

Abstract
Gain-of-function mutations in the Type I Bone Morphogenic Protein (BMP) receptor ACVR1 have been identified in two diseases: Fibrodysplasia Ossificans Progressiva (FOP), a rare autosomal dominant disorder characterized by genetically driven heterotopic ossification, and in 20–25% of diffuse intrinsic pontine gliomas (DIPGs), a pediatric brain tumor with no effective therapies and dismal median survival. While the ACVR1 mutation is causal for FOP, its role in DIPG tumor biology remains under active investigation. Here, we discuss cross-fertilization between the FOP and DIPG fields, focusing on the biological mechanisms and principles gleaned from FOP that can be applied to DIPG biology. We highlight our current knowledge of ACVR1 in both diseases, and then describe the growing opportunities and barriers to effectively investigate ACVR1 in DIPG. Importantly, learning from other seemingly unrelated diseases harboring similar mutations may uncover novel mechanisms or processes for future investigation.
Keywords: ACVR1, BMP, Diffuse Intrinsic Pontine Glioma, Fibrodysplasia Ossificans Progressiva, Mendelian disorders, Childhood cancers
1. Introduction
The bone morphogenetic protein (BMP) signaling cascade regulates diverse processes across multiple organ systems, including cell proliferation, tissue stem cell dynamics, organogenesis, tissue remodeling, and physiologic homeostasis[1]. Dysregulation of this signaling pathway has been implicated in a variety of human diseases, including vascular disorders, skeletal defects, congenital heart disease, and cancers[2, 3]. Of interest to this review is the rare autosomal dominant disorder Fibrodysplasia Ossificans Progressiva (FOP), a debilitating disease characterized by episodic and spontaneous heterotopic ossification (HO), the pathologic generation of bone in extraskeletal soft tissues. FOP is caused by moderately activating germline mutations in the Type I BMP receptor ACVR1[4]. To date, thirteen different ACVR1 mutations have been identified in FOP patients, the most common being the R206H amino acid substitution found in 95–97% of patients[5–10]. Since the identification of ACVR1 mutations in FOP over a decade ago, there has been remarkable progress in elucidating the mechanisms of mutant ACVR1 and its role in HO. These studies have highlighted the central role of dysregulated BMP signaling to FOP pathobiology and uncovered novel avenues for therapeutic intervention.
More recently, four independent studies reported seven somatic ACVR1 mutations in 20–25% of pediatric diffuse intrinsic pontine gliomas (DIPGs), a fatal brainstem tumor with a median survival of 8–10 months and no effective therapies. Of the seven identified DIPG-associated ACVR1 mutations, six have been described in FOP patients and the seventh also exhibiting moderate gain-of-function activity (Figure 1a)[11–14]. ACVR1 mutations previously have not been associated with other cancers, although recent reports reanalyzing The Cancer Genome Atlas (TCGA) noted that 3.4% of endometrial cancers harbored ACVR1 mutations and all endometrial cases with ACVR1 altercations appear mutually exclusive from other frequently mutated oncogenes[15, 16]. Notably, the frequency of discrete ACVR1 mutations differs between FOP and DIPGs. Whereas the R206H mutation is found in 97% of FOP patients, R206H mutations were found in only 20–25% of DIPG and instead there is a wider spectrum of ACVR1 mutations found in DIPG (Figure 1b)[17]. Interestingly, FOP patients have not been described to have a predisposition for cancer or display an increased incidence of brainstem gliomas, though there are reports of patients with neurological symptoms and asymptomatic brainstem lesions[18, 19]. These observations indicate that an ACVR1 mutation alone is not sufficient for oncogenesis. Nevertheless, given its prevalence in DIPGs, mutant ACVR1 likely provides a selective advantage to the brainstem tumor, though on which aspects of oncogenesis and tumor behavior is still under investigation.
Figure 1. Differences in ACVR1 mutations identified in Fibrodysplasia Ossificans Progressiva (FOP) and Diffuse Intrinsic Pontine Gliomas (DIPG).
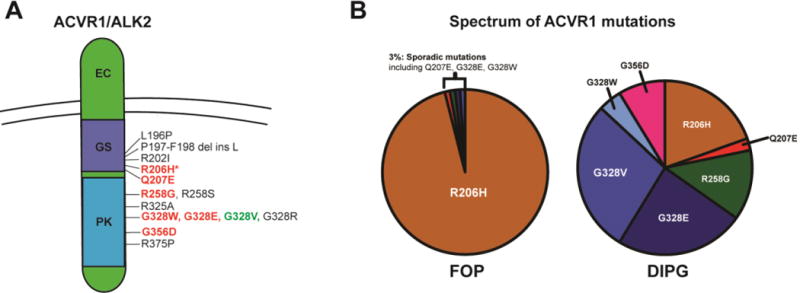
(A) ACVR1 mutations found in FOP (black), DIPG (green), or both diseases (red). All are located in the intracellular receptor domains, either the glycine-serine rich (GS, purple) or protein kinase (blue) domain. Of the seven DIPG-associated ACVR1 mutations, six have been identified in FOP. (B) Prevalence of ACVR1 mutations in FOP and DIPG taken from the four initial studies identifying ACVR1 in DIPG. While the R206H mutation occurs in 95–97% of FOP patients, the spectrum of ACVR1 mutations in DIPG is more varied.
Because ACVR1 mutations are found in both FOP and DIPG, there is growing collaboration and scientific cross talk between these two fields to gain insight into the shared mechanisms driving ectopic bone formation and tumor growth. In addition, the prevalence of shared mutations raises hope that similar targeted therapies could be utilized in both diseases. Here, we highlight this cross-fertilization between these two seemingly disparate pediatric diseases related by a common mutation, primarily focusing on the insights gleaned from FOP to inform DIPG biology. We describe some of our mechanistic understanding and therapeutic approaches for ACVR1-driven HO in FOP, and then discuss the opportunities and challenges of applying these principles to DIPG tumor biology and therapeutic targeting.
1.1 Brief primer on ACVR1 and BMP signaling pathway
ACVR1 (also known as ALK2) and its closely related BMP Type I receptors BMPR1A (ALK3), BMPRR1B (ALK6), and ACVR1L (ALK1) are part of the larger TGFβ superfamily of growth factor receptors. Extracellular BMP cytokines recruit two Type I receptors (ALK 1/2/3/6) and two Type II BMP receptors, mainly BMPR2, ACVR2A, and ACVR2B, to form heterotetrameric structures[20, 21]. Upon ligand interaction, the constitutively active Type II receptors phosphorylate the Type I receptor in the glycine-serine domain (GS). This GS-domain phosphorylation activates the Type I kinase (PK) domain, leading to downstream phosphorylation of SMAD proteins, primarily SMAD1, SMAD5, and SMAD8. These SMADs interact with regulatory SMADs, such as SMAD4, before translocating into the nucleus to control BMP target genes, including the inhibitor of differentiation (ID) genes, ID1–3. Regulation of BMP signaling occurs at multiple levels, including different BMP cytokine subtypes, the tissue-specific expression of BMP cytokines and BMP receptors, the specific combinations of Type I and II heterotetramers employed for receptor signaling, adaptor proteins that regulate Type I receptor activity such as FKBP1A, downstream inhibitory SMADs (primarily SMAD 6, 7), and extracellular antagonists that bind cytokines[22]. Type I BMP receptors can also activate SMAD-independent signaling cascades, including the p38-mitogen activated protein kinase (MAPK) pathway, JNK, Rho-like GTPases, and the PI3K/Akt pathway, in a cell type specific manner[23]. Overall, Type I BMP receptors like ACVR1 and its regulatory components coordinate a finely tuned complex network of intracellular signaling to regulate multiple phenotypes and cell types.
2. ACVR1 in Fibrodysplasia Ossificans Progressiva
2.1. Roles of ACVR1 in driving heterotopic ossification
Although BMPs are now considered multifunctional cytokines with wide-ranging effects, they were initially identified for their abilities to induce bone formation[24]. BMPs were shown to inhibit myogenic differentiation and promote osteogenic and chondrogenic differentiation of meschenymal progenitors[25–27]. Even prior to the identification of ACVR1 mutations, multiple studies examining FOP patient samples suggested deregulated BMP signaling as a candidate pathway[28–31].
Subsequent studies revealed that the R206H mutation moderately increased basal BMP pathway activation and enhanced chondrogenic and osteogenic differentiation when cells were exposed to BMPs or placed in permissive milieu[32–38]. A chimeric R206H knock-in mouse model reproduced many clinical features of FOP, such as the great toe congenital malformation and progressive and injury-induced HO, demonstrating the causality of mutant ACVR1 to disease patogenesis [39]. Furthermore, mice expressing a humanized conditional-on knock-in ACVR1-R206H allele under the control of Cre recombinase also develop spontaneous and injury-induce HO[40, 41]. Mechanistically, this mutation conferred leaky ligand independent BMP signaling and enhanced sensitivity to BMP ligands[4, 42]. A similar modest elevation in BMP signaling and enhanced responsiveness was observed across multiple ACVR1 mutations identified in both FOP and in DIPG[43]. Interestingly, ACVR1 appears to be required for early stages of chondrogenic differentiation of mouse embryonic fibroblasts, but becomes dispensable after initial chondrogenic induction[33]. This finding raises the possibility that therapy must target a particular window of opportunity in FOP and receptor inhibition after a particular time point during lesion formation will not halt/reverse disease progression. Together, these studies demonstrate a clear relationship between receptor activation, BMP signaling, and induction of HO.
