

Abstract
The charge density of DNA is a key parameter in strand hybridization and for the interactions occurring between DNA and molecules in biological systems. Due to the intricate structure of DNA, visualization of the surface charge density of DNA nanostructures under physiological conditions was not previously possible. Here, we perform a simultaneous analysis of the topography and surface charge density of DNA nanostructures using atomic force microscopy and scanning ion conductance microscopy. The effect of in situ ion exchange using various alkali metal ions is tested with respect to the adsorption of DNA origami onto mica, and a quantitative study of surface charge density reveals ion exchange phenomena in mica as a key parameter in DNA adsorption. This is important for structure‐function studies of DNA nanostructures. The research provides an efficient approach to study surface charge density of DNA origami nanostructures and other biological molecules at a single molecule level.
Keywords: adsorption, DNA origami, scanning ion conductance microscopy, scanning probe microscopy, surface charge density
DNA charge density: The charge density of DNA is a determining factor in strand hybridization and interactions with other molecules in biological systems. Here, the visualization of surface charge density of DNA origami nanostructures is presented under biological relevant conditions using scanning ion conductance microscopy. The findings can be used in the study of surface charge density of DNA origami nanostructure conjugates at the single molecule level.
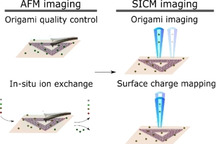
1. Introduction
The surface charge of biomolecules plays an important role in determining its biological responses.,[1,2] Especially in DNA, the phosphate backbone gives this important molecule a negative surface charge density (SCD), which affects strand hybridization in addition to the interaction of DNA with surfaces and molecular species ranging from small ions to large biomolecules such as antibodies.[ 3 , 4 , 5 ] However, the overall SCD of DNA is subject to change and can even be inverted by tuning the pH and composition of the surrounding electrolyte leading to different biophysical properties. [6] This has been shown to be the case in the development and study of DNA nanopore devices [7] that were based on the DNA origami method. [8] This DNA self‐assembly approach of arranging a long single‐strand into various well‐defined shapes by the use of complementary oligonucleotides has numerous applications in biophysics, molecular biology and drug delivery due to especially the highly controllable placement of molecules.[ 9 , 10 , 11 ] The tunability of the SCD is important to consider for the above applications and especially when applying DNA origami structures as delivery devices within biological systems due to the diverse ion conditions present.
In general, several methods are available for determining electrostatic or electrokinetic properties such as SCD and zeta potential. [12] The most common methods include electrophoretic light scattering (ELS) in the case of suspended particles and streaming current for large flat surfaces.,[13,14] Meanwhile, methods for the specific study of the SCD of DNA include electrophoresis, ELS and imaging of single‐stranded DNA under ambient conditions using Kelvin probe force microscopy.„[15,16,17] However, none of the aforementioned methods allow for surface charge imaging of individual structures with nanoscale resolution under biologically relevant conditions.
For nanoscale imaging of topography and SCD of water submersed structures, scanning ion conductance microscopy (SICM) is emerging as a powerful and versatile technique. [18] SICM operates by recording the ionic current through a nano‐ or micropipette according to a predetermined or current regulated path. The current is dependent on the pipette‐surface distance, and setting a current threshold enables constant‐height imaging or surface approaches. SICM allows topographical mapping on pore‐suspended membranes and membrane proteins of cells as well as many others in a true non‐contact fashion.[ 19 , 20 ] SICM topography profiles are dependent on the SCD of the sample and pipette. SCD effects lead to either a local increase or decrease in ion conductivity depending on the magnitude and polarity of the applied potential bias, named ion current rectification. [21] The arising change in the current‐distance behavior from ion current rectification effects can be used to quantify SCD.[ 22 , 23 ]
Currently, topographical study of DNA is optimally conducted using AFM‐based techniques under liquid conditions to avoid introducing capillary effects, which will change the dimensions and properties of DNA. [24] For such studies, mica is an ideal substrate for DNA deposition as it is atomically flat while also allowing for stable DNA adsorption in the presence of divalent cations such as Mg2+ and Ni2+ due to counter‐ion correlation effects.[ 25 , 26 ] Most importantly this approach does not induce structural alterations to DNA origami nanostructures and is therefore the preferred method for AFM imaging.[ 8 , 27 , 28 , 29 ] However, in the presence of Na+, the physiologically most abundant cation, DNA is reported to show poor adsorption, while investigations into the effects of other alkali metals require a careful and methodical approach. [26] The immobilization of DNA origami on mica can also be achieved by chemical approaches that are based on electrostatic interactions; these include covalent functionalization with positive species such as (3‐aminopropyl)triethoxysilane (APTES), 1‐3(3‐aminopropyl) silatrane (APS) and spermidine etc.[ 30 , 31 ] Chemical modifications introduce a possible source of artefacts, such as spatial heterogeneous surface coverage or chemically reactive surfaces.[ 30 , 32 ] This leads to a non‐definable reference point which can either complicate later AFM and SICM image interpretation or possibly even induce morphological changes of the DNA origami nanostructures. In addition, chemical deposition is more time consuming and requires either chemicals or synthesis that are not widely available. For these reasons, we avoid beforehand surface chemical modifications. Instead, we focus on the effects of modifying standard DNA buffers containing monovalent ions, while assuring stable DNA adsorption and structural integrity.
