

Abstract
Adenovirus-mediated combination gene therapies have shown promising results in vaccination or treating malignant and genetic diseases. Nevertheless, an efficient system for the rapid assembly and incorporation of therapeutic genes into high-capacity adenoviral vectors (HCAdVs) is still missing. In this study, we developed the iMATCH (integrated modular assembly for therapeutic combination HCAdVs) platform, which enables the generation and production of HCAdVs encoding therapeutic combinations in high quantity and purity within 3 weeks. Our modular cloning system facilitates the efficient combination of up to four expression cassettes and the rapid integration into HCAdV genomes with defined sizes. Helper viruses (HVs) and purification protocols were optimized to produce HCAdVs with distinct capsid modifications and unprecedented purity (0.1 ppm HVs). The constitution of HCAdVs, with adapters for targeting and a shield of trimerized single-chain variable fragment (scFv) for reduced liver clearance, mediated cell- and organ-specific targeting of HCAdVs. As proof of concept, we show that a single HCAdV encoding an anti PD-1 antibody, interleukin (IL)-12, and IL-2 produced all proteins, and it led to tumor regression and prolonged survival in tumor models, comparable to a mixture of single payload HCAdVs in vitro and in vivo. Therefore, the iMATCH system provides a versatile platform for the generation of high-capacity gene therapy vectors with a high potential for clinical development.
Keywords: high-capacity vectors, helper-dependent adenovirus, gutless adenovirus, gene therapy, cancer therapy, immunotherapy, gene vectors, PD-1, IL-12, IL-2
Graphical Abstract
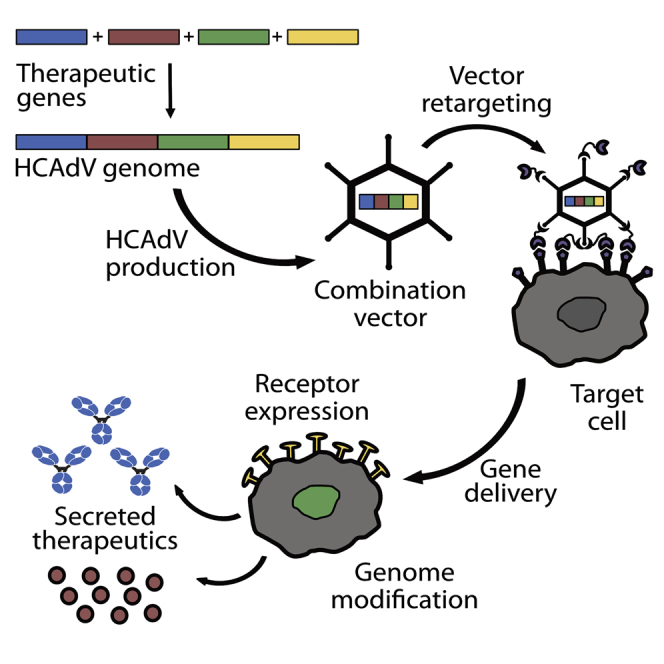
The iMATCH platform enables an efficient assembly of up to four orthogonal expression cassettes on a single plasmid from which high-capacity adenoviral vectors can be generated within 3 weeks in unprecedented purity. iMATCH vectors encoding immunotherapeutic combinations led to tumor regression and prolonged survival, comparable to mixtures of single payload HCAdVs.
Introduction
Gene therapy comprises viral, bacterial, and cellular vectors with cellular therapies, also including stem cell therapies.1,2 Viral gene therapy has come a long way since the early 1990s, when human adenoviruses (HAdVs) were first vectorized and applied in attempts to treat monogenic cystic fibrosis lung disease.3, 4, 5, 6, 7 Today, viral gene therapy with a variety of viruses is used in oncology, vaccination, and many other disciplines in clinical science.8, 9, 10, 11 Gene therapy vectors have been used preclinically for host genome editing and clinically for the expression of intracellular proteins, surface receptor presentation, and/or the secretion of therapeutics.12, 13, 14, 15 Recently, multiple gene delivery vectors have been developed in the expanding field of cancer immunotherapy.16 Cancer immunotherapy approaches modulate the activity of immune cells against cancer cells in various ways, e.g., by rendering the immunosuppressive state of the tumor microenvironment (e.g., by blocking the PD-1/PD-L1 axis) or by stimulating immune cell activation (e.g., by cytokine interleukin [IL]-12 or IL-2 release) to eradicate cancer cells.17
Despite numerous successes in different types of cancer such as melanoma, non-small cell lung carcinoma (NSCLC), urothelial carcinoma, and Hodgkin’s lymphoma,18, 19, 20 immunotherapies with checkpoint inhibitors have only been effective in a subset of patients,21 at least in part due to the complex immunosuppressive interactions of cancer cells within the tumor microenvironment.22 Certain combinations of immunotherapeutics have shown synergistic effects, e.g., anti-PD-1 with IL-12 or interferon (IFN)-γ,23 but also severe side effects upon systemic application, e.g., anti-cytotoxic T lymphocyte-associated protein 4 (anti-CTLA-4) with anti-PD-1.24
In contrast, immunotherapeutics delivered by viral or non-viral vectors to the tumor site have provided increased efficacy with less severe side effects due to the tumor-restricted expression of immunotherapeutic combinations.25, 26, 27 Non-replicative high-capacity adenoviral vectors (HCAdVs), also called helper-dependent vectors (HdVs) or gutless adenoviral vectors, are especially well suited for the gene delivery of multiple transgenes due to their high packaging capacity up to 36 kb,28 the efficient and long-lasting transgene expression of up to 7 years,29 and the attenuated immune clearance of transduced cells.30 The latter is largely a result of the absence of viral genes and their products.
Despite these advantages, the broad use of HCAdVs is currently hampered by the notoriously complex cloning and production schemes of HCAdVs.31, 32, 33 During HCAdV production, helper viruses (HVs) provide sufficient viral proteins in trans to achieve high HCAdV yields. HV particles are typically first-generation (FG) adenoviral vectors derived from HAdV-C5 lacking the E1 and E3 gene clusters.34 Due to the high immunogenicity of HVs and the potential interference of HV-expressed viral proteins with the target cells, HV must be removed from any HCAdV formulation. To suppress HV particle formation, the packaging signal of the HV DNA is typically flanked by loxP sites, which permits a Cre recombinase-mediated excision of the HV packaging signal in HCAdV producer cells, e.g., in the cell line 116.35 This conventional strategy reduces HV contamination to 0.01%–0.2% of the HCAdV, that is, 100–2,000 HV particles per million HCAdV particles (ppm).32,36
For gene therapy applications, the natural tropism of HAdV-C5 vectors for coxsackievirus and adenovirus receptor (CAR) has been ablated by genomic modifications to the adenovirus fiber knob.37 The tropism of adenoviral vectors has been redirected to defined biomarkers using genetically encoded tags, or exogenously added adapter proteins.37,38 While genetic modifications can lead to virion instability and low vector yields,39 exogenously added adapters do not affect vector production and thus allow for versatile use of the vector in different cell types.39 Previously, our group has developed a trimerized adapter that binds in a quasi-covalent manner to the HAdV-C5 knob by coupling the trimerizing SHP domain of the lambdoid phage 21 linked to a knob-binding designed ankyrin repeat protein (DARPin).38 Cell specificity is then mediated by a second retargeting DARPin coupled to the SHP domain. Retargeting DARPins against new proteins can be selected rapidly via ribosome display,40 produced efficiently in E. coli on a large scale, and the adapters constructed from them are then simply added to the virus.38 The receptor specificity of a given virus can thus be changed rapidly.
Off-target infection of HAdV-C5 vectors, especially liver infection, can further be reduced by genetic ablation of the hypervariable region 7 (HVR7) of the hexon, which facilitates blood factor X binding; however, HVR7 deletion also results in increased adenoviral depletion by neutralizing antibodies.41,42 To decrease the rapid clearance of modified or unmodified HAdV-C5 by neutralizing antibodies following systemic injection, our group has developed a trimerized single-chain variable fragment (scFv)-based shield that covers the adenoviral surface.43 The combined implementation of capsid mutation (ΔHVR7) and exogenously added shield and adapter proteins increases tumor specificity and transgene expression of non-replicative FG (E1 and E3 deleted) adenoviral vectors upon intravenous and intratumoral injection in murine models.43
To enhance the versatility of HAdV vectors, we have now designed a system that facilitates the rapid and efficient assembly of multiple transgenes into HCAdVs with very low HV contamination for gene therapy approaches. Our platform iMATCH (integrated modular assembly for therapeutic combination HCAdVs) combines a highly efficient assembly system with a novel purification strategy for generating combinations of HCAdVs, compatible with adenoviral retargeting and shielding strategies that can be used for the specific delivery of complex combinations of genes to target cells in a broad range of diseases. We validated the efficacy of the iMATCH system using reporter vectors as well as vectors harboring multiple immunotherapeutic genes for expression in cell culture and immune-competent murine tumor models.
