

Macrophages can be modulated with relaxin gene delivery to promote cancer fibrosis regression and T cell infiltration.
Abstract
Cancer fibrosis serves as a major therapeutic barrier in desmoplastic tumors. Relaxin (RLN; a systemic hormone) is efficacious to decrease fibrosis, but the in vivo mechanism of action is not clear. Considering the localization of relaxin family peptide receptor type 1 (RXFP1), the receptor for RLN, on macrophages, we hypothesize that macrophages can be modulated by RLN to ameliorate cancer fibrosis. Using KPC mouse model of pancreatic ductal adenocarcinoma (PDAC), here, we report locally expressed RLN with targeted gene delivery induces increased F4/80+CD206+ macrophages originating from Ly6C+ monocytes, promoting fibrosis depletion and cytotoxic T cell infiltration. Moreover, RLN gene delivery synergizes with PD-L1 blockade for tumor inhibition by enhancing T cell–mediated tumor cell killing and macrophage phagocytosis. Collectively, our results reveal previously unidentified insights into the modulation of macrophages to regulate tumor-associated fibrosis, providing a feasible strategy to reverse the immunosuppressive environment and improve the therapeutic outcome of checkpoint immunotherapies.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers in desmoplastic tumors, with a 5-year survival rate lower than 10% (1) due to the rapid development of advanced disease or metastasis. Combination chemotherapy is used as a standard of care for the treatment of PDAC, including FOLFIRINOX (2), or combination of gemcitabine and nanoparticle albumin-bound paclitaxel (3). Still, little benefit is observed in PDAC patients with these complex therapeutic strategies (4). Immune checkpoint blockade (ICB) therapies targeting programmed cell death 1 (PD-1) or programmed death ligand 1 (PD-L1) have emerged as an attractive strategy because of the relatively high specificity and long-term efficacy (5). Despite success in a large variety of cancer types and clinical trials (6), little satisfactory therapeutic efficacy of ICB therapies has been reported in PDAC. The limited response to chemotherapies and ICB therapies is mainly attributed to the highly desmoplastic microenvironment (7), which not only prevents the drug penetration but also limits the infiltration of T cells into the tumor tissue. Although therapeutic benefit has been observed with strategies targeting desmoplasia in PDAC (8, 9), the cellular and molecular mechanisms that are responsible for regulating the microenvironment remain to be determined.
The tumor microenvironment (TME) of PDAC contains plentiful extracellular matrix (ECM) components, including collagen, fibronectin, laminin, and hyaluronic acid (10). Besides, the fibrotic stroma also comprises a variety of cell types, such as macrophages, fibroblasts, pericytes, and nerve cells (11). Macrophages, as one of the most abundant stromal components in the TME, are associated with cancer fibrosis and negatively impact on responses to therapy (12). While macrophages can play a crucial role for both the progression and resolution of fibrosis for nonmalignant diseases (13, 14), tumor-associated macrophages promote cancer fibrosis by modulating the ECM (15). However, the phenotype of macrophages can be reprogrammed in response to signals received from the microenvironment to achieve antifibrotic activities (16). Classically, resident and infiltrating macrophages are usually defined as proinflammatory M1 and anti-inflammatory M2. However, the macrophage population in fibrotic tumor is much complex and cannot be simply classified as M1 or M2 by transcriptional analysis (14, 17). As a result, clearly revealing the characteristics of macrophages on the basis of function in cancer fibrosis is important for exploring the full spectrum of macrophage activation.
RLN is a potent antifibrotic hormone by inhibiting fibroblast activation after binding to its primary receptor RXFP1 (relaxin family peptide receptor type 1) (18). While previous studies on the breast cancer model reported that RLN enhanced tumor growth and metastasis (19), the therapeutic potential of RLN has been well established in pancreas cancer cell xenografts (9, 18). Moreover, we have shown that locally expressed RLN in metastatic liver serves as a regulator to deactivate hepatic stellate cells and reverses the stromal environment (20). However, macrophages also express plenty of RXFP1 (21), suggesting that fibroblast reeducating may not be the sole activity for RLN. Thus, an understanding of the in vivo mechanism of how RLN stimulates macrophages to modulate the microenvironment is critical to thoroughly elucidate the action of RLN.
In this report, we studied mouse models of KPC pancreatic cancer, BPD6 melanoma, and 4T1 breast cancer and demonstrated macrophages constituted the main source of RXFP1, instead of fibroblasts, in the tumor tissues. Using KPC mouse model, we found locally expressed RLN with targeted gene delivery abrogated fibrosis and facilitated T cell infiltration. We further investigated the mechanism by which macrophages regulate the tumor stroma. We found RLN gene delivery increased intratumoral F4/80+CD206+ macrophages originating from Ly6C+ monocytes. Depletion of these macrophages attenuated the antifibrotic activity of RLN, indicating the importance of macrophages for RLN to facilitate cancer fibrosis resolution. The combination of RLN gene delivery and PD-L1 blockade therapy achieved synergistic antitumor effect by enhancing T cell–mediated tumor cell killing. Besides, RLN gene delivery depleted macrophage PD-1 to enhance phagocytosis by cooperating with PD-L1 blockade, contributing to the overall anticancer efficacy.
RESULTS
Macrophages constituted the main source of RXFP1 in desmoplastic tumors
Previous studies mainly focus on the expression and function of RXFP1 on fibroblasts (22), but the distribution of RXFP1 in the TME remains undetermined. To investigate the expression and localization of RXFP1 in desmoplastic tumors, three tumor models with dense stroma were established, namely, orthotopic KPC pancreatic tumor, BPD6 melanoma, and 4T1 breast cancer. We first used immunofluorescence (IF) staining to monitor the localization of RXFP1 in the desmoplastic microenvironment. As shown in Fig. 1A, RXFP1 was labeled with anti-RXFP1 in red, and cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) in blue. Macrophages and fibroblasts were stained with anti-F4/80 (cyan) and anti–α-SMA (smooth muscle actin) (green), respectively. We observed that RXFP1 was expressed by both macrophages and fibroblasts. On the basis of quantification analysis, we further demonstrated that RXFP1 was mainly expressed by macrophages instead of fibroblasts (55.6 ± 5.5% versus 25.4 ± 9.1%, P < 0.001) in the KPC tumor tissue. Similar results were also observed in both BPD6 melanoma (55.5% ± 16.6% versus 32.0 ± 4.5%, P < 0.05) and 4T1 (60.1 ± 9.3% versus 30.2 ± 5.9%, P < 0.001) tumor models. Flow cytometry was further used to investigate the distribution of RXFP1 in KPC tumor tissue. Consistently, RXFP1 was mainly expressed by macrophages, occupying ~70% of all RXFP1-positive cells, with ~20% RXFP1 localized on fibroblasts (P < 0.001). Besides, more than 70% of cells expressed RXFP1 in both macrophage and fibroblast populations (Fig. 1B). Thus, macrophages in TME should play a major role in the response to RLN.
Fig. 1. The expression and distribution of RLN receptor RXFP1 in desmoplastic tumors.
