

Abstract
We describe an enantioselective synthesis of (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol which is a key subunit of darunavir, a widely used HIV-1 protease inhibitor drug for the treatment of HIV/AIDS patients. The synthesis was achieved in optically pure form utilizing commercially available sugar derivatives as the starting material. The key steps involve a highly stereoselective substrate-controlled hydrogenation, a Lewis acid catalyzed anomeric reduction of a 1,2-O-isopropylidene-protected glycofuranoside, and a Baeyer–Villiger oxidation of a tetrahydrofuranyl-2-aldehyde derivative. This optically active ligand alcohol was converted to darunavir efficiently.
Graphical Abstract
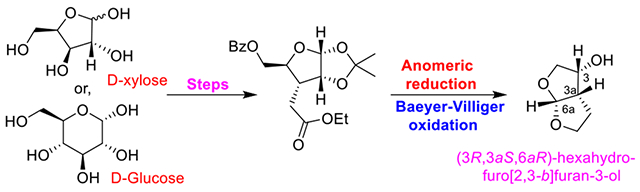
The development of combination antiretroviral therapies (cART) has transformed Acquired Immunodeficiency Syndrome (AIDS) from a fatal disease into a manageable chronic condition.1,2 The Human Immunodeficiency Virus Type-1 (HIV-1) protease inhibitor drugs are an important component of cART regimens.3,4 Darunavir (1, Figure 1) is the most recent FDA approved HIV-1 protease inhibitor drug for the treatment of patients with HIV-1 infection and AIDS.5,6 It is exceedingly potent and has exhibited broad-spectrum activity against highly multidrug-resistant HIV-1 variants.7,8 It received FDA approval in 2006 for the treatment of HIV/AIDS patients who are harboring multidrug-resistant HIV-1 variants and do not respond to other approved therapies. Darunavir received full approval in 2008 for all HIV/AIDS patients including pediatrics.5,9 Darunavir is a widely used protease inhibitor drug, and it has become the front-line therapy for treatment of HIV/AIDS. Darunavir was specifically designed to promote “backbone binding” through extensive hydrogen bonding interactions with the HIV-1 protease active site backbone atoms.8,10 One of the key features of darunavir is the stereochemically defined bicyclic (3R,3aS,6aR)-bis-tetrahydrofuran (bis-THF) heterocycle as the P2-ligand.7,10 Our extensive structure–activity studies and X-ray crystallographic studies established the bis-THF ligand as the privileged ligand for the S2 subsite of HIV-1 protease for a variety of very potent HIV-1 protease inhibitors with clinical potential.11–13
Figure 1.
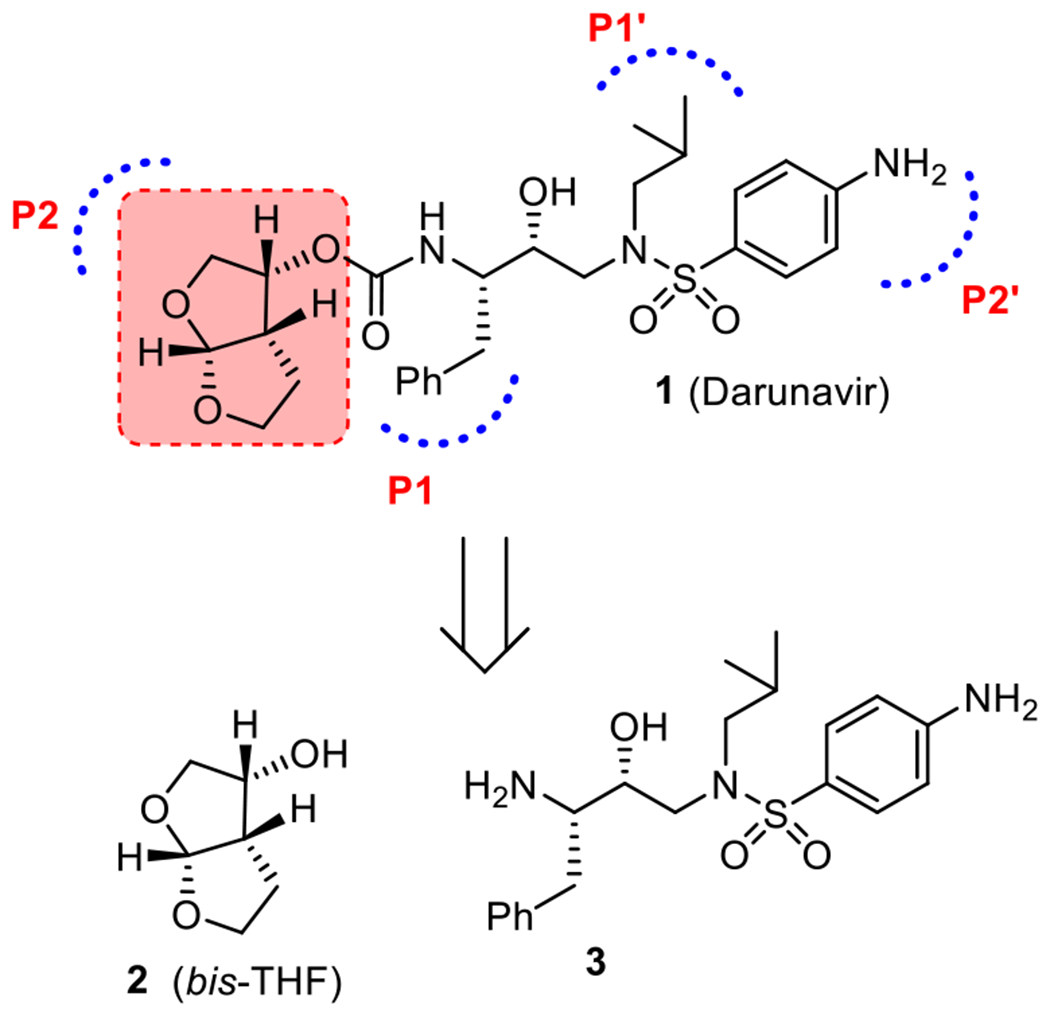
Structure of darunavir (1), bis-THF (2), and aminoalcohol 3.
