

Abstract
Cancer burden has been increasing worldwide, making cancer the second leading cause of death in the world. Over the past decades, various experimental models have provided important insights into the nature of cancer. Among them, the fruit fly Drosophila as a whole‐animal toolkit has made a decisive contribution to our understanding of fundamental mechanisms of cancer development including loss of cell polarity. In recent years, scalable Drosophila platforms have proven useful also in developing anti‐cancer regimens that are effective not only in mammalian models but also in patients. Here, we review studies using Drosophila as a tool to advance cancer study by complementing other traditional research systems.
Keywords: anti‐cancer drug, cancer, Drosophila, genetics, whole‐body platform
The manuscript is an overview on contributions that the fruit fly Drosophila has made thus far to advance cancer research. We aim to introduce useful applications of flies to scrutinize cancer mechanisms, model cancer patients, and accelerate drug discovery, which have become possible due to the accessible genetic and pharmacological toolkits that Drosophila offers.

Abbreviations
- A5
a sorafenib analog APS5‐16‐2; A6, a sorafenib analog APS6‐45
- btl
breathless
- CRC
colorectal cancer
- dEGFRλ
a constitutively active variant of Drosophila Egfr
- DFG
Asp‐Phe‐Gly
- DOHaD
Developmental Origins of Health and Disease
- dp110CAAX
a membrane‐targeted active Drosophila p110
- dRetM955T
an active M955T isoform of Drosophila Ret
- EMT
epithelial‐mesenchymal transition
- GBM
glioblastoma multiforme
- GEMM
genetically engineered mouse model
- GMR
Glass multiple reporter
- HDS
high dietary sugar
- KI
kinase inhibitor
- lgl
lethal giant larvae
- M
Minute
- MTC
medullary thyroid cancer
- NSCLC
non‐small cell lung cancer
- PTC
papillary thyroid cancer
- ptc
patched
- RNAi
RNA interference
- RTK
receptor tyrosine kinase
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- TC
thyroid cancer
- TCGA
The Cancer Genome Atlas
- UAS
Upstream Activation Sequence
1. INTRODUCTION
Thus far, experimental models including cancer cells and genetically engineered mouse models (GEMMs) have made significant contributions to advance cancer research. 1 , 2 Recently, in addition to these ‘traditional’ platforms, novel animal models have attracted much attention for their capacity to promote the field, such as zebrafish, the nematode Caenorhabditis elegans, and the fruit fly Drosophila.
We are interested in introducing particularly Drosophila in this review because they offer several advantages complementary to other model organisms in cancer research. Firstly, flies have conserved genes and signaling pathways with humans. Specifically, more than 70% of human genes whose abnormalities cause diseases have a functional ortholog in flies. 3 Secondly, Drosophila offers a powerful genetic toolkit including gene‐knockout and transgenic stocks. As we describe later, we can also generate flies with complex genotypes due to its advanced reverse genetics. Thirdly, Drosophila matures quickly and is highly reproductive without a need for massive lab equipment. For instance, their generation time is only 11‐12 d at 25°C, and an adult female can lay 400‐500 embryos within 10 d in laboratory vials. 4 These and other valuable features allow us to study fundamental interaction between genes, as well as distinct cells or tissues in not only developmental but also medical biology such as cancer modeling and drug discovery through complimentary use of Drosophila with mammals as we introduce below 4 , 5 (Figure 1).
FIGURE 1.
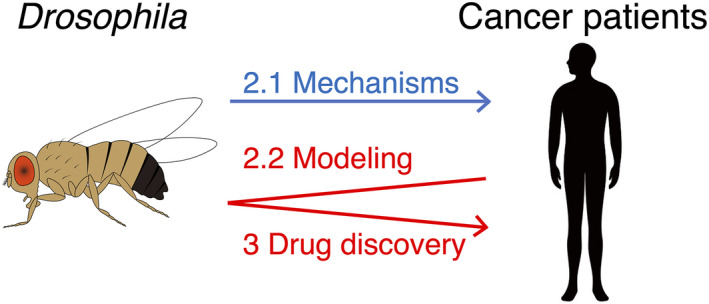
Elucidating tumorigenic mechanisms and developing new cancer treatments using Drosophila. Forward genetics revealed key tumorigenic mechanisms in Drosophila (blue arrow; Section 2.1). Conversely, reverse genetics generated various fly models mirroring patient genotypes (red arrow; Section 2.2). Following drug screening/derivatization in these models identified new therapeutic candidates (red arrow; Section 3)
2. STUDYING CANCER BIOLOGY WITH FLIES
Cancer originates as a localized disease but it can affect the whole body also, therefore we need whole‐body models to understand the mechanisms of its pathogenesis and to develop effective drugs with satisfactory therapeutic index. As one of such models, Drosophila has shown its value as genetic and pharmacological toolkits. Namely, their forward genetics allows phenotyping within or between tissues upon naturally occurring mutations while their reverse genetics enables modeling genetic alterations found in patients, which allows animal‐level drug explorations for specific genotypes. In this section, we describe cancer biology and candidate therapeutics that Drosophila studies have revealed.
2.1. Elucidating cancer mechanisms with flies
Early studies in the 1930s identified mutant Drosophila for the gene lethal giant larvae (lgl) to manifest gross disorganization and hyper‐proliferation of larval tissues including the brain and imaginal discs. 6 Upon transplantation into wild‐type hosts, lgl mutant cells invaded their surroundings to colonize. 6 Following genetic analyses in Drosophila pinpointed dlg and scrib to interact with lgl to regulate cell polarity whose loss occurs in ~80% of human cancers. 7 Convincingly, expression levels of their human orthologs are lower in several types of cancer compared with those in their normal counterparts. 4 , 8 These results collectively suggest functional conservation of lgl, dlg, and scrib as tumor suppressors across species.
