

The wild giant panda in their habitat in Liziping National Nature Reserve, Shimian. The copywight belongs to Liziping National Nature Reserve, Shimian.

Summary
The rise in infections by antibiotic‐resistant bacteria poses a serious public health problem worldwide. The gut microbiome of animals is a reservoir for antibiotic resistance genes (ARGs). However, the correlation between the gut microbiome of wild animals and ARGs remains controversial. Here, based on the metagenomes of giant pandas (including three wild populations from the Qinling, Qionglai and Xiaoxiangling Mountains, and two major captive populations from Yaan and Chengdu), we investigated the potential correlation between the constitution of the gut microbiome and the composition of ARGs across the different geographic locations and living environments. We found that the types of ARGs were correlated with gut microbiome composition. The NMDS cluster analysis using Jaccard distance of the ARGs composition of the gut microbiome of wild giant pandas displayed a difference based on geographic location. Captivity also had an effect on the differences in ARGs composition. Furthermore, we found that the Qinling population exhibited profound dissimilarities of both gut microbiome composition and ARGs (the highest proportion of Clostridium and vancomycin resistance genes) when compared to the other wild and captive populations studies, which was supported by previous giant panda whole‐genome sequencing analysis. In this study, we provide an example of a potential consensus pattern regarding host population genetics, symbiotic gut microbiome and ARGs. We revealed that habitat isolation impacts the ARG structure in the gut microbiome of mammals. Therefore, the difference in ARG composition between giant panda populations will provide some basic information for their conservation and management, especially for captive populations.
Introduction
Worldwide, the rise in infections by antibiotic‐resistant bacteria poses a serious public health problem (Stewart and Costerton, 2001; Ventola, 2015). Host diet and phylogeny are two main factors influencing animal gut microbiome composition and function (Ley et al., 2008; Muegge et al., 2011), and the gut microbiome of animals plays an important role in host immunity, development and health (Kinross et al., 2008; Spor et al., 2011; Wei et al., 2019). However, their gut microbiome is also considered to be a reservoir for antibiotic resistance genes (ARGs; Sommer et al., 2009; Looft et al., 2012; Zhou et al., 2012; Hu et al., 2013; Laxminarayan et al., 2013; Allen, 2014; Fitzpatrick and Walsh, 2016). Previous studies have revealed that the environment or habitat has a profound effect on the types of ARGs present in the microbiome (Forsberg et al., 2014; Pal et al., 2016). For example, different habitats (e.g. different human body sites, water, and soils) harbour different symbiotic microbiome communities and have a different composition of ARGs (Pal et al., 2016). In humans, the gut microbiome of people from different countries also shows a difference in ARGs composition, to some extent (Feng et al., 2018). Therefore, these findings indicate a potential correlation between the structure of the gut microbiome and the composition of ARGs. Moreover, the ARG structure in the gut microbiome within the same species may differ based on a geographic pattern. However, this hypothesis needs further supporting evidence.
The giant panda, which belongs to the Carnivora order, lives in six mountain regions in China (Fig. 1), including the Qinling Mountains, Minshan Mountains, Qionglai Mountains, Daxiangling Mountains, Xiaoxiangling Mountains and Liangshan Mountains (Schaller et al., 1985). Additionally, there are several captive populations, such as the Wolong Research Center (located in Wolong and Yaan) and Chengdu Breeding Center (located in Chengdu). The giant panda, along with the sympatric species the red panda, is a bamboo‐eating panda (herbivorous carnivorans; Schaller et al., 1985; Wei et al., 2000). There are two common findings in the giant panda gut microbiomes. First, previous studies have found a difference in the gut microbiome community between wild and captive populations of giant pandas (Zhu et al., 2011; Wei et al., 2015; Xue et al., 2015; Guo et al., 2019; Yao et al., 2019). Second, the gut microbiome of wild pandas in the Shaanxi region (only one wild population in the Qinling Mountains) harbours a high proportion of Clostridiaceae (Zhu et al., 2011; Wu et al., 2017). Considering the potential correlation between the community of the gut microbiome and the composition of ARGs, there are no large‐scale meta‐analyses of the gut microbiome of giant pandas and the composition ARGs across wild and captive populations from different geographic areas.
Fig. 1.
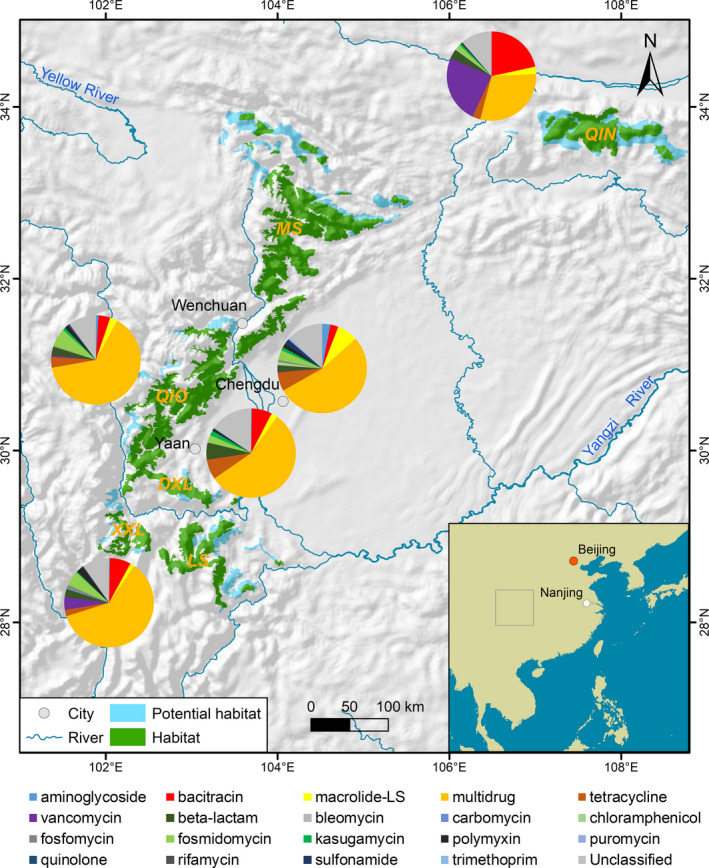
The wild giant panda habitats and antibiotic resistance genes (ARGs) structure (mean abundance). Six wild mountain giant panda populations: Qinling Mountains population (QIN), Minshan Mountains population (MS), Qionglai Mountains population (QIO), Daxiangling Mountains population (DXL), Xiaoxiangling Mountains population (XXL) and Liangshan Mountains population (LS). Two captive populations: Chengdu Breeding Center (CD) and the Giant Panda Research Center in Yaan (Yaan). In this study, the metagenomes from five populations (three wild populations: QIN, QIO and XXL; two captive populations: CD and Yaan) were analysed. Macrolide‐LS: macrolide–lincosamide–streptogramin.
