

Abstract
Background & Aims:
Patients with inflammatory bowel diseases (IBD) have intestinal barrier dysfunction. Creatine regulates energy distribution within cells and reduces the severity of colitis in mice. We studied the functions of the creatine transporter solute carrier family 6 member 8 (SLC6A8, also called CRT) in intestinal epithelial cells (IECs) and mice, and measured levels in mucosal biopsies from patients with IBD.
Methods:
Colon biopsies from patients with IBD (30 with Crohn’s disease and 27 with ulcerative colitis) and 30 patients without IBD (controls), and colon tissues from mice (with and without disruption of Crt), were analyzed by immunofluorescence, immunoblots, and/or quantitative reverse-transcription PCR (qPCR). CRT was knocked down or overexpressed in T84 cells, which were analyzed by immunofluorescence, immunoblots, high-performance liquid chromatography (to measure creatine levels), qPCR, transepithelial electrical resistance, barrier function, actin localization, wound healing, mitochondrial oxygen consumption, and glycolysis extracellular acidification rate assays. Organoids from colon cells of CRT-knockout mice and control mice were analyzed by qPCR, immunoblot and transepithelial electrical resistance.
Results:
CRT localized around tight junctions (TJs) of T84 IECs. In analyses of IECs with CRT knockdown or overexpression, we found that CRT regulates intracellular creatine, barrier formation, and wound healing. CRT-knockout organoids also had diminished barrier formation. In the absence of adequate creatine, IECs transition toward a stressed, glycolysis-predominant form of metabolism; this resulted in leaky TJs and mislocalization of actin and TJ proteins. Colon tissues from patients with IBD had reduced levels of CRT mRNA compared with controls.
Conclusions:
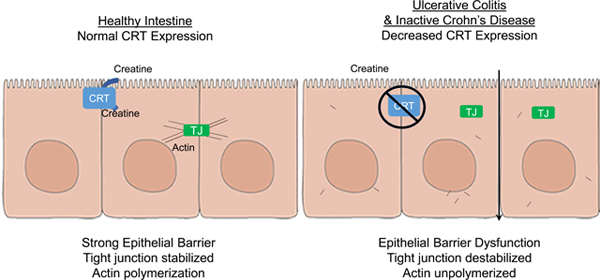
In an analysis of IEC cell lines and colonoids derived from CRT-knockout mice, we found that CRT regulates energy balance in IECs, and thereby epithelial integrity and barrier function. Mucosal biopsies from patients with ulcerative colitis and inactive Crohn’s disease have lower levels of CRT, which might contribute to the reduced barrier function observed in patients with IBD.
Keywords: CD, UC, claudin, actin
Graphical Abstract
Introduction:
The mucosal tissue microenvironment is tightly regulated in order to maintain homeostasis. Intestinal epithelial cells (IECs) act in the important role of maintaining a selectively permeable barrier that allows for absorption of nutrients while excluding luminal contents that promote inflammation. Epithelial barrier is maintained by regulation of the apical junction complex (AJC) composed of tight junctions (TJ) and adherens junctions (AJ)1. TJs and AJs are regulated and stabilized by the actin cytoskeleton2–5. Cycling of TJ proteins within the AJC is continuous and is required for the migration and change of shape that is necessary to maintain barrier in mucosal tissue6, 7. Additionally, endocytosis of TJ proteins has been demonstrated in response to many factors including proinflammatory cytokines that can result in impaired barrier, such as IFNγ and TNFα8, 9. Actin polymerization and myosin activity are necessary components of TJ regulation. The activities of actin and myosin are energetically demanding, as 20% of total available energy in IECs may be committed to maintenance of the actin cytoskeleton alone10.
The creatine-phosphocreatine cycle mediates an important mechanism of energy distribution. Creatine serves as an energetic buffer by carrying a phosphate bond as phosphocreatine that can be used to produce ATP at times of increased energy usage. Phosphocreatine diffuses more readily than ATP11, allowing for the distribution of energy to areas of increased need, such as those with active actin turnover10. Regulation of this process is mediated by adequate creatine uptake by the creatine transporter, SLC6A8 or CRT, and by metabolism through various creatine kinases12–14. Creatine kinases have been implicated in actin polymerization that is required for phagocytosis in macrophages15. Little is known about the role of CRT in epithelial cell function and tissue homeostasis. However, creatine activity has been demonstrated to be important in the mucosal environment as creatine supplementation was protective in mouse models of colitis16. Additionally, by random mutagenesis, an increased susceptibility to DSS colitis was identified in mice with mutations in GAMT, an enzyme that produces creatine using endogenous pathways17. To understand the role of creatine transport in intestinal epithelial function, we pursued targeted CRT loss and gain-of-function analysis that identified a role for CRT in epithelial barrier formation and wound healing. Of clinical significance, our studies identified a loss of CRT expression in patient biopsy samples from certain IBD patients that suggest a possible functional role for CRT in inflammatory mucosal disease.
Materials and Methods
Cell culture:
T84 IECs were grown in a 1:1 mixture of DMEM and Hams F-12 media supplemented with penicillin, streptomycin and 10% fetal calf serum as previously described18.
Cell culture immunofluorescence:
Immunolocalization and confocal imaging were performed as previously described16. Briefly, T84 cells that were grown on glass coverslips or transwell membranes (0.33-cm2, 0.4 μM permeable polyester) were fixed with 4% paraformaldehyde, followed by permeabilization with 0.2% Triton X-100. The cells were then blocked using 5% serum, followed by exposure to a primary antibody as noted below followed by a tagged species-specific secondary antibody. Cells were then stained with DAPI (Invitrogen) diluted 1:25,000 in PBS. Cells were covered with ProLong gold antifade (Invitrogen). Immunolabeling was visualized with a Zeiss Axiovert 200 M microscope. Primary antibodies for CRT (ab62196, Abcam), ZO-1 (617300, Lifetech), Claudin 1 (ab15098, Abcam), Claudin 2 (ab53032, Abcam) were used. Actin was stained using rhodamine tagged phalloidin (R415, Thermo Fisher Scientific).
