bulk electronic properties of low temperature synthesized InN microcrystals. J. Cryst. Growth 2015, 416, 154–158. 10.1016/j.jcrysgro.2015.01.010. [DOI] [Google Scholar]
- Miura A.; Takei T.; Kumada N. Synthesis of Wurtzite-Type InN Crystals by Low Temperature Nitridation of LiInO2 Using NaNH2 Flux. Cryst. Growth Des. 2012, 12, 4545–4547. 10.1021/cg3007266. [DOI] [Google Scholar]
- Chen L.; Huang T.; Qian Y.; Zhu L.; Gu Y. Nitridation of Silica to an α-Silicon Nitride Nanorod Using NaNH2 in the Autoclave at 700C. J. Am. Ceram. Soc. 2007, 90, 1243–1245. 10.1111/j.1551-2916.2007.01411.x. [DOI] [Google Scholar]
- Chen L.; Gu Y.; Shi L.; Yang Z.; Ma J.; Qian Y. A room-temperature approach to boron nitride hollow spheres. Solid State Commun. 2004, 130, 537–540. 10.1016/j.ssc.2004.03.009. [DOI] [Google Scholar]
- Miura A.; Takei T.; Kumada N. Low-Temperature Nitridation of Manganese and Iron Oxides Using NaNH2 Molten Salt. Inorg. Chem. 2013, 52, 11787–11791. 10.1021/ic401951u. [DOI] [PubMed] [Google Scholar]
- Laassiri S.; Zeinalipour-Yazdi C. D.; Catlow C. R. A.; Hargreaves J. S. The potential of manganese nitride based materials as nitrogen transfer reagents for nitrogen chemical looping. Appl. Catal., B 2018, 223, 60–66. 10.1016/j.apcatb.2017.04.073. [DOI] [Google Scholar]
- Chang F.; Guo J.; Wu G.; Wang P.; Yu P.; Chen P. Influence of alkali metal amides on the catalytic activity of manganese nitride for ammonia decomposition. Catal. Today 2017, 286, 141–146. 10.1016/j.cattod.2016.09.010. [DOI] [Google Scholar]
- Clark D. E. Peroxides and peroxide-forming compounds. Chem. Health Safety 2001, 8, 12–22. 10.1016/S1074-9098(01)00247-7. [DOI] [Google Scholar]
- Lagarec K.; Rancourt D. G.. Recoil—Mössbauer spectral analysis software for Windows, tech. rep.; Department of Physics, University of Ottawa: Ottawa, ON, Canada, 1998. [Google Scholar]
- Schneider C.; Rasband W.; Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergerhoff G.; Brown I. D. Inorganic Crystal Structure Database. Acta Crystallogr., Sect. A: Found. Crystallogr. 1981, 37, C342. 10.1107/S0108767381089411. [DOI] [Google Scholar]
- du Marchie van Voorthuysen E. H.; Boerma D. O.; Chechenin N. Low-temperature extension of the Lehrer diagram and the iron-nitrogen phase diagram. Metall. Mater. Trans. A 2002, 33, 2593–2598. 10.1007/s11661-002-0380-2. [DOI] [Google Scholar]
- Jacobs H.; Rechenbach D.; Zachwieja U. Structure determination of γ′-Fe4N and ε-Fe3N. J. Alloys Compd. 1995, 227, 10–17. 10.1016/0925-8388(95)01610-4. [DOI] [Google Scholar]
- Leineweber A.; Jacobs H.; Hüning F.; Lueken H.; Kockelmann W. Nitrogen ordering and ferromagnetic properties of ε-Fe3N1 + x (0.106 ≤ x ≤ 60.39) and ε-Fe3(N0.80C0.20)1.38. J. Alloys Compd. 2001, 316, 21–38. 10.1016/S0925-8388(00)01435-3. [DOI] [Google Scholar]
