

Abstract
In this study, a direct analysis of doping agents in urine samples with no sample preparation by a modified paper spray mass spectrometry (PS-MS) methodology has been demonstrated for the first time. We have described a paper surface treatment with trichloromethylsilane using a gas-phase reaction to increase the ionization of target compounds. This approach was applied for the analysis of two classes of banned substances in urine samples: anabolic agents (trenbolone and clenbuterol) and diuretics (furosemide and hydrochlorothiazide). Under optimized conditions, the developed methods presented satisfactory repeatability and analysis of variance showed linearity without lack-of-fit. Highly sensitive detections as low as sub-ng mL−1 which is below the minimum required performance levels proposed by the World Anti-Doping Agency has been reached using the hydrophobic PS-MS without any pre-concentration and clean-up step.
Keywords: Analytical methods, paper spray ionization, paper modification, doping, anabolic agent, diuretic
Graphical Abstract
INTRODUCTION
The use of performance-enhancing drugs for sports purposes is one component of a wider phenomenon known as doping, which is defined by the World Anti-Doping Agency (WADA)1 as the presence of prohibited substances (or their metabolites or markers) in an athlete’s samples. Between 1982 and 1995 elite athletes were asked about the possibility of taking a substance that guaranteed sporting victory but killed them in 5 years2. This study, now known as the Goldman Dilemma, showed a surprising 50% acceptance. Although recent studies have revealed the death outcome made in the Goldman Dilemma to be largely implausible3,4, high doping rates are still observed in modern sports. For example, >30% of athletes competing at the 2011 World Championships admitted to using banned substances during their careers5, representing a major challenge particularly because only <2% of tested athletes are usually caught6. One of the main reasons for the low sanction rates is attributed to a phenomenon known as microdoping in which athletes in track and field are able to attain marginal gains that can guarantee victory (by fractions of a second) by taking small quantities of illicit compounds7. Detecting doping with low doses of prohibited substances remain challenging. Here, we describe a paper-based sample collection platform capable of ultra-sensitive analysis of various doping agents from only 6 μL of raw urine directly from the paper substrate without sample pre-treatment.
The list of banned substances is published and reviewed annually by WADA and it includes all doping substances and methods prohibited in and out competitions8. The prohibited substances in all sports are classified in different classes (S0-S9), such as anabolic agents (S1), stimulants (S6), diuretics and masking agents (S5), and narcotics (S7), just to mention a few. The anti-doping control is done by accredited laboratories operating under the codes and specifications provided by WADA9. In order to guarantee that all laboratories can report the presence of doping substances in a uniform manner, a minimum detection and identification limit has been established that all the accredited laboratories must achieve. The minimum required performance level (MRPL) is the smallest concentration of a determined prohibited substance that analytical methods utilized in laboratories should be able to identify and detect with precision10. Among all the methods described in the literature, mass spectrometry (MS) is the gold-standard to analyze doping substances in biological samples11,12. Usually, MS is used as detection method following a separation stage, such as, liquid chromatography (LC-MS)13–16 and gas chromatography (GC-MS)17–20. These methodologies are very selective, sensitive, and have low limits of detection. However, direct sample examination is not possible; these hyphenated MS-based analytical platforms require extensive sample preparation and clean-up steps, including analyte derivatization, extraction and pre-concentration. As already pointed out in numerous studies21–23, these pre-analytical steps are usually the most laborious stages which are time-consuming, produce large volumes of waste and have the largest source of error in chemical analysis.
The high demand for doping testing has motivated scientists to develop fast and simple but still selective and sensitive methodologies minimizing the sample preparation, solvent consumption and waste production. Ambient ionization techniques are analytical methods that enable MS analysis to be performed with minimal or no samples preparation24,25, simplifying the analytical procedure, reducing equipment requirement, and the analysis time23,26. Paper spray (PS) ionization as an ambient ionization technique was introduced by Wang and co-workers in 201027 that integrates sample collection, separation and ionization in a single stage22. In PS-MS, the untreated sample (e.g., raw urine) is deposited on the paper substrate cut into a triangular tip. An appropriate solvent is added onto to the paper triangle containing the sample and the subsequent application of a DC high voltage (~2–5 kV) to the wet paper results in the formation of gas-phase ions via an electrospray-like mechanism. PS brings several advantages: 1) the fluid is transported by capillarity wicking, so no external pumping is required; 2) no nebulizer gas is needed either, further reducing the instrumentation requirements; 3) low volumes of sample and solvent are required; and 4) paper is inexpensive and can be safely disposed by incineration, decreasing the risk of biosafety for analyst. PS-MS has successfully been applied to analyze different compounds in biological fluids including metabolites28,29, drugs30–32, proteins33, enzyme activity34, and antigens for disease diagnostic35 without or with minimum sample preparation.
The minimum required performance level demands for analytical methods are usually low and, unfortunately, the limits of detection for PS-MS determination of compounds in biological samples are often inadequate30,36. These poor sensitivities have been attributed to inefficient analyte extraction from the sample matrix37. The extraction process is strongly dependent on the paper and spray solvent properties. For example, polar solvents can cause ionization suppression by extracting preferably more polar compounds and salts from the sample matrix27,31. Native paper is hydrophilic and aqueous biological samples wick to the core of the paper fibers increasing the interaction with the analytes and decreasing their extraction by the spray solvent and their transport to mass spectrometer38.
In this work, a modified hydrophobic paper surface30 is used to enhance extraction/ionization of analytes in urine. The optimized hydrophobic PS-MS method for detection of doping substances (e.g. trenbolone, clenbuterol, furosemide and hydrochlorothiazide) in raw urine samples is simple, fast, precise and accurate, allowing a sub-ng mL−1 sensitivity. This performance presents an increase in detectability > 3 orders of magnitude when compared with untreated hydrophilic paper. No sample preparation was required for these paper spray experiments, except the direct sample deposition on to the paper substrate and subsequent application of ethyl acetate spray solvent.
EXPERIMENTAL SECTION
Mass spectrometry
Mass spectra were acquired using a Thermo Fisher Scientific Finnigan LTQ linear ion trap mass spectrometer (San Jose, CA, USA). Data collection and processing was achieved using Thermo Fisher Scientific Xcalibur 2.2 SP1 software. The paper substrate was positioned with its tip parallel to the mass spectrometer inlet using a copper alligator clip, which was connected to an external high-voltage supply, as illustrated in Figure 1. MS parameters were as follows: ±30 V transfer capillary voltage; ±2 kV spray voltage; 3 microscans; 100 ms ion injection time; 3 mm distance from ion source to MS analyzer inlet for both positive- and negative-ion modes; 240°C transfer capillary temperature and 100 V capillary voltage for positive-ion mode; 250°C transfer capillary temperature and −120 V capillary voltage for negative-ion mode. Tandem MS with collision-induced dissociation (CID) was used for analyte identification and was optimized for each analyte.
Figure 1.
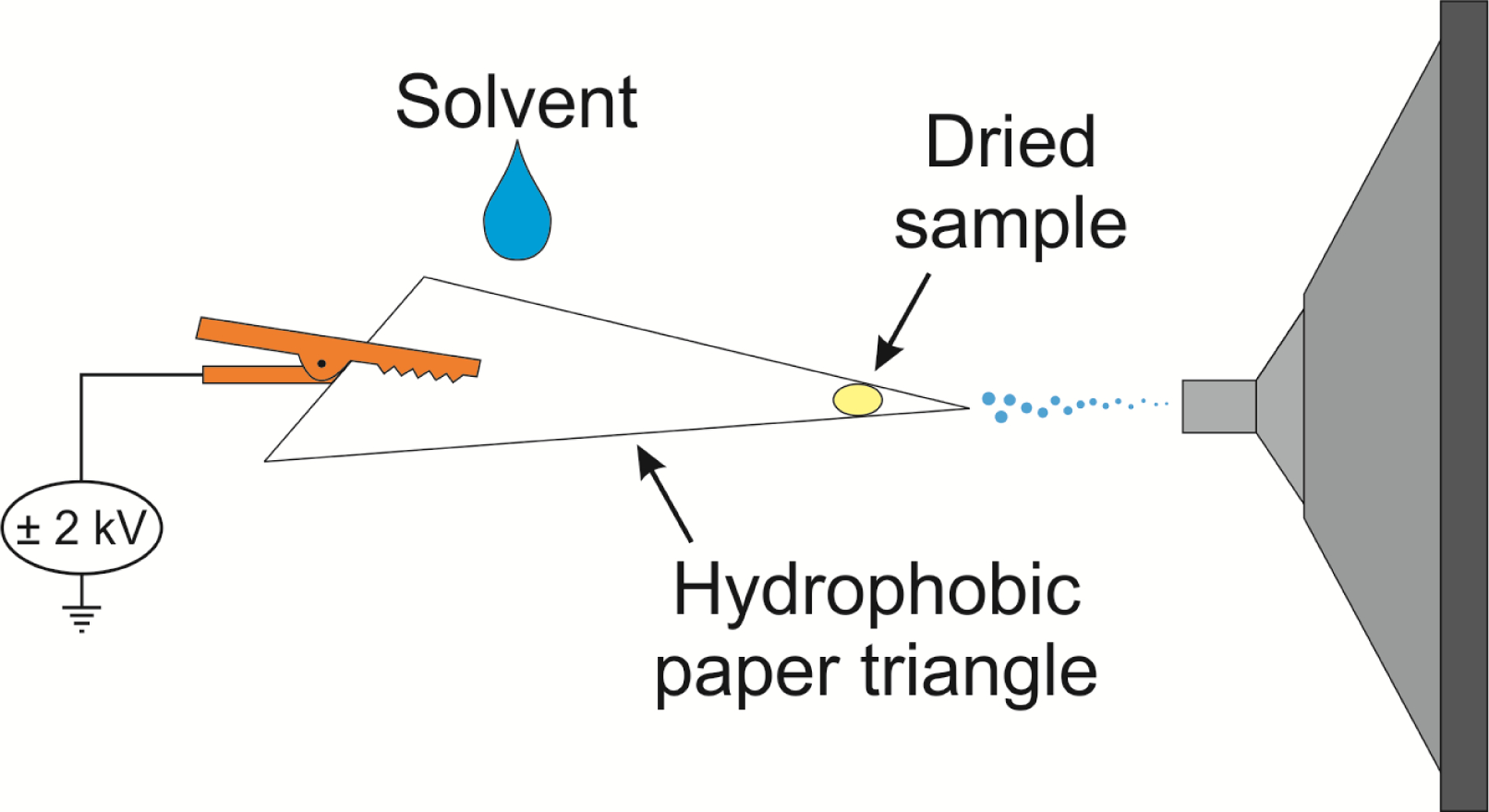
Schematic representation of the paper spray mass spectrometry experimental setup employed to analyze doping agents in raw urine samples.