Tissue Immunofluorescence:
Tissue was obtained from patients designated to be healthy by the physician obtaining the biopsy, in accordance with the IRB approved University of Colorado IBD biobank protocol. Mouse tissue was obtained from healthy BL6 mouse colons. Fresh tissue was fixed in 1:10 neutral buffered formalin, paraffin embedded and sectioned. Samples were deparaffinized with xylene and ethanol washes. Antigen retrieval was achieved at 125° C for 25 minutes in a pressure cooker. Tissue sections were blocked with 10% goat serum in PBS and treated with rabbit anti-human SLC6A8 (ab62196, Abcam) 20 ug/ml overnight and secondary antibody (AF562 anti-rabbit, Invitrogen) 1:500 dilution. Tissue was imaged using a Zeiss AX10 microscope.
Immunoblotting:
IECs, and mouse and human tissue samples were lysed using RIPA lysis buffer containing protease inhibitors, sonicated and centrifuged. The protein concentration was determined by BCA protein assay (Thermoscientific). Equal concentrations proteins were resolved using 10% SDS-PAGE and transferred to PVDF membrane. The membrane was blocked using 5% Milk in TBS-T, probed with primary antibody, CRT (ab62196, Abcam), Claudin 1 (ab180158, Abcam), Claudin 2 (ab53032) and Actin (ab8227, Abcam), and corresponding HRP-conjugated antibodies. Densitometry was measured using ImageJ comparing CRT bands to Actin bands.
Lentiviral CRT knockdown:
shRNA control and CRT targeted lentiviruses were added to media of T84 cells (SHC216, TRCN0000377577, Sigma-Aldrich). After 24 hours lentivirus containing media was removed. Transduced cells were then selected using 6ug/ml puromycin (Sigma-Aldrich).
RNA isolation, cDNA synthesis and qPCR:
RNA was isolated by phenol: chloroform extraction using TRIzol (Invitrogen), precipitated with isopropanol and resuspended in RNase-free water. cDNA synthesis was conducted using the iSCRIPT cDNA synthesis kit (Bio-Rad). qPCR expression of target mRNA was assessed using SYBR Green master mix (Applied Biosystems). Fold change in expression of target mRNA relative to beta-actin mRNA was calculated. Primer sequences: (F and R) CRT CCCCTACCTGTGCTACAAGA, TTGAACAGGGGACAGATGTT; hClaudin 1 CCAGTCAATGCCAGGTACGAAT, TTGGTGTTGGGTAAGAGGTTGTT; hClaudin 2 CTCCTGGGATTCATTCCTGTT, TCAGGCACCAGTGGTGAGTAGA; hZO-1 CAACATACAGTGACGCTTCACA, CACTATTGCCGTTTCCCCACTC; hJAM-A CCTGGGAATCTTGGTTTTTG, GGAATGACGAGGTCTGTTTG; hAGAT GGAAGGAGTGACAGTAAGGAGG, GCCCACAACTATCAGGATGTCTC; hGAMT TGGCACACTCACCAGTTCA, AAGGCATAGTAGCGGCAGTC; mAGAT TCACGCTTCTTGAGTACCG, TCAGTCGTCACGAACTTTCC; mGAMT TGGCACACTCACCAGTTCA, AAGGCATAGTAGCGGCAGTC
Cell proliferation assay:
Cells were plated in 100ul/well of media on 96 well plates. 10 ul of CCK-8 (Sigma-Aldrich) was added to each well. 10ul of 1% SDS was added to wells sequentially over 48 hours to obtain multiple time points for subsequent analysis. Absorbance was measured at 450nm in a microplate reader.
Brightfield image:
Cells were grown to confluence on tissue culture dishes. Images were obtained using a Nikon Diaphot microscope.
HPLC:
HPLC evaluations were performed as previously described10. Briefly, extractions were obtained from transwell permeable support inserts containing T84 cells which were submerged in 500 μl of 80% MeOH. The sample was then placed in liquid N2 until frozen. The extraction process was repeated twice more. The resulting 1.5 ml of extract was dried via an Eppendorf Vacufuge at room temperature. The dried extract was dissolved in 200 μl of HPLC mobile phase A (details below) and filtered (Whatman Puradisc 4, 0.45 μm, nylon) into vials for HPLC injection. Analyses were performed on an Agilent Technologies 1260 Infinity HPLC using a Phenomenex Luna C18(2) column.
Intracellular Creatine Assay:
T84 cells were grown in the presence of 0, 1 or 10 mM creatine (Sigma) in standard cell culture media. Cells were washed with PBS then creatine reaction mix (Sigma) was added to wells and incubated for 1 hour at 37° C. A standard curve was used for comparison. Fluorometric measurement was conducted at 535 nm.
Barrier measurement:
Epithelial barrier was measured by transepithelial electrical resistance (TEER) and FITC dextran flux assay. TEER was measured by growing IECs on permeable membranes, 0.4uM pore, 24 well plate using an epithelial voltmeter (model EVOM2, World Precision Instruments). Raw measurements were converted to Ohm*cm2. FITC-dextran flux was measured when cells were at maximal barrier as measured by TEER. Media was replaced with Hanks+ Buffer. The top well was filled with 80ul of 1.25 ug/ul of FITC-dextran 3KD in Hanks+ Buffer. Samples were obtained from the lower chamber every 30 minutes for 2 hours and measured relative to a standard curve. Flux was measured as ng/cm2/hour. Colonoid TEER experiments were conducted by dissociating colonoids in Trypsin-EDTA (Fisher Scientific) by vigorous pipetting. Cells were strained through a 70 um filter, counted and then plated in equal number per condition (approximately 25,000/well) on collagen coating solution (Sigma-Aldrich, 125–50) pretreated transwells. To collagen coat transwells, 0.4uM pore, 0.33cm2 transwells were incubated with 100ul of collagen coating solution for 12–24hrs prior to removal of excess by suction. Cells were grown in L-WRN cell conditioned enteroid media for 24 hours, followed by maturation media (L-WRN media diluted 1:10 with T84 media) for the remainder of the experiment. TEERs were obtained as above and cells were given fresh maturation media every 2–3 days.