- Jack K. H. Binary and Ternary Interstitial Alloys. I. The Iron-Nitrogen System: The Structures of Fe4N and Fe2N. Proc. R. Soc. London, Ser. A 1948, 195, 34–40. 10.1098/rspa.1948.0100. [DOI] [Google Scholar]
- Jepsen L. H.; Wang P.; Wu G.; Xiong Z.; Besenbacher F.; Chen P.; Jensen T. R. Thermal decomposition of sodium amide, NaNH2, and sodium amide hydroxide composites, NaNH2–NaOH. Phys. Chem. Chem. Phys. 2016, 18, 25257–25264. 10.1039/C6CP01604A. [DOI] [PubMed] [Google Scholar]
- Willis B. T. M.; Rooksby H. P. Change of structure of ferrous oxide at low temperature. Acta Crystallogr. 1953, 6, 827–831. 10.1107/S0365110X53002441. [DOI] [Google Scholar]
- Yamamoto A. Modulated structure of wustite (Fe1–xO) (three-dimensional modulation). Acta Crystallogr., Sect. B: Struct. Sci. 1982, 38, 1451–1456. 10.1107/S056774088200613X. [DOI] [Google Scholar]
- Jette E. R.; Foote F. An X-Ray Study of the Wüstite (FeO) Solid Solutions. J. Chem. Phys. 1933, 1, 29–36. 10.1063/1.1749215. [DOI] [Google Scholar]
- Voogt F. C.; Hibma T.; Smulders P.; Iesen L.; Fujii T. MBE-Growth of Iron and Iron Oxide Thin Films on MgO(100), Using NO2, NO, and N2O as Oxidising Agents. MRS Proc. 1997, 474, 211–216. 10.1557/PROC-474-211. [DOI] [Google Scholar]
- Smulders P.; Wijnja G.; Niesen L.; Fujii T.; Voogt F. C.; Smulders P. J. M.; Wijnja G. H.; James M. A.; Hibma T. NO2-assisted molecular-beam epitaxy of wustite like and magnetite like Fe oxynitride films on MgO(100). Phys. Revi. 2001, 63, 125409 10.1103/PhysRevB.63.125409. [DOI] [Google Scholar]
- Petitjean C.; Grafouté M.; Pierson J. F.; Rousselot C.; Banakh O. Structural, optical and electrical properties of reactively sputtered iron oxynitride films. J. Phys. D: Appl. Phys. 2006, 39, 1894–1898. 10.1088/0022-3727/39/9/026. [DOI] [Google Scholar]
- Grafouté M.; Petitjean C.; Rousselot C.; Pierson J.; Grenèche J. Chemical environment of iron atoms in iron oxynitride films synthesized by reactive magnetron sputtering. Scr. Mater. 2007, 56, 153–156. 10.1016/j.scriptamat.2006.09.012. [DOI] [Google Scholar]
- Petitjean C.; Grafouté M.; Rousselot C.; Pierson J. F. Reactive gas pulsing process: A method to extend the composition range in sputtered iron oxynitride films. Surf. Coat. Technol. 2008, 202, 4825–4829. 10.1016/j.surfcoat.2008.04.064. [DOI] [Google Scholar]
- Grafouté M.; Petitjean C.; Diama A.; Pierson J.; Greneche J.; Rousselot C. Structural investigations of iron oxynitride multilayered films obtained by reactive gas pulsing process. Surf. Coat. Technol. 2015, 272, 158–164. 10.1016/j.surfcoat.2015.04.010. [DOI] [Google Scholar]
- Suzuki K.; Morita H.; Kaneko T.; Yoshida H.; Fujimori H. Crystal structure and magnetic properties of the compound FeN. J. Alloys Compd. 1993, 201, 11–16. 10.1016/0925-8388(93)90854-G. [DOI] [Google Scholar]
