

Abstract
The beta hairpin motif is a ubiquitous protein structural motif that can be found in molecules across the tree of life. This motif, which is also popular in synthetically designed proteins and peptides, is known for its stability and adaptability to broad functions. Here, we systematically probe all 49,000 unique beta hairpin substructures contained within the Protein Data Bank (PDB) to uncover key characteristics correlated with stable beta hairpin structure, including amino acid biases and enriched interstrand contacts. We find that position specific amino acid preferences, while seen throughout the beta hairpin structure, are most evident within the turn region, where they depend on subtle turn dynamics associated with turn length and secondary structure. We also establish a set of broad design principles, such as the inclusion of aspartic acid residues at a specific position and the careful consideration of desired secondary structure when selecting residues for the turn region, that can be applied to the generation of libraries encoding proteins or peptides containing beta hairpin structures.
Keywords: beta hairpin, computational biology, PDB, protein design
1. INTRODUCTION
Beta hairpins, one of the simplest stable protein structural elements, consist of two antiparallel beta‐sheets joined by a short loop region. Despite their simplicity in form, beta hairpins are highly adaptable in function. Beta strands are known to participate in protein–protein interactions that are often facilitated by specific amino acid orientations 1 and beta hairpin motifs are no different. 2 , 3 , 4 Indeed, these motifs are a core feature in a diverse array of bioactive molecules, from large beta barrel proteins that transport cargo through cellular membranes 5 , 6 , 7 to substantially smaller antimicrobial peptides and peptide derivatives. 8 , 9 , 10 Whether through self‐aggregation, 11 , 12 target binding, 13 or amphipathic structure formation, 6 , 14 beta hairpin motifs facilitate a range of different biological functions.
In addition to its prevalence in nature, the beta hairpin motif is stable in even small structures and extensively adaptable to specific functions, making it a popular choice in engineered protein structures. Efforts to design such structures have benefited from several decades of research aimed at identifying how beta hairpins form 15 , 16 , 17 and what factors influence their stability and specific activity. 2 , 18 , 19 , 20 , 21 Examples of synthetic proteins that have successfully adapted the beta hairpin motif for specialized functions include hydrogels, 9 antimicrobial peptides, 22 and various molecules with material science applications. 8
Although largely successful, beta hairpin engineering efforts are typically limited to testing relatively small libraries involving derivatives of a stable scaffold structure or existing protein via peptidomimetics. 2 , 4 , 19 , 23 , 24 With the increasing availability of high throughput screening platforms to test for activity in large libraries of de novo sequences 25 , 26 , 27 there is an obvious need for broader design principles that can be applied to the generation of libraries with millions of diverse beta hairpin containing proteins. Knowledge of amino acid propensities throughout known beta hairpin sub‐structures could inform such design principles but existing catalogs are too broadly focused on beta sheets, outdated, or limited in scope. 16 , 20 , 28 , 29 , 30 , 31 An up‐to‐date characterization of amino acid distributions at specific positions within beta hairpins does not exist.
Using a systematic analysis of sequence and structural data from all beta hairpin containing proteins in the Protein Data Bank (PDB), we derived key sequence factors and patterns common to beta hairpins. Important features include amphipathic faces created by the periodic alternation of hydrophilic and hydrophobic amino acids within beta strands, the high prevalence of aspartic acid/asparagine caps at the N‐terminal end of beta strands, and specific residue contacts that are over (e.g., cysteine‐cysteine and salt bridges) and under (e.g., proline‐lysine) represented. These findings give us a broader understanding of naturally occurring beta hairpins and will aid future efforts in the design of bioactive molecules containing the beta hairpin motif.
2. RESULTS
2.1. General approach
To identify and classify motifs we used the following process (see Section 4 for further detail). We first collected all PDB structures 32 and their corresponding amino acid sequences filtered to 90% similarity. We then used DSSP‐derived secondary structure annotations 33 to identify potential beta hairpin substructures consisting of two antiparallel beta‐sheets joined by a short loop region (Figure 1). After determining contacting residues between beta strands, we excluded any structures with less than four contacts from further analysis. This process identified nearly 50,000 unique beta hairpin motifs from some 24,000 independent protein structures. Using these structures, we calculated average amino acid frequencies within structural regions and observed amino acid contacts between hairpin beta strands. We then classified and divided motif structures based on turn length and orientation of beta strand faces. Using these groupings, we determined average amino acid frequencies at each position of the beta hairpin motif.
FIGURE 1.
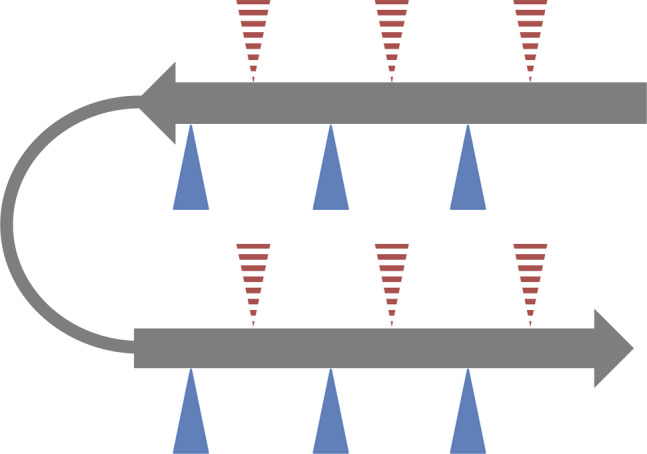
General beta hairpin structure. Beta hairpins consist of two anti‐parallel beta strands (grey arrows) linked with a flexible turn region (grey line). Beta strands typically have amphipathic characteristics conferred by alternating hydrophobic and hydrophilic residues. Triangles represent beta strand side amino acid side chains, with red indicating hydrophobic and blue indicating hydrophilic residues. Dashed triangles indicate side chains oriented away from the viewer while solid triangles indicate side chains oriented toward the viewer
2.2. Secondary structure explains average amino acid frequencies
It has long been known that different secondary structural elements tend to favor the inclusion of certain amino acids over others. 29 , 30 , 34 , 35 This is exactly what we see with our analysis of beta hairpin motifs (Figure 2), with a clear difference in average amino acid frequencies between beta strands, the turn region, and background levels (i.e., universal average frequencies for amino acids across all included protein structures). Our analysis agrees with previous work illustrating a strong preference for glycine, asparagine, and aspartic acid in flexible turn regions. 29 , 30 While proline is also more common in the turn region than in either beta strand, we see no difference in turn region prevalence when compared to background levels. This is in contrast to previous findings that saw significant enrichment of proline in turn regions. 8 , 36 , 37 , 38 This lack of proline enrichment and the relatively low average proline abundance in the turn region is particularly surprising given the known role of such residues in stabilizing beta turns. 36 , 39
FIGURE 2.