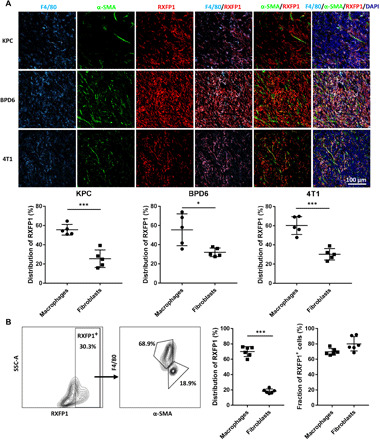
(A) The distribution of RXFP1 in three different desmoplastic tumors including KPC pancreatic cancer, BPD6 melanoma, and 4T1 breast cancer (n = 5 samples per group). Macrophages were stained with anti-F4/80 in cyan, and fibroblasts in the tumor tissue were stained with anti–α-SMA (green). RXFP1 was stained in red with the anti-RXFP1 antibody. Cell nuclei were stained with DAPI in blue. (B) The distribution of RXFP1 in KPC pancreatic tumor further evaluated by flow cytometry (n = 6). Statistical significance was calculated using t test. *P < 0.05, ***P < 0.001.
RLN gene delivery generated local expression of RLN in the tumor
The short circulation half-life and the potential off-target effect of RLN peptide restrict its direct use for therapy (18). However, targeted delivery of RLN plasmid (pRLN) into the tumor tissue might be a promising strategy by generating local expression of RLN. Aminoethyl anisamide (AEAA)–conjugated lipid-protamine-DNA (LPD) nanoparticles have shown high specificity for the delivery of macromolecules, including plasmid DNA, small interfering RNA, and mRNA to tumors by targeting the sigma-1 receptor (23). Therefore, we first evaluated the sigma-1 receptor expression in KPC pancreatic tumor and found a notable increase in the tumor tissue compared with normal pancreas (fig. S1A). AEAA-conjugated LPD nanoparticles for the encapsulation of pRLN were then prepared according to a standard protocol (fig. S1B) (24). The size of LPD nanoparticles was ~100 nm, and the zeta potential was ~30 mV according to dynamic light scattering measurements (fig. S1C).
The allografting KPC pancreatic tumor with dense stroma was then established by orthotopic injection of KPC cells into the tail of pancreas. To evaluate the biodistribution of LPD nanoparticles in vivo, approximately 0.1% of a fluorescence dye DiD was incorporated into the lipid membrane of LPD nanoparticles as a tracer to monitor the accumulation of nanoparticles in major organs and the tumor. After 14 days of tumor inoculation, DiD-labeled LPD nanoparticles were intravenously administrated. Mice were euthanized after 24 hours. Major organs and tumors were collected to analyze the biodistribution with the In Vivo Imaging System (IVIS) imaging system (fig. S1D). AEAA-conjugated LPD nanoparticles predominantly accumulated in the tumor tissue, which showed about twofold higher tumor accumulation compared with nontargeted LPD nanoparticles (fig. S1E). Local expression of RLN in the tumor after plasmid encoding green fluorescence protein (pGFP) or pRLN delivery was further investigated with enzyme-linked immunosorbent assay (ELISA) assay 48 hours after administration. An about fourfold increase was observed in pRLN-treated group compared with pGFP-treated mice (fig. S1F).
RLN gene delivery suppressed tumor growth and remodeled the TME
The KPC pancreatic tumor model was first used to investigate the therapeutic efficacy of pRLN delivery. KPC cells with a vector carrying mCherry red fluorescent protein (RFP) and firefly luciferase (Luc) (KPC-RFP/Luc) were orthotopically inoculated into the pancreas to generate the tumor model, and the dosing schedule was presented in Fig. 2A. Tumor growth was monitored by imaging the bioluminescence after injection of luciferin into the mice (Fig. 2B) and quantifying the number of photons emitted by the tumor (Fig. 2C). We found that there was no significant difference of tumor growth between phosphate-buffered saline (PBS)– and pGFP-treated mice. However, suppressed tumor progression was observed in pRLN-treated group (Fig. 2C). Decreased tumor weight at the end of the experiment in pRLN-treated mice further confirmed the significant antitumor efficacy of pRLN delivery compared with PBS- or pGFP-treated groups (Fig. 2D). In addition, the overall survival analysis demonstrated that pRLN delivery enhanced the median survival (47 days) compared with the administration of PBS (32 days) (P < 0.05) or pGFP (34 days) (P < 0.05) (Fig. 2E), indicating the relatively long-lasting therapeutic response induced by RLN gene delivery. As pGFP delivery did not show any therapeutic efficacy compared with PBS control, we chose PBS- and pRLN-treated groups for further analyses.
Fig. 2. RLN gene delivery suppressed tumor growth and modified TME in KPC pancreatic tumor model.
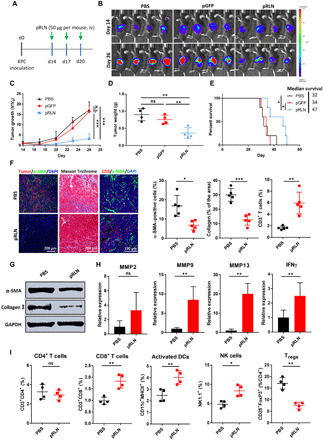
(A) The dosing schedule for the administration of pRLN. (B) Tumor growth monitored by IVIS imaging after different treatments and (C) the quantification of the imaging data (n = 4). (D) Tumor weight recorded on day 26 after the sacrifice of mice (n = 4). (E) Survival curves in each treatment group (n = 5). (F) Modification of the TME detected by IF staining and Masson Trichrome and the quantification of α-SMA, collagen expression, and CD3+ T cell infiltration (n = 5 samples per group). (G) Expression of α-SMA and collagen I detected with Western blot after the administration of PBS or pRLN (n = 3). (H) Relative expression of MMP2, MMP9, MMP13, and IFNγ detected by RT-PCR (n = 4). (I) Immune cell infiltration in the tumor tissue detected by flow cytometry (n = 4). Statistical significance was calculated in (C) using two-way analysis of variance (ANOVA) with multiple comparisons, in (D) using one-way ANOVA with multiple comparisons, in (E) using log rank test, and in (F), (H), and (I) using t test. *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant; Tregs, regulatory T cells.
IF staining and Masson Trichrome assay were performed to demonstrate the modification of TME after pRLN delivery. A decrease of α-SMA expression could be clearly observed in pRLN-treated group (6.4%) compared with PBS-treated mice (16.7%) (P < 0.05). The collagen content, as a major protein in the tumor stroma, decreased to 12 from 30% (P < 0.001) after pRLN treatment. The CD3+ T cell infiltration significantly increased (from 1.6 to 6.0%; P < 0.01) in response to the stromal microenvironment remodeling by the administration of pRLN (Fig. 2F). Reduced expression of α-SMA and collagen I after pRLN treatment was further confirmed by Western blot analysis (Fig. 2G and fig. S2). Reverse transcription polymerase chain reaction (RT-PCR) was used to detect the mRNA levels of matrix metalloproteinase (MMP) in the tumor, including MMP2, MMP9, and MMP13, which are responsible for the ECM degradation. As shown in Fig. 2H, pRLN delivery significantly increased the mRNA levels of MMP9 (8.5-fold) and MMP13 (20-fold) compared with PBS-treated group, but no obvious increase of MMP2 mRNA was detected. A 2.5-fold increase of interferon-γ (IFNγ) mRNA was also observed following RLN gene therapy (Fig. 2H). We subsequently used flow cytometry to analyze the immune microenvironment modulation in the tumor tissue. As shown in Fig. 2I, RLN gene delivery increased the infiltration of CD8+ T cells, activated dendritic cells (DCs), and natural killer (NK) cells but significantly (P < 0.01) decreased the level of regulatory T cells, suggesting that the immunosuppressive microenvironment has been reversed by RLN gene therapy.