Darunavir is readily synthesized by formation of the urethane between the bis-THF ligand alcohol 2 and amino-alcohol derivative 3.14 The bis-THF ligand structure contains three contiguous stereocenters. We and others reported a number of syntheses of bis-THF ligand alcohol 2 in optically active form.14 Our initial synthesis of bis-THF alcohol was achieved utilizing (3R)-diethyl malate as the key starting material.15 We reported an efficient racemic synthesis of bis-THF alcohol which was resolved by using a lipase-catalyzed enzymatic resolution to provide the optically active ligand.16 We also investigated the stereoselective photochemical route to bis-THF alcohol where 1,3-dioxolane was added to a chiral furanone derivative.17 These procedures provided access to optically active bis-THF ligand alcohol. However, the optical purity of ligand alcohol was in the range 92–96% ee. Quaedflieg and co-workers reported a large-scale synthesis of bis-THF ligand alcohol utilizing a diastereoselective Michael addition as the key step.18 Black and co-workers reported an asymmetric synthesis of bis-THF ligand alcohol using a Mukaiyama aldol reaction with a silyl ketene acetal.19 Yu and co-workers reported a large-scale synthesis of a racemic bis-THF ligand from glycolaldehyde dimer and 2,3-dihydrofuran using Yb(fod)3 catalyst.20 Xie and co-workers also reported a similar Lewis acid catalyzed synthesis of racemic bis-THF alcohol.21 For our continued interest in optically pure bis-THF ligand, we have investigated the feasibility of a carbohydrate-based synthesis of bis-THF alcohol. Herein, we report an optically active synthesis of bis-THF utilizing inexpensive d-xylofuranose or d-glucose as the starting materials. The overall route may furnish convenient access to quantities of optically pure bis-THF ligand alcohol.
Our chiron approach22,23 to the synthesis of optically active bis-THF alcohol 2 begins with commercially available 1,2-O-isopropylidine-α-d-xylofuranose 4 as shown in Scheme 1. Selective protection of the primary alcohol as a benzoate derivative with 1.1 equiv of benzoyl chloride in the presence of pyridine and a catalytic amount of DMAP in CH2Cl2 at 0 °C for 30 min furnished 5 in 95% yield. Swern oxidation of 5 provided the corresponding ketone which was subjected to Wittig olefination with commercially available (carbethoxy-methylene)triphenylphosphorane in CH2Cl2 at 23 °C for 24 h to afford α,β-unsaturated ester 6 along with a small amount of its isomer in a 8:1 mixture. The Z-isomer 6 was separated by silica gel chromatography in 85% yield over two steps. Catalytic hydrogenation of olefin 6 over 10% Pd/C in ethanol at 23 °C under a hydrogen-filled balloon afforded saturated derivative 7 as the only isolated product in 96% yield. Saturated ester 7 was converted to γ-lactone derivative 8 by exposure to BF3·OEt2 (8 equiv) followed by Et3SiH (3 equiv) in CH2Cl2 at −78 to 23 °C for 6 h.24,25 Bicyclic lactone 8 was isolated in 87% yield. Presumably, the reaction of the isopropylidene derivative with a Lewis acid resulted in the formation of an oxocarbenium ion intermediate. Silane reduction then provided the corresponding alcohol which formed the γ-lactone under the acidic conditions. The overall transformation is quite efficient. The reaction was carried out on gram scale to provide an excellent yield of γ-lactone 8. Hydrolysis of the benzoate ester using K2CO3 in MeOH at 23 °C for 15 min furnished bicyclic alcohol 9 in 85% yield. Alcohol 9 was converted to methyl acetal 10 in a three-step sequence involving (1) Dess-Martin oxidation of the primary alcohol to the corresponding aldehyde, (2) m-CPBA-promoted Baeyer–Villiger oxidation at 0 °C for 2 h, and (3) exposure of the resulting formate to 6% HCl in MeOH at 0 °C. Acetal 10 was obtained in 41% yield over three steps. Reduction of lactone 10 by LiAlH4 in THF at −78 to 23 °C for 1 h followed by exposure of the resulting diol to aqueous HCl at 0 to 23 °C furnished bis-THF alcohol 2 (c 0.73, MeOH; lit17 (c 1.16, MeOH)) in 63% yield over two steps.18 The optical purity of alcohol (−)-2 was determined after its conversion to p-nitrocarbonate 11.26 The reaction of (−)-2 with p-nitrophenylchloroformate in the presence of pyridine in CH2Cl2 at 0 to 23 °C for 12 h provided nitrocarbonate 11 in 92% yield. Chiral HPLC analysis of 11 on a CHIRALPAK OD-H column revealed an enantiomeric purity of 99% ee (please see Supporting Information).
Scheme 1.
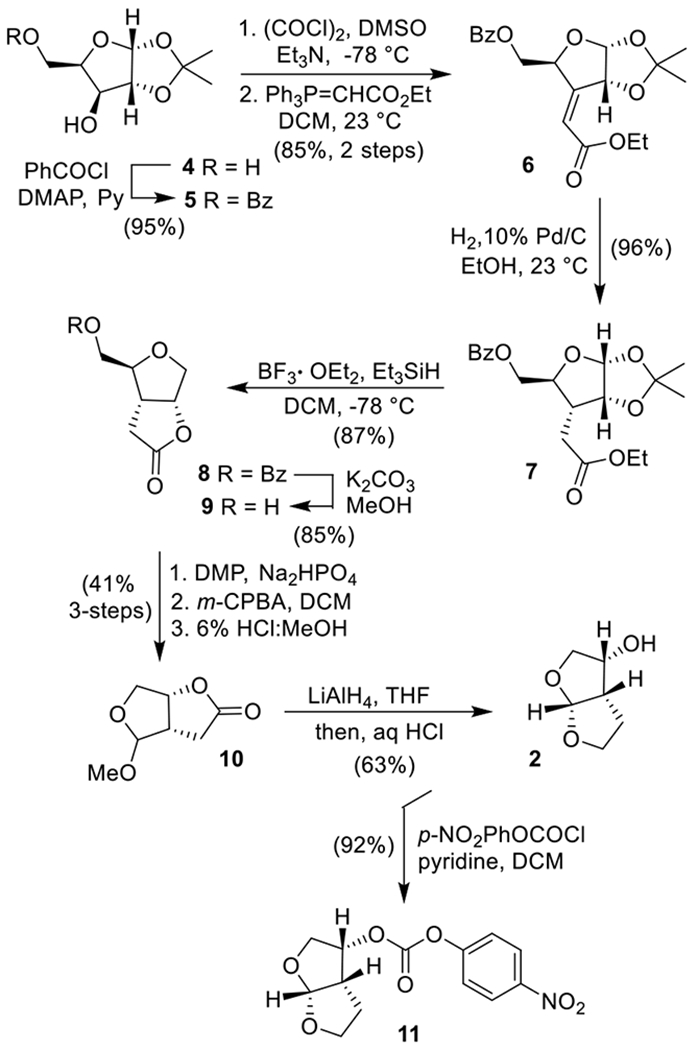
Synthesis of Optically Active bis-THF Ligand from d-Xylose
We have also converted commercially available 1,2:5,6-di-O-isopropylidene-α-d-glucofuranose 12 to bis-THF ligand alcohol 2. As shown in Scheme 2, Swern oxidation of 12 followed by Horner–Wadsworth–Emmons reaction of the resulting ketone provided α,β-unsaturated ester derivative 13 as a 4:1 Z/E mixture in 48% yield over two steps.27 The isomers were separated, and the Z-ester 13 was treated with 80% aqueous AcOH at 23 °C for 60 h to provide the corresponding diol.28 The resulting diol was hydrogenated over 10% Pd/C under a hydrogen-filled balloon at 23 °C for 24 h to afford the saturated diol 14 in 66% yield over two steps. Diol 14 was converted to alcohol 15 by exposure to NaIO4 in MeOH at 0 to 23 °C for 2 h.28 The resulting aldehyde was reacted with NaBH4 in MeOH at 0 °C for 1 h to furnish 15 in 88% yield over two steps.25 Protection of the primary alcohol as benzoate ester 7 was carried out with benzoyl chloride in the presence of Et3N. Benzoate derivative 7 has been converted to bis-THF alcohol 2 as described in Scheme 1. Alcohol 2 was converted to mixed activated carbonate 16 by treatment with N,N’-disuccinimidyl carbonate in the presence of Et3N in CH2Cl2 at 23 °C.17,29 The synthesis of darunavir is shown in Scheme 3. Commercially available epoxide 17 was reacted with isobutylamine in 2-propanol at 60 °C for 22 h. Reaction of the resulting amino alcohol with 4-nitrobenzenesulfonyl chloride in CH2Cl2 in the presence of Et3N at 23 °C for 5 h afforded Cbz-containing sulfonamide derivative 18 in 71% yield over two steps.30 For the synthesis of darunavir, the Cbz-derivative 18 was subjected to catalytic hydrogenation with mixed activated carbonate 16 in the presence of Et3N in THF under a hydrogen-filled balloon at 23 °C for 17 h to provide carbamate 1 (darunavir) in 53% yield. The hydrogenation condition accomplished deprotection of Cbz-group, reaction of the resulting amine with mixed succinimidyl carbonate to form the carbamate, and reduction of the aromatic nitro group to the amine.31
Scheme 2.