The stability and attachment of DNA in the presence of the different cations is relevant to the specific investigation of surface charge, but also for general in situ AFM studies where the study of conformational changes and dynamics are of interest. It is of even greater relevance if one aims to study molecules such as antibodies conjugated onto DNA origami frameworks as these often require the presence of specific ions for proper functioning. [33]
In this study, we created an experimental protocol, which has enabled SCD quantification and imaging of DNA origami structures using SICM and AFM. We determined the attachment of individual triangular DNA origami structures deposited on mica by in situ AFM imaging and investigated the effect of different alkali metals using liquid exchange. After optimizing the imaging conditions, we then present how SICM can provide insight into the adsorption of DNA by monitoring the changes in SCD of mica in the presence of different ions.
SICM images with spatial resolution comparable to standard AFM imaging quality revealed both detailed topographical and SCD information of the DNA origami nanostructures. The lateral resolution of SICM is normally considered significantly lower than AFM, [34] the obtained images are therefore of high‐resolution by SICM imaging standards and shows the promise of SICM for future imaging of single molecules such as DNA origami‐conjugates.
2. Results and Discussion
First, we investigated the optimal imaging conditions by preparing an experimental protocol (Figure 1), starting with AFM imaging of a two‐dimensional (2D) triangular DNA origami frame on mica in liquid. This particular structure is chosen due to its high popularity and use for different applications such as a delivery device and molecular pegboard.[ 35 , 36 ] An in situ ion exchange study followed, where the state of the DNA structures and DNA adsorption were carefully studied by AFM imaging. After finding optimal conditions that are compatible with standard SICM imaging, we performed imaging and SCD characterization of these structures.
Figure 1.
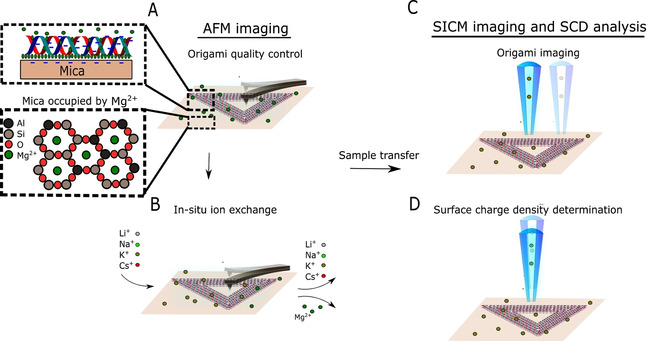
Schematic of the experimental workflow: DNA origami is studied using AFM under liquid conditions in folding buffer (A) and during ionic exchange (B). After finding the optimal conditions for liquid imaging, the sample is transferred to the SICM set‐up where the topography (C) and SCD (D) are studied.
2.1. AFM in situ Ion Exchange Reveals the Required Conditions for DNA Attachment
The structural state of the DNA origami was studied in its native folding buffer, consisting of 1×TAE (40 mM Tris, 20 mM acetic acid and 1 mM EDTA) and 12.5 mM Mg2+ , using liquid AFM operated in tapping mode. This revealed well‐folded triangular structures (Figure 2A). Imaging of structures under dry conditions was also performed to show the importance of using liquid conditions as dehydration results in deformed structures, which would complicate analysis of the structural stability. Afterwards, 10 mM Tris, acetic acid, pH 8.0 with either KCl (Figure 2C–E) or NaCl/Mg2+ (Figure 2F–H) was injected into the fluid chamber in order to study the influence of different ions on the stability and adsorption of DNA origami on mica. We omit the use of EDTA as it is a chelating agent that can interfere with electrolytes present in solution or on DNA. [37] Injection of NaCl/KCl was central for future SICM imaging and SCD determination studies. KCl and NaCl are commonly used and relevant electrolytes for various DNA studies.[ 38 , 39 , 40 ] The less commonly used alkali metal ions, Li+ and Cs+ were also studied (Figure S1 in the Supporting Information). Li+ and Cs+ are expected to affect the conformation of DNA due to a combination of their size and hydration properties.[ 41 , 42 ]
Figure 2.
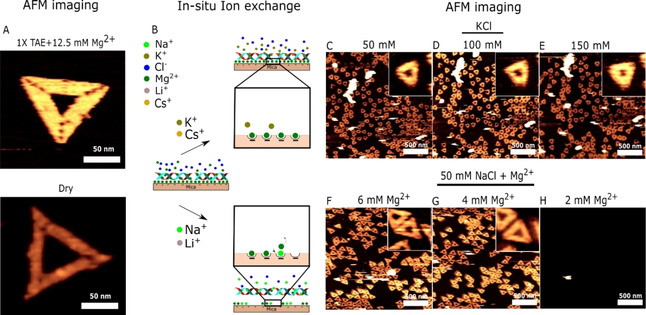
AFM study of DNA origami adsorption during ionic exchange: A) AFM image of DNA origami in standard folding buffer consisting of 1xTAE (40 mM Tris, 20 mM acetic acid and 1 mM EDTA) and 12.5 mM Mg2+ and in dry after washing with Milli‐Q water. B) Proposed pathway of ionic exchange in mica and the effects on DNA origami adsorption. AFM imaging during in situ buffer exchange with 10 mM Tris, acetic acid, pH 8.0 in addition to 50 (C), 100 (D) and 150 (E) mM KCl and 50/6 (F), 50,4 (G) and 50/2 (H) mM NaCl/Mg2+.