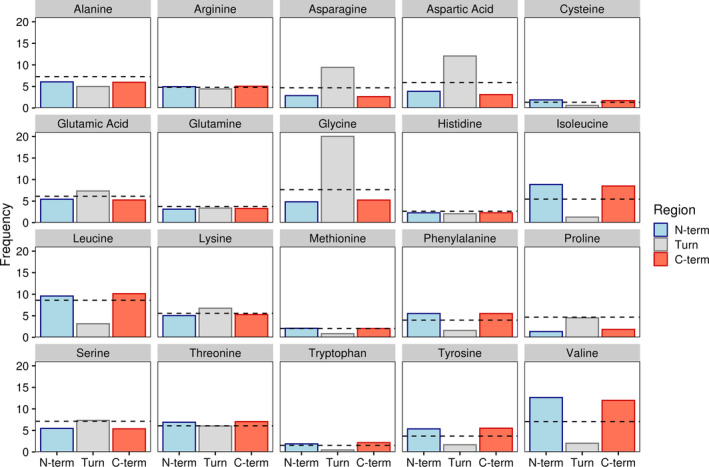
Amino acid frequencies by beta hairpin secondary structure region. Bars indicate average amino acid frequencies for each amino acid within a given region of all beta hairpins. The black dashed line indicates background amino acid frequencies for all sites in all proteins containing the beta hairpin motif. N‐term and C‐term refer to the N‐ and C‐terminal beta strands while turn denotes the turn region
When looking at amino acid levels in the beta strands, there appears to be little to no difference in prevalence between strands. Both strands show an increased occurrence of isoleucine, valine, and several other chiefly hydrophobic residues in beta sheet structures, supporting previous research. 40 Additionally, both strands show a greater tolerance for positively charged residues as is commonly observed with anti‐parallel beta strands as opposed to their parallel counterparts. 7 , 10 , 41 We further probed for differences across domains of life but saw no strong trends in individual amino acids (Figure S1a). There were, however, taxa specific differences in turn region preference for polar and negatively charged amino acids (Figure S1b).
2.3. Residue positional biases are linked to flexibility, stability, and hydrophobicity
Beta hairpins, especially those in membrane interacting structures such as beta barrels and some antimicrobial peptides, are known to incorporate amphipathic beta sheets that periodically alternate between hydrophilic and hydrophobic amino acids, creating two distinct faces 42 , 43 (Figure 1). To account for these faces in our analysis, we divided our dataset based on the presentation of an initial polar or hydrophobic face for both the N and C terminal beta strands (see Section 4). After accounting for these amphipathic faces as well as differences in turn region length, clear patterns emerged in all regions of the beta hairpin motif (Figure 3). The most obvious pattern observed was the alternating preference for charged/polar and hydrophobic residues in both beta strands (Figure 3a,b). While hydrophobic residues appear to be more favorable in either beta strand on average (Figure 2), polar and charged residues are well tolerated when oriented correctly.
FIGURE 3.
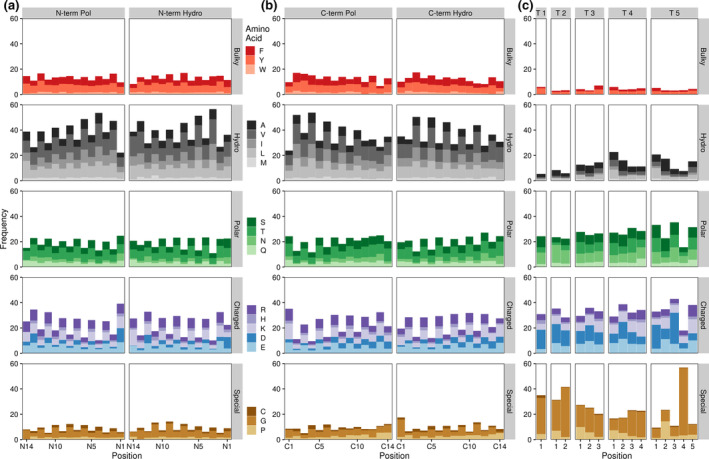
Amino acid frequencies by beta hairpin residue position. Bars indicate average amino acid frequencies for each amino acid at a given position across all beta hairpin structures. (a) and (b) N‐term and C‐term refer to the N‐ and C‐terminal beta strands. Pol refers to beta strands containing a polar face adjacent to the turn region, Hydro denotes a hydrophobic face at this position. Beta strand residues are numbered from the turn region, with residue 1 representing the residue closest to the turn. (c) T # denotes a turn region of a given length (e.g., T 3 indicates a three residue turn region). Turn residues are numbered from N‐terminal (residue 1) to C‐terminal
On a more granular level, we further surveyed for differences in amino acid frequencies at specific locations within the larger hairpin motif. In contrast to their average beta strand frequencies, hydrophobic amino acids are also less tolerated at the C‐terminal edge of either beta strand regardless of orientation. In their place, aspartic acid and (to a lesser extent) asparagine are over‐represented at these loci, with this effect being particularly strong for the N‐terminal beta strand where the last residue is one of these two amino acids in nearly 20% of observed hairpins. This frequency is roughly that observed for these two amino acids, on average, in the turn region (Figures 2 and 3c), although other common turn and cap‐associated residues, namely glycine and proline, do not show an over‐representation at these positions. Interestingly, aspartic acid residues at the C‐terminal end of either beta strand also correlate with increased frequencies of bulky aromatic amino acids (i.e., tyrosine, tryptophan, and phenylalanine) at the N‐terminally adjacent position and a preference for glycine at the first N‐terminal strand residue (Figure S2a).