Additionally, Drosophila studies uncovered a process ‘cell competition’ to eliminate cells with distinct characteristics. The first example came from flies carrying Minute (M), a mutant allele for a ribosomal gene. When genetic manipulation induced clones with M heterozygosity within wild‐type wing discs consisting of epithelial monolayers, apoptosis eliminated these clones keeping wing size and shape normal. 9 , 10 Curiously, cancer‐related genes also have key roles in cell competition. As a ‘supercompetitor,’ a cell overexpressing Myc kills surrounding wild‐type cells in developing wings. 11 , 12 Similarly, supercompetition occurs due to a variety of genetic abnormalities in Hippo, WNT/Wg, and JAK‐STAT pathways, suggesting a role for supercompetitor as a tumor seed. 13 , 14 , 15
Conversely, cell competition has an anti‐tumor role in different contexts. Namely, wild‐type cells eliminate a small population of oncogenic cells lacking lgl, dlg, or scrib, or those harboring SRC activation. 16 , 17 , 18 , 19 , 20 Besides, some of lgl mutant alleles cause proliferation rather than cell death (eg, a cleaned‐up allele of lgl4 or alleles of lgl27S3, lglE2S31, lgl23S9, or lglE6S), 21 , 22 indicating that lgl alleles cause distinct phenotypes. In addition to such genetic alterations, environmental factors also affect cell competition. For example, systemic hyperinsulinemia disturbs elimination of scrib mutant cells and promotes tumorigenesis in Drosophila. 23
Like Drosophila, mammals also execute cell competition. For instance, a non‐transformed epithelial monolayer in culture excludes apically a small population of cells expressing oncogenic RAS or SRC. 24 , 25 Also in mice, normal tissues eliminate cells with decreased Myc expression, or mutations in either a ribosomal protein, a cell polarity regulator, or Hippo pathway. 26 , 27 , 28 , 29 These pieces of evidence raise a fascinating possibility that cell competition works as an intrinsic mechanism preventing carcinogenesis.
2.2. Fly models for specific cancer types
Reverse genetics has allowed establishing Drosophila modeling cancer genotypes. One of the oldest and simplest methods to induce transgene artificially is to utilize a heat shock promoter by putting transgenic flies in a warm incubator. 30 However, heat shock induces transgene throughout the body, which can cause developmental abnormality. Also, there exists leak of transgene expression even without heat shock. 31
Complementing this tool, the GAL4/UAS system proved useful 31 (Figure 2). In principle, this method employs the yeast transcription factor GAL4 driven by cell type‐ or tissue‐specific enhancer/promoter and its target UAS integrated in the fly genome to allow spatial and/or temporal transgene regulation. 31 These and other powerful genetic tools make Drosophila an accessible tool to accelerate cancer research as we introduce in this section and Table 1.
FIGURE 2.
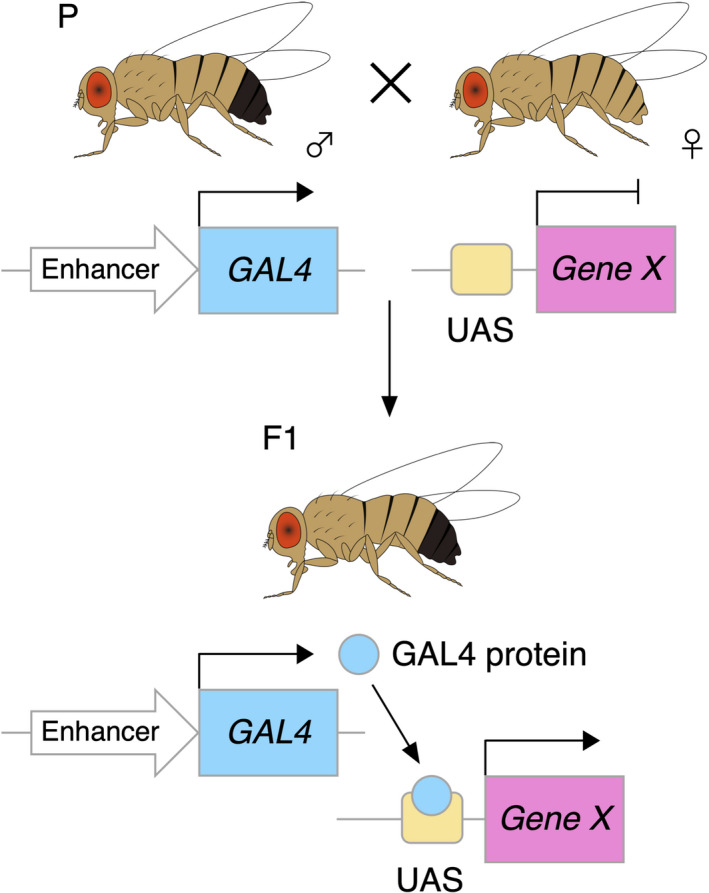
The GAL4/UAS system for regulating transgene in Drosophila. This system consists of 2 parts: the yeast transcription factor GAL4, driven by cell type‐specific or tissue‐specific enhancer/promoter, and its target UAS. Crossing parent flies carrying either Enhancer‐GAL4 or UAS‐X produces F1 offspring with X induction specifically in cells or tissues of interest
TABLE 1.
