

Abstract
Genetic therapies based on gene addition have witnessed a variety of clinical successes and the first therapeutic products have been approved for clinical use. Moreover, innovative gene editing techniques are starting to offer new opportunities in which the mutations that underlie genetic diseases can be directly corrected in afflicted somatic cells. The toolboxes underpinning these DNA modifying technologies are expanding with great pace. Concerning the ongoing efforts for their implementation, viral vector‐based gene delivery systems have acquired center‐stage, providing new hopes for patients with inherited and acquired disorders. Specifically, the application of genetic therapies using viral vectors for the treatment of inborn metabolic disorders is growing and clinical applications are starting to appear. While the field has matured from the technology perspective and has yielded efficacious products, it is the perception of many stakeholders that from the regulatory side further developments are urgently needed. In this review, we summarize the features of state‐of‐the‐art viral vector systems and the corresponding gene‐centered therapies they seek to deliver. Moreover, a brief summary is also given on emerging gene editing approaches built on CRISPR‐Cas9 nucleases and, more recently, nickases, including base editors and prime editors. Finally, we will point at some regulatory aspects that may deserve further attention for translating these technological developments into actual advanced therapy medicinal products (ATMPs).
Keywords: advanced therapy medicinal products, gene editing, genetic disease, genetic therapy, programmable nucleases, viral vectors
1. RECOMBINANT RETROVIRUSES FOR THE STABLE GENETIC MODIFICATION OF HUMAN CELLS
Research findings on virology and genetics provided biomedical scientists with an extensive toolbox that has permitted to identify and characterize genes and gene mutations that are causative of specific diseases. In the early 1970s, it was realized that it might be feasible to develop a dedicated gene transfer toolbox for ferrying functional genes into patient cells to remedy the consequences of genetic diseases. 1 This was and so remains the main concept of gene therapy. The identification of genes linked to human disorders and their smaller coding sequences started to become available in rapid succession from the 1980s onwards owing to steep developments in recombinant DNA techniques. 2 In parallel, these developments also contributed decisively to the making of efficient gene transfer vehicles that were primarily generated for stable genetic modification of human cells. Initially these vectors were mostly derived from retroviruses. 3 Simple and complex retroviruses, such as the gamma‐retrovirus named Moloney murine leukemia virus (M‐MLV) and the lentivirus named human immunodeficiency virus type 1 (HIV‐1), respectively, are particularly attractive for gene therapy in that, once engineered as vectors, they stably integrate their transgenic cargo into host cell chromosomes and, in doing so, provide a basis for long‐term therapeutic gene expression (Figure 1). During the process of generating a retroviral vector, all viral protein‐coding sequences are removed from the retrovirus genome and replaced by the coding sequence of a gene‐of‐interest (transgene). In addition to the transgene, the retroviral vector genome contains viral non‐coding sequences (cis‐acting elements) necessary for its replication and encapsidation into newly formed enveloped vector particles (Figure 2). These elements are the viral long terminal repeats (LTRs) and packaging signal (Ψ), respectively. Functional gamma‐retroviral vectors are made using dedicated packaging cell lines (Figure 2). These so‐called helper packaging cell lines provide in trans the products encoded by the genes that were deleted from the parental virus. The structural and some catalytic retroviral proteins assemble, together with viral vector genome transcripts (two RNA genomic copies per particle), to form fully mature vector particles that carry the transgene sequence and, importantly, no viral genes. Such viral vector particles are therefore capable of only a single round of infection (transduction) once target cells are exposed to them. Once inside the cell, the retroviral vector genome transcripts are reverse transcribed into double‐stranded complementary DNA (cDNA) copies that ultimately integrate into target‐cell chromosomes. The reverse transcription and chromosomal integration steps leading to the stable genetic modification of transduced cells are catalyzed by the vector particle‐associated reverse transcriptase and integrase proteins, respectively (Figure 1). Retroviral vectors (both simple and complex) are, therefore, particularly suitable for modifying stem cells since the integrated viral vector genome is not only stably maintained in the stem cells, but can also be passed on to all their daughter cells.
FIGURE 1.
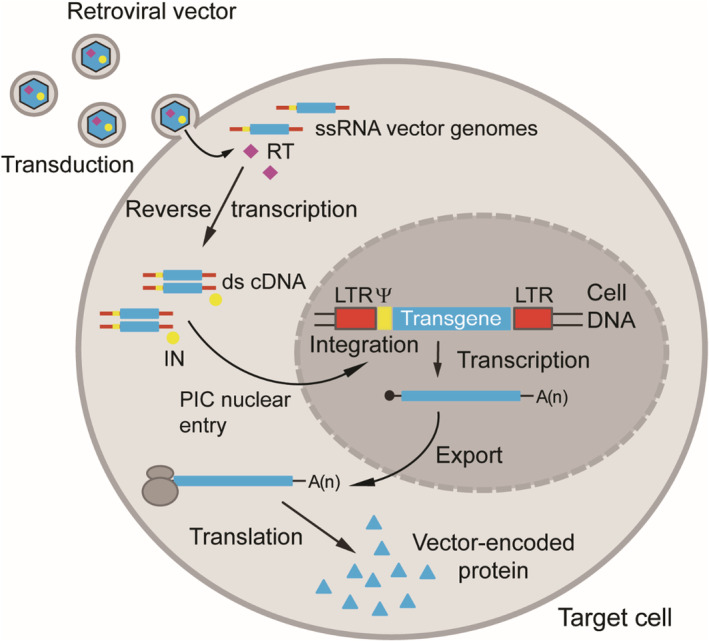
Schematics of the retroviral vector transduction process. The transgene sequence contained in the single‐stranded RNA (ssRNA) vector genome (two vector genome copies per vector particle) enters the cytoplasm of the target cell (transduction) after binding to specific cell surface receptors. This binding triggers fusion between the bilipid membranes of the vector envelope and the cell plasmalemma leading to the release of the ssRNA genomes in the cytoplasm of the target cell. Next, vector particle‐associated reverse transcriptase (RT) converts the incoming ssRNA vector genomes into double‐stranded complementary DNA (cDNA) copies via intermediate RNA–DNA heteroduplexes (not shown). The vector particle‐associated integrase (IN) together with cellular factors and the reverse transcribed vector genomes form a pre‐integration complex (PIC) responsible for establishing stable transduction of the target cell via the integration of the cDNA into the chromosomal DNA. Simple retroviruses, for example, the oncoretrovirus M‐MLV, have exclusively the basic retroviral get set (ie, gag, pro, pol, and env); Complex retroviruses, for example, the lentivirus HIV‐1, in addition to the basic retroviral gene set have genes encoding accessory proteins that play an auxiliary role at different stages of the infection process. Vectors based on simple retroviruses require nuclear envelope breakdown to access the cellular DNA; whereas vectors based on complex retroviruses can translocate their PICs through the pores of intact nuclear membranes. Thus, lentiviral vectors have the distinct advantage of being able to transduce non‐dividing cells. Transcription, messenger RNA export and translation yields stable and heritable vector‐encoded protein synthesis once vector cDNA integration takes place in a genomic region conducive for gene expression. IN, integrase; Red bars, retroviral long‐terminal repeat (LTR); Yellow bars, retroviral packaging signal (Ψ). Figure adapted from 2 Bioessays review
FIGURE 2.
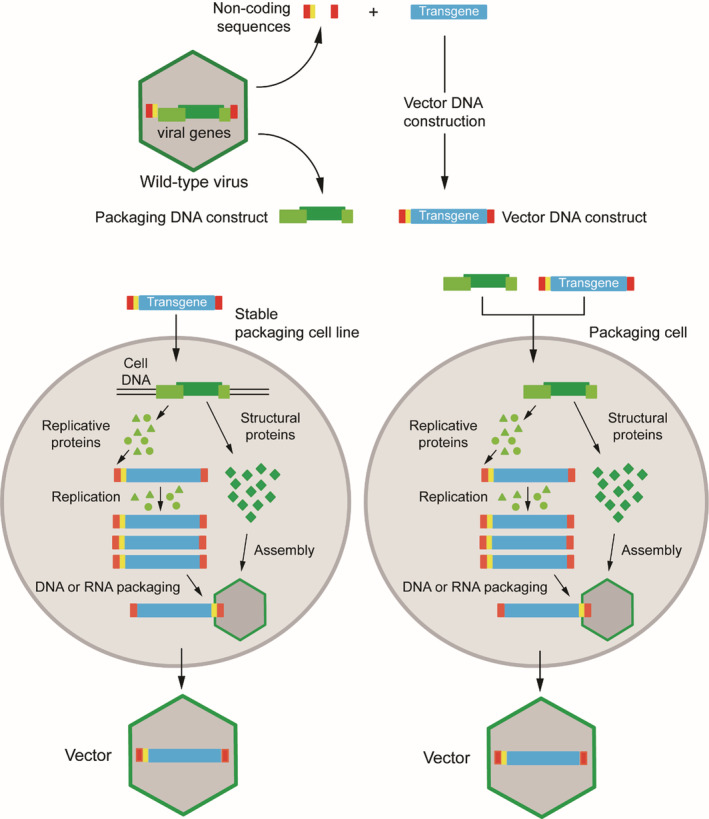
General strategy to engineer a replication‐defective vector from a replication‐competent virus. Coding trans‐acting viral sequences (green) are segregated from non‐coding cis‐acting elements containing the viral origins of replication (red) and viral genome packaging signals (yellow). The coding sequences (viral genes) specify the viral structural and non‐structural proteins (dark and light green, respectively). Viral gene removal, besides disabling the replication capacity of the vector in target cells, also creates room for a transgene expression unit (cyan) consisting of heterologous regulatory sequences (ie, promoter/enhancer elements) and a polyadenylation signal flanking an open reading frame of interest (not shown). Vector particles carrying the transgene cassette are assembled by introducing packaging and vector recombinant DNA constructs into produced cells (packaging cells). The viral gene products encoded by the packaging construct can be provided in specialized stable producer cell lines or can be introduced into vector‐producer cells by transient DNA transfection or helper vector transduction. The replicative, non‐structural, proteins (green triangles and green circles) recognize the cognate viral origins of replication resulting in the amplification of the vector genome (RNA or DNA depending on the type of vector); the structural proteins (green squares) assemble into viral particles. Finally, among the complex mixture of nuclei acids in the producer cell, the presence of packaging cis‐acting elements in the amplified vector genomes selects them for encapsidation resulting in the generation of functional vector particles. Crucially, the absence of packaging elements in helper/packaging constructs avoids their incorporation into viral particles. The vector particles are replication‐incompetent and hence can only undergo the initial cell entry stages of the parental virus “life cycle”. The resulting abortive infection (transduction) permits transgene delivery and expression in target cell nuclei without subsequent vector replication and spread to neighboring cells. Figure adapted from 2 Bioessays review