Injection of up to 150 mM KCl in the fluid chamber had no immediate apparent influence on either the continued attachment or the structural integrity of the DNA origami structures. The structures were unaffected even after 3 hours of imaging. The same result was also observed when adding Cs+ ions (see the Supporting Information Figure S1C–E). On the other hand, addition of 50 mM NaCl led to an instantaneous and complete desorption of even a fully covered origami surface (see Figure S2). To examine this in more detail, 50 mM NaCl accompanied by Mg2+ at various concentrations was added (Figure 2F–H). Continued attachment was observed in the presence of ≥4 mM Mg2+, while 2 mM Mg2+ led to DNA desorption. The same desorption tendency was also observed when adding Li+ ions under identical conditions (see Figure S1F–H). Subsequent replacement of the Mg2+ ions does not lead to detachment of DNA in the presence of K+, while the smaller alkali ions (Li+ and Na+) display a clear tendency to disrupt the DNA attachment. Finely tuning the ratio of Mg2+ to Na+ ion leads to origami diffusion on mica, which has been used in the self‐assembly of 2D DNA origami lattices.[ 43 , 44 ] However, under the conditions examined in Figure 2, no diffusion phenomenon is observed when DNA origami is on mica (see Figure S3), thereby showing that the conditions investigated provide strong DNA origami surface binding. Especially the continued adsorption of DNA origami in the presence of K+ and Cs+ ions is interesting and has to the best of our knowledge not been reported elsewhere. Supplementary Table 1 provides a comparison of selected literature examining ionic species used initial attachment and continued attachment/diffusion of DNA origami.[ 43 , 44 , 45 ] Based on the clear differences in DNA origami attachment under the different ionic conditions, we propose a likely pathway (Figure 2B) due to ion exchange phenomenon in the lattice sites of mica. Following ionic exchange, the morphology of individual DNA origami is also of interest, as the concentration of either divalent or monovalent ions in solution impacts the structural stability.[ 46 , 47 ] Figure S4 shows that adsorbed structures remain intact under the different ionic conditions studied. However, under high AFM imaging forces and in dry, DNA origami structures either become damaged or dehydrated thereby changing their morphology. The results emphasize the importance of optimal imaging conditions. Figure 2 shows that there are ionic conditions which do not support DNA adsorption on mica substrate. Chemically functionalized mica surface with species such as APTES can support DNA adsorption over wider ranges of ionic concentration, which can be of interest. [24] We examined the suitability of APTES chemically modified mica surface by performing liquid imaging in DNA origami folding buffer. Figure S5 contains time‐lapse imaging showing the formation of numerous particles after several hours. Particle formation on APTES surfaces during liquid imaging has previously been reported and is likely due to the hydrolysis of APTES and subsequent particle formation. [30] Therefore, such a surface can be problematic for later imaging. Mica on the other hand is an inert material and remains atomically flat under liquid conditions as also seen in Figure S5. This illustrates why mica is a highly attractive substrate for AFM imaging.
2.2. SICM Reveals Ion Exchange Phenomenon on Mica
The controllable adsorption of DNA origami based on the presence and concentration of various alkali metals is relevant to both SICM and AFM imaging of DNA nanostructures. To elucidate the mechanisms behind DNA adsorption, an investigation of the electric double layer (EDL) formed at the mica surface in the presence of ions in solution is required. The EDL can be divided into two regions: the Stern layer at close proximity to the surface where ions from the solution can be adsorbed and the diffuse layer where ions distribute due to uncompensated surface charge in accordance with the Poisson‐Boltzmann equation, and otherwise can diffuse vertically and laterally. [48] The structure and formation of the Stern layer can be complex and relies on parameters such as surface charge, surface topography, preferential bindings sites and interfacial hydrogen bonding arrangements. [49] Changes in the structure and composition of the Stern layer should result in a change of the effective SCD measurable by SICM.
Ion exchange phenomena at the mica surface, which are likely responsible for DNA adsorption, are herein studied by monitoring the SCD using SICM current‐distance curves. The relationship between the pipette distance and the current passing through the pipette tip is recorded as the pipette approaches the sample surface with an applied bias of +100 mV and −100 mV. [19] Diverging current‐distance curves is caused by ion current rectification effects due to surface charges on the sample surface. The SCD was estimated by first subtracting the vertical pipette positions at 99 % current obtained at −100 mV from that obtained at +100 mV. Based on the quantitative surface conductivity microscopy (QSCM) approach, [22] this value can be converted into SCD. The conversion to SCD is described in detail in Figure S7. Figure 3A shows raw data and a fit, where a difference of approximately 5 nm is found between the two curves at the 99 % current threshold. From this difference in pipette position, the SCD is estimated as −39 mC m−2 using the conversions previously mentioned. The negative SCD is explained by the exposure of the negative lattice sites of mica. [50] The adsorption of DNA on mica requires the presence of divalent cations such as Mg2+ and Ni2+ for screening the negative surface charge of mica as seen in Figure 2. The presence of these ions on the mica surface was investigated by studying the surface charge on mica in 150 mM NaCl and KCl imaging buffers. Three cases are of interest; freshly cleaved mica and mica pre‐treated with either Mg2+ or Ni2+ ions. Ni2+ ions are known to attach strongly to mica under various imaging conditions and is used for DNA imaging. [51] Figure 3A−C and E−G show representative SICM current‐distance curves, while Figure 3 D,H show the average SCD values extracted under each condition. Each average and standard deviation was calculated based on 10 measurements. Freshly cleaved mica in NaCl reveals an average surface charge density of −39.07 (±4.49) mC m−2 explained by the exposure of the negative lattice sites of mica. The found value is similar to other studies, yet the SCD and zeta potential of mica is dependent on environmental conditions such as pH.[ 52 , 53 ] A nearly identical SCD of −39.14 (±3.74) mC m−2 is observed for a mica surface pre‐treated with Mg2+, strongly indicating effective Mg2+ desorption. Meanwhile, for a Ni2+ pre‐treated surface, the SCD changes to +27.22 (±9.25) mC m−2 demonstrating the charge inversion facilitated by firmly attached Ni2+ ions. In general, Mg2+ ions are expected to be susceptible to thermal fluctuations in the mica lattices, while Ni2+ can undergo complex formation with mica preventing displacement in favor of monovalent ions. [26] Monolayers of Ni2+ have also been reported to form on mica theorized to be a result of metal or metal hydroxide precipitates. [54]
Figure 3.