The anticancer effect was also evaluated in BPD6 melanoma, another desmoplastic tumor model. Similarly, pRLN delivery inhibited the tumor growth with shrinking tumor size and reduced tumor weight compared with pGFP treatment. Consistently, decreased fibrotic stroma and improved immune microenvironment were observed (fig. S3).
RLN gene delivery induced macrophage-mediated fibrosis depletion and T cell infiltration
Macrophages in tumor tissue are polarized toward a proinflammatory (antitumor) M1 or anti-inflammatory (protumor) M2 phenotype, depending on various stimuli (25). Here, we first analyzed the phenotype of macrophages with widely used markers after treatments of PBS or pRLN. We found an increase of F4/80+CD11b+ total macrophages in the TME after pRLN administration. In addition, the population of F4/80+CD206+ (traditionally recognized as M2 phenotype) macrophages was also augmented (P < 0.001) following pRLN delivery. In contrast, the number of F4/80+CD206− macrophages did not change (Fig. 3A). RT-PCR analysis showed a shift of chemokines related to monocyte/macrophage recruitment, including CCL2 and CCL5 (Fig. 3B), suggesting enhanced recruitment of monocytes/macrophages. Consistent with the increase of F4/80+CD206+ macrophages, up-regulated mRNA levels of CD206 (MRC-1) were observed after pRLN delivery. Meanwhile, the mRNA expression of interleukin-1α (IL-1α) (responsible for the production of inflammation) also increased and FGF2 (fibroblast growth factor 2) remained unchanged (Fig. 3B). As PD-1 commonly correlates with CD206+ M2 macrophages (26), macrophage expressed PD-1 was further investigated. We found that RLN gene delivery significantly reduced PD-1 expression in F4/80+ total macrophages (Fig. 3C), especially in F4/80+CD206+ “M2-like” macrophages (P < 0.01) (Fig. 3D). These observations demonstrate that the F4/80+CD206+ macrophages induced by pRLN delivery were different from the traditional protumor M2 macrophages and may have distinct functions.
Fig. 3. RLN gene delivery altered macrophage populations in the tumor.
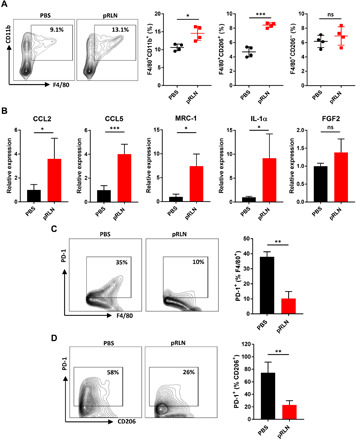
(A) Evaluation of the change of macrophage populations in the tumor after different treatments by detecting a macrophage pan-marker F4/80 and a traditional M2 marker CD206 with flow cytometry (n = 4). (B) Relative mRNA expression of cytokines in the tumor of mice bearing KPC tumor following PBS or pRLN treatment (n = 4). (C) PD-1 expression on F4/80+ macrophages and (D) F4/80+CD206+ macrophages in each treatment group (n = 3). Statistical significance was calculated using t test. *P < 0.05, **P < 0.01, and ***P < 0.001.
An in vitro experiment was further performed to evaluate RLN caused modulation of macrophages using RAW264.7 cells as a model. The results showed that RLN slightly increased the expression of Arg-1 (an M2 marker) mRNA (1.6-fold, P < 0.05) in IL-4–stimulated cells, without changing the mRNA levels of iNOS (an M1 marker) (fig. S5A). Instead, RLN showed the capacity to elevate iNOS (1.5-fold, P < 0.05) in LPS and IFNγ-treated cells but had no effect to alter the expression of Arg-1 (fig. S5B). These data indicate that RLN-induced macrophage modulation is beyond the phenotype transition between classical M1 and M2. Notably, RLN-treated RAW264.7 cells showed increased mRNA levels of MMP13 (fig. S5C), suggesting the potential role of modulated macrophages to degrade fibers in the tumor tissue.
Clodronate is a macrophage toxin if it is delivered inside the cells (27). We performed macrophage depletion by using clodronate encapsulated liposome (CEL) to determine the significance of modulated macrophages to regulate the TME. As shown in Fig. 4A, F4/80+CD11b+ total macrophages were down-regulated after the cotreatment of CEL and pRLN compared with pRLN delivery alone. Similarly, the amount of F4/80+CD206+ macrophages also declined in the presence of both CEL and pRLN. To determine whether the increased F4/80+CD206+ macrophages arise from infiltrated monocytes, we further measured Ly6C (a cell surface glycoprotein to identify murine monocytes) expression in the subset of CD206+ macrophages. While RLN gene delivery significantly increased the population of Ly6C+ macrophages, CEL depleted this subset (fig. S5). These results are consistent with the up-regulation of CCL2 (a monocyte chemokine) expression, indicating that RLN gene delivery stimulates macrophages originating from monocytes to infiltrate into the tumor tissue. Subsequently, we found that the mRNA levels of MMP13, IFNγ, and CXCL9 decreased in the presence of CEL (Fig. 4B). Masson Trichrome assay showed restored collagen deposition in the tumor following the treatment of CEL and pRLN (Fig. 4C), suggesting that the antifibrotic activity of pRLN was impaired because of the depletion of the macrophage subset. Correspondingly, the accumulation of CD3+ T cells, especially CD8+ T cells, decreased in CEL and pRLN cotreated group (Fig. 4, C and D). Further, an increase of tumor weight was observed in mice treated with both CEL and pRLN compared with pRLN delivery (Fig. 4E), indicating attenuated antitumor efficacy of RLN gene delivery by depleting the F4/80+CD206+ macrophages. These data illustrate the importance of F4/80+CD206+ macrophages originating from Ly6C+ monocytes for RLN to regulate fibrosis depletion and T cell infiltration. The dosing scheme of the treatment of CEL and pRLN is presented in Fig. 4F.
Fig. 4. RLN gene delivery caused macrophage-involved fibrosis regression and T cell infiltration.