The first clinical targets of gamma‐retroviral vectors were the primary immune deficiencies (PIDs) ADA‐SCID and SCID‐X1, caused by mutations in the adenosine deaminase gene (ADA) and the common gamma‐chain receptor gene (IL2RG), respectively. While initially T cells were genetically modified ex vivo for the treatment of ADA‐SCID, 4 subsequent studies focused instead on the ex vivo modification of CD34+ cell populations, in which a fraction consists of bona fide hematopoietic stem cells (HSCs). The first evidence of clinical efficacy of a gene therapy protocol was obtained by gamma‐retroviral vector transduction of CD34+ cells isolated from SCID‐X1 patients. This initial proof‐of‐concept for the whole field was published in 2000, and demonstrated the rescue of the disease phenotype in the first three SCID‐X1 patients, for a period of up to 30 months. 5 The SCID‐X1 patients enrolled in this study had no allogeneic HLA‐matched bone marrow available for transplantation. The treated patients recovered from the disease symptoms and had an excellent long‐term reconstitution of their immune functions. 6
In these initial studies, the therapeutic gene was inserted into a gamma‐retroviral vector and its expression was driven by the strong retroviral enhancer and promotor that is embedded in the LTRs (Figure 3). In subsequent gene therapy trials, it was this viral enhancer/promoter configuration that led to the occurrence of serious adverse events (SAEs) in 5 of 19 SCID‐X1 patients that had been treated. 7 , 8 , 9 In some cells, the therapeutic IL2RG transgene, carried by the gamma‐retroviral vector, had integrated in the vicinity of the proto‐oncogene LMO2, leading to its transcriptional activation. This insertional oncogenesis event, presumably together with mutations unrelated to the gene therapy, such as the deletion of the tumor suppressor gene CDKN2A, led to the expansion of T cell clones and eventually full‐fledged T cell leukemias. 9 In all but one of the patients that developed leukemia, chemotherapy led to disease remission and polyclonal restoration of transduced T cell populations. It is also noteworthy to mention that the induction of leukemias was not only seen in 5 of 19 SCID‐X1 patients, but also in 7 of 9 patients suffering from another PID named Wiskott‐Aldrich syndrome (WAS). 10 Moreover, in clinical trials using gamma‐retroviral vectors for treating the PID X‐linked chronic granulomatous disease (X‐CGD), 4 patients treated went on to develop SAEs that included myelodysplasia syndrome. 11 , 12 In contrast, none of the 42 ADA‐SCID patients receiving gamma‐retroviral gene therapy experienced leukemogenesis. 7 A recently described case of plasmablastic lymphoma in an ADA‐SCID patient that was treated with a gamma‐retroviral vector encoding ADA combined with enzyme‐replacement therapy, appears to have been associated with Epstein‐Barr virus infection, hence unrelated to the infusion of genetically modified T cells. 13 Taken together, these data suggest that in SCID‐X1, WAS, and X‐CGD, but not in ADA‐SCID, the transgene product delivered by gamma‐retroviral vectors, contributed to the cell transformation events.
FIGURE 3.
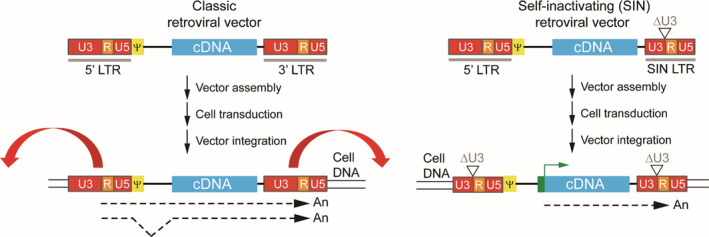
Schematic outline of classic vs SIN retroviral vectors. Transcripts of classic retroviral vectors start at the U3/R border in the 5′ LTR and end at the R/U5 border in the 3′ LTR. The transcripts corresponding to full‐length vector genomes are packaged into enveloped retroviral particles and are collected to transduce target cells. Classic retroviral vectors rely on the enhancer and promoter sequences embedded in the U3 region of the viral 5′ LTR for expression of the transgene. The enhancers embedded in the U3 regions of the LTRs can affect the expression of neighboring genes once retroviral vector genomes become integrated in target cell chromosomes. The outward red arrows indicate the enhancer activity that may affect the expression of flanking cellular genes. In self‐inactivating (SIN) retroviral vectors a deletion in the U3 region of the 3′LTR (ΔU3) removes the retroviral enhancer sequences inhibiting expression of flanking genes upon SIN retroviral vector chromosomal integration. An internal heterologous promotor is necessary to drive transgene expression in these SIN vectors (green box with broken arrow). In transduced cells, during reverse transcription of vector RNA genomes into double‐stranded cDNA, the U3 sequence in the 3′ LTR serves as template for the synthesis of the U3 region in both LTRs. As a consequence, in classic and SIN retroviral vectors the complete and partially deleted U3 regions of the 3′ LTRs, respectively, are duplicated and transferred to the 5′ LTRs. LTR, long terminal repeat; Ψ, packaging signal; cDNA
As aforementioned, the induction of leukemias was attributed to insertional oncogenesis resulting from the upregulation of nearby cell division‐promoting genes due to the enhancer/promoter activity of the LTRs (Figure 3). These SAEs are facilitated by the propensity of retroviral vectors derived from gamma‐retroviruses to integrate with a high preference into the regulatory regions of active genes, in particular into the transcription start sites of these genes. 14 , 15 To reduce the probability for cellular gene activation by viral promotor/enhancer insertions, so‐called self‐inactivating (SIN) retroviral vectors, were developed. 16 In SIN retroviral vector constructs enhancer sequences that are embedded in the LTR are deleted, essentially inactivating the LTR enhancer/promotor elements in the transduced cells which, in turn, minimizes vector genotoxicities (Figure 3). In SIN retroviral vectors, a heterologous internal promotor, normally derived from a cellular gene, is used to drive expression of the transgene. This genetic reconfiguration of retroviral vectors greatly reduces transcriptional activation of proto‐oncogenes limiting the risk for the development of SAEs, such as, clonal expansion and lymphoproliferative syndromes. 17 Indeed, application of SIN gamma‐retroviral vectors in SCID‐X1 patients demonstrated their improved safety profile, when compared to that of their classic counterparts, in terms of a significant reduction in the occurrence of SAEs. 18
Another important development in retroviral‐based clinical gene therapy was the introduction of gene transfer vectors derived from lentiviruses, of which HIV‐1‐based vectors became the most widely used. 19 Rather than a preference for integration of their cDNA genomes into the regulatory regions and transcription start sites as observed for gamma‐retroviruses, 15 lentiviruses have a preference for integration into the coding regions of transcriptionally active gene. 20 Besides the different chromosomal integration patterns, complex retroviruses (eg, HIV‐1) differ from simple retroviruses (eg, M‐MLV) in that they can infect non‐dividing cells owing to their capacity to access the host cell genome without mitosis‐dependent breakdown of the nuclear envelope. 19 This feature permits the efficient transduction of, for example, quiescent bona fide HSCs by HIV‐1‐based vectors which, in turn, has greatly contributed to strengthen the clinical gene therapy field. 21 Moreover, with the HIV‐1‐based SIN vectors, the transcriptional activation of genes near the integration site, and the resulting risks for cellular transformation, are greatly reduced as shown in cellular assays and in mice susceptible to tumorigenesis. 22 With this new HIV‐1 SIN vector design, improved safety and sustained clinical efficacy could be achieved in patients suffering from the PIDs SCID‐X1 18 , 23 , 24 , 25 and WAS. 26
To date, SIN lentiviral vectors are the state‐of‐the‐art for gene therapy involving stable genetic modification of HSCs for the treatment of blood and metabolic disorders. Indeed, in addition to the PIDs SCID‐X1 and WAS, the hemoglobinopathy β‐thalassemia, 27 and the demyelinating metabolic disorders X‐linked adrenoleukodystrophy (ALD), 28 and metachromatic leukodystrophy (MLD), 29 are being addressed by transducing HSCs with SIN HIV‐1 lentiviral vectors. In cases such as those of the metabolic disorders ALD and MLD, in which the hematopoietic compartment is not the primary affected tissue, HSC genetic modification is used as a treatment route where, upon homing and differentiation, HSC progeny locally secrete therapeutic gene products at affected tissues, such as those in the peripheral and central nervous systems. 30 Related with this approach, based on ectopic expression of secreted transgene products, preclinical studies suggest that overexpression of factor VIII in platelets might be used to treat hemophilia A in patients which developed blood clotting factor VIII‐blocking inhibitory antibodies. 31 , 32 Similarly, preclinical data indicate that genetic modification of HSCs can be effective in moderating the respiratory and cardiac defects resulting from Pompe, a lysosomal glycogen storage disorder. 33
To date, several ATMPs consisting of cells genetically modified by either gamma‐retroviral or lentiviral vectors have been registered (Table 1). 34 These ATMPs include Strimvelis, autologous CD34+ cells transduced with a gamma‐retroviral vector containing the ADA gene (Table 1) and Zalmoxis, T cells transduced with a gamma‐retroviral vector expressing herpes simplex virus thymidine kinase (HSV‐tk) and, for cell selection purposes, a truncated form of the low‐affinity nerve growth factor receptor (Table 1). 6 , 35 Besides its direct therapeutic application in Zalmoxis, the HSV‐tk protein mediates the conversion of a pro‐drug (eg, ganciclovir) to a cytotoxic product providing for a safety measure (suicide switch) in case graft eradication in blood‐cell cancer patients, transplanted with haploidentical HSCs, is necessary due to GVHD. Furthermore, CD34+ cells transduced with a lentiviral vector encoding a modified β‐globin chain (βA‐T87Q), form an ATMP marketed as Zynteglo used for the treatment of transfusion‐dependent β‐thalassemia patients (Table 1). 27 , 36 Yescarta and Kymriah, on the other hand, are chimeric antigen receptor (CAR) T cells transduced with lentiviral and gamma‐retroviral vectors, respectively, expressing chimeric T cell receptors directed to CD19 on B cell malignancies (Table 1). 37 , 38
TABLE 1.