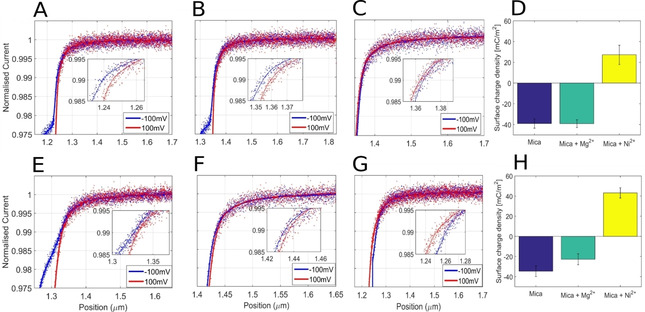
Surface charge of native mica, mica pre‐treated with Mg2+ or Ni2+: Representative SICM current‐distance curves performed in 150 mM NaCl on mica (A), mica pre‐treated with Mg2+ (B), and mica pre‐treated with Ni2+ (C) along with SCD statistics (D). Representative SICM current–distance curves performed in 150 mM KCl on mica (E), mica pre‐treated with Mg2+ (F), and mica pre‐treated with Ni2+ (G) along with SCD statistics (H).
Desorption of DNA origami was observed in NaCl. Identically prepared surfaces were subsequently imaged in KCl instead of NaCl. The SCD of untreated mica has a nearly identical value of −34.60 (±5.26) mC m−2 in KCl compared to NaCl. Yet, after exposure to divalent ions, several differences are apparent. First, the SCD for mica treated with Mg2+ has decreased to −22.72 (±5.45) mC m−2 due to partial coverage of Mg2+ ions showing higher affinity to the mica surface in the presence of KCl compared to NaCl. Secondly, mica treated with Ni2+ shows charge inversion as in the case with NaCl and thereby higher affinity compared to Mg2+, however here the SCD has increased to +43.15 (±5.05) mC m−2. These findings indicate that the affinity of divalent ions such as Mg2+ and Ni2+ is stronger in KCl compared to in NaCl.
Based on these findings, the adsorption of DNA in KCl imaging buffer can be partly explained by Mg2+ adsorption, which is then confined between mica and DNA. Since SCD of mica is not inverted under these conditions, other effects should also be discussed that go beyond considering simple electrostatic interactions. General factors that influence the efficiency and dynamics of Mg2+ binding could be incubation time, binding affinity based on ionic radii and enthalpy of hydration. [55] Furthermore, confinement effects leading to a higher concentration of Mg2+ ions between mica and DNA origami relative to that observed in the interface between mica and the solution should be considered.
Additionally, it is important to consider the general behavior of individual alkali metal ions on mica. The organization of alkali metals ions on mica have been studied by AFM and X‐ray reflectivity and have been found to behave differently.[ 56 , 57 ] Li+ and Na+ have multiple solvation states and higher mobility on mica, which results in an unstable organization that disrupts continued DNA adsorption by replacing Mg2+ on mica. On the other hand, K+ and heavier alkali metal ions are found to form a stable lateral organization of ions in the mica vacancies due to a combination of their ionic size and chaotropic nature, This minimal disruption of the interfacial water structure of mica leads to ion self‐assembly, which is primarily guided by the hydration energy minimization of mica and adsorbed ions. This should result in higher DNA adsorption stabilization which is observed in our experiments. The large interfacial surface area between DNA structures and mica could also enhance binding efficiency. This could be further studied by examining the adsorption efficiency of DNA origami structures of varying size.
2.3. Topography and SCD Information of DNA Nanostructures
Quantifying the SCD of DNA nanostructures and DNA can provide important biophysical information regarding their functionality. DNA origami on mica with high surface coverage was first investigated. A monolayer of DNA origami was deposited on mica using a 12.5 mM MgCl2 buffer and then replaced with a near‐physiological imaging buffer of 10 mM Tris, acetic acid, pH 8.0 and 150 mM KCl followed by AFM imaging (Figure 4A). Mg2+ over Ni2+ was studied for DNA adsorption, as Mg2+ has been shown to be competent for facilitating successful imaging and is the most commonly used divalent cation in general AFM studies of DNA origami.
Figure 4.
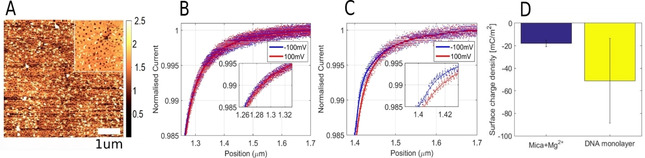
QSCM study of DNA origami monolayers: AFM images of a fully covered DNA layer (A) along with SICM current‐distance approach curves performed on mica surface pre‐treated with Mg2+ (B), mica surface fully covered with DNA (C), and SCD statistics (D).