Wound healing assay:
Cells were plated on a 96 well Incucyte plate the day prior to starting the assay. Confluent cells were wounded by the Incucyte wounder and images were collected every 2 hours for 3 days. Relative wound density was calculated by the Incucyte ZOOM software to calculate cell density in the wound area relative to the spatial cell density outside of the wound area.
Lentiviral CRT overexpression:
T84 cells were treated with Dharmacon Precision LentiORF lentiviral particles overexpressing CRT or control nonsense sequence with 8ug/ml of polybrene. Transduced cells were selected using 5ug/ml of blasticidin. After selection, cells were maintained in normal T84 media as above.
Seahorse assay:
Metabolic capabilities were assessed using the XF Cell Energy Phenotype Test that includes the metabolic stressors FCCP (final concentration 0.5 uM) and oligomycin (final concentration 1uM) in Seahorse XFe. Cells were plated on a 96 well plate in equal numbers. Analysis was done using Seahorse Wave Desktop Software.
Human biopsy biobank analysis:
Patient biopsy samples were collected for biobank repository at the University of Colorado IBD Center with IRB approval. Endoscopic disease status and location were noted by the physician who obtained the biopsies. Patient age, gender, current medications and duration of disease were also collected at that time. These mucosal biopsies were analyzed by rtPCR analysis, immunofluorescence and immunoblot.
Colonoid development:
Colonoids were developed using colon samples from CRT whole body knockout and control mice, generously donated by the lab of Dr. Matthew Skelton. Cells were isolated and grown as previously described18, 19. Briefly, upon obtaining the samples, colonic tissue was digested using LP dissociation kit (Miltenyi) with mechanical disruption using the GentleMACS C tube (Miltenyi) on intestine setting. Cells were strained through a 70-μm cell strainer, washed and resuspended in Matrigel Basement Membrane (Corning). L-WRN cell conditioned enteroid media (DMEM/F12 supplemented with 1% Pen/Strep, 1% glutamax, 20% FBS) was used to maintain the cells as previously described19. Cells were passaged 2 times per week involving dissociation by vigorous pipetting in the presence of Trypsin and T84 media. Cells then underwent 2 rounds of washes with T84 media, centrifugation at 200xg and then were resuspended in Matrigel and plated in spots on tissue culture plates.
Statistics:
Statistical analysis was conducted using Prism. Tests used include unpaired t test, ANOVA, mixed-effects analysis, one-way ANOVA and Wilson/Brown fraction of total analysis depending on data set. Error bars indicate standard deviation. Probability values of p<0.05 were considered to be statistically significant.
Results:
Creatine transporter in human and mouse colonic tissue localizes near the tight junction in epithelial cells
Creatine metabolism is an important component for protection against tissue damage in mouse models of colitis16, 17. Considering that creatine is acquired from both the diet and endogenous sources, we sought to define the relative contribution of creatine transport to energy homeostasis and function in IECs. As a starting point, we examined expression patterns of the creatine transporter (SLC6A8 or CRT) in the mucosa. In T84 human IECs, CRT was localized near the TJs as determined by close localization to the TJ protein ZO-1 (Figure 1A). In order to localize CRT in human primary cells, patient biopsy specimen were fixed, stained and imaged by immunofluorescence. Staining of healthy colonic biopsies revealed that numerous cells contained CRT, including IECs, that appeared to localize CRT to the tight junctional region (Figure 1B). Given the prior investigations in mouse models of colitis, we anticipated that mouse IECs would contain CRT in order to allow for creatine uptake. Indeed, CRT was localized to colonic IECs from healthy mouse colon (Figure 1C). We confirmed expression in healthy human colon biopsies and mouse colonic tissues by immunoblot (Figures 1D and 1E respectively). Interestingly creatine kinase localization in IECs was similarly observed coincident with adherens junctions, suggesting that localization of creatine machinery to the apical junction complex16 is important for function of the creatine pathway.
Figure 1. Expression of CRT in the colonic epithelium of humans and mice.

A. T84 human IECs were stained for immunofluorescence. Images were obtained by confocal microscopy(n=5). B. Healthy human biopsy tissue was fixed stained and immunostained for CRT(n=3). C. Healthy mouse colon tissue was fixed and stained for immunofluorescence labeling of CRT(n=5). D. Healthy human patient colonic biopsies were evaluated by immunoblot for CRT and Actin in 3 different patients. E. Healthy whole mouse colon tissue was evaluated by immunoblot for CRT and Actin in 3 different mice.
Targeted repression of CRT impairs creatine uptake, barrier function and wound healing
Given our understanding of the high energetic demand required to maintain barrier in IECs10, we hypothesized that CRT would be an integral component of epithelial cell function and homeostasis. To test this hypothesis, we selectively knocked down expression of CRT using lentiviral shRNA transduction in T84 IECs. Transduced cells were selected using puromycin and resulted in stable knockdown (KD) cell lines. CRT expression was assessed by qPCR of unstimulated cells and the most significantly knocked down line demonstrated a 80% reduction (CRT KD) (Figure 2A). CRT KD cells had a greater than 50% reduction in CRT protein as measured by immunoblot (Figure 2B). Given the important role of creatine in energy regulation within the cell, we evaluated CRT KD proliferative ability. The CRT KD cell line did not demonstrate a proliferation deficit as measured by CCK8 proliferation assay (Supplemental Figure 1A). When viewed by bright field microscopy, the CRT KD cell line appeared visually identical to the control cell line without visible deficits in plate adherence (Supplemental Figure 1B).
Figure 2. Influence of CRT knockdown on intracellular creatine, barrier function and wound healing in IECs.