Here, based on the published metagenomes of giant pandas [including three wild populations from the Qinling (Wu et al., 2017), Qionglai (Guo et al., 2019) and Xiaoxiangling Mountains (Zhu et al., 2018a), and two major captive populations from Wolong (Guo et al., 2019) and Chengdu (Zhang et al., 2018)] by both our group and other groups, we mainly aimed to investigate the potential correlation between the structure of the gut microbiome and the composition of ARGs across different geographic locations and living environments. In addition, we also integrated other published Carnivora metagenomes (including meat‐eating carnivorans (Zhu et al., 2018b) and omnivorous carnivorans (Guo et al., 2018; Zhu et al., 2018b)) to investigate the effect of diet on the ARGs in the gut microbiome.
Results and discussion
In this study, we analysed the metagenomes of 96 mammals: 19 meat‐eating carnivorans (CA), ten omnivorous carnivorans (OC), 55 bamboo‐eating carnivorans [49 giant panda samples: nine from the Qinling Mountains (QIN, wild), seven from the Qionglai Mountains (QIO, wild), 16 from the Xiaoxiangling Mountains (XXL, wild), seven from the Chengdu Breeding Center (CD, captive), ten from the Yaan research base of the Wolong Research Center (Yaan, captive) and six from red pandas in the Xiaoxiangling Mountains (wild)], and 12 herbivores (HE; Table S1).
The dissimilarity in both the gut microbiome community and ARG composition among the four diet groups
Our results showed a significant difference in both the gut microbiome community and ARG composition between the four diet groups (using all samples; Figs 2 and 3A,D, and Jaccard distance: PERMANOVA, P = 0.0001), and this difference was larger in regard to the ARG composition when compared to that in the gut microbiome community [PERMANOVA: F value (25.44) of ARGs > F value (10.98) of gut microbiome]. For example, at the phylum level (Fig. 2A), the dominant populations in the gut microbiome of CA included Bacteroidetes, Firmicutes, Proteobacteria and Fusobacteria. The main phyla in OC included Proteobacteria and Firmicutes, while the dominant populations in the gut microbiome of HE included Firmicutes and Bacteroidetes. Altogether, Proteobacteria and Firmicutes were the dominant populations in giant panda and red panda samples (Fig. 2A). In regard to the types of ARGs (Fig. 2B), the dominant ARGs in the gut microbiome of CA populations included macrolide–lincosamide–streptogramin, tetracycline, and beta‐lactam resistance genes. The main types of ARGs in OC populations included multidrug resistance genes and tetracycline resistance genes, whereas the dominant types of ARGs in HE populations included multidrug resistance genes, as well as macrolide–lincosamide–streptogramin and tetracycline resistance genes. Taken together, the mean abundance of multidrug resistance genes revealed them to be the dominant type of ARGs in giant panda and red panda samples (Fig. 2B). Therefore, combining with the previous findings on the difference in the gut microbiome among different diet mammal groups (e.g. Ley et al., 2008; Muegge et al., 2011; Zhu et al., 2018a), we here revealed the dissimilarity in ARG composition in their gut microbiome.
Fig. 2.
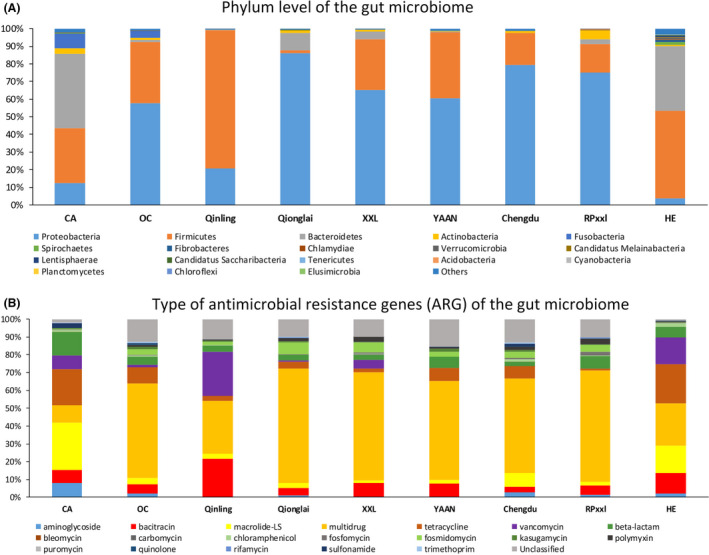
The gut microbiome composition and ARG patterns in giant pandas.
A. The phylum level of the gut microbiome identified in the 96 metagenomes included in the study.
B. The type of ARGs identified in the gut microbiome. CA, meat‐eating carnivorans. OC, omnivorous carnivorans. HE, herbivores. Qinling, wild Qinling giant panda populations. Qionglai, wild Qionglai giant panda population. XXL, wild Xiaoxiangling giant panda population. RPxxl, wild Xiaoxiangling red panda population. Yaan, the captive Yaan giant panda population. Chengdu, the captive Chengdu giant panda population. Macrolide‐LS: macrolide–lincosamide–streptogramin.