Hydrophobic paper preparation
Paper triangles were cut manually from filter paper (Whatman grade 1) with base width of 9.5 mm and height of 16.5 mm (paper area was approximately 80 mm2). Typically, the precut paper triangles were treated using 0.5 mL of trichloro(3,3,3-trifluoropropyl)silane or trichloromethylsilane in a vacuum desiccator. The untreated paper was not subject to this reaction.
Chemicals and reagents
Standard solution (1.0 mg mL−1) of trenbolone (TREN) was obtained from Cerilliant (Round Rock, TX). Clenbuterol (CLEN), furosemide (FUR), hydrochlorothiazide (HCT), trichloro(3,3,3-trifluoropropyl)silane (TCTFPS), acetonitrile (99.9%, HPLC grade), methanol (99.9%, HPLC grade), ethyl acetate (99.8%, anhydrous), ethylene glycol, dimethyl sulfoxide, quinoline, and cyclohexanol were all purchased from Sigma-Aldrich (St. Louis, MO). Trichloromethylsilane (TCMS) and acetone were supplied by Fisher Scientific (Pittsburgh, PA, USA). Whatman filter paper grade 1 (24 cm) was purchased from Whatman (Little Chalfont, England).
Doping in urine samples
Urine samples were collected from a healthy and drug-free volunteer to make sure that all the analytes tested in this work were absent in the original samples. A 6 μL aliquot of undiluted urine samples containing separately spiked trenbolone, clenbuterol, furosemide, and hydrochlorothiazide analytes were pipetted onto the triangle paper and allowed to dry at room temperature. Analysis of doping agents present in the dried urine sample was achieved using 20 μL of ethyl acetate.
RESULTS AND DISCUSSION
We aimed at developing a sensitive PS-MS platform for screening four different doping substances directly from urine: trenbolone (TREN), clenbuterol (CLEN), furosemide (FUR), and hydrochlorothiazide (HCT) in dried urine samples. We chose to examine these substances because they belong to two important classes of prohibited substances: anabolic agents and diuretics. Collectively, these two classes represent >59% of all occurrences in the most recent WADA report and the four selected analytes are the most reported substances in their classes (18% of all banned substances occurrences)39. Actually, diuretics (e.g., furosemide and hydrochlorothiazide) can be purchased without any prescription and are largely available to the general population for masking other doping substances. Urine samples are the main matrix employed for anti-doping analysis, representing approximately 91% of all samples analyzed in 201739. Moreover, urine sample is more advantageous than blood because it can be collected non-invasively and is available in a relatively large amounts40,41.
In the present work, we show that the MRPL proposed by WADA10 could be reached without any sample clean-up or pre-treatment steps using a simple and fast methodology. By using paper triangles treated with the vapor of trichloromethylsilane, it was possible to obtain lower limits of detection (LOD) and quantification (LOQ) when compared to untreated paper. The organosilane reacts with the surface OH groups in the paper substrate and changes it polarity from hydrophilic to hydrophobic30. This approach increases the transfer of analytes from the paper to the gas-phase. When using an organic solvent immiscible with water (e.g., ethyl acetate), inorganic salt components in the urine sample are negatively suppressed enabling the analytes, which are typically organic in nature, to be extracted, ionized and preferentially transferred to the mass spectrometer for a more sensitive detection.
Optimization of organosilane reagent for hydrophobic paper treatment
The properties of the paper substrate used as solid support for analyte extraction and ionization exhibited significant influence on the performance of paper spray experiment. When a native hydrophilic paper is used, the paper surface has hydroxyl groups (OH) that can form intermolecular bonding with analytes of interesting, decreasing their transfer/desorption from the paper to the mass spectrometer. In addition, the grade 1 filter paper typically used in PS experiments contains trace elements such as calcium (185 μg/g), sodium (160 μg/g), chlorine (130 μg/g), which can cause ion suppression during analysis42. Trichlorosilane derivatives react with the hydroxyl groups (OH) present in the paper, forming a monolayer that cover the paper surface (Figure S1) to prevent access to the pores of the paper that might contain the interfering ions. The silanized paper substrate has been proven to improve detectability and sensitivity in PS by converting the polar groups of the native paper into hydrophobic groups, decreasing the analyte-paper interactions43,44.
Generally, for a liquid-phase silanization reaction, the surface to be silanized is immersed in a solution of hexane containing the organosilane with concentrations ranging from 0.1 to 2 (v/v). After the reaction, the surface must be washed several times with different solvents, such as hexane and methanol, to remove the residual unreacted reagent45–47. Liquid-phase reaction can form agglomerates instead of monolayer during the silanization process, leading to a non-uniform coverage of the solid surface and an irreproducible paper modification48. The type of organosilane reagent available is limited to the compatibility with the solvent used in each step of the fabrication49. The vapor-phase silanization reaction is usually conducted under reduced pressure in a closed system. The reduced pressure increases the partial pressure of the silane regent, which in turn increases the efficiency of the reaction in a short amount to time to yield a more uniform hydrophobic surface30,44,49. The vapor-phase silanization reaction has several advantages over liquid-phase reaction: 1) no organic solvent is used during the silanization process and does not generate waste, meaning that this approach is safer for the operator and the environment; 2) it is simple (one step process)50; 3) less reagent is used for the process; 4) multilayers are less likely to be formed; 5) the monolayers formed are highly oriented and have typically higher-order and quality49,51.
Unlike any of our previous studies30,43,44, we chose to test the performance of two different organosilanes: trichloro(3,3,3-trifluoropropyl)silane (TCTFPS) and trichloromethylsilane (TCMS). Both silane reagents are highly volatile, facilitating vapor-phase silanization reactions without applying external heat. TCTFPS was selected based on previous preliminary studies30. We sought new reagents that will improve performance and enable incineration of the modified paper without generating harmful gases. The absence of the fluoro functional group in TCMS was attractive; plus, we anticipated the small methyl group will offer a more uniform and densely packed hydrophobic layer, which can increase extraction efficiency by reducing residual OH exposed at the paper surface after treatment. Reaction time for both reagents was kept at < 4 h to ensure that only hydroxyl groups at the surface of the paper are salinized, while leaving the OH groups located at the interior of the paper unreacted30. For the purposes of comparison, untreated hydrophilic paper substrates were also used for paper spray.
Figure 2 provides the results from PS-MS analysis of trenbolone and furosemide analytes using untreated (0 min), TCTFPS and TCMS modified paper triangles, treated for 15, 30, 60, 120 and 240 min. The analytes were spiked into urine and the dried samples were analyzed using ethyl acetate spray solvent. The results revealed 60 min TCMS treated paper to be superior to untreated and TCTFPS treated paper substrates when trenbolone was analyzed in the positive-ion mode (Figure 2a). Similar trend was observed in the negative-ion mode during the analysis of furosemide (Figure 2b) except that untreated paper gave a slightly better ion yield than TCTFPS treated paper. The optimum silane exposure time (60 min) observed here indicates that a more uniform monolayer of methyl groups at the paper surface is essential for analyte elution and ionization from the treated paper substrate. Before this optimum time, pockets of hydrophilic areas may remain in the paper and beyond the 60 min time, agglomerates may begin to form as opposed to monolayer coverage. Both effects (insufficient coverage and agglomeration) may lead to surface heterogeneity that decreases analytical performance. The higher overall ion signal derived from the TCMS treated paper, when compared to TCTFPS, is attributed to the reduced size of the methyl group in TCMS, which presents a smaller steric hindrance than the trifluoropropyl group in TCTFPS and thus promoting a more effective reaction between the surface hydroxyl groups of the paper and the TCMS reagent. The resultant TCMS treated paper has been determined to be more hydrophobic than TCTFPS treated paper (see Supporting Information, Table S2). Since wetting of the aqueous-based urine sample is limited on the denser and more uniform monolayer created by the methyl groups,51 the analyte become concentrated at a smaller area in the paper and facilitates extraction and transport to the mass spectrometer44,52. Surface heterogeneity (or roughness) can facilitate wetting, which dilutes the analyte by distributing the sample into larger area in the paper. The ion intensities recorded, in the negative-ion mode, from both untreated and TCTFPS treated paper substrates were lower when compared with TCMS treated paper for furosemide analyte (Figure 2b). This fact is probably related to the reduced onset of electrical discharge on the relatively more hydrophilic TCTFPS treated and untreated paper surfaces. Increased discharge is prevalent from hydrophilic surfaces27 especially in the presence of inorganic salts, which is known to annihilate analyte signal in the negative-ion mode (discussed in detail later).
Figure 2.
