

Summary
Epithelial-to-mesenchymal transition (EMT), an evolutionary conserved phenomenon, has been extensively studied to address the unresolved variable treatment response across therapeutic regimes in cancer subtypes. EMT has long been envisaged to regulate tumor invasion, migration, and therapeutic resistance during tumorigenesis. However, recently it has been highlighted that EMT involves an intermediate partial EMT (pEMT) phenotype, defined by incomplete loss of epithelial markers and incomplete gain of mesenchymal markers. It has been further emphasized that pEMT transition involves a spectrum of intermediate hybrid states on either side of pEMT spectrum. Emerging evidence underlines bi-directional crosstalk between tumor cells and surrounding microenvironment in acquisition of pEMT phenotype. Although much work is still ongoing to gain mechanistic insights into regulation of pEMT phenotype, it is evident that pEMT plays a critical role in tumor aggressiveness, invasion, migration, and metastasis along with therapeutic resistance. In this review, we focus on important role of tumor-intrinsic factors and tumor microenvironment in driving pEMT and emphasize that engineered controlled microenvironments are instrumental to provide mechanistic insights into pEMT biology. We also discuss the significance of pEMT in regulating hallmarks of tumor progression i.e. cell cycle regulation, collective migration, and therapeutic resistance. Although constantly evolving, current progress and momentum in the pEMT field holds promise to unravel new therapeutic targets to halt tumor progression at early stages as well as tackle the complex therapeutic resistance observed across many cancer types.
Subject areas: functional aspects of cell biology, cancer, bioengineering, tissue engineering
Graphical Abstract
Highlights
-
•
Partial EMT phenotype drives key hallmarks of tumor progression
-
•
Role of tumor microenvironment in pEMT phenotype via cellular signaling pathways
-
•
Engineering 3D in vitro models to study pEMT phenotype
-
•
Opportunities and challenges in understanding pEMT phenotype
Functional aspects of cell biology; cancer; bioengineering; tissue engineering
Introduction
Epithelial-to-mesenchymal transition (EMT), an evolutionary conserved mechanism, has long been known to facilitate invasion and metastasis of malignant epithelial (E) cells through reduced cell-cell adhesion and transition to mesenchymal (M) states (Kalluri and Weinberg, 2009; Ye and Weinberg, 2015) and chemoresistance during tumorigenesis (Shibue and Weinberg, 2017). Until now, EMT has been viewed as a binary process; however, it is increasingly evident that EMT involves a gradual transformation from epithelial to a partial EMT (pEMT) state without complete loss of epithelial and complete gain of mesenchymal phenotype (Dongre and Weinberg, 2019; Yang et al., 2020; Zhang and Weinberg, 2018). With presence of tumor cells in epithelial, mesenchymal, and hybrid E/M phenotypes simultaneously, tumors with pEMT also exhibit intra-tumoral heterogeneity and epithelial-mesenchymal plasticity (Figure 1). In addition to complete EMT and partial EMT phenotypes, current scientific evidence also supports the presence of intermediate hybrid states on either side of pEMT spectrum (Pastushenko et al., 2018; Bhatia et al., 2020). For example, early, intermediate, and late hybrid E/M states were identified by differential surface protein expression of CD106, CD61, and CD51 in skin and mammary primary tumor cells (Pastushenko et al., 2018). Although the proposed hybrid states affect tumor progression, metastasis, and drug resistance, the precise mechanisms of how cells transition in vivo through these intermediate states, their stability, and mechanistic regulation remain to be determined.
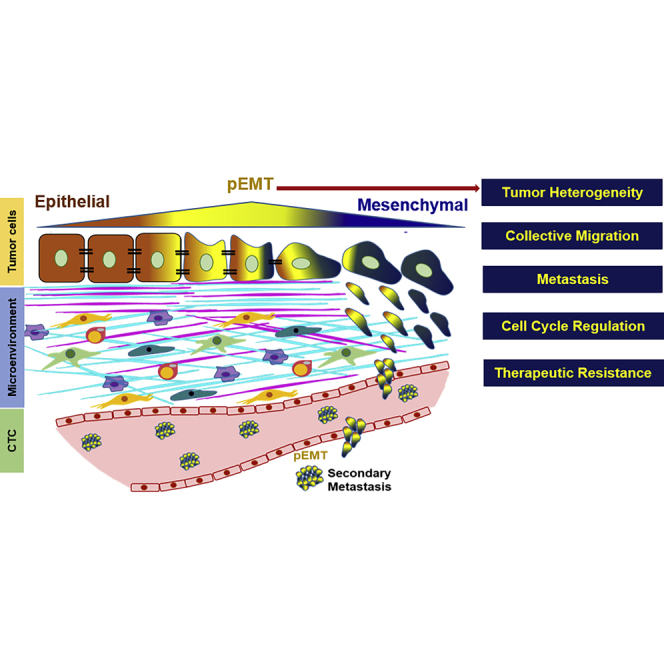
Figure 1.

Partial EMT (pEMT) phenotype involves a spectrum of changes between epithelial and mesenchymal phenotypes
The tumor cells expressing pEMT phenotype interact with surrounding extracellular matrix, which induces tumor heterogeneity. pEMT also regulates key processes in tumor progression: cell-cycle regulation, collective migration, metastasis, and therapeutic resistance.
The tumor microenvironment (TME) surrounding the tumor cells can contribute to the emergence, stability, and regulation of pEMT phenotype, consequently driving tumor progression (Bhatia et al., 2020). TME is heterogeneous, spatially organized yet complex amalgamation of tumor cells, fibroblasts, endothelial cells, immune cells, and other stromal cells recruited by tumor cells within the surrounding extracellular matrix (ECM). The phenotypic plasticity of tumor cells is dynamic and orchestrated by various factors in the stromal TME. The bilateral cross-talk between the pEMT+ tumor cells and TME leads to activation of paracrine signaling, further promoting hallmarks of tumor progression (Bhatia et al., 2020).
The concept of pEMT is of high clinical significance as it is associated with higher tumor grade, tumor relapse, and increased metastasis (Yagasaki et al., 1996; Haraguchi et al., 1999). The pEMT defined by co-expression of epithelial and mesenchymal markers has been observed in a subset of pancreatic, lung, colorectal, and breast cancers as well as non-small-cell lung carcinoma (NSCLC) and cutaneous carcinosarcoma (Bronsert et al., 2014; Kolijn et al., 2015; Zacharias et al., 2018; Paniz-Mondolfi et al., 2014). In oral squamous cell carcinoma patients, co-expression of keratin-14 (K14) and vimentin (VIM) was associated with poor prognosis (Dmello et al., 2017). Interestingly, in breast cancer cells concomitant expression of both epithelial and mesenchymal transcripts was also detected in the circulating tumor cells (CTCs) (Yu et al., 2013), metastatic pleural effusions (Donnenberg et al., 2018), and at the invading edges of primary carcinomas (Donnenberg et al., 2010). Recently, single-cell RNA sequencing identified a pEMT gene signature that was able to independently predict high tumor grade and nodal metastasis in head and neck squamous cell carcinoma (HNSCC) patients (Puram et al., 2017), further warranting mechanistic insights into pEMT biology.
In this review, we highlight the important crosstalk between tumor cells and microenvironmental factors that promote pEMT. We then summarize recent scientific knowledge on how pEMT regulates hallmarks of tumor progression. We note that majority of studies utilize two-dimensional (2D) cell culture approaches, which do not completely recapitulate the in vivo TME. Although tissue-engineered three-dimensional (3D) models better recapitulate in vivo microenvironment, the efforts in this area are lacking. Hence, we discuss how tumor-intrinsic factors drive pEMT through interactions with ECM and other stromal-derived factors with the hope to generate interest among tissue engineers to build innovative 3D models for studying pEMT phenotype.
Interplay between tumor microenvironment and pEMT
The TME consists of tumor cells, stromal cells along with their secreted factors, and surrounding ECM. TME is highly dynamic and both the tumor cells and TME co-evolve during tumor progression (Bussard et al., 2016). Here, we examine how TME contributes to pEMT. We further discern different dimensions of TME ranging from tumor-intrinsic factors, ECM-related factors, and stromal-related factors and their role in regulating pEMT phenotype (Figure 1, Table 1).
Table 1.