Fig. 3.
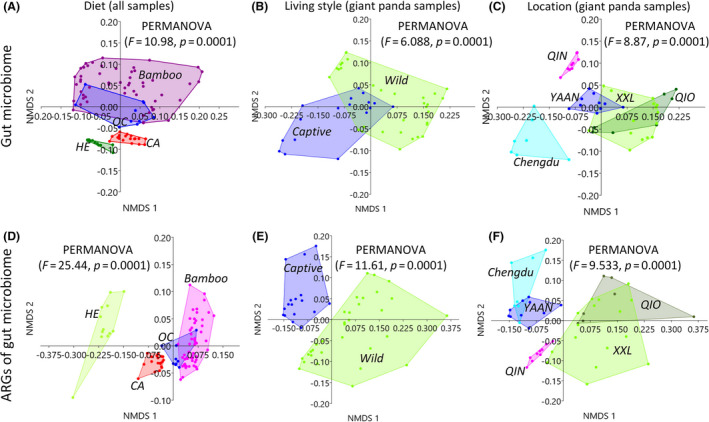
The differences in metagenome between groups were generated via the non‐metric multidimensional scaling (NMDS) method using the Jaccard distance.
A. The effect of diet on the similarities in gut microbiome communities between four groups (including 96 metagenomes, all samples used in this study) using the Jaccard distance of gut microbial genera abundance.
B. The effect of living environment (e.g. captivity) on the similarities in gut microbiome communities between two groups (only including giant panda metagenomes) using the Jaccard distance of gut microbial genera abundance.
C. The effect of geographic location on the similarities in gut microbiome communities between five groups (only including giant panda metagenomes) using the Jaccard distance of gut microbial genera abundance.
D. The effect of diet on the similarities in ARG composition between four groups (including 96 metagenomes, all samples used in this study) using the Jaccard distance of the ARG subtype abundance.
E. The effect of living environment (e.g. captivity) on the similarities in ARG composition between two groups (only including giant panda metagenomes) using the Jaccard distance of the ARG subtype abundance.
F. The effect of geographic location on the similarities in ARG composition between five groups (only including giant panda metagenomes) using the Jaccard distance of the ARG subtype abundance. CA, meat‐eating carnivorans. OC, omnivorous carnivorans. Bamboo, bamboo‐eating carnivorans (giant panda and red panda). HE, herbivores. Captive, the captive giant pandas including the Chengdu and Yaan populations. Wild, wild giant pandas including the Qinling, Qionglai, and Xiaoxiangling populations. QIN, the wild Qinling giant panda population. QIO, the wild Qionglai population. XXL, the wild Xiaoxiangling population. Yaan, the captive Yaan population. Chengdu, the captive Chengdu population.
The high proportion of tetracycline and macrolides resistance genes in captive mammals
We further discovered the high proportion of tetracycline and macrolides resistance genes in captive mammals in this study, especially in CA and HE groups (Figs 2B and 4). Tetracycline and macrolides are widely used to treat bacterial infections in various body systems (e.g. the skin, intestines and respiratory tract; Klein and Cunha, 1995; Anadon and Reevejohnson, 1999). They have been widely applied in many species (e.g. dogs, cats, cattle, sheep, swine, turkeys and chickens) for veterinary use in many places, such as zoos and farms, as well as for domestic animals (Stuart and Smith, 1992; Anadon and Reevejohnson, 1999; Winckler and Grafe, 2001; Li et al., 2018). As such, this may explain the higher proportion of tetracycline and macrolides resistance genes in captive mammals. However, the putative gut microbiome genera, which harboured tetracycline and macrolides resistance genes, were found to differ. In CA, the tetracycline resistance genes were mainly identified in Bacteroides and Escherichia, in OC and captive pandas mainly in Escherichia, and in HE mainly in Bacteroides and Prevotella (Fig. 5, Fig. S1). In CA, macrolide–lincosamide–streptogramin resistance genes were mainly in Escherichia, Blautia and Citrobacter, and in HE mainly in Bacteroides, Prevotella and Clostridium (Fig. 5, Figs S1‐S2). Therefore, the consensus pattern on the ARG composition in the captive mammal gut microbiome might be associated with the used antibiotics in the captivity, but the putative gut bacteria sources might be different given that the effect by the different host phylogeny position.
Fig. 4.
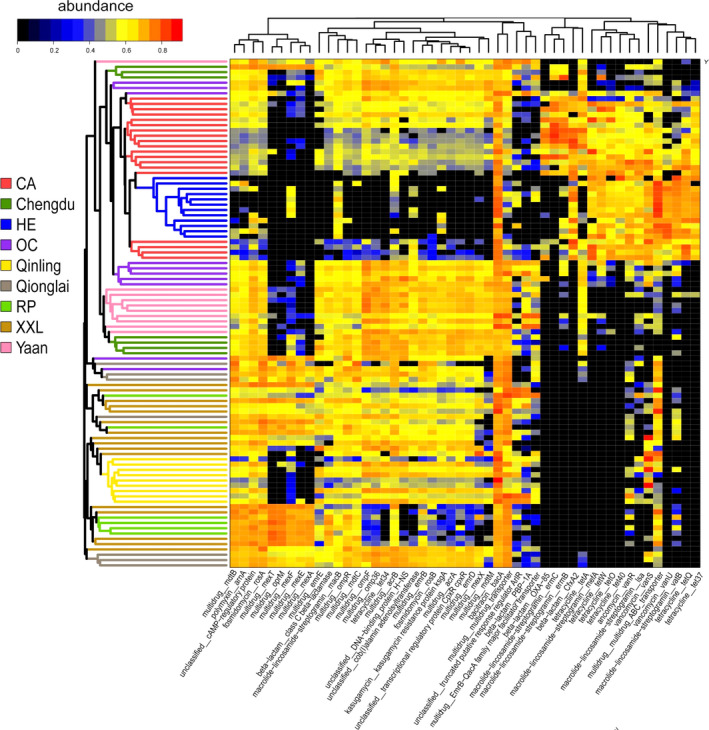
The heatmap of the top 50 ARG subtypes in the 96 metagenomes studied. The left tree is a neighbour‐joining tree constructed using the Jaccard distance of the ARG subtype abundance, with each colour representing one group. In the main figure, each row represents one metagenome. Each column represents one ARG subtype. In order to display the heatmap clearly, the abundance was transformed from the original abundance using the following formula: lg(original abundance x 1000).