Results
A modular assembly system for combing therapeutic genes
The generation and production of HCAdVs has so far been cumbersome, inefficient, and time-consuming.31 Overall, this is due to technical challenges in cloning of large (>28 kb) plasmids, which is required in standard HCAdV preparations. To enhance the cloning efficiency, we designed four size-optimized plasmids of 3–5 kb (pUniversal plasmids), each with an orthogonal expression cassette containing one of the following constitutive promoters: cytomegalovirus (CMV), EF1-α, SV40, or PGK (Figure 1A). The four promoters vary in the efficiency of transgene expression relative to each other, depending on the targeted cell.44 Expression levels of transgenes can thus be tuned relative to each other and relative to the chosen producer cell line. The expression cassettes were designed with minimal sequence similarity to each other, to the HCAdV backbone and to the HV genome, in order to reduce inter- and intra-vector homologous recombination events. Notably, the number of payloads encoded on the pUniversal plasmid is not limited by the number of promoters, as polycistronic gene expression elements (e.g., 2A peptides45 or internal ribosomal entry sites [IRESs]46) can be utilized within each open reading frame (ORF). Payloads encoding expression cassettes were flanked in each pUniversal plasmid by a unique pair of restriction sites and payload assembly overlaps (Figure 1A).
Figure 1.

Schematic overview of the iMATCH platform
(A) Four separate cloning plasmids (pUniversal 1–4) were constructed, each with an orthogonal expression cassette with a distinct promoter (CMV, SV40, EF1-α, or PGK). Unique restriction sites and payload assembly overlaps (shades of green) enable a rapid combination of payloads encoded on different pUniversal plasmids via ligation or Gibson assembly (dotted lines). (B) Assembled payload combinations (green), released from the pUniversal plasmids using a different set of unique restriction sites (Figures S2A and S2B), result in linear DNA fragments terminated by pC4HSU overlaps (red and shades of yellow), where pC4HSU is a HAdV backbone plasmid.35 Depending on the genomic size of the payload combination and the desired genome size of the HCAdV, different overlaps can be chosen (shades of yellow). In this manner, stuffer DNA (gray) of different sizes (0, 4.5, or 9 kb) is excised from the HCAdV genome backbone plasmid (pC4HSU). The pC4HSU overlaps of the pUniversal fragments and the linearized pC4HSU plasmid (dotted lines) thus allow the efficient generation of HCAdV genomes with defined genomic sizes via Gibson assembly. (C) HCAdV genomes (gray, green) can be packaged into HCAdV particles with defined capsid modifications, e.g., modification of the hexon (blue) or fiber (violet) protein by administering different helper virus (HV) vectors encoding the mutated capsid proteins during HCAdV production. (D) HCAdV particles can be retargeted to defined cells via a chosen surface biomarker by using a very tight-binding retargeting adapter that binds to the HAdV-C5 knob,38 here depicted in a simplified manner as bispecific DARPin retargeting adapters with two binding modules connected by a flexible linker: The first binder (knob binding DARPin, black) binds trivalently to the trimeric adenoviral fiber knob and simultaneously blocks HAdV-C5 from binding to its natural receptors (CAR). A new cell tropism is introduced by the second binding entity (retargeting DARPin, red, green, or yellow) that facilitates the specific binding to surface markers expressed on the cell of interest.
Upon restriction enzyme digestion, payloads encoded under the CMV, EF1-α, or PGK promoter are released and can be integrated into a linearized pUniversal plasmid of choice, which was beforehand linearized with the same restriction enzyme pair. Payload integration thus can be achieved either by cost-effective ligation or, alternatively, by Gibson assembly.47 The assembly of multiple expression cassettes was done sequentially or simultaneously, allowing for the assembly of up to four expression cassettes on a single pUniversal plasmid within 1 day. Plasmid-encoded, correctly assembled payloads were selected by antibiotic resistance and validated by sequencing, confirming a cloning efficacy of more than 80%. To verify this approach, reporter proteins were encoded within each expression cassette and combined on a single plasmid. Cells transfected with a combination of pUniversal plasmids containing all reporter expression cassettes showed similar or greater protein expression compared to a mixture of single reporter-encoding plasmid transfection (Figure S1).
Rapid integration of gene combinations into a HCAdV genome
Once the desired payload combination is assembled, an additional set of orthogonal restriction sites and overlaps (pC4HSU overlaps) on the pUniversal plasmids can be utilized for the integration of the payload combination into a well-characterized HCAdV backbone plasmid (pC4HSU)35,48 via Gibson assembly (Figure 1B). Depending on the payload size and the desired genome size of the HCAdV, the pUniversal plasmids can be digested with PmeI/PacI, PmeI/XmaI, or PmeI (Figure S2A). Each resulting pUniversal fragment (Figure S2B) can be integrated into a linearized pC4HSU plasmid with compatible overlaps that was generated by AscI, AscI/NotI, or AscI/SwaI restriction digest prior to assembly (Figure S2C). Linearization of the pC4HSU plasmid releases either 0, 4.5, or 9 kb of “stuffer DNA” (Figure S2D), which can be used to adapt the size of the resulting HCAdV. This enables production of HCAdV particles with the same genomic size and thus similar biophysical properties, independent of the payload size.
To increase cloning efficiency, a unique restriction site (FspI) was introduced into the antibiotic resistance gene of the pUniversal plasmid (Figure S2A). Backbone fragments of pUniversal plasmids that were used for payload release, but were re-circularized, can lead to false-positive clones due to the presence of an antibiotic resistance but a lack of payloads. For this reason, these fragments were digested with FspI in parallel to the unique restriction enzymes required for payload release. Upon Gibson assembly of the two fragments (linearized pUniversal and pC4HSU) and transformation of E. coli (Figure S2E), cloning efficiencies of up to 85% positive clones were obtained for a representative construct (Figure S2F).
HCAdVs with unique capsid modifications
The absence of viral genes on the HCAdV genome requires that the viral proteins are provided in trans by HVs for amplification of HCAdVs. We designed multiple HVs with different capsid modifications, including an HV with modifications in the HVR7 of the HAdV-C5 hexon to prevent factor X-mediated liver infection41 and an HV vector encoding the HVR7 modification with an additional deletion of the RGD motif from the HAdV-C5 penton that reduces integrin-mediated cell infection (off-target infection49). These different HVs readily enable the packaging of the HCAdV genome into a variety of different capsids (Figure 1C) without the need to genetically modify the HCAdV genome. The use of different capsid modifications can be advantageous to modulate viral clearance, uptake, or to reduce off-target transduction for gene therapy approaches.37 The desired target cell specificity of HCAdV particles with genetic capsid modifications was introduced by exogenously added retargeting adapters38 following amplification and purification of HCAdVs, again without the need to modify the original HCAdVs or HV vectors (Figure 1D). Retargeted HCAdV particles were additionally covered by a shield, formed by a trimerized scFv to prevent off-target infection and HCAdV clearance by neutralizing antibodies.43
Generation of HCAdVs with minimal HV contamination
HV contamination of HCAdV preparations must be kept as low as possible, due to the unavoidable viral gene expression from HV, which causes an increased adaptive immune response against HC-infected recipient cells.30 On the other hand, HVs are required for HCAdV production (Figure 2A), as they provide viral proteins in trans for the packaging of HCAdV genomes at the necessary balanced concentrations and in sufficient yields. To minimize HV contamination, we designed HVs that had minimal genomic similarity to the HCAdV, even including a modified encapsulation signal,35 which in addition to reduced genetic similarity, packaged at a lower efficiency than did the wild-type (WT) packaging signal of the HCAdV. Furthermore, our HV vectors were designed to have a genome size at the upper range of adenoviral packaging capacity (37.5 kb) to reduce the risk of generating fully replication-competent WT adenoviral vectors. The formation of competent WT adenoviral vectors can occur by E1/E3 recombination of HV vectors with E1 located on the chromosome of the producer cell line. The genomic size difference between the HCAdV (28–30 kb) and the HV (37.5 kb) also facilitates a sufficient separation of both vectors in subsequent CsCl density purification. Furthermore, we used the packaging cell line 116 for HCAdV amplification, which was specifically created to improve HCAdV production yield and purity.36 The cell line 116 was previously selected for increased HCAdV amplification and its high levels of Cre recombinase.35 Cre recombinase permits the removal of the loxP-flanked HV packaging signal and thus renders the HV DNA non-packageable.
Figure 2.

High-capacity adenoviral vector amplification and purification
(A) HCAdV producer cells, e.g., the cell line 116, were infected with HV and transfected with linearized HCAdV genomes (adenofection) in passage 0 (P0). During HCAdV amplification (P0–P4), HV amplification is inhibited by Cre-mediated excision of the HV packaging signal; thus, HV particles are re-administered to the cell lysate before infecting the next passage. After amplification, adenoviral particles are released from the 116 cell line and purified by two CsCl density purification steps. HCAdV (green curve) and HV (red curve) genomes are quantified by qPCR after amplification and purification. (B) Adenoviral particles (orange band) are separated from cell debris, cell lysate, and empty particles on a two-step CsCl gradient (left) and subsequently applied to a four-step gradient (right) for further separation of HCAdV particles (green band) from HV particles (red band). (C) A GFP-expressing HCAdV was purified. After the first centrifugation step (left), three distinct white bands are visible in the following order (from top-to-bottom): cell debris, empty viral particles, and mature viral particles. Notably, the cell lysate above the CsCl gradient is colored green from the presence of soluble HCAdV reporter (GFP). Following extraction of the band containing mature particles and application to the second CsCl gradient (right), a prominent upper band containing HCAdV particles (30 kb) is separated from a very faint lower HV band (37.5 kb).