Summary of pEMT markers
| E/M Markers used to characterize pEMT phenotypea | Proposed pEMT Markers | Cancer | Cell Lines | Microenvironmental Factor | Tumor Model (in vitro/in vivo) | Significance | Ref. |
|---|---|---|---|---|---|---|---|
| Tumor-intrinsic factors | |||||||
| ECAD, ZO1/SNAI1, VIM, β-catenin |
S100A6 | Breast cancer | MCF-7 | Acidic microenvironment (pH 6.5 2 months) | In vitro | Acidic-adaptation-induced pEMT phenotype with MCF-7 cells expressing high VIM and loss of β-catenin, ZO-1, SNAI1 while maintaining expression of ECAD. S100A6 proteins induced pEMT phenotype in acid-adapted MCF-7 cells. | Sadeghi et al., 2020 |
| ECAD/VIM | - | Breast cancer | MCF-7, T47D, MDA-MB-231, MDA-MB-468 |
Hypoxia (5% O2) | In vitro | pEMT cells had increased migration potential | Chen et al., 2018a |
| ECAD/VIM | - | Breast cancer | T47D | Hypoxia (Tumor—intrinsic) | In vitro | pEMT cells localized to leading edge of migratory tumors linking role of pEMT to collective migration | Singh et al., 2018a |
| ECAD/VIM | VEGF | Esophageal squamous cell carcinoma | KYSE140, KYSE180, KYSE510, KYSE520 | Hypoxia (1% O2) | In vitro | pEMT cells along with elevated VEGF generated invasive TME | Li et al., 2014 |
| ECAD/VIM | - | Pancreatic cancer | BxPc-3, Panc-1 | Hypoxia (0.1% O2 2 to 96 h) | In vitro | pEMT cells had increased migration potential | Lundgren et al., 2009 |
| ECAD/ZEB1 | NRF2 | Non-small-cell lung carcinoma, Bladder cancer | H1975 RT4 |
Tumor intrinsic metabolism | In vitro | NRF2 activated and stabilized pEMT phenotype | Bocci et al., 2019 |
| Extracellular matrix (ECM)-related factors | |||||||
| ECAD/VIM | - | Pancreatic cancer | BxPc-3 | Collagen and fibronectin matrix | In vitro | pEMT cells migrated with amoeboid mode or filopodium-like protrusions by ECM remodeling (collagen degradation and re-orientation of fibronectin matrix) | Kim et al., 2020 |
| ECAD/ FN1, MMP1 | L1CAM | Colorectal cancer | Caco-2 | L1CAM, α-2, α-5, and β-1 integrins, FN1 | In vitro | Loss of NEO1 induced pEMT through ECM remodeling i.e. upregulation of L1CAM, α-2, α-5, β-1 integrins, and FN1 | Chaturvedi et al., 2019 |
| ECAD/ VIM | Cathepsin B | Salivary adenoid cystic carcinoma | SACC-83 | Cathepsin B | In vitro | Leader cells expressed pEMT markers and cathepsin B, which facilitated ECM remodeling and tumor invasion | Wu et al., 2019 |
| ECAD/VIM, ZEB2 | COL2A1, FN1 | Hepatocellular carcinoma | Huh7 | FN1, COL2A1 | In vitro | pEMT cells in exosomal secretion expressed COL2A1 and FN1 | Karaosmanoglu et al., 2018 |
| ECAD/NCAD, ZEB2 | - | Breast cancer | EpH4 | Alginate matrix | In vitro | pEMT cells showed front-back polarity and aggressive phenotype | Bidarra et al., 2016 |
| ECAD/SNAI1, SNAI2 | Laminin 5 | Hepatocellular carcinoma | Hep3B | Laminin 5 | In vitro | Laminin 5 induced pEMT phenotype | Giannelli et al., 2005 |
| Stromal-cell-related factors | |||||||
| |||||||
| ECAD/ZEB1 | - | Breast cancer | MCF-7, NOG mice | Stromal fibroblasts | In vitro/in vivo | CAFs secreted SDF1 drive collective migration of pEMT cells | Matsumura et al., 2019 |
| ECAD/VIM | - | Pancreatic cancer | BxPc-3, Panc-1 | CAFs | In vitro | CAFs stabilized pEMT state and increased migration and invasion | Shan et al., 2017 |
| 100 pEMT gene signature | - | Head and neck cancer | SCC9 | CAFs | In vitro and patient samples | Paracrine interactions of CAFs and tumor cells promoted pEMT phenotype vai TGFB/TGFBI axis | Puram et al., 2017 |
| ECAD/ VIM | CD44 | NSCLC adenocarcinoma | HCC827, H3255, A549 | Stromal fibroblasts | In vitro | Fibroblasts in TME drive pEMT phenotype | Karacosta et al., 2019 |
| |||||||
| ECAD/CLDN7 | - | Breast cancer | MDA-MB-231, Hs578t | Adipocyte | In vitro | Mature adipocytes induced pEMT phenotype | Pallegar et al., 2019 |
| ECAD/VIM | - | Breast cancer | MCF-10A | Leptin (adipocyte-secreted hormone) | In vitro | Leptin induced pEMT at leading edge and induced collective migration | Villanueva-Duque et al., 2017 |
| |||||||
| ECAD/VIM, SNAI2 | - | Breast cancer | MCF-7, T47D | M1 TAMs | In vitro | M1 TAMs secretome derived pEMT phenotype, increased migration and invasion | Bednarczyk et al., 2018 |
Cadherin 2 (NCAD), claudin 7 (CLDN7), collagen type II alpha 1 (COL2A1), E-cadherin (ECAD), extracellular matrix (ECM), fibronectin 1 (FN1), L1 cell adhesion molecule (L1CAM), matrix metalloproteinase-1 (MMP1), partial EMT (pEMT), neogenin 1 (NEO1), NOD/Shi-scid IL2 null (NOG), non-small-cell lung carcinoma (NSCLC), snail family transcription repressor 1 (SNAI1), snail transcription repressor 2 (SNAI2), vascular endothelial growth factor (VEGF), transforming growth factor beta (TGFB), transforming growth factor beta induced (TGFBI), tumor microenvironment (TME), vimentin (VIM), zinc finfer E-box-binding homeobox 1 (ZEB1), zinc finger E-box-binding homeobox 2 (ZEB2), zonula occludens 1 (ZO1).
Few studies used multiple epithelial and mesenchymal markers to characterize pEMT phenotype, which are being listed using “/” between epithelial and mesenchymal markers.
Tumor-intrinsic factors
Tumor-intrinsic factors such as hypoxia may contribute to stabilization of cells in pEMT state (Figure 1). (Chen et al., 2018a; Singh et al., 2016, 2018a, 2019; Saxena et al., 2020) This is supported by data generated with commonly used breast and pancreatic cell lines (MCF-7, T47D, MDA-MB-231, MDA-MB-468, BxPc-3, and Panc-1) cultured under hypoxia, which resulted in expression of both epithelial (E-cadherin, ECAD) and mesenchymal markers (VIM) along with increased migration (Chen et al., 2018a; Lundgren et al., 2009). Mechanistically, it was shown that hypoxic esophageal squamous cell carcinoma (ESCC) cells expressed high levels of HIF1A and EIFA2 and exhibited pEMT phenotype with co-expression of ECAD and VIM. Together with high expression of VEGF in the TME, these cells generated highly invasive and vascularized aggressive tumor environment (Li et al., 2014). In a recent review (Saxena et al., 2020), it is proposed that cyclic or intermittent hypoxia observed in TME may play role in stabilization of pEMT phenotype through HIF1A stabilization and/or crosstalk between HIF1A and NRF2.
Unlike these initial studies in 2D cell monolayers exposed to hypoxic chambers, we have generated a unique 3D size-controlled microtumor model (Figures 2A and 2B) to recapitulate and study tumor-intrinsic hypoxia without any genetic manipulation or any artificial culture conditions (Aggarwal et al., 2020; Singh et al., 2015, 2016, 2018a, 2018b, 2019; Patel and Sant, 2016). Our 3D platform generates uniform microtumors, which recapitulate the tumor-intrinsic hypoxic environment in vitro (Singh et al., 2019). These microtumor models include (1) non-hypoxic, non-migratory small microtumors (≤150 μm) and (2) hypoxic, migratory large microtumors (≥500 μm) that migrate starting from day 3 to day 6 in culture (Figure 2D). (Singh et al., 2016) Interestingly, the microtumors generated from the same parent T47D cells showed tumor heterogeneity, with heterogeneous cell population expressing E, M, and E/M (pEMT) markers in large microtumors. Gene expression analysis of large migratory versus small non-migratory microtumors highlighted cell cycle as one of the biological functions of the differentially regulated genes enriched in the large hypoxic microtumors (unpublished data) (Figure 2C). The signaling pathways and genes associated with the progression of cell cycle such as cyclin A, B, D, and E, CDK1, and CDK2 were downregulated in large migratory microtumors while upregulating the cell cycle inhibitor p27. Cell cycle analysis demonstrated that 70% of the cells are in G0/G1 and G1/G2 on day 6 in the large hypoxic microtumors (Figure 2E). No changes were observed from day 1 to day 6 in the percentage of cells in S or G2/M suggestive of cell-cycle arrest (Singh et al., 2018a). The large hypoxic microtumors expressed HIF1A, SNAI1, SNAI2, VIM, and FN1 without loss of ECAD, which supports emergence of pEMT phenotype (Singh et al., 2018a, 2019). We also observed collective migration in these large hypoxic microtumors. Interestingly, the cells expressing pEMT were localized at the leading edge of the migrating tumors, which supports the notion that pEMT plays a role in collective migration (Figure 2F). Together, our unique 3D microtumor model recapitulates features of pEMT described in this review and is a unique tool to study the mechanistic link between hypoxia and pEMT and its association with tumor heterogeneity, cell-cycle arrest, and collective migration, which is currently under investigation.
Figure 2.

Three-dimensional (3D) microtumor models to study tumor-intrinsic hypoxia-driven migration
(A–F) (A) Size-controlled hydrogel microwell arrays (150 and 600 μm) used to generate the microtumors. (B) Photomicrographs show size-controlled 150 μm microtumors and 600 μm microtumors on day 1. (C) Hypoxia signature in large hypoxic 600 μm microtumors. (D) Hypoxia is absent in non-migrating 150 μm microtumors from day 1 to day 6. In large 600 μm microtumors, hypoxia is observed from day 1 to day 6 in the migrating 600 μm microtumors. Scale bars: 300 μm (top panel) and 250 μm (bottom panel). (E) Large microtumors show significant increase in sub G0/G1 phase and decrease in G0/G1 phase with no differences in S and G2/M phase compared with 2D and small microtumors (reproduced from Singh et al., 2018a). (F) Immunofluorescence images of microtumors showed uniform E-cad (green) staining in large microtumors, whereas VIM (red) was expressed only at the periphery in large microtumors.
The acidic TME has also been shown to drive the pEMT phenotype. It was shown that acidic microenvironments can cause a phenotypic switch leading to pEMT, providing evidence of tumor cell plasticity and the resulting heterogeneity among cancer cell populations undergoing acidic adaptation (Sadeghi et al., 2020). MCF-7 cells undergoing acid adaptation showed a phenotypic switch with high co-expression of mesenchymal and epithelial markers such as SNAI1, VIM, β-catenin, and ECAD and ZO-1. Integrative analysis of transcriptomics, proteomics, and spatial immunofluorescence of MCF-7 cells indicated that S100 proteins play a role in the acid-induced pEMT (Sadeghi et al., 2020). Survival analysis in ductal carcinoma in situ (DCIS) patients showed that higher expression of S100A6 correlated with worse survival, supporting the role of pEMT and acidic TME.
Tumor cells can also exhibit metabolic reprogramming/metabolic plasticity to survive under different microenvironments and in turn, can undergo transition from epithelial cells to pEMT state (Jia et al., 2018; Thomson et al., 2019). NRF2, a cellular metabolic reprogrammer, has been reported to activate and stabilize pEMT phenotype characterized by co-expression of ECAD and ZEB1 in both NSCLC and bladder cancer (Bocci et al., 2019). However, additional experimental evidence is needed to establish a causal association between metabolism and pEMT, which can further justify the use of metabolic inhibitors in pEMT targeting (Ramesh et al., 2020).