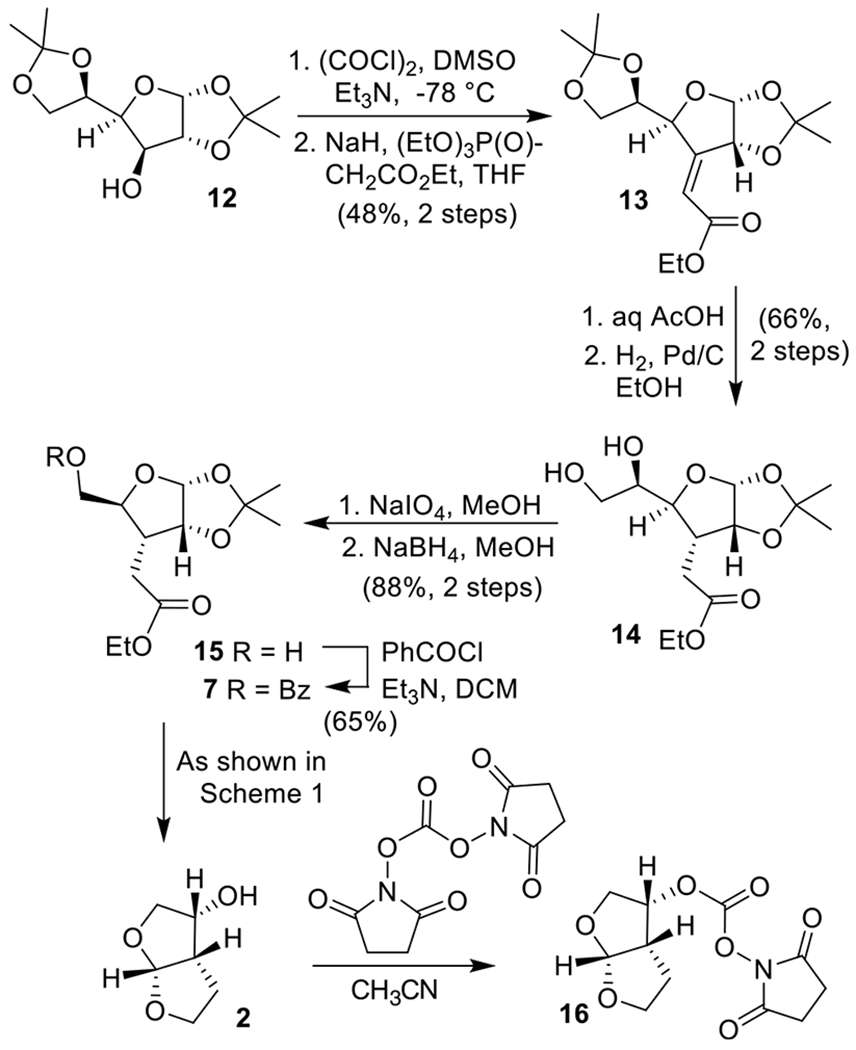
Synthesis of Optically Active bis-THF Ligand from d-Glucose
Scheme 3.