A. T84 IECs were transduced with lentiviral shRNA targeting CRT. Cells were selected and mRNA was obtained and analyzed using qPCR. B. Protein was isolated and analyzed by immunoblot(n=5). Densitometry was conducted using ImageJ software. C. CRT KD cell metabolites were isolated and analyzed by HPLC(n=3). D. CRT KD cells were grown in standard growth media with and without creatine supplementation. Cells were washed and intracellular creatine was evaluated using a creatine fluorometric assay(n=3). E. CRT KD cells were grown on a permeable membrane and evaluated for barrier formation by transepithelial electrical resistance (TEER)(n=5). F. CRT KD cells were grown to maximum TEER reading prior to FITC-dextran flux assay(n=3). G. CRT KD cells were grown to confluence and wounded with incucyte wounder. Wound closure was assessed over time by incucyte imager(n=4). H. Relative wound density was calculated Incucyte ZOOM software. I. Colonoids from CRT KO and control mice were evaluated by qPCR for relative CRT expression(n=3). J. Protein was isolated from the mouse CRT KO and control colonoids and analyzed by immunoblot(n=3). K. Barrier analysis by TEER was conducted as previously described using CRT KO and control colonoids(n=3). *p< 0.05, ** p< 0.01, ***p<0.001, ****p<0.0001 determined by unpaired T-test, one-way and two-way ANOVA
Given that CRT is the only known creatine transporter, we anticipated that loss of CRT would result in diminished intracellular creatine levels. We quantified intracellular creatine by high performance liquid chromatography (HPLC), using methods previously developed by our group10. This analysis revealed that CRT KD cells carried a 50% reduction of intracellular creatine compared to control cells in unstimulated conditions (Figure 2C). To evaluate the ability of CRT to augment creatine uptake based on extracellular creatine concentration, we incubated cells in the presence of extracellular creatine that was added to the media for 96 hours prior to evaluation by creatine assay. Baseline intracellular creatine in the CRT KD cells was reduced by 50%, similar to the HPLC findings. The addition of extracellular creatine (1mM and 10mM) significantly increased intracellular creatine in control cells but not in CRT KD cells (Figure 2D). These findings identify a limitation in creatine uptake by CRT based on relative CRT concentration.
Work in CRT-deficient mice has identified a compensatory increase in skeletal muscle of one of the intrinsic creatine production enzymes, AGAT, but not the other, GAMT20. We found no statistically significant change in AGAT and GAMT expression in the CRT knockdown IECs. However, in colonoids derived from CRT whole body knockout and control mice, we found that loss of CRT did result in increased AGAT mRNA expression compared to controls (Supplemental Figures 2A and 2B).
Recent work has revealed that adenylate energy balance contributes fundamentally to actin-based cytoskeletal functions10. Since barrier function is tightly linked to the cytoskeleton, we hypothesized that CRT deficiency would impact intestinal epithelial barrier function. To evaluate the development of transepithelial barrier over time, cells were plated in equal number and transepithelial electrical resistance (TEER) values were closely monitored. There was a direct correlation between CRT expression and barrier formation rate (Figure 2E). We also evaluated functional barrier activity using a FITC-dextran flux assay. By this measure of barrier function, CRT loss again resulted in reduced barrier function (Figure 2F). We used CRT KO mouse colonoids to confirm the cell line findings. We demonstrated that the cells were deficient in CRT by mRNA and protein (Figures 2G and 2H respectively). We then grew the cells in 2D monolayers and found a significant barrier deficit in the CRT KO colonoids as measured by TEER (Figure 2I).
Intestinal epithelial cell proliferation and movement are necessary in the wound healing response. This function requires significant energy expenditure given actin and myosin activity and we hypothesized that wound healing would also be impaired in the setting of reduced creatine concentration due to loss of CRT. Utilizing a scratch wound assay, cells were grown to confluence, then uniformly wounded and wound healing was monitored photographically every 2 hours over 72 hours. Figure 2J depicts a representative image obtained at 72 hours. A significant deficit in wound healing was identified in CRT KD cells as compared to controls. Additionally, when images were evaluated for relative wound density over time, the deficit was notable as early as 8 hours and maintained throughout the 72 hour period (Figure 2K). Wound closure and epithelial barrier deficits in CRT KD cells indicate that adequate CRT concentrations are vital to appropriate epithelial cell function.
CRT loss results in a leaky tight junction protein profile, mislocalization of tight junction protein and reduced actin polymerization
We have identified a profound barrier defect in IECs that lack adequate CRT expression. Epithelial barrier is mediated by the expression profile and localization of proteins in the junctional complex. We first profiled the TJ expression in the CRT KD cells, including target proteins that promote barrier (Claudin 1, ZO-1 and JAM-A)21–24 and a target that diminishes barrier (Claudin-2)25. There was a notable decrease in Claudin-1 and increase in Claudin-2 expression in the CRT KD cells relative to controls, a profile that represents a “leaky” TJ phenotype (Figure 3A). There were no differences noted in expression of ZO-1 or JAM-A between CRT KD and controls. Given the differences seen in expression in Claudin 1 and Claudin 2 mRNA, we next determined if this observation translated to the protein levels. Indeed, there was a significant loss of Claudin 1 protein, however increased expression of Claudin 2 was not observed by immunoblot (Figure 3B). In addition to expression levels, we evaluated localization of TJ proteins in the CRT KD cells relatives to controls. We noted that all of the TJ proteins evaluated (ZO-1, Claudin 1 and Claudin 2) were mislocalized in the CRT KD cell line as compared to controls, in that there is an absence of staining at the TJ and increased intracellular staining (Figure 3C). Thus, these results suggest that the phenotype (i.e. barrier dysfunction in CRT KD) is primarily explained by the loss of a “tight” Claudin as opposed to the introduction of a “leaky” Claudin as well as TJ mislocalization.
Figure 3. Impact of CRT knockdown on TJ phenotype, localization of TJ proteins and actin polymerization.