Although proline showed no enrichment in the average turn region compared to background levels (Figure 2), proline frequencies are slightly higher than background in the first residue of turns with three to four amino acids and substantially higher than background in the second residue of turns with five amino acids (Figure 3c). These findings largely agree with existing evidence on the prevalence and importance of prolines in the beginning of turn regions 44 , 45 , 46 but the nearly four‐fold enrichment for residue two prolines in hairpin structures with five amino acid long turn regions when compared to background levels is particularly surprising. In combination with the fact that over half of all fourth residues in five amino acid long turn regions are glycines, these findings suggest that beta hairpins with longer turn regions may have very specific physiochemical requirements that limit amino acid diversity.
2.4. Turn secondary structure provides further context for amino acid variations
Beta hairpin turn regions exhibit much more structural variability in comparison to their beta strand counterparts. Turns, as defined in our search parameters (see Section 4), can be comprised of hydrogen bonded turn residues or less well defined and more loosely structured bend or coil residues. 33 , 38 Turns can also incorporate any mixture of these residue types. As much of the existing research into turn residue amino acid propensities either lumps the separate DSSP classifications into one category as we have done above 30 , 37 , 47 or focuses on a limited number of protein structures, 38 we sought to clarify if beta hairpin turn regions exhibited different amino acid distributions based on residue type in our larger dataset.
In separating out turn regions by not only length, but also turn type we identified strong differences between turn regions that contained exclusively hydrogen bonded residues (bonded), all bend or coil residues (bend), and a mix of residues (mixed; Figure 4). At the turn‐wide level, the constraints imposed by specific secondary structure orientations lead to some striking trends in amino acid preferences. This is clearly seen with glycine propensities, with Mixed turns containing three to five residues showing drastically higher glycine preference toward the C‐terminal end of the turn and Bonded and Bend turns of the same length showing more even glycine levels. On a more granular level, amino acid differences at particular loci also abound. For instance, the first residue of both three and four amino acid long Bonded turns are enriched for proline residues while the same residues in Bend or Mixed turns are not. Taken altogether, these trends help to clarify the general averages established in Figure 3.
FIGURE 4.
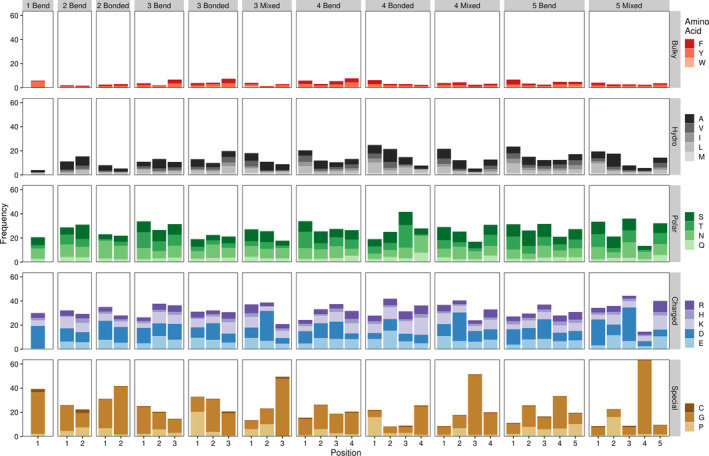
Amino acid frequencies by beta hairpin turn residue position, length, and type. Bars indicate average amino acid frequencies for each amino acid at a given position across all beta hairpin turn structures. Bonded refers to turn regions containing only hydrogen bonded turn residues, bend indicates turn regions with coil and/or bend residues, and mixed indicates a mixture or hydrogen bonded and coil and/or bend residues. Numbers preceding the turn type indicate the length of the turn region. Residue positions are numbered as in Figure 3. Frequencies are only shown for turn types with at least 100 representative structures in our dataset
Hydrogen bonded turns are a particularly active area of study with many specific subtypes characterized by both length and amino acid side chain psi and phi angles. 48 , 49 , 50 , 51 , 52 Using this existing nomenclature, we found that established class specific amino acid frequencies for beta and gamma turns 46 , 49 , 52 , 53 largely hold true in our dataset (Table S1). Additionally, our findings seem to agree with general amino acid frequencies previously observed in alpha turns, 54 although no published site specific amino acid frequencies across alpha turn subtypes exist for comparison.
2.5. Amino acid contacts between strands favor stabilizing interactions
As the overall beta hairpin structure is stabilized by interactions between the two beta strands, we sought to identify enriched amino acid pairings between strands to see if certain interactions were more common than expected. Pairings between residues with similar electrostatic properties, that is two hydrophobic residues or a polar residue and a polar/charged residue, were largely more common than expected (Figures 5 and S3). This data agrees with our previous findings regarding the grouping amino acids into beta strand faces based on similar physiochemical properties. In a similar vein to the pairing of electrochemically similar residues, oppositely charged residues tended to pair together in electrostatically favorable salt bridges that are known to stabilize protein structures. 55 , 56 , 57 , 58 Such salt bridges represented some of the most enriched amino acid pairings.
FIGURE 5.