Extracellular matrix-related factors
The crosstalk between tumor cells and ECM in regulating complete EMT is well established (Bussard et al., 2016). EMT has traditionally been known to be activated through collagen-dependent activation of EMT-transcription factors and through mechanotransduction by matrix stiffness, osmotic pressure, fluid flow, or tissue tension (Aiello and Kang, 2019). The pEMT is also modulated via ECM-mediated factors such as collagen, fibronectin, laminins, etc. (Table 1).
Fibrinogen gamma chain (FGG) along with post-transcriptionally modified collagen type II alpha 1 (COL2A1) and fibronectin 1 (FN1) were identified as biomarkers of pEMT (ECADhi/VIMlo/ZEB2lo) in exosomal secretion of SNAI2 overexpressing hepatocellular carcinoma (Huh7) cells (Karaosmanoglu et al., 2018). Interestingly, when stratified squamous epithelial cell sheets were grafted onto a hydrated type I collagen gel, the epithelial cells underwent transformation to transient pEMT and exhibited collective migration (Kasai et al., 2017). When co-cultured with pancreatic stellate cells in a collagen matrix, human BxPc-3 pancreatic cancer cells exhibited pEMT phenotype (ECAD+/VIM+) and migrated through filopodium-like protrusions or ameboid mode by activating focal adhesion kinase (FAK), elevated expression of integrin β1 (ITGB1), and ECM remodeling by collagen degradation, re-orientation of collagen fibers, and deposition of fibronectin matrix (Kim et al., 2020). In human colorectal carcinoma cells, loss of neogenin 1 (NEO1), a tumor suppressor gene, shifted their transcriptional profile to pEMT and contributed to increased motility through L1CAM upregulation, decreased F-actin stress fibers, and ECM remodeling (α-2, α-5, and β-1 integrins and FN1). NEO1 is known to localize to the adherence junctions in ECAD-dependent pathway, wherein it recruits ARP2/3 and WRC and promotes F-actin formation (Chaturvedi et al., 2019). Role of laminin 5 (LN5) has also been investigated in induction of pEMT where LN5 individually induced pEMT phenotype (ECADlo/Snailhi/Slughi) in hepatocellular carcinoma; however, together with TGFB1, LN5 synergistically drove complete EMT (Giannelli et al., 2005). Taken together, these studies highlight the role of ECM components in affecting pEMT and emphasize the importance of investigating how ECM components interact with tumor-specific signaling molecules to drive pEMT, tumor invasion, and metastasis.
Apart from 2D studies, role of ECM remodeling was shown in collective leader cell migration in 3D salivary adenoid cystic carcinoma-83 spheroids grown using three different methods, namely hanging drop technique, 96-well ultra-low attachment plates, and soft agar coated 96-well plates (Figures 3A and 3B). In this study, the leader cells expressing pEMT (ECAD+/VIM+) expressed cathepsin B (CTSB), which facilitated tumor invasion via ECM remodeling and upregulation of RhoA, ROCK1, ROCK2, FAK, and MMP9 (Wu et al., 2019). When EpH4 mouse mammary epithelial cells were cultured in 3D alginate matrices (Figure 3C) activated with RGD peptides, TGFB1 induced complete EMT generating mesenchymal-like cells. TGFβ1 removal resulted in phenotypic switching to an intermediate state, with cells expressing both epithelial and mesenchymal markers at gene/protein levels. These pEMT cells were able to form colonies with front-back polarity and aggressive phenotype (Bidarra et al., 2016). In an evaporation-based 3D paper pump model, type I collagen gel (Figures 3D and 3E) was used to generate epidermal growth factor (EGF) gradient, which induced pEMT and collective cell migration in MCF-7 spheroids demonstrating pseudopodia protrusions from the migratory cells (Kuo et al., 2014). The initial results from 3D models (Figure 3) elicit convincing analogy between the pEMT and collective migration and can potentially be promising tools to gain mechanistic insights on pEMT driving collective migration.
Figure 3.

Three-dimensional (3D) bioengineering platforms used to study partial EMT phenotype
(A) 96-well low attachment plate.
(B) Hanging drop spheroid culture method, adapted with permission Kuo et al. (2017).
(C) Soft-agar-coated spheroid model, adapted with permission from Chaicharoenaudomrung et al. (2019).
(D) Collagen matrix for studying microenvironment interactions with tumor cells, adapted with permission from Kasai et al., (2017).
(E) Microfluidic platform, reproduced from Kuo et al. (2014) with permission from The Royal Society of Chemistry.
(F) Breast tumor cells-adipocytes co-culture model, adapted with permission from cross-reference to Debnath et al. (2003).
Stromal cell-related factors
Tumor cells have also been reported to co-opt surrounding stromal cells to transition into tumor-associated stromal cells (TASCs). These TASCs express fibroblast-activating protein, alpha-smooth muscle actin, and MMPs and secrete pro-tumorigenic cytokines and chemokines such as SDF1A, MMPs, IL-6, IL-8, VEGF, and tenascin-C, which further modulate tumor progression (Bussard et al., 2016). The major stromal cells include cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs), adipose-tissue-derived stromal cells (ASCs), and tumor-associated macrophages (TAMs) (Table 1). In addition to stromal cells, stromal cell-derived cytokines or chemokines also contribute to pEMT regulation; however, experimental evidence is needed.
Cancer-associated fibroblasts
Fibroblasts are one of the most abundant cell type within the TME. The bi-directional crosstalk mediated through paracrine factors secreted from tumor cells and the microenvironment differentiate normal fibroblasts into CAFs, which further drive tumor progression via pEMT (Yoshida et al., 2019). Activation of CAFs is further linked to collective migration and stabilization of cells in the pEMT state (Table 1) (Yoshida, 2020). The pEMT tumor cells then interact with surrounding TME to evade therapy, promote chemoresistance (Yoshida et al., 2019), and enhance cell migration (Li et al., 2019a). In an indirect co-culture model of interstitial TME, paracrine activation of hedgehog signaling in CAFs stabilized pancreatic cancer cells (BxPc-3, Panc-1) in pEMT state, further promoting their migration and invasion potential (Shan et al., 2017).
Matsumura et al. used a co-implantation tumor xenograft model to determine the interactions between green fluorescent protein (GFP)-labeled tumor cells and human stromal fibroblasts, directly demonstrating that CAFs drive the tumor cell cluster formation and collective migration of both epithelial and pEMT cancer population (Matsumura et al., 2019). Further, CAFs secreted SDF1 and induced Src activation in tumor cells leading to collective migration, further contributing to increased invasion, metastasis, and presence of pEMT clusters in the circulation. Puram et al. (Puram et al., 2017) showed that paracrine interactions between tumor cells at the leading edge and surrounding CAFs promote a pEMT program via TGFB/TGFBI axes. The CAF-leading edge tumor interactions included secreted ligands from CAFs with expression of corresponding receptors on the tumor cells such as TGFB3-TGFBR2, FGF7-FGFR2, and CXCL12-CXCR7, all contributing to pEMT. Targeting these CAF-tumor cell interactions may represent a potential therapeutic approach.
Adipocyte tissue-derived stromal cells
Adipocytes are known to infiltrate the TME, and the ratio of ECM to adipocytes impacts tumor progression and metastases (Seo et al., 2015). In a novel 3D co-culture model recapitulating tumor cells, ECM, and adipocyte interactions (Figure 3F) in vivo, the pre-adipocytes were differentiated into mature adipocytes on chamber slides for 5 days, which were then overlaid on laminin-rich matrigel. These mature adipocytes elicited transition of mesenchymal MDA-MB-231 and Hs578t breast cancer cells to epithelial-like structure. Adipocytes induced significant expression of both ECAD and CLDN7 (pEMT) in MDA-MB-231 and Hs578 cells (Pallegar et al., 2019). Besides adipocytes, adipocyte-secreted hormone such as leptin was also reported to induce pEMT in MCF10A non-tumorigenic mammary cells via FAK-ERK dependent pathway (Villanueva-Duque et al., 2017). Leptin treatment induced change in sub-cellular localization of ECAD from plasma membrane to cytoplasm along with increased perinuclear VIM expression via elevated phosphorylation of FAK and ERK. This, in turn induced collective cell migration with fibroblast-like morphology for the cells at leading edge (Villanueva-Duque et al., 2017).
Tumor-associated macrophages
Tumor-associated macrophages (TAMs) are another major component of TME, and increased tumor infiltration of TAMs has been widely associated with poor prognosis. TAMs create an immunosuppressive TME, which facilitate metastatic cascade via secretion of chemokines, cytokines, and growth factors (Lin et al., 2019). The secretome of M1 TAMs has been shown to drive pEMT phenotype in MCF-7 and T47D cells, which was further correlated to increased tumor migration and invasion (Bednarczyk et al., 2018). Although TAMs and polarized phenotypes M1 and M2 have been extensively studied with respect to cancer progression, the experimental evidence of the role of TAMs in inducing pEMT remains to be explored. This association has been explored using in silico computational models, wherein polarized M1/M2 macrophages were shown to interact with tumor cells expressing pEMT phenotype (Li et al., 2019b) through interaction networks but remains to be validated using experimental models.
We note that the current understanding of pEMT phenotype is mainly driven by experimental approaches based on 2D culture studies, which are limited in their physiological relevance. Given the importance of TME in the pEMT phenotype, tissue engineering approaches offer timely opportunity to engineer controlled yet complex TME and tease out microenvironmental influence on pEMT driving aggressive tumor behaviors.
Significance/role of pEMT in tumor progression
Presence of pEMT phenotype entails presence of E, M, and E/M phenotype enhancing tumor heterogeneity and thus, variable treatment response to the established therapeutic treatments across cancer types (Donnenberg and Donnenberg, 2015). The following section highlights the role of pEMT in regulating key hallmarks of tumor progression.