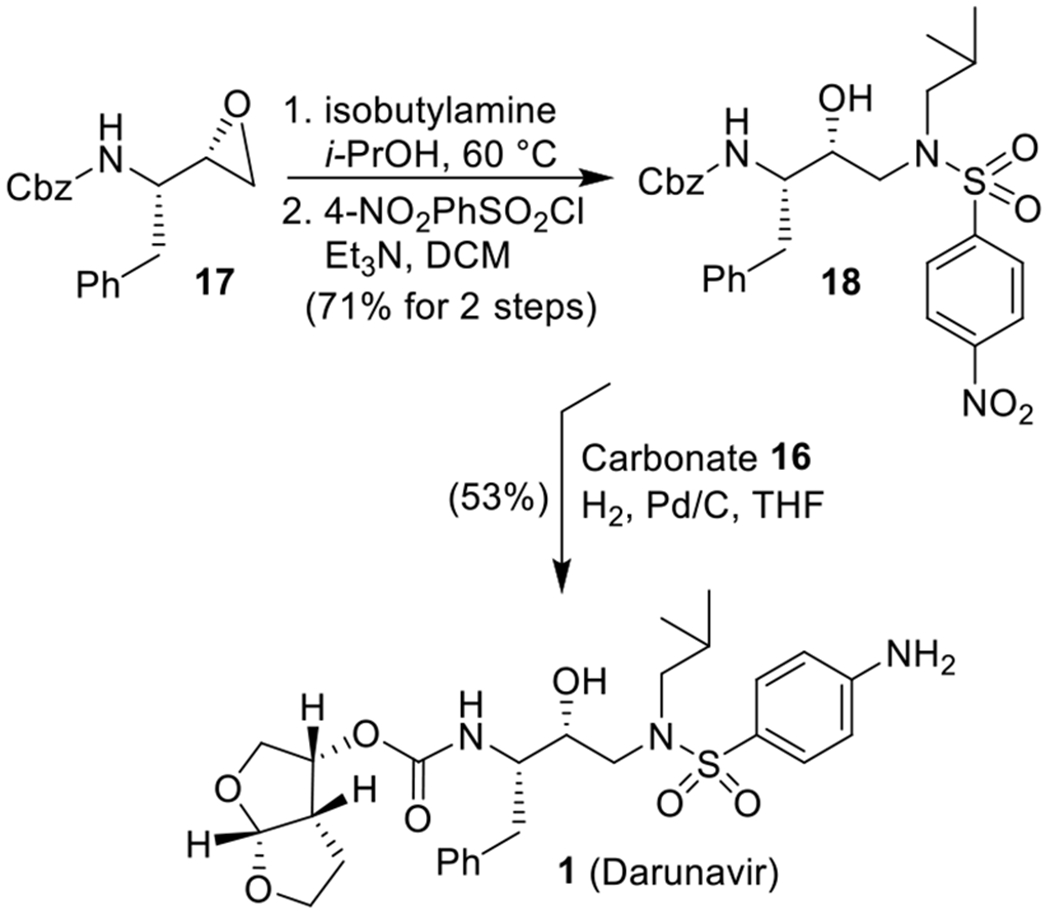
Synthesis of Darunavir (1)
In conclusion, we describe here a convenient synthesis of (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol (2) in optically pure form using commercially available glucose or xylose derivatives. The key synthetic steps involved a highly diastereoselective substrate controlled hydrogenation of an α,β-unsaturated ester, an efficient BF3·OEt2-catalyzed deprotection of an isopropylidene group and subsequent silane reduction, a Baeyer–Villiger oxidation, and an acid-catalyzed cyclization. Overall, the synthesis of the bis-THF ligand alcohol involves more steps than other published syntheses. However, the main advantage of the current route is the enantioselective synthesis using inexpensive sugars as the starting materials, efficient reaction steps, and high optical purity of the bis-THF alcohol. The route has the potential for scale-up. The bis-THF alcohol was converted to mixed activated succinimidyl carbonate. Catalytic hydrogenation of this carbonate with a Cbz-derivative of a dipeptide isostere furnished darunavir.
EXPERIMENTAL PROCEDURES
General Methods.
All chemical and reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. Solvents were purified as follows: CH2Cl2 was distilled from calcium hydride or purified using a solvent purification system; methanol was used without further purification; tetrahydrofuran was distilled from sodium/benzophenone. The flasks were fitted with rubber septa and kept under a positive pressure of argon. Heated reactions were ran using an oil bath on a hot plate equipped with a temperature probe. TLC analysis was conducted using glass-backed thin-layer silica gel chromatography plates (60 Å, 250 μm thickness, F-254 indicator). Flash chromatography was done using a 230–400 mesh, a 60 Å pore diameter silica gel. 1H NMR spectra were recorded on 400 and 500 MHz spectrometers. 13C NMR spectra were recorded at 100 MHz NMR. Chemical shifts are reported in parts per million and referenced to the deuterated residual solvent peak (CDCl3, 7.26 ppm for 1H and 77.16 ppm for 13C). NMR data are reported as δ value (chemical shift), J-value (Hz), and integration, where s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, p = quintet, m = multiplet, dd = doublet doublets, and so on. Optical rotations were recorded on a digital polarimeter. Low resolution mass spectra (LRMS) spectra were recorded using a quadrupole LCMS under positive electrospray ionization (ESI+). High-resolution mass spectrometry (HRMS) spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center. These experiments were performed under ESI+ and positive atmospheric pressure chemical ionization (APCI+) conditions using an Orbitrap XL Instrument.
((3aR,5R,6aR)-6-Hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl Benzoate (5).
A solution of commercially available 1,2-O-isopropylidene-α-d-xylofuranose 4 (3 g, 15.8 mmol) in dry CH2Cl2 (40 mL) was cooled to 0 °C. To the mixture were added 1.90 mL (23.6 mmol) pyridine and a catalytic amount (192 mg) of N,N-dimethylaminopyridine. The resulting mixture was stirred at 0 °C for 10 min, at which time 2 mL (17.3 mmol) benzoyl chloride were added to it dropwise over a period of 30 min. The reaction mixture was stirred at 0 °C for an additional 30 min and then quenched by the addition of 20 mL of a saturated solution of NH4Cl. The reaction was allowed to warm to 23 °C, the layers were separated, and the aqueous layer was extracted with CH2Cl2 (3×). The combined organic extracts were washed with an aqueous solution of CuSO4, water, and brine. The organic solution was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to afford 5 as an oil (4.43 g, 95%). Rf = 0.2 (50% EtOAc/hexanes, SiO2 plate). (c 0.15, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.08–8.01 (m, 2H), 7.62–7.54 (m, 1H), 7.49–7.40 (m, 2H), 5.95 (d, J = 3.6 Hz, 1H), 4.83–4.73 (m, 1H), 4.59 (d, J = 3.6 Hz, 1H), 4.43–4.34 (m, 2H), 4.18 (dd, J = 4.2, 2.3 Hz, 1H), 3.36–3.31 (m, 1H), 1.50 (s, H), 1.32 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 167.3, 133.5, 129.8 (2C), 129.1, 128.4 (2C), 111.8, 104.6, 84.9, 78.4, 74.3, 61.2, 26.7, 26.0. LRMS (ESI) m/z: [M + H]+ 295.1. HRMS (ESI) m/z: [M + Na]+ calcd C15H18O6Na 317.0996; found 317.0998.
(3aR,5R,6aS)-2,2-Dimethyl-6-oxotetrahydrofuro[2,3-d][1,3]-dioxol-5-yl)methyl Benzoate (6).
A solution of oxalyl chloride (2.42 mL, 28.5 mmol) in 40 mL of anhydrous CH2Cl2 was cooled to −78 °C under an argon atmosphere. To the mixture was added DMSO (4 mL, 57.1 mmol) dropwise over a period of 15 min. After the resulting solution had been stirred at the same temperature for 10 min, a solution of alcohol 5 (4.2 g, 14.2 mmol) in anhydrous CH2Cl2 (10 mL) was added to it dropwise over a period of 15 min. Stirring was continued at −78 °C for an additional 30 min. Then, Et3N (9.9 mL, 71.3 mmol) was added. The temperature of the reaction mixture was maintained at −78 °C for 10 min, and then the mixture was allowed to stir for 30 min while warming to 23 °C. The reaction was then quenched by 30 mL of water and extracted with (3×) CH2Cl2. The CH2Cl2 layer was washed with saturated aqueous NaHCO3 and then brine and was dried over anhydrous Na2SO4. Filtration and solvent removal under reduced pressure afforded crude product which was used in the next step without further purification.