Models and findings in cancer studies using Drosophila
| Cancer type | Patient genotype | Drosophila genotype | Drosophila phenotype | Therapeutic candidate | Description (Ref.) | |
|---|---|---|---|---|---|---|
| Thyroid cancer (TC) | Medullary TC (MTC) | RETM918T | GMR‐dRetM955T | ‘Rough eye’ by cell proliferation | Vandetanib as the first targeted therapy for MTC | Section 2.2.1 36 |
| ptc>dRetM955T | Proliferation and migration of transformed cells, and fly lethality | A novel kinase inhibitor (KI) drug lead AD80 | Section 2.2.1 33 | |||
| A novel KI drug lead A6 (APS6‐45) with improved therapeutic index in mouse xenografts of human MTC compared with its parent multikinase inhibitor drug sorafenib or cabozantinib, another FDA‐approved KI drug for MTC | Section 3.1 34 | |||||
| Papillary TC (PTC) |
CCDC6‐RET NCOA4‐RET |
ptc>CCDC6‐RET ptc>NCOA4‐RET |
Cell migration, delamination and EMT inducing fly lethality, with NCOA4‐RET causing more severe phenotypes than CCDC6‐RET | A combination of sorafenib and the WEE1 inhibitor AZD1775 | Section 2.2.1 38 | |
| Colorectal cancer (CRC) | KRASG12V, losses of TP53, PTEN, APC, and/or SMAD4 | byn>rasG12V,p53RNAi,ptenRNAi,apcRNAi, and/or smad4RNAi | Proliferation, EMT and/or distant metastasis of transformed cells in 32 multigenic flies, with rasG12V,p53RNAi,ptenRNAi,apcRNAi causing the most severe phenotypes | A two‐step therapy with the proteasome inhibitor bortezomib followed by the PI3K/mTOR inhibitor BEZ235 | Section 2.2.2 50 | |
| KRASG13A, biallelic losses of APC, TP53, and FBXW7, heterozygous mutations of SMARCA4, FAT4, MAPK14, and CDH1 (a refractory CRC patient) | byn>rasG12V,apcRNAi,p53RNAi,agoRNAi,putRNAi,brmRNAi,ftRNAi,p38aRNAi,shgRNAi | Expansion of the hindgut | A personalized combination therapy between the MEK inhibitor drug trametinib and the calcium metabolism modifier drug zoledronate being effective in the patient | Section 3.2 73 | ||
| Non‐small‐cell lung cancer (NSCLC) | KRASG12V, loss of PTEN | btl>rasG12V,ptenRNAi | Tracheal tissue proliferation and fly lethality | A combination of trametinib and the HMG‐CoA reductase inhibitor drug fluvastatin | Section 2.2.3 55 | |
| KIF5B‐RET | ptc>KIF5B‐RET | Proliferation, invasion and EMT of transformed cells harboring invadopodium‐like processes | A combination of sorafenib and the EGFR inhibitor drug erlotinib or the microtubule inhibitor drug paclitaxel | Section 2.2.3 56 | ||
| Glioblastoma multiforme (GBM) | Activation mutation or amplification of EGFR, activation mutation of PIK3CA | repo>dEGFRλ,dp110CAAX | Neoplastic, transplantable glial cells | mTOR, MYC, CCNG1‐CDKs and RB‐E2F pathways as candidate therapeutic targets | Section 2.2.4 58 | |
Genetic engineering has generated Drosophila stocks modeling human cancer genotypes, which accelerated identification of disease mechanisms and therapeutic candidates.
2.2.1. Thyroid cancer models
The case number of TC is increasing dramatically worldwide. In the United States, for example, there is a prediction that TC becomes the fourth most common type of cancer by 2030 replacing colorectal cancer (CRC), making it one of the most pressing health challenges. 32 TC subtypes include papillary TC (PTC) and relatively rare medullary TC (MTC). An active form of cell surface RTK RET is responsible for 90% < of MTC cases, but drug discovery for MTC treatment has been slow largely due to the lack of an efficient research platform.
To tackle this issue, we generated transgenic Drosophila models for MTC by inducing in epithelial tissues including eyes and wing discs an active M955T isoform of Drosophila Ret (dRetM955T) mimicking RETM918T in MTC patients 33 , 34 , 35 , 36 (Table 1). They served to validate the lead chemical ZD6474 to generate vandetanib as the first targeted therapy for MTC. 4 Furthermore, they made intensive chemical genetic screening possible, successfully generating novel lead compounds with much improved efficacy over sorafenib, the Food and Drug Administration (FDA)‐approved multikinase inhibitor drug 34 (described later).
In contrast to MTC, PTC accounts for ~85% of all TC cases. 37 PTC has subtypes with different genetic profiles for effectors in the RTK‐MAPK pathway including oncogenic RET fusion genes in 30% of PTC patients. 38 Although RET inhibitors show efficacy in this population, they also cause severe toxicity. 39 Among CCDC6‐RET and NCOA4‐RET fusions identified, the latter causes in patients more severe pathogenesis with undetermined mechanisms and therapeutics. 40
As with MTC, Drosophila became a powerful tool to tackle this cancer. Namely, flies expressing CCDC6‐RET or NCOA4‐RET driven by the patched (ptc) promoter displayed enhanced migration, delamination, and EMT of transformed cells. 38 In these fly models, the ptc promoter directs transgene expression in developing epithelia including wing, eye, and leg discs and other tissues. 33 Full kinome screening indicated that NCOA4‐RET signaled through kinases including WEE1 distinct from CCDC6‐RET. Targeting this NCOA4‐RET–WEE1 network by combining sorafenib with the WEE1 inhibitor AZD1775 suppressed above phenotypes, raising a novel candidate therapy for NCOA4‐RET–positive PTC 38 (Table 1).
2.2.2. Colorectal cancer models
CRC has the third highest incidence of cancer in both genders globally, with ~1.8 million new cases and 880,000 deaths in 2018, making it as the cancer type with the second highest mortality rate. 41
CRC harbors combinations of genetic abnormalities in RAS oncogenes (KRAS/NRAS/HRAS) and/or in tumor suppressor genes such as APC, TP53, SMAD4, and LLGL1. 42 To understand how such diversities affect CRC development, GEMMs for intestinal tumors have made pivotal contributions. 43 For example, we discovered CRC mechanisms including tumor‐promoting PGE2‐EP2 and NOTCH‐ABL‐TRIO‐RHO pathways, as well as the invasion/metastasis‐suppressing Aes gene to inhibit NOTCH signaling. 44 , 45 , 46 , 47
Unfortunately, GEMMs with complex genotypes require enormous efforts to generate and maintain. 48 Here, Drosophila CRC models proved to be complementary to mammals in scrutinizing quickly the CRC complexity regarding disease mechanisms and drug responses. To model CRC genotypes in flies, Bangi et al employed byn‐GAL4 active in the hindgut corresponding to the human colon 49 (Figure 3) as well as patient genomic data from TCGA. 50 Active rasG12V combined with RNA interference (RNAi) knockdown of tumor suppressors p53, pten, apc and/or smad4 recapitulated major CRC pathologies including cell proliferation, EMT and distant metastasis, with rasG12V,p53RNAi,ptenRNAi,apcRNAi causing the most severe phenotypes. Furthermore, each fly line showed distinct responses to anti‐cancer reagents, highlighting the importance of personalized medicine based on patient genotypes. 50
FIGURE 3.