Tumor heterogeneity
After malignant transformation, cancer cells remain dynamic and continue to respond to the intrinsic and extrinsic signals in the TME (Teeuwssen and Fodde, 2019). This constant change and evolution drives tumor heterogeneity with cell sub-populations exhibiting diverse molecular signatures as a consequence of genetic, transcriptomic, epigenetic, and phenotypic changes (Teeuwssen and Fodde, 2019; Tripathi et al., 2020). Such heterogeneity directly affects overall tumor progression and therapeutic response (Fischer et al., 2015; Donnenberg and Donnenberg, 2015; Donnenberg et al., 2007), allowing cells to respond and adapt quickly to diverse conditions including therapeutic treatments (Tripathi et al., 2020). Tumor heterogeneity can be influenced by various factors such as inflammation, anatomical location, presence of secreted factors, cell-cell communication, and ECM density and the pH of TME. Further, intrinsic factors such as the morphological and epigenetic changes also influence temporal heterogeneity (Teeuwssen and Fodde, 2019).
Through integrated computational-experimental framework (Bocci et al., 2019a), it was suggested that phenotypic heterogeneity arises from a gradient of EMT-inducing signals such as TGFB via Notch-Jagged pathway. The TGFB diffusion gradient generated heterogeneity by spatial segregation of mesenchymal-like cancer stem cells with complete EMT at the invasive front of the tumor, whereas cancer stem cells with pEMT were localized in the interior of the tumor. This model further revealed that Notch-Jagged signaling generated distinct pattern of pEMT states within the tumor interior surrounded by other tumor cells in different pEMT states, generating heterogeneous clusters (Bocci et al., 2019a). Similarly, it was predicted from in silico models that miR-200/ZEB1 behaves as a three-way decision-making switch enabling epithelial (E) to hybrid (E/M) to mesenchymal (M) transition in response to exogenous TGFB stimulation (Jia et al., 2017).
Metabolic states and epigenetic modifications also promote plasticity and heterogeneity. Chromatin remodeling complexes such as polycomb and NuRD lead to post-translational histone modifications and EMT-related transcription factors that epigenetically drive cells from epithelial to mesenchymal phenotypes (Teeuwssen and Fodde, 2019). EMT-related transcription factors such as NUMB, GRHL2, and OVOL act as phenotypic stability factors to stabilize pEMT (Teeuwssen and Fodde, 2019).
The stability of pEMT and tumor heterogeneity for extended periods also enables the acquisition of therapeutic resistance. Tripathi et al. (Tripathi et al., 2020) developed a computational model that predicted that heterogeneity arises from cell division-associated stochastic changes due to intrinsic noise, which is maintained over multiple cell passages and contributes to non-genetic heterogeneity. This behavior can contribute to metastatic aggressiveness and potential re-development of phenotypic complexity at distant organ sites (Tripathi et al., 2020; Donnenberg et al., 2013). Cells in pEMT state have superior survival strategies and can either be immune-resistant or chemo-tolerant, further promoting tumor heterogeneity (Tripathi et al., 2020).
pEMT and cell cycle regulation
The regulators of pEMT are also associated with cell-cycle regulation (Goetz et al., 2020; Denisov and Perelmuter, 2018). For instance, hypoxia can elevate TGFB1 levels (Topalovski et al., 2016), a transcription factor known to induce complete EMT in epithelial cells (Hao et al., 2019). The initiation of EMT caused by TGFB1 signaling pathway activates SNAI1 (Tran et al., 2011), which simultaneously upregulates the levels of the cyclin-dependent kinase inhibitor p21 causing cell-cycle arrest (Vega et al., 2004; Xing and Tian, 2019). The ERK is another regulator of EMT plasticity associated with the dysregulation of cell cycle during transient EMT states (Shin et al., 2019). Sustained ERK activation can suppress cell cycle via FOXO1 (Shin et al., 2019), enhancing expression of the cell cycle inhibitors p21 and p27 repressing cell proliferation (Diep et al., 2013; Schmidt et al., 2002). A number of studies in different cancer types such as melanoma (Carreira et al., 2006), epithelial adenocarcinoma (Svensson et al., 2003), colorectal (Rubio, 2008), pancreatic (Qian et al., 2002), gastric (Yano et al., 2014), hepatocellular (Iwasaki et al., 1995), and breast cancers (Wang et al., 2004; Goswami et al., 2004) have reported that invading cancer cells are predominantly in G0/G1 phase with repression of proliferation-related molecules such as cyclin D, cyclin E, and the overexpression of cell-cycle inhibitors p21 and p27 (Kohrman and Matus, 2017). The nascent evidence suggesting that pEMT is characterized by the overexpression of cell-cycle inhibitors and that invasive cancers are in cell-cycle arrest implies a link between pEMT, cell-cycle arrest, and invasiveness that needs to be further investigated.
pEMT and collective migration
Collective migration is observed in many aggressive cancers in which a group of cells maintain a coordinated movement guided by the cues from the microenvironment (Yang et al., 2019b). Contrary to the migratory single cells that exhibit complete loss of cell-cell adhesion, migratory cell clusters preserve cell-cell junctions principally through ECAD (Singh et al., 2016, 2019), gap junctions (Citi et al., 2014; Kuznik et al., 2016; Park et al., 2016; Leech et al., 2018; Upadhaya et al., 2019; Zhang et al., 2020), and surface adherent proteins from the immunoglobulin family (Friedl et al., 2012; Aiello et al., 2018).
Collective migratory clusters have different movement dynamics and morphologies ranging from cell strands, broad clusters that migrate together, and cell groups that form luminal structures (Friedl et al., 2012). These morphological arrangements depend on the adhesion system and cell-ECM relationships and the status of pEMT program in cell populations (Friedl et al., 2012). A recent study by Aiello et al. (Aiello et al., 2018) analyzed the migration pattern of pancreatic cancer cell spheroids with complete EMT or pEMT. The pEMT was defined in cluster of tumors that co-express epithelial markers (ECAD and CLDN7) and the mesenchymal markers (ETV1, PRRX1, ZEB1, TWIST1, SNAI1, SNAI2, and ZEB2). Authors observed that tumor spheres of pEMT cells retained cell-cell junctions and moved as a collective cluster. This is also supported by work from Friedl et al. and our group who reported that carcinomas expressing pEMT phenotype are present at the leading edge and promote collective migration while retaining cell-cell junctions (Friedl and Mayor, 2017; Donnenberg et al., 2010). In contrast, tumor spheres from cells representing complete EMT showed a spindle morphology and migrated as single cells (Aiello et al., 2018), supporting the notion that pEMT subtype impacts the migration pattern.
Gao et al. (Gao et al., 2014) observed that TGFB promoted collective motility of NIH-OVCAR3 cells without changes in the expression of ECAD and the increase in CLDN1 and N-cadherin (CDH2), a pattern characteristic of pEMT. Increased CLDN1 is further linked to collective migration in cells undergoing pEMT (Giampieri et al., 2009). Analogous results were obtained by Rypens et al. (Rypens et al., 2020) who studied the metastatic potential of invasive breast carcinoma (IBC) clusters migrating through the draining lymphatics (Rypens et al., 2020). Authors observed the presence of ECAD in collectively invading IBC, which is a characteristic of pEMT. Moreover, the study demonstrated that TGFB induced collective movement of IBC clusters through reduced SMAD3 expression and activation of MYC response program (Rypens et al., 2020; Kohn et al., 2012).
One of many different mechanisms of collective migration/invasion suggests role of leader and follower cells (Yang et al., 2019a, 2019b; Friedl and Gilmour, 2009; Zoeller et al., 2019). The leader cells locate to the invading front of the tumor, interact with the surrounding non-malignant cells, and facilitate the invasion of the migratory cluster, whereas the follower cells remain at the back of the cluster. Cells at the leading edge have more mesenchymal characteristics (Donnenberg et al., 2010) as compared with more epithelial, follower cells (Yang et al., 2019b). However, both leaders and followers show high expression of tight junction molecules, indicating that leader cells showing mesenchymal-like phenotype undergo pEMT (Yang et al., 2019a). Studies in basal-like breast cancer have shown that ΔNp63α stimulates collective invasion by initiating pEMT through expression of SNAI2 and Axl receptor tyrosine kinase (AXL) while silencing ZEB1 and ZEB2 to maintain ECAD adherence junctions (Chen et al., 2019). In luminal carcinomas, pEMT leader cells show high expression of CD44, whereas followers exhibit epithelial characteristics with no or low levels of CD44 expression; however, both cells can switch from high to low CD44 expression when MET occurs indicating phenotypic plasticity during seeding of collectively migrating cells (Yang et al., 2019a). The genetic heterogeneity of the collectively invading clusters was also shown in NSCLC where 14 mutations were selectively enriched in either leaders or follower cells. Specifically, ACTR3 found in the leader cells and KDM5B (Zoeller et al., 2019) in the follower cells were compared with a pEMT plasticity (da Silva-Diz et al., 2018; Cheung et al., 2013), supporting a direct relationship between collective migration, pEMT, and genetic as well as phenotypic differences in the leader/follower cells.
The “rear-wheel drive” collective migration mechanism emphasizes the contribution of the rear end of moving clusters to the directionality of collective migration (Shellard and Mayor, 2019; Shellard et al., 2018; Yamada and Sixt, 2019). In this mechanism, a supracellular contractile actomyosin belt that stretches across the cells at the back of the cluster pushes the cohort from behind resulting in a synchronized move (Shellard et al., 2018). The moving cluster acquires supracellular polarity in which the group, but not individual cells, acquires front-rear polarity (Shellard and Mayor, 2019; Shellard et al., 2018). The front of the cluster develops protrusions, whereas the back exhibits actomyosin contractility (Shellard and Mayor, 2019; Shellard et al., 2018). To acquire supracellular polarity, it is necessary that the cluster retain the cell-cell adhesions (Shellard and Mayor, 2019; Shellard et al., 2018; Venhuizen and Zegers, 2017; Yamada and Sixt, 2019). Although this collective migration mechanism has been described in clusters of migrating neural crest cells in Xenopus (Shellard et al., 2018), in border cells of Drosophila melanogaster (Combedazou et al., 2017), and in invading cancer cells (Hidalgo-Carcedo et al., 2011), the role of pEMT in supracellular polarity/"rear-wheel drive” migration remains to be determined.