A. TJ expression was assessed in CRT knockdown cells by qPCR (n=3). B. Control and CRT KD cells were evaluated by immunoblot for Claudin 1, Claudin 2 and actin (n=3). C. CRT knockdown and control cells were grown on a permeable membrane and stained by immunofluorescence for ZO-1, Claudin 1, Claudin 2 and polymerized actin with rhodamine phalloidin(n=3). **p<0.01, ***p<0.001 determined by two-way ANOVA.
Given the intimate connection between TJ proteins and the actin cytoskeleton as well as the energy dependence of actin polymerization, we hypothesized that the mislocalization of ZO-1 was likely related to dysregulation of actin polymerization. Indeed, polymerized actin staining with phalloidin identified an altered staining pattern as well as a reduction in total actin polymerization in the CRT KD cell lines compared to controls (Figure 3C).
Overexpression of CRT increases the rate and magnitude of epithelial barrier formation
Given the profound barrier deficit observed with targeted knockdown of CRT expression, we hypothesized that over-expression of CRT would enhance barrier formation. To evaluate this principle, T84 cells were transduced with a lentiviral CRT open reading frame (ORF) vector or empty vector control. Transduced cells were selected with blasticidin and qPCR analysis identified a 4-fold increase in CRT mRNA and a 2-fold increase in protein in CRT overexpressor (CRT OE) cells as compared to control cells (Vector) (Figure 4A and 4B). Functional analysis of intracellular creatine levels revealed a baseline 2-fold increase in intracellular creatine in CRT OE cells compared to Vector (Figure 4C). Addition of extracellular creatine did not significantly increase intracellular creatine in the CRT OE cells, suggesting that our culture conditions provide saturating creatine levels for CRT OE cells.
Figure 4. Influence of CRT overexpression on IECs intracellular creatine and barrier function.

A. T84 IECs were transduced with lentiviral CRT ORF. CRT expression was assessed by qPCR(n=4). B. CRT protein was assessed by immunoblot in Vector and CRT OE cells(n=4). Densitometry was completed using ImageJ software. C. Intracellular CRT was assessed by creatine fluorometric assay with and without creatine supplementation to media. D. Cells were plated on a permeable membrane for transepithelial electrical resistance measurements (TEER)(n=3). E. FITC-dextran flux was measured using cells grown to maximum TEER(n=3). F. Wound healing was evaluated using the Incucyte wounder as previously described(n=4). G. Relative wound density was calculated incucyte ZOOM software. H. TJ protein expression was evaluated using qPCR of unstimulated cells(n=3). **p<0.01, ****p<0.0001 determined by one-way and two-way ANOVA analysis.
We proceeded to evaluate barrier function of the CRT OE cells. Consistent with our hypothesis, TEER measurements revealed that both the rate and maximum barrier formation of CRT OE cells was enhanced relative to Vector (Figure 4D). Additionally, FITC-dextran flux assay identified a reduction in flux at maximum TEER reading (Figure 4E). These data support the finding that barrier is directly related to relative CRT expression. Wound healing density analysis demonstrated no statistically significant difference (Figure 4G), but images of the final wound identified a mild improvement in healing of CRT OE wounds (Figure 4F).
It is notable that an inverse pattern to the TJ profile was observed between loss and gain of CRT function cell lines. CRT OE cells expressed a profile consistent with “tighter” TJ expression with decreased “leaky” Claudin 2 and an increase in ZO-1 that acts a tether between other TJ proteins and the actin cytoskeleton (Figure 4H). Localization of ZO-1 and actin profile were not notably different between Vector and CRT OE (data not shown).
Loss of CRT results in metabolic stress as represented by an inability to increase mitochondrial respiration
Given the important role of creatine cycling in regulation of energy distribution within the cell, we hypothesized that loss of CRT would promote metabolic stress. To assess baseline and stressed phenotypes of CRT KD cells, we monitored mitochondrial oxygen consumption rates (OCR) and glycolysis extracellular acidification rates (ECAR) using the Seahorse XFe96 Analyzer. Energetic stress was induced using an ATP synthase inhibitor (oligomycin) and an uncoupler of oxidative phosphorylation (FCCP). Analysis of control cells revealed normal increases in ECAR and OCR in response to the stress condition. By stark contrast, CRT KD cells had an appropriate increase in glycolysis with stress but no increase in mitochondrial activity. In this analysis, the lack of reserve metabolic potential is clearly appreciated as the CRT KD cell OCR potential remains at baseline levels (Figure 5A). When measured over time, the OCR of CRT KD cells revealed an inability to increase mitochondrial respiration with stress induction (at 20 minute time point) compared to control cells (Figure 5A). There were minor differences in ECAR between the two cell lines, suggesting that glycolytic activity remains intact despite the loss of CRT expression. Metabolic analysis of the CRT OE and Vector control cells identified no significant differences in OCR or ECAR induction with stress (Figure 5B) suggesting that excess CRT expression does not augment the metabolic stress response.
Figure 5. Influence of CRT knockdown on IEC metabolic phenotype.

A. Control and CRT KD cells were grown on Seahorse analysis plates. Cells were then evaluated using the metabolic phenotype profiling protocol that measures metabolic activity with the addition of stressors FCCP and oligomycin. Measurements of OCR and ECAR were compared under baseline and stressed conditions. Metabolic potential was calculated based on ability to increase OCR and ECAR over baseline values. OCR and ECAR values plotted vs time with addition of stressors after third time point. B. The same metabolic analysis was conducted comparing the Vector and CRT OE cells. *P<0.05, ****p<0.0001 determined by two-way ANOVA.
Taken together, this metabolic profiling approach revealed a significant mitochondrial respiration deficit in the CRT KD cell line. As the role of creatine is to shuttle energy produced by the mitochondria, the inability to compensate during metabolic stress may contribute to cellular dysfunction in CRT deficient cells.