To a stirred solution of the above ketone (14.3 mmol) in dry CH2Cl2 (30 mL) was added (carbethoxymethylene)triphenyl-phophorane (5.96 g, 17.1 mmol). The reaction mixture was stirred under argon at 23 °C for 24 h. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica (5% EtOAc/hexanes to 10% EtOAc/hexane) to yield the ester as an 8:1(Z/E) mixture of separable isomers. Z-isomer of ester 6 (4.4 g, 85% over 2 steps), yellow oil. Rf = 0.4 (20% EtOAc/hexanes, SiO2 plate). (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.05–7.92 (m, 2H), 7.62–7.53 (m, 1H), 7.44 (dd, J = 8.4, 7.2 Hz, 2H), 6.03–5.95 (m, 2H), 5.78 (dt, J = 4.2, 1.5 Hz, 1H), 5.16 (ddt, J = 5.3, 3.7, 1.8 Hz, 1H), 4.57 (dd, J = 11.9, 3.5 Hz, 1H), 4.43 (dd, J = 11.9, 5.0 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H), 1.51 (s, 3H), 1.44 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 166.0, 164.5, 154.5, 133.2, 129.6 (2C), 129.4, 128.4 (2C), 117.1, 112.9, 105.0, 78.1, 77.8, 65.1, 60.8, 27.3, 27.0, 14.0. LRMS (ESI) m/z: [M + Na]+ 385.0. HRMS (ESI) m/z: [M + Na]+ calcd C19H22O7Na 385.1258; found 385.1247.
((3aR,5S,6R,6aR)-6-(2-Ethoxy-2-oxoethyl)-2,2-dimethyl-tetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl Benzoate (7).
To a solution of Z-ester 6 (4.2 g, 11.6 mmol) in anhydrous ethanol (70 mL) 10% Pd/C (147 mg, 5% w/w) was added. The resulting mixture was stirred under a hydrogen filled balloon for 6 h. Upon completion of the reaction, the mixture was filtered through Celite, and the filter cake was washed with EtOAc. Evaporation of solvent yielded a colorless oil that was purified by flash chromatography on silica (15% EtOAc/hexane) to provide ester 7 (4.02 g, 96%) as a colorless oil and as a single diastereomer. Rf = 0.4 (20% EtOAc/hexanes, SiO2 plate). (c 0.584, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.15–8.00 (m, 2H), 7.60–7.52 (m, 1H), 7.43 (t, J = 7.6 Hz, 2H), 5.87 (d, J = 3.7 Hz, 1H), 4.82 (t, J = 4.2 Hz, 1H), 4.56 (dd, J = 12.3, 2.9 Hz, 1H), 4.34 (dd, J = 12.3, 5.0 Hz, 1H), 4.19–4.08 (m, 3H), 2.75 (dd, J = 17.0, 9.8 Hz, 1H), 2.54–2.33 (m, 2H), 1.53 (s, 3H), 1.32 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 171.7, 166.3, 133.1, 129.7 (2C), 129.6, 128.3 (2C), 111.7, 104.8, 80.8, 78.6, 63.9, 60.7, 41.4, 29.6, 26.6, 26.2, 14.0. LRMS (ESI) m/z: [M + Na]+ 387.0. HRMS (ESI) m/z: [M + Na]+ calcd C19H24O7Na 387.1414; found 387.1421.
((3aR,4S,6aR)-2-Oxohexahydrofuro[3,4-b]furan-4-yl)methyl benzoate (8).
To a flask containing ester 7 (2.3 g, 6.3 mmol) in dry CH2Cl2 (20 mL) at −78 °C was added BF3·OEt2 (6.18 mL, 50.5 mmol) dropwise over 5 min, and then Et3SiH (3.01 mL, 18.9 mmol) was added. The reaction mixture was stirred at −78 to 23 °C for 6 h. The reaction mixture was cooled to 0 °C, quenched with a saturated solution of NaHCO3 (20 mL), and extracted with (3×) CH2Cl2. The combined organic layer was washed with water and brine. The organic solution was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The product was purified by silica gel column chromatography (50% EtOAc/hexane) to afford 8 as an oil (1.44 g, 87%). Rf = 0.3 (30% EtOAc/hexanes, SiO2 plate). (c 0.015, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.06–7.97 (m, 2H), 7.65–7.54 (m, 1H), 7.52–7.35 (m, 2H), 5.15 (ddd, J = 6.7, 4.8, 1.9 Hz, 1H), 4.53–4.35 (m, 2H), 4.24 (ddd, J = 11.2, 4.8, 0.5 Hz, 1H), 4.17–4.04 (m, 2H), 2.99–2.84 (m, 2H), 2.57 (dd, J = 17.8, 1.7 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 175.3, 166.1, 133.3, 129.6(2C), 129.3, 128.4(2C), 83.9, 82.9, 72.9, 64.6, 41.4, 33.0. LRMS (ESI) m/z: [M + H]+ 263.0. HRMS (ESI) m/z: [M + H]+ calcd C14H15O5 263.0914; found 263.0920.
(3aR,4S,6aR)-4-(Hydroxymethyl)tetrahydrofuro[3,4-b]-furan-2(3H)-one (9).
To a stirred solution of the lactone 8 (1.2 g, 4.6 mmol) in MeOH (15 mL) at 0 °C was added K2CO3 (695 mg, 5 mmol), and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to remove MeOH. The obtained residue was dissolved in water (10 mL) and CH2Cl2 (10 mL), and the aqueous layer was extracted with (3×) 10% MeOH/CH2Cl2. The combined organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by by silica gel column chromatography (10% MeOH/CH2Cl2) to afford 9 (690 mg, 85%) as a colorless oil. Rf = 0.2 (80% EtOAc/hexanes, SiO2 plate). (c 0.02, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.09 (ddd, J = 6.9, 4.8, 2.0 Hz, 1H), 4.17 (dd, J = 11.0, 4.7 Hz, 1H), 4.02 (dt, J = 11.4, 2.4 Hz, 1H), 3.86–3.70 (m, 2H), 3.64–3.56 (m, 1H), 2.98–2.88 (m, 1H), 2.82 (ddd, J = 18.1, 9.3, 1.4 Hz, 1H), 2.46 (dd, J = 18.1, 1.8 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 175.6, 85.2, 84.3, 72.7, 62.4, 40.0, 32.8. LRMS (ESI) m/z: [M + H]+ 159. HRMS (ESI) m/z: [M + Na]+ calcd C7H10O4Na 181.0471; found 181.0470.
(3aS,6aR)-4-Methoxytetrahydrofuro[3,4-b]furan-2(3H)-one (10).
