

Abstract
Endothelial dysfunction and diabetic vascular disease induced by chronic hyperglycemia involve complex interactions among high glucose, long non-coding RNAs (lncRNAs), microRNAs (miRNAs or miRs) and the Ser/Thr kinase AKT. However, the molecular mechanisms under-lying the regulatory crosstalk between these have not yet been completely elucidated. Thus, the present study aimed to explore the molecular mechanisms whereby high glucose (HG)-induced lncRNA MIR181A2HG modulates human umbilical vein endothelial cell (HUVEC) proliferation and migration by regulating AKT2 expression. The persistent exposure of HUVECs to HG resulted in MIR181A2HG down-regulation and thus reduced its ability to sponge miR-6832-5p, miR-6842-5p and miR-8056, subsequently leading to an increase in miR-6832-5p, miR-6842-5p and miR-8056 levels. Mechanistically, miR-6832-5p, miR-6842-5p and miR-8056 were found to target the 3′UTR of AKT2 mRNA in HUVECs, and the increase in their levels led to a decreased expression of AKT2. Thus, this also led to the suppression of HUVEC proliferation and migration, and the formation of capillary-like structures. Moreover, the suppression of HUVEC proliferation and migration induced by MIR181A2HG downregulation was accompanied by changes in glucose metabolism. On the whole, the present study demonstrates that the downregulation of lncRNA MIR181A2HG by HG impairs HUVEC proliferation and migration by dysregulating the miRNA/AKT2 axis. The MIR181A2HG/miRNA/AKT2 regulatory axis may thus be a potential therapeutic target for HG-induced endothelial dysfunction.
Keywords: high glucose, MIR181A2HG, miRNAs, AKT2, glucose metabolism, human umbilical vein endothelial cells
Introduction
Cardiovascular complications are the major causes of mortality for patients with type 2 diabetes mellitus (1). Endothelial cell injury and endothelial dysfunction play an important role in the development and progression of diabetic vascular disease (2). There is emerging evidence to indicate that chronic hyperglycemia is an independent risk factor for diabetic vascular disease in patients with type 1 diabetes and in those with type 2 diabetes mellitus (3). High glucose (HG) is well-known to exert multiple pathological effects on cardiovascular cells, including endothelial cells (ECs), smooth muscle cells and cardiomyocytes (4-6). In ECs, persistent exposure to HG induces the inflammatory response, oxidative stress, and cellular senescence and apoptosis, resulting in endothelial dysfunction in patients with diabetes (7-9). Thus, the pathogenesis of diabetic vascular complications is complex and multifactorial, and involves regulatory interactions between some important biological molecules. Despite advancements being made in the understanding of the toxic effects of HG on the cardiovascular system, the role of HG and the mechanisms of action of HG in mediating endothelial dysfunction are not yet fully understood.
AKT2 (GenBank Accession no. NM_001243027.3) is one of 3 closely related serine/threonine-protein kinases (AKT1, AKT2 and AKT3) and plays critical roles in cell survival, growth, proliferation, angiogenesis and cell metabolism (10,11). The aberrant loss or gain of AKT activation underlies the pathophysiological properties of a variety of complex diseases, including type 2 diabetes and cancer (10). The 3 AKT isoforms also play complex, yet critical roles in vascular health and vascular abnormalities (12). The activation of AKT1 by vascular endothelial growth factor (VEGF) promotes EC proliferation, migration and survival (13), and the loss of AKT1 in mouse ECs results in reduced nitric oxide (NO) release and impaired angiogenesis (14). In addition, AKT2 knockout mice exhibit insulin resistance and glucose intolerance and have type 2 diabetes (15,16), and dominant-negative mutations in AKT2 lead to the genetic development of severe diabetes in humans (17). Taken together, these findings suggest that AKT signaling plays a major role in normal EC physiological functions and the pathogenesis of type 2 diabetes. However, the mechanisms underlying the regulation of AKT2 by HG in ECs have not yet been completely elucidated.
Long non-coding RNAs (lncRNAs) regulate gene expression at the transcription, epigenetic or translation level, thereby altering cellular response to various stimuli (18). They have been implicated in the regulation of the angiogenic response of endothelial cells in vitro and vascularization in vivo (19). Aberrant lncRNA expression is associated with a number of human disorders, particularly with proliferative diseases (19,20). A previous study revealed that lncRNA-MIAT knockdown evidently ameliorated diabetes-induced retinal microvascular dysfunction in vivo, and inhibited endothelial cell proliferation, migration and tube formation in vitro (21). There is also evidence to indicate that lncRNA-ATB promotes the viability, migration and angiogenesis of human microvascular endothelial cells by sponging miR-195 (22). Conversely, lncRNA MEG3 has been shown to negatively regulate the proliferation and angiogenesis of vascular endothelial cells by sponging miR-9 (23). Although lncRNAs are shown to regulate endothelial cell function, there is currently little information to describe the function of lncRNAs induced by HG in vascular endothelial cells.
In the present study, the expression of lncRNAs in HG-exposed endothelial cells was characterized and the functional role of lncRNA MIR181A2HG in the regulation of the proliferation and migration of human umbilical vein endothelial cells (HUVECs) in response to HG was investigated.
Materials and methods
Cell culture and treatment
HUVECs (cat. no. 8000) were purchased from ScienCell Research Laboratories, and routinely cultured in ECM (cat. no. 1001; ScienCell Research Laboratories) supplemented with glucose (5.5 mM), 1% penicillin/streptomycin (PS, cat. no. 0503; ScienCell Research Laboratories), 5% fetal bovine serum (FBS, cat. no. 0025; ScienCell Research Laboratories) and 1% endothelial growth factor (EGF, cat. no. 1052; ScienCell Research Laboratories), in a humidified incubator at 37°C with 5% CO2. The growth medium was replaced every 2 days, and the cells were sub-cultured every 3 days at a ratio of 1 to 4 upon reaching 80% confluence. Human aortic smooth muscle cells (HASMCs, cat. no. 6110) were purchased from ScienCell Research Laboratories, and routinely cultured in smooth muscle cell medium (cat. no. 1101; ScienCell Research Laboratories) in a humidified incubator at 37°C with 5% CO2. D-glucose (Sigma-Aldrich; Merck KGaA) was added to the medium which contained 2% FBS, 1% PS and 1% EGF to generate hyperglycemic conditions. D-mannitol (30 mM; Sigma-Aldrich; Merck KGaA) was added to ensure a similar osmotic pressure. 293 cells stored in The Key Laboratory of Neural and Vascular Biology were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS. All cells were transfected using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Briefly, in 24-well plates, 1 μg plasmid or 20 pmol shRNA, miRNA mimcs or control and 1 μl Lipofectamine 2000 reagent were mixed gently in Opti-MEM™ (Thermo Fisher Scientific, Inc.) at room temperature. After 15 min, the mixture was incubated with the cells for 4 h and replaced with ECM medium. For luciferase reporter assay, 0.5 μg plasmid and 10 pmol miRNA mimics for miR-6832-5p, miR-6842-5p or miR-8056, shRNA or control were mixed to co-transfect cells with 1 μl Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, Inc.) together in a 24-well plate. AKT inhibitor (1 μM; MK2206; Beyotime Institute of Biotechnology) was used to treat the HUVECs for 72 h to block the AKT signaling pathway.