The prevalence of collective migration has been identified in invasive cancers and has been associated with poor clinical outcomes (Chung et al., 2016; Wang et al., 2020). The correlation between collective migration and pEMT and their contribution to distant metastasis was described in both invasive ductal carcinoma (IDC) and invasive lobular carcinoma (ILC) (Khalil et al., 2017). However, collective migration in IDC retained ECAD adherence junctions, whereas ILC, despite of the absence of ECAD, expressed CD44 at the cell-cell junction. Histological analysis of IDC and ILC indicated that both migratory groups retained epithelial characteristics and lacked complete EMT. Further, in both breast carcinoma types, the extent of collective invasion into the adjacent adipose tissue was positively correlated with metastasis (Khalil et al., 2017). Analogous results were found in 176 collectively invading breast cancer samples, where tumors at the primary and the metastatic sites exhibited pEMT (co-expression of ECAD and VIM) and had the worst disease-free survival (Yamashita et al., 2018; Donnenberg and Donnenberg, 2015). It is further shown pEMT cells form heterogeneous clusters and enter blood vessels as CTC clusters (Lecharpentier et al., 2011; Bocci et al., 2019b; Jia et al., 2019). The recent experimental models of collective migration and clinical evidence support a link between pEMT, emergence of leader and follower cells, collective migration, and metastasis. The identification of pEMT in early disease stages can be a possible diagnostic target of cancer aggressiveness.
Metastasis
Multiple pEMT states and phenotypes have been identified in various cell lines, and during metastasis in vivo and pEMT cancer cells are more prone to metastatic outgrowths (Donnenberg et al., 2018; Goetz et al., 2020; Karacosta et al., 2019; Pastushenko et al., 2018; Schliekelman et al., 2015; Stylianou et al., 2019). In a systematic mathematical analysis, Goetz et al. (Goetz et al., 2020) concluded that more the number of intermediate states, the more chances to metastasize. The simulations showed that by stabilizing one intermediate state, cells can be trapped for a much longer slowing down complete EMT.
Chen et al. (Chen et al., 2018b) used lineage tracing to monitor pEMT in genetically engineered mice that spontaneously develop pancreatic ductal carcinoma and identified that α-SMA and FSP-1 mediated pEMT in the primary tumor. In metastasis, only disseminated tumor clusters containing 3–5 cells (micrometastasis) presented a pEMT phenotype and expressed both α-SMA and FSP-1. However, in the same model, the established lung and liver metastases were predominantly composed of cancer cells that maintained epithelial phenotype without α-SMA and FSP-1 expression. One possible explanation is the emergence of larger metastatic nodules from small tumor clusters with pEMT, which may undergo MET leading to increased proliferation and formation of metastatic colonies.
Therapeutic resistance
Cellular reprogramming is essential to induce pEMT as cells reorganize cytoskeletal actin and alter their signaling and gene expression (Kalluri and Weinberg, 2009). This confers pEMT cells the phenotypic plasticity that is associated with stemness (Jia et al., 2019) and resistance to therapy (Gupta et al., 2019; Donnenberg and Donnenberg, 2005).
The study by Papadaki et al. (Papadaki et al., 2019) provides a link between pEMT, stemness, and therapeutic resistance. The CTCs expressing pEMT also exhibit cancer stem cell (CSC) markers (Donnenberg et al., 2010). These CSC+/pEMT+ CTC clusters co-expressing cytokeratin, nuclear TWIST1, and ALDH1 were identified in 27.7% of metastatic breast cancer patients (Papadaki et al., 2019). Post-chemotherapy, these CSC+/pEMT+ CTCs were identified as chemoresistant subpopulation and were increased in HER2-patients and non-responders following treatment (Papadaki et al., 2019). Similarly, pEMT+/programmed cell death ligand-1 (PD-L1)+ CTCs were resistant to anti-PD-1 treatment in patients with metastatic NSCLC (Raimondi et al., 2017).
The androgen receptor negative, docetaxel resistant PC-3, and DU145 cell lines were used to determine the effect of EMT drivers (Hanrahan et al., 2017). The PC-3 model exhibited a transition from epithelial colonies to a scattered cell phenotype similar to mesenchymal cells and corresponding to a complete EMT pattern. However, the DU145 model displayed a combination of compact epithelial and loose aggregates of quasi-mesenchymal cells exhibiting migration. These cells maintained higher ECAD expression despite upregulation of ZEB1 and ZEB2, suggesting pEMT. When treated with the CSC inhibitor salinomycin, PC-3 cells with complete EMT showed apoptotic death, whereas DU145 cells with pEMT were less sensitive to salinomycin despite the increased expression of CD44 (Hanrahan et al., 2017). This implies an additional level of therapeutic resistance conferred by pEMT alone, highlighting the complexity and challenges of treating cancer cells with phenotypic plasticity.
It has been observed that breast cancer patients with HER2 overexpression do not respond to chemotherapy with trastuzumab (Wu et al., 2012; Vogel et al., 2002). These patients have increased AKT expression and loss of phosphatase and tensin homolog (PTEN), resulting in accumulation of CTNNB1. It was shown that development of trastuzumab resistance in HER2 overexpressing breast cancer cells was accompanied by pEMT-like transition, activation of the WNT/CTNNB1 signaling pathway with overexpression of WNT3, and transactivation of EGFR (Wu et al., 2012).
Chemoresistance can also be dependent on the phenotypic state of cancer cells. Huang et al. (Huang et al., 2013) utilized a library of 43 ovarian cancer cell lines to explore relationship between EMT heterogeneity and sensitivity to the Src inhibitor, Saracatinib. The ovarian cancer cell library was characterized by four different EMT phenotypes where 20.9% of the cell lines were epithelial, 41.9% were intermediate epithelial, 18.6% intermediate mesenchymal, and 18.6% mesenchymal (Huang et al., 2013). When treated with Saracatinib, only cells presenting an intermediate mesenchymal phenotype underwent EMT reversal after treatment. Cell-based EMT functional studies revealed that intermediate mesenchymal cells are characterized by high CDH2 and ZEB1 expression and low ECAD and Erb-B2 receptor tyrosine kinase 3 (Erb-B3/HER3). Treatment with Saracatinib showed a dose-dependent effect on upregulation of ECAD and downregulation of SNAI1 and SNAI2, but no change in ZEB1, ZEB2, and TWIST. However, cells in the other three phenotypical EMT states were unaffected by the treatment with Saracatinib. This demonstrated that intermediate EMT states and phenotypic plasticity are major causes of therapeutic resistance (Huang et al., 2013).
Although the above discussion points to the role of pEMT in development of chemoresistant tumor subpopulation, there is still need for strong experimental evidence for establishing mechanistic patterns between pEMT and chemoresistance. Such knowledge will be instrumental for better management of patients with refractory and relapse profile and specifically, the cancer patients who do not respond to established chemotherapy/immunotherapy protocols.
Challenges and future prospects: pEMT as the driver of functional plasticity, therapy resistance, and metastasis
Epithelial to mesenchymal transition has been a well-established phenomenon, wherein the inherent plasticity of epithelial cells to undergo a mesenchymal transition explains several key features of the invasive and metastatic phenotypes in epithelial cancers. By activating this pathway, epithelial cells in the early phases of neoplastic transformation can detach, gain motility, and change their transcriptome in ways that favor invasion and spread through the lymphatic and circulatory systems. Upon reaching a hospitable niche, such cells could revert, wholly or in part, to an epithelial tumor phenotype through MET. Currently, this interpretation has been refined to include pEMT, as functional plasticity does not have to be inevitably tied to a particular differentiation pathway and may not be mutually exclusive (Donnenberg and Donnenberg, 2015) or required for tumor invasion and migration. With time, pEMT+ cells capable of functional plasticity may emerge, driven by intrinsic and extrinsic factors such as selection resulting from exposure to therapeutic agents. It is precisely the pEMT state that may mark the emergence of invasive and metastatic capacity.
One of the major challenges in the field is to standardize the characterization of pEMT based on co-expression of defined epithelial and mesenchymal markers. Presently, pEMT is defined based on co-expression of many different epithelial and mesenchymal markers without a clear consensus (Table 1). Further, emerging scientific evidence have highlighted presence of early, intermediate, and late pEMT states that cover the spectrum from pure epithelial to completely mesenchymal phenotypes. Emphasis should be placed on identifying molecular patterns and/or environmental factors that are capable of stabilizing cells in different stages of pEMT and consequence of such stabilization on tumor progression, therapeutic resistance, and halting tumor progression. Single-cell techniques should be exploited to determine how pEMT is regulated within heterogeneous tumors unlike the bulk tumor samples containing heterogeneous mixture of E, E/M, and M phenotypes (Chakraborty et al., 2020). Also, with the emerging evidence of the TME's role in activating and sustaining pEMT, it will be important to understand the cellular and molecular interactions of the tumor within its environment and the consequences on tumor progression and therapeutic resistance. Given their physiological relevance, tissue-engineered models will have a major impact into mechanistic insights of TME-pEMT pathobiology.
pEMT state provides an additional rationale for early intervention: (1) successful anti-pEMT therapies will need to simultaneously engage multiple targets including identifying and co-targeting environmental factors that are capable of stabilizing cells in different stages of pEMT; (2) the therapeutic benefit such as the prevention of metastasis, may not be immediate, and the therapeutic effects may not be measurable within a study time frame; (3) therapeutic trials of new approaches are typically carried out on end-stage heavily pre-treated patients who might not respond to pEMT-targeted treatment. Acknowledging the functional plasticity and the bi-directional nature of pEMT demand that therapy simultaneously target pEMT states, the permissive tumor microenvironment, and the processes by which pEMT occurs.
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the Lead Contact, Shilpa Sant (shs149@pitt.edu).
Materials availability
Not applicable.
Data and code availability
Not applicable.
Acknowledgments
The authors acknowledge the help from Ms. Oshin Miranda and Mr. Julio Aleman Hernandez for initial literature search. This work is supported by the National Institute of Health (NIH) [R37CA232209] to SS and VSD and by BC132245_W81XWH-14-0258 from the Department of Defense, METAvivor FP00002718, The Pennsylvania Breast Cancer Coalition to VSD.
Author contributions
S.S. and V.A. conceptualized the outline of the review. V.A. and C.A.M. investigated the literature. V.A. and C.A.M wrote the manuscript sections. V.A. and C.A.M. revised and edited the manuscript. V.A. and S.S. conceived the figure visualization. S.S. and V.S.D. supervised, reviewed, and revised the manuscript. All authors read and approved the final manuscript draft.
Declaration of interests
The authors declare no competing interests.