To a solution of alcohol 9 (200 mg, 1.3 mmol) in anhydrous CH2Cl2 (3 mL) were added Na2HPO4 (369 mg, 2.6 mmol) and DMP (1.10 g, 2.6 mmol) at 0 °C. The reaction mixture was stirred at 0 to 23 °C for 3 h and then cooled to 0 °C and quenched with a saturated solution of NaHCO3 (10 mL). The aqueous layer was extracted with CH2Cl2 (3×), and the organic layer was washed with water (10 mL) and brine. The organic solution was dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure to afford the aldehyde. The crude aldehyde was used in the next step without further purification.
To a stirred solution of the above crude aldehyde in anhydrous CH2Cl2 (10 mL) at 0 °C were added NaHCO3 (211 mg, 2.52 mmol) and m-CPBA (577 mg, 2.5 mmol). The reaction mixture was stirred for 2 h at 0 °C and quenched with a saturated aqueous solution of NaHCO3. The aqueous layer was extracted with (3×) CH2Cl2. The combined organic solvent was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the formate. The crude formate was used in the next step without further purification.
To a stirred solution of the above crude formate (1.2 mmol) in MeOH (5 mL) at 0 °C was added slowly 6% HCl/MeOH (5 mL), and the mixture was stirred for 12 h at 0 to 23 °C. The reaction mixture was then neutralized with a saturated solution of NaHCO3, concentrated under reduced pressure to remove MeOH, extracted with CH2Cl2, and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield a crude residue that was purified by flash chromatography on silica (50% ether/hexane) to yield lactone 10 (82 mg, 41% over 3 steps) as an amorphous solid with a 3:1 mixture of separable anomers. Major Anomer: Rf = 0.4 (40% EtOAc/hexanes, SiO2 plate). (c 0.05, CHCl3). 1H NMR (400 MHz, CDCl3) δ 5.13 (dd, J = 7.1, 3.9 Hz, 1H), 4.87 (s, 1H), 4.09 (d, J = 10.9 Hz, 1H), 3.94 (dd, J = 10.9, 3.9 Hz, 1H), 3.31 (s, 3H), 3.02 (ddd, J = 11.2, 7.1, 4.0 Hz, 1H), 2.83 (dd, J = 18.6, 11.3 Hz, 1H), 2.50 (dd, J = 18.6, 4.0 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 175.9, 109.9, 82.9, 70.6, 54.5, 45.0, 31.7. LRMS (ESI) m/z: [M + H]+ 159.1. HRMS (ESI) m/z: [M + Na]+ calcd C7H10O4Na 181.0471; found 181.0472.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol (2).17
To a flame-dried flask was added LiAlH4 (734 mg, 19.33 mmol). The flask was evacuated by vacuum and then flushed with argon. To the flask was added dry THF (38 mL). The mixture was stirred and cooled to −78 °C prior to the addition of a solution of lactone 10 (1.02 g, 6.44 mmol) in THF (38 mL). The reaction mixture was then allowed to warm to 23 °C over 1 h. The reaction mixture was cooled to 0 °C, and then H2O (0.7 mL), 2.0 M NaOH (0.7 mL), and H2O (2.1 mL) were added sequentially and slowly. The mixture was stirred vigorously at 0 °C for 1.5 h, and then it was filtered through Celite with MeOH. The filtrate was concentrated under reduced pressure to afford a residue containing the crude diol.
To the flask containing the above crude diol (6.44 mmol) was added 1.0 M HCl (15 mL). The mixture was cooled to 0 °C and stirred prior to slow addition of concentrated HCl (1.5 mL). The mixture was allowed to warm to 23 °C over 1 h, and then the mixture was cooled back down to 0 °C and neutralized by portionwise addition of solid Na2CO3. The aqueous layer was extracted with a 10% MeOH/CH2Cl2 solution (5 × 150 mL). The organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the crude ligand alcohol 2 that was purified by flash chromatography on SiO2 (70% ether/hexanes) to yield ligand alcohol 2 (533 mg, 63% over 2 steps) as a colorless oil. Rf = 0.56 (100% EtOAc, SiO2 plate). (c 0.732, MeOH). 1H NMR (400 MHz, CDCl3) δ 5.70 (d, J = 5.8 Hz, 1H), 4.45 (dtd, J = 8.0, 6.6, 5.2 Hz, 1H), 4.03–3.96 (m, 2H), 3.90 (ddd, J = 10.0, 8.6, 6.3 Hz, 1H), 3.64 (dd, J = 9.2, 7.0 Hz, 1H), 2.86 (dddd, J = 10.1, 7.9, 5.2, 2.5 Hz, 1H), 2.36–2.27 (m, 1H), 1.88 (dtd, J = 12.9, 9.9, 8.4 Hz, 1H), 1.79 (d, J = 5.3 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 109.7, 73.3, 71.1, 70.0, 46.7, 25.0.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl (4-nitrophenyl) Carbonate (11).26
To a flame-dried flask were added optically active bis-THF alcohol (−)−2 (6 mg, 0.046 mmol) and CH2Cl2 (1.0 mL) followed by pyridine (7.5 μL, 0.092 mmol). The mixture was stirred under argon and cooled to 0 °C. To the mixture was quickly added 4-nitrophenyl chloroformate (19 mg, 0.092 mmol), and the resulting reaction was stirred at 23 °C for 12 h. After this period, the mixture was concentrated under reduced pressure and purified by flash chromatography (20% EtOAc/hexanes) to yield carbonate 11 (12 mg, 92% yield) as an amorphous white solid. Rf = 0.15 (30% ethyl acetate/hexanes). 1H NMR (400 MHz, CDCl3) δ 8.34–8.25 (m, 2H), 7.43–7.35 (m, 2H), 5.77 (d, J = 5.1 Hz, 1H), 5.32–5.20 (m, 1H), 4.15 (dd, J = 10.0, 6.1 Hz, 1H), 4.05 (td, J = 8.4, 2.6 Hz, 1H), 3.97 (tt, J = 10.0, 6.1 Hz, 2H), 3.15 (dddd, J = 10.2, 7.9, 5.1, 2.4 Hz, 1H), 2.17 (ddt, J = 13.2, 5.4, 2.5 Hz, 1H), 2.00 (dtd, J = 13.2, 10.0, 8.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 155.1, 151.9, 145.5, 125.3, 121.6, 109.1, 77.5, 70.3, 69.5, 44.9, 25.9.
Ethyl 2-((3aR,5S,6aR)-5-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyldihydrofuro[2,3-d][1,3]dioxol-6(5H)-ylidene)-acetate (13).28
To a flame-dried flask was added CH2Cl2 (210 mL). The CH2Cl2 was stirred under argon and cooled to −78 °C prior to addition of oxalyl chloride (5.47 mL, 64.6 mmol). After 5 min, DMSO (9.18 mL, 129.2 mmol) was added dropwise to the reaction mixture. After 10 min, a solution of commercially available 1,2:5,6-di-O-isopropylidene-α-d-glucofuranose 12 (8.41 g, 32.3 mmol) in CH2Cl2 (20 mL) was added dropwise to the reaction mixture, and then the mixture was stirred at −78 °C for 1 h. At this time, Et3N (22.5 mL, 161.6 mmol) was added. The temperature of the reaction mixture was maintained at −78 °C for 10 min, and then the cooling bath was removed. The mixture was allowed to stir for 30 min while warming to 23 °C. The reaction mixture was then quenched with H2O and transferred to a separatory funnel. The organic layer was washed with saturated aqueous NaHCO3 and then with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the crude ketone as a brown oil. The crude ketone was used without further purification.