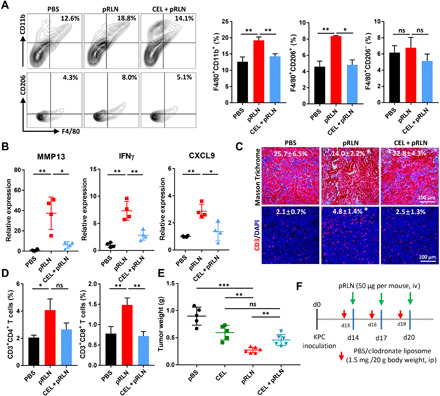
(A) Cotreatment of pRLN and CEL decreased the amount of F4/80+CD11b+ total macrophages and F4/80+CD206+ macrophages in the tumor tissue (n = 3). (B) Relative expression of MMP13, IFNγ, and CXCL9 detected by RT-PCR after different treatments (n = 4). (C) Collagen expression and CD3+ T cell infiltration in the tumor (n = 4 samples per group). (D) Tumor infiltrated CD3+CD4+ and CD3+CD8+ T cells quantified with flow cytometry (n = 3). (E) Tumor weight in each group after mice were euthanized on day 28. (F) The dosing scheme for the treatment of CEL and pRLN (n = 5 to 6). Statistical significance was calculated using one-way ANOVA with multiple comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001.
RLN gene delivery significantly improved therapy response to PD-L1 blockade
High levels of PD-L1 expression is associated with active response to PD-1/PD-L1 blockade therapy (28). However, PD-L1 blockade alone shows unsatisfactory antitumor activity in some desmoplastic tumors, such as PDAC, due to the “cold” immune microenvironment with insufficient T cell infiltration (6). The antifibrotic activity of pRLN delivery makes it a promising strategy to improve the therapy response to PD-L1 blockade via increasing T cell abundance in the tumor tissue. Therefore, a therapeutic strategy was proposed with the combination of pRLN and anti–PD-L1 (mAb) (Combo) for KPC tumor treatment (Fig. 5A). We found that RLN gene delivery alone induced remarkable tumor suppression according to the decreased tumor weight compared with PBS control group. No significant difference of tumor weight was observed between anti–PD-L1– and PBS-treated groups, which is consistent with the limited clinical therapeutic outcome of ICB therapies in PDAC. However, the Combo group achieved the lowest tumor weight, indicating a markedly improved therapy response to PD-L1 blockade by RLN gene delivery (Fig. 5, B and C). Besides, the combination therapy induced an about twofold increase in the median survival (65 days) compared with PBS (31 days)– or anti–PD-L1 (35 days)–treated groups (Fig. 5D), demonstrating that a long term anticancer efficacy has been generated in the Combo group. Subsequently, CD4+ and CD8+ T cells were depleted respectively with antibodies to determine the involvement of T cells in the combination therapy (fig. S6). As expected, the obvious decrease of tumor weight was compromised when CD8+ T cells were removed (Fig. 5E). Therefore, improved T cell infiltration in the tumor tissue is a major cause for the enhanced antitumor efficacy of the combination therapy. However, depletion of either CD4+ or CD8+ cells did not completely remove the therapy effect of the Combo group, suggesting the existence of other tumor cell killing mechanism(s).
Fig. 5. RLN gene delivery improved therapy response to PD-L1 blockade in KPC pancreatic tumor.
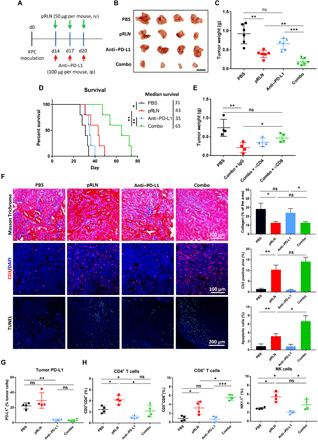
(A) Tumor inoculation and treatment scheme for the combination of pRLN and anti–PD-L1 therapy. (B) Representative images (scale bar, 1 cm) of tumors dissected from tumor bearing mice on day 28 and (C) weight of tumors quantified (n = 6). (D) Survival of mice after different treatments (n = 5). (E) Tumor weight of tumor bearing mice with the combination therapy and depletion of CD4+ and CD8+ T cells with antibodies (n = 4). (F) Collagen expression, CD3+ T cell infiltration, and apoptotic cell detection in the tumor tissue (n = 3 samples per group). Masson Trichrome assay was performed to detect collagen expression. IF staining and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay were used to evaluate T cell (red) infiltration and apoptotic cells (green) in the tumor, respectively. Cell nuclei were stained with DAPI in blue. (G) PD-L1 expression on cells in the tumor (n = 4). (H) Immune cell infiltration in the tumor detected with flow cytometry (n = 4). Statistical significance was calculated in (C), (E), (F), (G), and (H) using one-way ANOVA with multiple comparisons, and statistical significance in (D) was calculated using log rank test. *P < 0.05, **P < 0.01, and ***P < 0.001.
RLN gene delivery synergized with PD-L1 blockade to induce tumor cell apoptosis
We next investigated the mechanisms responsible for the enhanced anticancer efficacy in the combination therapy. We have demonstrated that RLN gene delivery facilitates fibrosis depletion in the tumor tissue. Consistently, a decrease of collagen expression was observed in both pRLN- and Combo-treated groups compared with PBS control group. In contrast, PD-L1 blockade alone showed comparable collagen deposition with PBS-treated group, indicating little efficiency in modulating tumor stroma by this antibody (Fig. 5F). T cell infiltration was subsequently evaluated by detecting CD3+ T cells with IF staining. While low infiltration of CD3+ T cells was observed in both PBS and anti-PD-L1–treated groups, the accumulation of CD3+ T cells significantly increased following the treatment of pRLN or Combo (Fig. 5F). Sufficient infiltration of T cells can achieve compromised anticancer efficacy due to PD-L1 expression on tumor cells (Fig. 5G). We therefore only found a marginal increase of apoptotic cells in the tumor after pRLN treatment, though a marked growth of T cell infiltration has been observed. The percentage of apoptotic cells significantly increased within the Combo group, suggesting an improved therapeutic outcome. PD-L1 expression on tumor cells was also detected by flow cytometry. Efficient blockade of tumor cell PD-L1 in both anti–PD-L1– and Combo-treated groups was observed (Fig. 5G). We next investigated the immune microenvironment modification induced by the combination therapy. The results showed that the immune microenvironment was not improved with PD-L1 blockade alone according to the constant number of CD8+ cytotoxic T cells and even decreased accumulation of CD4+ T cells and NK cells compared with PBS control. However, the combination therapy significantly increased CD8+ T cell and NK cell infiltration compared with PD-L1 blockade therapy (Fig. 5H). Overall, these findings (Fig. 5, F and H) demonstrate that pRLN delivery can improve the immune response to PD-L1 blockade therapy by facilitating T cell infiltration, and consequently, tumor cell killing.