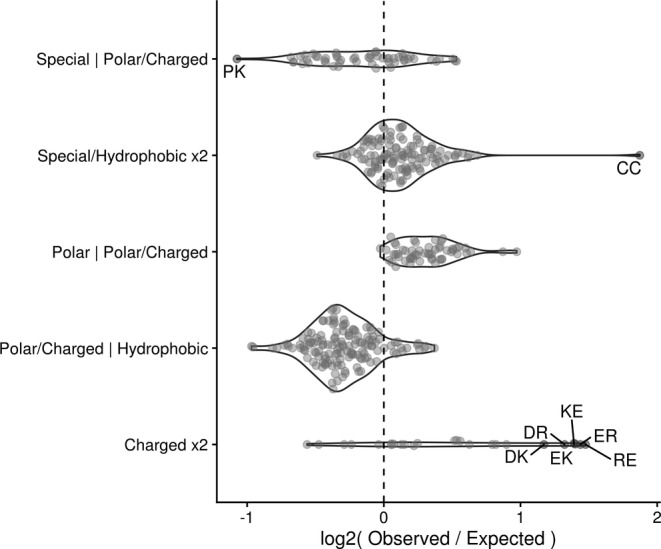
Grouped differences in observed versus expected residue contacts. Dots represent individual contacting pairs with red, labeled dots indicating contacts that are enriched or depleted at least two‐fold versus expected values. Residues are grouped as follows: Special refers to cysteine, proline, and glycine; Hydrophobic refers to valine, leucine, isoleucine, methionine, alanine, tryptophan, tyrosine, phenylalanine; Polar refers to glutamine, threonine, serine, and asparagine; Charged refers to arginine, histidine, lysine, aspartic acid, and glutamic acid
The most enriched amino acid pairing between beta strands is that of cysteine with itself to create a structurally stabilizing di‐sulfide bond. Such pairings are often used to stabilize engineered peptide structures 59 , 60 and cysteine coupling is so preferential in nature that many organisms possess a proteome‐wide bias toward even numbers of cysteine residues. 61
In contrast to enriched contact pairings, several classes of interactions, typically those between electrochemically dissimilar residues, were observed much less than expected. The low observance of interstrand contacts between polar/charged and hydrophobic amino acids (Figure 5) is intuitive given the strong repulsive nature between such residues which could destabilize overall protein structure.
2.6. Design principles
Taken altogether, our work provides a strong foundation of general principles that can be applied to the design of functionally diverse high throughput beta hairpin libraries (Table 1). First, libraries should seek to incorporate beta strands with amphipathic faces as seen in our analysis of beta strand positional biases (Figure 3a,b). Second, aspartic acid and asparagine should be favored at C‐terminal beta strand residues, especially in the beta strand preceding the turn region. Next, secondary structure‐based amino acid preferences should inform design choices, especially within the turn region. While residues in both hairpin beta strands show positionally specific frequency deviations from secondary structure averages (Figures 2 and 3a,b), there is a much stronger trend associated with specific secondary structure orientations in the turn region (Figure 4, Table S1). Last, stabilizing interactions should be favored between beta strands. Such interactions include salt bridges, disulfide bonds, and the pairing of certain biochemically similar residues (i.e., hydrophobic–hydrophobic and polar–polar pairings) (Figure 5). These simple guidelines are specific enough to inform design choices while flexible enough to allow for applications across broad research areas.
TABLE 1.
Design principles
| 1. Incorporate amphipathic beta strand faces |
| 2. Favor aspartic acid/asparagine at C‐terminal beta strand residues |
| 3. Account for secondary structure biases, especially in the turn region |
| 4. Favor salt bridges and di‐cysteine interactions to provide stability |
3. DISCUSSION
By analyzing the composition of beta hairpin motifs across all proteins within the PDB we have identified key characteristics of this versatile structure. Expanding on existing knowledge of secondary structure biases, we outline the preference for the amphipathic orientation of amino acids within beta strands to create two faces with different physiochemical properties. We further identify key positional preferences for specific amino acids in all regions of the hairpin motif with a detailed analysis of these preferences across different turn region types and subtypes. Lastly, we highlight the importance of stabilizing interactions between residues in the N and C terminal beta strands of the hairpin.
While previous works have characterized the amino acid frequencies common to specific secondary structures and protein turn motifs, 28 , 29 , 30 , 40 , 62 here we uncover amino acid propensities across all sub‐structures and residues of the specific beta hairpin motif. This work further builds upon prior research into beta hairpin classification 20 , 31 and beta hairpin scaffold design 23 , 24 , 44 , 63 by greatly expanding the number of beta hairpin structures considered from a maximum of few thousand to nearly 50,000. By analyzing 49,000 unique beta hairpin substructures we are empowered to provide a systematic framework and novel insights to describe the beta hairpin motif. We find that stable beta hairpin structures tend to possess site‐specific amino acid preferences and to incorporate amphipathic character in both hairpin beta strands. While existing secondary‐structure‐specific amino acid distributions 29 , 30 are accurate and informative, such averages prove inadequate to capture the inherent nuances of the beta hairpin motif. For instance, while our analysis finds that an average hairpin beta strand would consist of only hydrophobic residues (Figure 2), a beta hairpin containing two such average strands without any amphipathic character would be statistically improbable (Figure 3a,b) and highly unlikely to fold correctly, 21 let alone function biologically. 10
By breaking down our analysis by both residue location and secondary structure orientation, we have been able to uncover which position‐specific amino acid biases need to be considered to help form stable beta hairpin protein structures. Our observation that prolines are less enriched in turn regions (Figure 2) than previously observed 8 , 36 is perhaps best explained by the extreme position‐specific preference of proline residues in turn regions of a given length and secondary structure (Figures 3c and 4, Table S1). Thus, certain proline residues are enriched within and likely to stabilize hairpin turn regions even though there is no strong trend when averaged across all turn residues. This explanation would also hold true for glycine turn residues, which show the most extreme variability in propensities depending on location and secondary structure (Figure 4). Outside of the turn region, hairpin beta strands also exhibit amino acid biases at key loci as well as a strong proclivity to incorporate stabilizing interstrand contacts. We find that asparagine and aspartic acid residues are much more common at the C‐terminal end of either hairpin beta strand (Figures 3a,b and S2). These residues may participate in a beta capping phenomenon to block the continuation of beta structure into a turn region. 16 A beta capping role may also explain our observation of an increased prevalence of bulky aromatic residues preceding terminal aspartic acids (Figure S2) as aromatic residues are known to stabilize beta hairpin structures. 18 , 19
As expected, appropriate contacts between hairpin beta strands are imperative to provide structural stability. As an example, we identified cysteine pairings as being particularly enriched in beta hairpin substructures (Figure 5). Such pairings have long been used to stabilize engineered peptide structures 59 , 60 and are so preferential in nature that many organisms possess a proteome‐wide bias toward even numbers of cysteines. 61
While our analysis of amino acid preferences within beta hairpin secondary structures across the domains of life showed no strong differences (Figure S1a) there were some interesting minor trends as well as a notable difference in turn region composition between taxa (Figure S1b). Cysteines, which are fairly uncommon across proteins in general, appear twice as often in Eukaryotic beta hairpins than in Prokaryotic or Archaeaotic beta hairpins. This observation agrees with previous data showing the same trend of increasing cysteine occurrence in proteomes of more complex organisms. 64 , 65 , 66 , 67 Of greater note is the inverse relationship between polar and negative amino acid propensities within beta hairpin turn regions across taxa. Frequencies for negatively charged amino acids within the turn region decrease from Archaea to Bacteria, Eukarya, and finally Viruses while polar amino acids show the opposite trend. This difference is likely explained by protein adaptations to harsh environments in Archaea/Bacteria 68 that are less commonly encountered by Eukaryotic or viral proteins. This trend is not seen in either beta strand of the hairpin as turn structures are some of the most accessible protein regions 69 and would likely experience more selective pressure in harsh environments than less exposed beta strands.