Fig. 5.
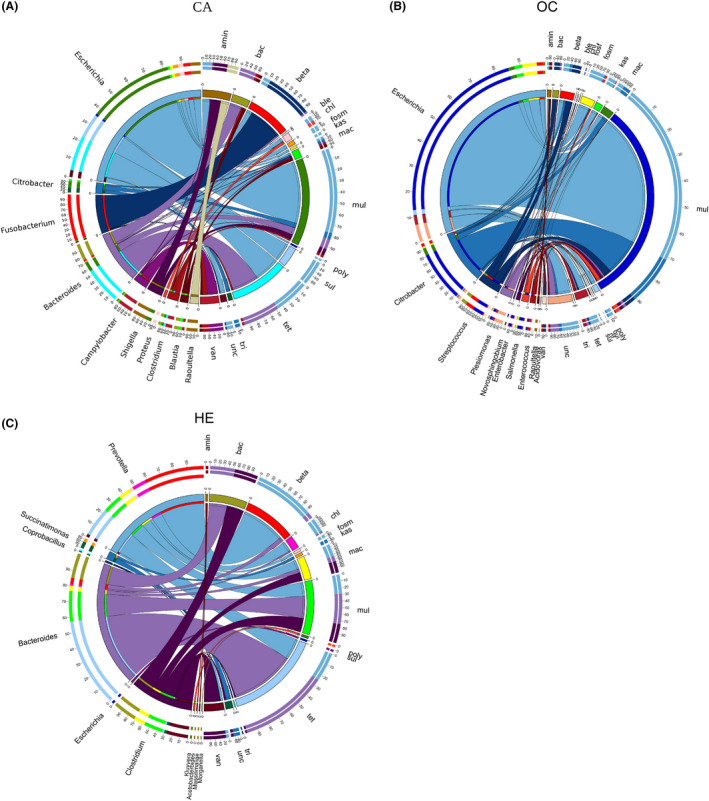
The distributions of ARG types and their abundance in the total annotated ARGs in the metagenomes [visualized by Circos (Krzywinski et al., 2009)]. The length of the bars on the outer ring represents the percentage of gut microbiome groups (genera level) for each ARG type. Each gut microbiome genus was represented by a specific ribbon colour and the width of each ribbon shows the abundance of each genus in the ARG type. A. CA (meat‐eating carnivorans). B. OC (omnivorous carnivorans). C. HE (herbivores). amin, aminoglycoside. bac, bacitracin. beta, beta‐lactam. ble, bleomycin. chl, chloramphenicol. fosm, fosmidomycin. kas, kasugamycin. mac, macrolide–lincosamide–streptogramin. mul, multidrug. poly, polymyxin. sul, sulfonamide. tet, tetracycline. tri, trimethoprim. unc, unclassified. van, vancomycin.
The captivity potentially resulting in the divergence in the ARG composition of the giant panda gut microbiome
Based on the meta‐analysis of the giant panda metagenome data, we confirmed the previous findings (mostly using the 16S rRNA data) on the difference in the gut microbiome composition (Wei et al., 2015; Xue et al., 2015; Guo et al., 2019; Yao et al., 2019). At the genus level, the dominant populations in the gut microbiome differed among giant panda populations (Table S2). The mean abundance of Escherichia (Proteobacteria) was highest in captive giant panda populations (0.34 ± 0.19 in Yaan, and 0.28 ± 0.21 in CD). In wild giant pandas, the mean abundance of Clostridium (Firmicutes) was highest in the QIN population (0.42 ± 0.35), and the mean abundance of Pseudomonas (Proteobacteria) was highest in non‐QIN populations (0.22 ± 0.19 in Qionglai, 0.44 ± 0.41 in XXL). Additionally, the mean abundance of Pseudomonas (Proteobacteria) was also highest in the XXL population of wild red pandas (0.61 ± 0.38). However, the new finding here was the dissimilarity in the ARG composition between the captive and wild giant panda gut microbiome.
By only taking into account the metagenomes of giant pandas, we also found a significant difference in both gut microbiome community and ARG composition between the captive and wild groups (Fig. 3B,E, Jaccard distance, PERMANOVA, P = 0.0001). This difference in ARG composition between the groups was larger than that in the gut microbiome community, with two clear clusters for ARGs (Fig. 3E, Captive vs. Wild). For example, the mean abundance of tetracycline resistance genes was higher in captive populations (0.07 ± 0.02 in Yaan and 0.07 ± 0.04 in CD) than that found in wild populations (0.03 ± 0.03 in QIN, 0.04 ± 0.02 in Qionglai, and 0.02 ± 0.02 in XXL). The gut microbiome of captive populations had a higher abundance of several ARG subtypes, including tetracycline_tet34, and multidrug_ompF. In captive CD populations, tetracycline resistance genes were mainly identified in Escherichia, multidrug resistance genes in Escherichia, Klebsiella, Citrobacter, and Pseudomonas, and macrolide–lincosamide–streptogramin resistance genes in Streptococcus (Fig. 6 and Fig. S2). In the captive Yaan giant panda population, tetracycline resistance genes were mainly found in Escherichia and Lactobacillus, multidrug resistance genes in Escherichia, Streptococcus, and Shigella, and macrolide–lincosamide–streptogramin resistance genes in Escherichia (Fig. 6 and Fig. S2). As mentioned above, some antibiotics (e.g. tetracycline, macrolides, gentamicin, azithromycin and cephalosporins) are used in the captive giant panda population, which might have the effect on the ARGs in the gut microbiome and resulted in the divergence with those in the wild giant pandas.