RLN gene delivery depleted macrophage PD-1 to enhance phagocytosis in the combination therapy
It has been shown that blocking CD47 (a do not–eat–me signal) unleashes macrophages to engulf tumor cells (29). Recently, macrophage-expressed PD-1 was also identified to correlate with compromised phagocytosis of tumor cells (26). PD-1/PD-L1 blockade can rescue the phagocytotic capacity of macrophages, leading to T cell independent killing of the tumor cells (26). Here, we demonstrated that macrophages, especially the F4/80+CD206+ phenotype, in KPC tumor expressed high level of PD-1. However, pRLN delivery significantly decreased PD-1 expression on macrophages, which may induce improved phagocytosis activity of these macrophages and offer additional therapeutic potential. Therefore, macrophage populations and phagocytosis in the combination therapy were analyzed with flow cytometry. Consistent with the administration of pRLN, the combination therapy also elevated the infiltration of F4/80+CD206+ macrophages in the tumor (fig. S7). We next examined the PD-1 expression on macrophages and found that both pRLN and combination therapy resulted in reduced amount of CD206+PD-1+ compared with PBS control group, whereas PD-L1 blockade showed no such effect (Fig. 6, A and B). A low positive rate of PD-1 on F4/80+CD206− macrophages (~20%) was observed in the PBS-treated group, and it can be also slightly down-regulated following the treatment of pRLN (~13%) (P < 0.05) as well as Combo (~10%) (Fig. 6C). We also noted that the population of F4/80−PD-1+ cells showed almost no change following pRLN delivery compared with PBS-treated group, suggesting a relative specificity for PD-1 regulation with RLN gene therapy (Fig. 6D). Given the strong effect in down-regulation of macrophage PD-1 with pRLN delivery, we examined in vivo phagocytosis to validate the potential therapeutic response. We generated orthotopic pancreatic tumor model with red fluorescence protein–positive (RFP+) KPC cells and assessed the total phagocytosis levels in the tumor by analyzing the percentage of macrophages that were also RFP+. A twofold increase of phagocytosis was observed in the Combo group. However, pRLN or anti–PD-L1 therapy alone showed little effect to improve the total phagocytosis levels (Fig. 6E), suggesting that only PD-1 depletion or PD-L1 blockade was insufficient to enhance phagocytosis for this tumor model. Phagocytosis was also evaluated in vitro by conducting coculture of KPC tumor cells (RFP+) and RLN-treated bone marrow–derived macrophages. An increase (P < 0.05) of phagocytic capacity was observed with the treatment of RLN peptide (fig. S8).
Fig. 6. RLN gene delivery depleted macrophage PD-1 to enhance phagocytosis in combination with PD-L1 blockade therapy.
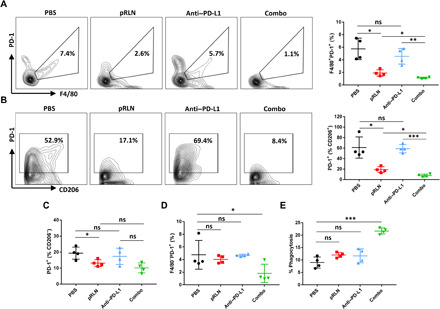
(A) Representative flow cytometric analysis images and quantification of PD-1 expression on F4/80+ macrophages and (B) F4/80+CD206+ macrophages in each treatment group (n = 4). (C) PD-1 expression on F4/80+CD206− macrophages (n = 4). (D) Quantification of F4/80−PD-1+ cells in the tumor following different treatments (n = 4). (E) Relative quantification of phagocytosis in F4/80+ macrophages by detecting F4/80+RFP+ cells and dividing all F4/80+ cells (n = 4). Statistical significance was calculated using one-way ANOVA with multiple comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001.
RLN gene delivery and combination therapy showed marginal toxicity
Hematoxylin and eosin (H&E) staining was performed to evaluate the histological abnormality of major organs including heart, liver, lung, spleen, and kidney after different treatments. No noticeable morphological changes in these organs were observed in each group (fig. S9A). However, what should be noted is that spontaneous metastasis can be found in the liver and spleen for both PBS and PD-L1 blockade therapy groups. Spleen metastasis was also observed in RLN gene therapy group, but no metastasis was observed in the Combo group (fig. S9A). The toxicity of different treatments was also evaluated by blood chemistry analysis (fig. S9B). We found that RLN gene delivery and the combination therapy caused slight decrease of platelet counts compared with PBS treatment, which is consistent with a previous study showing that RLN depresses platelet aggression (30). In addition to this, no abnormal changes of red blood cell counts and white blood cell counts were observed in each group. Consistently, the serum biochemical analysis showed that alanine aminotransferase (ALT), serum aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CREAT) remained constant within the normal ranges for all treatment groups, indicating that no severe toxicity was induced following these treatments.
DISCUSSION
Increasing therapeutic strategies by modulating the dense stroma have been developed for the treatment of PDAC (31, 32). RLN, as an endogenous hormone, is efficacious and safe to promote fibrosis resolution in both nonmalignant disorders and cancer (18, 20). Although the assessment of antifibrotic benefits has been well described, the in vivo antifibrotic mechanism of RLN in cancer fibrosis remains elusive. Previous studies demonstrated that RLN binds to its cognate receptor RXFP1 to inhibit fibroblast activation and reduce ECM production via intervening transforming growth factor–β signaling pathway (18, 33), leading to fibrosis regression. Consistently, we also found decreased α-SMA expression with RLN gene delivery, indicating suppressed fibroblast activation. Nevertheless, we found that macrophages, instead of fibroblasts, constitute the main source of RXFP1 in the microenvironment of desmoplastic tumors, suggesting a more predominant target of RLN. However, whether RLN ameliorates the cancer fibrosis by modulating macrophages remains to be determined. Our data now show that RLN gene delivery stimulated a subset of CD206+ macrophages originating from Ly6C+ monocytes to infiltrate the tumor, facilitating fibrosis depletion and T cell infiltration. Depletion of these macrophages with CEL attenuated the antifibrotic properties of RLN, indicating the important involvement of infiltrated macrophages to mediate fibrosis depletion in PDAC. MMP13 produced by macrophages is a critical enzyme during the resolution of murine liver fibrosis (16, 34). On the basis of our findings that the expression of MMP13 is closely related to infiltrated macrophages and fibrosis regression, we propose that a potential antifibrotic mechanism mediated by RLN modulated macrophages is inducing a shift of MMP profile to actively deplete fibrosis in PDAC.
In most human cancer types, infiltrated macrophages fuel the tumor progression and correlate with poor prognosis and disease outcome (12). Nevertheless, strategies by inhibiting monocyte/macrophage recruitment to tumor tissue or depleting macrophages are at risk to cause a rapid rebound of monocyte infiltration, inducing accelerated relapse (35). An alternative is to use the antifibrotic properties of macrophages to produce antitumor efficacy, such as CD40 agonists (16). In our study, we also showed that RLN gene delivery induced an anticancer activity by recruiting and modulating macrophages, indicating that infiltrated macrophages can be harnessed to generate improved therapeutic outcome, as long as RLN is produced locally in the TME. Macrophages in tumors always correlate with the acquisition of specific phenotypes driven by different environment factors. Classically, macrophages activated with LPS or IFNγ are regarded as proinflammatory M1 with antitumor activity, whereas macrophages stimulated with IL-4 are referred to anti-inflammatory M2 and viewed to promote tumor growth (12). Different markers have been used to identify the state of macrophages. For instance, Nos2 and IL12b are used to identify M1. In contrast, Arg-1, CD206 (MRC-1), and Ym1 are generally used to detect M2. Accumulating macrophage-targeting therapeutic strategies focus on the reprogramming of macrophages from a protumor M2 to an antitumor M1 state (36). However, increasing evidences suggest that the dualistic classification does not address the complex heterogeneity of macrophages in vivo (17), emphasizing the importance to unbiasedly describe the functions instead of the putative M1/M2 phenotypes. Macrophage expressed Nos2 (M1) may promote, rather than limit, tumor progression (37). We now demonstrate that macrophages with an M2 marker (CD206) can also present antitumor activities. Therefore, pro- or antitumor macrophages should be defined by specific activities rather than their repolarization state.