To a flame-dried flask was added NaH (60% in oil) (2.37 g, 59 mmol). The flask was evacuated and placed under argon prior to addition of dry THF (91 mL). The mixture was stirred and cooled to 0 °C prior to the dropwise addition of triethyl phosphonoacetate (12.3 mL, 62.2 mmol). After 15 min, a solution of the above crude ketone (31.1 mmol) in dry THF (31 mL) was added slowly at 0 °C. The reaction mixture was kept at a temperature of 0 °C for 30 min, and then the reaction mixture was allowed to warm to 23 °C over 30 min. The reaction was quenched by the addition of saturated aqueous NH4Cl, extracted with Et2O, and washed with brine. The organic layer was then dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield a brown oil that was purified by flash chromatography on SiO2 (15% ether/hexanes to 25% ether/hexanes) to yield α,β-unsaturated ester 13 as a 4:1 (Z/E) mixture of separable isomers (combined yield for mixture of isomers, 48% over 2 steps). Z-isomer of ester 13: (3.95 g, 39% over 2 steps), amorphous solid. Rf = 0.60 (50% ether/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 6.32 (dd, J = 2.2, 1.4 Hz, 1H), 5.82 (d, J = 4.2 Hz, 1H), 5.73 (dt, J = 4.2, 1.4 Hz, 1H), 4.66 (ddt, J = 6.0, 2.2, 1.4 Hz, 1H), 4.28–4.19 (m, 2H), 4.12–4.06 (m, 1H), 4.03–3.97 (m, 2H), 1.49 (s, 3H), 1.43 (s, 3H), 1.39 (s, 3H), 1.35 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.3, 155.8, 118.0, 112.9, 110.3, 105.0, 80.0, 78.5, 76.9, 67.4, 60.8, 27.4, 27.2, 26.8, 25.5, 14.3. E-isomer of ester 13E: (963 mg, 9% over 2 steps). Rf = 0.65 (50% ether/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 6.21 (t, J = 1.9 Hz, 1H), 5.92 (d, J = 4.8 Hz, 1H), 5.75 (q, J = 1.9 Hz, 1H), 5.9 (dt, J = 4.8, 1.9 Hz, 1H), 4.34 (ddd, J = 7.9, 6.3, 2.6 Hz, 1H), 4.17 (qt, J = 7.4, 3.8 Hz, 2H), 3.96 (dd, J = 8.8, 6.3 Hz, 1H), 3.56 (dd, J = 8.8, 7.9 Hz, 1H), 1.42 (s, 3H), 1.37 (s, 3H), 1.32–1.27 (m, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ 165.7, 158.1, 118.2, 113.8, 109.2, 103.9, 82.3, 80.1, 79.1, 65.5, 60.9, 27.99, 27.96, 26.2, 25.8, 14.3.
Ethyl-2-((3aR,5S,6R,6aR)-5-((R)1-,2-dihydroxyethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-yl)acetate (14).28
To a flask were added Z-ester 13 (1.77 g, 5.4 mmol) and 80% AcOH/H2O (20 mL). The mixture was stirred at 23 °C for 60 h. The reaction mixture was then concentrated under reduced pressure, and the resultant crude orange oil was purified by flash chromatography on SiO2 (50% EtOAc/hexanes) to yield the deprotected α,β-unsaturated ester (1.14 g, 74%) as a colorless oil. Rf = 0.45 (75% EtOAc/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 6.30 (dd, J = 2.2, 1.5 Hz, 1H), 5.86 (d, J = 4.2 Hz, 1H), 5.74 (dt, J = 4.2, 1.5 Hz, 1H), 4.79 (ddd, J = 6.6, 2.2, 1.4 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H), 3.80–3.67 (m, 3H), 2.95 (d, J = 6.6 Hz, 1H), 2.45 (s, 1H), 1.48 (s, 3H), 1.41 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 165.3, 155.7, 117.7, 113.0, 104.9, 80.0, 78.4, 73.5, 63.5, 60.9, 27.4, 27.2, 14.3.
To a flame-dried flask was added a solution of the above α,β-unsaturated ester (1.06 g, 3.68 mmol) in anhydrous ethanol (16 mL) followed by 10% Pd/C (53 mg, 5% w/w). The flask containing the mixture was evacuated by vacuum and flushed with argon three times, and then it was evacuated by vacuum and flushed with hydrogen three times. The reaction mixture was then left to stir under an atmosphere of hydrogen (1 atm) for 24 h. The reaction mixture was then filtered through Celite with EtOAc and concentrated under reduced pressure to yield a crude colorless oil that was purified by flash chromatography on SiO2 (3% MeOH/CH2Cl2) to yield the saturated diol 14 (950 mg, 89%) as a colorless syrup. Rf = 0.45 (75% EtOAc/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 5.76 (d, J = 3.7 Hz, 1H), 4.76 (dd, J = 4.8, 3.7 Hz, 1H), 4.13 (qd, J = 7.1, 3.2 Hz, 2H), 3.82–3.60 (m, 4H), 3.24 (d, J = 5.4 Hz, 1H), 2.91 (t, J = 5.6 Hz, 1H), 2.77–2.63 (m, 2H), 2.42–2.31 (m, 1H), 1.46 (s, 3H), 1.28 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 172.9, 111.9, 104.8, 81.6, 81.1, 73.7, 63.9, 60.8, 43.2, 30.5, 26.7, 26.4, 14.3.
(3aR,4S,6aR)-6-Hydroxy-4-(hydroxymethyl)tetrahydrofuro-[3,4-b]furan-2(3H)-one (15).28
To a flask were added saturated diol 14 (866 mg, 3.0 mmol) and MeOH (15 mL). The mixture was stirred and cooled to 0 °C prior to portionwise addition of NaIO4 (1.28 g, 6.0 mmol). The reaction mixture was then allowed to warm to 23 °C over 2 h while stirring vigorously. The reaction mixture was filtered through Celite with MeOH and concentrated under reduced pressure to yield a residue that was dissolved in H2O and CH2Cl2 and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the crude aldehyde that was used without further purification.