The combination of this HCAdV genome, HV vectors, and packaging cell line resulted in efficient amplification of the HCAdVs utilizing our platform (Figure S3). HCAdVs are traditionally purified via two density gradients, a first two-step gradient (1.25 and 1.35 g/cm3) in which the cell lysate, cell debris, and empty particles are separated from the viral particle, and a second continuous gradient (1.33 g/cm3) in which the HCAdV particles are separated from the HVs. Instead of utilizing a traditional continuous gradient32,36 as the second purification step, we used a discontinuous four-step gradient ranging from 1.29 to 1.35 g/cm3 (Figure 2B), which led to a visible separation of the desired HCAdV (30 kb) from the HV particles (37.5 kb; Figure 2C).
Purified HCAdVs with the capsid modifications described above encoding a reporter protein (GFP) showed dose-dependent reporter expression with non-detectable HV reporter protein expression (mCherry), even under very high multiplicities of infection (MOIs; Figures 3A–3D; Figure S4A). Sensitive quantification of HV contamination via quantitative polymerase chain reaction (qPCR) (Figure S4B) revealed unprecedented low HV contaminations of 0.00001%–0.00012%, or 0.1–1.2 ppm, of HV of HCAdVs generated with the iMATCH platform (Figure 3E). Other HCAdV production and purification protocols typically report 0.01%–0.2% HV contamination,32,36 which is 2–4 orders of magnitude higher than that obtained with the iMATCH platform.
Figure 3.

Production of HCAdVs in high purity and quantity
(A–C) A549 cells were infected (A) with HCAdVs carrying the wild-type HAdV-C5 capsid, (B) with a modified hexon-encoding a HVR7 mutation,41 or (C) with modified hexon and fiber proteins containing the HVR7 hexon mutation and the deletion of the RGD fiber motif. All HCAdVs showed titratable payload reporter (GFP) expression with no detectable HV reporter (mCherry) signal. (D) As a control, purified HV showed comparable reporter expression as HCAdVs. (E) Quantification of HCAdV and HV genomic particles via qPCR of purified HCAdVs revealed unprecedented purity of produced HCAdV particles of 0.00001%–0.00012%, or 0.1–1.2 ppm. Furthermore, HCAdVs were generated in high quantities of 0.3–1.5 × 1011 genomic HCAdV particles from 2–3 × 108 cells. (F) Transmission electron microscopy (TEM) micrographs analyzed with negative staining of purified HCAdVs showed intact HCAdV particles free from AAV and other contaminants. Error bars in (A)-(D) represent SEM with n = 3.
In addition to the high purity of the HCAdV particles, the iMATCH platform provided high yields of HCAdVs (Figure 3E) sufficient for multiple in vivo studies (see below). On average, 2 × 1011 genomic HCAdV particles per production from 3 × 108 cells with a ratio of 24 genomic HCAdV particles per transducing unit were obtained (averaged across 11 preparations). HCAdV yields were not affected by the number of transgenes encoded on the vectors, i.e., single payload HCAdVs yielded on average 1.98 × 1011 genomic HCAdV particles, similar to payload combination vectors (e.g., RMP1-14_IL-12_IL-2) with on average 2.02 × 1011 genomic HCAdV particles. Furthermore, negative staining of purified HCAdV viral particles with electron microscopy revealed intact HCAdV particles free from adeno-associated virus (AAV) and other contaminants (Figure 3F).
Cell- and organ-specific targeting of HCAdVs mediated by exogenously added shield and retargeting adapters
In order to test whether retargeting and shielding strategies previously developed for FG adenoviral vectors are compatible with HCAdVs generated by the iMATCH platform (Figure 4A), we transduced EMT6 cells that express HER2 (EMT6-HER2) in vitro (Figure 4B) or in vivo (Figures 4C–4E) with identical MOIs of HER2-retargeted and shielded FG or HCAdV reporter vectors encoding a firefly luciferase gene. All HCAdVs and FG adenoviral vectors used in this and the following in vitro and in vivo studies contained an HVR7 modification to reduce factor X binding.41 Transgene expression was only detected from cells infected with adenoviral vectors that were incubated prior to infection with retargeting adapters for HER243 and shield (Figure 4B). Cells transduced with naked virus, and those transduced with virus incubated with shield and adapters that lacked the retargeting DARPin but still contained the knob-binding DARPin (blocked vectors), showed no elevated transgene expression compared to PBS-treated cells. Notably, transgene expression mediated by the HER2-retargeted and shielded FG vector was equal in terms of transgene expression induced by the HER2-retargeted and shielded HCAdV.
Figure 4.

Retargeted and shielded HCAdVs showed comparable gene expression and biodistribution to FG vectors
(A) First-generation (FG) vectors and HCAdVs were retargeted via a trivalent knob-binding bispecific DARPin retargeting module (black and light brown) and coated with a trimeric scFv shield (gray). (B) EMT6-HER2 cells, infected in vitro by shielded and HER2-retargeted FG vectors or HCAdVs that encoded a luciferase reporter gene, both showed significantly elevated levels of luciferase activity. Only background levels of luciferase activity were detected from untreated cells (PBS), cells infected with untreated HCAdVs (naked virus), and cells infected with HCAdVs, which carry a blocking adapter (no HER2-retargeting DARPin) and the shield (blocked). No significant differences between luciferase activity mediated by FG vectors and by HCAdVs could be detected in vitro. Luciferase activity was measured from lysates of three separately transduced cell populations, which were measured in technical duplicates (n = 6). (C) EMT6-HER2 tumor-bearing mice were injected intratumorally with retargeted and shielded HCAdVs or FG vectors and luciferase activity was measured 24 h after infection with an in vivo imaging system after intraperitoneal injection of luciferin substrate. (D) Vector-treated mice showed detectable luciferase activity, while no luciferase signal was detected in untreated control mice. (E) Quantification of the luciferase signal from (D) revealed no significant differences of transgene expression mediated by retargeted and shielded vectors between the HCAdV or FG type. (F) Following organ harvest and lysis, luciferase signals were measured from multiple tissues, including tumor, draining lymph nodes (dLNs), non-draining lymph nodes (ndLNs), spleen, liver, kidney, lung, and heart as described for (D). Significantly elevated levels of luciferase activity could only be detected in tumor tissues. Three mice per group were used for studies presented in (D)–(F). Statistical analyses were done with a two-way ANOVA test. ∗p < 0.1, ∗∗p < 0.01, ∗∗∗p < 0.001. n.s., not significant (p > 0.9). All error bars represent SEM.
We further monitored gene delivery via retargeted and shielded FG vectors and HCAdVs in an immunocompetent mouse model. For this purpose, EMT6-HER2 cells were injected into the mammary gland of female BALB/c mice. Seven days later, tumor-bearing mice received an intratumoral application of virus with 3 × 108 transducing units of either HER2-retargeted and shielded FG vectors or HCAdVs (Figure 4C). Luciferase activity was measured in live animals using in vivo imaging 1 day post-injection (Figure 4D) following intraperitoneal injection of luciferin. The luciferase signal was only detectable within tumors treated with adenovirus but not in the untreated control animals. Quantification of the in vivo luciferase signals revealed no significant differences between FG vectors and HCAdVs (Figure 4E), in agreement with in vitro results. Following measurements in live animals, mice were sacrificed, and various tissues were harvested, lysed, and analyzed for luciferase activity. Transgene expression was indeed limited to the tumor, with no significant signal detectable in all other organs for both FG- and HCAdV-treated mice. No significant difference in luciferase activity could be detected between FG vectors and HCAdVs in the tumor (Figure 4F).
Expression of multiple therapeutic payloads from a single HCAdV
Next, we assessed payload transcription and expression from a single HCAdV encoding multiple therapeutic payloads and compared it to a mixture of HCAdVs encoding the same payloads as single payloads per vector. We generated an immunotherapeutic combination of three payloads, a murine anti-PD-1 antibody (RMP1-14) and two cytokines (IL-12 and IL-2). This combination was chosen due to the strong synergistic effects between anti-PD-1 and IL-12 and between anti-PD-1 and IL-2, as compared to single-agent treatments in previous preclinical and clinical studies.23,50 The number of total transducing viral particles was kept equal for each treatment group in the following in vitro and in vivo studies, in order to equalize the immunologic response to adenoviral particles, which was deemed a relevant factor in studies with non-human primates and human patients.51
Single payload-containing HCAdVs (Ad_RMP1-14, Ad_IL-12, or Ad_IL-2) and an HCAdV encoding all three payloads (triple vector) were generated via the iMATCH platform. According to the therapeutic windows of the payload, the antibody RMP1-14 was encoded under the control of a strong CMV promoter. In contrast, the two cytokines IL-12 and IL-2, which are highly effective already at low picomolar concentrations, were encoded under the control of a weaker SV40 promoter (Figure 5A). The payload design is described in further details in Materials and methods. Due to the toxicity of IL-2 at high concentrations,52 IL-2 was encoded under the translational control of an encephalomyocarditis virus (EMCV)-IRES53 in the triple and single payload HCAdV.
Figure 5.