Microarray analysis
HUVECs were exposed to 30 or 5.5 mM D-glucose for 24 h. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v12.1 software package (Agilent Technologies, Inc.). A NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc.) was used to measure the quantity and quality of the RNA. Arraystar Human lncRNA Microarray V3.0 was designed for the global profiling of human lncRNAs. Following the isolation of rRNA (using the mRNA-ONLY™ Eukaryotic mRNA Isolation kit, Epicentre), mRNA was purified from total RNA. The RNA was amplified and transcribed into fluorescent cRNA by utilizing a random priming method (Arraystar Flash RNA Labeling kit, Arraystar). Each labeled cRNA was fragmented by the addition of Blocking Agent and Fragmentation Buffer (Agilent Technologies, Inc.), and the mixture was then heated at 60°C for 30 min. The labeled cRNA was diluted by 2xGE Hybridization buffer (Agilent Technologies, Inc.). The hybridization solution was then dispensed into the gasket slide and assembled to the lncRNA expression microarray slide for 17 h at 65°C in Hybridization Oven (Agilent Technologies, Inc.). Finally, the hybridized arrays were washed, fixed and scanned with using the Agilent DNA Microarray Scanner (no. G2505C; Agilent Technologies, Inc.).
Isolation of RNA and RT-qPCR
Total RNA was extracted using the E.Z.N.A.® Total RNA kit I (Omega Bio-tek) according to the manufacturer's instructions. The quality of the RNA was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). For lncRNA and AKT2 mRNA, cDNA was synthesized using a M-MLV First-Strand cDNA Synthesis kit (Invitrogen; Thermo Fisher Scientific, Inc.). RT-PCR was performed at 65°C (10 min), on ice (2 min), 40°C (30 min) and 70°C (10 min). The amplification program was 50°C (2 min), 95°C (2 min), followed by 40 cycles of 95°C (15 sec) and 60°C (30 sec). For microRNAs (miRNAs or miRs), RT-PCR and RT-qPCR were performed using the microRNA reverse transcription and RT-qPCR kit (GenePharma, Inc.). qPCR was carried on a Bio-Rad CFX Manager (Bio-Rad Laboratories, Inc.). The reverse transcription program was 25°C (30 min), 42°C (30 min) and 85°C (30 min). The amplification program was 95°C (3 min), followed by 40 cycles of 95°C (12 sec) and 62°C (40 sec). A relative amount of transcripts was normalized using the 2−ΔΔCq method with β-actin for large mRNAs and U6 for miRNAs. The specific primers used are listed in Table I.
Table I.
Sequences of primes used in the present study.
| Primer name | Sequence |
|---|---|
| AKT2-qPCR-Forward | TGGTCGCCAACAGCCTCA |
| AKT2-qPCR-Reverse CC | GCCACTTCCATCTCCTCA |
| MIR181A2HG-qPCR-Forward C | GCGGTTCAATACCTCGTCT |
| MIR181A2HG-qPCR-Reverse | TGCTGTGGCTAGAGGACAAC |
| β-actin-qPCR-Forward | ACTCTTCCAGCCTTCCTTCC |
| β-actin-qPCR-Reverse C | GTACAGGTCTTTGCGGATG |
| shRNA-MIR181A2HG-Forward | GGGAUAGUAGAAAGUAACAGGCUCUTT |
| shRNA-MIR181A2HG-Reverse | AGAGCCUGUUACUUUCUACUAUCCCTT |
| MIR181A2HG probe 1 human-392 | CTTTCCACAGGACAGTTCGC |
| MIR181A2HG probe 2 human-377 | TTCGCCTTTCCTTCCTTTCCTCTGT |
| MIR181A2HG probe 3 human-19 | CCAGATTTCCTTGCAGCCC |
| miR-6832-5p mimics sense | AGUAGAGAGGAAAAGUUAGGGUC |
| miR-6832-5p mimics antisense | CCCUAACUUUUCCUCUCUACUUU |
| miR-6842-5p mimics sense | UGGGGGUGGUCUCUAGCCAAGG |
| miR-6842-5p mimics antisense | UUGGCUAGAGACCACCCCCAUU |
| miR-7110-5p mimics sense | UGGGGGUGUGGGGAGAGAGAG |
| miR-7110-5p mimics antisense | CUCUCUCCCCACACCCCCAUU |
| miR-6752-5p mimics sense | GGGGGGUGUGGAGCCAGGGGGC |
| miR-6752-5p mimics antisense | CCCCUGGCUCCACACCCCCCUU |
| miR-8056 mimics sense | CGUGGAUUGUCUGGAUGCAU |
| miR-8056 mimics antisense | GCAUCCAGACAAUCCACGUU |
| miR-181-3p mimics sense | ACCACTGACCGTTGACTGTAC |
| miR-181-3p mimics antisense | UACAGUCAACGGUCAGUGGUUU |
| miR-181-5p mimics sense | AACATTCAACGCTGTCGGTGAGT |
| miR-181-5p mimics antisense | UCACCGACAGCGUUGAAUGUUU |
| mimics Ctl sense | UUCUCCGAACGUGUCACGUTT |
| mimics Ctl antisense | ACGUGACACGUUCGGAGAATT |
| shRNA-AKT2-Forward C | GUGGUGAAUACAUCAAGATT |
| shRNA-AKT2-Reverse | UCUUGAUGUAUUCACCACGTT |
| miR-6832-5p-qPCR-Forward C | GCTGCGAGTAGAGAGGAAAAG |
| miR-6832-5p-qPCR-Reverse | TATGGTTCTTCACGACTGGTTCAC |
| miR7110-5p-qPCR-Forward C | TTTATTATGGGGGTGTGGGG |
| miR-7110-5p-qPCR-Reverse | TATGGTTGTTCACGAGTCCTTGTC |
| miR-8056-5p-qPCR-Forward CC | TCGCTCGTGGATTGTCT |
| miR-8056-5p-qPCR-Reverse | TATGGTTGTTCACCTCTCCTTCAC |
| miR-181-3p-qPCR-Forward C | TTTATTAACCACTGACCGT |
| miR-181-3p-qPCR-Reverse | TATGGTTCTTCACGACTGGTTCAC |
| miR-181-5p-qPCR-Forward C | TTTATTAAACATTCAACGC |
| miR-181-5p-qPCR-Reverse | TATGGTTCTTCACGACTGGTTCAC |
| U6-qPCR-Forward C | GCTTCGGCAGCACATATAC |
| U6-qPCR-Reverse | TTCACDAATTTGCGTGTCATC |
Plasmid construction and shRNA
Recombinant plasmids expressing lncRNA-MIR181A2HG and AKT2 were constructed by GENEWIZ. The MIR181A2HG sequences and the 3′UTR sequences of AKT2 mRNA containing wild-type (WT) and mutant binding sites for miR-6832-5p, miR-6842-5p or miR-8056 (synthesized by GENEWIZ) were inserted into pmirGLO Dual-Luciferase vector (Promega Corporation). shRNAs against the lncRNA-MIR181A2HG and AKT2 were designed and synthesized by GENEWIZ. The detailed sequences are listed in Table I.
Target prediction and luciferase reporter assay
The TargetScan prediction algorithm (http://www.targetscan.org/) to predict the binding between MIR181A2HG, miRNAs and AKT2. Luciferase reporter assay was performed as previously described (22). In brief, 293 cells were seeded into a 24-well plate and pmirGLO reporter plasmids (WT or mutant) or their empty plasmid were co-transfected with the miRNA mimic (GenePharma, Inc.) or miR-Ctl (GenePharma, Inc.) and pRL-TK. Following 24 h of transfection, luciferase activity was measured using a Dual-Glo Luciferase Assay System (Promega Corporation) with a Flash and Glow reader. The luciferase activity was expressed as the relative activity ratio of Firefly luciferase to Renilla luciferase. The sequences of MIR181A2HG are presented in Table SI.
RNA pulldown assay
Biotinylated lncRNA MIR181A2HG was used to perform RNA pull-down. HUVECs were cross-linked with 1% formaldehyde in PBS and quenched with 0.125 M glycine. HUVECs were dissolved in lysis buffer on ice for 10 min and then sonicated with a microtip ultrasonicator on ice. A total of 100 pmol biotinylated probes were added to the cell lysate diluted with hybridization buffer. Subsequently, 100 μl washed/blocked Streptavidin Dynabeads (Thermo Fisher Scientific, Inc.) blocked in lysis buffer containing yeast tRNA and BSA were added per 100 pmol of probes, and the whole mix was then rotated for 30 min at 37°C. Dynabeads were captured by magnets (Thermo Fisher Scientific, Inc.), washed with wash buffer and then eluted with elution buffer.