Fig. 6.
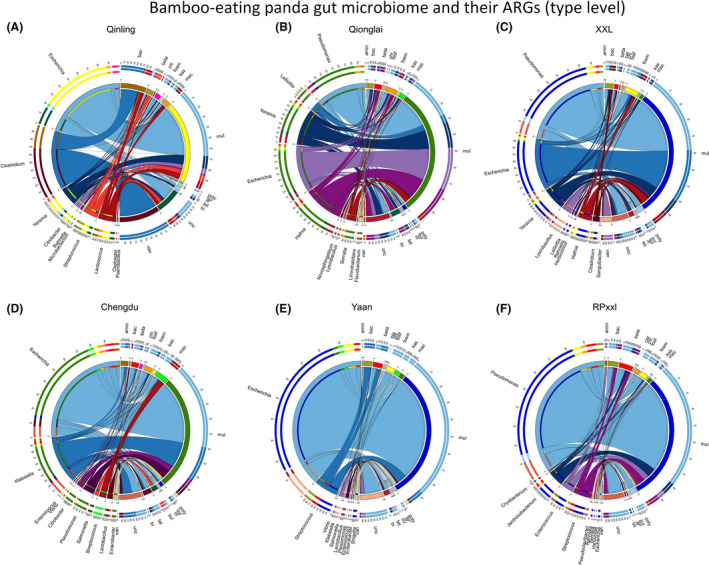
The distribution of ARG types and their abundance in the total annotated ARGs in giant panda and red panda metagenomes [visualized by Circos (Krzywinski et al., 2009)]. The length of the bars on the outer ring represents the percentage of gut microbiome groups (genera level) for each ARG type. Each gut microbiome genus was represented by a specific ribbon colour and the width of each ribbon indicates the abundance of each genus in the ARG type. A. Qinling (wild Qinling population). B. Qionglai (wild Qionglai population). C. XXL (wild Xiaoxiangling population). D. Chengdu (captive Chengdu population). E. Yaan (captive Yaan population). F. RPxxl (wild red panda Xiaoxiangling population). amin, aminoglycoside. bac, bacitracin. beta, beta‐lactam. ble, bleomycin. chl, chloramphenicol. fosm, fosmidomycin. kas, kasugamycin. mac, macrolide–lincosamide–streptogramin. mul, multidrug. poly, polymyxin. sul, sulfonamide. tet, tetracycline. tri, trimethoprim. unc, unclassified. van, vancomycin.
The geographic divergence in the composition of ARGs in gut microbiome among wild giant panda mountain populations
We then divided the giant panda metagenome samples into five groups based on their geographic locations and found a significant difference in both gut microbiome community and ARG composition (Fig. 3C,F, Jaccard distance, PERMANOVA, P = 0.0001). As such, our results revealed a clear divergence between QIN and non‐QIN populations (Qionglai and XXL populations) (Fig. 3C,F). Notably, the QIN population exhibited a unique pattern of ARG types, with the abundance of the vancomycin (0.25 ± 0.29) and bacitracin (0.22 ± 0.26) resistance genes being the highest when compared to the other wild or captive populations (Fig. 2B). In regard to the ARG subtypes (Fig. 6 and Table S3), the mean abundance of vancomycin_vanS (0.21 ± 0.30) and bacitracin_bacA (0.22 ± 0.26) was the highest in the gut microbiome of giant pandas from the QIN population when compared to the other wild or captive populations. In the QIN giant panda population (wild), the vancomycin resistance genes (e.g. vancomycin_vanS) were identified in Clostridium (Fig. 6 and Fig. S2). The mean abundance of several ARG subtypes from multidrug resistance genes type in the non‐QIN wild populations, such as multidrug_multidrug_ABC_transporter, multidrug_mexT, multidrug_oprM, multidrug_mexF, multidrug_mexE, and multidrug_mexA, was higher when compared to other giant panda populations (Fig. 6). And the multidrug resistance genes were mainly identified in Pseudomonas, Escherichia, Hafnia and Yersinia (Fig. 6 and Fig. S2). We deduced that the divergence in the ARG in the gut microbiome in QIN population might be associated their long‐term isolation with non‐QIN populations.
Giant panda genomic research uncovered three significant genetic clusters (QIN, Minshan, and Qionglai–Daxiangling–Xiaoxiangling–Lianshang clusters), with the QIN and non‐QIN populations diverging approximately 0.3 million years ago (Zhao et al., 2013). Interestingly, the results of the host genome and gut microbiome and their ARGs revealed some consensus patterns. The pandas in the QIN Mountains consume more bamboo leaves (containing high levels of alkaloids) when compared to non‐QIN populations, which may result in some positive selection of various taste genes (Zhao et al., 2013). Alkaloids can increase butanol concentrations and shorten its fermentation period by some Clostridium strains (Shao and Chen, 2015). Moreover, clostrindolin, a product of some Clostridium strain, is an antimycobacterial pyrone alkaloid (Schieferdecker et al., 2019). These findings indicate that Clostridium species may utilize alkaloids. Here, the gut microbiome of the QIN population was found to have the highest proportion of Clostridium species (Table S4), as well as vancomycin resistance genes when compared to other wild and captive populations, which were mainly found in Clostridium species. Vancomycin is also widely used to treat infections with Clostridium strains (Zar et al., 2007). Strictly anaerobic Clostridium species have the potential ability to produce bioactive compounds, including potent antimicrobials (Pahalagedara et al., 2020). Therefore, we speculated that the long‐term isolation, selection pressure from the diet, and inner characteristics of Clostridium species might result in the geographic pattern observed in current ARGs identified in the gut microbiome of giant pandas, such as the high proportion of Clostridium and vancomycin resistance genes in the QIN population. Therefore, we provide an example of a potential consensus pattern between host population genetics and its symbiotic gut microbiome. We revealed that habitat isolation also impacts the ARG structure in the gut microbiome of mammals.