CRT expression is reduced in IBD patient intestinal biopsies as compared to healthy controls
Given the significant barrier defects observed in IEC function with loss of CRT, we profiled CRT expression in a disease associated with barrier dysfunction, namely inflammatory bowel disease (IBD). We obtained biopsies from controls (n=30) and IBD patients (Crohn’s disease (CD) n=30, Ulcerative Colitis (UC) n=27) from the University of Colorado IBD biobank (see Table 1 for patient characteristics). Differences between the patient populations of these cohorts included a younger age among the CD active patients, female predominance among UC inactive patients and less anti-TNF exposure among UC patients (Table 1). RNA was isolated from endoscopic biopsies and processed for qPCR. This analysis revealed that CRT expression was significantly reduced in the UC patient cohort relative to control biopsies (Figure 6A). No differences were detected between the CD patient cohort and controls. When results were categorized based on endoscopic inflammation status, UC patients had reduced CRT expression in both active and inactive states compared to healthy controls. Only inactive CD patients had reduced CRT expression compared to healthy controls (Figure 6B). In CD, a similar expression pattern was observed in ileal and colonic biopsies, suggesting that intestinal location has minimal impact on CRT expression and that disease activity is a more important factor (Figure 6C). These results identify aberrant expression of CRT in IBD.
Table 1. IBD Patient biopsy demographics.
Control, Crohn’s disease (CD) and ulcerative colitis (UC) patient characteristics was tabulated. Statistics for mean age and duration of disease were calculated using one-way ANOVA.
| Control | CD Inactive | CD Active | UC Inactive | UC Active | |
|---|---|---|---|---|---|
| Mean Age (yrs.) | 59.6 | 48.79 | *35.46 | 49.57 | 50.31 |
| Sex (% Female) | 48.3 | 50 | 53.8 | *71.4 | 50 |
| Steroid exposure (%) | NA | 35.7 | 30.8 | 28.6 | 40 |
| Anti-TNF exposure (%) | NA | 50 | 46.2 | **14.3 | ***5.6 |
| Duration of Disease (yrs.) | NA | 19.4 | 10.2 | 9.7 | 11.56 |
| Colonic biopsies (%) | 100 | 64.3 | 38.5 | 100 | 100 |
p<0.0001 compared to control patients. Statistics for sex, medication exposure and % colonic biopsies were calculated using Wilson/Brown fraction of total analysis with 95% confidence interval.
indicates statistical significance relative to control patients
indicates statistical significance relative CD inactive patients
indicates statistical significance relative CD active patients.
Figure 6. CRT and TJ expression profile in intestinal biopsies from IBD patients.

A. CRT expression was measured relative to actin by qPCR from patient biopsy samples obtained from the University of Colorado IBD biobank. B and C. The data from these samples were then sorted based on disease activity and biopsy location respectively. D. Evaluation of Claudin 1, Claudin 2 and ZO-1 by qPCR from the same patients was also conducted. E. Relative CRT expression was plotted vs relative TJ mRNA expression for each patient and a linear regression analysis was conducted. *p<0.05, **p<0.01, ***p<0.001, determined by one-way ANOVA and Pearson correlation.
We also evaluated TJ protein expression in the biopsies and found a significant increase in Claudin 1 in CD patients with active disease as compared to those with inactive disease and increased Claudin 2 in UC patients with inactive disease as compared to control and active disease patients (Figure 6D). Prior studies have similarly seen an increase in Claudin 1 protein in IBD with increased disease activity26, 27. Additionally, another group has shown an increase in Claudin 2 in UC patients28, although they did not differentiate between patients with active vs inactive disease. When we compared expression of CRT with TJ proteins by linear regression, we found a statistical positive correlation between CRT and Claudin 1 and negative correlation between CRT and Claudin 2 (Figure 6E). This data validates our cell culture data and suggests that CRT and Claudin expression are related.
Discussion:
Given the highly ordered cytoskeletal network that supports mucosal surfaces, the maintenance of barrier function requires large amounts of energy. Diverse pro-inflammatory stimuli place significant metabolic stress on the mucosa, where IEC consume as much as 20% of overall ATP in barrier maintenance10. Given the energetic demands of IECs, we hypothesized a role for creatine transport in energy buffering and distribution. In the current work, we identify a role for CRT in IEC homeostasis and aberrant expression of CRT in IBD.
These studies identified several novel findings regarding the functional homeostasis in IECs. First, the observation that targeted repression of CRT in IECs resulted in a profound loss of epithelial barrier formation implicates intracellular creatine as a vital energy metabolite. This finding is supported by previous work that demonstrated a compelling role for creatine metabolism in the regulation of barrier function16, 17. Using loss and gain of CRT function approaches, we confirmed the importance of creatine uptake to barrier formation. Such findings support our recent metabolomic studies suggesting that creatine and phosphocreatine strongly parallel barrier formation and represent metabolites of “last resort” under severe stress of ATP depletion10. Second, these studies implicate extracellular creatine as the primary source of creatine for epithelial functional responses. A compensatory mechanism by intracellular creatine production is likely in human CRT KD IECs as evidenced by measurable intracellular creatine with minimal CRT and as was observed in mouse CRT KO colonoids with increased AGAT. Third, the adaptive phenotype elicited during CRT loss is insufficient to provide the energy necessary for functional barrier formation. Finally, the loss of CRT results in a stressed metabolic phenotype. Using energy phenotyping analysis, CRT KD cells were unable to increase mitochondrial respiration in response to metabolic stress. The inability to compensate for metabolic stress suggests that CRT deficient cells have limited energy reserve capacity, a phenotype observed in other metabolically stressed cells29. Such findings also support an important feedback loop between mitochondrial respiration and intracellular creatine levels.