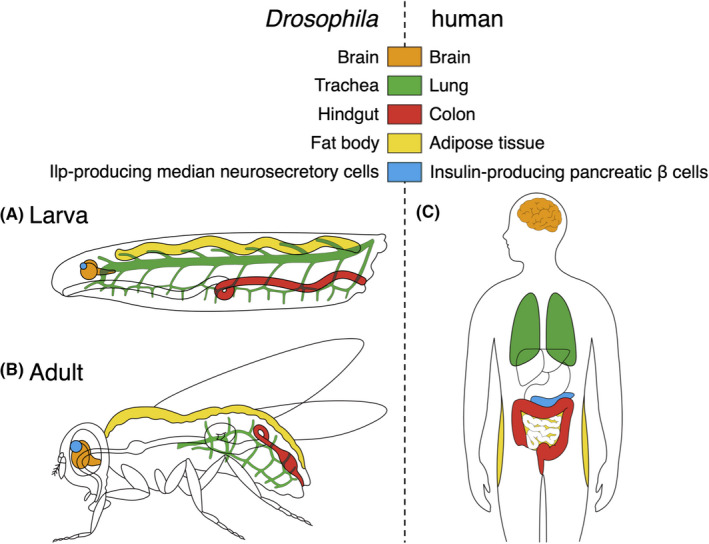
Tissue/organ similarity between Drosophila and human. Each color indicates functionally similar tissues/organs among Drosophila larva (A) and adult (B), and human (C). These similarities allow construction of fly models for human diseases including cancer affecting tissue/organ functions
Also, the authors identified ras activation and pten loss to inhibit mTORC1 as a mechanism for CRC resistance to PI3K/mTOR inhibitors such as BEZ235. Based on this finding, they found significant suppression of cancer traits in cultured human CRC cells, their xenografts, and Apc;Kras;Pten allograft from a CRC GEMM by treating them first with the mTORC1 activator SC79 or bortezomib followed by BEZ235 (Table 1). This study provided a rapid, large‐scale platform by combining flies with patient databases and mammalian models (also see Section 3.2).
In addition to exploring cancer complexity, Drosophila also serves as a quick platform to test hypotheses from epidemiological studies. Recently, a study demonstrated an association of social isolation with increased risk of cancer death, and indeed rats developed mammary tumors upon lifelong (≤ 18 mo) isolation. 51 Interestingly also in flies, social isolation accelerated progression of their gut tumors within 21 d, 52 highlighting their usefulness in studies on risk factors requiring long‐term observation when using mammals.
2.2.3. Lung cancer models
Globally, lung cancer has long had the highest mortality rate among all cancer types, with NSCLC accounting for 85% of all lung cancer diagnosis. 53 As the most commonly mutated oncogene in NSCLC, KRAS renders it resistance against adjuvant chemotherapy and EGFR inhibitors. 54
In raising therapeutic candidates for this KRAS‐positive NSCLC, Drosophila provided as a test tissue its tracheal system which develops similarly to the vertebrate lung (Figure 3). The breathless (btl)‐GAL4 targeted Drosophila rasG12V misexpression and pten knockdown to the trachea causing tumor‐like growths and lethality in early larval stages. 55 Following chemical screening for a library of 1192 FDA‐approved drugs identified the MEK inhibitor drug trametinib and the HMG‐CoA reductase inhibitor drug fluvastatin as candidates to generate a therapeutic cocktail. Indeed, they synergistically inhibited growth of A549 human NSCLC cells carrying active KRASG12S 55 (Table 1).
Drosophila also helped to create novel therapeutic strategies for patients with the KIF5B‐RET fusion oncogene, the most relevant fusion driver in NSCLC. 56 Namely, its product KIF5B‐RET activated multiple RTKs including EGFR to offer vulnerabilities to target by combinations of sorafenib with erlotinib or paclitaxel as candidate therapies for KIF5B‐RET‐positive NSCLC, which awaits validation in patients (Table 1).
2.2.4. Brain tumor models
Gliomas are the most common intracerebral tumors, with GBM as the most aggressive tumor with few effective therapies hence median patient survival being only 15 mo. Studies using GEMMs for GBM have revealed the mechanisms of its development and therapeutic resistance including EGFR‐PI3K signaling, but generating novel therapeutic strategies have remained extremely difficult for decades. 57
To overcome this situation, Read et al. established flies modeling GBM genotype by expressing activated isoforms of Drosophila Egfr (dEGFRλ) and p110 (dp110CAAX) using glia‐specific repo‐GAL4. 58 These transgenes induced glial proliferation, infiltration, and loss of cell polarity recapitulating human glioma to cause larval lethality. 58 These phenotypes were dependent on TOR, MYC, CCNG1‐CDKs and RB‐E2F pathways, suggesting them as novel targets for GBM therapy (Table 1). Therefore, flies offer a feasible platform to clarify signaling networks in cancer development.
2.2.5. Impact of metabolic disorders on cancers
Obesity and type 2 diabetes mellitus (T2DM) are representative metabolic disorders whose prevalence continues to rise worldwide. 59 Notably they are both risk factors established already for all cancer types, thus elucidating their pathogenesis can give clues for cancer prevention. 59
To this end, mouse, rat, and Drosophila models for obesity and T2DM have played substantial roles. Especially, feeding flies with HDS can rapidly generate a dietary‐induced obese/T2DM model. 60 Taking this advantage, Hirabayashi et al. unveiled that HDS enhanced tumor growth leading to emergent metastases in flies with increased RAS and SRC activity (rasG12V,csk‐/‐). 61 Curiously, WNT ortholog Wg upregulated insulin receptors to avoid insulin resistance, boosting tumor growth. Therapeutically, the authors successfully delineated anti‐tumor efficacy in these flies of a combination between 3 drugs; the T2DM drug acarbose, the WNT signaling inhibitor pyrvinium, and the RAS/SRC/mTOR signaling inhibitor AD81 of their own. 61 , 62
In addition to the T2DM studies described in this section, Drosophila can help studies also on type 1 diabetes mellitus (T1DM) due to depletion of insulin‐producing pancreatic β cells. 63 In flies, complete or partial ablation of insulin‐like peptides causes elevated sugar concentrations in larval hemolymph to induce T1DM‐like phenotypes, causing developmental delay 64 (Figure 3). To the best of our knowledge, no papers have examined association between T1DM and cancer using these flies. However, Drosophila can implement a valuable strategy to reveal causal relationships between cancers and these 2 DMs and even DOHaD, which all develop through complex interplay between genetic and acquired factors. 65
3. DRUG DISCOVERY WITH FLIES
In this section, we introduce 2 studies using Drosophila as a platform for chemical biology combined with genetics and as a ‘patient avatar’ to develop novel therapeutics. Thus far, mammalian models have discovered cancer mechanisms as partly described above. However, in drug discovery, cultured cells cannot fully recapitulate all aspects of cancer development and therapy such as inter‐cellular/organ interactions and pharmacokinetics. Also, GEMMs require enormous effort such as time and cost to evaluate novel therapeutic candidates. As a result, the success rate of clinical trials for cancer drug candidates has remained below 4%, demanding a paradigm shift in drug development pipeline. 66 Studies below demonstrate that Drosophila serves as a whole‐animal platform to advance the field.