One major limitation of our approach is that we were only able to establish broad general properties of beta hairpins that might influence overall structure or function. This is in contrast to prior work that has focused on identifying key design factors for specific beta hairpin scaffolds 23 , 24 , 44 , 63 or grouping beta hairpins and related structures into increasingly detailed classifications. 20 , 31 While the PDB dataset that we analyzed could be used to expand upon these highly focused areas of research, the broad applicability of our results would be compromised. For instance, existing beta hairpin classifications that group structures based on the number and types of hydrogen bonded residues within the turn region 20 , 31 have no way to classify half of our identified substructures that have some mix of hydrogen bond lacking bend and coil residues in this region.
In combination with prior research efforts, our simple design guidelines (Table 1) can be adapted to the creation of large‐scale protein or peptide libraries aimed at almost any functional purpose, from anticancer drugs to biosensors. For example, beta hairpin antimicrobial peptides are known to incorporate multiple disulfide bonds and favor an overall net positive charge while still maintaining amphipathic character. 10 , 13 Adapting our design principles with these properties in mind would facilitate the construction of a library of positively charged, disulfide stabilized peptides with presumptive beta hairpin structure to test for antimicrobial activity.
In summary, our findings are broadly adaptable to creating large libraries of beta hairpin containing molecules skewed toward a specific functionality and will help engineering efforts keep pace with the ever‐expanding capacity of screening assays.
4. MATERIALS AND METHODS
4.1. Identification of beta hairpin substructures
We defined the beta hairpin motif as an amino acid sequence containing two sets of four to fourteen extended beta strand residues joined by one to five turn, bend, or unannotated residues. A maximum beta strand length of 14 was selected based on the typical length of beta strands in monomeric beta barrel proteins 70 while the range of turn lengths was selected based on prior research into beta hairpins. 17 We searched DSSP 33 derived secondary structure annotations of all PDB proteins (downloaded from https://cdn.rcsb.org/etl/kabschSander/ss.txt.gz on July 22nd 2020) for this motif. We further filtered our dataset to include only IDs for representative structures clustered to within 90% sequence identity. Clusters were obtained from PDB on July 22nd 2020 using the RESTful Web Service Interface (https://www.rcsb.org/pdb/software/rest.do). Our analysis pipeline automatically retained only one copy of any sets of exactly duplicated beta hairpin sequences. Further manual filtering was applied to exclude overly similar hairpin sequences, largely from structures of nanobodies, antibodies, and their derivatives.
4.2. Identification of contacting residues
To ensure that our analyzed motifs possessed the correct beta hairpin 3D structure, we filtered our dataset to only include structures in which at least four amino acid side chain pairs formed contacts between the N and C terminal beta strands. We defined contacts as any pair of residues in which side‐chain beta carbons were within 8 Angstroms of one another, a common distance threshold derived from the CASP competition guidelines. 71 , 72 Determining contacts via the presence of backbone hydrogen bonds produced similar results (data not included). To calculate expected contact frequencies, individual amino acid frequencies were derived using the relative occurrence of each amino acid across all contact pairs. Values for amino acids in a pairing were then multiplied together to establish an expected frequency for every possible pairing of amino acids.
4.3. Grouping of beta hairpin substructures
To characterize the amphipathic faces of each beta strand, solvent accessibility was averaged across odd and even numbered amino acid residues with the first amino acid being the residue closest to the turn region. Strands in which the odd amino acid residues have a higher mean accessibility were categorized as polar while strands with the opposite phenotype were categorized as hydrophobic. Solvent accessibility was chosen in lieu of hydrophobicity or other metrics as PDB structures contain accessibility information and solvent accessibility is known to correlate with hydrophobicity. 69
4.4. Classification of turn regions
One residue turns have no existing classification structure and observed two and three residue turns did not exhibit the expected angle distributions of delta and gamma turns (data not shown). When we included the flanking residues at the C and N‐terminal ends of these turn regions, however, angle distributions for these turns did appear to agree with published ranges 46 , 49 , 52 , 53 (Table S1). As such, we have classified one, two, and three residue turns as three (gamma), four (beta), and five (alpha) residue turns, respectively. Four and five residue turns are classified as beta and alpha turns. These classification schemes rely on previously published distributions of Ramachandran angles. 48 , 49
4.5. Data and figures
All data was analyzed in R using the tidyverse family of packages 73 in combination with the data.table 74 and seqinr 75 packages. All figures were created using ggplot2 76 and cowplot. 77 Figure S3 additionally utilized the ggseqlogo package. 78 All processed data and analysis scripts are available at https://doi.org/10.5281/zenodo.4069580.
AUTHOR CONTRIBUTIONS
Cory D. DuPai: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; software; validation; visualization; writing‐original draft; writing‐review and editing. Bryan W. Davies: Conceptualization; funding acquisition; project administration; resources; supervision; writing‐review and editing. Claus O. Wilke: Conceptualization; funding acquisition; project administration; resources; supervision; writing‐review and editing.