Fluorescence in situ hybridization (FISH)
In situ hybridization was performed using fluorescence Cy3-labeled lncRNA-MIR181A2HG probes (50 nM) in hybridization buffer (Exiqon) by incubation at 55°C for 1 h. Following stringent washing with SSC buffer, non-specific binding sites were blocked with 10% normal goat serum (KPL). Images were acquired using a Leica microscope (Leica Microsystems GmbH) and digitized with a software of LAS AF software. The sequences of MIR181A2HG probes are presented in Table I.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde and pre-incubated with 10% normal goat serum (KPL) at room temperature and then incubated with primary antibody anti-AKT2 (mouse monoclonal, 1:100, ab175354, Abcam) at 4°C overnight. Secondary antibody was FITC-labeled antibody to mouse IgG (cat. no. 5230-0307, KPL, 1:50, US) and incubated at room temperature for 2 h. Antifading mounting medium (with DAPI) (Beijing Solarbio Science & Technology Co., Ltd.) was used for nuclear staining at room temperature and stored at 4°C. Images were captured using a confocal microscope (Leica Microsystems GmbH) and digitized using LAS AF software (v. 2.6.0, Leica Microsystems GmbH).
MTS assay
After the HUVECs were treated with plasmids and 5.5 or 30 mM glucose, the viability of the HUVECs cultured in 96-well plates was measured by MTS assay, as previously described (24). Following the addition of glucose 1-5 days later or transfection 24 later, the ECM medium of cultured HUVECs was replaced with 100 μl ECM medium containing 10 μl of CellTiter 96 AQueous One Solution (Promega Corporation). The plates were then incubated at 37°C for 4 h. The 96-well plate was then examined at 490 nm using a Multiskan spectrum spectrophotometer (Thermo Fisher Scientific, Inc.).
Scratch test
HUVECs were treated with plasmids and 5.5 or 30 mM glucose. when the cells were 90-95% confluent, a sterile 200 μl pipette tip was used to draw straight lines in the middle of the well, and serum-free ECM medium was used to wash the cells twice, and this was then replaced with ECM medium containing 2% FBS, 1% PS and 1% EGF, as previously described (25). Images were collected (Canon EOS 600d) at this time as a 0 h control. The culture was continued for a further 24 h at 37°C. The image data were collected after 24 h.
Transwell migration assay
A Transwell chamber was placed in a 24-well plate, and 800 μl of ECM medium containing 10% FBS was added to each well. A total of 2×104 cells were diluted in 200 μl of serum-free ECM medium and were added to the upper chamber. Following incubation at 37°C for 24 h, the upper surface of the membrane was wiped with a cotton-tipped applicator to remove non-migratory cells and the migratory cells on the under surface were fixed with 10% formaldehyde and stained with 2% crystal violet (Beijing Solarbio Science & Technology Co., Ltd.) solution for 10 min at room temperature. The membranes were mounted on glass slides and the number of migrated cells from 10 random microscopic fields were counted by manual counting after obtaining images. The image data were collected using a Canon EOS 600d camera (Canon EOS 600d).
3D-culture
Matrigel gel was placed in a 24-well plate at 40 μl per well in a 37°C incubator for 2 h. After the cells were properly treated, HUVECs were digested and the cell density was adjusted to 3×105/ml. The cell suspension was added to the surface of Matrigel gel and placed in a 37°C incubator. The degree of tube formation was evaluated using a microscope after 24 h incubation at 37°C and 5% CO2. The full capillary-like structures in the field were counted and collected using a Canon EOS 600d camera.
Measurement of total adenosine triphosphate (ATP) concentration
The total ATP level was detected using the ATP Assay kit (Beyotime Institute of Biotechnology) according to the manufacturer's instructions. A total of 100 μl working solution was added to a 10 μl diluted sample, and the luciferase activity was immediately evaluated by Modulus™ II Microplate multifunction tester. The ATP content was determined by comparison to a concurrent standard curve and was then normalized by cell number and expressed as μmol/ml.
Glucose uptake assay and glycogen synthesis assay
Before and after the cells were transfected with pGFPN1, pGFPN1/MIR181A2HG, pGFPN1/AKT2 or miRNA mimics, glucose in the cell culture medium was measured using Glucose Uptake Colorimetric assay kit (Sigma-Aldrich; MerckKGaA). Glucose uptake was calculated by the difference before and after the cells were treated. After the cells were treated, the glycogen level was detected using a glycogen assay kit (Nanjing Jiancheng Bioengineering Inc.), and the results were normalized to a standard curve and expressed as μg of glycogen, according to the manufacturer's instructions.
Western blot analysis
Cells were lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology). Equal amounts of protein (20 μg for total protein and for 60 μg for p-GSK3β) were run on 10% SDS-PAGE, and electro-transferred to a polyvinylidene fluoride (PVDF) membranes (EMD Millipore). PVDF membranes were blocked with 5% milk in TTBS at room temperature for 2 h and then incubated with primary anti-bodies, overnight at 4°C. The antibodies used were as follows: Anti-AKT2 (mouse monoclonal, 1:500, ab175354), anti-proliferating cell nuclear antigen (PCNA; rabbit monoclonal, 1:500, ab92552), anti-matrix metalloproteinase 2 (MMP2; rabbit polyclonal, 1:1,000, ab97779), anti-VE-cadherin (rabbit polyclonal, 1:500, ab33168), anti-GLUT1 (rabbit monoclonal, 1:1,000, ab150299), anti-glycogen synthase kinase 3β (GSK3β; mouse monoclonal, 1:500, ab93926), anti-p-GSK3β (rabbit polyclonal, 1:1,000, ab75745) or anti-β-actin (rabbit polyclonal, 1:1,000, ab8227 and mouse polyclonal, 1:1,000, ab8226) (all from Abcam). PVDF membranes were then incubated with the HRP-conjugated secondary antibody (goat anti-rabbit cat. no. 074-1506 or goat anti-mouse, cat. no. 074-1806 1:8,000, KPL) for 1 h at room temperature. The blots were treated using a high-sensitivity ECL chemiluminescence detection kit (Nanjing Vazyme Biotech Co., Ltd.) and exposed by films. The densitometry of blots was measured using ImageJ software (v.1.51j8, National Institutes of Health).
Statistical analysis
All statistical analyses were performed using the GraphPad Prism software (version 5.0; GraphPad Software, Inc.). Data are expressed as the means ± standard deviation. All data were tested for normality and equal variance and were proved to be consistent with variance with R studio software. The Student's t-test was used to compare the statistical differences between 2 groups. One-way ANOVA or two-way ANOVA followed by Tukey's post hoc test was used for comparisons among >2 groups. All experiments were repeated independently at least 3 times. A value of P≤0.05 was considered to indicate a statistically significant difference.