In vitro expression of a triple payload combination encoded on a triple vector or on different single payload HCAdVs
(A) RMP1-14 (CMV promoter), IL-12 (SV40 promoter), and IL-2 (SV40 promoter) were encoded each on a separate HCAdV (blue, violet, and light red) or together on a combination triple HCAdV (green). (B and C) Transcription (B) and secretion (C) levels of RMP1-14 (left panel), IL-12 (middle panel), and IL-2 (right panel) were measured in A549 cells 72 h after infection with 10 TU/cell. Cells were infected with either a single payload vector (left bar), the triple HCAdV (middle bar), or a mixture of single payload vectors (right bar). In all cases, the same total viral particle numbers were used. Transcription and translation levels of RMP1-14 (CMV promoter) were decreased only slightly upon co-expression of IL-12 and IL-2 from an SV40 promoter in the triple vector. In the mixture of single vectors, the gene dosage is one third, and this is reflected well in the transcript and protein level. Payload transcription and expression from the weaker SV40 construct was significantly reduced by the presence of the stronger CMV promoter independent of whether both promoters were located on a combination vector (triple vector) or on different vectors (a mixture of single payload vectors). The observed reduction of SV40-derived gene expression was in both conditions over-proportional to the number of transgenes delivered (more than a 3-fold reduction). As an exception, IL-2 transcription and expression from the triple vector was drastically increased, compared to the single Ad_IL-2 vector, due to the lack of a transgene upstream of the IRES in the single Ad_IL-2 vector. Overall, transcription and protein secretion levels of the encoded payload correlated well. In vitro protein secretion was determined from supernatants of three separately transduced cell populations, which were measured in duplicates in an antigen capture ELISA setup. Transcription levels were determined from A549 cell lysates of three separately transduced cell populations measured in duplicates via real-time PCR. Statistical analysis was done with a two-way ANOVA test. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n.s., not significant (p > 0.9). All error bars represent SEM.
Following infection of A549 cells, an overall correlation between transcript numbers (Figure 5B) and protein secretion (Figure 5C) could be observed. The triple HCAdV showed a small, albeit not significant, reduction of transgene transcription and expression levels for payloads encoded under the CMV promoter (RMP1-14), as compared to the single payload vector (Ad_RMP1-14) alone. The mixture of single payload vectors showed a 3-fold decrease in CMV-driven RMP1-14 expression, compared to the single Ad_RMP1-14 vector, which was proportional to the reduced copy numbers of the RMP1-14 transgene in the mixture. The CMV promoter generated an average of 6.5 CMV-driven mRNAs (RMP1-14) per GAPDH mRNA, compared to 0.7 SV40-driven (IL-12) mRNA per GAPDH in the single vector condition, i.e., a roughly 9-fold difference. This indicates that the CMV promoter has an increased transcriptional activity compared to the SV40 promoter in A549 cells. A decrease of SV40-driven transgene (IL-12 and IL-2) transcription and expression could be observed in the presence of the stronger CMV promoter in the triple HCAdV and in the mixture of single payload vectors. The absence of a transgene upstream of the EMCV-IRES and downstream of the SV40 promoter in the Ad_IL-2 HCAdV (Figure 5A) led to a decrease in IL-2 transgene transcription and expression compared to the triple HCAdV, which encoded IL-12 upstream of the EMCV-IRES.
We acknowledge that absence of a gene upstream of the IRES in front of IL-2 in the single vector might impede the comparison of the combination HCAdV and the mixture of single payload vectors. Therefore, we also analyzed transcription and expression of a second combination (RMP1-14 and IL-12) in the absence of IL-2 (Figure S5A). In analogy to the triple combination, a decrease of SV40 transgene (IL-12) expression in the presence of a CMV promoter was also observed in the dual combination (Figure S5B). CMV-mediated (RMP1-14) expression was not significantly reduced in the dual combination by SV40-driven co-expression of IL-12 (Figure S5C). Despite the small number of payloads in the tested combinations, we could observe that the transgene (RMP1-14 or IL-12) expression from a combination HCAdV was less reduced with increasing transgene numbers, compared to a mixture of single payload vectors (Figure S6), provided that the total viral particle numbers are constant. In conclusion, a HCAdV encoding multiple transgenes showed similar or even improved in vitro transgene transcription and protein secretion compared to a mixture of single payload HCAdVs.
In vivo efficacy of high-capacity adenovirus combination immunotherapy
To determine the in vivo efficacy of the combination triple HCAdV encoding RMP1-14, IL-12, and IL-2, we administered iMATCH vectors containing either a single payload or the combination of all three payloads intratumorally to immunocompetent C57BL/6 mice bearing subcutaneous B16-D5-HER2 tumors (Figure 6A). B16-D5-HER2 cells are highly aggressive melanoma cells that stably express HER2 and form immune-excluded tumors in C57BL/6 mice. The triple combination led to a successful tumor rejection, while mice treated with a single payload vector only showed a delay in tumor growth (Figure 6B). Tumor growth curves for individual mice of each treatment group are depicted in Figures S7 and S8. The improved tumor control of the triple vector resulted in prolonged survival of these mice compared to mice treated with single payload vectors or untreated mice (Figure 6C). IL-12 encoded on a single payload vector caused the second highest survival rate, while IL-2 as a single payload vector showed no improved survival compared to the untreated group.
Figure 6.

In vivo efficacy of a triple payload immunotherapy HCAdV
(A) Tumors were injected four times intratumorally with retargeted and shielded HCAdVs 10 days after injection of B16-HER2 cells at 2-day intervals. All treatments were given with the same total viral particle number (3 × 108 TU per injection). Tumor volumes were measured for 60 days after cell injection. (B–E) Mice treated with a triple vector showed improved tumor regression (B) and survival (C) compared to single payload vector treatment and compared to a mixture of single payload HCAdVs (D and E). (F) All payload-containing vectors except the Ad_IL-2 single vector showed a significant decrease of tumor volume with no significant differences between the triple vector and the mixture of single payload vectors 24 days after tumor cell injection. (G) The average survival was significantly improved for the Ad_IL-12 vector, the triple vector, and the mixture, compared to the untreated group, with no significant differences between the triple vector and the mixture. Treatment groups in (B) and (C) consisted of 5 mice, except for Ad_Empty and Ad_RMP1-14 (n = 4), and groups of (D) and (E) consisted of 11 mice. Data from (B) and (D) were pooled for (F), and data from (C) and (E) were pooled for (G). Statistical analysis of (F) and (G) was done with a two-way ANOVA test including all treatment groups. Indicated significances in (F) and (G) refer to the untreated control (upper lane) except for the separate comparison of the three samples Ad_IL-12, mixture of vectors, and triple vector (lower lane). ∗p < 0.1, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. n.s., not significant (p > 0.9). Error bars in (B), (D), and (F) represent SEM.
Next, we compared the triple combination vector against the mixture of single payload vectors. Both groups successfully decreased tumor growth (Figure 6D), compared to the untreated group, and showed prolonged survival (Figure 6E). Tumor regression and prolonged survival were increased in the triple vector treatment group compared to the mixture of single payload vectors. Overall, all mice treated with payload-containing HCAdVs, except for the IL-2 single payload vector, led to a significant reduction in tumor burden compared to the untreated group (Figure 6F). Although the treatment with the triple vector showed decreased tumor regression, it was not significantly different from tumor regression induced by IL-12 single vector treatment or treatment with a mixture of all three single payload vectors. The IL-12 single payload vector, the triple vector, and the mixture of single payload vectors led to similar improved survival compared to the untreated group (Figure 6G); that is, there was no significant difference between triple vector treatment and treatment with a mixture of single payload vectors or IL-12 single treatment. Neither treatment led to a significant change in the body and physical scoring of the mice (Figure S9).
Although the single IL-2 HCAdV showed no improvement in tumor control or survival, it is possible that the increased expression of IL-2 from the triple vector could positively influence the in vivo efficacy of the triple vector in comparison to a mixture of single payload vectors. For this reason, we performed an additional study with a dual vector encoding RMP1-14 and IL-12 and compared the dual vector with a mixture of single payload RMP1-14 and IL-12 vectors. The dual vector and the mixture of the two single payload vectors led to significantly decreased tumor growth (Figure S5D) and improved survival (Figure S5E) compared to the untreated group with no significant differences between the dual vector and the mixture of the single payload vectors. In conclusion, the mixtures of single payload vectors and the respective combination HCAdV showed virtually identical therapeutic efficacy in tumor regression and survival in vivo.
Discussion
So far, more than a dozen different gene therapy-based therapeutics have been approved in different countries for treating cancer, blindness, immune disorders, or neurological failure, and an increasing number of preclinical and clinical gene vector trials are currently ongoing. More than 400 adenoviral gene vectors have been tested in these trials.54 Nonetheless, a system for the rapid and efficient generation of high-capacity adenoviral gene vectors for cell-targeted gene delivery of therapeutic combinations is still missing.55
Here we designed the iMATCH platform, which enables the rapid assembly of multiple therapeutic genes, encoded under the control of four different constitutive promoters (CMV, SV40, EF1-α, or PGK) on a size-optimized pUniversal cloning plasmid. The cell type-specific transcription activity of these promoters allows for a defined transgene expression tailored to the targeted cell type and the required transgene concentration.44 In this highly modular system, each of these promoters could also be exchanged to tissue-specific or otherwise regulatable promoters. Promising combinations of immunotherapeutics, vaccination agents, or gene modifier can be generated with the presented modular assembly system rapidly and tested in vitro as plasmids, before starting time- and cost-intensive vector production. Moreover, because of the non-covalent nature of the targeting adapter, this extends to the search for the optimal surface receptor and its epitope, which can also be achieved without producing a new vector. Once the most suitable combination is found, the previously laborious and time-consuming integration of therapeutic combinations into HCAdVs31 can be avoided by the iMATCH system, with which cloning efficiencies of up to 85% were achieved.