References
- Aggarwal V., Miranda O., Johnston P.A., Sant S. Three dimensional engineered models to study hypoxia biology in breast cancer. Cancer Lett. 2020;490:124–142. doi: 10.1016/j.canlet.2020.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello N.M., Kang Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019;216:1016–1026. doi: 10.1084/jem.20181827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello N.M., Maddipati R., Norgard R.J., Balli D., Li J., Yuan S., Yamazoe T., Black T., Sahmoud A., Furth E.E. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell. 2018;45:681–695 e684. doi: 10.1016/j.devcel.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarczyk R.B., Tuli N.Y., Hanly E.K., Rahoma G.B., Maniyar R., Mittelman A., Geliebter J., Tiwari R.K. Macrophage inflammatory factors promote epithelial-mesenchymal transition in breast cancer. Oncotarget. 2018;9:24272–24282. doi: 10.18632/oncotarget.24917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S., Wang P., Toh A., Thompson E.W. New insights into the role of phenotypic plasticity and EMT in driving cancer progression. Front. Mol. Biosci. 2020;7:71. doi: 10.3389/fmolb.2020.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidarra S.J., Oliveira P., Rocha S., Saraiva D.P., Oliveira C., Barrias C.C. A 3D in vitro model to explore the inter-conversion between epithelial and mesenchymal states during EMT and its reversion. Sci. Rep. 2016;6:27072. doi: 10.1038/srep27072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci F., Tripathi S.C., Mercedes S.A.V., George J.T., Casabar J.P., Wong P.K., Hanash S.M., Levine H., Onuchic J.N., Jolly M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. (Camb) 2019;11:251–263. doi: 10.1093/intbio/zyz021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci F., Gearhart-Serna L., Boareto M., Ribeiro M., Ben-Jacob E., Devi G.R., Levine H., Onuchic J.N., Jolly M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. U S A. 2019;116:148–157. doi: 10.1073/pnas.1815345116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci F., Kumar Jolly M., Onuchic J.N. A Biophysical model uncovers the size distribution of migrating cell clusters across cancer types. Cancer Res. 2019;79:5527–5535. doi: 10.1158/0008-5472.CAN-19-1726. [DOI] [PubMed] [Google Scholar]
- Bronsert P., Enderle-Ammour K., Bader M., Timme S., Kuehs M., Csanadi A., Kayser G., Kohler I., Bausch D., Hoeppner J. Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer-host interface. J. Pathol. 2014;234:410–422. doi: 10.1002/path.4416. [DOI] [PubMed] [Google Scholar]
- Bussard K.M., Mutkus L., Stumpf K., Gomez-Manzano C., Marini F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84. doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S., Goodall J., Denat L., Rodriguez M., Nuciforo P., Hoek K.S., Testori A., Larue L., Goding C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaicharoenaudomrung N., Kunhorm P., Noisa P. Three-dimensional cell culture systems as an in vitro plaform for cancer and stem cell modeling. World J. Stem Cells. 2019;11:1065–1083. doi: 10.4252/wjsc.v11.i12.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty P., George J.T., Tripathi S., Levine H., Jolly M.K. Comparative study of transcriptomics-based scoring metrics for the epithelial-hybrid-mesenchymal spectrum. Front. Bioeng. Biotechnol. 2020;8:220. doi: 10.3389/fbioe.2020.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi V., Fournier-Level A., Cooper H.M., Murray M.J. Loss of Neogenin1 in human colorectal carcinoma cells causes a partial EMT and wound-healing response. Sci. Rep. 2019;9:4110. doi: 10.1038/s41598-019-40886-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Chen X., Li W., Shan T., Lin W.R., Ma J., Cui X., Yang W., Cao G., Li Y. Conversion of epithelial-to-mesenchymal transition to mesenchymal-to-epithelial transition is mediated by oxygen concentration in pancreatic cancer cells. Oncol. Lett. 2018;15:7144–7152. doi: 10.3892/ol.2018.8219. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chen Y., LeBleu V.S., Carstens J.L., Sugimoto H., Zheng X., Malasi S., Saur D., Kalluri R. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol. Med. 2018;10:e9085. doi: 10.15252/emmm.201809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.-j., Tang Y.-j., Tang Y.-l., Liang X.-h. What makes cells move: requirements and obstacles for leader cells in collective invasion. Exp. Cell Res. 2019;382:111481. doi: 10.1016/j.yexcr.2019.06.026. [DOI] [PubMed] [Google Scholar]
- Cheung K.J., Gabrielson E., Werb Z., Ewald A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y.C., Wei W.C., Hung C.N., Kuo J.F., Hsu C.P., Chang K.J., Chao W.T. Rab11 collaborates E-cadherin to promote collective cell migration and indicates a poor prognosis in colorectal carcinoma. Eur. J. Clin. Invest. 2016;46:1002–1011. doi: 10.1111/eci.12683. [DOI] [PubMed] [Google Scholar]
- Citi S., Guerrera D., Spadaro D., Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014;5:1–15. doi: 10.4161/21541248.2014.973760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combedazou A., Choesmel-Cadamuro V., Gay G., Liu J., Dupre L., Ramel D., Wang X. Myosin II governs collective cell migration behaviour downstream of guidance receptor signalling. J. Cell Sci. 2017;130:97–103. doi: 10.1242/jcs.179952. [DOI] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S.K., Brugge J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Denisov E.V., Perelmuter V.M. A fixed partial epithelial-mesenchymal transition (EMT) triggers carcinogenesis, whereas asymmetrical division of hybrid EMT cells drives cancer progression. Hepatology. 2018;68:807–810. doi: 10.1002/hep.29784. [DOI] [PubMed] [Google Scholar]
- Diep C., Charles N., Blake Gilks C., Kalloger S., Argenta P., Lange C.A. Progesterone receptors induce FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle. 2013;12:1433–1449. doi: 10.4161/cc.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmello C., Sawant S., Alam H., Gangadaran P., Mogre S., Tiwari R., D'Souza Z., Narkar M., Thorat R., Patil K. Vimentin regulates differentiation switch via modulation of keratin 14 levels and their expression together correlates with poor prognosis in oral cancer patients. PLoS One. 2017;12:e0172559. doi: 10.1371/journal.pone.0172559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- Donnenberg V.S., Donnenberg A.D. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J. Clin. Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- Donnenberg V.S., Donnenberg A.D. Stem cell state and the epithelial-to-mesenchymal transition: implications for cancer therapy. J. Clin. Pharmacol. 2015;55:603–619. doi: 10.1002/jcph.486. [DOI] [PubMed] [Google Scholar]
- Donnenberg V.S., Landreneau R.J., Donnenberg A.D. Tumorigenic stem and progenitor cells: implications for the therapeutic index of anti-cancer agents. J. Control. Release. 2007;122:385–391. doi: 10.1016/j.jconrel.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg V.S., Donnenberg A.D., Zimmerlin L., Landreneau R.J., Bhargava R., Wetzel R.A., Basse P., Brufsky A.M. Localization of CD44 and CD90 positive cells to the invasive front of breast tumors. Cytometry B. Clin. Cytom. 2010;78:287–301. doi: 10.1002/cyto.b.20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg A.D., Hicks J.B., Wigler M., Donnenberg V.S. The cancer stem cell: cell type or cell state? Cytometry A. 2013;83:5–7. doi: 10.1002/cyto.a.22208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg V.S., Zhang J.J., Moravcikova E., Meyer E.M., Lu H., Carson C.T., Donnenberg A.D. Antibody-based cell-surface proteome profiling of metastatic breast cancer primary explants and cell lines. Cytometry A. 2018;93:448–457. doi: 10.1002/cyto.a.23300. [DOI] [PubMed] [Google Scholar]
- Fischer K.R., Durrans A., Lee S., Sheng J., Li F., Wong S.T., Choi H., El Rayes T., Ryu S., Troeger J. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P., Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Friedl P., Mayor R. Tuning collective migration by cell-cell junction regulation. Cold Spring Harb. Perspect. Biol. 2017;9:a029199. doi: 10.1101/cshperspect.a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P., Locker J., Sahai E., Segall J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- Gao J., Zhu Y., Nilsson M., Sundfeldt K. TGF-β isoforms induce EMT independent migration of ovarian cancer cells. Cancer Cell Int. 2014;14:1–10. doi: 10.1186/s12935-014-0072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampieri S., Manning C., Hooper S., Jones L., Hill C.S., Sahai E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009;11:1287–1296. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli G., Bergamini C., Fransvea E., Sgarra C., Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- Goetz H., Melendez-Alvarez J.R., Chen L., Tian X.J. A plausible accelerating function of intermediate states in cancer metastasis. PLoS Comput. Biol. 2020;16:e1007682. doi: 10.1371/journal.pcbi.1007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S., Wang W., Wyckoff J.B., Condeelis J.S. Breast cancer cells isolated by chemotaxis from primary tumors show increased survival and resistance to chemotherapy. Cancer Res. 2004;64:7664–7667. doi: 10.1158/0008-5472.CAN-04-2027. [DOI] [PubMed] [Google Scholar]
- Gupta P.B., Pastushenko I., Skibinski A., Blanpain C., Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65–78. doi: 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan K., O'Neill A., Prencipe M., Bugler J., Murphy L., Fabre A., Puhr M., Culig Z., Murphy K., Watson R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017;11:251–265. doi: 10.1002/1878-0261.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Baker D., Ten Dijke P. TGF-beta-Mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019;20:2767. doi: 10.3390/ijms20112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi S., Fukuda Y., Sugisaki Y., Yamanaka N. Pulmonary carcinosarcoma: immunohistochemical and ultrastructural studies. Pathol. Int. 1999;49:903–908. doi: 10.1046/j.1440-1827.1999.00964.x. [DOI] [PubMed] [Google Scholar]