Management and conservation
Our study revealed that captivity might lead to a special combination of ARGs as the treatment of captive individuals during normal health management affects the composition of ARGs. Thus, the difference in the ARG structure among different mammal groups in this study would provide some basic information for their management and conservation, especially for captive populations. The normal treatment of the mammals in the zoos (e.g. CA) should consider their potential tetracycline and macrolides resistance genes of the gut microbiome. Furthermore, maintaining the health of the captive giant panda population is important for the translocation of some small and isolated wild populations. The management of the captive giant panda population should think of the antibiotics resistance by Escherichia. Moreover, the care for the wild injured giant pandas should assess their mountain source and the potential antibiotic resistance by Pseudomonas (e.g. in non‐QIN mountain populations) and vancomycin resistance (e.g. in QIN mountain population).
Experimental procedures
Data collection
Considering the phylogeny position of the giant panda (belonging to Carnivora order), we collected the published metagenomes (raw data) of giant pandas and other carnivorans (Table S1). The 49 giant panda metagenomes came from five populations: three wild populations from the Qinling (Wu et al., 2017), Qionglai (Guo et al., 2019) and Xiaoxiangling Mountains (Zhu et al., 2018a), and two major captive populations from Yaan (Guo et al., 2019) and Chengdu (Zhang et al., 2018). In addition, we also integrated other published Carnivora metagenomes, including meat‐eating carnivorans (Zhu et al., 2018b), omnivorous carnivorans (Guo et al., 2018; Zhu et al., 2018b) and bamboo‐eating red pandas (Zhu et al., 2018a)), as well as 12 herbivore metagenomes (Zhu et al., 2018b) to investigate the effect of diet on the ARGs in the gut microbiome. The total number of species (subspecies) involved in this study was about 39 (Table S1). Most of the metagenome data used were from our previously published data, which can decrease the bias regarding the sequencing depth.
Raw data treatment
Raw reads were filtered using Trimmomatic (Bolger et al., 2014) to remove (i) all read less than 50 bp in length, (ii) reads with degenerated bases (N's), and (iii) all duplicates defined as sequences whose initial 20 nucleotides were identical and shared an overall identity similarity of > 97% throughout the length of the shortest read. After blasting against the NR database in NCBI using diamond (Buchfink et al., 2014), we removed the putative host contamination. Megahit (Li et al., 2015) was used to assemble these clean reads into contigs. We used Prodigal (Hyatt et al., 2010) for gene prediction from the contigs and obtained the gene files per metagenome. Then, we used CD‐HIT (Li and Godzik, 2006) to construct non‐redundant gene sets with < 90% overlap and < 95% shared sequence identity from these gene files. Based on these gene profiles, we used salmon (Patro et al., 2015) to map the clean reads (keeping only the reads which theoretically belong to Prokaryotes) per metagenome to the clean non‐redundant gene profile and obtained the TPM (transcripts per million reads) abundance of these non‐redundant gene profiles in each metagenome. We also blasted these genes against the NR database in NCBI using diamond (Buchfink et al., 2014) and gained the putative taxon assignments of these genes per metagenome.
The annotation of ARGs
We blasted the identified genes against the ARDB database using SARG2.0 with the default parameters (e‐value: 1e‐7; the cut‐off of blastx alignment identity: 60%; the blastx score: 60; and the cut‐off of blastx alignment length: 50%) (Yin et al., 2018) and gained the putative ARG assignment of these genes per metagenome. Then, we used custom Perl scripts to gain the abundance (TPM) of ARG types and subtypes for each metagenome.
The relative abundance of gut microbiome communities and ARGs (type and subtype)
The abundance (TPM) of gut microbiome communities and ARGs per metagenome was transformed to relative abundance using STAMP (Parks et al., 2014). These relative abundance tables were then used for downstream analyses between groups, such abundance comparisons and cluster analysis.
The bacteria taxon annotation of ARGs
The putative sequences of ARGs were blasted against the NR database in NCBI using diamond with these parameters (e‐value: 1e‐5; the cut‐off of blastx alignment identity: 55%; the blastx score: 60; and the cut‐off of blastx alignment length: 50%) (Buchfink et al., 2014), which led to the putative taxon assignments of these ARGs per metagenome. Then, we used custom Perl scripts to gain the abundance (TPM) of the taxon for these ARG types and subtype in each metagenome. The abundance (TPM) of gut microbiome communities and annotated ARGs per metagenome was transformed to relative abundance using STAMP (Parks et al., 2014).
The contribution of taxon on these ARGs types and subtypes
Circos was used to visualized the contribution of bacteria taxon (at the genus level) regarding the ARG types and subtypes based on the relative abundance of bacteria genus for the annotated ARGs and the relative abundance of ARG types and subtypes in all annotated ARGs.
The differences in the gut microbiome and ARGs between groups
The Jaccard distance for gut microbiome genus and ARGs (types and subtypes) relative abundance was used to generate non‐metric multidimensional scaling (NDMS) in PAST3 (Hammer et al., 2001). First, when using all 96 metagenomes, we separated them into four groups based on their diet (CA: meat‐eating carnivorans; OC, omnivorous carnivorans, Bamboo‐eating pandas (giant panda and red panda); HE, herbivores). Second, we only used the giant panda samples, which were divided into two groups based on the living environment (Wild and Captive panda populations). Lastly, we further evaluated the effect of geographic location in the giant panda samples, which were divided into five groups: QIN, wild Qinling population; QIO, wild Qionglai population; XXL, wild Xiaoxiangling population; CD, captive Chengdu population; and Yaan, captive Yaan population.