Registered viral vector‐based gene therapy products
| Product name | Target disease | Parental virus | Transgene | Replication status | Reference |
|---|---|---|---|---|---|
| Rexin‐G | Solid tumors | Gamma‐retrovirus | Cyclin G1 | Replication defective | 167 |
| Strimvelis | ADA‐SCID | Gamma‐retrovirus | ADA | Replication defective | 6 |
| Zalmoxis | Leukemia | Gamma‐retrovirus | HSV‐thymidine kinase | Replication defective | 35 |
| Yescarta | Diffuse large B‐cell lymphoma | Gamma‐retrovirus | Chimeric T cell receptor | Replication defective | 38 |
| Invossa a | Osteoarthritis | Gamma‐retrovirus | TGF‐β1 | Replication defective | 168 |
| Kymriah | B‐cell acute lymphoblastic leukemia | Lentivirus | Chimeric T‐cell receptor | Replication defective | 37 |
| Zynteglo | Transfusion‐dependent beta‐thalassemia | Lentivirus | β‐globin | Replication defective | 27 |
| Gendicine | Head and neck squamous cell carcinoma | Adenovirus 5 | p53 | Replication defective | 59 |
| Oncorine (H101) | Nasopharyngeal carcinoma | Adenovirus 5 | None | Replication competent | 58 |
| Imlygic | Unresectable melanoma | Herpes simplex virus | GM‐CSF | Replication competent | 169 |
| Glybera b | Lipoprotein lipase deficiency | AAV1 | Lipoprotein lipase | Replication defective | 91 |
| Luxturna | Leber's congenital amaurosis | AAV2 | RPE65 | Replication defective | 104 |
| Zolgensma | Spinal muscular atrophy type I | AAV9 | SMN1 | Replication defective | 105 |
Permission revoked May 2019.
Product discontinued in 2017.
In common, these ATMPs consist of hematopoietic cells genetically modified by retroviral vectors ex vivo. However, for many inherited and acquired disorders (eg, certain cancers and infectious diseases), in vivo gene therapy strategies are more appropriate and, as a result, require alternative viral vector systems that, for instance, can transduce post‐mitotic cells without integrating into the target cell genome.
2. ADENOVIRAL VECTORS (ADVS) FOR in VIVO GENE TRANSFER
Adenoviruses are DNA viruses (Adenoviridae family) that are normally associated with mild pathologies in humans. 39 These viruses possess a linear double‐stranded DNA genome packaged in a non‐enveloped icosahedral capsid. After infection of a host cell, the adenovirus genome initiates an orchestrated program of expression of the viral genes that are generically divided in early (E), intermediated and late (L) sets. The early and late gene sets are roughly expressed before and after the onset of viral DNA replication, respectively. 40 The E1A gene is the first to be expressed with the resulting E1A proteins being responsible for regulating the expression of a number of viral and cellular genes that prepares the host cell for the preferential translation of viral transcripts and for the replication of the adenoviral genome. 41 , 42 Deletion of the early region 1 (E1), encompassing the E1A and E1B open reading frames, effectively cripples the virus rendering it replication‐incompetent except in helper packaging cell lines in which the E1‐encoded proteins complement in trans the E1‐deleted vector genomes. Importantly, the deletion of the E1A and E1B genes, besides making the vector replication‐incompetent in target cells, creates space for the insertion of transgenes. With these E1‐deleted AdVs, high‐level transgene expression can be obtained in murine and larger animal models; however, transgene expression commonly disappears within 2 to 3 weeks after gene transfer in immunocompetent animals. This has been attributed to the eradication of vector‐transduced cells by the interplay between the host's innate and adaptive immune systems in which CD8+ T cells take on a preponderant role. 39 Indeed, T cells found after AdV administration in vivo, recognize antigen peptides derived from both transgene and viral proteins. The latter are the result of residual (“leaky”) expression of the viral genes retained in the vector genome. 43 , 44
The residual synthesis of immunogenic adenoviral proteins has been remedied by the development of so‐called helper‐dependent AdVs (HD‐AdVs) (a.k.a. high‐capacity or “gutless” AdVs). 39 , 45 All adenoviral protein‐coding genes are removed from these vectors, with most of these proteins being provided in trans in specialized E1‐complementing packaging cell lines transduced with a helper E1‐deleted AdV (Figure 2). For the most part, the replicating helper genomes cannot be packaged owing to the selective removal of their packaging signals by site‐specific recombinases (eg, Cre or FLP). The HD‐AdVs retain from the parental adenovirus genome exclusively the packaging signal and the 103‐bp inverted terminal repeats (ITRs), in which the viral origins of replication are embedded. The removal of the entire protein‐coding regions endows HD‐AdVs with a vast transgene packaging capacity (ie, up to 36 kb). Moreover, with these HD‐AdVs, much longer transgene expression can be achieved in vivo when compared to that observed with E1‐deleted AdVs encoding the same transgene(s), (see, eg, References 46, 47). This is especially so in slowly‐dividing tissues, such as liver, or post‐mitotic tissues, such as brain and muscle. 48 , 49 , 50
Certain human adenovirus serotypes circulate commonly in the human population. In fact, approximately 65% to 80% of humans have detectable amounts of neutralizing antibodies against the prevalent human adenoviral serotypes 2 and 5. 51 These antibodies may affect the efficiency of gene delivery in vivo and have prompted the development of AdVs derived from more rare human adenoviral serotypes. 52 In humans, 103 serotypes have been identified so far on the basis of neutralizing assays or molecular analyses. These cluster in seven adenoviral subgroups or species (A through G). Normally, different adenoviruses use specific cell surface proteins or other molecules as primary cell entry receptors. 52 To date, human adenoviral receptors identified include CAR, CD46, DSG2, Scavenger receptor AII, polysialic acids, GD1, heparin sulfate proteoglycans, and integrins. 53 The natural diversity of adenoviral serotypes and their respective variegated receptor usage, allows for the generation of AdVs with a wide or narrow cellular tropism depending on the distribution of specific cell surface receptors on different target cell types and tissues.
Although AdVs were used initially in gene therapy for inherited diseases, currently they are predominantly used in indications where short‐term transgene expression is desired, such as, in cancer gene therapy, 54 and vaccination protocols. 55 Two AdV products have been registered for use in cancer treatment (Table 1) and various clinical trials are ongoing directed at tackling infectious agents (eg, HIV‐1 and Ebola) 56 , 57 . The anti‐cancer AdV products are Gendicine, a replication‐incompetent AdV type 5 vector carrying the TP53 gene coding for the tumor suppressor protein p53; and Oncorine, a replication‐competent oncolytic AdV lacking E1B‐55KD, intended for use in combination with chemotherapy. 58 , 59 Both of these ATMPs have been registered in China.
In what the treatment of monogenetic disorders is concerned, it is worth mentioning that an early clinical trial for the treatment of ornithine transcarbamylase deficiency (OTCD) resulted in the death of one among 18 patients receiving a replication‐incompetent AdV type 5 vector expressing the OTC transgene. An acute innate immune response against the vector was advanced as the culprit for the death of this patient, Jesse Gelsinger. 60 It was suggested that this severe immune response might have been caused by an immune memory against type 5 AdV, an immune response overactivation as a result of a prior viral infection, or a genetic predisposition related to, for instance, enhanced innate immunity. 61 , 62 This was an unexpected event since the pre‐clinical data did not show similar toxicities and another individual receiving a similar dose developed only mild adverse events. 61 The “Jesse Gelsinger case” took place in 1999 and has since raised safety concerns regarding liver‐directed application of AdVs for the treatment of genetic disorders. 63 Despite this case, in which in vivo application of AdVs showed potential safety issues, subsequent research in small and large animal models has demonstrated that, by substituting E1‐deleted AdVs for HD‐AdVs, long term successful phenotypic correction of monogenetic liver disorders is possible, including OTCD, 64 glycogen storage disease 1A, 65 Criggler‐Najjar syndrome, 66 and phenylketonuria, 67 and others. 68
3. ADENO‐ASSOCIATED VIRAL (AAV) VECTORS FOR in VIVO GENE TRANSFER
AAV vectors are attracting much attention as the parental virus is not associated with known pathologies in humans and, importantly, they can achieve prolonged transgene expression in small and large animal models in vivo following a single vector administration. 69 AAVs are small parvoviruses (circa 22 nm) of the dependoparvovirus genus which contain single‐stranded DNA genomes harboring Rep and Cap genes encoding sets of replicative and structural proteins, respectively. 2 , 70 The genus name reflects the observation that AAVs depend on other DNA viruses, so‐called helper viruses, such as herpes simplex viruses or adenoviruses to express gene products necessary for productive AAV replication. 71 A high proportion of the human population has been exposed to AAVs as evident by the relatively high prevalence of neutralizing immunity. Approximately 30% to 60% of individuals are seropositive with some variation observed depending on geographical origin and AAV serotype. 72 , 73 The prototypic AAV serotype 2 has a genome of only 4.9 kb. During the making of AAV vectors, the AAV Rep and Cap genes are replaced by transgene sequences (Figure 2). These transgene sequences are flanked by T‐shaped palindromic ITRs that are the non‐coding, cis‐acting, elements required for the replication and packaging of recombinant AAV genomes into AAV capsids. 2 , 70 Hence, in order to assemble AAV vectors, producer cells, often adenovirus E1‐expressing HEK293T cells, are co‐transfected with the ITR‐flanked transgene together with plasmids encoding the Rep and Cap gene products and helper adenoviral functions (ie, E4ORF6, E2A, VA‐I and VA‐II RNAs). Similarly to AdVs, there are various AAV serotypes that present specific cellular tropisms and on the basis of which AAV vectors can be generated. 74 Moreover, in addition to relaying on the natural AAV capsid diversity to transduce specific cell types, researchers are also exploiting new AAV capsid variants generated by site‐directed mutagenesis and directed evolution approaches. 75 Despite the small size of the AAV capsid (circa 22 nm), and of the packaged genome (4.9 kb), a wide variety of transgene cassettes have been inserted in these vectors.