Supporting information
Figure S1 Amino acid frequencies by beta hairpin secondary structure region and organism domain. (a) Bars indicate average amino acid frequencies for each amino acid within a given region of all beta hairpins broken down by Domain of origin. The black dashed line indicates background amino acid frequencies for all sites in all proteins containing the beta hairpin motif. N‐term and C‐term refer to the N and C‐terminal beta strands while turn denotes the turn region. (b) Same as in (a) with amino acid frequencies grouped into specific classes. Amino acids are grouped as follows: Bulky Aromatic—phenylalanine, tryptophan, tyrosine; Hydrophobic—alanine, isoleucine, leucine, methionine, valine; Negative—aspartic acid, glutamic acid; Polar—asparagine, glutamine, serine, threonine; Positive—arginine, histidine, lysine; Special—cysteine, glycine, and proline.
Figure S2 Motif logo of amino acid probabilities in hairpin beta strands. Amino acid letters are scaled to represent relative frequency at a given position in both beta strands, with more common amino acids listed first vertically. Color indicates the type of amino acid. (a). Logo motif for hairpin beta strands with a C‐terminal aspartic acid that start with either a hydrophobic (left) or polar (right) face relative to the turn region. (b). As in (a) but showing frequencies across all hairpin beta strands, not just those terminated with an aspartic acid residue. Amino acids are grouped as in Figure S1.
Figure S3 Differences in observed versus expected residue contacts Dots represent individual contacting pairs with labeled dots indicating contacts that are enriched or depleted at least two‐fold vs. expected values. Y‐axis labels denote the class of the first residue in the pair while facet headings denote the class of the second amino acid (e.g., the dot for DP would be in the Negative row of the Special column). Amino acids are grouped as in Figure S1.
Table S1: Supporting information
ACKNOWLEDGMENTS
This research was supported by NIH awards R01 AI125337 and R01 AI148419 as well as Welch Foundation award F‐1870. Funders had no role in study design. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DuPai CD, Davies BW, Wilke CO. A systematic analysis of the beta hairpin motif in the Protein Data Bank. Protein Science. 2021;30:613–623. 10.1002/pro.4020
Funding information Division of Intramural Research, National Institute of Allergy and Infectious Diseases, Grant/Award Numbers: R01 AI125337, R01 AI148419; Welch Foundation, Grant/Award Number: F‐1870
REFERENCES
- 1. Watkins AM, Arora PS. Anatomy of β‐strands at protein‐protein interfaces. ACS Chem Biol. 2014;9:1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robinson JA. β‐Hairpin peptidomimetics: Design, structures and biological activities. Acc Chem Res. 2008;41:1278–1288. [DOI] [PubMed] [Google Scholar]
- 3. Chakraborty K, Shivakumar P, Raghothama S, Varadarajan R. NMR structural analysis of a peptide mimic of the bridging sheet of HIV‐1 gp120 in methanol and water. Biochem J. 2005;390:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batalha IL, Lychko I, Branco RJF, Iranzo O, Roque ACA. β‐Hairpins as peptidomimetics of human phosphoprotein‐binding domains. Org Biomol Chem. 2019;17:3996–4004. [DOI] [PubMed] [Google Scholar]
- 5. Remmert M, Biegert A, Linke D, Lupas AN, Söding J. Evolution of outer membrane β‐barrels from an ancestral ββ hairpin. Mol Biol Evol. 2010;27:1348–1358. [DOI] [PubMed] [Google Scholar]
- 6. Chaturvedi D, Mahalakshmi R. Transmembrane β‐barrels: Evolution, folding and energetics. Biochim Biophys Acta Biomembr. 2017;1859:2467–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gupta K, Jang H, Harlen K, et al. Mechanism of membrane permeation induced by synthetic β‐hairpin peptides. Biophys J. 2013;105:2093–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marcelino AMC, Gierasch LM, Craik C. Roles of beta‐turns in protein folding: From peptide models to protein engineering. Biopolymers. 2008;89:380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong N, Ma Q, Shan A, Cao Y. Design and biological activity of beta‐hairpin‐like antimicrobial peptide. Sheng Wu Gong Cheng Xue Bao. 2012;28:243–250. [PubMed] [Google Scholar]
- 10. Edwards IA, Elliott AG, Kavanagh AM, Zuegg J, Blaskovich MAT, Cooper MA. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β‐hairpin peptides. ACS Infect Dis. 2016;2:442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mirecka EA, Shaykhalishahi H, Gauhar A, et al. Sequestration of a β‐hairpin for control of α‐synuclein aggregation. Angew Chem Int Ed. 2014;53:4227–4230. [DOI] [PubMed] [Google Scholar]
- 12. Larini L, Shea JE. Role of β‐hairpin formation in aggregation: The self‐assembly of the amyloid‐β(25‐35) peptide. Biophys J. 2012;103:576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Panteleev PV, Bolosov IA, Balandin SV, Ovchinnikova TV. Structure and biological functions of β‐hairpin antimicrobial peptides. Acta Naturae. 2015;7:37–47. [PMC free article] [PubMed] [Google Scholar]
- 14. Worthington P, Langhans S, Pochan D. β‐hairpin peptide hydrogels for package delivery. Adv Drug Deliv Rev. 2017;110–111:127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewandowska A, Ołdziej S, Liwo A, Scheraga HA. β‐Hairpin‐forming peptides; models of early stages of protein folding. Biophys Chem. 2010;151:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. FarzadFard F, Gharaei N, Pezeshk H, Marashi SA. β‐Sheet capping: Signals that initiate and terminate β‐sheet formation. J Struct Biol. 2008;161:101–110. [DOI] [PubMed] [Google Scholar]
- 17. Gunasekaran K, Ramakrishnan C, Balaram P. Beta‐hairpins in proteins revisited: Lessons for de novo design. Protein Eng Des Sel. 1997;10:1131–1141. [DOI] [PubMed] [Google Scholar]
- 18. Santiveri CM, Jiménez MA. Tryptophan residues: Scarce in proteins but strong stabilizers of β‐hairpin peptides. Pept Sci. 2010;94:779–790. [DOI] [PubMed] [Google Scholar]
- 19. Mahalakshmi R. Aromatic interactions in β‐hairpin scaffold stability: A historical perspective. Arch Biochem Biophys. 2019;661:39–49. [DOI] [PubMed] [Google Scholar]