We record SICM current‐distance curves on mica pre‐treated with Mg2+ and mica covered by a monolayer of DNA. The two samples are compared in Figure 4, where current‐distance curves obtained on a mica substrate (B) and a complete DNA origami layer (C), showing a large difference at the 99 % current threshold. Statistics of the SCD obtained based on 10 approach curves are shown in Figure 4D. The mica surface pre‐treated with Mg2+ is found to have a mean SCD of −17.88 (±3.01) mC m−2, which is higher than previous studies on pure untreated mica. [22] It should be noted that the SCD of mica varies depending on environmental conditions such as ion concentration and pH. [58] In this case, we perform imaging in KCl meanwhile also having pre‐treated with origami buffer containing Mg2+ ions, which partially neutralize the negative surface charge. On the other hand, the DNA treated mica shows a mean SCD of −51.1 (±37.6) mC m−2. Although having obtained a nearly complete DNA layer as seen from AFM imaging in Figure 4A, the DNA design consists of a triangular structure where the outer sides measure 120 nm in addition to a triangular hole of 40 nm, which exposes the underlying mica substrate. The pipette used has an estimated inner radius of 32.6 nm, which would allow for SCD contributions from both the DNA structure and the mica, which accounts for the large standard deviation. Mapping with multiple current distance curves could be used as an alternative method of quantifying the coverage of a monolayer, while also obtaining SCD information.
The large negative SCD recorded is due to the polyanionic nature of DNA. This is also specifically seen for DNA origami nanostructures as they have a reported zeta‐potential of up to −30 mV while in their native folding buffer. [16] The corresponding SCD can be estimated from the Grahame equation (detailed in Figure S8) to an estimated value of −19 mC m−2. However, this value is not directly comparable to the values obtained from SICM as the SCD is evaluated at different planes. In QSCM, the SCD is evaluated at the outer Helmholtz plane or Stern surface, which refers to the plane placed through the center of firmly adsorbed ions. On the other hand, zeta potential is evaluated at the slipping plane which separates mobile fluid from the fluid that remains attached to charged species on the surface. [59] QSCM also allows for studying the SCD of DNA at various electrolyte concentrations, with the only limitation being successful DNA adsorption. The theoretical SCD of the DNA origami can be estimated by first assuming that all charges of the phosphate backbone of the m13 plasmid [60] and staple strands (14498 nucleotides) are equally distributed on the origami surface (11085 nm2). This gives a SCD of −209.55 mC m−2. The calculation is detailed in Supplementary note 3 (in the Supporting Information). This value is significantly higher than the SCD recorded by SICM due to the accumulation of positively charged Mg2+ and K+ counter‐ions. Generally, the composition of salts will have an intricate effect on the apparent SCD of DNA due to differences in electric charge and specific adsorption. [61]
Characterization of individual DNA nanostructures can provide new insight into the structure‐function relationship of this critical molecule. We deposit a low concentration of DNA origami on mica using Mg2+ and image the individual structures using AFM (Figure 5A) and SICM (Figure 5C) in an imaging buffer of 10 mM Tris and 150 mM KCl, pH 8. Using these optimized conditions imaging of DNA nanostructures becomes possible. The lateral resolution can be found based on resolved features of the DNA origami structure. The estimated resolution is <8 nm for AFM and <20 nm for SICM. From height histograms obtained from SICM data (Figure 5D), the DNA structures have a height of 6.91±0.71 nm, which is well above the expected physical height of 2 nm for this structure, as verified by AFM imaging and height histograms where a height of 1.9±0.13 nm was measured (Figure 5A,B). This height difference is in line with previous reports on QSCM.[ 19 , 20 ] This leads to a convolution effect whereby SCD information is stored within the height data. This large apparent height while scanning at +100 mV suggests that the DNA origami structures have a significantly more negative SCD than the mica substrate in accordance with the findings presented in Figure 4D. Figure 5E shows a schematic illustration of a SICM scan profile at +100 mV depending on the SCD as compared to the AFM scan profile. From our findings presented in Figure 3, we propose that the mica surface is partially covered by Mg2+ ions situated in the mica lattice plane. On the other hand, DNA origami has a more complicated and flexible geometrical structure with a continuously twisting negatively charged phosphate backbone leading to an intricate placement of screening Mg2+ ions. We find that DNA origami remains overall negatively charged and therefore does not undergo charge inversion under regular folding conditions. It is also important to acknowledge that Mg2+ ions stabilizing the DNA structures may have been replaced partially or fully by K+, hereby lowering the effective SCD. Based on prior knowledge of the topography of the DNA nanostructures through AFM imaging, imaging using SICM can be applied to study the SCD of various DNA nanostructures as well as DNA‐molecule conjugates with nanoscale resolution. This approach can be used under the mild ionic conditions identified in this paper, but it can also be extended to other conditions by using chemically functionalized surfaces and covalent attachment of the DNA. [62]
Figure 5.
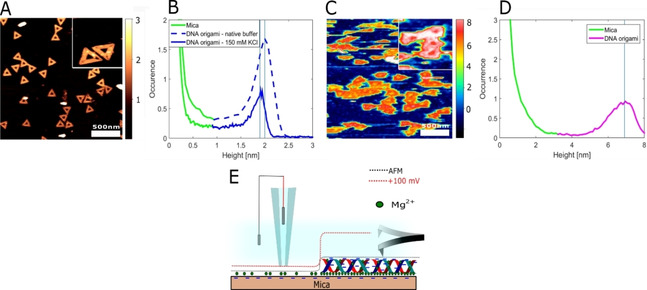
Images of DNA origami obtained by AFM and SICM: Height imaging and histograms of DNA nanostructures obtained by AFM (A, B) and SICM (C, D). E) Schematic illustration of scan profiles obtained by AFM and SICM at +100 mV.