- Hidalgo-Carcedo C., Hooper S., Chaudhry S.I., Williamson P., Harrington K., Leitinger B., Sahai E. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 2011;13:49–58. doi: 10.1038/ncb2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.Y., Wong M., Tan T., Kuay K., Ng A., Chung V., Chu Y., Matsumura N., Lai H., Lee Y. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530) Cell Death Dis. 2013;4:e915. doi: 10.1038/cddis.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T., Shinkai K., Mukai M., Yoshioka K., Fujii Y., Nakahara K., Matsuda H., Akedo H. Cell-cycle-dependent invasion in vitro by rat ascites hepatoma cells. Int. J. Cancer. 1995;63:282–287. doi: 10.1002/ijc.2910630223. [DOI] [PubMed] [Google Scholar]
- Jia D., Jolly M.K., Tripathi S.C., Hollander P.D., Huang B., Lu M., Celiktas M., Ramirez-Pena E., Ben-Jacob E., Onuchic J.N. Distinguishing mechanisms underlying EMT tristability. Cancer Converg. 2017;1:2. doi: 10.1186/s41236-017-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Park J.H., Jung K.H., Levine H., Kaipparettu B.A. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells. 2018;7:21. doi: 10.3390/cells7030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Li X., Bocci F., Tripathi S., Deng Y., Jolly M.K., Onuchic J.N., Levine H. Quantifying cancer epithelial-mesenchymal plasticity and its association with stemness and immune response. J. Clin. Med. 2019;8:725. doi: 10.3390/jcm8050725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karacosta L.G., Anchang B., Ignatiadis N., Kimmey S.C., Benson J.A., Shrager J.B., Tibshirani R., Bendall S.C., Plevritis S.K. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 2019;10:5587. doi: 10.1038/s41467-019-13441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaosmanoglu O., Banerjee S., Sivas H. Identification of biomarkers associated with partial epithelial to mesenchymal transition in the secretome of slug over-expressing hepatocellular carcinoma cells. Cell Oncol (Dordr) 2018;41:439–453. doi: 10.1007/s13402-018-0384-6. [DOI] [PubMed] [Google Scholar]
- Kasai Y., Takeda N., Kobayashi S., Takagi R., Yamato M. Cellular events and behaviors after grafting of stratified squamous epithelial cell sheet onto a hydrated collagen gel. FEBS Open Bio. 2017;7:691–704. doi: 10.1002/2211-5463.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A.A., Ilina O., Gritsenko P.G., Bult P., Span P.N., Friedl P. Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin. Exp. Metastasis. 2017;34:421–429. doi: 10.1007/s10585-017-9858-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.K., Jang S.D., Kim H., Chung S., Park J.K., Kuh H.J. Phenotypic heterogeneity and plasticity of cancer cell migration in a pancreatic tumor three-dimensional culture model. Cancers (Basel) 2020;12:1305. doi: 10.3390/cancers12051305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn E.A., Yang Y.-a., Du Z., Nagano Y., Van Schyndle C.M., Herrmann M.A., Heldman M., Chen J.-Q., Stuelten C.H., Flanders K.C. Biological responses to TGF-β in the mammary epithelium show a complex dependency on Smad3 gene dosage with important implications for tumor progression. Mol. Cancer Res. 2012;10:1389–1399. doi: 10.1158/1541-7786.MCR-12-0136-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrman A.Q., Matus D.Q. Divide or conquer: cell cycle regulation of invasive behavior. Trends Cell Biol. 2017;27:12–25. doi: 10.1016/j.tcb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolijn K., Verhoef E.I., van Leenders G.J. Morphological and immunohistochemical identification of epithelial-to-mesenchymal transition in clinical prostate cancer. Oncotarget. 2015;6:24488–24498. doi: 10.18632/oncotarget.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C.T., Liu H.K., Huang G.S., Chang C.H., Chen C.L., Chen K.C., Huang R.Y., Lin C.H., Lee H., Huang C.S. A spatiotemporally defined in vitro microenvironment for controllable signal delivery and drug screening. Analyst. 2014;139:4846–4854. doi: 10.1039/c4an00936c. [DOI] [PubMed] [Google Scholar]
- Kuo C.T., Wang J.Y., Lin Y.F., Wo A.M., Chen B.P.C., Lee H. Three-dimensional spheriod culture targeting versatile tissue bioassays using a PDMS-based hanging drop array. Sci. Rep. 2017;7:4363. doi: 10.1038/s41598-017-04718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznik B.I., Linkova N.S., Kolchina N.V., Kukanova E.O., Khavinson V.K. The JAM family of molecules and their role in the regulation of physiological and pathological processes. Usp. Fiziol. Nauk. 2016;47:76–97. [PubMed] [Google Scholar]
- Lecharpentier A., Vielh P., Perez-Moreno P., Planchard D., Soria J.C., Farace F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer. 2011;105:1338–1341. doi: 10.1038/bjc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech A.O., Vellanki S.H., Rutherford E.J., Keogh A., Jahns H., Hudson L., O'Donovan N., Sabri S., Abdulkarim B., Sheehan K.M. Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings. Breast Cancer Res. 2018;20:140. doi: 10.1186/s13058-018-1064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Fu L., Li J.B., Qin Y., Zeng T.T., Zhou J., Zeng Z.L., Chen J., Cao T.T., Ban X. Increased expression of EIF5A2, via hypoxia or gene amplification, contributes to metastasis and angiogenesis of esophageal squamous cell carcinoma. Gastroenterology. 2014;146:1701–1713 e1709. doi: 10.1053/j.gastro.2014.02.029. [DOI] [PubMed] [Google Scholar]
- Li N., Babaei-Jadidi R., Lorenzi F., Spencer-Dene B., Clarke P., Domingo E., Tulchinsky E., Vries R.G.J., Kerr D., Pan Y. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis. 2019;8:13. doi: 10.1038/s41389-019-0125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Jolly M.K., George J.T., Pienta K. Computational modeling of the crosstalk between macrophage polarization and tumor cell plasticity in the tumor microenvironment. Front. Oncol. 2019;9:10. doi: 10.3389/fonc.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Xu J., Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019;12:76. doi: 10.1186/s13045-019-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren K., Nordenskjold B., Landberg G. Hypoxia, Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br. J. Cancer. 2009;101:1769–1781. doi: 10.1038/sj.bjc.6605369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y., Ito Y., Mezawa Y., Sulidan K., Daigo Y., Hiraga T., Mogushi K., Wali N., Suzuki H., Itoh T. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial-mesenchymal plasticity. Life Sci. Alliance. 2019;2:e201900425. doi: 10.26508/lsa.201900425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallegar N.K., Garland C.J., Mahendralingam M., Viloria-Petit A.M., Christian S.L. A novel 3-dimensional Co-culture method reveals a partial mesenchymal to epithelial transition in breast cancer cells induced by adipocytes. J. Mammary Gland Biol. Neoplasia. 2019;24:85–97. doi: 10.1007/s10911-018-9420-4. [DOI] [PubMed] [Google Scholar]
- Paniz-Mondolfi A., Singh R., Jour G., Mahmoodi M., Diwan A.H., Barkoh B.A., Cason R., Huttenbach Y., Benaim G., Galbincea J. Cutaneous carcinosarcoma: further insights into its mutational landscape through massive parallel genome sequencing. Virchows Arch. 2014;465:339–350. doi: 10.1007/s00428-014-1628-0. [DOI] [PubMed] [Google Scholar]
- Papadaki M.A., Stoupis G., Theodoropoulos P.A., Mavroudis D., Georgoulias V., Agelaki S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol. Cancer Ther. 2019;18:437–447. doi: 10.1158/1535-7163.MCT-18-0584. [DOI] [PubMed] [Google Scholar]
- Park J.A., Atia L., Mitchel J.A., Fredberg J.J., Butler J.P. Collective migration and cell jamming in asthma, cancer and development. J. Cell Sci. 2016;129:3375–3383. doi: 10.1242/jcs.187922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I., Brisebarre A., Sifrim A., Fioramonti M., Revenco T., Boumahdi S., Van Keymeulen A., Brown D., Moers V., Lemaire S. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- Patel A., Sant S. Hypoxic tumor microenvironment: opportunities to develop targeted therapies. Biotechnol. Adv. 2016;34:803–812. doi: 10.1016/j.biotechadv.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624 e1624. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L.-W., Mizumoto K., Urashima T., Nagai E., Maehara N., Sato N., Nakajima M., Tanaka M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 2002;8:1223–1227. [PubMed] [Google Scholar]
- Raimondi C., Carpino G., Nicolazzo C., Gradilone A., Gianni W., Gelibter A., Gaudio E., Cortesi E., Gazzaniga P. PD-L1 and epithelial-mesenchymal transition in circulating tumor cells from non-small cell lung cancer patients: a molecular shield to evade immune system? Oncoimmunology. 2017;6:e1315488. doi: 10.1080/2162402X.2017.1315488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh V., Brabletz T., Ceppi P. Targeting EMT in cancer with repurposed metabolic inhibitors. Trends. Cancer. 2020;6:942–950. doi: 10.1016/j.trecan.2020.06.005. [DOI] [PubMed] [Google Scholar]
- Rubio C.A. Arrest of cell proliferation in budding tumor cells ahead of the invading edge of colonic carcinomas. A preliminary report. Anticancer Res. 2008;28:2417–2420. [PubMed] [Google Scholar]
- Rypens C., Marsan M., Van Berckelaer C., Billiet C., Melis K., Lopez S.P., van Dam P., Devi G.R., Finetti P., Ueno N.T. Inflammatory breast cancer cells are characterized by abrogated TGFβ1-dependent cell motility and SMAD3 activity. Breast Cancer Res. Treat. 2020;180:385–395. doi: 10.1007/s10549-020-05571-z. [DOI] [PubMed] [Google Scholar]
- Sadeghi M., Ordway B., Rafiei I., Borad P., Fang B., Koomen J.L., Zhang C., Yoder S., Johnson J., Damaghi M. Integrative analysis of breast cancer cells reveals an epithelial-mesenchymal transition role in adaptation to acidic microenvironment. Front. Oncol. 2020;10:304. doi: 10.3389/fonc.2020.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena K., Jolly M.K., Balamurugan K. Hypoxia, partial EMT and collective migration: emerging culprits in metastasis. Tranl. Oncol. 2020;13:100845. doi: 10.1016/j.tranon.2020.100845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliekelman M.J., Taguchi A., Zhu J., Dai X., Rodriguez J., Celiktas M., Zhang Q., Chin A., Wong C.-H., Wang H. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015;75:1789–1800. doi: 10.1158/0008-5472.CAN-14-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M., de Mattos S.F., van der Horst A., Klompmaker R., Kops G.J.L., Lam E.W.-F., Burgering B.M., Medema R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo B.R., Bhardwaj P., Choi S., Gonzalez J., Andresen Eguiluz R.C., Wang K., Mohanan S., Morris P.G., Du B., Zhou X.K. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015;7:301ra130. doi: 10.1126/scitranslmed.3010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan T., Chen S., Chen X., Lin W.R., Li W., Ma J., Wu T., Ji H., Li Y., Cui X. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int. J. Oncol. 2017;50:121–128. doi: 10.3892/ijo.2016.3779. [DOI] [PubMed] [Google Scholar]