- 20. Milner‐White EJ, Poet R. Four classes of beta‐hairpins in proteins. Biochem J. 1986;240:289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popp A, Wu L, Keiderling TA, Hauser K. Effect of hydrophobic interactions on the folding mechanism of β‐hairpins. J Phys Chem B. 2014;118:14234–14242. [DOI] [PubMed] [Google Scholar]
- 22. Rughani RV, Schneider JP. Molecular design of beta‐hairpin peptides for material construction. MRS Bull. 2008;33:530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Natale C, La Manna S, Avitabile C, et al. Engineered β‐hairpin scaffolds from human prion protein regions: Structural and functional investigations of aggregates. Bioorg Chem. 2020;96:103594. [DOI] [PubMed] [Google Scholar]
- 24. Cochran AG, Tong RT, Starovasnik MA, et al. A minimal peptide scaffold for β‐turn display: Optimizing a strand position in disulfide‐cyclized β‐hairpins. J Am Chem Soc. 2001;123:625–632. [DOI] [PubMed] [Google Scholar]
- 25. Wu C‐H, Liu I‐J, Lu R‐M, Wu H‐C. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016;23:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tucker AT, Leonard SP, DuBois CD, et al. Discovery of next‐generation antimicrobials through bacterial self‐screening of surface‐displayed peptide libraries. Cell. 2018;172:618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wójcik M, Telzerow A, Quax WJ, Boersma YL. High‐throughput screening in protein engineering: Recent advances and future perspectives. Int J Mol Sci. 2015;16:24918–24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhattacharjee N, Biswas P. Position‐specific propensities of amino acids in the strand. BMC Struct Biol. 2010;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujiwara K, Toda H, Ikeguchi M. Dependence of α‐helical and β‐sheet amino acid propensities on the overall protein fold type. BMC Struct Biol. 2012;12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otaki JM, Tsutsumi M, Gotoh T, Yamamoto H. Secondary structure characterization based on amino acid composition and availability in proteins. J Chem Inf Model. 2010;50:690–700. [DOI] [PubMed] [Google Scholar]
- 31. Sibanda BL, Blundell TL, Thornton JM. Conformation of β‐hairpins in protein structures. A systematic classification with applications to modelling by homology, electron density fitting and protein engineering. J Mol Biol. 1989;206:759–777. [DOI] [PubMed] [Google Scholar]
- 32. Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. [DOI] [PubMed] [Google Scholar]
- 33. Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 34. Koehl P, Levitt M. Structure‐based conformational preferences of amino acids. Proc Natl Acad Sci U S A. 1999;96:12524–12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geisow MJ, Roberts RDB. Amino acid preferences for secondary structure vary with protein class. Int J Biol Macromol. 1980;2:387–389. [Google Scholar]
- 36. Trevino SR, Schaefer S, Scholtz JM, Pace CN. Increasing protein conformational stability by optimizing beta‐turn sequence. J Mol Biol. 2007;373:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Costantini S, Colonna G, Facchiano AM. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem Biophys Res Commun. 2006;342:441–451. [DOI] [PubMed] [Google Scholar]
- 38. Malkov SN, Živković MV, Beljanski MV, Hall MB, Zarić SD. A reexamination of the propensities of amino acids towards a particular secondary structure: classification of amino acids based on their chemical structure. J Mol Model 2008;14:769–775. 10.1007/s00894-008-0313-0. [DOI] [PubMed] [Google Scholar]
- 39. Melnikov S, Mailliot J, Rigger L, et al. Molecular insights into protein synthesis with proline residues. EMBO Rep. 2016;17:1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsutsumi M, Otaki JM. Parallel and antiparallel beta‐strands differ in amino acid composition and availability of short constituent sequences. J Chem Inf Model. 2011;51:1457–1464. [DOI] [PubMed] [Google Scholar]
- 41. Bowerman CJ, Nilsson BL. Self‐assembly of amphipathic β‐sheet peptides: Insights and applications. Biopolymers. 2012;98:169–184. [DOI] [PubMed] [Google Scholar]
- 42. Sivanesam K, Kier BL, Whedon SD, Chatterjee C, Andersen NH. Hairpin structure stability plays a role in the activity of two antimicrobial peptides. FEBS Lett. 2016;590:4480–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang XC, Han L. How does a β‐barrel integral membrane protein insert into the membrane? Protein Cell. 2016;7:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blandl T, Cochran AG, Skelton NJ. Turn stability in beta‐hairpin peptides: Investigation of peptides containing 3:5 type I G1 bulge turns. Protein Sci. 2003;12:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang H, Varady J, Ng L, Sung S‐S. Molecular dynamics simulations of β‐hairpin folding. Proteins. 1999;37:325–333. [DOI] [PubMed] [Google Scholar]
- 46. Shapovalov MI, Vucetic S, Dunbrack RL. A new clustering and nomenclature for beta turns derived from high‐resolution protein structures. PLoS Comput Biol. 2019;15:e1006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chemmama IE, Chapagain PP, Gerstman BS. Pairwise amino acid secondary structural propensities. Phys Rev E Stat Nonlinear Soft Matter Phys. 2015;91:042709. [DOI] [PubMed] [Google Scholar]
- 48. Chou KC. Prediction of tight turns and their types in proteins. Anal Biochem. 2000;286:1–16. [DOI] [PubMed] [Google Scholar]
- 49. Guruprasad K, Rajkumar S. β‐ and γ‐turns in proteins revisited: A new set of amino acid turn‐type dependent positional preferences and potentials. J Biosci. 2000;25:143–156. [PubMed] [Google Scholar]
- 50. Toniolo C, Crisma M, Moretto A, et al. Peptide δ‐turn: Literature survey and recent progress. Chem A Eur J. 2015;21:13866–13877. [DOI] [PubMed] [Google Scholar]
- 51. Sapse A‐M. Sapse A‐M tight turns in proteins Molecular orbital calculations for amino acids and peptides. Boston: Birkhäuser, 2000; p. 113–123. [Google Scholar]
- 52. De Brevern AG. Extension of the classical classification of β‐turns. Sci Rep. 2016;6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koch O, Klebe G. Turns revisited: A uniform and comprehensive classification of normal, open, and reverse turn families minimizing unassigned random chain portions. Proteins. 2009;74:353–367. [DOI] [PubMed] [Google Scholar]