- Walter C.; Menezes P. W.; Orthmann S.; Schuch J.; Connor P.; Kaiser B.; Lerch M.; Driess M. A Molecular Approach to Manganese Nitride Acting as a High Performance Electrocatalyst in the Oxygen Evolution Reaction. Angew. Chem., Int. Ed. 2018, 57, 698–702. 10.1002/anie.201710460. [DOI] [PubMed] [Google Scholar]
- Schaaf P.; Illgner C.; Niederdrenk M.; Lieb K. Characterization of laser-nitrided iron and sputtered iron nitride films. Hyperfine Interact. 1995, 95, 199–225. 10.1007/BF02146315. [DOI] [Google Scholar]
- Prieto P.; Marco J. F.; Sanz J. M. Synthesis and characterization of iron nitrides. An XRD, Mössbauer, RBS and XPS characterization. Surf. Interface Anal. 2008, 40, 781–785. 10.1002/sia.2658. [DOI] [Google Scholar]
- Borsa D.; Boerma D. Phase identification of iron nitrides and iron oxy-nitrides with Mössbauer spectroscopy. Hyperfine Interact. 2003, 151/152, 31–48. 10.1023/B:HYPE.0000020403.64670.02. [DOI] [Google Scholar]
- Chen G.; Jaggl N.; Butt J.; Yeh E.; Schwartz L. Mössbauer and magnetic studies of ε-FexN, 2 < x < 3. J. Phys. Chem. A 1983, 87, 5326–5332. 10.1021/j150644a005. [DOI] [Google Scholar]
- Schaaf P. Laser nitriding of metals. Prog. Mater. Sci. 2002, 47, 1–161. 10.1016/S0079-6425(00)00003-7. [DOI] [Google Scholar]
- Qaim S. M. Recoil-free fractions of the 14.4 keV Mössbauer gamma line of 57Fe in various host lattice. J. Phys. F: Met. Phys. 1971, 1, 320–327. 10.1088/0305-4608/1/3/313. [DOI] [Google Scholar]
- Kurian S.; Gajbhiye N. S. Magnetic and Mössbauer study of ε-FeyN (2 < y < 3) nanoparticles. J. Nanopart. Res. 2010, 12, 1197–1209. 10.1007/s11051-009-9793-9. [DOI] [Google Scholar]
- Litasov K. D.; Shatskiy A.; Ponomarev D. S.; Gavryushkin P. N. Equations of state of iron nitrides ε-Fe3Nx and γ-Fe4Ny to 30 GPa and 1200 K and implications for nitrogen in the Earth’s core. J. Geophys. Res.:Solid Earth 2017, 122, 3574–3584. 10.1002/2017JB014059. [DOI] [Google Scholar]
- Oda K.; Umezu K.; Ino H. Interaction and arrangement of nitrogen atoms in FCC γ-iron. J. Phys.: Condens. Matter 1990, 2, 10147–10158. 10.1088/0953-8984/2/50/018. [DOI] [Google Scholar]
- Imbihl R.; Behm R. J.; Ertl G.; Moritz W. The structure of atomic nitrogen absorbed on Fe(100). Surf. Sci. 1982, 123, 129–140. 10.1016/0039-6028(82)90135-2. [DOI] [Google Scholar]
- Fisher R. A.; Campbell A. J.; Shofner G. A.; Lord O. T.; Dera P.; Prakapenka V. B. Equation of state and phase diagram of FeO. Earth Planet. Sci. Lett. 2011, 304, 496–502. 10.1016/j.epsl.2011.02.025. [DOI] [Google Scholar]
- Rissanen L.; Neubauer M.; Lieb K.; Schaaf P. The new cubic iron-nitride phase FeN prepared by reactive magnetron sputtering. J. Alloys Compd. 1998, 274, 74–82. 10.1016/S0925-8388(98)00594-5. [DOI] [Google Scholar]
- Rissanen L.; Schaaf P.; Neubauer M.; Lieb K.-P.; Keinonen J.; Sajavaara T. The production of the new cubic FeN phase by reactive magnetron sputtering. Appl. Surf. Sci. 1999, 138–139, 261–265. 10.1016/S0169-4332(98)00404-8. [DOI] [Google Scholar]