Moreover, to evaluate the effect of these factors on the composition of gut microbiota or ARG profiles, we performed one‐way PERMANOVA for Jaccard dissimilarities in species abundance using PAST3 (Hammer et al., 2001).
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
LZ designed the research and wrote the manuscript, with input from QD, XG, XY and ZY. LZ, HC, TH, QD and ZZ conducted the metagenome data analysis. All authors read and approved the final manuscript.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Supporting information
Table S1. Information regarding the samples used in this study.
Table S2. The genera summary of the nine groups formed from these 96 metagenomes.
Table S3. The mean abundance (%) of main ARG subtypes in the gut microbiome of giant pandas.
Table S4. The mean abundance (%) of the dominant putative Clostridium species in the gut microbiome of giant pandas.
Fig. S1. The distributions of dominant ARG subtypes and their abundances in the total annotated ARGs subtypes in the metagenomes.
Fig. S2. The distributions of ARG subtypes and their abundances in the total annotated ARGs subtypes in giant panda and red panda metagenomes.
Acknowledgements
We thank for the help by Xu Yang from Chengdu Xing'ai Information and Technology Company Ltd. We thank for the language editing by the editor from Wiley Editing Serveces.
Microbial Biotechnology (2021) 14(1), 186–197
Funding informationThis project was supported by the National Natural Science Foundation of China (31222009), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the basic research program of the gut microbiome of the translocated giant pandas in Sichuan (5118242019000288).
Data availability statement
Not applicable.
References
- Allen, H.K. (2014) Antibiotic resistance gene discovery in food‐producing animals. Curr Opin Microbiol 19: 25–29. [DOI] [PubMed] [Google Scholar]
- Anadon, A. , and Reevejohnson, L. (1999) Macrolide antibiotics, drug interactions and microsomal enzymes: implications for veterinary medicine. Res Vet Sci 66: 197–203. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. , and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink, B. , Xie, C. , and Huson, D.H. (2014) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59. [DOI] [PubMed] [Google Scholar]
- Feng, J. , Li, B. , Jiang, X. , Yang, Y. , Wells, G.F. , Zhang, T. , and Li, X. (2018) Antibiotic resistome in a large‐scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ Microbiol 20: 355–368. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, D. , and Walsh, F. (2016) Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol Ecol 92: fiv168. [DOI] [PubMed] [Google Scholar]
- Forsberg, K.J. , Patel, S. , Gibson, M.K. , Lauber, C.L. , Knight, R. , Fierer, N. , and Dantas, G. (2014) Bacterial phylogeny structures soil resistomes across habitats. Nature 509: 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, S. , Hu, J. , Pan, W. , and Zhu, J. (1985) Giant Pandas of Wolong. Chicago, IL, USA and London, UK: University of Chicago Press. [Google Scholar]
- Guo, W. , Mishra, S. , Zhao, J. , Tang, J. , Zeng, B. , Kong, F. , et al (2018) Metagenomic study suggests that the gut microbiota of the giant panda (Ailuropoda melanoleuca) may not be specialized for fiber fermentation. Front Microbiol 9: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Mishra, S. , Wang, C. , Zhang, H. , Ning, R. , Kong, F. , et al (2019) Comparative study of gut microbiota in wild and captive giant pandas (Ailuropoda melanoleuca). Genes 10: 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, Ø. , Harper, D. , and Ryan, P. (2001) PAST‐Palaeontological statistics. Palaeontologia Electronica. 4(1). https://www.uv.es/~pardomv/pe/2001_1/past/pastprog/past.pdf. [Google Scholar]
- Hu, Y. , Yang, X. , Qin, J. , Lu, N. , Cheng, G. , Wu, N. , et al (2013) Metagenome‐wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 4: 2151. [DOI] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P.F. , Land, M.L. , Larimer, F.W. , and Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinross, J.M. , von Roon, A.C. , Holmes, E. , Darzi, A. , and Nicholson, J.K. (2008) The human gut microbiome: implications for future health care. Curr Gastroenterol Rep 10: 396–403. [DOI] [PubMed] [Google Scholar]
- Klein, N.C. , and Cunha, B.A. (1995) Tetracyclines. Med Clin North Am 79: 789–801. [DOI] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J.E. , Birol, I. , Connors, J.M. , Gascoyne, R.D. , Horsman, D. , et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxminarayan, R. , Duse, A. , Wattal, C. , Zaidi, A.K. , Wertheim, H.F. , Sumpradit, N. , et al (2013) Antibiotic resistance—the need for global solutions. Lancet Infect Dis 13: 1057–1098. [DOI] [PubMed] [Google Scholar]
- Ley, R.E. , Hamady, M. , Lozupone, C.A. , Turnbaugh, P.J. , Ramey, R.R. , and Bircher, J.S. (2008) Evolution of mammals and their gut microbes. Science 320: 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , and Godzik, A. (2006) Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li, D. , Liu, C.‐M. , Luo, R. , Sadakane, K. , and Lam, T.‐W. (2015) MEGAHIT: an ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31: 1674–1676. [DOI] [PubMed] [Google Scholar]
- Li, W. , Drucker, A.M. , Cho, E. , Laden, F. , Vopham, T. , Li, S. , et al (2018) Tetracycline use and risk of incident skin cancer: a prospective study. Br J Cancer 118: 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft, T. , Johnson, T.A. , Allen, H.K. , Bayles, D.O. , Alt, D.P. , Stedtfeld, R.D. , et al (2012) In‐feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci USA 109: 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge, B.D. , Kuczynski, J. , Knights, D. , Clemente, J.C. , Gonzalez, A. , and Fontana, L. (2011) Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahalagedara, A.S.N.W. , Flint, S. , Palmer, J. , Brightwell, G. , and Gupta, T.B. (2020) Antimicrobial production by strictly anaerobic Clostridium species. Int J Antimicrob Agents 55: 105910. [DOI] [PubMed] [Google Scholar]