More recently, the TGFβ related cytokine, activin A, was identified a critical regulator of HO in FOP. While wild-type ACVR1 was known to bind activin, its interaction does not induce BMP signaling and bone formation, but rather acts as a competitive antagonist to BMP cytokines[44]. The mutant receptor in contrast conferred novel responsiveness and aberrant activin-mediated BMP pathway activation and functional studies demonstrated that activin A was both necessary and sufficient for HO [40, 41, 45]. Additional studies from our group and others have found that aberrant responsiveness to activin A is shared across the spectrum of gain-of-function ACVR1 mutations found in DIPG and FOP[45, HJH and ACR unpublished data]. This novel mechanism highlights the growing number of ways by which gain-of-function ACVR1 mutations deregulate BMP signaling.
Beyond altered intrinsic receptor properties impacting mesenchymal cells, there are also significant interactions between local cells and the lesion microenvironment that further augment heterotopic ossification. Early lesions prior to chondrogenesis are hypoxic, leading to stabilization of HIF1α. Two recent studies noted that hypoxia and HIF1α contributes to HO through HIF1α-mediated regulation of ACVR1 endosomal trafficking [46, 47]. Importantly, inhibiting HIF1α reduced HO. In addition to hypoxia, the immune system also impacts ectopic bone formation. Inflammatory triggers, such as trauma, illness, or vaccinations, are often antecedents of FOP flares[48]. Patients receive symptomatic benefit from immunosuppressive therapies such as corticosteroids[48] and depletion of mast cells and macrophages reduced HO in a BMP4-overexpression FOP mouse model[49, 50]. Interestingly, clinical specimens and murine studies demonstrated significant immune infiltration in early lesion formation, including mast cells found at ten to hundred fold higher number in FOP lesions compared to ACVR1 wild-type specimens[51–53]. While the ACVR1 mutation in FOP is germline and present in all cell types, this exaggerated immunologic response raises the possibility that mutant immune cells display enhanced response to inflammation and/or that mutant mesenchymal cells themselves contribute to over-recruitment of immune cells to lesion sites. Finally, in animal studies, both ACVR1 wild-type and mutant cells are recruited to ectopic cartilage and bone formation, indicating that mutant cells may also impact their neighboring wild-type cells [39, 40].
2.2 Targeted therapeutic approaches
Currently, there are no curative treatments for FOP patients. Given the central role of ACVR1 and deregulated BMP signaling to FOP, current targeted strategies focus on dysregulated BMP signaling and mutant ACVR1. Dorsomorphin, and its more potent derivative LDN-193189, was initially identified as a small molecule inhibitor of the Type I BMP receptors ALK2, ALK3, and ALK6[54, 55]. Dorsomorphin specifically blocked SMAD-dependent BMP receptor signaling and did not inhibit p38-MAPK signaling. Derivative compounds DMH1, 1LWY (also known as ML-347), and LDN-212184, exhibited increased specificity towards ALK2 and nanomolar inhibition of SMAD 1/5/8 phosphorylation with little effect on TGFβ signaling[56–58]. More recently, a new class of ALK2 inhibitors with a 2-aminopyridine backbone, such as K02288 and LDN-214117, emerged showing higher ALK2 specificity compared to dorsomorphin and LDN-193189, though their effects on SMAD-independent signaling remains unclear[59, 60]. Several of these inhibitors reduced HO in FOP patient-derived cells and in mouse models expressing either ACVR1-R206H or the artificial, constitutively active Q207D-ACVR1 mutation [35, 54, 58]. However, some of the small molecule kinase inhibitors have off-target effects, particularly the vascular endothelial growth factor receptor (VEGFR) and receptor-interacting serine/threonine kinase 2 (RIPK2). Furthermore, none of these agents are used in the clinic. Interestingly, several clinically available kinase inhibitors, such as mometinib, dasatinib, and saracatinib, exhibit off-target activity against ALK2, highlighting potential drug repurposing opportunities[61–63]. Beyond small molecules, allele specific siRNA and antisense oligonucleotides against ACVR1 have also been shown to be effective in cell based FOP model systems[64, 65]. Finally, approaches targeting other mediators of HO, such as hypoxia, immune cells, and downstream differentiation, have also been examined. In particular, inhibition of the retinoic acid receptor (RAR) signaling cascade is required for chondrogenesis. Palovarotene, an RARγ agonist, was efficacious at reducing HO in FOP animal models and is currently in Phase II/III clinical trials[66, 67].
3. ACVR1 in Diffuse Intrinsic Pontine Gliomas
3.1 Genomic analysis and co-mutations in DIPG
While ACVR1 in FOP is the causal mutation for HO in FOP patients, somatic ACVR1 mutations in DIPGs often co-occur with other mutations, adding a layer of complexity for DIPG biology. The most common mutations identified in all DIPGs are somatic lysine-to-methionine amino acid substitutions at residue 27 (K27M) in genes encoding Histone 3 variants, most commonly H3F3A and HIST1H3B[68, 69]. K27M mutations were identified in ~80% of DIPG patient samples (60% harbor H3F3A and 18–20% harbor HIST1H3B). K27M mutations were associated with a worse prognosis compared to patients who were wild-type H3F3A/HIST1H3B[69–72]. Mutant Histone 3 alters the distribution of repressive H3K27 trimethylation across the genome, resulting in a global reduction in K27 trimethylation on Histone 3 and widespread transcriptional dysregulation [73–75]. Epigenome modifying agents, like the histone demethylase inhibitor GSKJ4[76], Polycomb repressive complex 2 (PRC2) inhibitors [77, 78], and histone deacetylase inhibitor panobinostat[79] and agents that inhibit efficient RNA polymerase II transcription, such as the bromodomain and extra-terminal (BET) protein inhibitors[78, 80, 81] and the cyclin-dependent kinase 7 inhibitor THZ1[81], have been shown to be efficacious in DIPG preclinical models.
Importantly, H3K27M mutations alone are insufficient to promote oncogenic transformation[75, 82]. Instead, these oncohistones frequently co-occur with other mutations, including alterations in known cancer-associated pathways such as TP53 loss, PPMD1 mutations, and amplifications of the platelet-derived growth factor receptor alpha gene (PDGFRA)[14, 83–86]. Many of these mutations co-occur with specific histone variant subtypes, suggesting that these co-mutations require a specific cellular context and partner histone mutations to promote oncogenesis (Table 1).
Table 1.
Comparing H3.3 and H3.1 mutations in DIPG
| H3.3 K27M | H3.1 K27M | |
|---|---|---|
| Genes | H3F3A | HIST1H3B/C |
| Locations | Pons Midline (Thalamus, Cerebellum, Spine) |
Restricted to pons |
| % DIPG showing mutations | 60–70% | 20% |
| Median Overall Survival (OS) | 11 mo | 15 mo |
| Secondary mutations | PDGFRA amplifications TP 53 loss |
ACVR1 PI3KR1 PIK3CA |
| Epigenetic features | Global H3 K27me3 loss | Global H3 K27me3 loss |
ACVR1 mutations co-segregated more commonly with HIST1H3B K27M alterations instead of the more prevalent H3F3A variant[11–14]. These tumors did not have TP53 loss, rarely expressed amplifications in PDGFRA, and had mutations in PI3-kinase signaling pathway components in ~60% of cases. Clinically, patients with ACVR1-mutant tumors had an earlier age of diagnosis, longer survival time, and occurred more frequently in females. One study analyzing the clonal evolution of DIPGs found that ACVR1 mutations were present at the earliest tumor clone alongside HIST1H3B K27M mutations, suggesting that both mutations are needed for oncogenesis[87]. Given these discrete clinical and genomic associations, it is thought that ACVR1-mutant DIPGs form a distinct tumor subgroup that have a unique biology and oncogenic dependencies compared to ACVR1 wild-type tumors.
3.2 Current understanding of the functional role of ACVR1 in DIPG
Studies from the FOP field have laid significant groundwork for investigating the role of ACVR1 in DIPG. Recent studies demonstrate that ACVR1-mutant DIPGs have increased BMP signaling compared to control brain and ACVR1-WT DIPG, as evidenced by phosphorylated SMAD 1/5/8 immunohistochemical staining[12]. Furthermore, similar to FOP, these mutations increased p-SMAD 1/5/8 signal and downstream BMP target gene expression when compared to WT-ACVR1 patient-derived cultures in vitro or when the mutation is expressed in human or mouse astrocytes [11–14]. Finally, we have found that patient-derived ACVR1-mutant DIPGs, but not WT-ACVR DIPG cells, aberrantly activate the BMP signaling cascade upon activin exposure [HJH, ACR unpublished results]. These studies demonstrate that two signaling mechanisms that drive HO, elevated BMP signaling and aberrant responsiveness to activin, are also present in DIPG.