- Shellard A., Mayor R. Supracellular migration - beyond collective cell migration. J. Cell Sci. 2019;132:jcs226142. doi: 10.1242/jcs.226142. [DOI] [PubMed] [Google Scholar]
- Shellard A., Szabo A., Trepat X., Mayor R. Supracellular contraction at the rear of neural crest cell groups drives collective chemotaxis. Science. 2018;362:339–343. doi: 10.1126/science.aau3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S., Buel G.R., Nagiec M.J., Han M.-J., Roux P.P., Blenis J., Yoon S.-O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. U S A. 2019;116:2967–2976. doi: 10.1073/pnas.1811923116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva-Diz V., Lorenzo-Sanz L., Bernat-Peguera A., Lopez-Cerda M., Muñoz P. Cancer cell plasticity: impact on tumor progression and therapy response. Semin. Cancer Biol. 2018;53:48–58. doi: 10.1016/j.semcancer.2018.08.009. [DOI] [PubMed] [Google Scholar]
- Singh M., Close D.A., Mukundan S., Johnston P.A., Sant S. Production of uniform 3D microtumors in hydrogel microwell Arrays for measurement of viability, morphology, and signaling pathway activation. Assay Drug Dev. Technol. 2015;13:570–583. doi: 10.1089/adt.2015.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Mukundan S., Jaramillo M., Oesterreich S., Sant S. Three-dimensional breast cancer models mimic hallmarks of size-induced tumor progression. Cancer Res. 2016;76:3732–3743. doi: 10.1158/0008-5472.CAN-15-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Venkata Krishnan H., Ranganathan S., Kiesel B., Beumer J.H., Sreekumar S., Sant S. Controlled three-dimensional tumor microenvironments recapitulate phenotypic features and differential drug response in early vs advanced stage breast cancer. ACS Biomater. Sci. Eng. 2018;4:421–431. doi: 10.1021/acsbiomaterials.7b00081. [DOI] [PubMed] [Google Scholar]
- Singh M., Warita K., Warita T., Faeder J.R., Lee R.E.C., Sant S., Oltvai Z.N. Shift from stochastic to spatially-ordered expression of serine-glycine synthesis enzymes in 3D microtumors. Sci. Rep. 2018;8:9388. doi: 10.1038/s41598-018-27266-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Tian X.-J., Donnenberg V.S., Watson A.M., Zhang J., Stabile L.P., Watkins S.C., Xing J., Sant S. Targeting the temporal dynamics of hypoxia-induced tumor-secreted factors halts tumor migration. Cancer Res. 2019;79:2962–2977. doi: 10.1158/0008-5472.CAN-18-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianou N., Lehman M.L., Wang C., Fard A.T., Rockstroh A., Fazli L., Jovanovic L., Ward M., Sadowski M.C., Kashyap A.S. A molecular portrait of epithelial–mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene. 2019;38:913–934. doi: 10.1038/s41388-018-0488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson S., Nilsson K., Ringberg A., Landberg G. Invade or proliferate? Two contrasting events in malignant behavior governed by p16INK4a and an intact Rb pathway illustrated by a model system of basal cell carcinoma. Cancer Res. 2003;63:1737–1742. [PubMed] [Google Scholar]
- Teeuwssen M., Fodde R. Cell heterogeneity and phenotypic plasticity in metastasis formation: the case of colon cancer. Cancers (Basel) 2019;11:1368. doi: 10.3390/cancers11091368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson T.M., Balcells C., Cascante M. Metabolic plasticity and epithelial-mesenchymal transition. J. Clin. Med. 2019;8:967. doi: 10.3390/jcm8070967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalovski M., Hagopian M., Wang M., Brekken R.A. Hypoxia and transforming growth factor beta cooperate to induce fibulin-5 expression in pancreatic cancer. J. Biol. Chem. 2016;291:22244–22252. doi: 10.1074/jbc.M116.730945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran D.D., Corsa C.A., Biswas H., Aft R.L., Longmore G.D. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol. Cancer Res. 2011;9:1644–1657. doi: 10.1158/1541-7786.MCR-11-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S., Chakraborty P., Levine H., Jolly M.K. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLoS Comput. Biol. 2020;16:e1007619. doi: 10.1371/journal.pcbi.1007619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhaya P., Barhoi D., Giri A., Bhattacharjee A., Giri S. Joint detection of claudin-1 and junctional adhesion molecule-A as a therapeutic target in oral epithelial dysplasia and oral squamous cell carcinoma. J. Cell. Biochem. 2019;120:18117–18127. doi: 10.1002/jcb.29115. [DOI] [PubMed] [Google Scholar]
- Vega S., Morales A.V., Ocana O.H., Valdes F., Fabregat I., Nieto M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venhuizen J.H., Zegers M.M. Making heads or tails of it: cell-cell adhesion in cellular and supracellular polarity in collective migration. Cold Spring Harb. Perspect. Biol. 2017;9:a027854. doi: 10.1101/cshperspect.a027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva-Duque A., Zuniga-Eulogio M.D., Dena-Beltran J., Castaneda-Saucedo E., Calixto-Galvez M., Mendoza-Catalan M.A., Ortuno-Pineda C., Navarro-Tito N. Leptin induces partial epithelial-mesenchymal transition in a FAK-ERK dependent pathway in MCF10A mammary non-tumorigenic cells. Int. J. Clin. Exp. Pathol. 2017;10:10334–10342. [PMC free article] [PubMed] [Google Scholar]
- Vogel C.L., Cobleigh M.A., Tripathy D., Gutheil J.C., Harris L.N., Fehrenbacher L., Slamon D.J., Murphy M., Novotny W.F., Burchmore M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- Wang W., Goswami S., Lapidus K., Wells A.L., Wyckoff J.B., Sahai E., Singer R.H., Segall J.E., Condeelis J.S. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- Wang S.J., Chao D., Wei W., Nan G., Li J.Y., Liu F.L., Li L., Jiang J.L., Cui H.Y., Chen Z.N. CD147 promotes collective invasion through cathepsin B in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020;39:145. doi: 10.1186/s13046-020-01647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Ginther C., Kim J., Mosher N., Chung S., Slamon D., Vadgama J.V. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012;10:1597–1606. doi: 10.1158/1541-7786.MCR-12-0155-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.S., Li Z.F., Wang H.F., Yu X.H., Pang X., Wu J.B., Wang S.S., Zhang M., Yang X., Cao M.X. Cathepsin B defines leader cells during the collective invasion of salivary adenoid cystic carcinoma. Int. J. Oncol. 2019;54:1233–1244. doi: 10.3892/ijo.2019.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J., Tian X.J. Investigating epithelial-to-mesenchymal transition with integrated computational and experimental approaches. Phys. Biol. 2019;16:031001. doi: 10.1088/1478-3975/ab0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagasaki R., Noguchi M., Minami M., Earashi M. Clinical significance of E-cadherin and vimentin co-expression in breast cancer. Int. J. Oncol. 1996;9:755–761. doi: 10.3892/ijo.9.4.755. [DOI] [PubMed] [Google Scholar]
- Yamada K.M., Sixt M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019;20:738–752. doi: 10.1038/s41580-019-0172-9. [DOI] [PubMed] [Google Scholar]
- Yamashita N., Tokunaga E., Iimori M., Inoue Y., Tanaka K., Kitao H., Saeki H., Oki E., Maehara Y. Epithelial paradox: clinical significance of coexpression of E-cadherin and vimentin with regard to invasion and metastasis of breast cancer. Clin. Breast Cancer. 2018;18:e1003–e1009. doi: 10.1016/j.clbc.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Yang C., Cao M., Liu Y., He Y., Du Y., Zhang G., Gao F. Inducible formation of leader cells driven by CD44 switching gives rise to collective invasion and metastases in luminal breast carcinomas. Oncogene. 2019;38:7113–7132. doi: 10.1038/s41388-019-0899-y. [DOI] [PubMed] [Google Scholar]
- Yang Y., Zheng H., Zhan Y., Fan S. An emerging tumor invasion mechanism about the collective cell migration. Am. J. Transl. Res. 2019;11:5301–5312. [PMC free article] [PubMed] [Google Scholar]
- Yang J., Antin P., Berx G., Blanpain C., Brabletz T., Bronner M., Campbell K., Cano A., Casanova J., Christofori G. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020;21:341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S., Miwa S., Mii S., Hiroshima Y., Uehara F., Yamamoto M., Kishimoto H., Tazawa H., Bouvet M., Fujiwara T. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle. 2014;13:953–960. doi: 10.4161/cc.27818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X., Weinberg R.A. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida G.J. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020;39:112. doi: 10.1186/s13046-020-01611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida G.J., Azuma A., Miura Y., Orimo A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 2019;20:2256. doi: 10.3390/ijms20092256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Bardia A., Wittner B.S., Stott S.L., Smas M.E., Ting D.T., Isakoff S.J., Ciciliano J.C., Wells M.N., Shah A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias M., Brcic L., Eidenhammer S., Popper H. Bulk tumour cell migration in lung carcinomas might be more common than epithelial-mesenchymal transition and be differently regulated. BMC Cancer. 2018;18:717. doi: 10.1186/s12885-018-4640-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Weinberg R.A. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. Med. 2018;12:361–373. doi: 10.1007/s11684-018-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., He R., Chen S., Zhang L., Cao G., Yang W., Li J. The JAM-B/c-src/MMP9 pathway is associated with progression and regulates the invasion of pancreatic cancer. J. Cancer. 2020;11:3246–3255. doi: 10.7150/jca.40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeller E.L., Pedro B., Konen J., Dwivedi B., Rupji M., Sundararaman N., Wang L., Horton J.R., Zhong C., Barwick B.G. Genetic heterogeneity within collective invasion packs drives leader and follower cell phenotypes. J. Cell Sci. 2019;132:jcs231514. doi: 10.1242/jcs.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.