Results
Persistent exposure of HUVECs to HG increases miR-6832-5p, miR-6842-5p and miR-8056 expression levels by downregulating MIR181A2HG
As lncRNAs are known to be involved in diabetes mellitus-induced vascular dysfunction (21), the present study determined the effects of the persistent exposure of HUVECs to HG on lncRNA expression. HUVECs were exposed to 5.5 mM (normal glucose) and 30 mM glucose (HG) for different periods of time. It was found that HG significantly increased cell viability during the first 2 days, whereas after 3 days of exposure to HG, the viability of the HG-exposed HUVECs decreased significantly compared with the cells treated with normal glucose or 30 mM mannitol, which was used to eliminate the possible effects of osmotic pressure (Fig. 1A). Subsequently, lncRNA microarray analysis was performed in the HUVECs exposed to 5.5 or 30 mM glucose for 24 h. The results revealed that 709 or 457 lncRNAs were upregulated or downregulated, respectively, in the HUVECs exposed to HG for 24 h compared to those treated with 5.5 mM glucose (Fig. 1B and Table SII). As the expression level of MIR181A2HG was highest in the HUVECs exposed to HG for 24 h compared with those of the other lncRNAs, lncRNA MIR181A2HG (GenBank Accession no. NR_038975.1) was selected to validate the microarray data by RT-qPCR. The results revealed that MIR181A2HG expression was markedly increased after 1 day of stimulation with HG, but significantly decreased 4 days later (Fig. 1C). Similar results were obtained by FISH using probes against MIR181A2HG (Fig. 1D). These results suggest that the expression of MIR181A2HG may be associated with the viability of HUVECs exposed to HG.
Figure 1.

Persistent exposure of HUVECs to HG increases miR-6832-5p, miR-6842-5p and miR-8056 expression by downregulating MIR181A2HG. (A) MTS assay detected the viability of HUVECs treated with different doses of D-glucose for the indicated times. **P<0.01 and ***P<0.001 vs. day 0. (B) Heatmap representation of differential expression of lncRNAs between HUVECs treated with 5.5 mM (L5.5) and 30 mM D-glucose (H30) for 24 h. (C) RT-qPCR was used to detect the relative expression of MIR181A2HG in HUVECs cultured in medium containing 30 mM D-glucose for the indicated times. *P<0.05 and ***P<0.001 vs. day 0. (D) FISH was used to detect the location and expression of MIR181A2HG in HUVECs exposed to 5.5 or 30 mM D-glucose for the indicated times. (E) Predicted binding sites of the miRNAs on MIR181A2HG. (F) Luciferase reporter assay was used to verify the direct binding between MIR181A2HG and miRNAs. *P<0.05 and **P<0.01 vs. miR-Ctl. (G) RT-qPCR was used to detect the relative expression of the indicated miRNAs in HUVECs cultured in medium containing 30 mM D-glucose for 96 h. *P<0.05 and **P<0.01 vs. Ctl. (H) RNA pulldown assay was performed to detect the direct binding between MIR181A2HG and the 3 miRNAs. **P<0.01 vs. Ctl. (I and J) RT-qPCR was used to detect the relative expression of (I) MIR181A2HG and (J) miRNAs in HASMCs cultured in medium containing 5.5 or 30 mM D-glucose. (K and L) RT-qPCR and western blot analysis detected the relative expression of AKT2 mRNA and protein in HASMCs transfected with miR-181-3p mimics, miR-181-5p mimics or their corresponding control. HUVECs, human umbilical vein endothelial cells; HASMCs, human aortic smooth muscle cells.
As a number of lncRNAs have been known to act as competing endogenous RNAs (ceRNA) by interacting with miRNAs (26,27), the present study investigated whether MIR181A2HG could interact with miRNAs in HUVECs. Using the TargetScan prediction algorithm (http://www.targetscan.org/), 5 miRNAs were predicted (miR-6832-5p, miR-6842-5p, miR-7110-5p, miR-6752-5p and miR-8056) targeting sites on MIR181A2HG (Fig. 1E). Subsequently, MIR181A2HG was cloned, which contains WT or the mutated binding sites for the predicted 5 miRNAs, into pm i r GLO plasm id (pm i r GLO -MI R181A2HG or pmirGLO-MIR181A2HG-mut) and examined the effects of the predicted 5 miRNAs on MIR181A2HG-directed luciferase activity. After the successful transfection of these 5 miRNA mimics into HUVECs was confirmed by RT-qPCR (Fig. S1A), it was found that co-transfection of miR-6832-5p, miR-6842-5p or miR-8056, but not miR-6752-5p and miR-7110-5p, with pmirGLO-MIR181A2HG significantly reduced the luciferase activities directed by the WT reporter vector. The mutation of binding sites for miR-6832-5p, miR-6842-5p or miR-8056 on MIR181A2HG (pmirGLO-MIR181A2HG-mut) abolished this inhibition (Fig. 1F). In parallel with these findings, RT-qPCR revealed that the levels of miR-6832-5p, miR-6842-5p and miR-8056 expression were significantly increased in the HUVECs exposed to HG for 4 days compared with that of the cells exposed to normal glucose (Fig. 1G). To further validate the direct association of MIR181A2HG with these 3 miRNAs, RNA pull-down assay was performed, demonstrating that miR-6832-5p, miR-6842-5p and miR-8056 were enriched in the precipitates of 3′-biotinylated MIR181A2HG (Fig. 1H). These results suggested that MIR181A2HG specifically associated with these 3 miRNAs, and that the prolonged exposure of HUVECs to HG downregulated MIR181A2HG expression, abolishing the interaction between MIR181A2HG and miR-6832-5p, miR-6842-5p or miR-8056 in HUVECs.
The present study also examined that the effects of HG on the expression of MIR181A2HG and the 3 miRNAs in HASMCs. The results revealed that the exposure of HASMCs to HG did not significantly affect the expression of MIR181A2HG (Fig. 1I) and the 3 miRNAs (Fig. 1J), suggesting that the effect and regulatory mechanisms of HG on MIR181A2HG/miRNAs in HUVECs was specific, which is different from the HASMCs. Furthermore, considering that lncRNA MIR181A2HG is the MIR181A2 (including miR-181-3p and miR-181-5p) host gene, the present study investigated whether miR-181-3p and miR-181-5p directly target AKT2. After the successful transfection of miR-181-3p and miR-181-5p into HUVECs was verified (Fig. S1B), RT-qPCR and western blot analysis were performed and it was revealed that the transfection of HUVECs with these 2 miRNAs did not affect the expression of AKT2 mRNA and protein (Fig. 1K and L), indicating that lncRNA MIR181A2HG-derived miR181-3p and miR181-5p do not directly target the AKT2 gene in HUVECs. miR-6832-5p, miR-6842-5p and miR-8056 target the AKT2 3′UTR and regulate its expression in HUVECs. As the downregulation of lncRNA MIR181A2HG induced by HG increased the miR-6832-5p, miR-6842-5p and miR-8056 levels, the bioinformatics database, TargetScan, was used to predict the targets of miR-6832-5p, miR-6842-5p and miR-8056. It was found that the AKT2 3′UTR contained the potential binding sites of these 3 miRNAs (Fig. 2A). The AKT downstream substrates GSK3β and GLUT1 contained no binding sites for these miRNAs within their 3′UTR. Subsequently, luciferase reporter plasmids which contained the WT (pmirGLO-AKT2 3′UTR) or mutated (pmirGLO-AKT2 3′UTR-mut) binding sites for these miRNAs in the AKT2 3′UTR were constructed. Luciferase reporter assay revealed that co-transfection with miR-6832-5p, miR-6842-5p or miR-8056 and pmirGLO-AKT2 3′UTR reduced the luciferase activity, respectively, by 67.59, 58.27 and 45.58%, while no obvious changes were observed when these 3 miRNAs were co-transfected with pmirGLO-AKT2 3′UTR-mut (Fig. 2B). After the successful MIR181A2HG knockdown and overexpression were confirmed via RT-qPCR (Fig. S1C), it was found that the knockdown of MIR181A2HG decreased, whereas the overexpression of MIR181A2HG increased the luciferase activity directed by pmirGLO-AKT2 3′UTR, but not pmirGLO-AKT2 3′UTR-mut (Fig. 2C).
Figure 2.