Yet, although both disorders share similar signaling responses, the functional role of ACVR1 in DIPG remains limited and somewhat puzzling. Overexpression of ACVR1 mutations in normal human astrocytes increased BrdU incorporation and growth rate in vitro, but when expressed in p53-null mouse astrocytes implanted into brain parenchyma, ACVR1 mutations were unable to promote tumorigenesis, highlighting the requirement of co-mutations in the oncogenic process[11, 14]. Pharmacologic inhibition of ACVR1 using LDN-193189 resulted in dose-dependent cytotoxicity across all tested DIPG cell lines, including WT-ACVR1 tumor cells [13]. However, LDN-193189 was previously shown to have off-target effects[57, 88]. Our own preliminary studies using a panel of ACVR1 inhibitors in patient-derived DIPG cell lines revealed a more heterogeneous effect in vitro where some inhibitors exhibited cytotoxicity whereas others displayed little cytotoxic activity [HJH, ACR unpublished results]. Furthermore, while dasatinib exhibited an effect on DIPG cell viability in vitro, 72-hour incubation of saracatinib against a large panel of ACVR1 mutant cell lines did not show significant cytotoxic response[63, 79]. Notably, dasatinib also inhibits PDGFR signaling, a cascade associated with a known gene amplification in DIPGs, confounding the viability effect. Together, these latter findings indicate that inhibiting ACVR1 may not have a cytotoxic effect in vitro and raise the possibility that unlike in FOP where inhibition of ACVR1 appears effective in HO, targeting ACVR1 in DIPG may instead be limited. However, there are many caveats to the in vitro experimental approach used that should be considered, including the addition of growth factors to maintain in vitro cell lines and differences in oncogenic dependencies that emerge in vitro versus in vivo[89]. Nevertheless, these data indicate that unlike kinase activating mutations that can be effectively targeted by inhibitors with dramatic therapeutic response in other cancers such as BRAF V600E in melanoma, ACVR1 likely has a more complex role in brain tumorigenesis than simply activating a pathway to promote oncogenic growth.
4. The challenges of applying FOP principles to DIPG biology
Despite the foundation provided by FOP studies, applying principles from FOP to DIPG is not as straightforward as anticipated. Why might the FOP field have made tremendous progress in understanding the role of ACVR1 in disease pathogenesis while its function in DIPG remains unclear? While time likely plays a significant role (it has been ~3 years since the reports of ACVR1 in DIPG, ~11 years for FOP), there are additional challenges that impede our understanding of ACVR1 in DIPG, mainly the complexity of modeling DIPG. In FOP, clinically relevant and reproducible model systems span the full spectrum of FOP lesion progression and animal models develop congenital phenotypes[39–41]. Conversely, the reported DIPG model systems harboring ACVR1 mutations do not fully capture the entire tumorigenic process (Figure 2). Several established patient-derived DIPG cell lines harbor ACVR1 mutations, but many of these lines are derived from patients whom previously received chemotherapies and radiation[79]. In addition, patient-derived cell lines, though powerful tools, have their own caveats, including the addition of supplemental growth factors to maintain lines in vitro and differences in oncogenic dependencies compared to the original tumor in vivo. Furthermore, these patient-derived cells represent already-formed tumors and it is possible that the role of ACVR1 may be earlier in the tumorigenic timeline. A second approach is to develop cell-based models using immortalized astrocytes, NPCs, or human embryonic stem cells that express DIPG-associated mutations and examine oncogenic transformation. However, there are no reported studies successfully generating heterologous cell-based tumor models with mutant ACVR1. Finally, while there are several genetically engineered mouse models of DIPG harboring a combination of H3K27M, PDGF-B, and/or p53−/− alterations[75, 90, 91], there are no reported animal models harboring ACVR1 mutations. As such, we currently do not have laboratory models that recapitulate the full spectrum of tumor formation and growth. This inability to effectively model DIPG stems from multiple factors: (1) heterogeneous response of central nervous system (CNS) derived cells to BMP signaling, (2) unclear role of ACVR1 in the developing central nervous system, (3) additional mutations that co-occur with ACVR1, and (4) unclear phenotype and pathways for analysis.
Figure 2. Model systems for Fibrodysplasia Ossificans Progressiva (FOP) and Diffuse Intrinsic Pontine Gliomas (DIPGs) to study ACVR1.
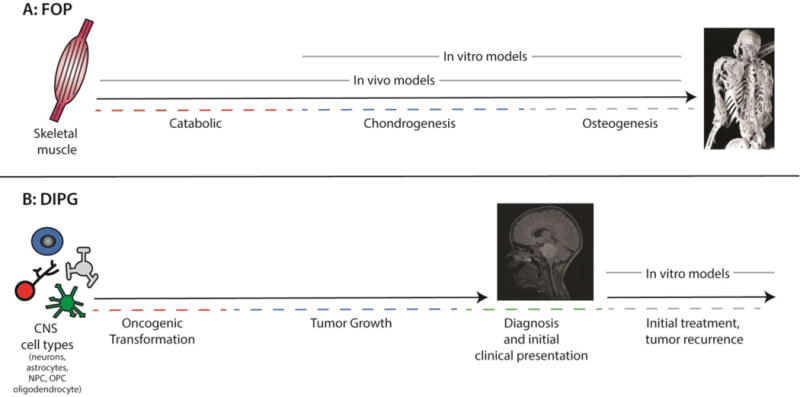
(A) In FOP, lesions initially undergo muscle breakdown and infiltration of immune cells prior to the formation of a chondrogenic scaffold and subsequent bone formation. Genetically engineered mouse models expressing the R206H mutation recapitulate this full lesion progression. Furthermore, in vitro R206H cell-based models faithfully recapitulate the chondrogenic and osteogenic phases of heterotopic ossification when placed in permissive differentiation media. Thus, the full spectrum of lesion progression is recapitulated in laboratory model systems. (B) In contrast, current DIPG models that harbor ACVR1 mutations consist mainly of patient-derived DIPG tumor cell lines, many of which are derived from patients whom already received chemoradiation. There are few reported cell-based and transgenic animal models for DIPG, though generation of brainstem tumors after expression of ACVR1 and co-mutations has yet to be reported. Given these limitations, our ability to effectively study ACVR1 is hampered by our inability to fully model the oncogenic process. FOP skeleton image courtesy of Dr. Frederick Kaplan, University of Pennsylvania; T2 FLAIR MRI image courtesy of Dr. Angela Waanders, Children’s Hospital of Philadelphia.
4.1 Heterogeneity of BMP signaling in CNS-derived cells
Cellular heterogeneity is seen across all organ systems, with the CNS being the most complex. Even within the main three CNS lineages, neurons, astrocytes, and oligodendrocytes, there is significant heterogeneity within each population[92–96]. CNS progenitors exhibit spatial and temporal diversity as they integrate the multitude of developmental signaling gradients, such as FGF, PDGF, Sonic hedgehog, Wnt, and BMPs, which regulate CNS patterning and the balance of neurogenesis, gliogenesis, and oligodendrogenesis[96–98]. Importantly, while BMP signaling promotes osteogenesis in mesenchymal precursors[99], how CNS-derived cells respond to BMP signaling depends on when during neural development and in which cell subtype the pathway is activated[97, 100, 101]. For example, during early neurodevelopment, neural progenitor cells (NPCs) proliferate and self-renew upon BMP pathway activation. At later stages, NPCs differentiate into neurons and astrocytes in response to BMPs. The pleotropic response to BMP signaling is also seen in adult glioblastomas. Secreted BMPs have a tumor suppressive effect on the majority of patient-derived gliomas stem cells (GSCs) and BMPs are being considered as a potential therapeutic avenue for adult GBM[102–105]. However, a subset of adult GSCs (~20%) instead proliferate and display enhanced oncogenicity in response to BMPs[106]. Interestingly, this differential response seen in adult GSCs and during neurodevelopment is associated with the relative expression of the Type 1 BMP receptors ALK3 (BMPR1A) and ALK6 (BMPR1B) [100, 106, 107]. Given this heterogeneity towards BMP signaling seen in adult GSCs, a similar heterogeneous response may also occur in DIPG tumor cells where a particular tumor subgroup harboring specific mutations proliferate upon BMP pathway activation whereas another subgroup suppresses growth when the pathway is turned on. As such, different tumors may have unique responses to BMP cytokines. To circumvent this issue, it might be beneficial to compare DIPG tumor cells’ gene expression profile to temporally and spatially defined precursor populations as this might provide insight what “age” these cancer cells are and how they may respond to BMP cytokines.