- 54. Pavone V, Gaeta G, Lombardi A, Nastri F, Maglio O, Saviano M. Discovering protein secondary structures: Classification and description of isolated a‐turns. Biopolymers. 1996;38:705–721. [DOI] [PubMed] [Google Scholar]
- 55. Pylaeva S, Brehm M, Sebastiani D. Salt bridge in aqueous solution: Strong structural motifs but weak enthalpic effect. Sci Rep. 2018;8:13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ciani B, Jourdan M, Searle MS. Stabilization of β‐hairpin peptides by salt bridges: Role of preorganization in the energetic contribution of weak interactions. J Am Chem Soc. 2003;125:9038–9047. [DOI] [PubMed] [Google Scholar]
- 57. Donald JE, Kulp DW, DeGrado WF. Salt bridges: Geometrically specific, designable interactions. Proteins. 2011;79:898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bosshard HR, Marti DN, Jelesarov I. Protein stabilization by salt bridges: Concepts, experimental approaches and clarification of some misunderstandings. J Mol Recognit. 2004;17:1–16. [DOI] [PubMed] [Google Scholar]
- 59. Sussman D, Westendorf L, Meyer DW, et al. Engineered cysteine antibodies: An improved antibody‐drug conjugate platform with a novel mechanism of drug‐linker stability. Protein Eng Des Sel. 2018;31:47–54. [DOI] [PubMed] [Google Scholar]
- 60. Dombkowski AA, Sultana KZ, Craig DB. Protein disulfide engineering. FEBS Lett. 2014;588:206–212. [DOI] [PubMed] [Google Scholar]
- 61. Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A. 2008;105:11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Madan B, Seo SY, Lee S‐G. Structural and sequence features of two residue turns in beta‐hairpins. Proteins. 2014;82:1721–1733. [DOI] [PubMed] [Google Scholar]
- 63. Pastor MT, López de la Paz M, Lacroix E, Serrano L, Pérez‐Payá E. Combinatorial approaches: A new tool to search for highly structured β‐hairpin peptides. Proc Natl Acad Sci U S A. 2002;99:614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brooks DJ, Fresco JR. Increased frequency of cysteine, tyrosine, and phenylalanine residues since the last universal ancestor. Mol Cell Proteomics. 2002;1:125–131. [DOI] [PubMed] [Google Scholar]
- 65. Wiedemann C, Kumar A, Lang A, Ohlenschläger O. Cysteines and disulfide bonds as structure‐forming units: Insights from different domains of life and the potential for characterization by NMR. Front Chem. 2020;8:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000;17:1232–1239. [DOI] [PubMed] [Google Scholar]
- 67. Tsuji J, Nydza R, Wolcott E, et al. The frequencies of amino acids encoded by genomes that utilize standard and nonstandard genetic codes. Bios. 2010;81:22–31. [Google Scholar]
- 68. Reed CJ, Lewis H, Trejo E, Winston V, Evilia C. Protein adaptations in archaeal extremophiles. Archaea. 2013;2013:373275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lins L, Thomas A, Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003;12:1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tamm LK, Hong H, Liang B. Folding and assembly of β‐barrel membrane proteins. Biochim Biophys Acta Biomembr. 2004;1666:250–263. [DOI] [PubMed] [Google Scholar]
- 71. Adhikari B, Cheng J. Protein residue contacts and prediction methods. Methods Mol Biol. 2016;1415:463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schaarschmidt J, Monastyrskyy B, Kryshtafovych A, Bonvin AMJJ. Assessment of contact predictions in CASP12: Co‐evolution and deep learning coming of age. Proteins. 2018;86:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wickham H, Averick M, Bryan J, et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4:1686. [Google Scholar]
- 74. Dowle M, Srinivasan A. data.table: Extension of ‘data.frame’. 2020.
- 75. Charif D, Lobry JR. SeqinR 1.0–2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis In: Bastolla U, Porto M, Roman HE, Vendruscolo M, editors. Structural approaches to sequence evolution: Molecules, networks, populations. Berlin, Heidelberg: Springer Berlin Heidelberg, 2007; p. 207–232. [Google Scholar]
- 76. Wilkinson L. ggplot2: Elegant graphics for data analysis by Wickham, H. Biometrics. 2011;67:678–679. [Google Scholar]
- 77. Wilke CO. cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2.” 2019.
- 78. Wagih O. Ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics. 2017;33:3645–3647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Amino acid frequencies by beta hairpin secondary structure region and organism domain. (a) Bars indicate average amino acid frequencies for each amino acid within a given region of all beta hairpins broken down by Domain of origin. The black dashed line indicates background amino acid frequencies for all sites in all proteins containing the beta hairpin motif. N‐term and C‐term refer to the N and C‐terminal beta strands while turn denotes the turn region. (b) Same as in (a) with amino acid frequencies grouped into specific classes. Amino acids are grouped as follows: Bulky Aromatic—phenylalanine, tryptophan, tyrosine; Hydrophobic—alanine, isoleucine, leucine, methionine, valine; Negative—aspartic acid, glutamic acid; Polar—asparagine, glutamine, serine, threonine; Positive—arginine, histidine, lysine; Special—cysteine, glycine, and proline.
Figure S2 Motif logo of amino acid probabilities in hairpin beta strands. Amino acid letters are scaled to represent relative frequency at a given position in both beta strands, with more common amino acids listed first vertically. Color indicates the type of amino acid. (a). Logo motif for hairpin beta strands with a C‐terminal aspartic acid that start with either a hydrophobic (left) or polar (right) face relative to the turn region. (b). As in (a) but showing frequencies across all hairpin beta strands, not just those terminated with an aspartic acid residue. Amino acids are grouped as in Figure S1.
Figure S3 Differences in observed versus expected residue contacts Dots represent individual contacting pairs with labeled dots indicating contacts that are enriched or depleted at least two‐fold vs. expected values. Y‐axis labels denote the class of the first residue in the pair while facet headings denote the class of the second amino acid (e.g., the dot for DP would be in the Negative row of the Special column). Amino acids are grouped as in Figure S1.
Table S1: Supporting information