3. Conclusions
We have created an experimental protocol that allows routine SCD characterization and topographical imaging of DNA origami nanostructures using SICM for the first time. The fast acquisition time of this methodology makes it a useful tool for SCD quantification for in situ measurement of most materials, demonstrated here by the investigation of the ion exchange of mica necessary for DNA deposition. Initially, we studied the adsorption of DNA origami in the presence of Na+ and K+, used for standard SICM imaging. DNA origami deposited on mica using Mg2+ ions remains attached when imaged in 50–150 mM KCl buffers, while imaging in NaCl buffers was not possible due to ion‐substitution events leading to DNA desorption. Proper DNA deposition is critical to AFM imaging of DNA nanostructures under liquid conditions. To explain the differences in DNA adsorption, we studied ion‐exchange events in the Stern layer of mica by monitoring the SCD using the SICM mode, QSCM. We found that changes in the SCD of mica is associated with binding of divalent ions, showing that divalent ions have greater affinity to mica in the presence of KCl compared to NaCl. Demonstrated here, SICM QSCM allows quantification of SCD down to the single‐molecule level and topography imaging with resolution comparable to AFM. We believe that simultaneously quantification of SCD and topography of biological nanostructures will allow for future studies of DNA origami‐molecule conjugates such as proteins and antibodies. The presented results show the prospects of applying SICM QSCM to study topography and SCD of other biological molecules at the single molecule level under physiologically relevant conditions.
Experimental Section
DNA Origami Synthesis
The DNA nanostructure used was a two‐dimensional (2D) triangular DNA Origami Frame (tDOF) which has sides that measure 120 nm. DNA origami samples were assembled using single‐stranded M13 DNA (10 nM, Thermo Scientific) as the scaffold strand, staple strands (100 nM, IDT), Mg(OAc)2 (12.5 mM), 1X TAE (Thermo Fisher) (40 mM Tris, 20 mM acetic acid and 1 mM EDTA) over a 90 min thermal annealing ramp from 80 °C to 20 °C. DNA origami was purified by removing excess staples using IllustraMicroSpin columns S‐400 HR (GE Healthcare) equilibrated with DNA origami assembly buffer.
AFM Imaging and Data Analysis
For AFM measurements of a partial DNA origami covered surface, 2 ul of a DNA origami solution (2 nM) was deposited on freshly cleaved mica for 2 min followed by rinsing with 1 ml of imaging/folding buffer. Complete coverage was achieved by increasing the origami concentration (5 nM) and incubation time to 10 min.
The general state of the origami structure and the coverage/adsorption on mica was studied in liquid using tapping mode AFM (MultiMode VIII, Bruker, Santa Barbara, CA, USA). Mica was temporarily glued onto a stainless‐steel puck using silicon grease. Tapping in liquid was performed using triangular silicon nitride probes (TR400PSA, OLYMPUS, USA, nominal resonance frequency: 6 kHz, nominal spring constant: 0.08 N/m, and tip radius: 15 nm). A fluid cell (MTFML, Bruker) was mounted onto the setup and allowed for a closed and sealed environment for buffer exchange of Tris (10 mM), acetic acid, pH 8.0 and various alkali metals. In situ AFM imaging started 30 min after DNA origami preparation on mica. Liquid exchange with various alkali metals occurred at time intervals of 30 min hereafter due to thermal equilibration of the AFM equipment. Buffer was exchanged at a rate of approximately 100 ul s−1 with a total exchange of 2 ml. As the liquid cell can contain ∼30 ul of liquid, all previous buffer solution was replaced. Images were collected with a scan‐rate of 1 Hz and 256 lines. All other parameters were optimized for best image quality. Obtained AFM images were flattened using SPIP software (Image Metrology ApS, Lyngby, Denmark). Rearrangement/diffusion studies were performed by using the “Align” and “Subtract” function in SPIP software (Image Metrology ApS, Lyngby, Denmark).
SICM Imaging
After imaging of DNA using AFM, the sample was moved to an XE‐Bio system (Park Systems, Suwon, South Korea) setup to record SICM images. An imaging buffer consisting of Tris (10 mM), acetic acid, pH 8.0 and KCl (150 mM) was used throughout all experiments. Imaging was conducted at +100 mV (with respect to the bath electrode) with a set‐point of 99 % of the free current (338 pA).
SICM Current–Distance Curves
Current distance curves were recorded at an approach and withdraw speed of 100 nm s−1. The pipette was moved towards the surface until a current of 97 % of the free current was measured. 10 curves were obtained at 10 locations at both ±100 mV for statistical analysis where the mean and the standard deviation were calculated. Curve fitting was performed in MATLAB (MathWorks, USA) by using a moving average filter. The inner radius of a pipette was estimated by measuring the free current and comparing it to simulation. Simulations were performed in COMSOL Multiphysics. Details are provided in the Supporting Information. One single pipette with an estimated inner radius of 18.9 nm was used for studying the SCD of mica. For the measurement of SCD of DNA monolayers, a pipette with an estimated inner radius of 32.6 nm was used.
Nanopipette Fabrication
Single‐barrel nanopipettes were pulled from borosilicate capillaries (Sutter Instruments, Novato, CA, USA) using a CO2‐laser puller system (P2000, Sutter Instruments, Novato, CA, USA).