The heterogeneous response of CNS cells to BMP signaling may also impact which cell types are more susceptible to oncogenic transformation. While the origin of DIPG remains unknown, several studies identified discrete proliferating populations in the postnatal pons that might serve as potential cells of origin. Monje and colleagues noted two Nestin+ NPCs populations, one in the pontine parenchyma and one located in the floor of the fourth ventricle in the dorsal pons[108]. Half of the pontine parenchymal NPCs expressed Olig2, a transcription factor in NPCs that primarily give rise to oligodendrocyte precursor cells (OPCs). Tate et al., found proliferating pontine cells during early postnatal life that dramatically decreased by age 2, of which 60% of cells were Olig2+[109]. Finally, using various Cre driver mouse lines and careful stereological analysis, Lindquist et al. identified Olig2+, Sox2+ OPCs and astrocytes as the proliferative populations in the mouse pons[110]. Olig2 is expressed in >90% of DIPGs[111] and was recently shown to be required for DIPG proliferation[112]. In addition, one recent study profiling DIPG super-enhancers (SEs), large clusters of enhancer elements that display high amounts of activation marks like H3K27ac and transcriptional complex binding, found that many SEs were associated with genes previously connected with oligodendrocyte lineage identity[81]. It is unclear how these different proliferating progenitor populations within the pons respond to BMP signaling. Nevertheless, given their location and timing in relation to DIPG age of onset, it will be crucial to examine how each of these specific proliferating progenitor populations, in particular OLIG2+ cells and OPCs, are impacted by ACVR1 mutations and co-occurring alterations (discussed below). Furthermore, cell-based models using infected immortalized astrocytes, mouse neural stem cells, or directed differentiation of human embryonic stem cells may not represent the right “signaling and cellular milieu” to stimulate oncogenic transformation and growth. Identifying the cell origin will likely provide a platform to develop cell-based models that better represent the oncogenic process.
4.2 Unclear role of ACVR1 in the CNS
Studies examining the role of ACVR1 in the developing nervous system are limited. Knocking out ACVR1 impacts hippocampal dentate gyrus development[113] and ACVR1 loss or expression of the constitutive active Q207D receptor within forebrain mouse neurons did not impact gross anatomic morphology or neuronal survival[114]. However, its role in the hindbrain remains unclear. Interestingly, although FOP patients do not have an increased prevalence of brain tumors, there is 20-times greater prevalence of neuropathic pain in FOP patients and these patients also report other sensory abnormalities, including numbness, tingling, allodynia, and temperature-dependent sensitivities[19]. Histologic analysis of the knock-in ACVR1-R206H mouse found increased demyelination throughout the brain and spinal cord. FOP patients have hyperintense T2 lesions in the dorsal pons, cerebellum and spinal cord on MRI, suggestive of demyelination[115]. During neurodevelopment, BMPs inhibited rodent neurospheres and oligodendrocyte precursor cells (OPCs) from differentiating into an oligodendrocyte lineage. Instead, BMPs skewed OPCs towards astrocytic differentiation in vitro and robustly increased astrocyte number with mild region-specific oligodendrocyte loss[98, 116]. These findings support the hypothesis that ACVR1 may regulate progenitor cell fate, similar to its role in chondrogenesis and osteogenesis.
4.3 Additional mutations that co-occur with ACVR1
Unlike FOP where ACVR1 is the clear causal genetic alteration, for DIPG there are two or more collaborating mutations that appear to be essential for oncogenesis: ACVR1, HIST1H3B K27M, and possibly PI3K pathway alterations. How does each mutation independently impact the CNS and how do they synergize in promoting tumorigenesis and tumor growth? K27M mutations are thought to act as an epigenetic differentiation block as the gene expression profiles of H3.3 K27M mutant cells reflect a primitive earlier progenitor state when compared to their Histone 3.3 WT cells[82]. In addition, although H3K27M mutations are not sufficient to form overt tumors, they do increase proliferation in vivo[75]. Is it possible that these two mutations together collaborate and trap glioma precursors in a “progenitor” state that are primed for oncogenic transformation? Additionally, how might the microenvironment, including secreted cytokines such as BMPs and activin, vasculature, immune cells, and hypoxia, impact progenitor behavior? Like BMPs, activin also have multiple roles in cell fate, including regulation of OPC differentiation and maturation, cortical interneuron differentiation, and NPC viability [117–119]. Given that activin alone can activate both the BMP and TGF beta cascades in mutant ACVR1 DIPGs, how might this combination of signaling pathways impact glioma precursors in vivo compared to single pathway activation? Thus, understanding if ACVR1 alters progenitor cell fate decisions and how this decision is perturbed when H3K27M is co-expressed is a excellent starting point to investigate the developmental origins of DIPG.
4.4 Unclear phenotypes, pathways, and processes for analysis
Finally, whereas there is a clear reproducible phenotype in FOP model systems (bone formation and HO in permissive media in vitro or in vivo), the DIPG field has yet to identify a phenotype impacted by mutant ACVR1. Current studies examining ACVR1 in DIPG, including our own, have focused on the mutant receptor’s role in promoting proliferation in vitro, its inhibition as a therapeutic target in vitro, or its ability to form tumor by co-expressing multiple mutations in discrete NPC populations in vivo. Though promoting oncogenic growth is one selective benefit that the mutation could provide to DIPGs, ACVR1 and BMP signaling can also contribute to other tumor-associated phenotypes (Figure 3). While proliferation and cytotoxicity are relatively easy to examine, ACVR1 and BMP signaling might also regulate cancer stem cell self-renewal as seen in adult gliomas. Inhibition of ACVR1 may not cause cytotoxicity, but rather cellular senescence and slowed growth. For example, while a prior study noted no significant cytotoxicity of the EZH2 inhibitor Tazemetostat after three days, more recent studies noted its potential therapeutic value against DIPG, though the inhibitory effects required >6 days to show a significant difference [77, 78, 120]. Rather than promoting apoptosis and cell death, EZH2 inhibition instead induced senescence. In addition to proliferation and tumor initiation in specific progenitors as discussed above, we should also consider ACVR1’s role in regulating the invasive behavior of DIPG cells, in mediating resistance to therapies, and in its protective effects against the microenvironment (i.e. nutrient availability, immune surveillance). In other cancers, BMP signaling enhances migration and invasive capabilities[121, 122]. Furthermore, increased BMP pathway activation through increased BMPRII expression made lung squamous cell carcinomas resistant to an EGFR targeted kinase inhibitor[123]. Whether these same mechanisms occur within DIPG remains to be investigated.
Figure 3. Multiple phenotypes for analysis in Diffuse intrinsic pontine gliomas (DIPGs).
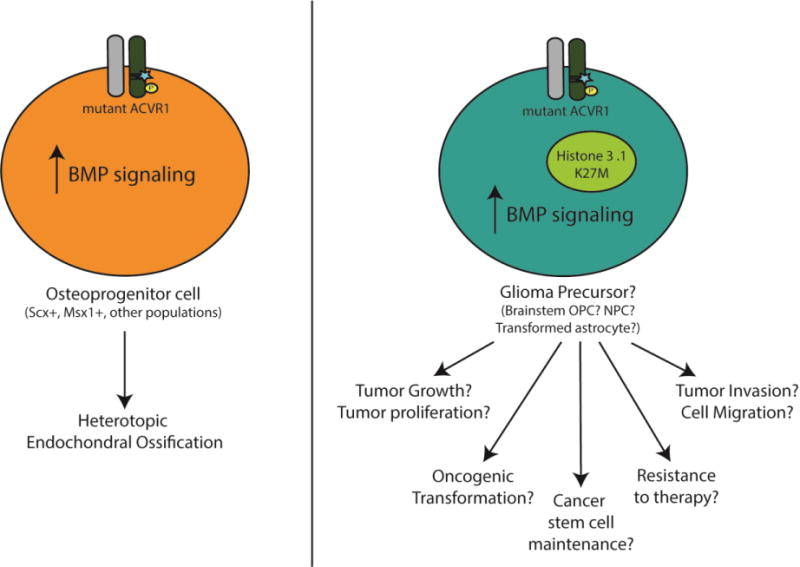
(Left) In FOP, ACVR1 and elevated BMP signaling is both necessary and sufficient for heterotopic ossification (HO). This has led to the development of multiple model systems with a consistent, reproducible phenotype. Although the cell type origin of HO remains unclear, recent studies identified Scleraxis+ and Msx1+ cells as sufficient to form heterotopic bone in the presence of ACVR1 mutations. (Right) In contrast, the role of ACVR1 mutations in DIPG remains unclear. In particular, we have yet to identify a tumor-associated phenotype(s) impacted by ACVR1 mutations. While tumor growth and tumor proliferation remains a key phenotype for cancers, there are other important oncogenic features that should be examined. Importantly, understanding the role of ACVR1 in DIPG is complicated by co-mutations present in ACVR1 mutant DIPGs, such as K27M substitution in Histone 3 variants. In addition, the cell of origin of DIPG remains unclear and each potential cell type origin has pleotropic responses to BMP signaling.