- Pal, C. , Bengtsson‐Palme, J. , Kristiansson, E. , and Larsson, D.J. (2016) The structure and diversity of human, animal and environmental resistomes. Microbiome 4: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D.H. , Tyson, G.W. , Hugenholtz, P. , and Beiko, R.G. (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30: 3123–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , and Kingsford, C. (2015) Salmon: accurate, versatile and ultrafast quantification from RNA‐seq data using lightweight‐alignment. Biorxiv 021592. [Google Scholar]
- Schieferdecker, S. , Shabuer, G. , Knuepfer, U. , and Hertweck, C. (2019) Clostrindolin is an antimycobacterial pyrone alkaloid from Clostridium beijerinckii . Org Biomol Chem 17: 6119–6121. [DOI] [PubMed] [Google Scholar]
- Shao, M. , and Chen, H. (2015) Feasibility of acetone–butanol–ethanol (ABE) fermentation from Amorphophallus konjac waste by Clostridium acetobutylicum ATCC 824. Process Biochem 50: 1301–1307. [Google Scholar]
- Sommer, M.O. , Dantas, G. , and Church, G.M. (2009) Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325: 1128–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spor, A. , Koren, O. , and Ley, R. (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9: 279–290. [DOI] [PubMed] [Google Scholar]
- Stewart, P.S. , and Costerton, J.W. (2001) Antibiotic resistance of bacteria in biofilms. Lancet 358: 135–138. [DOI] [PubMed] [Google Scholar]
- Stuart, A. , and Smith, D. (1992) Use of the fluorochromes xylenol orange, calcein green, and tetracycline to document bone deposition and remodeling in healing fractures in chickens. Avian Dis 447–449. [PubMed] [Google Scholar]
- Ventola, C.L. (2015) The antibiotic resistance crisis: part 1: causes and threats. P T 40: 277. [PMC free article] [PubMed] [Google Scholar]
- Wei, F. , Feng, Z. , Wang, Z. , and Hu, J. (2000) Habitat use and separation between the giant panda and the red panda. J Mammal 81: 448–455. [Google Scholar]
- Wei, F. , Wang, X. , and Wu, Q. (2015) The giant panda gut microbiome. Trends Microbiol 23: 450–452. [DOI] [PubMed] [Google Scholar]
- Wei, F. , Wu, Q. , Hu, Y. , Huang, G. , Nie, Y. , and Yan, L. (2019) Conservation metagenomics: a new branch of conservation biology. Sci China Life Sci 62: 168–178. [DOI] [PubMed] [Google Scholar]
- Winckler, C. , and Grafe, A. (2001) Use of veterinary drugs in intensive animal production. J Soils Sediments 1: 66. [Google Scholar]
- Wu, Q. , Wang, X. , Ding, Y. , Hu, Y. , Nie, Y. , Wei, W. , et al (2017) Seasonal variation in nutrient utilization shapes gut microbiome structure and function in wild giant pandas. Proc R Soc B Biol Sci 284: 20170955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Z. , Zhang, W. , Wang, L. , Hou, R. , Zhang, M. , Fei, L. , et al (2015) The bamboo‐eating giant panda harbors a carnivore‐like gut microbiota, with excessive seasonal variations. mBio 6: e00022‐00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, R. , Xu, L. , Hu, T. , Chen, H. , and Zhu, L. (2019) The “wildness” of the giant panda gut microbiome and its relevance to effective translocation. Glob Ecol Conserv 18: e00644. [Google Scholar]
- Yin, X. , Jiang, X. , Chai, B. , Li, L. , Yang, Y. , Cole, J.R. , et al (2018) ARGs‐OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34: 2263–2270. [DOI] [PubMed] [Google Scholar]
- Zar, F.A. , Bakkanagari, S.R. , Moorthi, K. , and Davis, M.B. (2007) A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile–associated diarrhea, stratified by disease severity. Clin Infect Dis 45: 302–307. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Liu, W. , Hou, R. , Zhang, L. , Schmitz‐Esser, S. , Sun, H. , et al (2018) Age‐associated microbiome shows the giant panda lives on hemicelluloses, not on cellulose. ISME J 12: 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Zheng, P. , Dong, S. , Zhan, X. , Wu, Q. , Guo, X. , et al (2013) Whole‐genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet 45: 67. [DOI] [PubMed] [Google Scholar]
- Zhou, W. , Wang, Y. , and Lin, J. (2012) Functional cloning and characterization of antibiotic resistance genes from the chicken gut microbiome. Appl Environ Microbiol 78: 3028–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. , Wu, Q. , Dai, J. , Zhang, S. , and Wei, F. (2011) Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci USA 108: 17714–17719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. , Yang, Z. , Yao, R. , Xu, L. , Chen, H. , and Gu, X. , et al (2018a) Potential mechanism of detoxification of cyanide compounds by gut microbiomes of bamboo‐eating pandas. mSphere 3: e00229‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. , Wu, Q. , Deng, C. , Zhang, M. , Zhang, C. , Chen, H. , et al (2018b) Adaptive evolution to a high purine and fat diet of carnivorans revealed by gut microbiomes and host genomes. Environ Microbiol 20: 1711–1722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information regarding the samples used in this study.
Table S2. The genera summary of the nine groups formed from these 96 metagenomes.
Table S3. The mean abundance (%) of main ARG subtypes in the gut microbiome of giant pandas.
Table S4. The mean abundance (%) of the dominant putative Clostridium species in the gut microbiome of giant pandas.
Fig. S1. The distributions of dominant ARG subtypes and their abundances in the total annotated ARGs subtypes in the metagenomes.
Fig. S2. The distributions of ARG subtypes and their abundances in the total annotated ARGs subtypes in giant panda and red panda metagenomes.
Data Availability Statement
Not applicable.