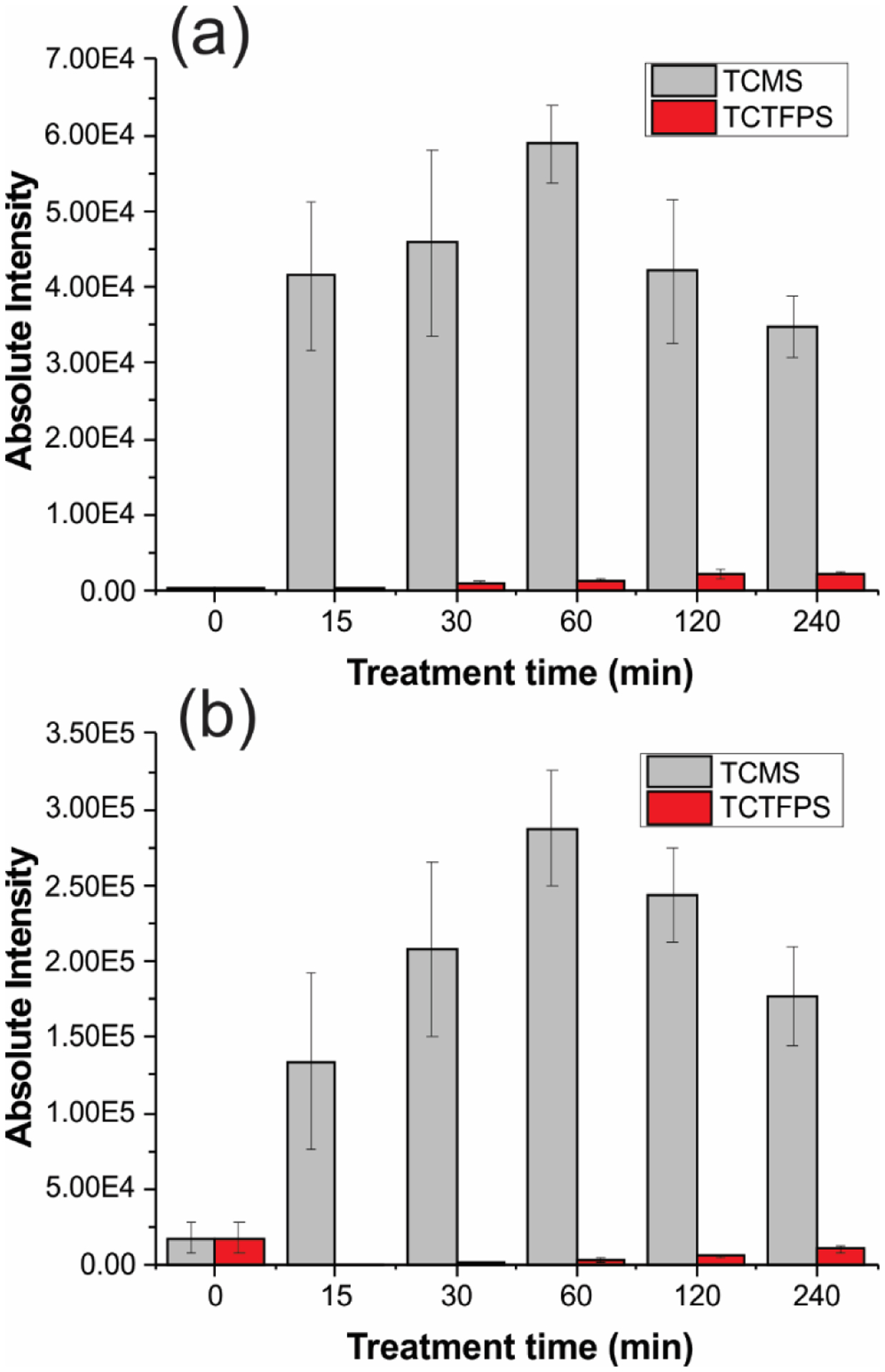
Optimization of paper treatment reagent and time. Absolute intensity of (a) 500 ng mL−1 trenbolone and (b) 12.5 μg mL−1 furosemide spiked in urine samples. Characteristic fragments of protonated trenbolone (271 → 253) and deprotonated furosemide (329 → 285) were used for analysis. Paper triangles were treated with TCMS (light gray) and TCTFPS (red) for 15, 30, 60, 120 and 240 min.
Optimization of ionization and ion transmission variables
Our initial optimization experiments involved the characterization of analytes in both positive- and negative-ion modes. While positive-ion mode analysis of trenbolone (MW 270 Da) and clenbuterol (MW 276 Da) predominantly produced (M+H)+ ions in high abundance (Figures 3a and b), the ion signal in the negative-ion mode for furosemide (MW 331 Da) and hydrochlorothiazide (MW 298 Da) was found to spread into many different ion types, including (M–H)−, (M+Cl) − and fragments (Figures 3c and d). The structures of protonated analytes were confirmed by tandem MS via multistage collision-induced dissociation (CID) experiments. For example, protonated trenbolone precursor ion fragmented primarily through the elimination of H2O to give a product ion at m/z 253. A competitive fragmentation pathway involved the elimination of neutral ethanal (CH3CH=O; MW 44 Da) to give m/z 227, which subsequently dissociated to yield fragment ion at m/z 199 via ethylene (CH2=CH2; MW 28 Da) loss. Likewise, the structure of protonated clenbuterol was characterized using CID tandem MS experiments in which the precursor ion at m/z 277 also fragmented via H2O to give the main ion peak at m/z 259, followed by loss of an isobutylene (MW 56 Da) to form the product ion at m/z 203. The product-ion mass spectra for protonated trenbolone and clenbuterol are shown as inserts in Figures 3a and b, respectively. External calibration curves for trenbolone and clenbuterol were constructed by measuring the intensity of the diagnostic ions m/z 227 and 203, respectively.
Figure 3.
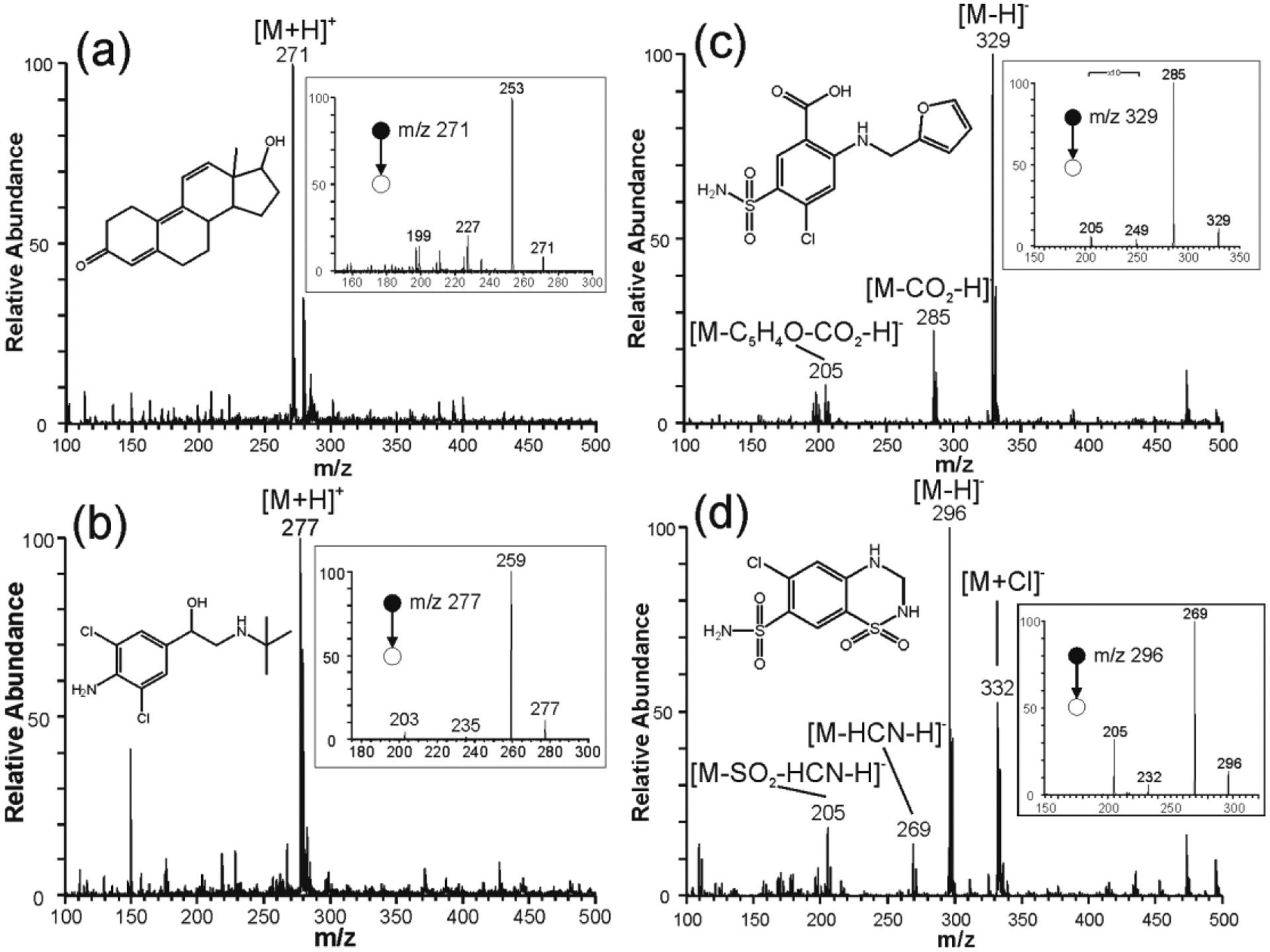
Positive-ion mode paper spray mass spectra recorded for (a) trenbolone and (b) clenbuterol, and in negative-ion mode paper spray for (c) furosemide and (d) hydrochlorothizaide prepared in MeOH:H2O (1:1) using treated paper with trichloro(3,3,3-trifluoropropyl)silane for 120 min. Inserts show CID MS2 spectra and structures of the analytes.
The identity of the fragment ions observed during the full MS analysis of negative ions derived from furosemide and hydrochlorothiazide were confirmed in MS/MS experiments. For example, mass-selected gas-phase CID fragmentation of deprotonated furosemide ions at m/z 329 yielded product ions at m/z 285 via the loss of CO2 (MW 44 Da), which in turn dissociated by losing sulfonic amide (MW 80 Da) to give ion at m/z 205. Competitive dissociation involved m/z 329 → 249 via the loss of sulfonic amide directly from the (M–H)– ion. As can be observed in Figure 3c, these fragment ions were observed in the full MS analysis indicating in-source fragmentation had occurred. Similar comparison between the full mass spectrum and the MS/MS product-ion spectrum for hydrochlorothiazide recorded in the negative-ion mode revealed in-source fragmentation was also prevalent for hydrochlorothiazide (Figure 3d). Although the in-source fragmentations detected here can enable structural characterization in a single-stage MS experiment, it is undesirable in quantitative analysis. Therefore, it was necessary to establish experimental conditions that will enable the production of one major analyte species without significant in-source fragmentation. This included the optimization of tube lens voltage and transfer capillary temperature, and spray solvent and voltage.
Tube lens voltage and capillary temperature
Aside from in-source fragmentation, Cl– adduction leading to (M+Cl)– formation was also observed for hydrochlorothiazide at m/z 332 (Figure 3d). Chlorine ions are commonly present in biological samples such as urine and blood, and when working in negative-ion mode can form adduct with organic compounds, as observed for the hydrochlorothiazide. The deliberate Cl– adduct formation has been used to improve the ionization of organic molecules in negative-ion mode53–55. Unfortunately, tandem MS tend not to give informative fragmentation patterns. In addition, like in-source fragmentation, an adduct in mass spectra decreases the intensity of the deprotonated ion impairing sensitivity of the analytical method31.
Therefore, the influence of tube lens voltage and capillary temperature on adduct formation as well as in-source fragmentation were investigated using a central composite design. The design matrix for furosemide and hydrochlorothiazide is described in Table S3. For furosemide, the most important aspects were to avoid in-source fragmentation and to increase the (M-H)− ion intensity. The adduct intensity for furosemide was 100X lower when a capillary temperature >200 °C was used. For the hydrochlorothiazide, the best condition was obtained for a higher intensity of (M-H)− and a lower intensity of the (M+Cl)−. The in-source fragmentation was not investigated because ion dissociation was less prevalent below 275 °C. As summarized in Figures S2a–d, capillary temperature of 250°C and tube lens voltage of −120 V presented the highest intensity for (M-H)− ions and lowest intensity for the in-source fragmentation and chlorine adducts for both analytes. It was determined that the capillary temperature is the most influential factor for the formation of adducts; this is presumably because the temperature increases the internal energy of the chloride adduct leading to the dissociation and formation of (M-H)− and hydrochloric acid as a byproduct55. The tube lens voltage was found to be the most significant factor for driving in-source fragmentation: decreasing tube lens voltage, in absolute value, accelerates the ions in a region where the pressure is in the millibar range, allowing the ions to collide with the residual gas molecules to give enough energy to induce fragmentation56.
After learning that the intensity of the deprotonated (M–H)– ions is significantly dependent on the capillary temperature and the tube lens voltage, we optimized these two parameters for trenbolone and clenbuterol in the positive-ion mode. The design matrix for this study is described in Table S4, where we identified capillary temperature of 240°C and tube lens voltage of −100 V as the best parameters for high ion intensities in MS/MS product–ion data collection for trenbolone and clenbuterol (Figure S2e,f).
Spray solvent and voltage
The binary mixture MeOH/H2O is the most common solvent selected for bioanalysis when using PS. However, unlike the situation in the traditional electrospray ionization, binary solvent mixture comprising of solvents with different vapor pressures evaporate at different rates, which consequently cause changes in solvent composition during analyte solubilization and ionization over a short time period57.
One significant difference between positive- and negative-ion modes is spray stability. In negative mode, stable spray is difficult to establish with a solvent mixture containing water; the current is typically higher than for positive-ion mode and a glowing electrical discharge that degrades ion signal are usually observed27. The onset of discharge can contribute to in-source fragmentation. Different studies have shown that hydrophobic surfaces are capable of reducing corona discharge when using negative-ion mode31,58. When a pure organic solvent is used, such as methanol, a stable Taylor cone and analyte signal could be obtained. Thus, the spray solvent should preferably be a pure organic solvent to avoid electrical discharge and must have three proprieties: suitable for electrospray, must have surface tension less than that of the hydrophobic paper to allow wetting, and a good solubilizing power for the target analytes30. Acetone, acetonitrile, ethyl acetate and methanol satisfy these criteria. Urine samples spiked with trenbolone and furosemide were chosen to study the best spray solvent. Ethyl acetate presented the best ionization enhancement for the analytes, followed by acetonitrile and acetone. The ion intensities obtained when using ethyl acetate was 7X and 5X higher than that derived from acetonitrile, during the analysis of trenbolone and furosemide in positive- and negative-ion modes, respectively. Methanol presented the poorest intensity of all the solvents used (Figure 4).
Figure 4.
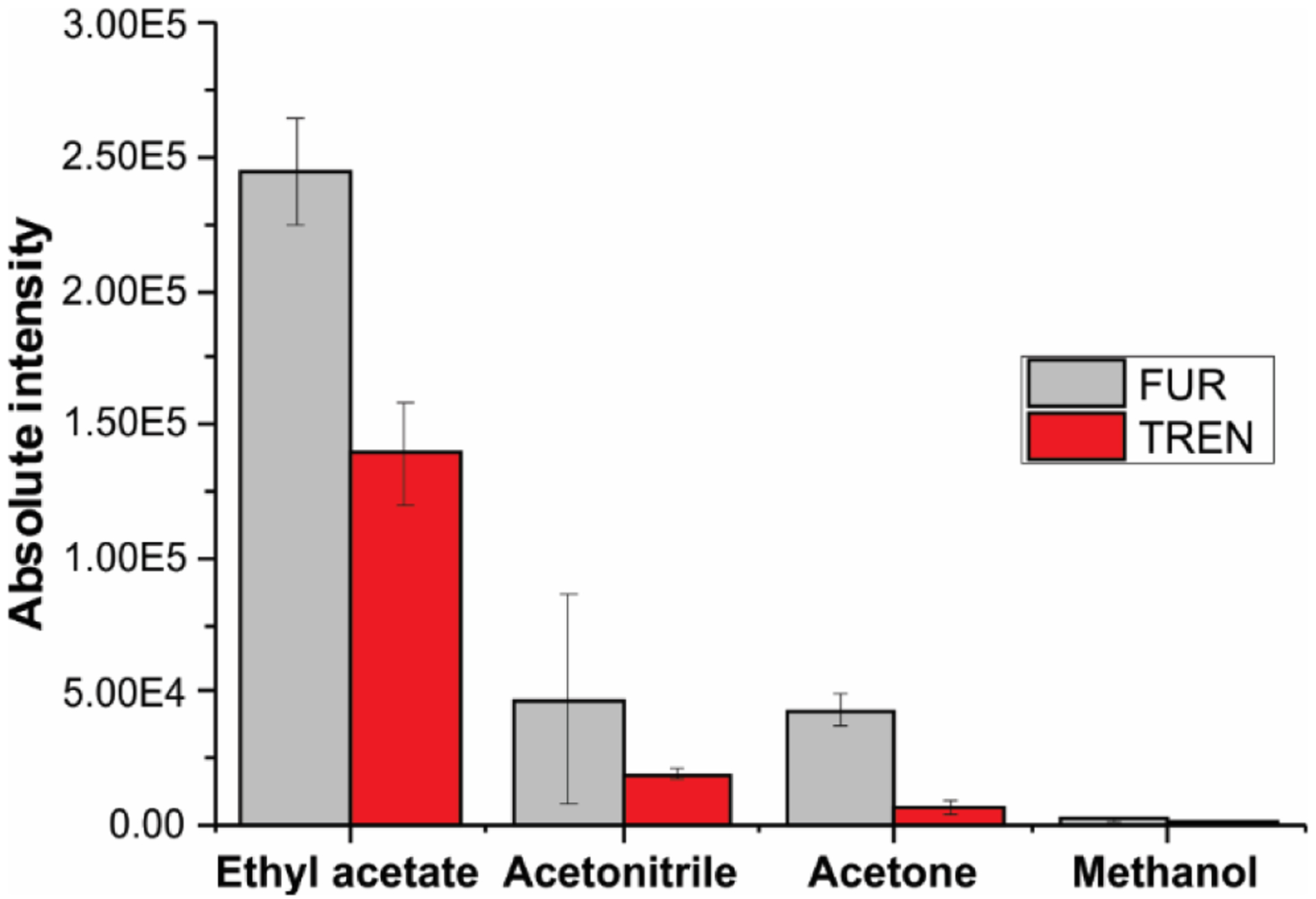
Optimization of spray solvent employed during the paper spray analysis. Absolute intensity of 12.5 μg mL−1 furosemide (light gray) and 1000 ng mL−1 trenbolone spiked in urine samples (red). Characteristic fragments of protonated trenbolone (271 → 253) and deprotonated furosemide (329 → 285) were used for analysis.
During the paper spray analysis, solvent performance is influenced by different properties of the solvent, such as surface tension, dielectric constant (or relative permittivity), conductivity, and chemical properties. Ethyl acetate is weakly polar, it has a high eluent strength (0.58) and immiscible in water, all permitting the efficient extraction of polar organic compound while leaving behind inorganic salts present in the urine sample and in the paper substrate. For instance, biological samples have a high concentration of inorganic ions (e.g., Na+, K+, Ca2+, Mg2+, Cl−, SO42−, CO32−and PO43−) and contain different polar organic compounds (e.g., urea, creatinine, uric acid, citric acid, and lactic acid), which are known to induce ionization suppression of the analytes. Such matrix interference not only affect detectability but it can also affect the accuracy and precision of analytical measurements59. The eluent strength of a solvent to solubilize an ionic salt is dependent on the dielectric constant. Solvents with higher dielectric constant can separate charges (or ions) more effectively than a solvent with a smaller dielectric constant. Consequently, solvents with lower dielectric constant, or lower relative permittivity, solubilizes fewer salts and therefore, the intensity of the salt-induced ionization suppression is reduced31,60,61. Ethyl acetate showed the best results because it has the lowest dielectric constant (Table S5), decreasing ion suppression caused by inorganic salt and it is an aprotic solvent and less polar than the other solvents, decreasing the suppression caused by highly polar compounds found in urine. Acetonitrile was supposed to be the worst solvent for PS-MS since it has the highest dielectric constant of all the organic solvents tested, but polarity also is an important property in the solubilization/ionization step60. Methanol is a protic solvent with the highest polarity among all solvents tested; it presented the poorest performance because it has the capacity to increase the salt and organic composition of the spray.
In addition to solvent optimization, we investigated the effect of spray voltage on ion yield using trenbolone furosemide in positive- and negative-ion modes, respectively. The results are summarized in Figure S3, in which voltages ranging from 1.5 to 5.0 kV were applied to the paper triangle. The ion signal was strongly dependent on spray voltage. The optimized spray voltage was 2 kV for positive-ion mode and –2 kV for negative-ion mode. Lower ion signals were observed for voltages less than ±2 kV, while spray voltages higher than 5 kV in negative-ion mode generate corona discharge62,63, producing highly unstable spray current (> 7 μA)64 and no ion signal of interest was observed.
Direct detection of doping agents in urine samples
Determination of doping compounds is a highly demanding task. The most recent anti-doping exercise reported by WADA showed that more than 320,000 samples were analyzed in 2017, which represents ~7.1% increase from 2016; of these, 4,596 samples registered the presence of various illicit compounds39. Unlike chromatographic methods, the paper spray MS method reported here requires no sample preparation. In a typical experiment, we spiked fresh urine samples separately with trenbolone, clenbuterol, furosemide, and hydrochlorothiazide with concentrations ranging from 1 – 25,000 ng mL−1 (see Table S6 for standard concentrations used for a specific analyte). A 6 μL aliquot of the samples were deposited onto the hydrophobic paper treated with TCMS (60 min). After the samples were dried, ± 2 kV DC high voltage was applied to the paper triangle positioned in front of the mass spectrometer. Concomitant application of 20 μL of ethyl acetate spray solvent allowed MS/MS analysis of analyte for identification.
Calibration curves (Figure S4) were constructed for each analyte based on the absolute analyte signal obtained in tandem MS experiments. Data were analyzed using the least square method and analysis of variance (ANOVA) where we determined regression equation, linear range, coefficient of determination and precision (Figure S6). The data showed excellent linearity (R2 > 0.997), acceptable precision (indicated by tight error bars, Figure S4) and accuracy (RSD < 10%). Additionally, we further characterized the quantitation capabilities of the method by performing quality control standard addition and recovery experiments. The results from quality control standard addition and recovery experiments suggest minimal matrix effects during the application of the proposed method for the determination of selected doping agents65. The results (Table 1) strongly support the absence of uncontrolled experimental variations. The ANOVA analysis further demonstrated linearity without lack-of-fit. These results suggest that anti-doping analysis can be achieved effectively using PS-MS at a much faster speed than the conventional MS-based methods that utilize prior separation step.
Table 1.
Precision and recovery for trenbolone, clenbuterol, furosemide and hydrochlorothiazide in urine samples using hydrophobic PS-MS.
| Trenbolone | 50 | 3.8 | 101 ± 3 |
| 500 | 6.3 | 99 ± 3 | |
| Clenbuterol | 50 | 4.4 | 101 ± 2 |
| 500 | 3.9 | 97 ± 2 | |
| Furosemide | 2000 | 4.5 | 96 ± 1 |
| 12500 | 3.9 | 102 ± 3 | |
| Hydrochloro-thiazide | 2000 | 7.0 | 100 ± 5 |
| 12500 | 6.0 | 103 ± 3 |
Limits of detection (LODs) and limits of quantification (LOQs) obtained using our proposed PS-MS methodology are smaller (i.e., more sensitive) compared to required MRPL (Table 2), which is the performance level established by WADA10. As shown in Table 2, the analytical merits derived from untreated hydrophilic paper substrate satisfied the WADA requirement for only one of the tested compounds (hydrochlorothiazide; LOQ 3.44 ng mL−1). Sensitivity (LOQ) for trenbolone recorded from hydrophilic paper was >100X above the limits set by WADA. On the contrary, there was an obvious improvement in sensitivity and detectability for all four compounds when hydrophobic paper was used. The LODs and LOQs obtained for PS-MS utilizing treated hydrophobic paper were 2 orders of magnitude lower for clenbuterol and hydrochlorothiazide and 3 orders of magnitude lower for trenbolone and furosemide, when comparing to untreated hydrophilic paper, which achieved sub-ng mL−1 sensitivity levels. Once deposited onto the hydrophobic paper, the urine sample bead and formed a spherical drop due to the reduced surface energy of the silanized paper resulting in a limited wettability. After drying, the urine sample formed a concentrated yellowish spot close to the tip of the hydrophobic paper triangle, with decreased analyte/paper interaction. This prevented the penetration of the sample into the core of the fibrous paper and facilitated analyte extraction and transfer to the mass spectrometer. In contrast, the urine sample was completely absorbed into the hydrophilic paper when 6 μL was applied; this effect increased strong interactions between the analyte(s) and the cellulose substrate, decreasing analyte transfer to the gas-phase.
Table 2.
LODs and LOQs for trenbolone, clenbuterol, furosemide, and hydrochlorothiazide in urine samples using hydrophobically treated paper and hydrophilic untreated paper.
| Treated hydrophobic paper | Untreated hydrophilic paper | ||
|---|---|---|---|
| Trenbolone | 5 | 0.21 (0.42) | 226 (576) |
| Clenbuterol | 0.2 | 0.041 (0.076) | 1.75 (4.24) |
| Furosemide | 200 | 0.82 (1.65) | 126 (321) |
| Hydrochlorothiazide | 200 | 0.058 (0.12) | 1.37 (3.44) |
MRPL = Minimum Required Performance Levels by WADA10
LODs and LOQs were calculated using the slope from respective calibration curves using the intensity signal corresponding to (Sblank + 3 × σblank) and (Sblank + 10 × σblank), respectively; where Sblank is the average blank signal and σ blank is the standard deviation of the signal from ten replicates
In Tables S7 and S8, we provide LOD/LOQ values (and sample preparation requirements) for trenbolone, clenbuterol, furosemide and hydrochlorothiazide in urine samples as determined by various different analytical methods. These included electrochemical66, spectrophotometric67 and fluorimetric68 methodologies, which are typically applied to detect one substance or one class of substance in urine. That is, they are not capable of detecting compounds from different classes of doping agents. Take hydrochlorothiazide analysis as an example, LODs from voltammetry66 and LC-Vis69 were 6 and 4 ng mL−1, respectively, after sample was purified via centrifugation, filtration and dilution. For furosemide, centrifugation followed by fluorescence analysis68 yielded LOD 6 ng mL−1 whereas liquid-liquid extraction followed by UV-Vis analysis67 afforded LOD 110 ng mL−1 (LOQ was 280 ng mL−1). The most common analytical methods employed to analyze doping agents are LC-MS and GC-MS. Highest reported sensitivities for hydrochlorothiazide and furosemide are LOD 0.85 and 0.24 ng mL−1 from LC-MS14, respectively, and LOD 50 ng mL−1 each from GC-MS20. These sensitivities were achievable only with extensive sample preparation and clean-up steps such as, liquid-liquid extraction15,70,71 and solid-phase extraction13,70,72. In addition, analysis times can be long (>3 h total). The PS-MS methodology presented here simplifies the instrumentation requirements and reduces the time of analysis by combining sample collection, separation and ionization in a single step. The observed detection levels of sub-ng mL−1, achieved without any sample preparation, are comparable or even lower than LC-MS methods. The recent study by Kasperkiewicz and co-authors73 utilizing coated blade spray for quantification of trenbolone and clenbuterol in urine samples is the only example of ambient MS analysis of doping agents reported in the literature procedure, however, the associated LOQs were higher than the MRPL although additional extraction steps are required in this procedure.
Storage study
Paper substrate is one the efficient means of collecting, storing and transporting biofluids. Though not as common as dried blood spot74, paper-based urine collection kits are commercially available for a variety of tests that is based on the analysis of dried urine spot (DUS). To investigate the stability of selected doping agents in DUS samples, we spiked urine separately with trenbolone (1 μg mL−1), clenbuterol (1 μg mL−1), furosemide (2.5 μg mL−1) and hydrochlorothiazide (2.5 μg mL−1). Then, 6 μL aliquots of the spiked samples were spotted on both TCMS treated hydrophobic and untreated hydrophilic paper substrates. The DUS samples presents on the pre-cut paper triangles were stored under two different conditions: in ambient air at room temperature and at 4°C cold conditions. The stored samples were analyzed daily in the first week, followed by a five-day analysis interval for a maximum 25 days. The results of this stability studies are summarized in Figure 5, which showed marked reduction in analytes stability for storage done at room temperature compared to samples stored at 4°C. The larger decrease in ion signal at higher temperatures is expected due to the accelerated degradation promoted by the elevated temperature.
Figure 5.
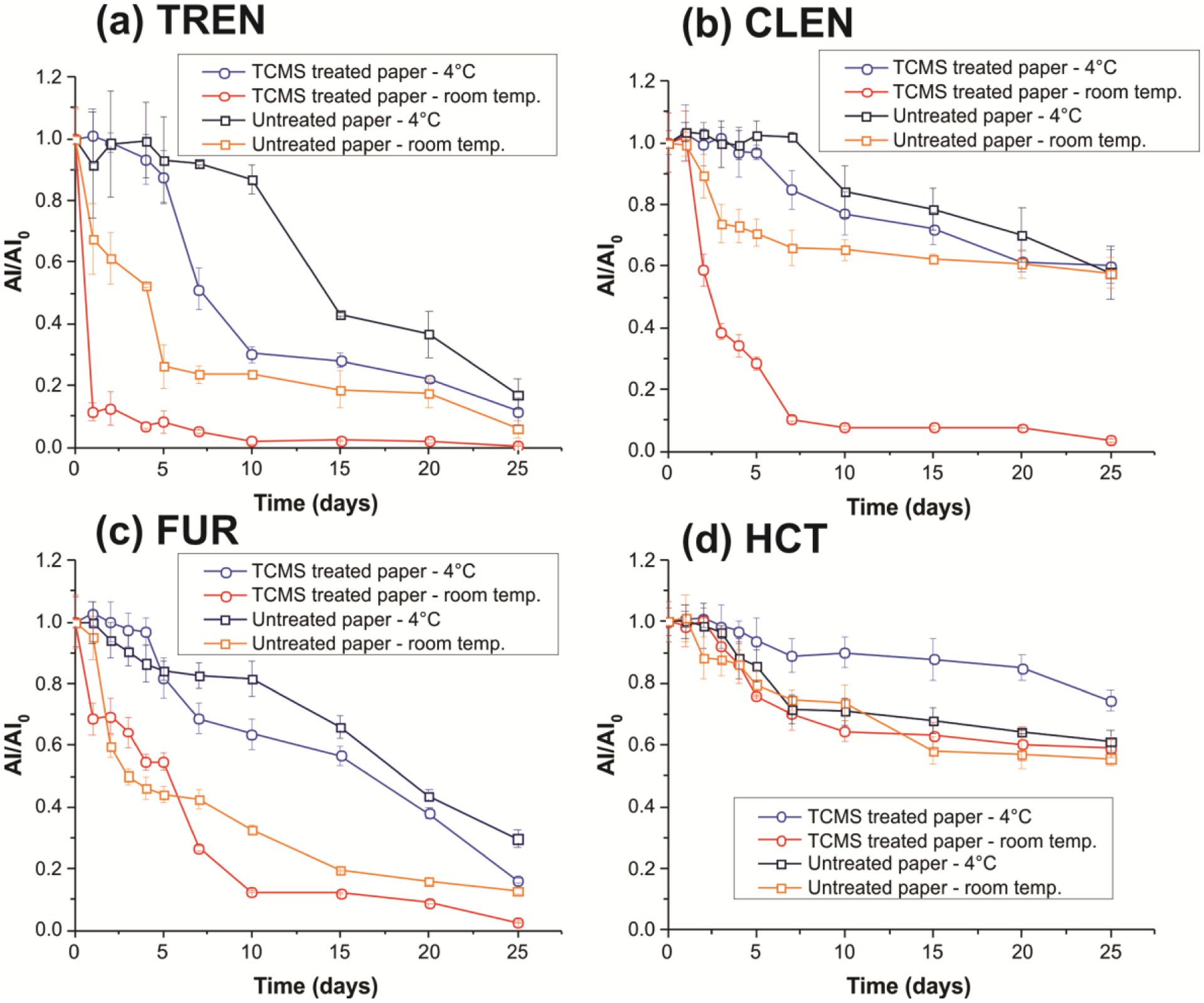
Stability of (a) trenbolone (TREN, 1 μg mL−1), (b) clenbuterol (CLEN, 1 μg mL−1), (c) furosemide (FUR, 2.5 μg mL−1), and (d) hydrochlorothiazide (HCT, 2.5 μg mL−1) spiked in urine samples deposited onto TCMS treated and untreated paper over 25 days stored under room temperature and at 4°C. Error bars represent the standard deviations of analyses for three replicates with independent paper triangles.
Of all the analytes tested, hydrochlorothiazide exhibited better stability in DUS, where >55% was still intact after 25-days of storage on both hydrophobic and hydrophilic paper and under room temperature and cold conditions. While hydrochlorothiazide showed lower degradation rates when stored on treated versus untreated paper substrates, we observed higher stability for trenbolone, clenbuterol and furosemide in untreated hydrophilic paper compared to hydrophobic paper. We ascribe this effect to the fact that urine samples stored on treated paper are more exposed to the environment, since the resultant DUS rests at the surface of the hydrophobic paper. Being non-viscous, urine samples dry as 2D disk. This is contrary to blood samples which dry to yield 3D dried blood spheroids43 due to its high viscosity derived from the presence of erythrocytes, proteins and other macromolecules. In this case, analytes present in the interior of the 3D dried blood spheroid prepared in the hydrophobic paper are protected by critical radius of insulation by the cellular content43. For untreated paper, the non-viscous urine sample penetrates into the core of the paper where the individual fibers of the cellulose paper substrate can serve to protect the labile analyte from oxidative stress. It is important to point out that the degradation effect detected for treated hydrophobic paper substrates is most pronounce under room temperature storage conditions. Analyte stability can be restored by keeping the DUS samples present on the hydrophobic paper at 4 °C. For example, 60% of clenbuterol was detected after the 25th day when stored on hydrophobic paper and kept 4 °C compared with 5% when the same DUS sample was left in the open air at room temperature. These results imply that DUS prepared on hydrophobic can be used as a sample collection and storage platform for doping agents for at least 25 days if preserved at 4°C. Considering the same degradation rate, samples with concentration of MRPL can be detected by the proposed method. For untreated paper, however, only samples containing hydrochlorothiazide with MRPL concentrations can be detected after 25 days storage at 4°C.
CONCLUSIONS
In summary, we have demonstrated direct mass spectrometry method for screening doping agents in raw urine samples using chemically treated paper. The hydrophobic paper modified with TCMS allows a direct and highly sensitive detection of anabolic agents and diuretics in urine samples. The proposed hydrophobic PS-MS methodology shows some advantages over LC-MS and GC-MS platforms: 1) it integrates sample collection, separation and ionization in a single stage, 2) it decreases the solvent consumption, waste production and analysis time, and 3) no sample preparation, other than its deposition on the surface, is required. Surface modification with trimethylsilane effectively reduces the analyte-paper interactions to increase the analyte transfer to the mass spectrometer. The utilization of a hydrophobic paper combined with a weakly polar ethyl acetate organic solvent increased salt tolerance of the method, which in turn limited possible salt-induced discharge in the negative-ion mode as well as matrix effects. Collectively, these resulted in a more selective extraction of target compounds, enabling sub-ng mL−1 sensitivities, which are significantly lower than the World Anti-Doping Agency’s minimum required performance levels for the tested compounds.
Supplementary Material
ACKNOWLEDGEMENT
This research was supported by NIH grant R01-AI-143809. ELR and HRP are grateful for the financial support provided by the São Paulo Research Foundation (FAPESP, grant #2018/05993-1).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Gas-phase process for silanization of paper, surface energy of treated paper, details of central composite design experiment, spray solvent properties, effect of spray voltage, calibration curved and linear regression analysis, comparison of analytical performance for conventions methods. (PDF)
REFERENCES
- (1).World Anti-doping Agency. World Anti-Doping code https://www.wada-ama.org/sites/default/files/resources/files/wada_anti-doping_code_2019_english_final_revised_v1_linked.pdf (accessed Aug 30, 2019).
- (2).Goldman BR; Bush PJ; Klatz R Death in the Locker Room: Steroids & Sports, 1st ed; Icarus Press: South Bend, Indiana, 1984. [Google Scholar]
- (3).Woolf J; Mazanov J; Connor J The Goldman Dilemma Is Dead: What Elite Athletes Really Think about Doping, Winning, and Death. Int. J. Sport Policy 2017, 9 (3), 453–467. [Google Scholar]
- (4).González JM; Johnson FR; Fedoruk M; Posner J; Bowers L Trading Health Risks for Glory: A Reformulation of the Goldman Dilemma. Sport. Med 2018, 48 (8), 1963–1969. [DOI] [PubMed] [Google Scholar]
- (5).Sport B Doping: More than 30% of athletes at 2011 Worlds admit to doping https://www.bbc.com/sport/athletics/41084175 (accessed Nov 7, 2019).
- (6).Ulrich R; Pope HG; Cléret L; Petróczi A; Nepusz T; Schaffer J; Kanayama G; Comstock RD; Simon P Doping in Two Elite Athletics Competitions Assessed by Randomized-Response Surveys. Sport. Med 2018, 48 (1), 211–219. [DOI] [PubMed] [Google Scholar]
- (7).Mullen J; Börjesson A; Hopcraft O; Schulze JJ; Ericsson M; Rane A; Lehtihet M; Ekström L Sensitivity of Doping Biomarkers after Administration of a Single Dose Testosterone Gel. Drug Test. Anal 2018, 10 (5), 839–848. [DOI] [PubMed] [Google Scholar]
- (8).World Anti-doping Agency. Prohibited list https://www.wada-ama.org/sites/default/files/wada_2019_english_prohibited_list.pdf (accessed Jan 30, 2019).
- (9).World Anti-doping Agency. International standard for laboratories https://www.wada-ama.org/sites/default/files/resources/files/isl_nov2019.pdf (accessed Aug 25, 2019).
- (10).World Anti-doping Agency. Minimum required performance levels for detection and identification of non-threshold substances https://www.wada-ama.org/sites/default/files/resources/files/td2019mrpl_eng.pdf (accessed Apr 30, 2019).
- (11).Ozgur E; Roberts KE; Ozgur EO; Gin AN; Bankhead JR; Wang Z; Su J Ultrasensitive Detection of Human Chorionic Gonadotropin Using Frequency Locked Microtoroid Optical Resonators. Anal. Chem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Krone N; Hughes BA; Lavery GG; Stewart PM; Arlt W; Shackleton CHL Gas Chromatography/Mass Spectrometry (GC/MS) Remains a Pre-Eminent Discovery Tool in Clinical Steroid Investigations Even in the Era of Fast Liquid Chromatography Tandem Mass Spectrometry (LC/MS/MS). J. Steroid Biochem. Mol. Biol 2010, 121 (3–5), 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sardela VF; Martucci MEP; de Araújo ALD; Leal EC; Oliveira DS; Carneiro GRA; Deventer K; Van Eenoo P; Pereira HMG; Aquino Neto FR Comprehensive Analysis by Liquid Chromatography Q-Orbitrap Mass Spectrometry: Fast Screening of Peptides and Organic Molecules. J. Mass Spectrom 2018, 53 (6), 476–503. [DOI] [PubMed] [Google Scholar]
- (14).Domínguez-Romero JC; García-Reyes JF; Lara-Ortega FJ; Molina-Díaz A Screening and Confirmation Capabilities of Liquid Chromatography-Time-of-Flight Mass Spectrometry for the Determination of 200 Multiclass Sport Drugs in Urine. Talanta 2015, 134, 74–88. [DOI] [PubMed] [Google Scholar]
- (15).He G; Wu Y; Lu J Doping Control Analysis of 13 Steroids and Structural-like Analytes in Human Urine Using Quadrupole-Orbitrap LC–MS/MS with Parallel Reaction Monitoring (PRM) Mode. Steroids 2018, 131 (August 2017), 1–6. [DOI] [PubMed] [Google Scholar]
- (16).Abushareeda W; Vonaparti A; Saad K. Al; Almansoori M; Meloug M; Saleh A; Aguilera R; Angelis Y; Horvatovich PL; Lommen A; et al. High Resolution Full Scan Liquid Chromatography Mass Spectrometry Comprehensive Screening in Sports Antidoping Urine Analysis. J. Pharm. Biomed. Anal 2018, 151, 10–24. [DOI] [PubMed] [Google Scholar]
- (17).Gansbeke Van W; Polet M; Hooghe F; Devos C; Eenoo Van P Improved Sensitivity by Use of Gas Chromatography-Positive Chemical Ionization Triple Quadrupole Mass Spectrometry for the Analysis of Drug Related Substances. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2015, 1001, 221–240. [DOI] [PubMed] [Google Scholar]
- (18).Abushareeda W; Lyris E; Kraiem S; Wahaibi A. Al;Alyazidi S; Dbes N; Lommen A; Nielen M; Horvatovich PL; Alsayrafi M; et al. Gas Chromatographic Quadrupole Time-of-Flight Full Scan High Resolution Mass Spectrometric Screening of Human Urine in Antidoping Analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2017, 1063, 74–83. [DOI] [PubMed] [Google Scholar]
- (19).Yang S; Liu X; Xing Y; Zhang D; Wang S; Wang X; Xu Y; Wu M; He Z; Zhao J Detection of Clenbuterol at Trace Levels in Doping Analysis Using Different Gas Chromatographic-Mass Spectrometric Techniques. J. Chromatogr. Sci 2013, 51 (5), 436–445. [DOI] [PubMed] [Google Scholar]
- (20).Zaporozhets O; Tsyrulneva I; Ischenko M Determination of 8 Diuretics and Probenecid in Human Urine by Gas Chromatography-Mass Spectrometry: Confirmation Procedure. Am. J. Anal. Chem 2012, 03 (04), 320–327. [Google Scholar]
- (21).Swiner D; Jackson S; Burris B; Badu-Tawiah AK Applications of Mass Spectrometry for Clinical Diagnostics: The Influence of Turnaround Time. Anal. Chem 2019, 92, 183–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Frey BS; Damon DE; Badu‐Tawiah AK Emerging Trends in Paper Spray Mass Spectrometry: Microsampling, Storage, Direct Analysis, and Applications. Mass Spectrom. Rev 2019, No. i, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Badu-Tawiah AK; Eberlin LS; Ouyang Z; Cooks RG Chemical Aspects of the Extractive Methods of Ambient Ionization Mass Spectrometry. Annu. Rev. Phys. Chem 2013, 64 (1), 481–505. [DOI] [PubMed] [Google Scholar]
- (24).Takáts Z; Wiseman JM; Gologan B; Cooks RG Mass Spectrometry Sampling under Ambient Conditions with Desorption Electrospray Ionization. Science. 2004, 306 (5695), 471–473. [DOI] [PubMed] [Google Scholar]
- (25).Cooks RG; Ouyang Z; Takats Z; Wiseman JM Ambient Mass Spectrometry. Science. 2006, 311 (5767), 1566–1570. [DOI] [PubMed] [Google Scholar]
- (26).Feider CL; Krieger A; Dehoog RJ; Eberlin LS Ambient Ionization Mass Spectrometry: Recent Developments and Applications. Anal. Chem 2019, 91 (7), 4266–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang H; Liu J; Graham Cooks R; Ouyang Z Paper Spray for Direct Analysis of Complex Mixtures Using Mass Spectrometry. Angew. Chemie - Int. Ed 2010, 49 (5), 877–880. [DOI] [PubMed] [Google Scholar]
- (28).Wan L; Gong G; Liang H; Huang G In Situ Analysis of Unsaturated Fatty Acids in Human Serum by Negative-Ion Paper Spray Mass Spectrometry. Anal. Chim. Acta 2019, 1075, 120–127. [DOI] [PubMed] [Google Scholar]
- (29).Wang H; Ren Y; McLuckey MN; Manicke NE; Park J; Zheng L; Shi R; Graham Cooks R; Ouyang Z Direct Quantitative Analysis of Nicotine Alkaloids from Biofluid Samples Using Paper Spray Mass Spectrometry. Anal. Chem 2013, 85 (23), 11540–11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Damon DE; Davis KM; Moreira CR; Capone P; Cruttenden R; Badu-Tawiah AK Direct Biofluid Analysis Using Hydrophobic Paper Spray Mass Spectrometry. Anal. Chem 2016, 88 (3), 1878–1884. [DOI] [PubMed] [Google Scholar]
- (31).Liu J; He Y; Chen S; Ma M; Yao S; Chen B New Urea-Modified Paper Substrate for Enhanced Analytical Performance of Negative Ion Mode Paper Spray Mass Spectrometry. Talanta 2017, 166 (October 2016), 306–314. [DOI] [PubMed] [Google Scholar]
- (32).Borges MMC; Santos H; Vasconcelos GA; Nascimento TA; Dutra FVA; Pires BC; Allochio Filho JF; Aquije GMFV; Borges WS; Lacerda V; et al. The Use of Conductive Polymers as a Substrate for Paper Spray Ionization Mass Spectrometry. Anal. Methods 2019, 11 (27), 3388–3400. [Google Scholar]
- (33).Riboni N; Quaranta A; Motwani HV; Österlund N; Gräslund A; Bianchi F; Ilag LL Solvent-Assisted Paper Spray Ionization Mass Spectrometry (SAPSI-MS) for the Analysis of Biomolecules and Biofluids. Sci. Rep 2019, 9 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Carmany DO; Mach PM; Rizzo GM; Dhummakupt ES; McBride EM; Sekowski JW; Benton B; Demond PS; Busch MW; Glaros T On-Substrate Enzymatic Reaction to Determine Acetylcholinesterase Activity in Whole Blood by Paper Spray Mass Spectrometry. J. Am. Soc. Mass Spectrom 2018, 29 (12), 2436–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen S; Wan Q; Badu-Tawiah AK Mass Spectrometry for Paper-Based Immunoassays: Toward on-Demand Diagnosis. J. Am. Chem. Soc 2016, 138 (20), 6356–6359. [DOI] [PubMed] [Google Scholar]
- (36).Zhang C; Manicke NE Development of a Paper Spray Mass Spectrometry Cartridge with Integrated Solid Phase Extraction for Bioanalysis. Anal. Chem 2015, 87 (12), 6212–6219. [DOI] [PubMed] [Google Scholar]
- (37).Manicke NE; Abu-Rabie P; Spooner N; Ouyang Z; Cooks RG Quantitative Analysis of Therapeutic Drugs in Dried Blood Spot Samples by Paper Spray Mass Spectrometry: An Avenue to Therapeutic Drug Monitoring. J. Am. Soc. Mass Spectrom 2011, 22 (9), 1501–1507. [DOI] [PubMed] [Google Scholar]
- (38).Wong MYM; Tang HW; Man SH; Lam CW; Che CM; Ng KM Electrospray Ionization on Porous Spraying Tips for Direct Sample Analysis by Mass Spectrometry: Enhanced Detection Sensitivity and Selectivity Using Hydrophobic/Hydrophilic Materials as Spraying Tips. Rapid Commun. Mass Spectrom 2013, 27 (6), 713–721. [DOI] [PubMed] [Google Scholar]
- (39).World Anti-doping Agency. 2017. Laboratory Testing Figures https://www.wada-ama.org/sites/default/files/resources/files/2017_anti-doping_testing_figures_en_0.pdf (accessed Apr 30, 2019).
- (40).Lin CC; Tseng CC; Chuang TK; Lee DS; Lee Bin G. Urine Analysis in Microfluidic Devices. Analyst 2011, 136 (13), 2669–2688. [DOI] [PubMed] [Google Scholar]
- (41).Protti M; Mandrioli R; Mercolini L Perspectives and Strategies for Anti-Doping Analysis. Bioanalysis 2019, 11 (3), 149–152. [DOI] [PubMed] [Google Scholar]
- (42).Zhang Y; Ju Y; Huang C; Wysocki VH Paper Spray Ionization of Noncovalent Protein Complexes. Anal. Chem 2014, 86 (3), 1342–1346. [DOI] [PubMed] [Google Scholar]
- (43).Damon DE; Yin M; Allen DM; Maher YS; Tanny CJ; Oyola-Reynoso S; Smith BL; Maher S; Thuo MM; Badu-Tawiah AK Dried Blood Spheroids for Dry-State Room Temperature Stabilization of Microliter Blood Samples. Anal. Chem 2018, 90 (15), 9353–9358. [DOI] [PubMed] [Google Scholar]
- (44).Swiner DJ; Jackson S; Durisek GR; Walsh BK; Kouatli Y; Badu-Tawiah AK Microsampling with Cotton Thread: Storage and Ultra-Sensitive Analysis by Thread Spray Mass Spectrometry. Anal. Chim. Acta 2019, 1082, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bambauer TP; Maurer HH; Weber AA; Hannig M; Pütz N; Koch M; Manier SK; Schneider M; Meyer MR Evaluation of Novel Organosilane Modifications of Paper Spray Mass Spectrometry Substrates for Analyzing Polar Compounds. Talanta 2019, 204 (March), 677–684. [DOI] [PubMed] [Google Scholar]
- (46).Wang J; Wong JXH; Kwok H; Li X; Yu HZ Facile Preparation of Nanostructured, Superhydrophobic Filter Paper for Efficient Water/Oil Separation. PLoS One 2016, 11 (3), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).He Q; Ma C; Hu X; Chen H Method for Fabrication of Paper-Based Microfluidic Devices by Alkylsilane Self-Assembling and UV/O3-Patterning. Anal. Chem 2013, 85 (3), 1327–1331. [DOI] [PubMed] [Google Scholar]
- (48).Bunker BC; Carpick RW; Assink RA; Thomas ML; Hankins MG; Voigt JA; Sipola D; De Boer MP; Gulley GL Impact of Solution Agglomeration on the Deposition of Self-Assembled Monolayers. Langmuir 2000, 16 (20), 7742–7751. [Google Scholar]
- (49).Glass NR; Tjeung R; Chan P; Yeo LY; Friend JR Organosilane Deposition for Microfluidic Applications. Biomicrofluidics 2011, 5 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Glavan AC; Martinez RV; Subramaniam AB; Yoon HJ; Nunes RMD; Lange H; Thuo MM; Whitesides GM Omniphobic “RF Paper” Produced by Silanization of Paper with Fluoroalkyltrichlorosilanes. Adv. Funct. Mater 2014, 24 (1), 60–70. [Google Scholar]
- (51).Ulman A Formation and Structure of Self-Assembled Monolayers. Chem. Rev 1996, 96 (4), 1533–1554. [DOI] [PubMed] [Google Scholar]
- (52).Basuri P; Baidya A; Pradeep T Sub-Parts-per-Trillion Level Detection of Analytes by Superhydrophobic Preconcentration Paper Spray Ionization Mass Spectrometry (SHPPSI MS). Anal. Chem 2019, 91 (11), 7118–7124. [DOI] [PubMed] [Google Scholar]
- (53).Bridoux MC; Schwarzenberg A; Schramm S; Cole RB Combined Use of Direct Analysis in Real-Time/Orbitrap Mass Spectrometry and Micro-Raman Spectroscopy for the Comprehensive Characterization of Real Explosive Samples. Anal. Bioanal. Chem 2016, 408 (21), 5677–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rannulu NS; Cole RB Novel Fragmentation Pathways of Anionic Adducts of Steroids Formed by Electrospray Anion Attachment Involving Regioselective Attachment, Regiospecific Decompositions, Charge-Induced Pathways, and Ion-Dipole Complex Intermediates. J. Am. Soc. Mass Spectrom 2012, 23 (9), 1558–1568. [DOI] [PubMed] [Google Scholar]
- (55).Zhu J; Cole RB Formation and Decompositions of Chloride Adduct Ions, [M+Cl]-, in Negative Ion Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom 2000, 11 (11), 932–941. [DOI] [PubMed] [Google Scholar]
- (56).Hoffman de E; Stroobant V Mass Spectrometry: Principles and Applications, 3rd ed; Wiley: Chichester, 2007. [Google Scholar]
- (57).Tsai CW; Tipple CA; Yost RA Application of Paper Spray Ionization for Explosives Analysis. Rapid Commun. Mass Spectrom 2017, 31 (19), 1565–1572. [DOI] [PubMed] [Google Scholar]
- (58).Higashiyama Y; Yanase S; Sugimoto T DC Corona Discharge from Water Droplets on a Hydrophobic Surface. J. Electrostat 2002, 55 (3–4), 351–360. [Google Scholar]
- (59).Kulyk DS; Miller CF; Badu-Tawiah AK Reactive Charged Droplets for Reduction of Matrix Effects in Electrospray Ionization Mass Spectrometry. Anal. Chem 2015, 87 (21), 10988–10994. [DOI] [PubMed] [Google Scholar]
- (60).Xin N; Sun Y; He M; Radke CJ; Prausnitz JM Solubilities of Six Lithium Salts in Five Non-Aqueous Solvents and in a Few of Their Binary Mixtures. Fluid Phase Equilib. 2018, 461, 1–7. [Google Scholar]
- (61).Khalil Mutasim I.. The Role of Dielectric Constant in Sodium Chloride Solution Chemistry: Magnitude of Super Saturation. Int. J. Phys. Sci 2012, 7 (4), 578–583. [Google Scholar]
- (62).Kulyk DS; Swiner DJ; Sahraeian T; Badu-Tawiah AK Direct Mass Spectrometry Analysis of Complex Mixtures by Nanoelectrospray with Simultaneous Atmospheric Pressure Chemical Ionization and Electrophoretic Separation Capabilities. Anal. Chem 2019, 91, 11562–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kulyk DS; Sahraeian T; Wan Q; Badu-Tawiah AK Reactive Olfaction Ambient Mass Spectrometry. Anal. Chem 2019, 91, 6790–6799. [DOI] [PubMed] [Google Scholar]
- (64).McKenna J; Dhummakupt ES; Connell T; Demond PS; Miller DB; Michael Nilles J; Manicke NE; Glaros T Detection of Chemical Warfare Agent Simulants and Hydrolysis Products in Biological Samples by Paper Spray Mass Spectrometry. Analyst 2017, 142 (9), 1442–1451. [DOI] [PubMed] [Google Scholar]
- (65).Taverniers I; De Loose M; Van Bockstaele E Trends in Quality in the Analytical Laboratory. II. Analytical Method Validation and Quality Assurance. TrAC - Trends Anal. Chem 2004, 23 (8), 535–552. [Google Scholar]
- (66).Beitollahi H; Hamzavi M; Torkzadeh-Mahani M Electrochemical Determination of Hydrochlorothiazide and Folic Acid in Real Samples Using a Modified Graphene Oxide Sheet Paste Electrode. Mater. Sci. Eng. C 2014, 52, 297–305. [DOI] [PubMed] [Google Scholar]
- (67).Tharpa K; Basavaiah K; Vinay KB Use of a Diazocoupling Reaction for Sensitive and Selective Spectrophotometeric Determination of Furosemide in Spiked Human Urine and Pharmaceuticals. Chem. Pap 2010, 64 (4), 415–423. [Google Scholar]
- (68).Tsyrulneva I; Zaporozhets O Simple and Rapid Determination of Diuretics by Luminescent Method. Pharmacol. Pharm 2013, 04 (07), 520–527. [Google Scholar]
- (69).Aydoǧmuş Z Simultaneous Determination of Aliskiren, Amlodipine and Hydrochlorothiazide in Spiked Human Plasma and Urine by High Performance Liquid Chromatography. J. Anal. Chem 2015, 70 (4), 502–509. [Google Scholar]
- (70).Cha E; Kim S; Kim HW; Lee KM; Kim HJ; Kwon OS; Lee J Relationships between Structure, Ionization Profile and Sensitivity of Exogenous Anabolic Steroids under Electrospray Ionization and Analysis in Human Urine Using Liquid Chromatography-Tandem Mass Spectrometry. Biomed. Chromatogr 2016, 30 (4), 555–565. [DOI] [PubMed] [Google Scholar]
- (71).Jeong ES; Kim SH; Cha EJ; Lee KM; Kim HJ; Lee SW; Kwon OS; Lee J Simultaneous Analysis of 210 Prohibited Substances in Human Urine by Ultrafast Liquid Chromatography/Tandem Mass Spectrometry in Doping Control. Rapid Commun. Mass Spectrom 2015, 29 (4), 367–384. [DOI] [PubMed] [Google Scholar]
- (72).Arabi M; Ghaedi M; Ostovan A Development of a Lower Toxic Approach Based on Green Synthesis of Water-Compatible Molecularly Imprinted Nanoparticles for the Extraction of Hydrochlorothiazide from Human Urine. ACS Sustain. Chem. Eng 2017, 5 (5), 3775–3785. [Google Scholar]
- (73).Kasperkiewicz A; Gómez-Ríos GA; Hein D; Pawliszyn J Breaching the 10 Second Barrier of Total Analysis Time for Complex Matrices via Automated Coated Blade Spray. Anal. Chem 2019, 91 (20), 13039–13046. [DOI] [PubMed] [Google Scholar]
- (74).Peake RWA A Case of Increased C5-OH Acylcarnitine. Clin. Chem 2016, 62 (9), 1278–1279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.