In addition to SMAD-dependent ACVR1 signaling, we should also consider the potential role of SMAD-independent signaling pathways. Several recent studies identified efficacy of PI3K inhibition and MAPK targeting both as single agent and in combination against DIPG in vitro[79, 81, 124] and genes in several key signaling cascades were enriched for H3K27ac enhancer peaks, including TGF-beta, p38-MAPK, and Hedgehog signaling[81]. Is it possible that ACVR1 mediates tumorigenic effects through these alternative pathways? Furthermore, while the role of BMPs in gliomas has been explored, activin has not been extensively investigated. Activin A promoted cell proliferation in the human glioma U87 cell line that was blocked using the naturally occurring inhibitor Follistatin[125], but its role in GSCs, transformed CNS glioma cell or murine models has not been characterized. TGFβ and its associated downstream signaling has been implicated in multiple glioma phenotypes, including angiogenesis, invasion and migration, and proliferation. Finally, we should also consider the receptor’s interactions with its microenvironment, including cell-extrinsic processes that modulate HO such as hypoxia and immune cells[126]. Macrophage and microglia infiltrate DIPGs, similar to the catabolic inflammatory response seen in FOP lesions[127, 128]: do these cells cross-talk with ACVR1 mutant tumor cells and how do they influence tumor activity? Similarly, do we see upregulation of BMP signaling in DIPG tumor cells under hypoxic conditions as we do with hypoxic mesenchymal cells? These processes that contribute to HO are also associated with cancers and present additional avenues for investigation into DIPG tumor biology.
4.5 Additional challenges translating targeted ACVR1 therapies into the CNS
Beyond the challenges of studying the functional role of ACVR1 in DIPG, we also need to investigate delivery and poor penetrance of drugs into the relatively protected CNS. The blood-brain barrier (BBB) limits the influx of 98% of small and 100% large molecules into the CNS[129]. Given this highly selective barrier, we need to ensure a balance between reaching sufficient drug concentration in the CNS with systemic side effects and toxicities. Modulating the integrity of the BBB through ultrasound or hydrophobic disruption or engineering molecules that can better penetrate the CNS through modifications in solubility or liposome packaging, are under active investigation[130, 131]. Alternatively, direct infusion into the CNS or local tumor, such as convection-enhanced delivery, is emerging as a promising means of bypassing the BBB and directly treating the tumor[132].
5. Perspectives and Conclusions
Overall, DIPG research has greatly benefitted from the lesson learned in FOP pathogenesis, including potential new mechanisms, signaling cascades, and processes that could contribute to DIPG biology. Currently, data suggest that although DIPG cells share similar biochemical responses with FOP, receptor inhibition may not impact tumor cell viability, raising initial questions about the role or clinical efficacy of receptor targeting. Although the data thus far are inconclusive, ACVR1 remains a viable target given the wide range of mechanisms and processes that still require investigation. Despite the challenges of investigating DIPG in the laboratory, there is a lot the field can glean from the mechanisms driving endochondral bone in FOP, in particular potential pathways, processes, and signaling mechanisms that can influence tumor behavior.
More broadly, transdisciplinary studies across FOP and DIPG presage untapped opportunities for shared data-driven discovery that both harness and empower the rare disease research community. Especially in pediatric cancer where developmental biology serves as the substrate for shared research interests, the growing collaboration and cross talk between FOP and DIPG, two seemingly unrelated pediatric diseases, represents just one example highlighting the value of Mendelian disorders informing cancer biology and vice versa. Recent studies overlaying datasets from Mendelian disorders and cancer genomics have uncovered novel candidate driver mutations that would likely not emerge without increased sample size[15, 16, 133]. However, overlying these datasets is only the entry point. To fully understand the role of ACVR1 in DIPG, we must integrate the lessons learned from FOP with the complex role of BMP signaling within neurodevelopment. Furthermore, we should also recognize other phenotypes besides tumor growth and proliferation could provide a selective benefit for DIPG, such as susceptible cell type origin, invasive capacity, and collaboration with other signaling cascades. Not until we determine what selective benefit ACVR1 provides DIPG can we truly understand whether ACVR1 is a viable therapeutic target for this deadly tumor. Nevertheless, the expanded platform of collaboration across FOP and DIPG can only help in these efforts.
HIGHLIGHTS.
Fibrodysplasia Ossificans Progressiva (FOP) and Diffuse Intrinsic Pontine Gliomas (DIPGs) share ACVR1 mutations.
While ACVR1 mutations underlie FOP, their functional role in DIPG remains unclear.
Developmental biology provides a context of shared biology among diverse diseases.
Intersecting Mendelian disorders and cancer genomics can elucidate potential cancer genes.
Acknowledgments
We thank and acknowledge the many children and families affected by FOP and DIPG for their continual support and generous contribution to our research. We thank Mateusz Koptyra and Sneha Narasimhan for critical reading of the manuscript. We apologize to those authors whose relevant work has not been included in this review article.
FUNDING: This work was supported by the National Institutes of Health [grants R01NS085336 and R01NS0916220 to ACR], the Kortney Rose Foundation [ACR], NIH training grant TL1R000138 to HJH [PI: G. Fitzgerald], a St. Baldrick’s Foundation Student Fellowship [HJH], and an Alex’s Lemonade Stand Foundation Student Fellowship [HJH].
ABBREVIATIONS
- BMP
bone morphogenetic protein
- CNS
Central nervous system
- DIPG
diffuse intrinsic pontine glioma
- FOP
Fibrodysplasia Ossificans Progressiva
- GBM
glioblastoma
- GSC
glioma stem cells
- HO
heterotopic ossification
- NPC
neural progenitor cells
- OPC
oligodendrocyte precursor cells
- SE
Superenhancers
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waite KA, Eng C. From developmental disorder to heritable cancer: it’s all in the BMP/TGF-beta family. Nat Rev Genet. 2003;4(10):763–73. doi: 10.1038/nrg1178. [DOI] [PubMed] [Google Scholar]
- 2.MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-beta Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harbor perspectives in biology. 2017 doi: 10.1101/cshperspect.a022269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massague J. TGFbeta in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525–7. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009;30(3):379–90. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bocciardi R, Bordo D, Di Duca M, Di Rocco M, Ravazzolo R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet. 2009;17(3):311–8. doi: 10.1038/ejhg.2008.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregson CL, Hollingworth P, Williams M, Petrie KA, Bullock AN, Brown MA, Tobias JH, Triffitt JT. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone. 2011;48(3):654–8. doi: 10.1016/j.bone.2010.10.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan FS, Kobori JA, Orellana C, Calvo I, Rosello M, Martinez F, Lopez B, Xu M, Pignolo RJ, Shore EM, Groppe JC. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1 c.772G>A; R258G): A report of two patients. Am J Med Genet A. 2015;167A(10):2265–71. doi: 10.1002/ajmg.a.37205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG, Brown MA, Wordsworth BP, Triffitt JT. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PloS one. 2009;4(3):e5005. doi: 10.1371/journal.pone.0005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whyte MP, Wenkert D, Demertzis JL, DiCarlo EF, Westenberg E, Mumm S. Fibrodysplasia ossificans progressiva: middle-age onset of heterotopic ossification from a unique missense mutation (c.974G>C, p.G325A) in ACVR1. J Bone Miner Res. 2012;27(3):729–37. doi: 10.1002/jbmr.1473. [DOI] [PubMed] [Google Scholar]
- 11.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, Barszczyk M, Chornenkyy Y, Bourgey M, Bourque G, Montpetit A, Cordero F, Castelo-Branco P, Mangerel J, Tabori U, Ho KC, Huang A, Taylor KR, Mackay A, Bendel AE, Nazarian J, Fangusaro JR, Karajannis MA, Zagzag D, Foreman NK, Donson A, Hegert JV, Smith A, Chan J, Lafay-Cousin L, Dunn S, Hukin J, Dunham C, Scheinemann K, Michaud J, Zelcer S, Ramsay D, Cain J, Brennan C, Souweidane MM, Jones C, Allis CD, Brudno M, Becher O, Hawkins C. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–6. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, Johann P, Tomita T, Goldman S, Nagib M, Bendel A, Goumnerova L, Bowers DC, Leonard JR, Rubin JB, Alden T, Browd S, Geyer JR, Leary S, Jallo G, Cohen K, Gupta N, Prados MD, Carret AS, Ellezam B, Crevier L, Klekner A, Bognar L, Hauser P, Garami M, Myseros J, Dong Z, Siegel PM, Malkin H, Ligon AH, Albrecht S, Pfister SM, Ligon KL, Majewski J, Jabado N, Kieran MW. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46(5):462–6. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46(5):457–61. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, Lu C, Nagahawatte P, Hedlund E, Rusch M, Pounds S, Lin T, Onar-Thomas A, Huether R, Kriwacki R, Parker M, Gupta P, Becksfort J, Wei L, Mulder HL, Boggs K, Vadodaria B, Yergeau D, Russell JC, Ochoa K, Fulton RS, Fulton LL, Jones C, Boop FA, Broniscer A, Wetmore C, Gajjar A, Ding L, Mardis ER, Wilson RK, Taylor MR, Downing JR, Ellison DW, Zhang J, Baker SJ, P. St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–50. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma M, Wang C, Glicksberg BS, Schadt EE, Li SD, Chen R. Identify cancer driver genes through shared Mendelian disease pathogenic variatns and cancer somatic mutations, Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing. 2016;22:473–484. doi: 10.1142/9789813207813_0044. [DOI] [PubMed] [Google Scholar]