The genomic size of adenoviral vectors can influence packaging efficiency and thus the yield of functional virions, but also their biophysical properties, for instance thermostability.56 To exclude that suboptimal vector size influences the efficacy of combinational HCAdVs, the iMATCH system was designed to generate HCAdVs with defined genomic sizes by exploring unique sets of restriction enzymes and overlaps for Gibson assembly. Utilizing this versatile, efficient cloning system, HCAdVs with multiple therapeutic genes can be cloned, amplified, and purified from existing transgene constructs with the iMATCH system within 3 weeks. This reduces the production time for therapeutic HCAdVs by half, compared to previously reported protocols (43 days),32 enabling a faster screening of therapeutic combinations, and allowing the production of several HCAdVs in parallel.
HV contamination of purified HCAdVs should be minimized due to the high immunogenicity of HV particles and the potential interference with the target cells.30 Although multiple systems have been developed to reduce HV contamination of HCAdV preparations in the past,32,35,36,57 HV contamination is still a major issue in many applications, limiting the broad use of HCAdVs. We achieved unprecedented low amounts of HV contamination of 0.00001%–0.00012%, or 0.1–1.2 ppm, HCAdV particles, using optimized HVs and purification protocols, which is an improvement of 100- to 2,000-fold to previously published HCAdV purification protocols, which have reported 0.2%–0.01% HV contamination.32,36 We hypothesize that the residual absolute HV contamination level in our HCAdV preparations is constant under the established conditions (<100 HV genomes per mL) and that the relative contamination therefore varies, depending on the HCAdV yield obtained. Due to the reduced production time and the repeatedly low HV contamination of the iMATCH platform, this approach is adaptable and straightforward, and it might thus be widely used to achieve HCAdV production of the highest quality.
Furthermore, our results indicate that transgene combinations of interest can be systematically screened as combinations of FG vectors or HCAdVs encoding single payloads. Promising combinations can then be encoded on a single HCAdV with a packaging capacity of up to 36 kb. We suggest this procedure as the most efficient strategy, based on the following findings: (1) retargeted and shielded HCAdVs and FG vectors showed similar protein expression in vitro and in vivo as well as similar biodistribution; (2) expression and transcription from a combination vector was equal to or higher than from a mixture of single payload vectors when comparing the same total particle numbers; (3) transgene expression from a combination vector decreased less drastically with increasing numbers of transgenes, compared to a mixture of single payload vectors; and (4) combination HCAdVs gave rise to comparable in vivo tumor regression and survival than the mixture of the respective single payload vectors, notably in an aggressive syngeneic immune-excluded tumor model (B16-HER2 in C57BL/6 mice).
We noticed that the concentration of the most effective single payload (IL-12 encoded under a SV40 promoter) was reduced by the presence of a transcriptionally more active promoter (CMV), independent of whether the transgenes were encoded on a combination vector or administered as a mixture. The downregulation of one promoter by a second cis-encoded promoter was also previously observed for retroviral vectors.58 As a consequence, we recommend adapting the choice of promoter for each of the payloads after an initial in vitro or in vivo assessment with the single payload vectors. Utilizing this procedure, it may be possible to further increase the potency of the tested, synergistic RMP1-14, IL-12, and IL-2 combination. Nevertheless, we emphasize that the presented studies were designed to investigate the suitability of combination HCAdVs for the delivery of complex immunotherapy combinations, rather than establish a pre-clinical argument for the particular anti-PD1, IL-12, and IL-2 combination and their promoters and expression levels.
The clinical approval of combination treatments such as in immunotherapies in a variety of different indications, for instance in metastatic melanoma, advanced renal cell carcinoma, and metastatic colorectal cancer,59 emphasizes that the preclinical testing of combination HCAdVs in multiple different cancer or disease models would be beneficial. However, the systemic administration of some of these combinations as systemically administered proteins has led to adverse side effects,24 highlighting the need for cell- or organ-specific production of these agents. This has been a key motivation for developing the iMATCH platform. Cell-specific transduction of adenoviral vectors can be achieved via genetic modifications,37 which requires the reconstruction and reproduction of the same combination HCAdV for each disease model and can lead to virion instability and reduced production yields.39 Furthermore, many strategies would not be able to generically target any surface receptor of choice.
For this reason, HCAdVs generated by the iMATCH system were designed to be compatible with any exogenously added retargeting adapter for HAdV-C5 vectors. The large library of existing DARPins, the rapid selection of new DARPin adapters against surface markers,40 and the efficient production of DARPin-based retargeting adapters in bacteria38 are advantages unique to the retargeted strategy presented herein. In addition, due to the high abundance of pre-existing antibodies against HAdV-C560 in the population of many countries, covering of iMATCH-generated HAdV-C5 vectors with the previously described shield that has been shown to reduce neutralizing antibody binding to HAdV-C5 FG vectors43 would be beneficial for therapeutic uses. Alternatively, HCAdVs of other serotypes with a lower pre-existing immunity can now be developed for clinical purposes4 based on the strategy described herein.
In conclusion, we think that the rapid production of combination HCAdVs in high quantity and purity, the compatibility with established cell-specific targeting strategies, and the broad variety of payload molecules that can be encoded on HCAdVs make the iMATCH platform an attractive vector-developing tool for multiple research fields.
Materials and methods
Plasmid, cells, and mice strains
The plasmid pAdEasy-1 was obtained from Agilent Technologies61 and modified as described below. The pC4HSU plasmid was purchased from Microbix and originally developed by Sandig et al.35 The murine cell line EMT6-HER2 was generated as previously described.62 Briefly, murine EMT6 cells were modified for constitutive expression of HER2. The B16-D5-HER2 murine cell line was kindly provided by Louis Weiner.63 A poorly immunogenic subclone of murine B16 cells, D5, was modified to constitutively produce HER2. The human cell line 116 was kindly provided by Philip Ng and cultured as recommended.36 The remaining cell lines were purchased from the vendor ATCC and cultivated according to the provider’s recommendations. Cells were passaged no more than 15 times from the original stocks. C57BL/6 and BALB/c mice were bred in-house at the University Hospital of Basel (Basel, Switzerland). Animals were housed under specific pathogen-free conditions. All animal experiments were performed in accordance with Swiss federal regulations.
Plasmid constructions
The helper viral vector pmCherry-HV plasmid was generated by replacing the natural packaging signal of the pAdEasy-1 vector with a loxP-flanked modified encapsulation signal derived from Sandig et al.35 and a mCherry reporter under the control a PGK promoter. The plasmid pmCherry-HVR7 was derived from pmCherry-HV by inserting four mutations into the hypervariable loop 7 of the hexon (I421G, T423N, E424S and L426Y) as previously described.43 Further deletion of the RGD region of the fiber protein generated the plasmid vector pmCherry-HVR7-ΔRGD. All cloning steps involving helper viral plasmids were carried out by homologous recombination as reported in previous studies.43 The pUniversal plasmids were generated de novo using Gibson assembly47 by combining the pUniversal backbone fragment that encodes the origin of replication and ampicillin resistance derived from the pcDNA3.1(+) plasmid (Invitrogen) with gene fragments containing expression cassettes synthesized by GeneArt (Thermo Fisher Scientific). Each expression cassette consists of a unique pair of promoters and terminators (e.g., SV40 promoter + SV40 terminator, CMV promoter + bovine growth hormone terminator, EF1-α promoter + human growth hormone terminator, or PGK promoter + human β-globin terminator), each with a set of unique restriction sites for payload insertion (e.g., EcoRI and BamHI or EcoRI and XbaI).
Payload design
The rat anti-PD1 antibody clone RMP1-14, an immunoglobulin of type IgG2aκ, whose sequence was identified from hybridoma sequencing (Bio X Cell), was converted to a chimeric mouse IgG2aκ antibody with rat variable domains. The mouse IgG2a∗01 heavy chain (HC; IMGT: IGHG2A∗01; accession no. V00825)64 and the κ light chain (LC; IMGT: IGKC∗01; accession no. V00807) were used as scaffolds. The following mutations were included in the HC: (1) the CH2 glycosylation site was mutated to alanine (N297A) to impair FcγR binding; (2) effector functions were further ablated by introducing L234A, L235A, and P329G mutations to the CH2 domain;65,66 and (3) a cloning site was added that introduces a K115S mutation into the CH1 domain. Optimized H5 and L1 leader sequences were used for HC and LC secretion,67 respectively. Chains were expressed using a F2A68,69 sequence with an optimized furin site (RKRR)70 for expression from a single open reading frame, with the orientation HC-F2A-LC as described previously.71
The murine IL-12 gene was generated from translated GenBank cDNA sequences for IL-12B/p40 (GenBank: BC103608.1) and IL-12B/p35 (GenBank: BC146595.1) connected by F2A peptide as above, and the murine IL-2 gene was created from the translated GenBank cDNA sequence (GenBank: NM_008366.3). The cytokine genes included their native signal sequences. All payload constructs were codon-optimized for mouse expression and synthesized by GeneArt (Thermo Fisher Scientific).