Sufficient T cell infiltration is important for ICB therapies to achieve the optimum antitumor efficacy (8). Therefore, RLN gene delivery provides a feasible approach to improve the therapy response to PD-L1 blockade by modulating the immune microenvironment with increased T cell and NK cell accumulation. The combination of pRLN delivery and PD-L1 blockade significantly improved the anticancer effect compared with pRLN delivery or PD-L1 blockade alone. Moreover, PD-1 has been found on activated T cells, B cells, NK cells, DCs, and macrophages (38, 39). Despite the function of PD-1 in inhibiting T cell activation and proliferation is well understood, PD-1 expressed by macrophages can have different activities due to their differences of innate and adaptive immune cells. Gordon et al. (26) showed PD-1+ macrophages express increased levels of M2 marker CD206 and show significantly inhibited engulfment of cancer cells compared with PD-1− macrophages in a colon cancer model. In our study, RLN gene delivery down-regulated macrophage PD-1 expression with an undetermined mechanism, but enhanced phagocytosis was not observed after macrophage PD-1 depletion. However, combination of PD-1 depletion and PD-L1 blockade produced significant increase of phagocytosis and contributed to the overall anticancer efficacy, suggesting that simultaneous blockade of macrophage PD-1 and tumor cell PD-L1 is required to remarkably restrain the “do not–eat–me” signal for the KPC tumor model. More studies are still required to clearly address the pathway in which RLN depletes macrophage PD-1 and the molecular mechanism of PD-1–mediated phagocytosis. In addition, with the identification of emerging phagocytosis checkpoints, including CD47–signal regulatory protein α (SIRPα) axis, PD-1–PD-L1, and major histocompatibility complex class I–leukocyte immunoglobulin-like receptor 1 (LILRB1) axes (29), the mechanistical correlations between different phagocytosis regulators remain to be elucidated.
In summary, our findings demonstrate that RLN gene delivery modulates infiltrated macrophages in the KPC mouse model of PDAC to promote fibrosis regression and facilitate T cell infiltration, identifying a therapeutic approach to harness recruited macrophages. The strategy based on the combination of antifibrosis and checkpoint immunotherapy may be applied to other desmoplastic tumors and achieve significant antitumor potential.
MATERIALS AND METHODS
Materials
Plasmid encoding relaxin 1 (RLN) was obtained from Sino Biological Inc. (Beijing, China). The pGFP with the cytomegalovirus promoter was purchased from Bayou Biolabs (Harahan, LA). N-(methoxypolyethylene oxycarbonyl)-1,2-distearoryl-sn-glycero-3-phosphoethanolamine (DSPE-PEG) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol)-2000] (DSPE-PEG-NH2) were purchased from NOF Corporation (Tokyo, Japan). N-(2-aminoethyl)-4-methoxybenzamide conjugated (DSPE-PEG-AEAA) was synthesized according to a previously established protocol (40). Cholesterol and protamine were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other lipids were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Clodronate and control liposomes were purchased from Encapsula Nano Sciences (Brentwood, TN). InVivoMab anti-mouse PD-L1, anti-mouse CD4, anti-mouse CD8, and polyclonal immunoglobulin G (IgG) were purchased from BIOXCELL (NH, USA). Recombinant mouse relaxin 1 peptide was obtained from LifeSpan BioSciences (Seattle, USA). All other chemicals were obtained from Sigma-Aldrich unless specifically mentioned.
Cell lines and animals
The primary KPC cell line was derived from a genetically engineered mouse model (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre, syngeneic to C57BL/6 strain) and provided by S. Kozlov (National Cancer Institute, Center for Advanced Preclinical Research). The primary KPC cells were stably transfected with the lentiviral vector carrying mCherry RFP and firefly luciferase (Luc) (KPC-RFP/Luc). The KPC cells were cultured in Dulbecco’s modified Eagle’s medium: nutrient mixture F-12 (DMEM/F-12), further supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (PS) and maintained at 37°C and 5% CO2 in a humidified atmosphere. Murine melanoma cell line BPD6 (BRAFV600E, PTEN−/−) was provided by B. Hanks (Duke Cancer Institute). Murine breast cancer cell line 4T1 and murine monocytes RAW264.7 were obtained from Tissue Culture Facility—UNC Lineberger Comprehensive Cancer Center. BPD6 and 4T1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% PS (Invitrogen, Carlsbad, CA) at 37°C and 5% CO2. RAW264.7 cells were cultured in DMEM (high glucose, Gibco) supplemented with 10% FBS and 1% PS at 37°C and 5% CO2. Eight-week-old female C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). All animal regulations and procedures were accepted by Institutional Animal Care and Use Committee of University of North Carolina at Chapel Hill.
Mouse models
Orthotopic pancreatic tumor model was established by orthotopic injection of 1 × 106 KPC cells into the tail of the pancreas. Briefly, 8-week-old C57BL/6 female mice were anesthetized by inhalation of isoflurane and placed in supine position. A midline incision was made to exteriorize the spleen and pancreas, and 1 × 106 cells in 40 μl were injected into the pancreas. Then, the abdominal wall and skin were closed.
To establish the BPD6 melanoma model, 8-week-old C57BL/6 female mice were inoculated subcutaneously on the lower flank with 1 × 106 BPD6 cells. After the inoculation, tumor growth was measured by a digital caliper where volume = 0.5 × length × width × height. Mice were randomized into different groups, and treatments were started when tumor volume reached ∼200 mm3. Orthotopic allografting 4T1 model was established by orthotopic injection of 1 × 106 4T1 cells on the mammary fat pad of the 6-week old Balb/c female mice.
Tumor growth inhibition and survival analysis
Mice bearing KPC or KPC-RFP/Luc allografts were randomized into different treatment groups, and treatments were initiated on day 14. PBS, pGFP LPD (50-μg pDNA per mouse, iv), pRLN LPD (50-μg pDNA per mouse, iv), anti–PD-L1 (100 μg per mouse, ip), and Combo (50-μg pRLN, iv + 100 μg anti–PD-L1, ip) were given as scheduled. For mice bearing KPC-RFP/Luc allografts, tumor growth was monitored by an IVIS imaging system (PerkinElmer) every 3 days. d-luciferin (100 mg/kg of body weight) was intraperitoneally injected into anesthetized mice, and the bioluminescence was recorded. The increase of tumor volume was calculated as the radiance of the intensities and standardized with the initial tumor volume (Vt/V0). For mice bearing KPC allografts, the tumor weight was recorded after sacrifice of mice. To deplete T cells in vivo, Combo + anti-mouse CD8 (100 μg per mouse, ip), Combo + anti-mouse CD4 (100 μg per mouse, ip), and Combo + polyclonal rat IgG (100 μg per mouse, ip) were given at respective schedules. Long-term survival was also monitored on mice bearing the KPC-RFP/Luc allografts with different treatments (n = 5). The end point achieved when one of the following conditions applied: Drastic weight gain or loss greater than 10% within 1 week or clear signs of distress were detected, such as dehydration, inactivity, and lethargy.