3.1. Creating new drug leads through rational balancing of polypharmacology
To treat cancer, chemotherapy has been one of the primary options for decades. However, chemotherapy elicits significant toxicity such as bone marrow suppression in patients, which often hampers therapeutic protocols.
To overcome this issue, the idea of targeted therapy emerged 3 decades ago aiming to target molecules present primarily in cancer tissues to reduce systemic side effects. 67 Among such molecules, kinases turned out to be fascinating targets, because cancer genomes frequently carry their alterations that cause their deregulation hence cell transformation. Due to these and other reasons, generating KI drugs has been one of the most active areas in drug development. 68 Indeed, the US FDA has approved so far 70 < KIs including ABL inhibitor imatinib with tremendous effects in patients with ABL‐positive chronic myeloid leukemia. Unfortunately, however, even approved KI drugs frequently cause unacceptable adverse effects in patients. For example, the multikinase inhibitor drug sorafenib has shown promise in patients with liver or kidney cancer or MTC as well as in a Drosophila MTC model ptc>dRetM955T where the ptc promoter drives expression of dRetM955T (Figure 4A), but sorafenib causes new tumors or even fatal toxicity in patients. 69
FIGURE 4.
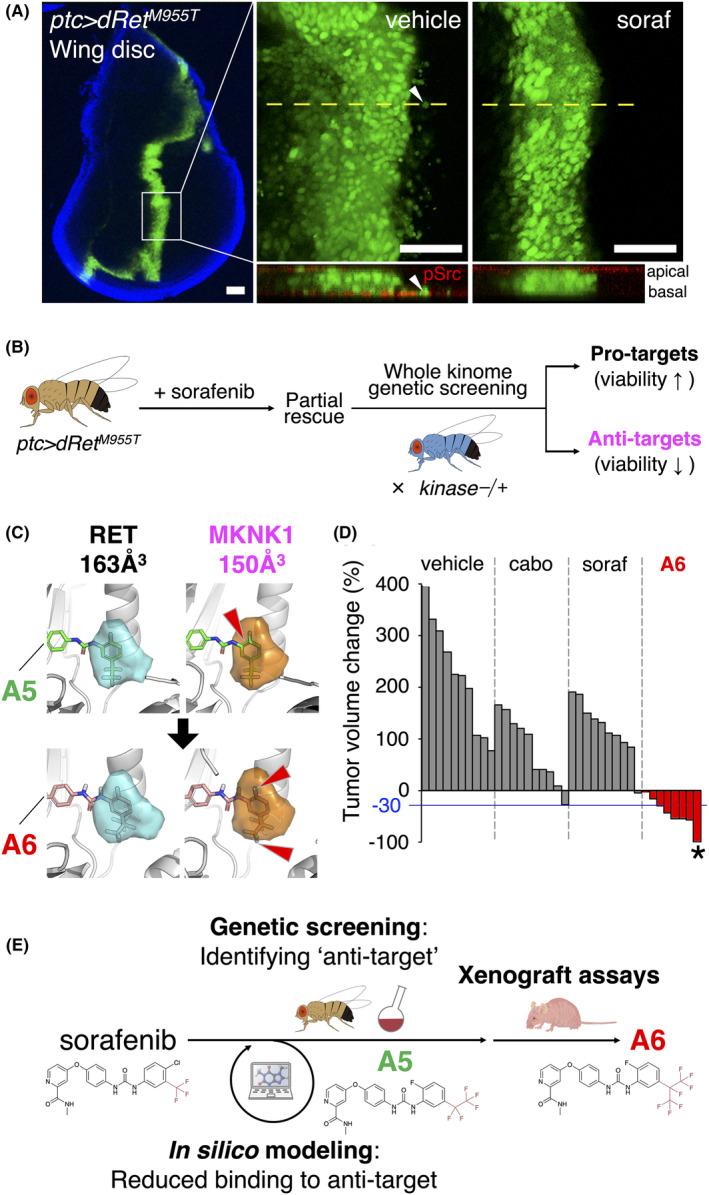
A whole‐animal platform to create novel drug leads through rational balancing of polypharmacology. A, In vivo cell migration assay using a Drosophila model for MTC. In these ptc>dRetM955T flies, the patched (ptc) promoter drives expression of an M955T isoform of Drosophila Ret (dRetM955T) modeling RETM918T mutation in MTC patients. Left panel, a larval wing disc harboring GFP‐labeled, dRetM955T‐expressing transformed cells among wild‐type cells (black). Blue, DAPI staining outlining the disc margin. Middle top and right top, basal images in DMSO vehicle (middle top; inset box in left panel) or the KI drug sorafenib (soraf)‐treated flies (right top; 400 μmol/L in fly food), respectively. Bottom, confocal z‐stack images derived from the plane indicated by dotted lines in top panels. Arrowhead, example of basally migrating cell expressing phospho(p)‐Src (red). Scale bars, 50 μm. B, Chemical genetic screening identified ‘anti‐target’ kinases whose inhibition by sorafenib accounted for its toxicity. We introduced a heterozygous mutation of each kinase into ptc>dRetM955T flies and fed sorafenib to their offspring. As a result, heterozygosity of 9 genes including a sorafenib target MKNK1 reduced significantly the efficacy of sorafenib decreasing fly viability. Besides, we identified ‘pro‐target’ kinases whose heterozygous mutation enhanced the efficacy of sorafenib increasing fly viability. C, In silico prediction of a sorafenib derivative A6 (APS6‐45) as a less potent binder compared with another derivative A5 (APS5‐16‐2) to MKNK1, an anti‐target of sorafenib. Compared with sorafenib (not shown) and A5 (top), A6 with a larger modification group (‐isoC3F7) has reduced binding capacity to the DFG pocket in MKNK1 due to steric clashes (arrowhead), although it can still bind to RET with larger pocket size compared with MKNK1. D, A6 suppresses human MTC xenografts. A 4‐wk oral administration of A6 reduced the volume of existing tumors more effectively than its seed sorafenib (soraf) or cabozantinib (cabo), another standard of care for MTC treatment. Each bar represents tumor volume change in 1 mouse. Asterisk, complete remission. E, Stepwise evolution of sorafenib. First, we performed genetic screening in Drosophila for all of 252 kinases in the entire kinome in the presence of sorafenib to pinpoint MKNK1 as an anti‐target of sorafenib. A sorafenib derivative A5 with predictable reduced binding capacity to MKNK1 showed higher efficacy than sorafenib in ptc>dRetM955T flies. We further inflated A5 to generate A6, achieving reduced toxicity hence improved therapeutic index in human MTC xenografts
Therefore, we intended to create an alternative approach to generate novel anti‐cancer drugs: to improve an existing KI drug, we evolved it toward a unique network of kinase targets to lower its toxicity through complementary use of Drosophila with mammals. Picking MTC and sorafenib as models of cancer type and KI drug, respectively, first we performed chemical genetic screening in ptc>dRetM955T flies for all of 252 kinases in the entire kinome in the presence of sorafenib. Introducing a heterozygous mutation of each kinase into ptc>dRetM955T flies and feeding sorafenib to them discovered that heterozygosity of 9 genes significantly reduced the efficacy of sorafenib decreasing fly viability. Therefore, we defined these genes as ‘anti‐targets’ of sorafenib whose inhibition by sorafenib accounted for sorafenib toxicity (Figure 4B). Among these anti‐targets, we especially focused on a sorafenib target MKNK1, as its inhibition diminished sorafenib efficacy completely and we successfully modeled in silico the interaction of its allosteric DFG pocket with sorafenib based on ‘DFGmodel’ platform that we had reported. 70