HCAdV generation
Helper viral vectors were generated in the human cell line HEK293 as previously described; they originate from the HV plasmids pmCherry, pmCherry-HVR7, and pmCherry-HVR7-ΔRGD.61 HCAdVs were amplified as described in detail by Ehrke-Schulz et al.72 Following three wash steps with PBS, the collected cells from 15 × 15-cm dishes were lysed by three freeze-thaw cycles using a 37°C water bath and liquid nitrogen. The cell lysate was cleared by 8 min of centrifugation at 500 × g, 4°C. The cell supernatant was applied on the first CsCl gradient consisting of two steps, 1.25 and 1.25 g/cm3 CsCl. The remaining cell pellet was washed with 2 mL of PBS, and the supernatant was additionally applied on the first CsCl gradient. The lower viral band of the first CsCl gradient was extracted after 2 h of centrifugation at 12°C, 226,000 × g with a syringe and transferred to a second, four-step CsCl gradient, consisting of four steps ranging from 1.29 to 1.35 g/cm3. The lower-density HCAdV particles formed an upper band and were extracted from the second gradient after 18–24 h of centrifugation at 12°C, 226,000 × g. Following dialysis in dialysis buffer of 20 mM HEPES (pH 8.1), 150 mM NaCl, and 1 mM MgCl2, glycerol was supplied to the viral solution reaching a final concentration of 10% prior to viral storage at −80°C.
Genomic and transducing particle determination
For transducing particle determination, 0.5 × 105 A549 cells per well were seeded in a 24-well plate 24 h prior to infection. Cells were transduced with 3 μL of purified HCAdVs and incubated for 2 h at 37°C. The infection medium was aspirated, and the cells were washed with 0.5 mL of PBS. Following trypsin digest, detached cells were washed once with 1 mL of PBS, 1,000 × g, 5 min, 4°C. For viral genome quantification during HCAdV amplification, viral genomes were extracted from collected cells using a DNA isolation kit (Genekam, SB0071), following the manufacturer’s protocol. Viral genomes of purified HCAdV samples were quantified directly from the viral solution following a 5-min inactivation step at 80°C. HCAdV genomes were quantified by qPCR with specific primers (5′-TCTGCTGGTTCACAAACTGG-3′, 5′-TCCTCCCTTCTGTCCAAATG-3′) and a specific probe (5′-CGCCTTCTCCAGCATCCCGA-3′) in a multiplex reaction with HV-specific primers (5′-GAATAACAAGTTTAGAAACCCCACGGTGG-3′, 5′-GTTTGACCTTCGACACCTATTTGAATACCC-3′) and probe (5′-TGACATCCGCGGCGTGCTGGACAGG-3′). All reactions were performed using PrimeTime gene expression master mix (Integrated DNA Technologies, 105571), and HCAdV and HV signals were normalized to the passive dye rhodamine-X (ROX). All qPCR primer and probes were generated with the double quench technology by Integrated DNA Technologies. The qPCR reaction was performed and analyzed as described previously.72
Reporter quantification in HEK293 cells
HEK293 cells were seeded at 10,000 cells per well in a 96-well plate, 24 h prior to infection. Cells were infected with a defined number of viral particles and harvested 72 h after infection. Following three wash steps with 1× PBS, fluorescence protein expression was quantified with a BD LSRFortessa cytometer. The Alexa Fluor 488 channel was used for detecting GFP expression, and the phycoerythrin (PE)-Texas red channel was used for detecting tdTomato expression after gating on single cells.
Electron microscopy analyzed with negative staining of high-capacity adenoviral particles
Purified HCAdV (encoding no transgene) was diluted to a concentration of 1 μg/μL. Next, 10 μL of droplet HCAdV samples was spotted on copper grids (300-mesh, glow-discharged) for 30 s, then overlaid with 10 μL of droplets of 2% uranyl acetate for 1 min. The excess uranyl acetate was removed with filter paper. A CM100 transmission electron microscope (Thermo Fisher Scientific) with an acceleration voltage of 80 kV and an Orius 1000 digital camera (Gatan) were used to examine the grids.
In vitro retargeting of adenoviral vectors
EMT6-HER2 cells were seeded with 1 × 105 cells per well of a 24-well plate, 24 h prior to infection. FG adenoviral vectors or HCAdVs encoding firefly luciferase under the control of a CMV promoter were incubated either with a retargeting G3 adapter38 containing human HER2 specificity and a shield (termed “retargeted”), a blocking adapter that only contained the knob-binding DARPin but no retargeting DARPin and a shield (“blocked”), or without any retargeting adapter or shield (“naked virus”) for 1 h at 4°C. The ratio of viral knobs to retargeting adapter and shield to hexon was 1:5 as previously described43 with an MOI of 10 transducing units per cell (TU/cell). Viral particle-containing supernatants were removed 3 h after infection and replaced by fresh culture medium. Luciferase activity was determined by a luciferase assay (Promega, E1500) according to the manufacturer’s instructions 72 h after infection.
In vivo biodistribution study (EMT6-HER2 model)
1 × 106 EMT6-HER2 cells, suspended in phenol red-free DMEM (without supplements), were injected into the mammary gland of 8- to 12-week-old BALB/c WT mice. Once the tumors reached an average volume of 40–60 mm3, luciferase-encoding retargeted and shielded HCAdV or FG viruses (3 × 108 TU per mouse) were injected intratumorally. The luciferase signal was determined in live animals 1 day after virus injection and 10 min after intraperitoneal injection of 150 mg/kg d-luciferin (PerkinElmer) using the in vivo imaging system NightOWL II LB 983 (Berthold Technologies). Following live imaging, luciferase activity was determined in isolated tumors and organs (draining and non-draining lymph node, spleen, liver, kidney, lung, and heart). The overlay of the real image and the luminescence representation allowed the localization and measurement of luminescence emitted from xenografts. The signal intensities from manually derived regions of interest (ROIs) were obtained and data were expressed as photon flux (photons/s). All measurements were performed under the same conditions, including camera settings, exposure time (60 s), distance from lenses to the animals, and ROI size.
In vitro quantification of payload expression
For quantification of payload expression, A549 cells were infected 24 h after seeding (1 × 105 cells per well of a 24-well plate) at an MOI of 10 TU/cell for single and triple payload-containing viruses. A549 cells infected with a mixture of viruses were infected with the same total MOI of 10 TU/cell, i.e., an MOI of 3.33 TU/cell per single payload vector. Infected cells and cell supernatant were harvested 72 h after infection. Infected cells were stored at −80°C before mRNA levels were quantified (see below). Cytokine secretion was determined from supernatants using a mouse IL-12-p70 sandwich ELISA assay (Invitrogen, 88-7121) for IL-12 and a mouse IL-2 sandwich ELISA kit (Invitrogen, 88-7024) for IL-2, following the manufacturer’s instructions. RMP1-14 expression was quantified via an antigen capture ELISA. A protein-binding 96-well plate (Thermo Fisher Scientific, 44-2404-21) was coated with a 20 nM murine PD-1 (Amsbio, AMS.PD1-M82F4-25UG) solution at 4°C overnight (100 μL/well). Following blocking with 1× casein (Merck, B6429), the cell supernatant or a standard of recombinantly produced and purified RMP1-14 antibodies were applied. Following three wash steps with PBS + 0.1% Tween 20, an alkaline phosphatase-coupled anti-mouse κ-LC detection antibody (SouthernBiotech, 1050-04) was added. After wash, para-nitrophenylphosphate (p-NPP) was used as a substrate, and absorbance at 406 nm was measured with an Infinite M1000 microplate reader (Tecan). Antibody concentrations were determined from a standard curve of recombinantly produced RMP1-14 protein using a sigmoidal four parameter fit.
mRNA quantification by RT-PCR
To quantify mRNA levels of the expressed payloads by quantitative reverse transcriptase PCR (qRT-PCR), RNA was extracted from cells used for quantification of payload expression, according to the manufacturer’s instructions (Zymo Research, R1054). 500 ng of extracted RNA was reverse-transcribed into cDNA using a QuantiTect reverse transcription kit (QIAGEN, 205311) with the supplied mix of oligo(dT) and random primers. Following RT-PCR, cDNA samples were analyzed by qPCR using specific primers for RMP1-14 (5′-CGCTTCTGTCGTGTGCTTCC-3′, 5′-TTGTGGGTAGCCTCGCATGT-3′), IL-12 (5′-CCTGGCGAGACAGTGAACCT-3′, 5′-CCGCCCTTGTGACAGGTGTA-3′), IL-2 (5′-GCCACCGAGCTGAAGGATCT-3′, 5′-ATGCTCTGGCAGAAGGCGAT-3′), and GAPDH (5′-AGCCACATCGCTCAGACAC-3′, 5′-GCCCAATACGACCAAATCC-3′) in single reactions with a SYBR Green dye for quantification (Applied Biosystems, ANF00117457).
In vivo efficacy study (B16-HER2 model)
C57BL/6N WT mice were injected subcutaneously into the right flank with 5 × 105 syngeneic B16-D5-HER2 cells suspended in phenol red-free DMEM (without additives). Mice bearing palpable B16-HER2 tumors (average volume of 20–30 mm3) received intra-tumoral injection of 3 × 108 TU of HER2-targeted and shielded HCAdVs, coding for the indicated payloads, on days 11, 13, 15, and 18 after tumor challenge. The tumor volume was calculated according to the formula: V = ½[D (mm) × d2 (mm)], with D and d being the longest and shortest tumor diameter, respectively. Bodyweight and physiological score, including appearance, behavior, and respiration, were monitored during the whole experiment to detect signs of toxicity.