miR-6832-5p, miR-6842-5p and miR-8056 target AKT2 3′UTR in HUVECs. (A) The predicted binding sites between miR-6832-5p, miR-6842-5p or miR-8056 and AKT2 3′UTR. Red color indicates the sequence of mutated sites. (B) Luciferase reporter assay was performed in HUVECs co-transfected with miRNAs, pRL-TK or pmirGLO-AKT2 3′ UTR (wild-type or mut). *P<0.05 and **P<0.01 vs. miR-Ctl. (C) Luciferase reporter assay was performed in HUVECs co-transfected with pmirGLO-AKT2 3′ UTR (wild-type or mut) and pcDNA3.1-MIR181A2HG or shR-MIR181A2HG. **P<0.01 vs. pcDNA3.1; #P<0.01 vs. shR-Ctl. (D-F) RT-qPCR, western blot analysis and immunofluorescence staining detected the relative expression of AKT2 mRNA and protein in HUVECs transfected with miR-6832-5p, miR-6842-5p, or miR-8056 mimics. *P<0.05 and **P<0.01 vs. miR-Ctl. (G-I) RT-qPCR, western blot analysis and immunofluorescence staining detected the relative expression of AKT2 mRNA and protein in HUVECs treated with 5.5 or 30 mM D-glucose for different time periods. **P<0.01 vs. 5.5 mM D-glucose. (J) Immunofluorescence staining detected the expression of AKT2 in MIR181A2HG-overexpressing or depleted HUVECs. (K and L) RT-qPCR and western blot analysis detected the expression of AKT2 mRNA and protein in MIR181A2HG-depleted or AKT2-overexpressing HUVECs. **P<0.01 vs. shR-Ctl+pGFP-N1; ##P<0.01 vs. shR-MIR181A2HG+pGFP-N1. HUVECs, human umbilical vein endothelial cells; ns, not significant.
To provide further evidence that MIR181A2HG increased the expression of AKT2 by sponging miR-6832-5p, miR-6842-5p and miR-8056, HUVECs were transfected with these miRNAs, respectively. RT-qPCR and western blot analysis revealed that miR-6832-5p, miR-6842-5p and miR-8056 overexpression significantly decreased AKT2 expression at both the mRNA and protein level (Fig. 2D and E). A similar result was also obtained by the immunofluorescence staining of AKT2, demonstrating that these 3 miRNAs evidently reduced AKT2 expression compared to the miR-Ctl (Fig. 2F). Simultaneously, the expression of AKT2 mRNA and protein was significantly suppressed in the HUVECs exposed to HG, but not in those exposed to normal glucose or mannitol, for 4 days, as shown by RT-qPCR, western blot and immunofluorescence staining (Fig. 2G-I). In addition, immunofluorescence staining revealed that MIR181A2HG overexpression enhanced, whereas its knockdown attenuated the protein expression of AKT2 in the HUVECs relative to their corresponding control (Fig. 2J). After the successful overexpression of AKT2 was confirmed in HUVECs (Fig. S1D), rescue experiments showed that the inhibition of AKT2 mRNA and protein expression induced by MIR181A2HG knockdown could be rescued by overexpression of AKT2 (Fig. 2K and L). These results suggest that the prolonged exposure of HUVECs to HG resulted in MIR181A2HG downregulation and thus in its reduced association with miR-6832-5p, miR-6842-5p and miR-8056, subsequently leading to an inhibition of AKT2 expression.
Knockdown of MIR181A2HG inhibits the proliferation, migration and capillary-like structures of HUVECs
The above-mentioned results led us to investigate the biological functions of MIR181A2HG in HUVECs. Using loss- and gain-of function approaches, it was found that overexpression of MIR181A2HG promoted, whereas the knockdown of MIR181A2HG inhibited the proliferation of HUVECs, as shown by MTS assay (Fig. 3A). A scratch test and Transwell migration assay were then performed to determine the effects of MIR181A2HG on the migration of HUVECs. The overexpression or knockdown of MIR181A2HG promoted or inhibited the migration of HUVECs, respectively, compared with their corresponding control (Fig. 3B and C). Subsequently, 3D-culture was performed to examine the ability of MIR181A2HG to form capillary-like structures. The results revealed that the knockdown of MIR181A2HG significantly attenuated the formation of capillary-like structures, while the overexpression of MIR181A2HG increased this capacity (Fig. 3D). Taken together, these results indicated that MIR181A2HG facilitated the proliferation, migration and capillary-like structure formation in HUVECs.
Figure 3.

Knockdown of MIR181A2HG inhibits proliferation, migration and capillary-like structures of HUVECs. (A-C) HUVECs were transfected with pGFP-N1-MIR181A2HG, shR-MIR181A2HG or their corresponding control. (A) MTS assay, (B) scratch test, and (C) Transwell migration assay were used to detect the cell viability and migration. *P<0.05 and **P<0.01 vs. pGFP-N1; ##P<0.01 vs. shR-Ctl. (D) 3D-culture was used to detect the formation of capillary-like structures. *P<0.05 vs. pGFP-N1; ##P<0.01 vs. shR-Ctl. (E-H) HUVECs were transfected with shR-MIR181A2HG or shR-Ctl and exposed to 5.5 or 30 mM D-glucose. (E) MTS assay, (F) scratch test, (G) Transwell migration, and (H) 3D-culture were used to detect the cell viability, migration and the formation of capillary-like structures. **P<0.01 vs. shR-Ctl+5.5 mM D-glucose; ##P<0.01 vs. shR-Ctl+30 mM D-glucose. (I) Western blot analysis was used to detect PCNA, MMP2 and VE-cadherin expression in HUVECs transfected with pGFP-N1-MIR181A2HG, shR-MIR181A2HG or their corresponding control. HUVECs, human umbilical vein endothelial cells.
To further corroborate the causal role of MIR181A2HG in HUVEC proliferation and migration, HUVECs were exposed to HG for 24 h to stimulate HUVEC proliferation and migration, and the effects of MIR181A2HG depletion on HUVEC biological behavior were observed. As was anticipated, the exposure of HUVECs to HG for 24 h significantly enhanced cell proliferation, whereas the knockdown of MIR181A2HG greatly abolished this effect (Fig. 3E). Similarly, exposure to HG for 24 h combined with MIR181A2HG knockdown also abrogated the inducing effects of HG on HUVEC migration, as shown by scratch wound healing assay and Transwell migration assay (Fig. 3F and G). Moreover, MIR181A2HG depletion markedly reduced the formation of capillary-like structures induced by exposure to HG for 24 h (Fig. 3H). To further verify the functions of MIR181A2HG, cell proliferation and migration-related markers, including PCNA, MMP2 and VE-cadherin were detected. It was found that MIR181A2HG promoted the expression of PCNA, MMP2 and VE-cadherin in HUVECs (Fig. 3I).
MIR181A2HG/miRNAs/AKT2 axis regulates proliferation, migration and capillary-like structure formation in HUVECs
Subsequently, the present study investigated whether these 3 miRNAs and AKT2 could exert effects on HUVEC proliferation, migration and capillary-like structure formation. The transfection of miR-6832-5p, miR-6842-5p or miR-8056 significantly inhibited the proliferation and migration of HUVECs, compared with the control group, as examined by MTS assay, scratch test and Transwell migration assay (Fig. 4A-C). Correspondingly, 3D-culture revealed that these miRNAs significantly decreased the formation of the capillary-like structures in HUVECs (Fig. 4D). Consistent with the above-mentioned results, miR-6832-5p, miR-6842-5p and miR-8056 markedly suppressed the expression levels of PCNA, MMP2 and VE-cadherin (Fig. 4E). Moreover, western blot analysis revealed that the expression of PCNA, MMP2 and VE-cadherin was markedly inhibited by the AKT inhibitor, MK2206, in the HUVECs (Fig. 4F).
Figure 4.