In comparison of sorafenib binding to MKNK1 or RET, in silico modeling uncovered ~10% smaller size of the MKNK1 allosteric pocket compared with that in RET, indicating that ‘inflated’ sorafenib would cause steric clash with MKNK1. Convincingly, sorafenib derivatives A5 (APS5‐16‐2) and A6 (APS6‐45) with enlarged warhead showed tremendously reduced toxicity hence improved efficacy as compared to parent drug sorafenib and cabozantinib, another KI drug for MTC, in human MTC xenografts 34 , 71 (Table 1, Figure 4C‐E).
Collectively this work demonstrates a rational path for balancing polypharmacology in a drug, which enables a sophisticated attack on cellular networks, complementing targeted therapy to open up a new avenue for deriving novel cancer therapeutics. By leveraging this powerful multidisciplinary approach between Drosophila, computation, medicinal chemistry and mammals, we are now tackling cancer types for which drug discovery has been problematic, such as pancreatic cancer.
3.1.1. Personalizing CRC therapy with flies
Cancer patients can benefit from a standard of care early in treatment, but cancers often acquire drug resistance, one of the biggest long‐standing issues in treatment. Such resistance can establish through systemic changes including alterations in drug metabolism, genomic sequence of therapeutic targets and/or cancer traits affecting multiple organs, with CRC as an example to have a 5‐y survival rate of only 13.3% despite intensive treatment. 72
To solve this issue, Bangi et al generated a personalized Drosophila model for a CRC patient with chemotherapy‐resistant liver and lung metastases. 73 Analyzing primary tumors and blood as control in the patient revealed 132 somatic and 965 rare germline variants. Of these, the authors focused on 9 gene alterations due to their potential relevance, including an oncogenic KRASG13A mutation, biallelic losses of APC, TP53 and FBXW7, and heterozygous mutations in TGFBR2, SMARCA4, FAT4, MAPK14 and CDH1. Targeting these alterations to the hindgut epithelium by byn‐GAL4 caused its expansion similarly to their previous CRC model flies. 50
Subsequent screening for 121 FDA‐approved anti‐cancer drugs in this model with fly lethality as a readout revealed efficacy of a combination between trametinib and the bisphosphonate class drug zoledronate. Notably, this combination significantly decreased volume of patient tumors by ~45% and controlled their growth for 11 mo 73 (Table 1). These results strongly suggested that this Drosophila approach provides a personalized treatment option for patients with refractory cancer.
4. CONCLUSIONS AND PERSPECTIVES
We have described examples of Drosophila contributions to cancer research made thus far. Flies are particularly useful not only as a ‘hypothesis‐testing’ tool but also as a quick and inexpensive ‘hypothesis‐building’ tool by offering genetic and pharmacologic toolkits established during more than 100‐y history. 4 In the past decades, there have been marked advances in technologies for cancer research including next‐generation sequencing, in vivo imaging, CRISPR‐Cas editing and omics analyses. Also, new ideas for treatment are emerging such as immune checkpoint inhibitor drug, sophisticated drug delivery systems, and gut microbiome which affects human diseases including various cancer types. 74 , 75 Combining these and other powerful modalities with Drosophila will accelerate further cancer research by offering a comprehensive framework for exploring the disease complexity.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We apologize to those whose work we could not quote because of space limitation. We thank Madoka Sato for artwork, members in the Sonoshita laboratory and Ross L. Cagan, Arvin C. Dar, Avner Schlessinger, and Yoichiro Tamori for discussion. The Sonoshita laboratory was supported in part by the Joint Usage/Research Center for Genetic Medicine, Hokkaido University and the Photo‐excitonix Project in Hokkaido University, and by grants from the MEXT/JSPS KAKENHI (Grant Number 19H05412, 19K22478, 20H03524, and 20K07558), AMED (Grant Number JP20ck0106548, JP20cm0106273, and The Translational Research program; Strategic PRomotion for practical application of INnovative medical Technology (TR‐SPRINT)), MSD Life Science Foundation, SGH Foundation, Foundation for Promotion of Cancer Research in Japan, Project Mirai Cancer Research Grants, The Ichiro Kanehara Foundation, The Princess Takamatsu Cancer Research Fund, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, The Suhara Memorial Foundation, The Tokyo Biochemical Research Foundation, Japan Foundation for Applied Enzymology, Takeda Science Foundation, Suzuken Memorial Foundation, The Akiyama Life Science Foundation and All Japan Coffee Association and The Pharmacological Research Foundation, Tokyo.