- Frątczak E. Z.; Prieto J.; Moneta M. Magnetic study of α″ and γ″-phases of iron nitride thin films. Acta Phys. Pol., A 2014, 126, 214–215. 10.12693/APhysPolA.126.214. [DOI] [Google Scholar]
- Hinomura T.; Nasu S. A study of Fe–N alloy systems. Hyperfine Interact. 1998, 111, 221–226. 10.1023/A:1012614019538. [DOI] [Google Scholar]
- Murad E. Mössbauer spectroscopy of clays, soils and their mineral constituents. Clay Miner. 2010, 45, 413–430. 10.1180/claymin.2010.045.4.413. [DOI] [Google Scholar]
- Widenmeyer M.; Hansen T. C.; Meissner E.; Niewa R. Formation and Decomposition of Iron Nitrides Observed by in situ Powder Neutron Diffraction and Thermal Analysis. Z. anorg. allg. Chem. 2014, 640, 1265–1274. 10.1002/zaac.201300676. [DOI] [Google Scholar]
- Rognerud E. G.; Rom C. L.; Todd P. K.; Singstock N. R.; Bartel C. J.; Holder A. M.; Neilson J. R. Kinetically Controlled Low-Temperature Solid-State Metathesis of Manganese Nitride Mn3N2. Chem. Mater. 2019, 31, 7248–7254. 10.1021/acs.chemmater.9b01565. [DOI] [Google Scholar]
- Zeman O. E. O.; von Rohr F. O.; Neudert L.; Schnick W. Facile One-step Synthesis of Zn1–xMnxSiN2 Nitride Semiconductor Solid Solutions via Solid-state Metathesis Reaction. Z. anorg. allg. Chem. 2020, 646, 228–233. 10.1002/zaac.201900315. [DOI] [Google Scholar]
- Matanovic I.; Garzon F. H. Assessing Stability of Transition Metal Nitrides in Aqueous Environments: The Case of Molybdenum, Iron, Vanadium and Nickel Nitride. J. Electrochem. Soc. 2020, 167, 046518 10.1149/1945-7111/ab7a8b. [DOI] [Google Scholar]
- Sun W.; Dacek S. T.; Ong S. P.; Hautier G.; Jain A.; Richards W. D.; Gamst A. C.; Persson K. A.; Ceder G. The thermodynamic scale of inorganic crystalline metastability. Sci. Adv. 2016, 2, e1600225 10.1126/sciadv.1600225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W.; Holder A. M.; Orvañanos B.; Arca E.; Zakutayev A.; Lany S.; Ceder G. Thermodynamic Routes to Novel Metastable Nitrogen-Rich Nitrides. Chem. Mater. 2017, 29, 6936–6946. 10.1021/acs.chemmater.7b02399. [DOI] [Google Scholar]
- Sun W.; Bartel C. J.; Arca E.; Bauers S. R.; Matthews B.; Orvañanos B.; Chen B.-R.; Toney M. F.; Schelhas L. T.; Tumas W.; Tate J.; Zakutayev A.; Lany S.; Holder A. M.; Ceder G. A map of the inorganic ternary metal nitrides. Nat. Mater. 2019, 18, 732–739. 10.1038/s41563-019-0396-2. [DOI] [PubMed] [Google Scholar]
- Hyatt N. C.; Corkhill C. L.; Stennett M. C.; Hand R. J.; Gardner L. J.; Thorpe C. L. The HADES facility for high activity decommissioning engineering & science: part of the UK national nuclear user facility. IOP Conf. Ser.: Mater. Sci. Eng. 2020, 818, 012022 10.1088/1757-899X/818/1/012022. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.