- 16.Zhao B, Pritchard JR. Inherited Disease Genetics Improves the Identification of Cancer-Associated Genes. PLoS genetics. 2016;12(6):e1006081. doi: 10.1371/journal.pgen.1006081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor KR, Vinci M, Bullock AN, Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res. 2014;74(17):4565–70. doi: 10.1158/0008-5472.CAN-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Severino M, Bertamino M, Tortora D, Morana G, Uccella S, Bocciardi R, Ravazzolo R, Rossi A, Di Rocco M. Novel asymptomatic CNS findings in patients with ACVR1/ALK2 mutations causing fibrodysplasia ossificans progressiva. Journal of medical genetics. 2016;53(12):859–864. doi: 10.1136/jmedgenet-2016-104076. [DOI] [PubMed] [Google Scholar]
- 19.Kitterman JA, Strober JB, Kan L, Rocke DM, Cali A, Peeper J, Snow J, Delai PL, Morhart R, Pignolo RJ, Shore EM, Kaplan FS. Neurological symptoms in individuals with fibrodysplasia ossificans progressiva. Journal of neurology. 2012;259(12):2636–43. doi: 10.1007/s00415-012-6562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiramongkolchai P, Owens P, Hong CC. Emerging roles of the bone morphogenetic protein pathway in cancer: potential therapeutic target for kinase inhibition. Biochem Soc Trans. 2016;44(4):1117–34. doi: 10.1042/BST20160069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012;586(14):1846–59. doi: 10.1016/j.febslet.2012.02.043. [DOI] [PubMed] [Google Scholar]
- 22.Brazil DP, Church RH, Surae S, Godson C, Martin F. BMP signalling: agony and antagony in the family. Trends Cell Biol. 2015;25(5):249–64. doi: 10.1016/j.tcb.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Zhang YE. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harbor perspectives in biology. 2017;9(2) doi: 10.1101/cshperspect.a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urist MR. A morphogenetic matrix for differentiation of bone tissue. Calcif Tissue Res. 1970;(Suppl):98–101. doi: 10.1007/BF02152373. [DOI] [PubMed] [Google Scholar]
- 25.Katagiri T, Yamaguchi A, Ikeda T, Yoshiki S, Wozney JM, Rosen V, Wang EA, Tanaka H, Omura S, Suda T. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein-2. Biochemical and biophysical research communications. 1990;172(1):295–9. doi: 10.1016/s0006-291x(05)80208-6. [DOI] [PubMed] [Google Scholar]
- 26.Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127(6 Pt 1):1755–66. doi: 10.1083/jcb.127.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi A, Katagiri T, Ikeda T, Wozney JM, Rosen V, Wang EA, Kahn AJ, Suda T, Yoshiki S. Recombinant human bone morphogenetic protein-2 stimulates osteoblastic maturation and inhibits myogenic differentiation in vitro. J Cell Biol. 1991;113(3):681–7. doi: 10.1083/jcb.113.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gannon FH, Kaplan FS, Olmsted E, Finkel GC, Zasloff MA, Shore E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Human pathology. 1997;28(3):339–43. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan FS, Fiori J, LS DLP, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann N Y Acad Sci. 2006;1068:54–65. doi: 10.1196/annals.1346.008. [DOI] [PubMed] [Google Scholar]
- 30.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. The New England journal of medicine. 1996;335(8):555–61. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 31.Virdi AS, Shore EM, Oreffo RO, Li M, Connor JM, Smith R, Kaplan FS, Triffitt JT. Phenotypic and molecular heterogeneity in fibrodysplasia ossificans progressiva. Calcified tissue international. 1999;65(3):250–5. doi: 10.1007/s002239900693. [DOI] [PubMed] [Google Scholar]
- 32.Billings PC, Fiori JL, Bentwood JL, O’Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2008;23(3):305–13. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells. 2014;32(5):1289–300. doi: 10.1002/stem.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, Endo K, Awakura T, Fukushi J, Nakashima Y, Chiyonobu T, Kawara A, Nishida Y, Wada I, Akita M, Komori T, Nakayama K, Nanba A, Maruki Y, Yoda T, Tomoda H, Yu PB, Shore EM, Kaplan FS, Miyazono K, Matsuoka M, Ikebuchi K, Ohtake A, Oda H, Jimi E, Owan I, Okazaki Y, Katagiri T. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. The Journal of biological chemistry. 2009;284(11):7149–56. doi: 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto Y, Hayashi Y, Schlieve CR, Ikeya M, Kim H, Nguyen TD, Sami S, Baba S, Barruet E, Nasu A, Asaka I, Otsuka T, Yamanaka S, Conklin BR, Toguchida J, Hsiao EC. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet journal of rare diseases. 2013;8:190. doi: 10.1186/1750-1172-8-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, Shore EM. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009;119(11):3462–72. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Dinther M, Visser N, de Gorter DJ, Doorn J, Goumans MJ, de Boer J, ten Dijke P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J Bone Miner Res. 2010;25(6):1208–15. doi: 10.1359/jbmr.091110. [DOI] [PubMed] [Google Scholar]
- 38.Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, Ryoo HM. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. The Journal of biological chemistry. 2010;285(29):22542–53. doi: 10.1074/jbc.M109.094557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012;27(8):1746–56. doi: 10.1002/jbmr.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, Vonner AJ, Shen Y, Mohedas AH, Lee A, Eekhoff EM, van Schie A, Demay MB, Keller C, Wagers AJ, Economides AN, Yu PB. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016;8(366):366ra163. doi: 10.1126/scitranslmed.aaf1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, Economides AN. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7(303):303ra137. doi: 10.1126/scitranslmed.aac4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groppe JC, Shore EM, Kaplan FS. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- 43.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. The Journal of biological chemistry. 2012;287(44):36990–8. doi: 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, Waage A, Sundan A, Holien T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal. 2015;13:27. doi: 10.1186/s12964-015-0104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, Toguchida J. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A. 2015;112(50):15438–43. doi: 10.1073/pnas.1510540112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agarwal S, Loder SJ, Cholok D, Peterson J, Li J, Breuler C, Cameron Brownley R, Hsin Sung H, Chung M, Kamiya N, Li S, Zhao B, Kaartinen V, Davis TA, Qureshi AT, Schipani E, Mishina Y, Levi B. Scleraxis-Lineage Cells Contribute to Ectopic Bone Formation in Muscle and Tendon. Stem Cells. 2016 doi: 10.1002/stem.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, Mangiavini L, Groppe JC, Shore EM, Schipani E, Kaplan FS, Pignolo RJ. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J Bone Miner Res. 2016;31(9):1652–65. doi: 10.1002/jbmr.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, Kaplan FS. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J Bone Miner Res. 2016;31(3):650–6. doi: 10.1002/jbmr.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kan L, Liu Y, McGuire TL, Berger DM, Awatramani RB, Dymecki SM, Kessler JA. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells. 2009;27(1):150–6. doi: 10.1634/stemcells.2008-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kan L, Lounev VY, Pignolo RJ, Duan L, Liu Y, Stock SR, McGuire TL, Lu B, Gerard NP, Shore EM, Kaplan FS, Kessler JA. Substance P signaling mediates BMP-dependent heterotopic ossification. Journal of cellular biochemistry. 2011;112(10):2759–72. doi: 10.1002/jcb.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Convente MR, Wang H, Pignolo RJ, Kaplan FS, Shore EM. The immunological contribution to heterotopic ossification disorders. Curr Osteoporos Rep. 2015;13(2):116–24. doi: 10.1007/s11914-015-0258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gannon FH, Glaser D, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Human pathology. 2001;32(8):842–8. doi: 10.1053/hupa.2001.26464. [DOI] [PubMed] [Google Scholar]
- 53.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1998;(346):19–25. [PubMed] [Google Scholar]