Older studies identified a creatine shuttle and functional coupling between creatine and myosin in isolated brush borders, predominantly at the circumferential actomyosin ring30, 31. The present studies identified CRT localization to the TJ region of confluent IEC. It is notable that creatine kinases (CKs) also localize to adherens junction region of IEC16. In addition to phosphocreatine generation via oxidative phosphorylation coupling, a subset of cytosolic CKs have also been shown to associate with glycolytic enzymes and to mediate phosphocreatine repletion from ATP generated through glycolysis32. Given the importance of TJ integrity to epithelial homeostasis, co-localization of CRT and CK isoforms may promote epithelial function through localized phosphocreatine production, particularly under conditions of energetic stress.
As part of our analysis, we examined CRT expression in patients with active and inactive IBD, a disease widely accepted to involve barrier dysfunction33. The expression pattern of CRT in IBD patient samples revealed a profound loss of CRT in all UC samples but a more selective loss of CRT in patients with inactive CD. Regulation of CRT expression has not been well characterized previously. Our prior studies identified induction of CRT expression in the setting of hypoxia treatment16. It is possible that the increase in CRT expression in active disease in CD is related to hypoxia, however other possible modulators include extracellular creatine concentration34, hypertonic media35, metabolic regulators PGC-1α and PGC-1β36 and AMP-kinase37. All of the aforementioned mediators have been implicated in CRT expression. Beyond expression alone, the osmotic sensor kinases, SPAK and OSR1, and cytokine signaling molecules JAK2 and JAK3 were also found to negatively regulate CRT activity38, 39. The above kinases have all been identified to be upregulated in IBD. The difference in CRT expression in CD vs UC may be due to differential activation of JAK signaling40, 41. It is also notable that early JAK inhibitors (e.g. tofacitinib) appear to be more effective in UC 42, although more specific JAK inhibitors (e.g. filgotinib) may provide benefit in CD 43. These clinical trials suggest that the mucosal milieu could be fundamentally different between UC and CD.
CRT expression in IECs correlated with TJ protein expression in cell culture as well as in patient samples, with loss of CRT resulting in a leakier phenotype. This phenotype is observed in patients with both CD and UC44, 45. Given the decrease in expression of CRT in IBD patient biopsy samples, it is possible that lack of creatine is contributing to TJ regulation in IBD with implications on baseline barrier function. Interestingly, 35% of individuals who have an X-linked creatine deficiency self-reported GI symptoms suggesting that loss of CRT may result in a digestive tract that is more prone to disease46. Further studies to identify possible regulators of CRT expression and activity may provide opportunities to understand CRT function in IBD patient.
Current medical therapies in IBD have focused primarily on immune regulation. No current therapies are targeting barrier function, which would have the benefit of avoiding systemic immune dysregulation that can increased risk of infection and malignancy. Creatine and CRT regulation are attractive therapeutic targets. Creatine supplementation has been used for other disease processes with a favorable side effect profile47, 48. Moreover, there is a single case report of creatine monotherapy improving endoscopic and symptomatic disease in Crohn’s ileitis49, which is promising. Further studies to test the utility of creatine in IBD treatment will be necessary.
In summary, these findings identify an important role for creatine transport in epithelial energetics and barrier formation. Results from these studies demonstrate that CRT expression correlates directly with intestinal epithelial creatine concentration, barrier formation and wound healing responses. Loss of CRT results in a metabolically stressed and dysfunctional epithelial cell and these findings may provide important insight into the pathophysiology of IBD given the profound loss of CRT expression in IBD patients.
Supplementary Material
What you need to know:
Background and Context:
Creatine regulates energy distribution within cells and reduces the severity of colitis in mice. This study investigated the functions of the creatine transporter CRT in intestinal epithelial cells (IECs) and maintenance of the intestinal epithelial barrier.
New Findings:
In an analysis of IEC cell lines and colonoids from CRT-knockout mice, we found CRT to regulate energy balance in IECs, along with epithelial integrity and barrier function. Mucosal biopsies from patients with IBD have lower levels of CRT, which might contribute to the reduced barrier function observed in patients with Crohn’s disease or ulcerative colitis.
Limitations:
This study was performed in cell lines, mice, and human tissue samples — further in vivo studies in mice and humans are needed.
Impact:
Strategies to restore creatine to IECs might be developed for treatment of IBD.
Lay Summary:
The authors found that a protein that transports creatine across intestinal cells (CRT) is required for maintenance of the intestinal epithelial barrier, which is disrupted in some patients with IBD. Colon tissues from patients with IBD had lower levels of CRT.
Acknowledgments
Grant Support: This work was supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR002535 (CHTH), NIH Grants 2T32DK067009 (CHTH), HD080910 (MRS), DK1047893 (SPC), DK50189(SPC), DK095491(SPC), DK103639(SPC) and VA Merit BX002182(SPC).