Yamamura R, Ooshio T, Sonoshita M. Tiny Drosophila makes giant strides in cancer research. Cancer Sci. 2021;112:505–514. 10.1111/cas.14747
Ryodai Yamamura and Takako Ooshio authors contributed equally to this work.
REFERENCES
- 1. Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136(3):780‐798. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 3. Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63(2):411‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sonoshita M, Cagan R. Modeling human cancers in Drosophila In: Pick L., eds. Current Topics in Developmental Biology, Vol. 121 Amsterdam: Elsevier; 2017;287‐309. [DOI] [PubMed] [Google Scholar]
- 5. Mirzoyan Z, Sollazzo M, Allocca M, Valenza AM, Grifoni D, Bellosta P Drosophila melanogaster: A model organism to study cancer. Front Genet. 2019;10:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gateff E. Malignant neoplasms of genetic origin in Drosophila melanogaster . Science. 1978;200(4349):1448‐1459. [DOI] [PubMed] [Google Scholar]
- 7. Huang L, Muthuswamy SK. Polarity protein alterations in carcinoma: a focus on emerging roles for polarity regulators. Curr Opin Genet Dev. 2010;20(1):41‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pearson HB, Perez‐Mancera PA, Dow LE, et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest. 2011;121(11):4257‐4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morata G, Ripoll P Minutes: Mutants of Drosophila autonomously affecting cell division rate. Dev Biol. 1975;42(2):211‐221. [DOI] [PubMed] [Google Scholar]
- 10. Moreno E, Basler K, Morata G. Cells compete for Decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416(6882):755‐759. [DOI] [PubMed] [Google Scholar]
- 11. de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA Drosophila Myc Regulates Organ Size by Inducing Cell Competition. Cell. 2004;117(1):107‐116. [DOI] [PubMed] [Google Scholar]
- 12. Moreno E, Basler K. dMyc Transforms Cells into Super‐Competitors. Cell. 2004;117(1):117‐129. [DOI] [PubMed] [Google Scholar]
- 13. Tyler DM, Baker NE. Expanded and fat regulate growth and differentiation in the Drosophila eye through multiple signaling pathways. Dev Biol. 2007;305(1):187‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vincent J‐P, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger Myc‐independent competitive cell interactions. Dev Cell. 2011;21(2):366‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodrigues AB, Zoranovic T, Ayala‐Camargo A, et al. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development. 2012;139(21):4051‐4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agrawal N, Joshi S, Kango M, Saha D, Mishra A, Sinha P. Epithelial hyperplasia of imaginal discs induced by mutations in Drosophila tumor suppressor genes: growth and pattern formation in genetic mosaics. Dev Biol. 1995;169(2):387‐398. [DOI] [PubMed] [Google Scholar]
- 17. Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila . EMBO J. 2003;22(21):5769‐5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Igaki T, Pastor‐Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila . Dev Cell. 2009;16(3):458‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vidal M, Larson DE, Cagan RL. Csk‐deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell. 2006;10(1):33‐44. [DOI] [PubMed] [Google Scholar]
- 20. Enomoto M, Igaki T. Src controls tumorigenesis via JNK‐dependent regulation of the Hippo pathway in Drosophila . EMBO Rep. 2013;14(1):65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grzeschik NA, Amin N, Secombe J, Brumby AM, Richardson HE. Abnormalities in cell proliferation and apico‐basal cell polarity are separable in Drosophila lgl mutant clones in the developing eye. Dev Biol. 2007;311(1):106‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr Biol. 2010;20(7):573‐581. [DOI] [PubMed] [Google Scholar]
- 23. Sanaki Y, Nagata R, Kizawa D, Léopold P, Igaki T. Hyperinsulinemia Drives Epithelial Tumorigenesis by Abrogating Cell Competition. Dev Cell. 2020;53(4):379‐389. [DOI] [PubMed] [Google Scholar]
- 24. Hogan C, Dupré‐Crochet S, Norman M, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11(4):460‐467. [DOI] [PubMed] [Google Scholar]
- 25. Kajita M, Hogan C, Harris AR, et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src‐transformed cells. J Cell Sci. 2010;123(2):171‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliver ER, Saunders TL, Tarlé SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute . Development. 2004;131(16):3907‐3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Norman M, Wisniewska KA, Lawrenson K, et al. Loss of Scribble causes cell competition in mammalian cells. J Cell Sci. 2012;125(1):59‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hashimoto M, Sasaki H. Epiblast Formation by TEAD‐YAP‐Dependent Expression of Pluripotency Factors and Competitive Elimination of Unspecified Cells. Dev Cell. 2019;50(2):139‐154. [DOI] [PubMed] [Google Scholar]
- 29. Clavería C, Giovinazzo G, Sierra R, Torres M. Myc‐driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500(7460):39‐44. [DOI] [PubMed] [Google Scholar]
- 30. Ashburner M, Bonner JJ. The induction of gene activity in Drosophila by heat shock. Cell. 1979;17(2):241‐254. [DOI] [PubMed] [Google Scholar]
- 31. Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401‐415. [DOI] [PubMed] [Google Scholar]
- 32. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913‐2921. [DOI] [PubMed] [Google Scholar]
- 33. Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti‐targets for cancer polypharmacology. Nature. 2012;486(7401):80‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sonoshita M, Scopton AP, Ung PMU, et al. A whole‐animal platform to advance a clinical kinase inhibitor into new disease space. Nat Chem Biol. 2018;14(3):291‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014;14(3):173‐186. [DOI] [PubMed] [Google Scholar]
- 36. Vidal M, Wells S, Ryan A, Cagan R. ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res. 2005;65(9):3538‐4351. [DOI] [PubMed] [Google Scholar]