- 54.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nature medicine. 2008;14(12):1363–9. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4(1):33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engers DW, Frist AY, Lindsley CW, Hong CC, Hopkins CR. Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: the discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg Med Chem Lett. 2013;23(11):3248–52. doi: 10.1016/j.bmcl.2013.03.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS chemical biology. 2010;5(2):245–53. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS chemical biology. 2013;8(6):1291–302. doi: 10.1021/cb300655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohedas AH, Wang Y, Sanvitale CE, Canning P, Choi S, Xing X, Bullock AN, Cuny GD, Yu PB. Structure-activity relationship of 3,5-diaryl-2-aminopyridine ALK2 inhibitors reveals unaltered binding affinity for fibrodysplasia ossificans progressiva causing mutants. J Med Chem. 2014;57(19):7900–15. doi: 10.1021/jm501177w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, Wang Y, Triffitt JT, Cuny GD, Yu PB, Hill CS, Bullock AN. A new class of small molecule inhibitor of BMP signaling. PloS one. 2013;8(4):e62721. doi: 10.1371/journal.pone.0062721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, Seifert M, Posch W, Nairz M, Maciejewski P, Fowles P, Burns CJ, Smith G, Wagner KU, Weiss G, Whitney JA, Theurl I. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production and ameliorates anemia of chronic disease in rodents. Blood. 2017 doi: 10.1182/blood-2016-09-740092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis TC, Prywes R. Serum regulation of Id1 expression by a BMP pathway and BMP responsive element. Biochim Biophys Acta. 2013;1829(10):1147–59. doi: 10.1016/j.bbagrm.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini-Rousseau L, Cornilleau G, Le Dret L, Richon C, Lacroix L, Puget S, Geoerger B, Vassal G, Ostman A, Grill J. Preclinical evaluation of dasatinib alone and in combination with cabozantinib for the treatment of diffuse intrinsic pontine glioma. Neuro Oncol. 2015;17(7):953–64. doi: 10.1093/neuonc/nou330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene therapy. 2012;19(7):786–90. doi: 10.1038/gt.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi S, Cai J, de Gorter DJ, Sanchez-Duffhues G, Kemaladewi DU, Hoogaars WM, Aartsma-Rus A, t Hoen PA, ten Dijke P. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PloS one. 2013;8(7):e69096. doi: 10.1371/journal.pone.0069096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, Pacifici M, Iwamoto M, Shore EM. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J Bone Miner Res. 2016;31(9):1666–75. doi: 10.1002/jbmr.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, Chandraratna RA, Mishina Y, Enomoto-Iwamoto M, Pacifici M, Iwamoto M. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nature medicine. 2011;17(4):454–60. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 69.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ, P. St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica. 2012;124(3):439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA, Zheludkova O, Milde T, Witt O, Kulozik AE, Reifenberger G, Jabado N, Perry A, Lichter P, von Deimling A, Pfister SM, Jones DT. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta neuropathologica. 2015;129(5):669–78. doi: 10.1007/s00401-015-1405-4. [DOI] [PubMed] [Google Scholar]
- 72.Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R, Ling C, Pajovic S, Yu M, Rubin JB, Hukin J, Steinbok P, Bartels U, Bouffet E, Tabori U, Hawkins C. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta neuropathologica communications. 2016;4(1):93. doi: 10.1186/s40478-016-0353-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Hojfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Seker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, von Deimling A, Jabado N, Puget S, Grill J, Helin K, Korshunov A, Lichter P, Monje M, Plass C, Cho YJ, Pfister SM. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24(5):660–72. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 74.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, Sarkaria J, Zhang Z. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & development. 2013;27(9):985–90. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (New York, NY) 2013;340(6134):857–61. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, Solomon D, Mueller S, Paris PL, Zhang Z, Petritsch C, Gupta N, Waldman TA, James CD. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nature medicine. 2014;20(12):1394–6. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, Tvardovskiy A, Jensen ON, Olaciregui NG, Lavarino C, Sunol M, de Torres C, Mora J, Carcaboso AM, Helin K. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nature medicine. 2017 doi: 10.1038/nm.4293. [DOI] [PubMed] [Google Scholar]
- 78.Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, Misharin AV, Abshiru NA, Lulla RR, Saratsis AM, Kelleher NL, James CD, Shilatifard A. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nature medicine. 2017 doi: 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, Ponnuswami A, Chen S, Johung TB, Sun W, Kogiso M, Du Y, Qi L, Huang Y, Hutt-Cabezas M, Warren KE, Le Dret L, Meltzer PS, Mao H, Quezado M, van Vuurden DG, Abraham J, Fouladi M, Svalina MN, Wang N, Hawkins C, Nazarian J, Alonso MM, Raabe EH, Hulleman E, Spellman PT, Li XN, Keller C, Pal R, Grill J, Monje M. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature medicine. 2015;21(6):555–9. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor IC, Hutt-Cabezas M, Brandt WD, Kambhampati M, Nazarian J, Chang HT, Warren KE, Eberhart CG, Raabe EH. Disrupting NOTCH Slows Diffuse Intrinsic Pontine Glioma Growth, Enhances Radiation Sensitivity, and Shows Combinatorial Efficacy With Bromodomain Inhibition. J Neuropathol Exp Neurol. 2015;74(8):778–90. doi: 10.1097/NEN.0000000000000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, Ma J, Hulleman E, Swigut T, Wysocka J, Tang Y, Monje M. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017 doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (New York, NY) 2014;346(6216):1529–33. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, Ligon KL, Horbinski C, Venneti S, Zadeh G, Santi M, Zhou S, Appin CL, Sioletic S, Sullivan LM, Martinez-Lage M, Robinson AE, Yong WH, Cloughesy T, Lai A, Phillips HS, Marshall R, Mueller S, Haas-Kogan DA, Molinaro AM, Perry A. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain pathology (Zurich, Switzerland) 2013;23(5):565–73. doi: 10.1111/bpa.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, Roujeau T, Dessen P, Richon C, Lazar V, Le Teuff G, Sainte-Rose C, Geoerger B, Vassal G, Jones C, Grill J. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PloS one. 2012;7(2):e30313. doi: 10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28(8):1337–44. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 86.Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, Yang S, Jones S, Wang S, Zhou W, Zhu H, Killela PJ, Zhang J, Wu Z, Li G, Hao S, Wang Y, Webb JB, Friedman HS, Friedman AH, McLendon RE, He Y, Reitman ZJ, Bigner DD, Yan H. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet. 2014;46(7):726–30. doi: 10.1038/ng.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho CY, Kambhampati M, Hwang EI, Faury D, Siu A, Papillon-Cavanagh S, Bechet D, Ligon KL, Ellezam B, Ingram WJ, Stinson C, Moore AS, Warren KE, Karamchandani J, Packer RJ, Jabado N, Majewski J, Nazarian J. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nature communications. 2016;7:11185. doi: 10.1038/ncomms11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal. 2011;23(11):1831–42. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 89.Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006;66(8):4215–22. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- 90.Halvorson KG, Barton KL, Schroeder K, Misuraca KL, Hoeman C, Chung A, Crabtree DM, Cordero FJ, Singh R, Spasojevic I, Berlow N, Pal R, Becher OJ. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PloS one. 2015;10(3):e0118926. doi: 10.1371/journal.pone.0118926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Misuraca KL, Hu G, Barton KL, Chung A, Becher OJ. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia. 2016;18(1):60–70. doi: 10.1016/j.neo.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.John Lin CC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, Mohila CA, Ahmed N, Patel AJ, Arenkiel BR, Noebels JL, Creighton CJ, Deneen B. Identification of diverse astrocyte populations and their malignant analogs. Nature neuroscience. 2017;20(3):396–405. doi: 10.1038/nn.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505(7483):318–26. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bergles DE, Richardson WD. Oligodendrocyte Development and Plasticity. Cold Spring Harbor perspectives in biology. 2015;8(2):a020453. doi: 10.1101/cshperspect.a020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bayraktar OA, Fuentealba LC, Alvarez-Buylla A, Rowitch DH. Astrocyte development and heterogeneity. Cold Spring Harbor perspectives in biology. 2014;7(1):a020362. doi: 10.1101/cshperspect.a020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martynoga B, Drechsel D, Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harbor perspectives in biology. 2012;4(10) doi: 10.1101/cshperspect.a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bond AM, Bhalala OG, Kessler JA. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Developmental neurobiology. 2012;72(7):1068–84. doi: 10.1002/dneu.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grinspan JB. Bone Morphogenetic Proteins: Inhibitors of Myelination in Development and Disease. Vitam Horm. 2015;99:195–222. doi: 10.1016/bs.vh.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 99.Katagiri T, Tsukamoto S. The unique activity of bone morphogenetic proteins in bone: a critical role of the Smad signaling pathway. Biological chemistry. 2013;394(6):703–14. doi: 10.1515/hsz-2012-0310. [DOI] [PubMed] [Google Scholar]