Abbreviations:
- CK
creatine kinase
- CRT
creatine transporter
- IBD
Inflammatory bowel disease
- IEC
intestinal epithelial cell
- KD
knockdown
- TEER
transepithelial electrical resistance
- TJ
tight junction
- UC
ulcerative colitis
Footnotes
Disclosures: The authors declare that no conflict of interests exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Capaldo CT, Nusrat A. Claudin switching: Physiological plasticity of the Tight Junction. Semin Cell Dev Biol. 2015;42:22–29. [DOI] [PubMed] [Google Scholar]
- 2.Yu D, Marchiando AM, Weber CR, et al. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc Natl Acad Sci U S A 2010;107:8237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J 2002;16:1835–7. [DOI] [PubMed] [Google Scholar]
- 4.Ivanov AI. Actin motors that drive formation and disassembly of epithelial apical junctions. Front Biosci 2008;13:6662–81. [DOI] [PubMed] [Google Scholar]
- 5.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol 2010;177:512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda M, Kubo A, Furuse M, et al. A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci 2004;117:1247–57. [DOI] [PubMed] [Google Scholar]
- 7.Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell 2003;112:19–28. [DOI] [PubMed] [Google Scholar]
- 8.Utech M, Mennigen R, Bruewer M. Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol 2010;2010:484987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang F, Graham WV, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol 2005;166:409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JS, Wang RX, Alexeev EE, et al. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J Biol Chem 2018;293:60396051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guimaraes-Ferreira L Role of the phosphocreatine system on energetic homeostasis in skeletal and cardiac muscles. Einstein (Sao Paulo) 2014;12:126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallimann T, Tokarska-Schlattner M, Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011;40:1271–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bessman SP, Geiger PJ. Transport of energy in muscle: the phosphorylcreatine shuttle. Science 1981;211:448–52. [DOI] [PubMed] [Google Scholar]
- 14.Wallimann T, Wyss M, Brdiczka D, et al. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J 1992;281 ( Pt 1):21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuiper JW, Pluk H, Oerlemans F, et al. Creatine kinase-mediated ATP supply fuels actin-based events in phagocytosis. PLoS Biol 2008;6:e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glover LE, Bowers BE, Saeedi B, et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proceedings of the National Academy of Sciences of the United States of America 2013;110:19820–19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turer E, McAlpine W, Wang KW, et al. Creatine maintains intestinal homeostasis and protects against colitis. Proc Natl Acad Sci U S A 2017;114:E1273–E1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curtis VF, Cartwright IM, Lee JS, et al. Neutrophils as sources of dinucleotide polyphosphates and metabolism by epithelial ENPP1 to influence barrier function via adenosine signaling. Mol Biol Cell 2018;29:2687–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 2013;8:2471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell AP, Ghobrial L, Wright CR, et al. Creatine transporter (SLC6A8) knockout mice display an increased capacity for in vitro creatine biosynthesis in skeletal muscle. Front Physiol 2014;5:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umeda K, Matsui T, Nakayama M, et al. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J Biol Chem 2004;279:44785–94. [DOI] [PubMed] [Google Scholar]
- 22.Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 2007;204:3067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furuse M, Hata M, Furuse K, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 2002;156:1099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saeedi BJ, Kao DJ, Kitzenberg DA, et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell 2015;26:2252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenthal R, Milatz S, Krug SM, et al. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci 2010;123:1913–21. [DOI] [PubMed] [Google Scholar]
- 26.Weber CR, Nalle SC, Tretiakova M, et al. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest 2008;88:1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poritz LS, Harris LR, Kelly AA 3rd, et al. Increase in the tight junction protein claudin-1 in intestinal inflammation. Dig Dis Sci 2011;56:2802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshima T, Miwa H, Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol 2008;23 Suppl 2:S146–50. [DOI] [PubMed] [Google Scholar]
- 29.Sriskanthadevan S, Jeyaraju DV, Chung TE, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015;125:2120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon PV, Keller TC, 3rd. Functional coupling to brush border creatine kinase imparts a selective energetic advantage to contractile ring myosin in intestinal epithelial cells. Cell Motil Cytoskeleton 1992;21:38–44. [DOI] [PubMed] [Google Scholar]
- 31.Keller TC, 3rd, Gordon PV. Discrete subcellular localization of a cytoplasmic and a mitochondrial isozyme of creatine kinase in intestinal epithelial cells. Cell Motil Cytoskeleton 1991;19:169–79. [DOI] [PubMed] [Google Scholar]
- 32.Wallimann T, Tokarska-Schlattner M, Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011;40:1271–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 2009;9:799–809. [DOI] [PubMed] [Google Scholar]
- 34.Loike JD, Zalutsky DL, Kaback E, et al. Extracellular creatine regulates creatine transport in rat and human muscle cells. Proc Natl Acad Sci U S A 1988;85:807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alfieri RR, Bonelli MA, Cavazzoni A, et al. Creatine as a compatible osmolyte in muscle cells exposed to hypertonic stress. J Physiol 2006;576:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown EL, Snow RJ, Wright CR, et al. PGC-1alpha and PGC-1beta increase CrT expression and creatine uptake in myotubes via ERRalpha. Biochim Biophys Acta 2014;1843:293743. [DOI] [PubMed] [Google Scholar]
- 37.Darrabie MD, Arciniegas AJ, Mishra R, et al. AMPK and substrate availability regulate creatine transport in cultured cardiomyocytes. Am J Physiol Endocrinol Metab 2011;300:E870–6. [DOI] [PubMed] [Google Scholar]
- 38.Fezai M, Elvira B, Borras J, et al. Negative regulation of the creatine transporter SLC6A8 by SPAK and OSR1. Kidney Blood Press Res 2014;39:546–54. [DOI] [PubMed] [Google Scholar]
- 39.Yan Y, Laroui H, Ingersoll SA, et al. Overexpression of Ste20-related proline/alanine-rich kinase exacerbates experimental colitis in mice. J Immunol 2011;187:1496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 1996;157:1261–70. [PubMed] [Google Scholar]
- 41.Sakuraba A, Sato T, Kamada N, et al. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology 2009;137:1736–45. [DOI] [PubMed] [Google Scholar]
- 42.Sandborn WJ, Su C, Panes J. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017;377:496–7. . [DOI] [PubMed] [Google Scholar]
- 43.Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-tosevere Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389:266–275. . [DOI] [PubMed] [Google Scholar]
- 44.Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007;56:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005;129:550–64. [DOI] [PubMed] [Google Scholar]
- 46.van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 2013;50:463–72. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi AP, Solis MY, Sapienza MT, et al. Efficacy and safety of creatine supplementation in childhood-onset systemic lupus erythematosus: a randomized, double-blind, placebo-controlled, crossover trial. Lupus 2014;23:1500–1511. [DOI] [PubMed] [Google Scholar]
- 48.Bohnhorst B, Geuting T, Peter CS, et al. Randomized, controlled trial of oral creatine supplementation (not effective) for apnea of prematurity. Pediatrics 2004;113:e303–7. [DOI] [PubMed] [Google Scholar]
- 49.Roy A, Lee D. Dietary Creatine as a Possible Novel Treatment for Crohn’s Ileitis. ACG Case Rep J 2016;3:e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.