Pipettes for imaging of individual DNA origami nanostructures were produced from the following pulling parameters Heat: 330 – Fil: 4 – Vel: 25 – Delay:150 – Pull: 150, producing a pipette with a free current of 338 pA in 150 mM KCl at +100 mV.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by grants from the Sino‐Danish Center for Education and Research, Danish Council for Independent Research (grant no. 6108‐00396B), EU H2020 Marie Sklodowska‐Curie Actions (MNR4SCELL no. 734174, SENTINEL no. 812398), the National Natural Science Foundation of China (Grant No. 21929401, 21773050), Sichuan Science and Technology Foundation (20YYJC3895), Fundamental Research Funds for the Central Universities, China (YJ201893) and State Key Lab of Advanced Metals and Materials, China (Grant No. 2019‐Z03). L.H.K. acknowledges support from the Carlsberg Foundation.
S. Møller Sønderskov, L. Hyldgaard Klausen, S. Amland Skaanvik, X. Han, M. Dong, ChemPhysChem 2020, 21, 1474.
Contributor Information
Dr. Lasse Hyldgaard Klausen, Email: lassehk@stanford.edu
Prof. Xiaojun Han, Email: hanxiaojun@hit.edu.cn.
Prof. Mingdong Dong, Email: dong@inano.au.dk.
References
- 1. Michelow I. C., Dong M., Mungall B. A., Yantosca L. M., Lear C., Ji X., Karpel M., Rootes C. L., Brudner M., Houen G., et al., J. Biol. Chem. 2010, 285, 24729–24739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong M., Xu S., Oliveira C. L. P., Pedersen J. S., Thiel S., Besenbacher F., Vorup-jensen T., J. Immunol. 2007, 178, 3016–3022. [DOI] [PubMed] [Google Scholar]
- 3. Ouldridge T. E., Sulc P., Romano F., Doye J. P. K., Louis A. A., Nucleic Acids Res. 2013, 41, 8886–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almaqwashi A. A., Paramanathan T., Rouzina I., Williams M. C., Nucleic Acids Res. 2016, 44, 3971–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Podestà A., Indrieri M., Brogioli D., Manning G. S., Milani P., Guerra R., Finzi L., Dunlap D., Biophys. J. 2005, 89, 2558–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guo Z., Wang Y., Yang A., Yang G., Soft Matter 2016, 12, 6669–6674. [DOI] [PubMed] [Google Scholar]
- 7. Hernández-Ainsa S., Keyser U. F., Nanoscale 2014, 6, 14121–14132. [DOI] [PubMed] [Google Scholar]
- 8. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 9. Puchkova A., Vietz C., Pibiri E., Wünsch B., Paz M. S., Acuna G. P., Tinnefeld P., Nano Lett. 2015, 15, 8354–8359. [DOI] [PubMed] [Google Scholar]
- 10. Ketterer P., Ananth A. N., Trip D. S. L., Mishra A., Bertosin E., Ganji M., v d Torre J., Onck P., Dietz H., Dekker C., Nat. Commun. 2018, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Udomprasert A., Kangsamaksin T., Cancer Sci. 2017, 108, 1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delgado A. V., González-Caballero F., Hunter R. J., Koopal L. K., Lyklema J., J. Colloid Interface Sci. 2007, 309, 194–224. [DOI] [PubMed] [Google Scholar]
- 13. Ware B. R., Adv. Colloid Interface Sci. 1974, 4, 1–44. [Google Scholar]
- 14. Werner C., Zimmermann R., Kratzmüller T., Colloids Surf. A 2001, 192, 205–213. [Google Scholar]
- 15. Stellwagen N. C., J. Phys. Chem. B 2017, 121, 2015–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeng Y., Liu J., Yang S., Liu W., Xu L., Wang R., J. Mater. Chem. B 2018, 6, 1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leung C., Maradan D., Kramer A., Howorka S., Mesquida P., Hoogenboom B. W., Appl. Phys. Lett. 2010, 97, 203703. [Google Scholar]
- 18. Hansma P. K., Drake B., Marti O., Gould S. A. C., Prater C. B., Science 1989, 243, 641–643. [DOI] [PubMed] [Google Scholar]
- 19. Böcker M., Muschter S., Schmitt E. K., Steinem C., Schäffer T. E., Langmuir 2009, 25, 3022–3028. [DOI] [PubMed] [Google Scholar]
- 20. Shevchuk A. I., Frolenkov G. I., Sánchez D., James P. S., Freedman N., Lab M. J., Jones R., Klenerman D., Korchev Y. E., Angew. Chem. Int. Ed. 2006, 45, 2212–2216; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2270–2274. [Google Scholar]
- 21. Sa N., Lan W. J., Shi W., Baker L. A., ACS Nano 2013, 7, 11272–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klausen L. H., Fuhs T., Dong M., Nat. Commun. 2016, 7, 12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuhs T., Klausen L. H., Sønderskov S. M., Han X., Dong M., Nanoscale 2018, 10, 4538–4544. [DOI] [PubMed] [Google Scholar]
- 24. Lyubchenko Y. L., Shlyakhtenko L. S., Proc. Natl. Acad. Sci. USA 1997, 94, 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bezanilla M., Manne S., Laney D. E., Lyubchenko Y. L., Hansma H. G., Langmuir 1995, 11, 655–659. [Google Scholar]
- 26. Pastré D., Piétrement O., Fusil S., Landousy F., Jeusset J., David M., Hamon L., Cam E. L., Zozime A., Biophys. J. 2003, 85, 2507–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang F., Jiang S., Wu S., Li Y., Mao C., Liu Y., Yan H., Nat. Nanotechnol. 2015, 10, 779–784. [DOI] [PubMed] [Google Scholar]