AAV vectors can be produced with relative ease and concentrated to high titers after introducing into producer cells constructs containing the recombinant AAV DNA, the trans‐complementing Rep and Cap genes and the aforementioned adenovirus helper functions. These components are mostly delivered into producer cells via either transient plasmid transfections in mammalian cells or via transduction of producer insect cells with baculoviral vectors (Figure 2). 69 , 76 However, generating the amounts of functional AAV vector particles needed for in vivo administration to large animal models and patients remains a substantial hurdle in the AAV‐based gene therapy field. 69 , 76
Upon in vivo transduction of non‐dividing or slowly dividing tissues most AAV vector genomes persist long‐term in an episomal (ie, chromosomally non‐integrated) form that can evolve into linear and circular multimers. 77 , 78 A fraction of these AAV vector genomes can also integrate into the genome of target cells in vitro and in vivo at chromosomal double‐stranded DNA breaks (DSBs), that occur due to DNA damage or DNA metabolic processes. 79 , 80 , 81 , 82 , 83 Moreover, AAV vector genomes can equally integrate into site‐specific DSBs created by programmable nucleases. 84 , 85 , 86 , 87 The potential genotoxicity of AAV vectors has also been a subject of controversy due to several liver‐directed gene therapy experiments in rodents in which AAVs were implied in the induction of hepatocellular carcinoma (HCC) upon genomic integration. 88 , 89 However, the limited amount of human clinical data does not seem to reflect the rodent results in terms of the association between AAV vector transductions and tumor formation. 90 Thus, whether sporadic chromosomal insertion of recombinant AAV genomes contributes to cellular transformation remains a subject requiring further investigation in larger animal models and humans. 90 For example, reminiscent of retroviral vector systems, it has been suggested that the composition of regulatory elements in AAV vector genomes may themselves play a role in the development of genotoxic side effects. 90
One of the disorders that can be treated by AAV‐based gene therapy is familial lipoprotein lipase (LPL) deficiency. This is a genetic metabolic disorder that prevents patients from digesting triglycerides which in turn result in the accumulation of chylomicrons (lipoprotein particles) in the circulation. This accumulation can cause episodes of abdominal pain and recurrent pancreatitis. AAV vector‐mediated transfer of a gene encoding a hyperactive LPL enzyme variant (LPLS447X) reduced the number of pancreatitis events up to 6 years following gene transfer. 91 The AAV vector harboring the modified LPL gene, marketed as Glybera, was approved in Europe in 2012. However, due to a lack of demand, the product was removed from the market in 2017 (Table 1). Apart from the development of Glybera, genetic therapies for the treatment of inborn metabolic disorders, such as lysosomal storage diseases, glycogen storage diseases, urea cycle disorders, and phenylketonuria, are getting more attention in recent years and will presumably progress toward clinical translation in the relative near future. 92 , 93 , 94 , 95 , 96 , 97 Although AAV vectors are emerging as prime candidates for gene therapy of metabolic liver disorders, other viral vector systems, such as adenoviral and lentiviral vectors, are being investigated as well for this purpose. 68 , 98 However, important challenges need to be addressed, such as minimizing genotoxicity and immunogenicity of gene therapy systems, before safer clinical applications are implemented. 68 , 98
Besides metabolic disorders, AAV vectors have also been tested for the treatment of a variety of other diseases such as hemophilia A, 99 , 100 hemophilia B, 101 , 102 , 103 Leber's congenital amaurosis, 104 and spinal muscular atrophy type 1 (SMA1). 105 In hemophilia A and hemophilia B patients the levels of clotting factors VIII and IX, respectively, were consistently increased after gene therapy and resulted in a strong reduction in the number of bleeding episodes, as well as in the use of prophylactic clotting‐factor protein concentrates. These clotting factor levels were maintained for several years. 103 , 106 In a number of patients, an asymptomatic increase in serum aminotransferase levels was observed, which could be resolved by a short‐term prednisone treatment. 99 , 100 , 103 Importantly, the AAV‐based gene therapies for Leber's congenital amaurosis (Luxturna) and for SMA1 (Zolgensma) have been approved for clinical use by regulatory authorities (Table 1). 104 , 105 In the case of the latter ATMP, pre‐clinical studies in animal models revealed an unexpected capacity of AAV serotype 9 (AAV9) particles to pass through the blood‐brain barrier and to transduce motor neurons in the spinal cord. 107 Subsequent clinical evaluation in 15 patients demonstrated that a single intravenous infusion of an AAV9 vector encoding SMN1 resulted in longer infant survival and improved motor function compared to historical cohorts. 105 As in the aforementioned hemophilia gene therapy clinical trials, some SMA1 patients required corticosteroid treatment to suppress the transient elevation of serum aminotransferase levels. Next to these significant clinical outcomes in patients, expanding clinical application of AAV vectors to an increasing number of conditions affecting several organs (eg, eye, muscle, brain and liver) is expected to provide further valuable insights on the challenges posed by immune responses against AAV vector components and/or the transgene product itself. 108 Finally, it is noteworthy mentioning that viral vectors currently being applied for treating human disorders via transgene delivery, have also started to be investigated as delivery agents for DNA editing tools. 109
4. THE NEW KID ON THE BLOCK: DNA EDITING
With the appearance of precise DNA editing tools, that is, sequence‐specific programmable nucleases, the prospect for modifying specific nucleotide sequences among the 6.4 billion base pairs that make up a diploid human genome has become a reality. 110 Indeed, by designing and constructing programmable nucleases, one can now modify specific genes of interest in order to either study their function or correct genetic defects. DNA editing, or gene editing, directed at treating human disorders is currently in early experimental stages; however, an increasing number of clinical studies are starting to build‐up using these technologies. 111 At the moment, the primary fields of application are in oncology (eg, CAR‐T cell therapies) followed by infectious and monogenetic disorders. In this section, we will briefly touch upon the different gene editing platforms in a chronological fashion and provide an overview of the translation of these techniques into the clinical arena.
Before diving into the different gene editing techniques as such it is important to understand the mechanisms through which the main gene editing tools operate. Typically, gene editing relies on the formation of targeted chromosomal DSBs by programmable nucleases. The subsequent activation of endogenous cellular DNA repair pathways involved in fixing the DNA damage is exploited to bring about specific DNA editing outcomes (eg, gene knockouts or gene knock‐ins). 112 To explain briefly, after the induction of a DSB the cell will respond by recruiting specific DNA repair machineries. There is a variety of mechanisms through which the cell can repair the DSB. 113 The principal DSB repair mechanisms in mammalian cells are homology‐directed repair (HDR) and non‐homologous end joining (NHEJ) pathways. The classic NHEJ (cNHEJ) pathway is the most prevalent and active of the DSB repair mechanisms and normally results in the direct ligation of chromosomal termini. 113 DSB repair by NHEJ pathways can lead to chromosomal ligation products containing small insertion and deletions (indels). These indels can emerge due to end‐processing by exonucleases prior to end‐to‐end ligation 113 or, in case of DSBs made by a programmable nuclease, due to cycles of re‐cleavage until an indel disrupts the target site and becomes installed in the target cell population. If occurring within a gene coding sequence, the latter indels, varying in size and nucleotide composition, can lead to frame‐shifts that effectively result in target gene knock‐out. 110 When relying on HDR, a more precise edit can be made after the introduction of an exogenous DNA donor template. Normally, upon chromosomal DSB formation, sister chromatid sequences, available during the S and late G2 phases of the cell cycle, are recruited to serve as DSB repair templates, resulting in the reconstitution of the original sequence. In a gene editing setup, an exogenous donor DNA template contains the intended edit and, by taking over the role of the sister chromatid, allows for the precise insertion of the foreign genetic information into the genomic region initially subjected to the site‐specific chromosomal DSB. 110 , 112 This being said, there are presently different gene editing approaches emerging that are less strictly dependent on the induction of a DSB. These approaches and attendant tools will be discussed later on.
5. ZINC‐FINGER NUCLEASES (ZFNS)
The development of ZFNs started the era of targeted genome modifications. 110 , 114 The classical zinc‐finger (ZF) motif Cys2His2 and its nucleic acid binding properties were discovered in 1985. 115 , 116 This ZF is a short structural protein motif whose coding sequence is found in approximately 3% of the human genes. The nucleotide sequences recognized by ZF motifs are, normally, three base‐pairs (bps) in length (triplets). When multiple ZF motifs are rationally designed and connected to each other to form an array, they can be used for the recognition of specific larger genomic sequences. Most commonly, 3 to 4 ZF motifs are assembled for binding to 9‐bp and 12‐bp nucleotide sequences, respectively. 110 , 117 As described earlier, in order to establish a genetic modification at a predefined genomic sequence, the induction of a site‐specific DSB is required to trigger the aforementioned endogenous DNA repair mechanisms (ie, NHEJ and HDR). To this end, ZF arrays are fused to the catalytic domain of the restriction enzyme FokI. The FokI nuclease domain requires dimerization in order to induce a DSB. Therefore, ZFNs must work as dimers in which one ZFN monomer binds to a target site separated by a short sequence (spacer) from the target site of another ZFN monomer. The local binding of ZFN pairs to their respective target sites brings the bound FokI nuclease domains in close proximity leading to their catalytic activation and, ultimately, to the induction of a site‐specific DSB. 110 , 117 Currently, ZFNs are being investigated for the treatment of inherited and acquired disorders. 111 In fact, the first utilization of gene editing tools in a clinical setting involved the ex vivo adenoviral vector transduction of a ZFN pair designed to knockout the HIV‐1 co‐receptor gene CCR5 in CD4+ T cells isolated from AIDS patients. 118
6. TRANSCRIPTION ACTIVATOR‐LIKE EFFECTOR NUCLEASES (TALENS)
Transcription activator‐like effector (TALE) proteins found in certain phytopathogenic bacteria, for example, Xanthomonas sp., 119 provide a customizable DNA‐binding scaffold for a class of programmable nucleases named TALE nucleases (TALENs). 110 , 120 , 121 TALENs, are more easy‐to‐design than ZFNs in that there is a one‐to‐one direct relationship between the binding of each of their DNA‐binding units, called TALE repeats, and specific nucleotides. Typically, each TALE repeat consists of 33 to 35 conserved amino acids with polymorphic residues, named repeat variable di‐residues (RVDs), located at positions 12 and 13 of the repeat. The TALE repeat‐to‐nucleotide binding code is governed by these polymorphic residues embedded within each repeat. 122 , 123 For instance, RVDs NI, NG, and HD recognize preferentially A, T, and C, respectively.