In vivo macrophage depletion with clodronate liposome
Mice bearing KPC allografts were injected intraperitoneally with clodronate liposome (CEL, 1.5 mg/20 g body weight) or with an equivalent volume of PBS liposome for three times as scheduled. Macrophage depletion in tumor tissue was analyzed 2 days later of the final injection of pRLN LPD nanoparticles by flow cytometry.
Fabrication of LPD nanoparticles and the biodistribution
LPD nanoparticles were prepared according to a well-established protocol (24). Briefly, 1,2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP) and cholesterol (1:1, mol/mol) were dissolved in chloroform, and the solvent was removed by evaporation to form a uniform lipid film. The film was then hydrated with distilled water to make the final concentration of 10 mM cholesterol and DOTAP. Then, the liposome was sequentially extruded through 200- and 100-nm polycarbonate membranes (Millipore, MA) to form unilamellar liposomes. The LPD polyplex cores were formulated by mixing 100 μl of protamine (200 μg/ml) in distilled water with equal volume of 50 μg of plasmid. The mixture was incubated at room temperature for 10 min, and then 60 μl of cholesterol/DOTAP liposomes was added. DSPE-PEG (10 μl, 10 mg/ml) and DSPE-PEG-AEAA (10 μl, 10 mg/ml) were further added into the mixture, and the resulted liquid was incubated at 60°C for 15 min followed by the addition of 20 μl of 20% glucose. The size and zeta potential of the nanoparticles were determined by a Malvern ZetaSizer Nano series (Westborough, MA).
To monitor the biodistribution of LPD nanoparticles, approximately 0.1% of a hydrophobic dye DiD was incorporated into DOTAP liposomes to formulate the DiD-labeled LPD nanoparticles. Twenty-four hours after intravenous injection of the DiD-labeled LPD nanoparticles, mice were euthanized and major organs and tumors were collected. The distribution of LPD nanoparticles was quantitatively visualized with an IVIS imaging system (PerkinElmer, CA). The excitation wavelength was set at 640 nm, and the emission wavelength was set at 670 nm.
Flow cytometry assay
Tumors were harvested and made into single-cell suspensions. Tumor-infiltrating immune lymphocytes were analyzed by flow cytometry. Briefly, tumors were digested with collagenase A and deoxyribonuclease at 37°C for 1 hour. After red blood cell lysis with ACK buffer, cells were resuspended in fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 5% FBS and 2 mM EDTA). For intracellular marker staining, the cells were penetrated with penetration buffer (BD Biosciences) following the manufacturer’s instructions. Different immune lymphocytes (5 × 106/ml) were stained by the addition of a cocktail of fluorescence-conjugated antibodies. Then, cells were fixed with 4% paraformaldehyde (PFA) and analyzed via FACS (BD LSR II). Antibodies used for flow cytometry are listed in table S1.
IF staining and Masson Trichrome assay
Frozen sections of organs or tumors were used for IF staining. Tissues for frozen sections were first rinsed with PBS and placed in 4% PFA for fixation overnight at 4°C. Then, tissues were dehydrated sequentially with 15 and 30% sucrose solution overnight at 4°C. Tissues were frozen in O.C.T. (Thermo Fisher Scientific). To perform the IF staining, frozen sections were permeabilized with 0.1% Triton X-100 in PBS for 15 min and blocked in 5% goat serum for 1 hour. Primary antibodies were incubated at 4°C overnight, and fluorescent secondary antibodies were incubated at 37°C for 1 hour. Cell nuclei were counterstained with DAPI containing mounting medium (Thermo Fisher Scientific). All antibodies were diluted according to the manufacturer’s manual. Images were acquired using fluorescence microscopy (Zeiss LSM 700). Antibodies used for IF staining are listed in table S1. The Masson Trichrome assay was performed to detect collagen among tumor tissue. Tumor slides were stained by the UNC Tissue Procurement Core.
Western blot and ELISA assay
To assess the expression of α-SMA and collagen I in the tumor tissue, tumor samples were lysed with radioimmunoprecipitation assay buffer, and protein concentration was determined using a bicinchoninic acid assay (Invitrogen). The protein solution was diluted with lysis buffer and heated at 95°C for 5 min. Protein was separated by 4 to 12% SDS–polyacrylamide gel electrophoresis (Invitrogen) and then transferred to polyvinylidene difluoride membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk in TBST for 1 hour and then incubated with primary antibodies overnight at 4°C. The membranes were rinse in TBST and further incubated with a secondary antibody (appropriate diluted) at room temperature for 1 hour and detected using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the control. ELISA assay was performed according to the manufacturer’s protocols after appropriate titration.
Quantitative RT-PCR
Total RNA was extracted from the tumor tissues using an RNeasy kit (Qiagen). Reverse transcription was conducted using the iScript cDNA Synthesis Kit (BIO-RAD). One hundred nanograms of cDNA was amplified with TaqMan Gene Expression Master Mix. All primers for RT-PCR reactions are listed in table S2. GAPDH was used as the endogenous control. PCR reactions were conducted using the 7500 real-time PCR system, and the data were analyzed with the 7500 software.
TUNEL assay
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assays were performed using a DeadEnd Fluorometric TUNEL system (Promega) according to the manufacturer’s instructions. Fragmented DNAs of apoptotic cells were fluorescently stained with FITC and defined as TUNEL-positive nuclei. Slides were mounted, and cell nuclei were stained with Prolong Diamond antifade mountant with DAPI (Thermo Fisher Scientific). Images were acquired with confocal microscopy (Zeiss LSM 700).
Blood chemistry analysis and H&E staining
At the experiment end point, both whole blood and serum were collected. Whole blood cellular components were counted and compared. CREAT, BUN, AST, and ALT in the serum were assayed as indicators of renal and liver functions. Major organs including heart, liver, lung, kidney, and spleen were collected and fixed for H&E staining at the University of North Carolina at Chapel Hill histology facility to evaluate the organ-specific toxicity.
Statistical analysis
Data are presented as the means ±SD. Statistical analysis was performed by a two-tailed Student’s t test or a one-way analysis of variance (ANOVA) for comparing two groups or larger than two groups, respectively. For survival analyses, log rank test was used for comparison. For comparison between multiple groups, ordinary two-way ANOVA with multiple comparisons adjusted by Šidák correction was used. Symbols *, **, *** denote P < 0.05, P < 0.01, and P < 0.001, respectively, and were considered significant and documented in figures or figure legends.