Acknowledgments
We acknowledge Prof. Anja Ehrhardt (University of Witten/Herdecke) and Dr. Maren Schiwon (University of Witten/Herdecke) for providing fruitful help and discussions in establishing the HCAdV system in our group. Furthermore, we want to thank Dr. Philip Ng (Baylor College of Medicine) for providing the cell line 116, Dr. Louis Weiner for providing the cell lines B16-HER2, Dr. Annemarie Honegger (University of Zurich) for assisting in antibody design, Dr. Abhishek Kashyap (University of Basel) and Karen Patricia Hartmann (University of Zurich) for valuable input, Lea Flühler (University of Zurich) for assistance in FG-HV genomic assembly, and Polina Zaytseva (University of Zurich) for providing shield protein. This work was supported by SNF Sinergia grant 170929 (to A.P., A.Z., and U.G.), the National Cancer Institute of the National Institutes of Health under award number F32CA189372 (to S.N.S.), and by the University of Zurich Forschungskredit 2017 ID 3761 (to D.B.).
Author contributions
D.B., A.P., M.S., and S.N.S. designed the project; S.N.S. designed antibody and cytokine constructs; D.B., C.K., J.S., and N.S. designed, cloned and generated all pUniversal plasmid and HCAdV genomes; D.B., J.S., and N.S. generated and characterized all HCAdVs; D.B., M.S., P.C.F., and J.K. generated all FG vectors; P.C.F. and S.N.S. generated shield protein; S.N.S. generated retargeted adapters; D.B., J.K., and F.W. performed qPCR and RT-PCR analysis; N.K. performed all in vivo experiments; D.B., A.P., and A.Z. coordinated the project; and D.B., N.K., S.N.S., and A.P. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2021.01.002.
Supplemental information
References
- 1.Sedighi M., Zahedi Bialvaei A., Hamblin M.R., Ohadi E., Asadi A., Halajzadeh M., Lohrasbi V., Mohammadzadeh N., Amiriani T., Krutova M. Therapeutic bacteria to combat cancer; current advances, challenges, and opportunities. Cancer Med. 2019;8:3167–3181. doi: 10.1002/cam4.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Luca M., Aiuti A., Cossu G., Parmar M., Pellegrini G., Robey P.G. Advances in stem cell research and therapeutic development. Nat. Cell Biol. 2019;21:801–811. doi: 10.1038/s41556-019-0344-z. [DOI] [PubMed] [Google Scholar]
- 3.Baker A.T., Aguirre-Hernández C., Halldén G., Parker A.L. Designer oncolytic adenovirus: coming of age. Cancers (Basel) 2018;10:201. doi: 10.3390/cancers10060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao J., Mese K., Bunz O., Ehrhardt A. State-of-the-art human adenovirus vectorology for therapeutic approaches. FEBS Lett. 2019;593:3609–3622. doi: 10.1002/1873-3468.13691. [DOI] [PubMed] [Google Scholar]
- 5.Lundstrom K. Viral vectors in gene therapy. Diseases. 2018;6:42. doi: 10.3390/diseases6020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greber U.F., Flatt J.W. Adenovirus entry: from infection to immunity. Annu. Rev. Virol. 2019;6:177–197. doi: 10.1146/annurev-virology-092818-015550. [DOI] [PubMed] [Google Scholar]
- 7.Greber U.F. Adenoviruses–infection, pathogenesis and therapy. FEBS Lett. 2020;594:1818–1827. doi: 10.1002/1873-3468.13849. [DOI] [PubMed] [Google Scholar]
- 8.Ura T., Okuda K., Shimada M. Developments in viral vector-based vaccines. Vaccines (Basel) 2014;2:624–641. doi: 10.3390/vaccines2030624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Björklund T. Repairing the brain: gene therapy. J. Parkinsons Dis. 2018;8(s1):S123–S130. doi: 10.3233/JPD-181485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon K., Del Medico A., Sander I., Kumar A., Hamad B. Gene therapies in ophthalmic disease. Nat. Rev. Drug Discov. 2019;18:415–416. doi: 10.1038/d41573-018-00016-1. [DOI] [PubMed] [Google Scholar]
- 11.Wirth T., Ylä-Herttuala S. Gene therapy used in cancer treatment. Biomedicines. 2014;2:149–162. doi: 10.3390/biomedicines2020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehrke-Schulz E., Schiwon M., Leitner T., Dávid S., Bergmann T., Liu J., Ehrhardt A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017;7:17113. doi: 10.1038/s41598-017-17180-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer D.J., Turner D.L., Ng P. A single “all-in-one” helper-dependent adenovirus to deliver donor DNA and CRISPR/Cas9 for efficient homology-directed repair. Mol. Ther. Methods Clin. Dev. 2020;17:441–447. doi: 10.1016/j.omtm.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubberstey A.V., Pavliv M., Parks R.J. Cancer therapy utilizing an adenoviral vector expressing only E1A. Cancer Gene Ther. 2002;9:321–329. doi: 10.1038/sj.cgt.7700436. [DOI] [PubMed] [Google Scholar]
- 15.Ruan M.Z.C., Cerullo V., Cela R., Clarke C., Lundgren-Akerlund E., Barry M.A., Lee B.H.L. Treatment of osteoarthritis using a helper-dependent adenoviral vector retargeted to chondrocytes. Mol. Ther. Methods Clin. Dev. 2016;3:16008. doi: 10.1038/mtm.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neshat S.Y., Tzeng S.Y., Green J.J. Gene delivery for immunoengineering. Curr. Opin. Biotechnol. 2020;66:1–10. doi: 10.1016/j.copbio.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma P., Allison J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020;20:75–76. doi: 10.1038/s41577-020-0275-8. [DOI] [PubMed] [Google Scholar]
- 18.Ansell S.M., Lesokhin A.M., Borrello I., Halwani A., Scott E.C., Gutierrez M., Schuster S.J., Millenson M.M., Cattry D., Freeman G.J. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Carvajal R.D., Sosman J.A., Atkins M.B. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garon E.B., Rizvi N.A., Hui R., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L., KEYNOTE-001 Investigators Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 21.Haslam A., Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open. 2019;2:e192535. doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galluzzi L., Chan T.A., Kroemer G., Wolchok J.D., López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018;10:459. doi: 10.1126/scitranslmed.aat7807. [DOI] [PubMed] [Google Scholar]
- 23.Garris C.S., Arlauckas S.P., Kohler R.H., Trefny M.P., Garren S., Piot C., Engblom C., Pfirschke C., Siwicki M., Gungabeesoon J. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49:1148–1161.e7. doi: 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan K.K., Bass A.R. Autoimmune complications of immunotherapy: pathophysiology and management. BMJ. 2020;369:m736. doi: 10.1136/bmj.m736. [DOI] [PubMed] [Google Scholar]
- 25.Huang K.W., Hsu F.F., Qiu J.T., Chern G.J., Lee Y.A., Chang C.C., Huang Y.T., Sung Y.C., Chiang C.C., Huang R.L. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci. Adv. 2020;6:eaax5032. doi: 10.1126/sciadv.aax5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter C.E., Rosewell Shaw A., Jung Y., Yip T., Castro P.D., Sandulache V.C., Sikora A., Gottschalk S., Ittman M.M., Brenner M.K., Suzuki M. Oncolytic adenovirus armed with BiTE, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol. Ther. 2020;28:1251–1262. doi: 10.1016/j.ymthe.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakao S., Arai Y., Tasaki M., Yamashita M., Murakami R., Kawase T., Amino N., Nakatake M., Kurosaki H., Mori M. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl. Med. 2020;12:526. doi: 10.1126/scitranslmed.aax7992. [DOI] [PubMed] [Google Scholar]
- 28.Rosewell A., Vetrini F., Ng P. Helper-dependent adenoviral vectors. J. Genet. Syndr. Gene Ther. 2011;(Suppl 5):001. doi: 10.4172/2157-7412.s5-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brunetti-Pierri N., Ng T., Iannitti D., Cioffi W., Stapleton G., Law M., Breinholt J., Palmer D., Grove N., Rice K. Transgene expression up to 7 years in nonhuman primates following hepatic transduction with helper-dependent adenoviral vectors. Hum. Gene Ther. 2013;24:761–765. doi: 10.1089/hum.2013.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muruve D.A., Cotter M.J., Zaiss A.K., White L.R., Liu Q., Chan T., Clark S.A., Ross P.J., Meulenbroek R.A., Maelandsmo G.M., Parks R.J. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004;78:5966–5972. doi: 10.1128/JVI.78.11.5966-5972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee D., Liu J., Junn H.J., Lee E.J., Jeong K.S., Seol D.W. No more helper adenovirus: production of gutless adenovirus (GLAd) free of adenovirus and replication-competent adenovirus (RCA) contaminants. Exp. Mol. Med. 2019;51:1–18. doi: 10.1038/s12276-019-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jager L., Hausl M.A., Rauschhuber C., Wolf N.M., Kay M.A., Ehrhardt A. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat. Protoc. 2009;4:547–564. doi: 10.1038/nprot.2009.4. [DOI] [PubMed] [Google Scholar]
- 33.Dormond E., Kamen A.A. Manufacturing of adenovirus vectors: production and purification of helper dependent adenovirus. Methods Mol. Biol. 2011;737:139–156. doi: 10.1007/978-1-61779-095-9_6. [DOI] [PubMed] [Google Scholar]
- 34.Zhou H.S., Zhao T., Rao X.M., Beaudet A.L. Production of helper-dependent adenovirus vector relies on helper virus structure and complementing. J. Gene Med. 2002;4:498–509. doi: 10.1002/jgm.301. [DOI] [PubMed] [Google Scholar]