- 28. Kabiri Y., Ravelli R. B. G., Lehnert T., Qi H., Katan A. J., Roest N., Kaiser U., Dekker C., Peters P. J., Zandbergen H., Sci. Rep. 2019, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aslan H., Krissanaprasit A., Besenbacher F., Gothelf K. V., Nanoscale 2016, 8, 15233–15240. [DOI] [PubMed] [Google Scholar]
- 30. Shlyakhtenko L. S., Gall A. A., Lyubchenko Y. L., Methods Mol. Biol. 2013, 931, 295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suzuki Y., Higuchi Y., Hizume K., Yokokawa M., Yoshimura S. H., Yoshikawa K., Takeyasu K., Ultramicroscopy 2010, 110, 682–688. [DOI] [PubMed] [Google Scholar]
- 32. Crampton N., Bonass W. A., Kirkham J., Thomson N. H., Langmuir 2005, 21, 7884–7891. [DOI] [PubMed] [Google Scholar]
- 33. Zhao H., J. Chem. Technol. Biotechnol. 2016, 91, 25–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rheinlaender J., Schäffer T. E., J. Appl. Phys. 2009, 105, 94905. [Google Scholar]
- 35. Song L., Jiang Q., Liu J., Li N., Liu Q., Dai L., Gao Y., Liu W., Liu D., Ding B., Nanoscale 2017, 9, 7750–7754. [DOI] [PubMed] [Google Scholar]
- 36. Lu Z., Wang Y., Xu D., Pang L., Chem. Commun. 2017, 53, 941–944. [DOI] [PubMed] [Google Scholar]
- 37. Flora S. J. S., Pachauri V., Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pasi M., Maddocks J. H., Lavery R., Nucleic Acids Res. 2015, 43, 2412–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rehm C., Holder I. T., Groß A., Wojciechowski F., Urban M., Sinn M., Drescher M., Hartig J., Chem. Sci. 2014, 5, 2809–2818. [Google Scholar]
- 40. Tan Z., Chen S., Biophys. J. 2006, 90, 1175–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Savelyev A., A. D. Mackerell Jr , J. Phys. Chem. Lett. 2015, 6, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen X., Ding K. K., Zhang F. S., Chem. Phys. Lett. 2013, 574, 100–105. [Google Scholar]
- 43. Rafat A. A., Pirzer T., Scheible M. B., Kostina A., Simmel F. C., Angew. Chem. Int. Ed. 2014, 53, 7665–7668; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7797–7801. [Google Scholar]
- 44. Woo S., Rothemund P. W. K., Nat. Commun. 2014, 5, 1–11. [DOI] [PubMed] [Google Scholar]
- 45. Kielar C., Ramakrishnan S., Fricke S., Grundmeier G., Keller A., ACS Appl. Mater. Interfaces 2018, 10, 44844–44853. [DOI] [PubMed] [Google Scholar]
- 46. Kielar C., Xin Y., Shen B., Kostiainen M. A., Grundmeier G., Linko V., Keller A., Angew. Chem. 2018, 130, 9614–9618; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2018, 57, 9470–9474. [DOI] [PubMed] [Google Scholar]
- 47. Ramakrishnan S., Ijäs H., Linko V., Keller A., Comput. Struct. Biotechnol. J. 2018, 16, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bourg I. C., Lee S. S., Fenter P., Tournassat C., J. Phys. Chem. C 2017, 121, 9402–9412. [Google Scholar]
- 49. Siretanu I., Ebeling D., Andersson M. P., Stipp S. L. S., Philipse A., Stuart M. C., v d Ende D., Mugele F., Sci. Rep. 2014, 4, 4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bampoulis P., Sotthewes K., Siekman M. H., Zandvliet H. J. W., Sci. Rep. 2017, 7, 43451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pastré D., Piétrement O., Fusil S., Landousy F., Jeusset J., David M. O., Hamon L., Cam E. L., Zozime A., Biophys. J. 2003, 85, 2507–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nosrati A., Addai-Mensah J., Skinner W., Chem. Eng. J. 2009, 152, 406–414. [Google Scholar]
- 53. Scales P. J., Grieser F., Healy T. W., Langmuir 1990, 6, 582–589. [Google Scholar]
- 54. Hsueh C., Chen H., Gimzewski J. K., Reed J., Abdel-Fattah T. M., ACS Appl. Mater. Interfaces 2010, 2, 3249–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hansma H. G., Laney D. E., Biophys. J. 1996, 70, 1933–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ricci M., Spijker P., Voïtchovsky K., Nat. Commun. 2014, 5, 4400. [DOI] [PubMed] [Google Scholar]
- 57. Lee S. S., Fenter P., Nagy K. L., Sturchio N. C., Langmuir 2012, 28, 8637–8650. [DOI] [PubMed] [Google Scholar]
- 58. Perrott K. W., Geoderma 1981, 26, 311–322. [Google Scholar]
- 59. Bhattacharjee S., J. Controlled Release 2016, 235, 337–351. [DOI] [PubMed] [Google Scholar]
- 60. Yang Y., Han D., Nangreave J., Liu Y., Yan H., ACS Nano 2012, 6, 8209–8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Black J. M., Zhu M., Zhang P., Unocic R. R., Guo D., Okatan M. B., Dai S., Cummings P. T., Kalinin S. V., Feng G., et al., Sci. Rep. 2016, 6, 32389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shlyakhtenko L. S., Gall A. A., Weimer J. J., Hawn D. D., Lyubchenko Y. L., Biophys. J. 1999, 77, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary