

Abstract
Horizontal transfer (HT) alters the repertoire of symbiosis genes in rhizobial genomes and may play an important role in the on-going evolution of the rhizobia–legume symbiosis. To gain insight into the extent of HT of symbiosis genes with different functional roles (nodulation, N-fixation, host benefit and rhizobial fitness), we conducted comparative genomic and selection analyses of the full-genome sequences from 27 rhizobial genomes. We find that symbiosis genes experience high rates of HT among rhizobial lineages but also bear signatures of purifying selection (low Ka : Ks). HT and purifying selection appear to be particularly strong in genes involved in initiating the symbiosis (e.g. nodulation) and in genome-wide association candidates for mediating benefits provided to the host. These patterns are consistent with rhizobia adapting to the host environment through the loss and gain of symbiosis genes, but not with host-imposed positive selection driving divergence of symbiosis genes through recurring bouts of positive selection.
Keywords: lateral gene transfer, mutualism, coevolution, microbial evolution, plant–microbe, generax
1. Background
In bacterial populations, horizontal transfer (HT) is an important mechanism for introducing potentially adaptive genetic variation [1]. The potential advantage of horizontally transferred genes is that they can confer an adaptive phenotype more rapidly than might be possible through the accumulation of mutations within a single lineage. Such phenotypes may even be inaccessible through step-wise mutational processes, either because the population might go extinct in the time it would take for the new phenotype to evolve or because acquiring the phenotype would require a population to cross an adaptive valley [2].
There is strong evidence for the between-rhizobia transfer of at least some of the genes needed to establish a functional symbiosis between rhizobial bacteria and legume hosts [3]. Rhizobia are soil bacteria that form a facultative (i.e. can survive without the host) resource-based mutualism with legume host plants by infecting the roots of compatible legume species. Infected roots form structures called nodules, and inside of the nodules rhizobia convert atmospheric nitrogen into a plant-usable form. In exchange for this nitrogen, the bacteria obtain carbon resources from the host plant. However, not all rhizobia can form an effective symbiosis with all legumes. Rather, the ability of a rhizobium to form a symbiosis with a particular host is determined by a series of host- and rhizobia-specific nodulation molecules [4,5]. Functional analyses also have identified and characterized many of the genes that are needed for N-fixation and resource exchange after a nodule is formed [6].
Strict coevolution of rhizobia and legumes would result in parallel cladogenesis. However, host and bacterial phylogenies reveal evidence for extensive host switching. Rhizobia from some phylogenetically close lineages can form a symbiosis with distantly related plant hosts, and members of some distantly related bacterial lineages can form a symbiosis with the same host [7]. The non-parallel cladogenesis of legumes and rhizobia is suggestive of HT of symbiosis genes. This has been confirmed by multiple examples of individual symbiosis genes having relatedness that differs from that of the rhizobial species in which they are found [8–13]. Although there are many examples of the HT of symbiosis genes, we know relatively little about the evolutionary dynamics of HT among rhizobia, beyond that it occurs and can be important for shifting the host range.
Here, we use comparative genomic analyses to gain insight into the evolution of symbiosis genes and, by extension, the evolution of symbiosis. We had three main objectives. The first objective was to characterize the extent of HT of symbiosis genes. Certainly, rhizobia acquire some of their repertoire of symbiosis genes from other lineages [3]. Given that forming a symbiosis is expected to confer a fitness advantage, that HT potentially offers an opportunity for a rhizobium to expand its range of compatible hosts, and that many nodulation genes are found near insertion sequences or on plasmids [3,14–17], we expect that symbiosis genes are transferred at a higher rate than non-symbiosis genes. However, systematic genome-wide estimates of the HT rates of symbiosis relative to non-symbiosis genes across a wide range of rhizobial lineages are lacking.
Our second objective was to determine whether HT affects the evolution of some aspects of the symbiosis more than others. If HT primarily enables a symbiont to expand or alter its host range, then we would expect symbiosis genes acquired from different lineages would be over-represented by genes that are central to nodule formation. Alternatively, if post-establishment processes (e.g. fixation of nitrogen) are most affected by HT, then we would expect to see stronger evidence of HT among genes involved in N-fixation or in the exchange of benefits between host and bacteria.
Our third objective was to examine how selection has contributed to the divergence of symbiosis genes. Symbiosis genes might experience repeated bouts of positive selection in response to selection imposed by hosts to exclude less beneficial symbionts [18,19]. If this is the case, we would expect that symbiosis genes harbour signatures of positive selection having driven their divergence. Alternatively, symbiotic partners might be at an evolutionary stasis [18,20] because they are at or near a selective optimum. In this case, symbiosis genes would be expected to experience purifying selection, as has been found in the nod region in Ensifer medicae [9] and type III effector proteins in Bradyrhizobium japonicum and E. fredii [19].
To address these objectives, we analysed publicly available full-genome sequences from 27 species of alphaproteobacterial rhizobia. We examine four sets of symbiosis genes: those annotated as having a role in nodule formation, those annotated as involved in N-fixation, genes identified by genome-wide association (GWA) analyses as candidates underlying variation in the benefits plant hosts obtain from rhizobia, and GWA-identified candidates underlying variation in host-associated rhizobial fitness. For each group of genes, we estimate the phylogenetic signal of gene presence, the extent of HT among lineages, and past selection as measured by the rate of non-synonymous to synonymous nucleic acid changes (Ka : Ks).
2. Methods
We identified genomes for analysis by searching the NCBI Genome database for genomes from the named species or genera in the alpha- and betaproteobacterial rhizobial clades (species names from [21]). Because our objectives are to estimate the extent of HT among rhizobia, we include genomes from a widely related set of rhizobia species. We then used the NCBI ‘CladeIDs’ to identify a representative strain from every clade in these species or genera. In cases with different named species or biovars sharing the same CladeID (E. americanum CCGM7 and E. fredii NGR234; R. phaseoli N161, R. esparanzae N561, and biovars of R. etli; biovars of R. leguminosarum; and the three Mesorhizobium species), we included a representative of each of the named taxa (electronic supplementary material, table S1). We also included two genomes obtained from MaGe (mage.genoscope.cns.fr), Ensifer terangae (USDA4894) which was not in NCBI and E. meliloti USDA1106 because it was used in previous GWA analyses [22]. Our initial search identified 74 genomes. After running OrthoFinder through the ‘orthogroup' stage (see below), we excluded 43 genomes because manual inspection revealed that their assemblies were incomplete. Because we identified only three betaproteobacterial genomes, and beta and alpha lineages are highly diverged (core gene mean protein distances between Alphaproteobacteria strains = 0.36, and between Alpha- and Betaproteobacteria strains = 0.98), we limited our analyses to Alphaproteobacteria. After filtering, we were left with assemblies of 27 genomes (21 named species and 4 strains identified only to genus, electronic supplementary material, table S1), representing the major clades of alphaproteobacterial rhizobia (figure 1). These species include E. fredii NGR234, which can form nodules with members of 112 legume genera [23], and WSM2073, which resulted from the HT from a commercial inoculant to a native strain [24] and forms nodules with two species [25].
Figure 1.

Relationships among analysed strains and per genome number of symbiosis genes (one gene per column). The phylogenetic tree was constructed from concatenated single-copy core gene protein sequences using IQ-tree with the JTT + G4 model of evolution, scale bar is amino acid substitutions per site. Genus names abbreviated (in order from top to bottom: M. Methylobacterium, A. Azorhizobium, B. Bradyrhizobium, E. Ensifer, A. Agrobacterium, N. Neorhizobium, R. Rhizobium, M. Mesorhizobium). Genes in multiple categories are marked with a black oval. (Online version in colour.)
We identified homologous protein-coding genes by using OrthoFinder (v. 2.2.7, [26]) with the default settings except that the MCL (Markov Cluster algorithm [27]) inflation factor was set to 4. OrthoFinder groups genes by running all-versus-all BLAST (using diamond [28]), then using MCL [27] to form ‘orthogroups' on the basis of sequence similarity and then uses rooted, species-tree-reconciled gene trees (constructed using distance-based algorithms in FastME v. 2.1.5 [29] and STride [30] to group sequences. This analysis identified 31 853 phylogenetic clusters, hereafter referred to as genes. We aligned the amino acid sequences of each gene using Muscle v. 3.8.31 [31], then converted alignments to nucleotide sequences. The NCBI annotation indicated that 6878 sequences were pseudogenes and we removed these before subsequent analyses, leaving 29 671 genes and 176 566 sequences.
(a). Symbiosis genes
We identified four groups of symbiosis-related genes—two based on functional annotation and two based on genome-wide association (GWA) studies linking bacterial genotype to the benefit that either plants or rhizobia derived from symbiosis. We identified the annotated genes by searching the annotated gene names and product descriptions for: nod, noe, nol, nop, nfe, nodul, nif, fix, fixation and nitrogenase (see electronic supplementary material, table S2 for references and information on search terms). These searches identified 148 genes: 65 annotated nodulation genes, 55 annotated nitrogen fixation genes and 28 genes we removed because their relationship to symbiosis was unclear (details in electronic supplementary material, table S3).
The GWA candidates were from two studies with E. meliloti bacteria and M. truncatula hosts [22,32]. From these studies, we identified the 10 candidates showing the strongest statistical support for contributing to among-strain variation in the benefits plant hosts obtained in single-strain inoculations [32] (hereafter referred to as ‘host benefit’ genes) or contributing to variation in nodule-associated rhizobial fitness [22] (hereafter referred to as ‘rhizobia fitness’ genes). Due to linkage disequilibrium in the Ensifer panel used for the GWAS, the 10 rhizobia fitness candidates comprise 21 candidate genes (table 1; five rhizobia fitness candidates identified through GWA analyses were not included because they were in large LD groups, each of which harboured more than six genes). We note that the function of the genes identified by GWA may differ among lineages and may not be related to symbiosis outside of Ensifer. The rhizobia fitness genes and host benefit genes partially overlapped with the nodulation and fixation genes, but did not overlap with each other (table 1). Electronic supplementary material, table S3 contains the full list of the analysed symbiosis genes.
Table 1.
Number of genes in the 27 representative genomes and number of symbiosis genes.
| all genes | nodulation | N-fixation | host benefit | rhizobial fitness | |
|---|---|---|---|---|---|
| number of genes | 29 671 | 65 | 55 | 10 | 21 |
| proportion genes that are single-copy | 0.86 | 0.54 | 0.51 | 0.40 | 0.43 |
| median family size | 2 | 8 | 27 | 19.5 | 18 |
| proportion of genes in all genomes | 0.04 | 0.17 | 0.40 | 0.50 | 0.24 |
| prop. of genes found in one genome | 0.48 | 0.05 | 0.07 | 0.10 | 0.05 |
| median number of genomes | 2 | 6 | 22 | 19 | 17 |
| mean copies/genome | 1.07 | 1.22 | 1.26 | 1.37 | 1.45 |
| number of genes also found in | |||||
| nodulation | — | 0 | 2 | 0 | |
| N-fixation | — | 4 | 2 | ||
| host benefit | — | 0 | |||
(b). Phylogenetic distribution and horizontal gene transfer
We used three approaches to characterize the extent of HT. First, we examined the correlation between the amino acid divergence of symbiosis genes to the average inter-species phylogenetic distance, estimated as the median amino acid divergence (pairwise protein distances estimated using the JTT model in FastME v. 2.1.5 [29]) of 830 single-copy genes found in all 27 genomes. For these analyses, if a symbiosis gene was not found in a lineage, that lineage was excluded from the analysis. For lineages that contained multiple gene copies we included pairwise distances between all copies between species, but because we are interested in inter-species distance we did not calculate the pairwise distances between copies within a species. A high correlation between protein distance and species distance would be consistent with no HT or HT occurring only between closely related lineages. By contrast, a low correlation between gene and species distance would be consistent with extensive HT or highly variable evolutionary rates within lineages.
Second, we used the δ statistic of [33] to estimate the strength of the phylogenetic signal of gene presence. δ provides insight into whether the presence, or number of copies, of a gene is phylogenetically conserved but is not a specific test of HT; low δ could mean that genes have been lost from multiple strains, independent of those strains' relatedness whereas high δ could result from extensive loss and gain.
Finally, we used GeneRax [34] to specifically estimate the rate of HT of each gene. GeneRax is a species-tree aware method that estimates the rates of gene duplication, transfer and loss. For both δ and GeneRax analyses, the species tree was constructed using IQ-tree v. 1.6.10 [35] with the JTT + G4 model of protein evolution. IQ-tree was run on the concatenated alignments of the single-copy core genes that were trimmed with trimAl v. 1.4.1 [36]. The tree was manually rooted using FigTree v. 1.4.2 based on the positions of lineages in the OrthoFinder species tree that used Betaproteobacteria as an outgroup.
(c). Evolutionary rates
We estimated the rate of non-synonymous to synonymous site divergence (Ka : Ks) for each pair of sequences, including pairs found in the same genome using Comeron's method [37] implemented in the gestimator program in libsequence (v. 0.8.2, [38]). Sequences were excluded if Ks was too high for gestimator to estimate the number of substitutions (399 620 pairs in 7556 genes out of 2 076 943 pairs in 15 636 genes) or Ks = 0 (4977 pairs in 2225 genes); the sequence pairs with unestimated Ks values also had substantially higher Ka values (median = 0.71) than other sequence pairs (median = 0.35). In the text, we report Ka : Ks values only for genes with greater than or equal to 3 comparisons, although results were qualitatively similar when genes with only a single pair of sequences were included (electronic supplementary material, table S4). Summary statistics and annotations for every gene can be found in electronic supplementary material, table S5.
(d). Comparison data
We compared the phylogenetic distribution and evolutionary rates for each set of symbiosis genes to randomly selected sets of the same number of genes as well as to samples of non-symbiosis genes that were ‘matched' to be found in the same number of strains and have a similar number of copies per strain as the symbiosis genes. Using this matched set accounts for differences that might arise due to variation in gene representation. Comparison to randomly selected sets of genes, for which there is no control for gene occurrence, are shown in electronic supplementary material, table S4.
3. Results
The annotated protein-coding sequences in the 27 alphaproteobacterial genomes we analysed were grouped by OrthoFinder into 29 671 genes, 1216 of which were found in all 27 genomes, and 14 249 of which were found in only one genome (table 1). Relative to the entire genome, the symbiosis genes (electronic supplementary material, table S3) were more likely to be present in all 27 genomes (37 of 143 symbiosis genes, Fisher's exact test p < 0.001), less likely to be found in only a single genome (9 of 143, p < 0.001), and found in higher copy number in the genomes in which they were found (1.26 copies of symbiosis genes versus 1.1 copies of other genes per genome, p < 0.001; table 2). The higher mean copy number was driven by a few genes with very high copy number; both the symbiosis and non-symbiosis genes had a median of one copy per genome.
Table 2.
Symbiosis genes tend to be present in more genomes, have higher copy number, higher rates of horizontal transfer than other genes and less phylogenetic signal in gene presence/absence (lower δ) than non-symbiosis genes. Median values are shown. Duplication and transfer are estimates from GeneRax for genes with at least four genomes represented and at least six total copies. δ and Ka : Ks calculated only for genes with at least three sequences (numbers of gene in parentheses; results including all genes were similar—see electronic supplementary material, table S4). The Welch p-value is from a t-test comparing nodulation to fixation genes. The lower part of the table shows the probability that values are greater than a random sample of genes found in the same number of genomes and having approximately the same number of copies as the symbiosis genes.
| strains | copies/genome | duplication | transfer | median pairwise Ka : Ks | R2 | δ | |
|---|---|---|---|---|---|---|---|
| non-symbiosis | 2 | 1 | 10−7 | 0.08 | 0.22 (n = 11 152) | 0.76 | 3.3 (n = 9737) |
| nodulation | 6 | 1 | 10−7 | 0.24 | 0.20 (n = 48) | 0.23 | 1.6 (n = 35) |
| fixation | 22 | 1 | 10−7 | 0.14 | 0.20 (n = 47) | 0.54 | 2.2 (n = 23) |
| p-value Welch | <0.001 | 0.55 | 0.17 | <0.001 | 0.9 | 0.007 | 0.56 |
| benefit | 19 | 1.08 | 0.002 | 0.20 | 0.15 (n = 7) | 0.29 | 1.1 (n = 3) |
| fitness | 17 | 1.13 | 10−7 | 0.18 | 0.23 (n = 19) | 0.19 | 1.9 (n = 12) |
| probability of the estimated values being less than random samples of genes (%) | |||||||
| nodulation | 0.2 | 0 | 99.7 | 99.7 | 99.9 | ||
| fixation | 0 | 0 | 99.2 | 88.6 | 99.2 | ||
| benefit | 6.7 | 0.6 | 99.6 | 96.5 | 95.9 | ||
| fitness | 0.8 | 4.1 | 73.4 | 85.5 | 94.5 | ||
Among the symbiosis genes, N-fixation genes were more likely to be found in all genomes than nodulation genes (tables 1 and 2; figure 1), about as likely to be found in only a single genome (4 fixation, 3 nodulation, p = 0.70), and harboured a similar number of copies in the genomes in which they were found (means of 1.26 fixation, 1.22 nodulation, p = 0.55; table 2). The number of nodulation gene copies in a genome was positively correlated with genome size (r = 0.56, p = 0.002) and the total number of annotated genes (r = 0.55, p = 0.003), but the number of fixation genes was not (r = 0.09, 0.17, respectively, both p > 0.3). The genome size and nodulation gene count correlation may reflect among-lineage variation in the efficiency of removing non-essential/non-advantageous genes. The host benefit and rhizobia fitness candidates were more likely to be found in all 27 strains than randomly selected genes (table 1; both p ≤ 0.001). They also tended to be found in greater copy number in the genomes in which they were present (tables 1 and 2; electronic supplementary material, table S4).
(a). Extent of HT
If genes are either not transferred between lineages or lost from multiple strains, we would expect gene presence to have a strong phylogenetic signal and divergence at the sequence level to be strongly correlated with genome-wide divergence. We used the δ statistic [33], to estimate the strength of the phylogenetic signal in gene presence. All four classes of symbiosis genes had lower median δ values than comparable sets of non-symbiosis genes (table 2; electronic supplementary material, table S4; figure 2). The amino acid divergence of symbiosis genes also tended to be less strongly correlated with genome-wide between species divergence than non-symbiosis genes (table 2).
Figure 2.
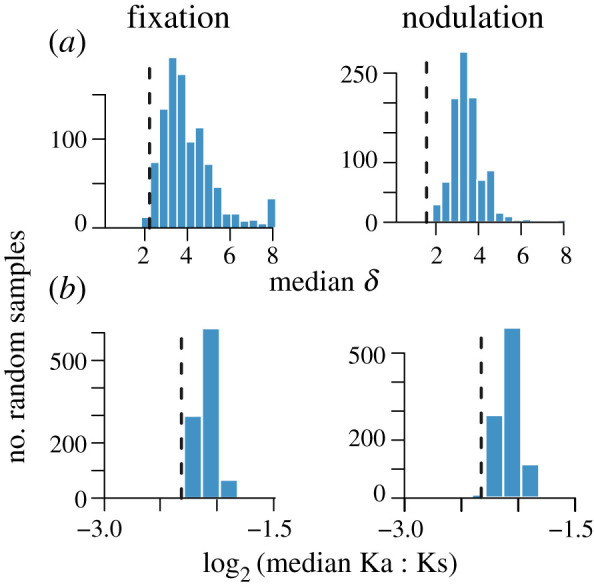
Distribution of median δ (a; only genes with greater than or equal to 3 sequences) and median log2 Ka/Ks (b; only genes with greater than or equal to 3 comparisons) of 1000 randomly sampled sets of genes (bars) compared to the median values for symbiosis genes (dashed lines). Random samples are of genes found in the same number of strains and having similar copy number as symbiosis genes. δ is a measure of phylogenetic signal in the distribution among genomes with greater numbers indicating greater signal. (Online version in colour.)
The weaker phylogenetic signal of gene presence and the weaker correlations between gene and species divergences are both suggestive of symbiosis genes experiencing more HT than non-symbiosis genes. Estimates of the rate of HT, obtained from GeneRax, are consistent with this suggestion: median HT estimates of symbiosis genes were more than twice the median estimated rate of HT of non-symbiosis genes (table 2). The estimated HT rate of annotated nodulation genes was nearly twice as great as the rate obtained for annotated fixation genes (t-test, p < 0.001). Moreover, only four nodulation genes, of the 38 for which HT could be estimated, had rates of HT below the median of non-symbiosis genes and nearly 25% of the nodulation genes with estimated transfer rates were among the upper 10% of the non-symbiosis genes. The estimated duplication rates of symbiosis genes are also greater than that of non-symbiosis genes that are found in similar numbers of strains and copies as the symbiosis genes (table 2; electronic supplementary material, table S4).
(b). Evolutionary rates
The low phylogenetic signal, high estimates of HT, and high rates of duplication of annotated symbiosis genes (nodulation and N-fixation), suggest that these genes have highly dynamic evolutionary histories, at least from the perspective of their loss and gain from genomes. To determine whether these genes also experience atypical evolutionary rates at the sequence level, we examined the ratio of non-synonymous substitutions per non-synonymous site to synonymous substitutions per synonymous site, Ka : Ks. We found that symbiosis genes tend to harbour signatures of purifying selection that are stronger than the non-symbiosis genes (lower median Ka : Ks, p < 0.01 for all but the rhizobial fitness genes; table 2; electronic supplementary material, table S4). However, the mean Ka : Ks of symbiosis genes was slightly greater than the mean of the empirical null expectations (electronic supplementary material, table S4), although this difference was not unlikely by chance. The lower median, yet slightly higher mean, is consistent with most symbiosis genes having experienced stronger than average purifying selection but some genes having evolved under more relaxed or possibly even positive selection (electronic supplementary material, figure S2). In fact, four symbiosis genes have Ka : Ks values that are among the highest 0.2%: noeE (ortholog ID OG0006225), nolE (OG0014065), a nitrogenase (OG0014063) and fixK (OG0014068). The lower median Ka : Ks of the symbiosis genes is not because symbiosis genes are more likely to be transferred and transferred genes tend to be more highly constrained. Consistent with [39], genome-wide there is a positive correlation between the rate of transfer and Ka : Ks (rN = 7068 = 0.14) indicating that genes with higher transfer rates tend to have weaker signatures of purifying selection.
4. Discussion
HT is an important mechanism by which bacterial lineages gain adaptive genetic variants. In rhizobial bacteria, many symbiosis genes are on plasmids or flanked by insertion elements [3,14–17], suggesting that these genes experience high rates of HT. This expectation is supported by comparative phylogenetic analyses, sequence analyses of a handful of symbiosis genes, and genomic analyses of closely related lineages (e.g. [3,13]). Here, we expand upon those studies by estimating the extent of HT among a wide range of rhizobial lineages and determining whether different stages of the rhizobia–legume symbiosis are differentially affected by HT. Using publicly available, fully assembled and annotated genome sequences from 27 rhizobial strains, we found that symbiosis genes show a low correlation between the divergence of gene sequences and the genomes in which those genes are found, weak phylogenetic signal of occurrence, and high rates of HT among genomes. The genes annotated as involved in nodule formation are particularly dynamic, showing rates of transfer nearly three times that of non-symbiosis genes, more than 50% greater than that of N-fixation genes, and more than 20% greater than rhizobia genes that GWA analyses identified as affecting the benefits hosts derive from symbiosis and host-associated rhizobial fitness.
The high rates of HT of symbiosis genes suggests that symbiosis genes, particularly nodulation genes, experience environmentally dependent selection. Given that the function of at least some symbiosis genes is host-specific [40–43], environmentally dependent selection may not be unexpected. In some environments, presumably when specific hosts are present, these genes probably confer a selective advantage because they allow rhizobia to form a symbiosis (e.g. transfer of a symbiosis island from a commercial inoculant to M. australicum WSM2073 [24]). When compatible hosts are absent, however, the symbiosis genes may be lost (e.g. [44–46]), either through drift or because these genes are environmentally costly in the absence of a compatible host. The changes in host availability may change from site to site [20], from season to season in locations with mixed-species communities [47], and over longer time periods as plant ranges shift [48,49]. High rates of HT also may result from a combination of the selective advantage of losing symbiotic capability in certain abiotic environments or when hosts are not present (e.g. [44,45]) and the selective advantage of regaining symbiotic ability when host plants are present. Two population genetic studies provide support for environmentally dependent selection acting on symbiosis genes; a frequently introgressed region containing several symbiosis genes bears a signature of partial sweeps in clover-nodulating R. leguminosarum in Europe [13] and a genomic region in R. leguminosarum that bears a strong signature of adaptation in response to long-term N addition harbours several symbiosis genes as well as several presence–absence variants [45].
(a). Nodulation genes experience stronger purifying selection
Whereas symbiosis genes are clearly evolutionarily labile, in the sense that they are transferred among lineages at a high rate, most are conserved at the nucleotide level and bear a signature of purifying selection that is slightly stronger than the genome-wide average. Although identifying the sources of selection acting on rhizobial genes would require experimental work, it seems likely that host-imposed selection shapes the evolution of these genes. Purifying selection on nodulation genes likely reflects constraints related to the signalling molecules exchanged between hosts and rhizobia during nodule formation [4,5,43] (i.e. mutations that affect proteins involved in nodule formation may reduce the probability of forming a nodule). Similarly, purifying selection on the N-fixation and plant benefit genes suggests that mutations in genes that alter the exchange of benefits plants receive are selected against, which would be consistent with the action of sanctions [50] against rhizobia with reduced benefits.
There has been debate about whether symbiotic partners impose positive selection on one another, due to an arms-race scenario in which hosts impose selection for more beneficial symbionts or whether there is evolutionary stasis due to symbiotic partners being at local optima [18]. Our analyses do not allow us to make inferences about the fitness of symbionts or hosts, but they do show that the vast majority of genes we examined have not diverged in response to positive selection. Our data, therefore, seem to argue against arms-race coevolution being important in shaping the evolution of most genes involved in the legume–rhizobia symbiosis. The lack of positive selection on symbiosis genes is consistent with several studies that have compared signatures of selection in mutualistic and pathogenic lineages: an outer membrane protein that acts as an antigen in Rickettsiaceae showed evidence of positive selection in pathogenic but not in mutualistic lineages [51]; both the distribution among strains and the amino acid sequences of type III secretion system and effector proteins, which are involved in host interaction, are more conserved in Bradyrhizobium and Ensifer rhizobia than Pseudomonas plant pathogens [19]; and Rhizobium sp. sym plasmid genes appear to experience high rates of HT and purifying selection [12].
Although we do not find an overall signal of positive selection having driven the evolution of symbiosis genes, four symbiosis genes harbour Ka : Ks values that exceeded all but 0.2% of the non-symbiosis genes (electronic supplementary material, figure S2), suggesting that host-imposed positive selection may drive adaptation in these genes. Of course, we also cannot exclude the possibility that positive selection is important in driving the evolution of symbiosis genes not included in our analyses. Many of the genes we analysed were identified through forward genetic screens and likely play central roles in the establishment of a functional symbiosis. It is possible that other genes play important roles in shaping symbiont fitness and bear signatures of selection distinct from those characterized here. Finally, it is important to acknowledge that Ka : Ks will only identify genes that have experienced repeated bouts of adaptation [52]. The signal of purifying selection from Ka : Ks is, therefore, not incompatible with occasional bouts of positive selection within a population that drive a rapid increase in the frequency of alleles recently introgressed from other lineages (e.g. [13]). Due to their frequent transfer, the evolution of symbiosis genes may be de-coupled from the evolution of the populations of rhizobia in which they are found. Nevertheless, we found little evidence for antagonistic coevolution between legumes and rhizobia.
Although we have only looked at the symbiont here, a full understanding of host-symbiont coevolution would also consider the host. There is some evidence for selection on legume symbiosis genes from intraspecific studies in M. truncatula, but these signatures do not appear to be pervasive [53–55]. Moreover, the patterns we find may be dependent on the scope of sampling. We examined a wide range of rhizobial species with broad host ranges. A more narrow sampling of rhizobial diversity, or rhizobia that are all found on a single legume host might reveal different patterns of HT. For example, Tian et al. [56] found that symbiosis gene content overall is reflective of phylogeny, although even their restricted sample revealed evidence of HT of some symbiosis genes (e.g. nodC) and variation of symbiosis gene copies, with many symbiosis genes found in only a few genomes.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
This work was supported by computing resources provided by the Minnesota Supercomputing Institute (MSI) at the University of Minnesota. We thank four anonymous reviewers for their valuable comments on the manuscript.
Data accessibility
Code and data are available in the electronic supplementary material and from the Dryad Digital Repository: https://doi.org/10.5061/dryad.2fqz612n2 [57].
Authors' contributions
Both B.E. and P.T. were actively engaged in all aspects of this work.
Competing interests
We declare that we have no competing interests.
Funding
This work was supported by the National Science Foundation under grant nos. IOS-1724993 and IOS-1856744.
References
- 1.Wilmes P, Simmons SL, Denef VJ, Banfield JF. 2008. The dynamic genetic repertoire of microbial communities. FEMS Microbiol. Rev. 33, 109–132. ( 10.1111/j.1574-6976.2008.00144.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vos M 2009. Why do bacteria engage in homologous recombination? Trends Microbiol. 17, 226–232. ( 10.1016/j.tim.2009.03.001) [DOI] [PubMed] [Google Scholar]
- 3.Andrews M, De Meyer S, James EK, Stępkowski T, Hodge S, Simon MF, Young JPW. 2018. Horizontal transfer of symbiosis genes within and between rhizobial genera: occurrence and importance. Genes 9, 321 ( 10.3390/genes9070321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stacey G, Libault M, Brechenmacher L, Wan J, May GD. 2006. Genetics and functional genomics of legume nodulation. Curr. Opin Plant Biol. 9, 110–121. ( 10.1016/j.pbi.2006.01.005) [DOI] [PubMed] [Google Scholar]
- 5.Cooper JE 2007. Early interactions between legumes and rhizobia: disclosing complexity in a molecular dialogue. J. Appl. Microbiol. 103, 1355–1365. ( 10.1111/j.1365-2672.2007.03366.x) [DOI] [PubMed] [Google Scholar]
- 6.Udvardi M, Poole PS. 2013. Transport and metabolism in legume–rhizobia symbioses. Annu. Rev. Plant Biol. 64, 781–805. ( 10.1146/annurev-arplant-050312-120235) [DOI] [PubMed] [Google Scholar]
- 7.Doyle JJ 1998. Phylogenetic perspectives on nodulation: evolving views of plants and symbiotic bacteria. Trends Plant Sci. 3, 473–478. ( 10.1016/S1360-1385(98)01340-5) [DOI] [Google Scholar]
- 8.Sun S, Guo H, Xu J. 2006. Multiple gene genealogical analyses reveal both common and distinct population genetic patterns among replicons in the nitrogen-fixing bacterium Sinorhizobium meliloti. Microbiology 152, 3245–3259. ( 10.1099/mic.0.29170-0) [DOI] [PubMed] [Google Scholar]
- 9.Bailly X, Olivieri I, De Mita S, Cleyet-Marel J-C, Béna G.. 2006. Recombination and selection shape the molecular diversity pattern of nitrogen-fixing Sinorhizobium sp. associated to Medicago. Mol. Ecol. 15, 2719–2734. ( 10.1111/j.1365-294X.2006.02969.x) [DOI] [PubMed] [Google Scholar]
- 10.Epstein B, et al. 2012. Population genomics of the facultatively mutualistic bacteria Sinorhizobium meliloti and S. medicae. PLoS Genet. 8, e1002868 ( 10.1371/journal.pgen.1002868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parker MA 2012. Legumes select symbiosis island sequence variants in Bradyrhizobium. Mol. Ecol. 21, 1769–1778. ( 10.1111/j.1365-294X.2012.05497.x) [DOI] [PubMed] [Google Scholar]
- 12.Pérez Carrascal OM, VanInsberghe D, Juárez S, Polz MF, Vinuesa P, González V. 2016. Population genomics of the symbiotic plasmids of sympatric nitrogen-fixing Rhizobium species associated with Phaseolus vulgaris. Environ. Microbiol. 18, 2660–2676. ( 10.1111/1462-2920.13415) [DOI] [PubMed] [Google Scholar]
- 13.Cavassim MIA, Moeskjær S, Moslemi C, Fields B, Bachmann A, Vilhjálmsson BJ, Schierup MH, Young JPW, Andersen SU. 2020. Symbiosis genes show a unique pattern of introgression and selection within a Rhizobium leguminosarum species complex. Microb. Genom. 6, e000351 ( 10.1099/mgen.0.000351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brewin NJ, Beringer JE, Johnston AWB. 1980. Plasmid-mediated transfer of host-range specificity between two strains of Rhizobium leguminosarum. Microbiology 120, 413–420. ( 10.1099/00221287-120-2-413) [DOI] [Google Scholar]
- 15.Sullivan JT, Ronson CW. 1998. Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc. Natl Acad. Sci. USA 95, 5145–5149. ( 10.1073/pnas.95.9.5145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnett MJ, et al. 2001. Nucleotide sequence and predicted functions of the entire Sinorhizobium meliloti pSymA megaplasmid. Proc. Natl Acad. Sci. USA 98, 9883–9888. ( 10.1073/pnas.161294798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneko T, et al. 2002. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9, 189–197. ( 10.1093/dnares/9.6.189) [DOI] [PubMed] [Google Scholar]
- 18.Sachs JL, Essenberg CJ, Turcotte MM. 2011. New paradigms for the evolution of beneficial infections. Trends Ecol. Evol. 26, 202–209. ( 10.1016/j.tree.2011.01.010) [DOI] [PubMed] [Google Scholar]
- 19.Kimbrel JA, Thomas WJ, Jiang Y, Creason AL, Thireault CA, Sachs JL, Chang JH. 2013. Mutualistic co-evolution of type III effector genes in Sinorhizobium fredii and Bradyrhizobium japonicum. PLoS Pathog. 9, e1003204 ( 10.1371/journal.ppat.1003204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker MA 1999. Mutualism in metapopulations of legumes and rhizobia. Am. Nat. 153, S48–S60. ( 10.1086/303211) [DOI] [PubMed] [Google Scholar]
- 21.Tak A, Gehlot P, Pathak R, Singh SK. 2017. Species diversity of rhizobia. In Rhizobium biology and biotechnology (eds Hansen AP, Choudhary DK, Agrawal PK, Varma A), pp. 215–245. Cham, Switzerland: Springer International Publishing. [Google Scholar]
- 22.Burghardt LT, Epstein B, Guhlin J, Nelson MS, Taylor MR, Young ND, Sadowsky MJ, Tiffin P. 2018. Select and resequence reveals relative fitness of bacteria in symbiotic and free-living environments. Proc. Natl Acad. Sci. USA 115, 2425–2430. ( 10.1073/pnas.1714246115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pueppke SG, Broughton WJ. 1999. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant Microbe Interact. 12, 293–318. ( 10.1094/MPMI.1999.12.4.293) [DOI] [PubMed] [Google Scholar]
- 24.Haskett TL, Terpolilli JJ, Bekuma A, O'Hara GW, Sullivan JT, Wang P, Ronson CW, Ramsay JP. 2016. Assembly and transfer of tripartite integrative and conjugative genetic elements. Proc. Natl Acad. Sci. USA 113, 12268–12273. ( 10.1073/pnas.1613358113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nandasena KG, O'Hara GW, Tiwari RP, Sezmiş E, Howieson JG. 2007. In situ lateral transfer of symbiosis islands results in rapid evolution of diverse competitive strains of mesorhizobia suboptimal in symbiotic nitrogen fixation on the pasture legume Biserrula pelecinus L. Environ. Microbiol. 9, 2496–2511. ( 10.1111/j.1462-2920.2007.01368.x) [DOI] [PubMed] [Google Scholar]
- 26.Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 ( 10.1186/s13059-015-0721-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dongen S 2000. A cluster algorithm for graphs Amsterdam, The Netherlands: National Research Institute for Mathematics and Computer Science. [Google Scholar]
- 28.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. ( 10.1038/nmeth.3176) [DOI] [PubMed] [Google Scholar]
- 29.Lefort V, Desper R, Gascuel O. 2015. FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. ( 10.1093/molbev/msv150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emms DM, Kelly S. 2017. STRIDE: species tree root inference from gene duplication events. Mol. Biol. Evol. 34, 3267–3278. ( 10.1093/molbev/msx259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. ( 10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epstein B, Abou-Shanab RAI, Shamseldin A, Taylor MR, Guhlin J, Burghardt LT, Nelson M, Sadowsky MJ, Tiffin P. 2018. Genome-wide association analyses in the model rhizobium Ensifer meliloti. mSphere 3, e00386-18 ( 10.1128/mSphere.00386-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borges R, Machado JP, Gomes C, Rocha AP, Antunes A. 2018. Measuring phylogenetic signal between categorical traits and phylogenies. Bioinformatics 35, 1862–1869. ( 10.1093/bioinformatics/bty800) [DOI] [PubMed] [Google Scholar]
- 34.Morel B, Kozlov AM, Stamatakis A, Szöllősi GJ. 2020. GeneRax: a tool for species-tree-aware maximum likelihood-based gene family tree inference under gene duplication, transfer, and loss. Mol. Biol. Evol. 37, 2763–2774. ( 10.1093/molbev/msaa141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2014. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. ( 10.1093/molbev/msu300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. ( 10.1093/bioinformatics/btp348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comeron JM 1995. A method for estimating the numbers of synonymous and nonsynonymous substitutions per site. J. Mol. Evol. 41, 1152–1159. ( 10.1007/BF00173196) [DOI] [PubMed] [Google Scholar]
- 38.Thornton K 2003. Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics 19, 2325–2327. ( 10.1093/bioinformatics/btg316) [DOI] [PubMed] [Google Scholar]
- 39.Davids W, Zhang Z. 2008. The impact of horizontal gene transfer in shaping operons and protein interaction networks: direct evidence of preferential attachment. BMC Evol. Biol. 8, 23 ( 10.1186/1471-2148-8-23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masson-Boivin C, Giraud E, Perret X, Batut J. 2009. Establishing nitrogen-fixing symbiosis with legumes: how many rhizobium recipes? Trends Microbiol. 17, 458–466. ( 10.1016/j.tim.2009.07.004) [DOI] [PubMed] [Google Scholar]
- 41.Staehelin C, Krishnan HB. 2015. Nodulation outer proteins: double-edged swords of symbiotic rhizobia. Biochem. J. 470, 263–274. ( 10.1042/BJ20150518) [DOI] [PubMed] [Google Scholar]
- 42.Galardini M, et al. 2011. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genom. 12, 235 ( 10.1186/1471-2164-12-235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Triplett E, Sadowsky M. 1992. Genetics of competition for nodulation of legumes. Annu. Rev. Microbiol. 46, 399–428. ( 10.1146/annurev.mi.46.100192.002151) [DOI] [PubMed] [Google Scholar]
- 44.Hollowell AC, et al. 2016. Epidemic spread of symbiotic and non-symbiotic Bradyrhizobium genotypes across California. Microb. Ecol. 71, 700–710. ( 10.1007/s00248-015-0685-5) [DOI] [PubMed] [Google Scholar]
- 45.Klinger CR, Lau JA, Heath KD. 2016. Ecological genomics of mutualism decline in nitrogen-fixing bacteria. Proc. Biol. Sci. 283, 20152563 ( 10.1098/rspb.2015.2563) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gano-Cohen KA, Wendlandt CE, Al Moussawi K, Stokes PJ, Quides KW, Weisberg AJ, Chang JeffH, Sachs JL.. 2020. Recurrent mutualism breakdown events in a legume rhizobia metapopulation. Proc. R. Soc. B 287, 20192549 ( 10.1098/rspb.2019.2549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siefert A, Zillig KW, Friesen ML, Strauss SY. 2019. Mutualists stabilize the coexistence of congeneric legumes. Am. Nat. 193, 200–212. ( 10.1086/701056) [DOI] [PubMed] [Google Scholar]
- 48.La Pierre KJ, Simms EL, Tariq M, Zafar M, Porter SS.. 2017. Invasive legumes can associate with many mutualists of native legumes, but usually do not. Ecol. Evol. 7, 8599–8611. ( 10.1002/ece3.3310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison TL, Simonsen AK, Stinchcombe JR, Frederickson ME. 2018. More partners, more ranges: generalist legumes spread more easily around the globe. Biol. Lett. 14, 20180616 ( 10.1098/rsbl.2018.0616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiers ET, Rousseau RA, West SA, Denison RF. 2003. Host sanctions and the legume-rhizobium mutualism. Nature 425, 78–81. ( 10.1038/nature01931) [DOI] [PubMed] [Google Scholar]
- 51.Jiggins FM, Hurst GDD, Yang Z. 2002. Host-symbiont conflicts: positive selection on an outer membrane protein of parasitic but not mutualistic Rickettsiaceae. Mol. Biol. Evol. 19, 1341–1349. ( 10.1093/oxfordjournals.molbev.a004195) [DOI] [PubMed] [Google Scholar]
- 52.Nielsen R 2005. Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218. ( 10.1146/annurev.genet.39.073003.112420) [DOI] [PubMed] [Google Scholar]
- 53.De Mita S, Ronfort J, McKhann HI, Poncet C, El Malki R, Bataillon T.. 2007. Investigation of the demographic and selective forces shaping the nucleotide diversity of genes involved in nod factor signaling in Medicago truncatula. Genetics 177, 2123 ( 10.1534/genetics.107.076943) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Mita S, Chantret N, Loridon K, Ronfort J, Bataillon T.. 2011. Molecular adaptation in flowering and symbiotic recognition pathways: insights from patterns of polymorphism in the legume Medicago truncatula. BMC Evol. Biol. 11, 229 ( 10.1186/1471-2148-11-229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paape T, Bataillon T, Zhou P, JY Kono T, Briskine R, Young ND, Tiffin P. 2013. Selection, genome-wide fitness effects and evolutionary rates in the model legume Medicago truncatula. Mol. Ecol. 22, 3525–3538. ( 10.1111/mec.12329) [DOI] [PubMed] [Google Scholar]
- 56.Tian CF, et al. 2012. Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc. Natl Acad. Sci. USA 109, 8629–8634. ( 10.1073/pnas.1120436109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Epstein B, Tiffin P. 2021. Data from: Comparative genomics reveals high rates of horizontal transfer and strong purifying selection on rhizobial symbiosis genes Dryad Digital Repository. ( 10.5061/dryad.2fqz612n2) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Epstein B, Tiffin P. 2021. Data from: Comparative genomics reveals high rates of horizontal transfer and strong purifying selection on rhizobial symbiosis genes Dryad Digital Repository. ( 10.5061/dryad.2fqz612n2) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Code and data are available in the electronic supplementary material and from the Dryad Digital Repository: https://doi.org/10.5061/dryad.2fqz612n2 [57].