MIR181A2HG/miRNAs/AKT2 axis regulates proliferation, migration and capillary-like structures of HUVECs. (A-D) MTS assay, scratch test, Transwell migration, and 3D-culture were used to detect the cell viability, migration, and the formation of capillary-like structures. *P<0.05, **P<0.01 and ***P<0.001 vs. miR-Ctl. (E) Western blot analysis detected the expression of PCNA, MMP2 and VE-cadherin in HUVECs transfected with the different miRNAs. (F) Western blot analysis detected the expression of PCNA, MMP2 and VE-cadherin in HUVECs treated with the AKT2 inhibitor, MK2206. (G) MTS assay, (H) scratch test, (I) Transwell migration, and (J) 3D-culture were used to detect the cell viability, migration and the formation of capillary-like structures in HUVECs transfected with pGFP-N1-MIR181A2HG alone or together with shR-AKT2. **P<0.01 and ***P<0.001 vs. pGFP-N1+shR-Ctl; #P<0.05 and ##P<0.01 vs. pGFP-N1/MIR181A2HG+shR-Ctl. HUVECs, human umbilical vein endothelial cells.
To further determine whether AKT2 is required for the MIR181A2HG-mediated effects, AKT2 was knocked down using shRNA and MIR181A2HG was overexpressed by plasmid vector in HUVECs (Fig. S1C and D). As shown in Fig. 4G-J, the knockdown of AKT2 in HUVECs significantly abrogated the inducing effects of MIR181A2HG on proliferation, migration and capillary-like structure formation. Taken together, these results indicated that the prolonged exposure of HUVECs to HG impaired cell functions by dysregulating the MIR181A2HG/miRNAs/AKT2 axis.
The promotion of HUVEC proliferation and migration by MIR181A2HG is accompanied by changes in glucose metabolism
As AKT2 is known to be a central protein in many cellular processes, such as cell survival, proliferation, glucose uptake and metabolism and angiogenesis (28,29), the present study then sought to examine whether MIR181A2HG, which upregulates the expression of AKT2, also affects glucose metabolism in HUVECs. It was found that the overexpression or knockdown of MIR181A2HG increased or decreased, respectively, the ATP content, glucose uptake and glycogen synthesis compared with their corresponding control in HUVECs (Fig. 5A). The ATP content, glucose uptake and glycogen synthesis were also significantly decreased in the HUVECs transfected with miR-6832-5p, miR-6842-5p or miR-8056 (Fig. 5B). Likewise, AKT2 also enhanced the ATP content, glucose uptake and glycogen synthesis (Fig. 5C).
Figure 5.

The promotion of proliferation and migration by MIR181A2HG is accompanied by changes of glucose metabolism. (A) ATP content, glucose uptake and glycogen synthesis were detected by luminescence assay, the glucose oxidase (GOD) method and glycogen colorimetric assay in HUVECs transfected with pGFP-N1-MIR181A2HG, shR-MIR181A2HG or their corresponding control. *P<0.05 and **P<0.01 vs. pGFP-N1, #P<0.05 vs. shR-Ctl. (B) ATP content, glucose uptake and glycogen were measured in HUVECs transfected with miR-6832-5p, miR-6842-5p and miR-8056. *P<0.05 and **P<0.01 vs. miR-Ctl. (C) ATP content, glucose uptake and glycogen were measured in HUVECs transfected with pGFP-N1-AKT2, shR-AKT2 and their corresponding control. *P<0.05 and **P<0.01 vs. pGFP-N1; #P<0.05 and ##P<0.01 vs. shR-Ctl. (D-F) Western blot analyiss was used to detect the expression of GLUT1, p-GSK3β and GSK3β in HUVECs treated as in (A-C). *P<0.05 and **P<0.01 vs. pGFP-N1 or miR-Ctl, #P<0.05 and ##P<0.01 vs. shR-Ctl. HUVECs, human umbilical vein endothelial cells.
Considering the influence of MIR181A2HG downregulation on glucose metabolism, the present study then examined whether MIR181A2HG-miRNAs-AKT2 can regulate the expression of glucose metabolism-related genes, such as GLUT1 and GSK3β. The results revealed that the overexpression of MIR181A2HG enhanced GLUT1 expression and the phosphorylation of GSK3β. By contrast, the knockdown of MIR181A2HG reduced GLUT1 expression and GSK3β phosphorylation in HUVECs (Fig. 5D). Moreover, the transfection of miR-6832-5p, miR-6842-5p and miR-8056 inhibited GLUT1 expression and GSK3β phosphorylation (Fig. 5E). Furthermore, AKT2 promoted GLUT1 expression and GSK3β phosphorylation in HUVECs (Fig. 5F). Taken together, these results indicated that the injury of vascular endothelial cells induced by HG may be attributed to the downregulation of MIR181A2HG, which leads to the energy scarcity and the disruption of glucose metabolism.
Discussion
Previous studies have demonstrated that HG can damage vascular ECs by inducing inflammation (30), oxidative stress (31), apoptosis and cellular senescence (9). lncRNAs regulate EC function and vessel growth (19). However, it remains largely unclear whether and how HG-induced lncRNAs impair EC function. The main findings of the present study are the following: i) The persistent exposure of HUVECs to HG downregulated lncRNA MIR181A2HG expression; ii) MIR181A2HG downregulation significantly suppressed the proliferation, migration and angiogenesis of HUVECs by decreasing its sponge for miR-6832-5p, miR-6842-5p and miR-8056; iii) miR-6832-5p, miR-6842-5p and miR-8056 target the 3′UTR of AKT2, and their increase elicited by MIR181A2HG downregulation attenuated expression of AKT2, and thus suppressed HUVEC proliferation, migration and the formation of the capillary-like structures.
It is well known that the endothelial cell monolayers located in the inner lining of blood vessels play a protective role by blocking direct contact between blood cells and the underlying tissue. Small defined areas of endothelial damage are rapidly repaired by the migration and proliferation of ECs in the vicinity (32). Thus, EC proliferation and migration are a key process in wound repair occurring on the luminal surface of blood vessels. Endothelial dysfunction may alter EC proliferation and migration, resulting in delayed endothelium repair with an increased risk of thrombosis (33). Moreover, several studies have demonstrated that the prevention of restenosis may be achieved by promoting endothelial regeneration through the use of growth factors (34), EC seeding (35), and vessel reconstruction with autologous EC/fibrin matrix (36). In the present study, it was demonstrated that the viability of HUVECs was significantly decreased in response to HG and that the toxic effects of HG on HUVECs were increased with the duration of HG stimulation. Specifically, the persistent exposure of HUVECs to HG resulted in an obvious downregulation of lncRNA MIR181A2HG. Although MIR181A2HG has been registered in the GenBank under the Accession no. 100379345, its function had not yet been reported. As the function of lncRNAs, as competitive endogenous RNAs (ceRNA) to regulate gene expression by sponging some miRNAs, is well established, we identified putative miRNAs associated with MIR181A2HG by using bioinformatics analyses. Luciferase reporter assays and the affinity pull-down of miRNAs by MIR181A2HG revealed that miR-6832-5p, miR-6842-5p and miR-8056 could be sponged by MIR181A2HG. Furthermore, opposite to the changes in MIR181A2HG expression in HUVECs exposed to HG for 4 days, the expression of miR-6832-5p, miR-6842-5p and miR-8056 was significantly enhanced in chronic HG-stimulated cells. These findings suggest that there is a specific interaction between these 3 miRNAs and MIR181A2HG, and the HG-induced downregulation of MIR181A2HG results in increased level of miR-6832-5p, miR-6842-5p and miR-8056 by reducing the association between MIR181A2HG and the 3 miRNAs.