From the above it follows that by forming an array of linked TALE repeats (normally 17.5), one can construct a DNA‐binding domain that, upon linkage to the FokI nuclease domain, yields a TALEN monomer that targets specific sequences in the genomic DNA. Similarly, to ZFNs, induction of a site‐specific DSB can take place through two TALEN monomers that bind to DNA in close proximity and, in doing so, assemble a catalytically active FokI nuclease domain dimer. The discovery of the TALE code and the ensuing development of TALENs were revolutionary developments as they unlocked the possibility to alter the genome with high specificity and higher flexibility than that offered by the ZFN platform. 124 Although the designing and validation of ZFNs and, to a lesser extent, TALENs are time consuming processes, they can, once optimized, display high specificities. Programmable nuclease reagents displaying high specificities induce no or low frequencies of DSBs at unintended sequences located elsewhere in the genome (off‐target sites). The ability to generate ZFNs and TALENs with high specificities results, in large part, from the need for coordinated binding of two monomers which have to be in the correct orientation and properly spaced from each other. 110 , 117 , 124 Clearly, highly specific programmable nucleases are paramount for clinical translation. In this regard, TALENs have also entered clinical testing in the form of reagents for editing CAR‐T cells for treating an infant leukemia. 125
7. CRISPR‐CAS9 NUCLEASES
Following the appearance of ZFNs and TALENs by the mid‐1990s 110 , 114 and late 2000s, 120 respectively, engineered CRISPR‐Cas9 nucleases emerged in 2013. 126 , 127 , 128 , 129 These RNA‐guided programmable nucleases are nowadays the most widely used gene editing platform. What sets engineered CRISPR‐Cas9 nucleases apart from their programmable nuclease predecessors, and decisively contributes to their wider dissemination, is their versatility underpinned by a protein engineering‐free mode of construction as target site specificity is ultimately governed by RNA‐DNA hybridizations as opposed to protein‐DNA interactions. 110 , 112 , 130
Engineered CRISPR‐Cas nucleases are derived from clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR‐associated protein (Cas) loci found in the genomes of prokaryotes. 130 Together these components form an adaptive immune defense mechanism against bacteriophages and foreign plasmids in these organisms 131 of which prototypic examples are the CRISPR‐Cas9 systems from Streptococcus pyogenes 132 and Streptococcus thermophilus. 133 Essentially, the adaptation of the S. pyogenes CRISPR‐Cas9 system into a programmable nuclease platform for editing the genomes of eukaryotic cells involved: (a) assembling single‐guide RNAs (sgRNAs) by fusing sequence‐tailored CRISPR RNAs (crRNAs) to an invariant trans‐activating CRISPR RNA scaffold (tracrRNA); (b) codon‐optimizing the Cas9 open reading frame; and (c) adding nuclear localization signals to the Cas9 nuclease. 132 , 134
Once introduced into target cells, sgRNAs and engineered Cas9 nucleases form binary ribonucleoprotein complexes that enter the nucleus and start screening the genome for so‐called protospacer‐adjacent motifs (PAMs). PAMs are short DNA sequences recognized by the PAM‐interacting domain of Cas9 nucleases. 135 In the case of the prototypic Cas9 protein from S. pyogenes the PAM sequence is NGG; where N can be any of the four deoxyribonucleotides. Cas proteins derived from CRISPR systems evolved in other microorganisms engage other PAMs. 136 , 137 A target site for a RNA‐guided nuclease based on the S. pyogenes CRISPR‐Cas9 system typically consists of NGG adjoined to a 20 nt‐long nucleotide sequence (protospacer) complementary to the 5′ end of the sequence‐variable crRNA portion of the sgRNA (spacer). The binding of a Cas9:sgRNA complex to the PAM of its target site triggers local melting of the DNA double helix and subsequent RNA‐DNA annealing. RNA‐DNA hybridization activates the two nuclease domains of Cas9 (ie, HNH and RuvC) ultimately leading to targeted DSB formation, normally three base pairs away from the PAM. At this point, it is noteworthy mentioning that this series of events ending with DSB formation can also take place at sequences bearing mismatches to the 5′ end of the sgRNA (see, eg, References 138, 139, 140). Hence, similarly to the earlier programmable nuclease platforms (ie, ZFNs and TALENs), the specificity of RNA‐guided CRISPR‐Cas9 nucleases is not absolute in that DNA cleavage can occur at off‐target sequences. In fact, CRISPR‐Cas9 nuclease off‐target activities can be substantial, surpassing in some instances their activity at the intended targets site (see, eg, References 140, 141). Importantly, substantial research efforts are ongoing to minimize the off‐target activities of programmable nucleases, including those based on CRISPR systems. In this regard, CRISPR systems are being adapted for developing a more diverse and potentially safer array of gene editing tools such as high‐fidelity nucleases, nickases, base editors and prime editors. 136 , 142 , 143
8. HIGH‐FIDELITY CRISPR‐CAS9 NUCLEASES
As mentioned before, the original engineered CRISPR‐Cas9 nucleases can induce substantial off‐target activities. In recent years, this platform has been subject to rigorous optimization by numerous groups through studying alterations to both the sgRNA (tracrRNA and crRNA) and Cas9 nuclease components in order to improve gene editing outcomes. 136 A growing number of high‐fidelity CRISPR‐based nucleases have been published showing remarkable improvements in reducing off‐target DNA cleaving activities. These protein variants were obtained either through structure‐guided rational design or directed evolution approaches and bear specific point mutations that confer reduced off‐target effects to the respective ribonucleoprotein complexes. Possible mechanisms for preferential cleavage at target sites over off‐target genomic positions by these Cas9 variants are their higher energy thresholds for conformational activation of the catalytic HNH domain 144 or lowering nonspecific protein‐DNA interactions. 145 , 146 Moreover, sgRNAs have been subjected to optimization such as 5′ end truncations, sequence optimization, such as gRNA duplex extension, and chemical modifications. 136 , 147 , 148 , 149 As aforementioned, besides these research efforts directed toward optimizing the precision of DSB‐based gene editing, other CRISPR‐based approaches are being developed which, instead of relying on mutagenic DSB formation, depend on less disruptive single‐stranded DNA breaks (SSBs), or nicks.
9. SEQUENCE‐ AND STRAND‐SPECIFIC NUCLEASES (NICKASES)
Nickases based on CRISPR‐Cas9 complexes are generated by disabling one of the two catalytic domains of the Cas9 nuclease (ie, HNH or RuvC) through site‐directed mutagenesis. 130 Paired chromosomal SSBs formed by two nicking CRISPR‐Cas9 complexes with gRNAs targeting sequences on opposite DNA strands located near to each other, can generate a targeted DSB. 150 , 151 Crucially, with this dual CRISPR‐Cas9 nickase strategy, off‐target activities are greatly reduced in that SSBs made elsewhere in the genome are mostly resolved through conservative DNA repair processes.
When compared to DSBs, SSBs are significantly less disruptive to the genome as these lesions are not substrates for mutagenic NHEJ pathways. Unfortunately, SSBs are poor stimuli for HDR. Recent research has, however, uncovered that coordinated nicking of target and donor DNA templates (in trans paired nicking) by CRISPR‐Cas9 nickases can increase the efficiency of nick‐induced gene editing. 141 , 152 , 153 , 154 In addition, the specificity and accuracy of HDR‐mediated knock‐in of whole transgenes via in trans paired nicking was shown to be superior to those achieved by CRISPR‐Cas9 nucleases. 141 , 152
10. BASE EDITING
A base editor is a Cas9 nickase covalently linked to an enzyme capable of inducing targeted substitutions, in particular, either the transition C → T or A → G within a narrow, so‐called, editing window of around 4‐bp located at a defined distance from the PAM. 155 , 156 The enzymes responsible for C → T and A → G transitions are cytidine and adenine deaminases, respectively. Base editors, as for regular Cas9 proteins, are addressed to specific genomic sequences via their coupling to sgRNAs. Once at the target sequence, cytidine deaminase base editors chemically convert C·G into U·G base‐pairs, after which mismatch repair mechanisms convert these U·G into U·A pairs. Finally, after DNA replication or repair, a T·A base pair change is established (Figure 4). 156 Adenine deaminase base editors operate similarly to their cytidine deaminase counterparts except that they chemically convert A·T to inosine·T base‐pairs that are subsequently processed to the final G·C base pair (Figure 4). 155
FIGURE 4.
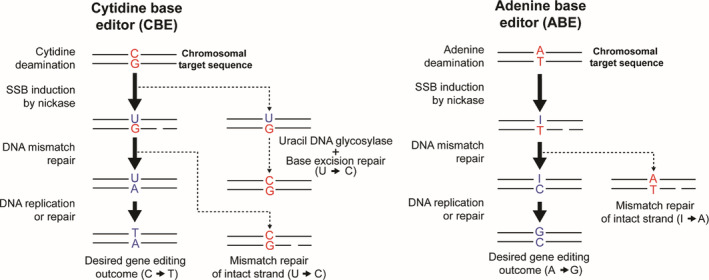
Schematic representation of the reactions induced by base editors. Upon binding of the cytidine deaminase base editor to the chromosomal target sequence, the cytidine deaminase enzyme converts the cytidine base to a uridine intermediate. Subsequently, DNA repair pathways favor the repair of the mismatched base (G) on the strand containing the SSB induced by the CRISPR‐Cas9 nickase, causing a G → A replacement on the nicked strand. Through further DNA processing, by either DNA replication or repair, the uridine is replaced by thymine, effectively resulting in the desired C → T edit. Of note, the U → C reversion, by cellular uracil DNA glycosylase (UG) and subsequent base excision repair, can abort the desired C → T transition, resulting in an unaltered chromosomal sequence. For this reason, cytidine base editors also incorporate as fusion partner an UG inhibitor (UGI). Adenine deaminase base editors operate through a similar series of DNA processing steps except that conversion of adenine to an inosine intermediate takes place at the beginning. After DNA mismatch repair and DNA replication this conversion results in the final A → G transition
Similar to the original Cas9 nucleases, base editors are also being subjected to incremental optimization, such as broadening their genomic space coverage by altering their PAM preferences and reducing their off‐target activities that can occur at the levels of the genome and transcriptome. 143 , 157 , 158 , 159
11. PRIME EDITING
Prime editing is one of the most recent developments in the gene editing field. 142 Although this system still requires further testing and optimization, when compared to base editors, it allows a broader range of small genomic edits. Importantly, similarly to base editing, primer editing does not require the catalytic induction of DSBs and the delivery of separate donor DNA templates. The prime editor 2 (PE2) system consists of a Cas9 nickase covalently linked to an engineered M‐MLV reverse transcriptase (RT) whose five mutations confer enhanced thermostability and enzyme processivity. 142 Crucially, the sgRNA is restructured into a so‐called prime editor gRNA (pegRNA). This pegRNA is a 3'‐end prolonged sgRNA which has, in addition to the conventional crRNA and tracrRNA sequences, a RT primer binding site (PBS) and a RT template sequence containing the desired edit. Upon binding of the PE:pegRNA complex to the target DNA sequence, the RT copies the information from the RT template into the target genomic locus resulting in the introduction of the desired edit. Prime editing permits installing all transitions and transversions (totally 12 base‐to‐base conversions) as well as small insertions and deletions (i.e., up to 80 bp). 142