- 35.Sandig V., Youil R., Bett A.J., Franlin L.L., Oshima M., Maione D., Wang F., Metzker M.L., Savino R., Caskey C.T. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc. Natl. Acad. Sci. USA. 2000;97:1002–1007. doi: 10.1073/pnas.97.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer D., Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Barry M.A., Rubin J.D., Lu S.C. Retargeting adenoviruses for therapeutic applications and vaccines. FEBS Lett. 2020;594:1918–1946. doi: 10.1002/1873-3468.13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dreier B., Honegger A., Hess C., Nagy-Davidescu G., Mittl P.R., Grütter M.G., Belousova N., Mikheeva G., Krasnykh V., Plückthun A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc. Natl. Acad. Sci. USA. 2013;110:E869–E877. doi: 10.1073/pnas.1213653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poulin K.L., McFall E.R., Chan G., Provost N.B., Christou C., Smith A.C., Parks R.J. Fusion of large polypeptides to human adenovirus type 5 capsid protein IX can compromise virion stability and DNA packaging capacity. J. Virol. 2020;94:e01112–e01120. doi: 10.1128/JVI.01112-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dreier B., Plückthun A. Rapid selection of high-affinity binders using ribosome display. Methods Mol. Biol. 2012;805:261–286. doi: 10.1007/978-1-61779-379-0_15. [DOI] [PubMed] [Google Scholar]
- 41.Xu Z., Qiu Q., Tian J., Smith J.S., Conenello G.M., Morita T., Byrnes A.P. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat. Med. 2013;19:452–457. doi: 10.1038/nm.3107. [DOI] [PubMed] [Google Scholar]
- 42.Allen R.J., Byrnes A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019;593:3449–3460. doi: 10.1002/1873-3468.13649. [DOI] [PubMed] [Google Scholar]
- 43.Schmid M., Ernst P., Honegger A., Suomalainen M., Zimmermann M., Braun L., Stauffer S., Thom C., Dreier B., Eibauer M. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018;9:450. doi: 10.1038/s41467-017-02707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin J.Y., Zhang L., Clift K.L., Hulur I., Xiang A.P., Ren B.Z., Lahn B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS ONE. 2010;5:e10611. doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chng J., Wang T., Nian R., Lau A., Hoi K.M., Ho S.C.L., Gagnon P., Bi X., Yang Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs. 2015;7:403–412. doi: 10.1080/19420862.2015.1008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renaud-Gabardos E., Hantelys F., Morfoisse F., Chaufour X., Garmy-Susini B., Prats A.C. Internal ribosome entry site-based vectors for combined gene therapy. World J. Exp. Med. 2015;5:11–20. doi: 10.5493/wjem.v5.i1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 48.Flynn R., Buckler J.M., Tang C., Kim F., Dichek D.A. Helper-dependent adenoviral vectors are superior in vitro to first-generation vectors for endothelial cell-targeted gene therapy. Mol. Ther. 2010;18:2121–2129. doi: 10.1038/mt.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shayakhmetov D.M., Eberly A.M., Li Z.Y., Lieber A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005;79:1053–1061. doi: 10.1128/JVI.79.2.1053-1061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rafei-Shamsabadi D., Lehr S., von Bubnoff D., Meiss F. Successful combination therapy of systemic checkpoint inhibitors and intralesional interleukin-2 in patients with metastatic melanoma with primary therapeutic resistance to checkpoint inhibitors alone. Cancer Immunol. Immunother. 2019;68:1417–1428. doi: 10.1007/s00262-019-02377-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brunetti-Pierri N., Palmer D.J., Beaudet A.L., Carey K.D., Finegold M., Ng P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum. Gene Ther. 2004;15:35–46. doi: 10.1089/10430340460732445. [DOI] [PubMed] [Google Scholar]
- 52.Dutcher J.P., Schwartzentruber D.J., Kaufman H.L., Agarwala S.S., Tarhini A.A., Lowder J.N., Atkins M.B. High dose interleukin-2 (aldesleukin)—expert consensus on best management practices-2014. J. Immunother. Cancer. 2014;2:26. doi: 10.1186/s40425-014-0026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mizuguchi H., Xu Z., Ishii-Watabe A., Uchida E., Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 54.Wold W.S., Toth K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013;13:421–433. doi: 10.2174/1566523213666131125095046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goswami R., Subramanian G., Silayeva L., Newkirk I., Doctor D., Chawla K., Chattopadhyay S., Chandra D., Chilukuri N., Betapudi V. Gene therapy leaves a vicious cycle. Front. Oncol. 2019;9:297. doi: 10.3389/fonc.2019.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith A.C., Poulin K.L., Parks R.J. DNA genome size affects the stability of the adenovirus virion. J. Virol. 2009;83:2025–2028. doi: 10.1128/JVI.01644-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parks R.J., Chen L., Anton M., Sankar U., Rudnicki M.A., Graham F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. USA. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emerman M., Temin H.M. Genes with promoters in retrovirus vectors can be independently suppressed by an epigenetic mechanism. Cell. 1984;39:449–467. [PubMed] [Google Scholar]
- 59.Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019;38:255. doi: 10.1186/s13046-019-1259-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fausther-Bovendo H., Kobinger G.P. Pre-existing immunity against Ad vectors: humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014;10:2875–2884. doi: 10.4161/hv.29594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo J., Deng Z.L., Luo X., Tang N., Song W.X., Chen J., Sharff K.A., Luu H.H., Haydon R.C., Kinzler K.W. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2007;2:1236–1247. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
- 62.D’Amico L., Menzel U., Prummer M., Müller P., Buchi M., Kashyap A., Haessler U., Yermanos A., Gébleux R., Briendl M. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer. 2019;7:16. doi: 10.1186/s40425-018-0464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang S., Astsaturov I.A., Bingham C.A., McCarthy K.M., von Mehren M., Xu W., Alpaugh R.K., Tang Y., Littlefield B.A., Hawkins L.D. Effective antibody therapy induces host-protective antitumor immunity that is augmented by TLR4 agonist treatment. Cancer Immunol. Immunother. 2012;61:49–61. doi: 10.1007/s00262-011-1090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giudicelli V., Duroux P., Ginestoux C., Folch G., Jabado-Michaloud J., Chaume D., Lefranc M.P. IMGT/LIGM-DB, the IMGT comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Res. 2006;34:D781–D784. doi: 10.1093/nar/gkj088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arduin E., Arora S., Bamert P.R., Kuiper T., Popp S., Geisse S., Grau R., Calzascia T., Zenke G., Kovarik J. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol. Immunol. 2015;63:456–463. doi: 10.1016/j.molimm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 66.Lo M., Kim H.S., Tong R.K., Bainbridge T.W., Vernes J.M., Zhang Y., Lin Y.L., Chung S., Dennis M.S., Zuchero Y.J.Y. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J. Biol. Chem. 2017;292:3900–3908. doi: 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haryadi R., Ho S., Kok Y.J., Pu H.X., Zheng L., Pereira N.A., Li B., Bi X., Goh L.T., Yang Y., Song Z. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS ONE. 2015;10:e0116878. doi: 10.1371/journal.pone.0116878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fang J., Qian J.J., Yi S., Harding T.C., Tu G.H., VanRoey M., Jooss K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005;23:584–590. doi: 10.1038/nbt1087. [DOI] [PubMed] [Google Scholar]
- 69.Simmons A.D., Moskalenko M., Creson J., Fang J., Yi S., VanRoey M.J., Allison J.P., Jooss K. Local secretion of anti-CTLA-4 enhances the therapeutic efficacy of a cancer immunotherapy with reduced evidence of systemic autoimmunity. Cancer Immunol. Immunother. 2008;57:1263–1270. doi: 10.1007/s00262-008-0451-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang J., Yi S., Simmons A., Tu G.H., Nguyen M., Harding T.C., VanRoey M., Jooss K. An antibody delivery system for regulated expression of therapeutic levels of monoclonal antibodies in vivo. Mol. Ther. 2007;15:1153–1159. doi: 10.1038/sj.mt.6300142. [DOI] [PubMed] [Google Scholar]
- 71.Brücher D., Franc V., Smith S.N., Heck A.J.R., Plückthun A. Malignant tissues produce divergent antibody glycosylation of relevance for cancer gene therapy effectiveness. MAbs. 2020;12:1792084. doi: 10.1080/19420862.2020.1792084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ehrke-Schulz E., Zhang W., Schiwon M., Bergmann T., Solanki M., Liu J., Boehme P., Leitner T., Ehrhardt A. Cloning and large-scale production of high-capacity adenoviral vectors based on the human adenovirus type 5. J. Vis. Exp. 2016;107:e52894. doi: 10.3791/52894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.