Recently, some lncRNAs have been identified in ECs treated with different stimuli, including TNF-α (37), serum deprivation (38), hypoxia (39) and rapamycin (40). These lncRNAs regulate the cell viability, proliferation, migration and angiogenesis of ECs by sponging miRNA-21 and miR-199a-5p, as well as by modulating VEGFR2, ERK1/2, respectively. The present study then wished to determine the mechanisms through which MIR181A2HG-regulated miR-6832-5p, miR-6842-5p and miR-8056 affects HUVEC proliferation, migration and the formation of the capillary-like structures. First, the targets of miR-6832-5p, miR-6842-5p and miR-8056 we predicted by bioinformatics databases, and it was identified that AKT2 contains the potential binding sites of these 3 miRNAs in the 3′UTR of its mRNA. Luciferase reporter assays, RT-qPCR and western blot analysis confirmed that the overexpression of miR-6832-5p, miR-6842-5p and miR-8056 enhanced, whereas their knockdown attenuated the expression of AKT2 protein. Moreover, the persistent exposure of HUVECs to HG significantly attenuated the expression of AKT2 mRNA and protein, consistent with the above-mentioned observations that HG downregulated the MIR181A2HG expression level, and thus decreased its sponging for miR-6832-5p, miR-6842-5p and miR-8056, subsequently facilitating the suppression of AKT2 expression by miRNAs.
The serine/threonine kinase AKT, also known as protein kinase B (PKB), is a central node in cell signaling downstream of growth factors, cytokines, and other cellular stimuli (10). Mouse and human genetic studies have revealed physiological roles for the AKT network in nearly every organ system, and AKT also has particularly important roles in diverse pathological processes, including cardiovascular disease, insulin resistance and type 2 diabetes (12). Accumulating evidence has indicated shown that AKT expression is regulated at the post-transcriptional level by miRNAs. For instance, miR-373-3p targets the 3′UTR of AKT1 and inhibits its expression, and its downregulation promotes the proliferation of prostate cancer cells (41). miR-124 suppresses proliferation, glycolysis and energy metabolism in non-small cell lung cancer cells by targeting AKT1/2 (42). miR-16 inhibits cell proliferation and induces apoptosis in oral squamous cell carcinoma through decreasing AKT3 (43). However, little is known about whether AKT is regulated in ECs by miRNAs. In the present study, it was found that miR-6832-5p, miR-6842-5p and miR-8056 targeted, respectively, their corresponding sites at the 3′UTR of AKT2 mRNA and regulated AKT2 expression by enhancing its mRNA degradation and inhibiting translation. Among these 3 miRNAs, the inhibitory effect of miR-6832-5p on AKT2 expression was much more potent than the other 2 miRNAs. Thus, miR-6832-5p may have a greater impact on AKT2-mediated HUVEC proliferation and migration. These findings provide novel insight into the regulation of AKT2 at the post-transcriptional level by miRNAs.
Considering that AKT-mediated phosphorylation of its substrates, such as FOXO, GSK3, p27, eNOS, BAD and TSC2, contributes to activation of the various cellular processes, including survival, growth, proliferation, angiogenesis, glucose uptake and metabolism (10), it is reasonable to conclude that increased miR-6832-5p, miR-6842-5p and miR-8056 by MIR181A2HG downregulation attenuated the expression of AKT2 and thus impaired HUVEC proliferation, migration and angiogenesis (Fig. 6). These findings are consistent with previous evidence demonstrating that AKT signaling is attenuated in metabolic tissues in the insulin-resistant state that underlies type 2 diabetes (44). This newly identified MIR181A2HG-miRNAs-AKT2 regulatory axis may be a potential therapeutic target for HG-induced endothelial dysfunction.
Figure 6.
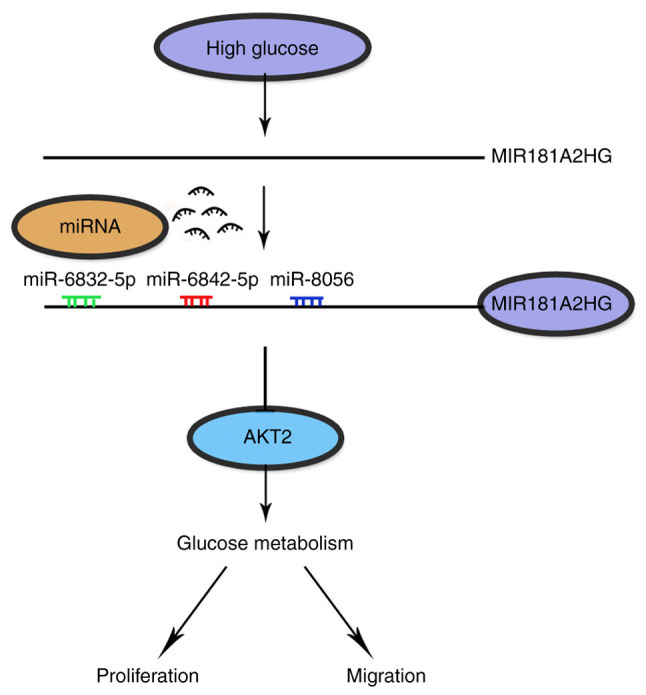
Schematic diagram of the regulation of HUVEC proliferation and migration by high glucose-mediated MIR181A2HG-miRNAs-AKT2 axis. HUVECs, human umbilical vein endothelial cells.
Supplementary Data
Acknowledgments
Not applicable.
Funding Statement
The present study was supported by grants from the National Natural Science Foundation of China (nos. 31871152 and 8177085) and the Graduate Innovation Program of Hebei Province (no. 2015).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
SW performed most of the experiments and wrote the original draft of the manuscript. HZ performed cell cultures and Transwell migration assay. XZ and YL performed the 3D-culture and contributed to the acquisition of data and statistical analyses. JW and BZ were involved in the study design, supervised the study, obtained funding for the study and revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Gregg EW, Gu Q, Cheng YJ, Narayan KM, Cowie CC. Mortality trends in men and women with diabetes 1971 to 2000. Ann Intern Med. 2007;147:149–155. doi: 10.7326/0003-4819-147-3-200708070-00167. [DOI] [PubMed] [Google Scholar]
- 2.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428. [PMC free article] [PubMed] [Google Scholar]
- 3.Madonna R, De Caterina R. Cellular and molecular mechanisms of vascular injury in diabetes-part I: Pathways of vascular disease in diabetes. Vascul Pharmacol. 2011;54:68–74. doi: 10.1016/j.vph.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Pei-Yuan Z, Yu-Wei L, Xiang-Nan Z, Song T, Rong Z, Xiao-Xiao H, Sheng-Shuai S, Kun W, Cheng-Yun L. Overexpression of Axl reverses endothelial cells dysfunction in high glucose and hypoxia. J Cell Biochem. 2019 Mar 8; doi: 10.1002/jcb.28462. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5.Zhang ML, Zheng B, Tong F, Yang Z, Wang ZB, Yang BM, Sun Y, Zhang XH, Zhao YL, Wen JK. iNOS-derived peroxynitrite mediates high glucose-induced inflammatory gene expression in vascular smooth muscle cells through promoting KLF5 expression and nitration. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2821–2834. doi: 10.1016/j.bbadis.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Jiang P, Zhang D, Qiu H, Yi X, Zhang Y, Cao Y, Zhao B, Xia Z, Wang C. Tiron ameliorates high glucose-induced cardiac myocyte apoptosis by PKCδ-dependent inhibition of osteopontin. Clin Exp Pharmacol Physiol. 2017;44:760–770. doi: 10.1111/1440-1681.12762. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Zhou Q, Pei C, Liu B, Li M, Fang L, Sun Y, Li Y, Meng S. Hyperglycemia and advanced glycation end products regulate miR-126 expression in endothelial progenitor cells. J Vasc Res. 2016;53:94–104. doi: 10.1159/000448713. [DOI] [PubMed] [Google Scholar]