12. WHERE DO WE GO FROM HERE?
The pace with which gene transfer and gene editing technologies are developing is impressive. These technological advances offer new treatment avenues for serious genetic diseases for which there are no therapies available. The prospects for patients with genetic diseases, such as SCID‐X1, SMA1, and Leber's congenital amaurosis affecting the immune system, nervous system and the eye, respectively, have already been dramatically improved. However, there are challenges. The pricing of these ATMPs is very high and currently at a level that limits their affordability by many governments and other stakeholders. In order to create a system in which genetic therapies become financially available to patients, rapid adaptation of reimbursement models is required in which participation of multiple stakeholders is key. 160
Besides genetic disorders afflicting vast populations (eg, hemoglobinopathies and cancer), for which commercial implementation is a priori more feasible, the emerging DNA modifying techniques are also starting to open the perspective for new therapies directed at rare and ultrarare diseases, for example, certain PIDs. The DNA modification technologies facilitate the development of genotype‐specific therapeutics. A powerful example of such an approach is the development of a personalized ex vivo gene therapy for a patient with junctional epidermolysis bullosa. A homozygous mutation in the patient's LAMB3 gene led to blistering and epidermal loss of over 60% of the total body surface area due to the absence of laminin‐332. Within 7 months after diagnosis, a gene therapy product, consisting of autologous epidermal grafts generated from keratinocytes transduced with a gamma‐retroviral vector encoding the LAMB3 protein, could be developed and grafted onto the patient. This particular ex vivo gene therapy was able to regenerate a fully functional epidermis in the patient. 161
While this is a powerful example of the flexibility and robustness that gene therapy protocols can achieve, it also imposes the question of how one can ensure the development of commercially viable personalized treatments. For example, can such genetic therapies for individual patients, or very small patient cohorts, be registered as an ATMP? Currently, the genetic therapies developed for individual patients or very small cohorts are incompatible with the conventional registration procedures that involve phased clinical studies, normally requiring increasing numbers of patients. Yet, European, US and Japanese regulatory authorities seem inclined to take on a more flexible approach to the approval of gene and cell therapy products by early indications of clinical risks/benefits assessments of disorders with unmet medical need status in combination with substantial post‐marketing risk management. 162
Generally, with new technologies also come new dilemmas. These dilemmas require continuous and transparent ethical debate in order to reach consensus between the different stakeholders. While applications of somatic gene therapy and somatic gene editing for the treatment of severe diseases have found broad acceptance, there are concerns in Society for their unbound application. For example, the use of genetic modification for enhancing an individual's traits is deemed ethically questionable by many. There are already endeavors to develop tests capable of detecting the use of gene therapy technology for “gene doping”. Testing athletes in competitive sports should impede the illicit use of gene therapy techniques for expressing transgenes such as erythropoietin, insulin‐like growth factors, or growth hormones. 163 , 164 Moreover, germline gene modifications offer the prospect for correcting faulty alleles in all cells of an individual as well as in his or her offspring and subsequent generations. However, it is generally perceived that the current state of the technology, linked to the unpredictable and far‐reaching impact of germline modifications on future generations, warrants broad discussion, oversight and regulation. 165 Highlighting the pressing need for guidelines and oversight on the application of human germline gene modification, is the recent episode involving CCR5 gene disablement in two girls with the aim of conferring them with resistance to HIV‐1 infection. 166 Preeminent issues related with the application of gene editing technologies to the human germline include, besides off‐target effects and embryo mosaicism, a considerable lack of knowledge about the possible consequences arising from altering specific genomic sequences and associated developmentally‐regulated epigenetic marks.
In this review, we provided an introduction to gene therapies based on retroviral, adenoviral and adeno‐associated viral vector technologies together with an overview of the state‐of‐the‐art gene editing technologies. Next to this, we have also briefly covered the potential of this research on genetic therapies for treating in an effective and safe manner both inherited and acquired disorders. Despite the variety of issues touched upon, stretching from the scientific and economic to the ethical, it is sensible to expect that the registration of novel genetic therapies will continue to grow as well as the efforts to improve their performance and accessibility to patients.
CONFLICT OF INTEREST
Rob C. Hoeben is named inventor on patent applications claiming new adenoviruses for use as oncolytic agents. Hidde A. Zittersteijn and Manuel A.F.V. Gonçalves declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Hidde A. Zittersteijn, Manuel A.F.V. Gonçalves, and Rob C. Hoeben designed the work, collected literature data, and drafted the initial manuscript. All authors approved the final manuscript as submitted. All authors are responsible for the accuracy and integrity of the manuscript.
ACKNOWLEDGMENTS
The authors thank Josephine M. Janssen (Department of Cell and Chemical Biology, Leiden University Medical Center, Leiden, The Netherlands) for her detailed comments and critical reading of the manuscript. This work was performed with support of the Leiden University Medical Center. The gene therapy research in our laboratory is supported by the Netherlands Commission on Genetic Modification (COGEM 21334), the Utrecht Technology Foundation STW (STW 15414), the Prinses Beatrix Spierfonds (W.OR16‐13), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement 765269 (IMGENE—Improving genome editing efficiency), and the Duchenne Parent Project NL (17.012).
Zittersteijn HA, Gonçalves MAFV, Hoeben RC. A primer to gene therapy: Progress, prospects, and problems. J Inherit Metab Dis. 2021;44:54–71. 10.1002/jimd.12270
Communicating Editor: Robin Lachmann
Funding information Duchenne Parent Project, Grant/Award Number: 17.012; H2020 Marie Skłodowska‐Curie Actions, Grant/Award Number: 765269; Netherlands Commission on Genetic Modification, Grant/Award Number: COGEM 21334; Prinses Beatrix Spierfonds, Grant/Award Number: W.OR16‐13; Utrecht Technology Foundation STW, Grant/Award Number: STW 15414
REFERENCES
- 1. Friedmann T, Roblin R. Gene therapy for human genetic disease? Science. 1972;175:949‐955. [DOI] [PubMed] [Google Scholar]
- 2. Goncalves MA. A concise peer into the background, initial thoughts and practices of human gene therapy. Bioessays. 2005;27:506‐517. [DOI] [PubMed] [Google Scholar]
- 3. Miller AD, Jolly DJ, Friedmann T, Verma IM. A transmissible retrovirus expressing human hypoxanthine phosphoribosyltransferase (HPRT): gene transfer into cells obtained from humans deficient in HPRT. Proc Natl Acad Sci U S A. 1983;80:4709‐4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blaese RM, Culver KW, Miller AD, et al. T lymphocyte‐directed gene therapy for ADA‐SCID: initial trial results after 4 years. Science. 1995;270:475‐480. [DOI] [PubMed] [Google Scholar]
- 5. Cavazzana‐Calvo M, Hacein‐Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)‐X1 disease. Science. 2000;288:669‐672. [DOI] [PubMed] [Google Scholar]
- 6. Cicalese MP, Ferrua F, Castagnaro L, et al. Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short‐ and medium‐term safety. Mol Ther. 2018;26:917‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fischer A, Hacein‐Bey Abina S, Touzot F, Cavazzana M. Gene therapy for primary immunodeficiencies. Clin Genet. 2015;88:507‐515. [DOI] [PubMed] [Google Scholar]
- 8. Hacein‐Bey‐Abina S, Von Kalle C, Schmidt M, et al. LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐X1. Science. 2003;302:415‐419. [DOI] [PubMed] [Google Scholar]
- 9. Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID‐X1 patients. J Clin Invest. 2008;118:3143‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott‐Aldrich syndrome—long‐term efficacy and genotoxicity. Sci Transl Med. 2014;6:227ra33. [DOI] [PubMed] [Google Scholar]
- 11. Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X‐linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1‐EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401‐409. [DOI] [PubMed] [Google Scholar]
- 12. Stein S, Ott MG, Schultze‐Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198‐204. [DOI] [PubMed] [Google Scholar]
- 13. Migliavacca M, Assanelli A, Ponzoni M, et al. First occurrence of Plasmablastic lymphoma in adenosine deaminase‐deficient severe combined immunodeficiency disease patient and review of the literature. Front Immunol. 2018;9:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Felice B, Cattoglio C, Cittaro D, et al. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS One. 2009;4:e4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749‐1751. [DOI] [PubMed] [Google Scholar]
- 16. Zufferey R, Dull T, Mandel RJ, et al. Self‐inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873‐9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thornhill SI, Schambach A, Howe SJ, et al. Self‐inactivating gammaretroviral vectors for gene therapy of X‐linked severe combined immunodeficiency. Mol Ther. 2008;16:590‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hacein‐Bey‐Abina S, Pai SY, Gaspar HB, et al. A modified gamma‐retrovirus vector for X‐linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407‐1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263‐267. [DOI] [PubMed] [Google Scholar]
- 20. Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV‐1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521‐529. [DOI] [PubMed] [Google Scholar]
- 21. Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long‐term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682‐686. [DOI] [PubMed] [Google Scholar]
- 22. Modlich U, Navarro S, Zychlinski D, et al. Insertional transformation of hematopoietic cells by self‐inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Ravin SS, Wu X, Moir S, et al. Lentiviral hematopoietic stem cell gene therapy for X‐linked severe combined immunodeficiency. Sci Transl Med. 2016;8:335ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hacein‐Bey‐Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X‐linked severe combined immunodeficiency. N Engl J Med. 2010;363:355‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low‐dose Busulfan in infants with SCID‐X1. N Engl J Med. 2019;380:1525‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott‐Aldrich syndrome. Science. 2013;341:1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion‐dependent beta‐thalassemia. N Engl J Med. 2018;378:1479‐1493. [DOI] [PubMed] [Google Scholar]