- 100.Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Developmental neuroscience. 2000;22(1–2):74–85. doi: 10.1159/000017429. [DOI] [PubMed] [Google Scholar]
- 101.Hegarty SV, O’Keeffe GW, Sullivan AM. BMP-Smad 1/5/8 signalling in the development of the nervous system. Progress in neurobiology. 2013;109:28–41. doi: 10.1016/j.pneurobio.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 102.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444(7120):761–5. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 103.Chirasani SR, Sternjak A, Wend P, Momma S, Campos B, Herrmann IM, Graf D, Mitsiadis T, Herold-Mende C, Besser D, Synowitz M, Kettenmann H, Glass R. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain. 2010;133(Pt 7):1961–72. doi: 10.1093/brain/awq128. [DOI] [PubMed] [Google Scholar]
- 104.Zhou Z, Sun L, Wang Y, Wu Z, Geng J, Miu W, Pu Y, You Y, Yang Z, Liu N. Bone morphogenetic protein 4 inhibits cell proliferation and induces apoptosis in glioma stem cells. Cancer Biother Radiopharm. 2011;26(1):77–83. doi: 10.1089/cbr.2010.0857. [DOI] [PubMed] [Google Scholar]
- 105.Srikanth M, Kim J, Das S, Kessler JA. BMP signaling induces astrocytic differentiation of clinically derived oligodendroglioma propagating cells. Molecular cancer research : MCR. 2014;12(2):283–94. doi: 10.1158/1541-7786.MCR-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, Kim M, Totonchy M, Cusack T, Ene C, Ma H, Su Q, Zenklusen JC, Zhang W, Maric D, Fine HA. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13(1):69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes & development. 2001;15(16):2094–110. doi: 10.1101/gad.894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, Fisher PG, Weissman IL, Rowitch DH, Vogel H, Wong AJ, Beachy PA. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–8. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tate MC, Lindquist RA, Nguyen T, Sanai N, Barkovich AJ, Huang EJ, Rowitch DH, Alvarez-Buylla A. Postnatal growth of the human pons: a morphometric and immunohistochemical analysis. J Comp Neurol. 2015;523(3):449–62. doi: 10.1002/cne.23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lindquist RA, Guinto CD, Rodas-Rodriguez JL, Fuentealba LC, Tate MC, Rowitch DH, Alvarez-Buylla A. Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nature communications. 2016;7:11628. doi: 10.1038/ncomms11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ballester LY, Wang Z, Shandilya S, Miettinen M, Burger PC, Eberhart CG, Rodriguez FJ, Raabe E, Nazarian J, Warren K, Quezado MM. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol. 2013;37(9):1357–64. doi: 10.1097/PAS.0b013e318294e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anderson JL, Muraleedharan R, Oatman N, Klotter A, Sengupta S, Waclaw RR, Wu J, Drissi R, Miles L, Raabe EH, Weirauch ML, Fouladi M, Chow LM, Hoffman L, DeWire M, Dasgupta B. The transcription factor Olig2 is important for the biology of diffuse intrinsic pontine gliomas. Neuro-Oncology. 2017 doi: 10.1093/neuonc/now299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choe Y, Pleasure SJ, Mira H. Control of Adult Neurogenesis by Short-Range Morphogenic-Signaling Molecules. Cold Spring Harbor perspectives in biology. 2015;8(3):a018887. doi: 10.1101/cshperspect.a018887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Israelsson C, Lewen A, Kylberg A, Usoskin D, Althini S, Lindeberg J, Deng CX, Fukuda T, Wang Y, Kaartinen V, Mishina Y, Hillered L, Ebendal T. Genetically modified bone morphogenetic protein signalling alters traumatic brain injury-induced gene expression responses in the adult mouse. J Neurosci Res. 2006;84(1):47–57. doi: 10.1002/jnr.20856. [DOI] [PubMed] [Google Scholar]
- 115.Kan L, Kitterman JA, Procissi D, Chakkalakal S, Peng CY, McGuire TL, Goldsby RE, Pignolo RJ, Shore EM, Kaplan FS, Kessler JA. CNS demyelination in fibrodysplasia ossificans progressiva. Journal of neurology. 2012;259(12):2644–55. doi: 10.1007/s00415-012-6563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255(1):164–77. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- 117.Dutta DJ, Zameer A, Mariani JN, Zhang J, Asp L, Huynh J, Mahase S, Laitman BM, Argaw AT, Mitiku N, Urbanski M, Melendez-Vasquez CV, Casaccia P, Hayot F, Bottinger EP, Brown CW, John GR. Combinatorial actions of Tgfbeta and Activin ligands promote oligodendrocyte development and CNS myelination. Development. 2014;141(12):2414–28. doi: 10.1242/dev.106492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 118.Cambray S, Arber C, Little G, Dougalis AG, de Paola V, Ungless MA, Li M, Rodriguez TA. Activin induces cortical interneuron identity and differentiation in embryonic stem cell-derived telencephalic neural precursors. Nature communications. 2012;3:841. doi: 10.1038/ncomms1817. [DOI] [PubMed] [Google Scholar]
- 119.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ. C. ffrench-Constant, M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature neuroscience. 2013;16(9):1211–8. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wiese M, Schill F, Sturm D, Pfister S, Hulleman E, Johnsen SA, Kramm CM. No Significant Cytotoxic Effect of the EZH2 Inhibitor Tazemetostat (EPZ-6438) on Pediatric Glioma Cells with Wildtype Histone 3 or Mutated Histone 3.3. Klin Padiatr. 2016;228(3):113–7. doi: 10.1055/s-0042-105292. [DOI] [PubMed] [Google Scholar]
- 121.Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis. 2009;30(2):238–48. doi: 10.1093/carcin/bgn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Deng H, Ravikumar TS, Yang WL. Overexpression of bone morphogenetic protein 4 enhances the invasiveness of Smad4-deficient human colorectal cancer cells. Cancer Lett. 2009;281(2):220–31. doi: 10.1016/j.canlet.2009.02.046. [DOI] [PubMed] [Google Scholar]
- 123.Wang Z, Shen Z, Li Z, Duan J, Fu S, Liu Z, Bai H, Zhang Z, Zhao J, Wang X, Wang J. Activation of the BMP-BMPR pathway conferred resistance to EGFR-TKIs in lung squamous cell carcinoma patients with EGFR mutations. Proc Natl Acad Sci U S A. 2015;112(32):9990–5. doi: 10.1073/pnas.1510837112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu YL, Maachani UB, Schweitzer M, Singh R, Wang M, Chang R, Souweidane MM. Dual Inhibition of PI3K/AKT and MEK/ERK Pathways Induces Synergistic Antitumor Effects in Diffuse Intrinsic Pontine Glioma Cells. Transl Oncol. 2017;10(2):221–228. doi: 10.1016/j.tranon.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang DF, Li XG, Su LJ, Meng QL. Expression of activin A and follistatin in glioblastoma and their effects on U87 in vitro. J Int Med Res. 2010;38(4):1343–53. doi: 10.1177/147323001003800416. [DOI] [PubMed] [Google Scholar]
- 126.Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017;31(3):326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caretti V, Sewing AC, Lagerweij T, Schellen P, Bugiani M, Jansen MH, van Vuurden DG, Navis AC, Horsman I, Vandertop WP, Noske DP, Wesseling P, Kaspers GJ, Nazarian J, Vogel H, Hulleman E, Monje M, Wurdinger T. Human pontine glioma cells can induce murine tumors. Acta neuropathologica. 2014;127(6):897–909. doi: 10.1007/s00401-014-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nazarian J, Mason GE, Ho CY, Panditharatna E, Kambhampati M, Vezina LG, Packer RJ, Hwang EI. Histological and molecular analysis of a progressive diffuse intrinsic pontine glioma and synchronous metastatic lesions: a case report. Oncotarget. 2016;7(27):42837–42842. doi: 10.18632/oncotarget.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF. Getting into the brain: approaches to enhance brain drug delivery. CNS drugs. 2009;23(1):35–58. doi: 10.2165/0023210-200923010-00003. [DOI] [PubMed] [Google Scholar]
- 130.Dasgupta A, Liu M, Ojha T, Storm G, Kiessling F, Lammers T. Ultrasound-mediated drug delivery to the brain: principles, progress and prospects. Drug discovery today Technologies. 2016;20:41–48. doi: 10.1016/j.ddtec.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rip J. Liposome technologies and drug delivery to the CNS. Drug discovery today Technologies. 2016;20:53–58. doi: 10.1016/j.ddtec.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 132.Zhou Z, Singh R, Souweidane MM. Convection-Enhanced Delivery for Diffuse Intrinsic Pontine Glioma Treatment. Current neuropharmacology. 2017;15(1):116–128. doi: 10.2174/1570159X14666160614093615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Melamed RD, Emmett KJ, Madubata C, Rzhetsky A, Rabadan R. Genetic similarity between cancers and comorbid Mendelian diseases identifies candidate driver genes. Nature communications. 2015;6:7033. doi: 10.1038/ncomms8033. [DOI] [PMC free article] [PubMed] [Google Scholar]