- 37. Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med. 2016;375(11):1054‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levinson S, Cagan RL Drosophila cancer models identify functional differences between ret fusions. Cell Rep. 2016;16(11):3052‐3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xing M, Haugen BR, Schlumberger M. Progress in molecular‐based management of differentiated thyroid cancer. Lancet. 2013;381(9871):1058‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155(5):645‐653. [DOI] [PubMed] [Google Scholar]
- 41. The International Agency for Research on Cancer . World Cancer Report: Cancer Research for Cancer Prevention. Lyon: World Health Organization; 2020. [Google Scholar]
- 42. Raskov H, Søby JH, Troelsen J, Bojesen RD, Gögenur I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann Surg. 2020;271(1):75‐85. [DOI] [PubMed] [Google Scholar]
- 43. Taketo MM. Roles of stromal microenvironment in colon cancer progression. J Biochem. 2012;151(5):477‐481. [DOI] [PubMed] [Google Scholar]
- 44. Sonoshita M, Aoki M, Fuwa H, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell. 2011;19(1):125‐137. [DOI] [PubMed] [Google Scholar]
- 45. Sonoshita M, Itatani Y, Kakizaki F, et al. Promotion of colorectal cancer invasion and metastasis through activation of NOTCH‐DAB1‐ABL‐RHOGEF protein TRIO. Cancer Discov. 2015;5(2):198‐211. [DOI] [PubMed] [Google Scholar]
- 46. Itatani Y, Sonoshita M, Kakizaki F, et al. Characterization of Aes nuclear foci in colorectal cancer cells. J Biochem. 2016;159(1):133‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kakizaki F, Sonoshita M, Miyoshi H, et al. Expression of metastasis suppressor gene AES driven by a Yin Yang (YY) element in a CpG island promoter and transcription factor YY2. Cancer Sci. 2016;107(11):1622‐1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winters IP, Murray CW, Winslow MM. Towards quantitative and multiplexed in vivo functional cancer genomics. Nat Rev Genet. 2018;19(12):741‐755. [DOI] [PubMed] [Google Scholar]
- 49. Takashima S, Mkrtchyan M, Younossi‐Hartenstein A, Merriam JR, Hartenstein V. The behaviour of Drosophila adult hindgut stem cells is controlled by Wnt and Hh signalling. Nature. 2008;454(7204):651‐655. [DOI] [PubMed] [Google Scholar]
- 50. Bangi E, Murgia C, Teague AG, Sansom OJ, Cagan RL. Functional exploration of colorectal cancer genomes using Drosophila . Nat Commun. 2016;7(1):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fleisch Marcus A, Illescas AH, Hohl BC, Llanos AA. Relationships between social isolation, neighborhood poverty, and cancer mortality in a population‐based study of US adults. PLoS One. 2017;12(3):e0173370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dawson EH, Bailly TPM, Dos Santos J, et al. Social environment mediates cancer progression in Drosophila . Nat Commun. 2018;9(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Riely GJ, Marks J, Pao W KRAS mutations in non–small cell lung cancer. Proc Am Thorac Soc. 2009;6(2):201‐205. [DOI] [PubMed] [Google Scholar]
- 55. Levine Benjamin D, Cagan RL Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Rep. 2016;14(6):1477‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Das TK, Cagan RL. KIF5B‐RET oncoprotein signals through a multi‐kinase signaling hub. Cell Rep. 2017;20(10):2368‐2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15(11):1311‐3133. [DOI] [PubMed] [Google Scholar]
- 58. Read RD, Cavenee WK, Furnari FB, Thomas JB. A Drosophila model for EGFR‐Ras and PI3K‐dependent human glioma. PLoS Genet. 2009;5(2):e1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. World Health Organization . Global Report on Diabetes. Geneva: World Health Organization; 2016. [Google Scholar]
- 60. Musselman LP, Kühnlein RP. Drosophila as a model to study obesity and metabolic disease. J Exp Biol. 2018;221(Suppl 1):jeb163881. [DOI] [PubMed] [Google Scholar]
- 61. Hirabayashi S, Baranski TJ, Cagan RL. Transformed Drosophila cells evade diet‐mediated insulin resistance through wingless signaling. Cell. 2013;154(3):664‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hirabayashi S, Cagan RL. Salt‐inducible kinases mediate nutrient‐sensing to link dietary sugar and tumorigenesis in Drosophila . eLife. 2015;4:e08501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383(9911):69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rulifson EJ, Kim SK, Nusse R. Ablation of insulin‐producing neurons in flies: growth and diabetic phenotypes. Science. 2002;296(5570):1118‐1120. [DOI] [PubMed] [Google Scholar]
- 65. Bianco‐Miotto T, Craig JM, Gasser YP, van Dijk SJ, Ozanne SE. Epigenetics and DOHaD: from basics to birth and beyond. J Dev Orig Health Dis. 2017;8(5):513‐519. [DOI] [PubMed] [Google Scholar]
- 66. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hollingsworth SJ. Precision medicine in oncology drug development: a pharma perspective. Drug Discov Today. 2015;20(12):1455‐1463. [DOI] [PubMed] [Google Scholar]
- 68. Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10(2):130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ung PM‐U, Schlessinger A. DFGmodel: Predicting Protein Kinase Structures in Inactive States for Structure‐Based Discovery of Type‐II Inhibitors. ACS Chem Biol. 2015;10(1):269‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ung PMU, Sonoshita M, Scopton AP, Dar AC, Cagan RL, Schlessinger A. Integrated computational and Drosophila cancer model platform captures previously unappreciated chemicals perturbing a kinase network. PLoS Comput Biol. 2019;15(4):e1006878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019;2:141‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bangi E, Ang C, Smibert P, et al. A personalized platform identifies trametinib plus zoledronate for a patient with KRAS‐mutant metastatic colorectal cancer. Sci. Adv. 2019;5(5):eaav6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell. 2018;33(4):570‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yamamura R, Nakamura K, Kitada N, et al. Associations of gut microbiota, dietary intake, and serum short‐chain fatty acids with fecal short‐chain fatty acids. Biosci Microbiota Food Health. 2020;39(1):11‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]