Acknowledgments
We are grateful to M. Das for assisting with the establishment of KPC orthotopic tumor model. Funding: The work was supported by NIH grant CA198999, NSFC (51773176 and 21975218), and China Scholarship Council. Author contributions: X.Z., M.H., X.L., and L.H. conceived and designed the research. X.Z., Y.L., and M.W. performed the experiments and analyzed data. X.Z., X.L., and L.H. wrote the manuscript. Competing interests: L.H. is a consultant for PDS Biotechnology, Samyang Biopharmaceuticals, Stemirna, and Beijing Inno Medicine. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabb6596/DC1
REFERENCES AND NOTES
- 1.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., Adenis A., Raoul J.-L., Gourgou-Bourgade S., de la Fouchardière C., Bennouna J., Bachet J.-B., Khemissa-Akouz F., Péré-Vergé D., Delbaldo C., Assenat E., Chauffert B., Michel P., Montoto-Grillot C., Ducreux M.; Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup , FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., Seay T., Tjulandin S. A., Ma W. W., Saleh M. N., Harris M., Reni M., Dowden S., Laheru D., Bahary N., Ramanathan R. K., Tabernero J., Hidalgo M., Goldstein D., Van Cutsem E., Wei X., Iglesias J., Renschler M. F., Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnittert J., Bansal R., Prakash J., Targeting pancreatic stellate cells in cancer. Trends Cancer 5, 128–142 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.-J., Rutkowski P., Lao C. D., Cowey C. L., Schadendorf D., Wagstaff J., Dummer R., Ferrucci P. F., Smylie M., Hogg D., Hill A., Márquez-Rodas I., Haanen J., Guidoboni M., Maio M., Schöffski P., Carlino M. S., Lebbé C., McArthur G., Ascierto P. A., Daniels G. A., Long G. V., Bastholt L., Rizzo J. I., Balogh A., Moshyk A., Hodi F. S., Wolchok J. D., Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381, 1535–1546 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Galon J., Bruni D., Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Pandey V., Storz P., Targeting the tumor microenvironment in pancreatic ductal adenocarcinoma. Expert Rev. Anticancer Ther. 19, 473–482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valkenburg K. C., de Groot A. E., Pienta K. J., Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung K. H., Choi I.-K., Lee H.-S., Yan H. H., Son M. K., Ahn H. M., Hong J. W., Yun C. O., Hong S. S., Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett. 396, 155–166 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Neoptolemos J. P., Kleeff J., Michl P., Costello E., Greenhalf W., Palmer D. H., Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 15, 333–348 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zhan H.-X., Zhou B., Cheng Y.-G., Xu J.-W., Wang L., Zhang G.-Y., Hu S.-Y., Crosstalk between stromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett. 392, 83–93 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Ostuni R., Kratochvill F., Murray P. J., Natoli G., Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 36, 229–239 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Wynn T. A., Vannella K. M., Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braga T. T., Agudelo J. S. H., Camara N. O. S., Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 6, 602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeNardo D. G., Ruffell B., Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long K. B., Gladney W. L., Tooker G. M., Graham K., Fraietta J. A., Beatty G. L., IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. 6, 400–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosser D. M., Edwards J. P., Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mardhian D. F., Storm G., Bansal R., Prakash J., Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J. Control. Release 290, 1–10 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Binder C., Chuang E., Habla C., Bleckmann A., Schulz M., Bathgate R., Einspanier A., Relaxins enhance growth of spontaneous murine breast cancers as well as metastatic colonization of the brain. Clin. Exp. Metastasis 31, 57–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu M., Wang Y., Xu L., An S., Tang Y., Zhou X., Li J., Liu R., Huang L., Relaxin gene delivery mitigates liver metastasis and synergizes with check point therapy. Nat. Commun. 10, 2993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horton J. S., Yamamoto S. Y., Bryant-Greenwood G. D., Relaxin modulates proinflammatory cytokine secretion from human decidual macrophages. Biol. Reprod. 85, 788–797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unemori E. N., Pickford L. B., Salles A. L., Piercy C. E., Grove B. H., Erikson M. E., Amento E. P., Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J. Clin. Investig. 98, 2739–2745 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song W., Shen L., Wang Y., Liu Q., Goodwin T. J., Li J., Dorosheva O., Liu T., Liu R., Huang L., Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 9, 2237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao L., Li J., Liu Q., Feng R., Das M., Lin C. M., Goodwin T. J., Dorosheva O., Liu R., Huang L., Transient and local expression of chemokine and immune checkpoint traps to treat pancreatic cancer. ACS Nano 11, 8690–8706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P., Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S. R., Maute R. L., Dulken B. W., Hutter G., George B. M., McCracken M. N., Gupta R., Tsai J. M., Sinha R., Corey D., Ring A. M., Connolly A. J., Weissman I. L., PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griesmann H., Drexel C., Milosevic N., Sipos B., Rosendahl J., Gress T. M., Michl P., Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 66, 1278–1285 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Zou W. P., Wolchok J. D., Chen L. P., PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng M., Jiang W., Kim B. Y. S., Zhang C. C., Fu Y.-X., Weissman I. L., Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bani D., Bigazzi M., Masini E., Bani G., Sacchi T. B., Relaxin depresses platelet aggregation: In vitro studies on isolated human and rabbit platelets. Lab. Invest. 73, 709–716 (1995). [PubMed] [Google Scholar]
- 31.Bastea L. I., Liou G. Y., Pandey V., Fleming A. K., von Roemeling C. A., Doeppler H., Li Z., Qiu Y., Edenfield B., Copland J. A., Tun H. W., Storz P., Pomalidomide alters pancreatic macrophage populations to generate an immune-responsive environment at precancerous and cancerous lesions. Cancer Res. 79, 1535–1548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elahi-Gedwillo K. Y., Carlson M., Zettervall J., Provenzano P. P., Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res. 79, 372–386 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bathgate R. A. D., Halls M. L., van der Westhuizen E. T., Callander G. E., Kocan M., Summers R. J., Relaxin family peptides and their receptors. Physiol. Rev. 93, 405–480 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Fallowfield J. A., Mizuno M., Kendall T. J., Constandinou C. M., Benyon R. C., Duffield J. S., Iredale J. P., Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 178, 5288–5295 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Bonapace L., Coissieux M.-M., Wyckoff J., Mertz K. D., Varga Z., Junt T., Bentires-Alj M., Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Georgoudaki A. M., Prokopec K. E., Boura V. F., Hellqvist E., Sohn S., Östling J., Dahan R., Harris R. A., Rantalainen M., Klevebring D., Sund M., Brage S. E., Fuxe J., Rolny C., Li F., Ravetch J. V., Karlsson M. C. I., Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15, 2000–2011 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hofseth L. J., Saito S., Hussain S. P., Espey M. G., Miranda K. M., Araki Y., Jhappan C., Higashimoto Y., He P., Linke S. P., Quezado M. M., Zurer I., Rotter V., Wink D. A., Appella E., Harris C. C., Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. U.S.A. 100, 143–148 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharpe A. H., Pauken K. E., The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lim T. S., Chew V., Sieow J. L., Goh S., Yeong J. P.-S., Soon A. L., Ricciardi-Castagnoli P., PD-1 expression on dendritic cells suppresses CD8+ T cell function and antitumor immunity. Onco. Targets. Ther. 5, e1085146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee R., Tyagi P., Li S., Huang L., Anisamide-targeted stealth liposomes: A potent carrier for targeting doxorubicin to human prostate cancer cells. Int. J. Cancer 112, 693–700 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabb6596/DC1