- 8.Di Stefano V, Cencioni C, Zaccagnini G, Magenta A, Capogrossi MC, Martelli F. p66ShcA modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc Res. 2009;82:421–429. doi: 10.1093/cvr/cvp082. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF, Kamalanathan S, Hattori Y, Ignarro LJ, Iguchi A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA. 2006;103:17018–17023. doi: 10.1073/pnas.0607873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manning BD, Toker A. AKT/PKB Signaling: Navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemmings BA, Restuccia DF. The PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2015;7:a026609. doi: 10.1101/cshperspect.a026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 16.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Wei G, Luo H, Wu W, Skogerbø G, Luo J, Chen R. The long noncoding RNA SNHG1 promotes tumor growth through regulating transcription of both local and distal genes. Oncogene. 2017;36:6774–6783. doi: 10.1038/onc.2017.286. [DOI] [PubMed] [Google Scholar]
- 19.Michalik KM, You X, Manavski Y, Doddaballapur A, Zörnig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- 20.Yuan SX, Yang F, Yang Y, Tao QF, Zhang J, Huang G, Yang Y, Wang RY, Yang S, Huo XS, et al. Long noncoding RNA associated with microvascular invasion in hepatocellular carcinoma promotes angiogenesis and serves as a predictor for hepatocellular carcinoma patients' poor recurrence-free survival after hepatectomy. Hepatology. 2012;56:2231–2241. doi: 10.1002/hep.25895. [DOI] [PubMed] [Google Scholar]
- 21.Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–1156. doi: 10.1161/CIRCRESAHA.116.305510. [DOI] [PubMed] [Google Scholar]
- 22.Zhu AD, Sun YY, Ma QJ, Xu F. lncRNA-ATB promotes viability, migration, and angiogenesis in human microvascular endothelial cells by sponging microRNA-195. J Cell Biochem. 2019;120:14360–14371. doi: 10.1002/jcb.28692. [DOI] [PubMed] [Google Scholar]
- 23.He C, Yang W, Yang J, Ding J, Li S, Wu H, Zhou F, Jiang Y, Teng L, Yang J. Long noncoding RNA MEG3 negatively regulates proliferation and angiogenesis in vascular endothelial cells. DNA Cell Biol. 2017;36:475–481. doi: 10.1089/dna.2017.3682. [DOI] [PubMed] [Google Scholar]
- 24.Zhang LJ, Chen L, Lu Y, Wu JM, Xu B, Sun ZG, Zheng SZ, Wang AY. Danshensu has anti-tumor activity in B16F10 melanoma by inhibiting angiogenesis and tumor cell invasion. Eur J Pharmacol. 2010;643:195–201. doi: 10.1016/j.ejphar.2010.06.045. [DOI] [PubMed] [Google Scholar]
- 25.Wei N, Yu SP, Gu XH, Taylor TM, Song D, Liu XF, Wei L. Delayed intranasal delivery of hypoxic-preconditioned bone marrow mesenchymal stem cells enhanced cell homing and therapeutic benefits after ischemic stroke in mice. Cell Transplant. 2013;22:977–991. doi: 10.3727/096368912X657251. [DOI] [PubMed] [Google Scholar]
- 26.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Häggblad Sahlberg S, Mortensen AC, Haglöf J, Engskog MK, Arvidsson T, Pettersson C, Glimelius B, Stenerlow B, Nestor M. Different functions of AKT1 and AKT2 in molecular pathways, cell migration and metabolism in colon cancer cells. Int J Oncol. 2017;50:5–14. doi: 10.3892/ijo.2016.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muslin AJ. Akt2: A critical regulator of cardiomyocyte survival and metabolism. Pediatr Cardiol. 2011;32:317–322. doi: 10.1007/s00246-010-9879-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhu DD, Tang RN, Lv LL, Wen Y, Liu H, Zhang XL, Ma KL, Liu BC. Interleukin-1β mediates high glucose induced phenotypic transition in human aortic endothelial cells. Cardiovasc Diabetol. 2016;15:42. doi: 10.1186/s12933-016-0358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, et al. The hyperglycemia-induced inflammatory response in adipocytes: The role of reactive oxygen species. J Biol Chem. 2005;280:4617–4626. doi: 10.1074/jbc.M411863200. [DOI] [PubMed] [Google Scholar]
- 32.Kishimoto T, Oguri T, Abe M, Kajitani H, Tada M. Inhibitory effect of methylmercury on migration and tube formation by cultured human vascular endothelial cells. Arch Toxicol. 1995;69:357–361. doi: 10.1007/s002040050184. [DOI] [PubMed] [Google Scholar]
- 33.Tang R, Zhang G, Chen SY. Smooth muscle cell proangiogenic phenotype induced by cyclopentenyl cytosine promotes endothelial cell proliferation and migration. J Biol Chem. 2016;291:26913–26921. doi: 10.1074/jbc.M116.741967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kipshidze N, Dangas G, Tsapenko M, Moses J, Leon MB, Kutryk M, Serruys P. Role of the endothelium in modulating neointimal formation: vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions. J Am Coll Cardiol. 2004;44:733–9. doi: 10.1016/j.jacc.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 35.Kipshidze N, Ferguson JJ, III, Keelan MH, Jr, Sahota H, Komorowski R, Shankar LR, Chawla PS, Haudenschild CC, Nikolaychik V, Moses JW. Endoluminal reconstruction of the arterial wall with endothelial cell/glue matrix reduces restenosis in an atherosclerotic rabbit. J Am Coll Cardiol. 2000;36:1396–1403. doi: 10.1016/S0735-1097(00)00848-2. [DOI] [PubMed] [Google Scholar]
- 36.Kipshidze N, Dangas G, Tsapenko M, Moses J, Leon MB, Kutryk M, Serruys P. Role of the endothelium in modulating neointimal formation: Vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions. J Am Coll Cardiol. 2004;44:733–739. doi: 10.1016/j.jacc.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 37.Halimulati M, Duman B, Nijiati J, Aizezi A. Long noncoding RNA TCONS_00024652 regulates vascular endothelial cell proliferation and angiogenesis via microRNA-21. Exp Ther Med. 2018;16:3309–3316. doi: 10.3892/etm.2018.6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou J, Wang L, Wu Q, Zheng G, Long H, Wu H, Zhou C, Guo T, Zhong T, Wang L, et al. Long noncoding RNA H19 upregulates vascular endothelial growth factor A to enhance mesenchymal stem cells survival and angiogenic capacity by inhibiting miR-199a-5p. Stem Cell Res Ther. 2018;9:109. doi: 10.1186/s13287-018-0861-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruan W, Zhao F, Zhao S, Zhang L, Shi L, Pang T. Knockdown of long noncoding RNA MEG3 impairs VEGF-stimulated endothelial sprouting angiogenesis via modulating VEGFR2 expression in human umbilical vein endothelial cells. Gene. 2018;649:32–39. doi: 10.1016/j.gene.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 40.Sun H, Wang S, Song M. Long non-coding RNA SENCR alleviates the inhibitory effects of rapamycin on human umbilical vein endothelial cells. Mol Med Rep. 2018;18:1405–1414. doi: 10.3892/mmr.2018.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu HW, Jin Y, Cui ZL, Jin XB. MicroRNA-373-3p inhibits prostate cancer progression by targeting AKT1. Eur Rev Med Pharmacol Sci. 2018;22:6252–6259. doi: 10.26355/eurrev_201810_16032. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Lu C, Chu W, Zhang B, Zhen Q, Wang R, Zhang Y, Li Z, Lv B, Li H, Liu J. MicroRNA-124 suppresses proliferation and glycolysis in non-small cell lung cancer cells by targeting AKT-GLUT1/HKII. Tumour Biol. 2017;39:1010428317706215. doi: 10.1177/1010428317706215. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Li GH. MicroRNA-16 functions as a tumor-suppressor gene in oral squamous cell carcinoma by targeting AKT3 and BCL2L2. J Cell Physiol. 2018;233:9447–9457. doi: 10.1002/jcp.26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–1496. doi: 10.7150/ijbs.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.