- 28. Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science. 2009;326:818‐823. [DOI] [PubMed] [Google Scholar]
- 29. Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. [DOI] [PubMed] [Google Scholar]
- 30. Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med. 2019;11(e9958):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi Q. Platelet‐targeted gene therapy for Hemophilia. Mol Ther Methods Clin Dev. 2018;9:100‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi Q, Wilcox DA, Fahs SA, et al. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high‐titer inhibitory antibodies. J Clin Invest. 2006;116:1974‐1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Til NP, Stok M, Aerts Kaya FS, et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood. 2010;115:5329‐5337. [DOI] [PubMed] [Google Scholar]
- 34. Shahryari A, Saghaeian Jazi M, Mohammadi S, et al. Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet. 2019;10:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vago L, Oliveira G, Bondanza A, et al. T‐cell suicide gene therapy prompts thymic renewal in adults after hematopoietic stem cell transplantation. Blood. 2012;120:1820‐1830. [DOI] [PubMed] [Google Scholar]
- 36. Schuessler‐Lenz M, Enzmann H, Vamvakas S. Regulators' advice can make a difference: European medicines agency approval of Zynteglo for Beta thalassemia. Clin Pharmacol Ther. 2019:107(3):492–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic Leukemia. N Engl J Med. 2018;378:439‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377:2531‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goncalves MA, de Vries AA. Adenovirus: from foe to friend. Rev Med Virol. 2006;16:167‐186. [DOI] [PubMed] [Google Scholar]
- 40. Hoeben RC, Uil TG. Adenovirus DNA replication. Cold Spring Harb Perspect Biol. 2013;5:a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dyson N, Harlow E. Adenovirus E1A targets key regulators of cell proliferation. Cancer Surv. 1992;12:161‐195. [PubMed] [Google Scholar]
- 42. Flint J, Shenk T. Adenovirus E1A protein paradigm viral transactivator. Annu Rev Genet. 1989;23:141‐161. [DOI] [PubMed] [Google Scholar]
- 43. Jager L, Ehrhardt A. Emerging adenoviral vectors for stable correction of genetic disorders. Curr Gene Ther. 2007;7:272‐283. [DOI] [PubMed] [Google Scholar]
- 44. Nevins JR, Imperiale MJ, Kao HT, Strickland S, Feldman LT. Detection of an adenovirus E1A‐like activity in mammalian cells. Curr Top Microbiol Immunol. 1984;113:15‐19. [DOI] [PubMed] [Google Scholar]
- 45. Gao J, Mese K, Bunz O, Ehrhardt A. State‐of‐the‐art human adenovirus vectorology for therapeutic approaches. FEBS Lett. 2019;593:3609‐3622. [DOI] [PubMed] [Google Scholar]
- 46. Brunetti‐Pierri N, Ng T, Iannitti D, et al. Transgene expression up to 7 years in nonhuman primates following hepatic transduction with helper‐dependent adenoviral vectors. Hum Gene Ther. 2013;24:761‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morral N, O'Neal W, Rice K, et al. Administration of helper‐dependent adenoviral vectors and sequential delivery of different vector serotype for long‐term liver‐directed gene transfer in baboons. Proc Natl Acad Sci U S A. 1999;96:12816‐12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brunetti‐Pierri N, Ng P. Gene therapy with helper‐dependent adenoviral vectors: lessons from studies in large animal models. Virus Genes. 2017;53:684‐691. [DOI] [PubMed] [Google Scholar]
- 49. Dudley RW, Lu Y, Gilbert R, et al. Sustained improvement of muscle function one year after full‐length dystrophin gene transfer into mdx mice by a gutted helper‐dependent adenoviral vector. Hum Gene Ther. 2004;15:145‐156. [DOI] [PubMed] [Google Scholar]
- 50. Junyent F, Kremer EJ. CAV‐2‐why a canine virus is a neurobiologist's best friend. Curr Opin Pharmacol. 2015;24:86‐93. [DOI] [PubMed] [Google Scholar]
- 51. Vogels R, Zuijdgeest D, van Rijnsoever R, et al. Replication‐deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J Virol. 2003;77:8263‐8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alonso‐Padilla J, Papp T, Kajan GL, et al. Development of novel adenoviral vectors to overcome challenges observed with HAdV‐5‐based constructs. Mol Ther. 2016;24:6‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stasiak AC, Stehle T. Human adenovirus binding to host cell receptors: a structural view. 209 Med Microbiol: Immunol; 2019;325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaw AR, Suzuki M. Immunology of adenoviral vectors in Cancer therapy. Mol Ther Methods Clin Dev. 2019;15:418‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barry M. Single‐cycle adenovirus vectors in the current vaccine landscape. Expert Rev Vaccines. 2018;17:163‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barouch DH, Tomaka FL, Wegmann F, et al. Evaluation of a mosaic HIV‐1 vaccine in a multicentre, randomised, double‐blind, placebo‐controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13‐19). Lancet. 2018;392:232‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shukarev G, Callendret B, Luhn K, Douoguih M. A two‐dose heterologous prime‐boost vaccine regimen eliciting sustained immune responses to Ebola Zaire could support a preventive strategy for future outbreaks. Hum Vaccin Immunother. 2017;13:266‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liang M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr Cancer Drug Targets. 2018;18:171‐176. [DOI] [PubMed] [Google Scholar]
- 59. Zhang WW, Li L, Li D, et al. The first approved gene therapy product for Cancer ad‐p53 (Gendicine): 12 years in the clinic. Hum Gene Ther. 2018;29:160‐179. [DOI] [PubMed] [Google Scholar]
- 60. Raper SE, Yudkoff M, Chirmule N, et al. A pilot study of in vivo liver‐directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum Gene Ther. 2002;13:163‐175. [DOI] [PubMed] [Google Scholar]
- 61. Raper SE, Chirmule N, Lee FS, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148‐158. [DOI] [PubMed] [Google Scholar]
- 62. Wilson JM. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96:151‐157. [DOI] [PubMed] [Google Scholar]
- 63. Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. Gene therapy clinical trials worldwide to 2017: an update. J Gene Med. 2018;20:e3015. [DOI] [PubMed] [Google Scholar]
- 64. Mian A, Lee B. Urea‐cycle disorders as a paradigm for inborn errors of hepatocyte metabolism. Trends Mol Med. 2002;8:583‐589. [DOI] [PubMed] [Google Scholar]
- 65. Koeberl DD, Sun B, Bird A, Chen YT, Oka K, Chan L. Efficacy of helper‐dependent adenovirus vector‐mediated gene therapy in murine glycogen storage disease type Ia. Mol Ther. 2007;15:1253‐1258. [DOI] [PubMed] [Google Scholar]
- 66. Schmitt F, Pastore N, Abarrategui‐Pontes C, et al. Correction of hyperbilirubinemia in gunn rats by surgical delivery of low doses of helper‐dependent adenoviral vectors. Hum Gene Ther Methods. 2014;25:181‐186. [DOI] [PubMed] [Google Scholar]
- 67. Cerreto M, Mehdawy B, Ombrone D, et al. Reversal of metabolic and neurological symptoms of phenylketonuric mice treated with a PAH containing helper‐dependent adenoviral vector. Curr Gene Ther. 2012;12:48‐56. [DOI] [PubMed] [Google Scholar]
- 68. Baruteau J, Waddington SN, Alexander IE, Gissen P. Gene therapy for monogenic liver diseases: clinical successes, current challenges and future prospects. J Inherit Metab Dis. 2017;40:497‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno‐associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kotterman MA, Schaffer DV. Engineering adeno‐associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Geoffroy MC, Salvetti A. Helper functions required for wild type and recombinant adeno‐associated virus growth. Curr Gene Ther. 2005;5:265‐271. [DOI] [PubMed] [Google Scholar]
- 72. Harrington EA, Sloan JL, Manoli I, et al. Neutralizing antibodies against adeno‐associated viral capsids in patients with Mut Methylmalonic Acidemia. Hum Gene Ther. 2016;27:345‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kruzik A, Fetahagic D, Hartlieb B, et al. Prevalence of anti‐adeno‐associated virus immune responses in international cohorts of healthy donors. Mol Ther Methods Clin Dev. 2019;14:126‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Srivastava A. In vivo tissue‐tropism of adeno‐associated viral vectors. Curr Opin Virol. 2016;21:75‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Davidsson M, Wang G, Aldrin‐Kirk P, et al. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc Natl Acad Sci U S A. 2019:116(52):27053–27062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Clement N, Grieger JC. Manufacturing of recombinant adeno‐associated viral vectors for clinical trials. Mol Ther Methods Clin Dev. 2016;3:16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen ZY, Yant SR, He CY, Meuse L, Shen S, Kay MA. Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol Ther. 2001;3:403‐410. [DOI] [PubMed] [Google Scholar]
- 78. Yan Z, Zak R, Zhang Y, Engelhardt JF. Inverted terminal repeat sequences are important for intermolecular recombination and circularization of adeno‐associated virus genomes. J Virol. 2005;79:364‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Miller DG, Petek LM, Russell DW. Adeno‐associated virus vectors integrate at chromosome breakage sites. Nat Genet. 2004;36:767‐773. [DOI] [PubMed] [Google Scholar]
- 80. Miller DG, Rutledge EA, Russell DW. Chromosomal effects of adeno‐associated virus vector integration. Nat Genet. 2002;30:147‐148. [DOI] [PubMed] [Google Scholar]
- 81. Nakai H, Iwaki Y, Kay MA, Couto LB. Isolation of recombinant adeno‐associated virus vector‐cellular DNA junctions from mouse liver. J Virol. 1999;73:5438‐5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet. 2003;34:297‐302. [DOI] [PubMed] [Google Scholar]
- 83. Porteus MH, Cathomen T, Weitzman MD, Baltimore D. Efficient gene targeting mediated by adeno‐associated virus and DNA double‐strand breaks. Mol Cell Biol. 2003;23:3558‐3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Anguela XM, Sharma R, Doyon Y, et al. Robust ZFN‐mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283‐3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hanlon KS, Kleinstiver BP, Garcia SP, et al. High levels of AAV vector integration into CRISPR‐induced DNA breaks. Nat Commun. 2019;10:4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li H, Haurigot V, Doyon Y, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nelson CE, Wu Y, Gemberling MP, et al. Long‐term evaluation of AAV‐CRISPR genome editing for Duchenne muscular dystrophy. Nat Med. 2019;25:427‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chandler RJ, LaFave MC, Varshney GK, et al. Vector design influences hepatic genotoxicity after adeno‐associated virus gene therapy. J Clin Invest. 2015;125:870‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Donsante A, Miller DG, Li Y, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. [DOI] [PubMed] [Google Scholar]
- 90. Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV‐mediated in vivo gene therapy. Mol Ther Methods Clin Dev. 2018;8:87‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gaudet D, Stroes ES, Methot J, et al. Long‐term retrospective analysis of gene therapy with alipogene tiparvovec and its effect on lipoprotein lipase deficiency‐induced pancreatitis. Hum Gene Ther. 2016;27:916‐925. [DOI] [PubMed] [Google Scholar]
- 92. Chandler RJ, Venditti CP. Gene therapy for metabolic diseases. Transl Sci Rare Dis. 2016;1:73‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Grisch‐Chan HM, Schwank G, Harding CO, Thony B. State‐of‐the‐art 2019 on gene therapy for phenylketonuria. Hum Gene Ther. 2019;30:1274‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kishnani PS, Sun B, Koeberl DD. Gene therapy for glycogen storage diseases. Hum Mol Genet. 2019;28:R31‐r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ohashi T. Gene therapy for lysosomal storage diseases and peroxisomal diseases. J Hum Genet. 2019;64:139‐143. [DOI] [PubMed] [Google Scholar]
- 96. Piccolo P, Brunetti‐Pierri N. Gene therapy for inherited diseases of liver metabolism. Hum Gene Ther. 2015;26:186‐192. [DOI] [PubMed] [Google Scholar]
- 97. Soria LR, Mew NA, Brunetti‐Pierri N. Progress and challenges in development of new therapies for urea cycle disorders. Hum Mol Genet. 2019;28:R42‐r48. [DOI] [PubMed] [Google Scholar]
- 98. Ginocchio VM, Ferla R, Auricchio A, Brunetti‐Pierri N. Current status on clinical development of adeno‐associated virus‐mediated liver‐directed gene therapy for inborn errors of metabolism. Hum Gene Ther. 2019;30:1204‐1210. [DOI] [PubMed] [Google Scholar]
- 99. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med. 2020;382:29‐40. [DOI] [PubMed] [Google Scholar]
- 100. Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377:2519‐2530. [DOI] [PubMed] [Google Scholar]
- 101. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high‐specific‐activity factor IX variant. N Engl J Med. 2017;377:2215‐2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kay MA, Manno CS, Ragni MV, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24:257‐261. [DOI] [PubMed] [Google Scholar]
- 103. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2‐hRPE65v2) in patients with RPE65‐mediated inherited retinal dystrophy: a randomised, controlled, open‐label, phase 3 trial. Lancet. 2017;390:849‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mendell JR, Al‐Zaidy S, Shell R, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713‐1722. [DOI] [PubMed] [Google Scholar]
- 106. Batty P, Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum Mol Genet. 2019;28:R95‐r101. [DOI] [PubMed] [Google Scholar]
- 107. Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a Long journey to successful gene transfer. Mol Ther. 2020;28:723‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chen X, Goncalves MA. Engineered viruses as genome editing devices. Mol Ther. 2016;24:447‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol. 2016;428:963‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Foss DV, Hochstrasser ML, Wilson RC. Clinical applications of CRISPR‐based genome editing and diagnostics. Transfusion. 2019;59:1389‐1399. [DOI] [PubMed] [Google Scholar]
- 112. Maggio I, Goncalves MA. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol. 2015;33:280‐291. [DOI] [PubMed] [Google Scholar]
- 113. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol. 2017;18:495‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010;79:213‐231. [DOI] [PubMed] [Google Scholar]
- 116. Miller J, McLachlan AD, Klug A. Repetitive zinc‐binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985;4:1609‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rahman SH, Maeder ML, Joung JK, Cathomen T. Zinc‐finger nucleases for somatic gene therapy: the next frontier. Hum Gene Ther. 2011;22:925‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tebas P, Stein D, Tang WW, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gurlebeck D, Thieme F, Bonas U. Type III effector proteins from the plant pathogen Xanthomonas and their role in the interaction with the host plant. J Plant Physiol. 2006;163:233‐255. [DOI] [PubMed] [Google Scholar]
- 120. Christian M, Cermak T, Doyle EL, et al. Targeting DNA double‐strand breaks with TAL effector nucleases. Genetics. 2010;186:757‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Miller JC, Tan S, Qiao G, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143‐148. [DOI] [PubMed] [Google Scholar]
- 122. Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL‐type III effectors. Science. 2009;326:1509‐1512. [DOI] [PubMed] [Google Scholar]
- 123. Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. [DOI] [PubMed] [Google Scholar]
- 124. Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol. 2012;23:644‐650. [DOI] [PubMed] [Google Scholar]
- 125. Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B‐ALL after infusion of universal TALEN gene‐edited CAR T cells. Sci Transl Med. 2017;9(374):1–8. [DOI] [PubMed] [Google Scholar]
- 126. Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA‐guided endonuclease. Nat Biotechnol. 2013;31:230‐232. [DOI] [PubMed] [Google Scholar]
- 127. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA‐programmed genome editing in human cells. Elife. 2013;2:e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mali P, Yang L, Esvelt KM, et al. RNA‐guided human genome engineering via Cas9. Science. 2013;339:823‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR‐Cas9. Science. 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 131. Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA‐interference‐based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9‐crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579‐E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature. 2011;471:602‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM‐dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chen X, Goncalves M. DNA, RNA, and protein tools for editing the genetic information in human cells. iScience. 2018;6:247‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fonfara I, Le Rhun A, Chylinski K, et al. Phylogeny of Cas9 determines functional exchangeability of dual‐RNA and Cas9 among orthologous type II CRISPR‐Cas systems. Nucleic Acids Res. 2014;42:2577‐2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta‐globin and CCR5 genes have substantial off‐target activity. Nucleic Acids Res. 2013;41:9584‐9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome‐wide detection of DNA double‐stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fu Y, Foden JA, Khayter C, et al. High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat Biotechnol. 2013;31:822‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chen X, Tasca F, Wang Q, et al. Expanding the editable genome and CRISPR‐Cas9 versatility using DNA cutting‐free gene targeting based on in trans paired nicking. Nucleic Acids Res. 2020;48:974‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Anzalone AV, Randolph PB, Davis JR, et al. Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature. 2019;576:149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Chen JS, Dagdas YS, Kleinstiver BP, et al. Enhanced proofreading governs CRISPR‐Cas9 targeting accuracy. Nature. 2017;550:407‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kleinstiver BP, Pattanayak V, Prew MS, et al. High‐fidelity CRISPR‐Cas9 nucleases with no detectable genome‐wide off‐target effects. Nature. 2016;529:490‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Dang Y, Jia G, Choi J, et al. Optimizing sgRNA structure to improve CRISPR‐Cas9 knockout efficiency. Genome Biol. 2015;16:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR‐Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ryan DE, Taussig D, Steinfeld I, et al. Improving CRISPR‐Cas specificity with chemical modifications in single‐guide RNAs. Nucleic Acids Res. 2018;46:792‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Mali P, Aach J, Stranges PB, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chen X, Janssen JM, Liu J, et al. In trans paired nicking triggers seamless genome editing without double‐stranded DNA cutting. Nat Commun. 2017;8:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hyodo T, Rahman ML, Karnan S, et al. Tandem paired nicking promotes precise genome editing with scarce interference by p53. Cell Rep. 2020;30:1195‐207.e7. [DOI] [PubMed] [Google Scholar]
- 154. Nakajima K, Zhou Y, Tomita A, Hirade Y, Gurumurthy CB, Nakada S. Precise and efficient nucleotide substitution near genomic nick via noncanonical homology‐directed repair. Genome Res. 2018;28:223‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017;551:464‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature. 2016;533:420‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Grunewald J, Zhou R, Iyer S, et al. CRISPR DNA base editors with reduced RNA off‐target and self‐editing activities. Nat Biotechnol. 2019;37:1041‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Koblan LW, Doman JL, Wilson C, et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol. 2018;36:843‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zafra MP, Schatoff EM, Katti A, et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol. 2018;36:888‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Elverum K, Whitman M. Delivering cellular and gene therapies to patients: solutions for realizing the potential of the next generation of medicine. Gene Ther. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hirsch T, Rothoeft T, Teig N, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551:327‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Coppens DGM, de Wilde S, Guchelaar HJ, et al. A decade of marketing approval of gene and cell‐based therapies in the United States, European Union and Japan: an evaluation of regulatory decision‐making. Cytotherapy. 2018;20:769‐778. [DOI] [PubMed] [Google Scholar]
- 163. de Boer EN, van der Wouden PE, Johansson LF, van Diemen CC, Haisma HJ. A next‐generation sequencing method for gene doping detection that distinguishes low levels of plasmid DNA against a background of genomic DNA. Gene Ther. 2019;26:338‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Friedmann T, Koss JO. Gene transfer and athletics‐ an impending problem. Mol Ther. 2001;3:819‐820. [DOI] [PubMed] [Google Scholar]
- 165. van Dijke I, Bosch L, Bredenoord AL, Cornel M, Repping S, Hendriks S. The ethics of clinical applications of germline genome modification: a systematic review of reasons. Hum Reprod. 2018;33:1777‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Normile D. Shock greets claim of CRISPR‐edited babies. Science. 2018;362:978‐979. [DOI] [PubMed] [Google Scholar]
- 167. Chawla SP, Bruckner H, Morse MA, Assudani N, Hall FL, Gordon EM. A phase I‐II study using Rexin‐G tumor‐targeted Retrovector encoding a dominant‐negative cyclin G1 inhibitor for advanced pancreatic Cancer. Mol Ther Oncolytics. 2019;12:56‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Cherian JJ, Parvizi J, Bramlet D, Lee KH, Romness DW, Mont MA. Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF‐beta1 in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthr Cartil. 2015;23:2109‐2118. [DOI] [PubMed] [Google Scholar]
- 169. Collichio F, Burke L, Proctor A, et al. Implementing a program of Talimogene laherparepvec. Ann Surg Oncol. 2018;25:1828‐1835. [DOI] [PubMed] [Google Scholar]