The above crude aldehyde was dissolved in MeOH (15 mL) and cooled to 0 °C prior to portionwise addition of NaBH4 (226 mg, 6.0 mmol). The reaction mixture was stirred at 0 °C for 1 h. The mixture was then concentrated under reduced pressure to remove MeOH. Saturated aqueous NH4Cl was added, and the organics were extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield a colorless oil that was purified by flash chromatography on SiO2 (40% EtOAc/hexanes) to yield ester 15 (683 mg, 88% over 2 steps) as a colorless oil and as a single diastereomer. Rf = 0.25 (40% EtOAc/hexanes, SiO2 plate). (c 1.31, CHCl3) 1H NMR (400 MHz, CDCl3) δ 5.81 (d, J = 3.8 Hz, 1H), 4.77 (t, J = 3.9 Hz, 1H), 4.15 (dttd, J = 10.8, 7.4, 3.7, 1.2 Hz, 2H), 3.91–3.83 (m, 2H), 3.60–3.52 (m, 1H), 2.69 (ddd, J = 16.8, 8.3, 1.1 Hz, 1H), 2.48–2.33 (m, 2H), 2.05 (s, 1H), 1.48 (s, 3H), 1.31 (s, 3H), 1.26 (td, J = 7.1, 1.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 172.4, 111.9, 104.8, 81.7, 81.5, 61.5, 60.9, 39.8, 29.9, 26.8, 26.4, 14.3.
2,5-Dioxopyrrolidin-1-yl ((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl) Carbonate (16).17
To a flame-dried flask were added bis-THF ligand alcohol 2 (84 mg, 0.65 mmol) and acetonitrile (2.5 mL). To the mixture was added triethylamine (180 μL, 1.29 mmol) followed by commercially available N,N’-disuccinimidyl carbonate (248 mg, 0.97 mmol). The mixture was stirred under argon at 23 °C for 16 h, and then saturated aqueous NaHCO3 was added. The organics were extracted with a solution of 10% MeOH/CH2Cl2, dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography on SiO2 using EtOAc to yield 16 (51 mg, 29% yield (quantitative yield brsm)) as a white solid. Rf = 0.85 (EtOAc, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 5.71 (d, J = 5.1 Hz, 1H), 5.22 (dt, J = 8.2, 6.0 Hz, 1H), 4.13–4.05 (m, 1H), 4.00 (td, J = 8.4, 2.5 Hz, 1H), 3.91 (ddd, J = 10.6, 8.8, 5.9 Hz, 2H), 3.16–3.04 (m, 1H), 2.82 (s, 4H), 2.11 (ddt, J = 13.5, 5.4, 2.4 Hz, 1H), 1.95 (dddd, J = 13.4, 10.3, 9.5, 8.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 168.6, 151.3, 109.3, 79.7, 70.1, 69.7, 45.2, 26.0, 25.5.
((3aR,5S,6R,6aR)-6-(2-Ethoxy-2-oxoethyl)-2,2-dimethyl-tetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methyl Benzoate (7, from 12).
To a flame-dried flask was added a solution of 15 (16 mg, 0.1 mmol) in CH2Cl2. To the mixture was added triethylamine (42 μL, 0.3 mmol), and the mixture was then cooled to 0 °C prior to addition of benzoyl chloride (35 μL, 0.3 mmol). The mixture was then allowed to warm to 23 °C, and it was stirred under argon for 18 h. The mixture was then diluted with water, extracted with CH2Cl2, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography on SiO2 with 20% EtOAc/hexanes provided the desired benzoate ester 7 (15 mg, 65% yield) as a colorless oil. Rf = 0.30 (20% EtOAc/hexanes, SiO2 plate). (c 0.55, CHCl3). 1H NMR (400 MHz, CDCl3) δ 8.15–8.00 (m, 2H), 7.60–7.52 (m, 1H), 7.43 (t, J = 7.6 Hz, 2H), 5.87 (d, J = 3.7 Hz, 1H), 4.82 (t, J = 4.2 Hz, 1H), 4.56 (dd, J = 12.3, 2.9 Hz, 1H), 4.34 (dd, J = 12.3, 5.0 Hz, 1H), 4.19–4.08 (m, 3H), 2.75 (dd, J = 17.0, 9.8 Hz, 1H), 2.54–2.33 (m, 2H), 1.53 (s, 3H), 1.32 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 171.7, 166.3, 133.1, 129.7 (2C), 129.6, 128.3 (2C), 111.7, 104.8, 80.8, 78.6, 63.9, 60.7, 41.4, 29.6, 26.6, 26.2, 14.0. LRMS (ESI) m/z: [M + Na]+ 387.0. HRMS (ESI) m/z: [M + Na]+ calcd C19H24O7Na 387.1414; found 387.1421.
Benzyl-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-nitrophenyl)-sulfonamido)-1-phenylbutan-2-yl)carbamate (18).17
To a flask were added commercially available benzyl (1S)-1-[(2S)-2-oxiranyl]-2-phenylethylcarbamate (17) (200 mg, 0.7 mmol) and isopropanol (2.8 mL). To the mixture was added isobutylamine (0.43 mL, 4.26 mmol). A reflux condenser was attached, and the mixture was stirred and heated at 60 °C for 22 h. The mixture was concentrated under reduced pressure to afford the corresponding aminoalcohol as an amorphous solid that was used in the next step without further purification.
The crude aminoalcohol (0.71 mmol) was dissolved in CH2Cl2 (7 mL) and transferred to a flame-dried flask. The mixture was cooled to 0 °C prior to the addition of triethylamine (0.3 mL, 2.1 mmol) and then 4-nitrobenzenesulfonyl chloride (236 mg, 1.1 mmol). The mixture was placed under argon and stirred at 0 °C for 5 min. The cooling bath was removed, and the reaction was warmed to 23 °C and stirred for 5 h. The mixture was transferred to a separatory funnel, and 1.0 M HCl was added. The organics were extracted with CH2Cl2, dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography on SiO2 using 20% EtOAc/hexane and then 30% EtOAc/hexane as the eluent to yield the desired CBz-protected amine 18 (231 mg, 71% over 2 steps) as an amorphous solid. Rf = 0.30 (30% EtOAc/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 8.35–8.24 (m, 2H), 7.92 (d, J = 8.5 Hz, 2H), 7.38–7.12 (m, 10H), 4.99 (td, J = 18.2, 17.1, 10.6 Hz, 3H), 3.92–3.80 (m, 2H), 3.62 (s, 1H), 3.26–3.11 (m, 2H), 3.05–2.84 (m, 4H), 1.85 (dt, J = 13.8, 6.8 Hz, 1H), 0.86 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3) δ 156.7, 150.1, 144.8, 137.3, 136.3, 129.5, 128.8, 128.7, 128.6, 128.3, 128.0, 126.9, 124.4, 72.3, 67.0, 57.9, 55.6, 52.8, 35.6, 27.1, 20.1, 19.9.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl-((2S,3R)-4-((4-amino-N-isobutylphenyl)-sulfonamido)-3-hydroxy-1-phenyl-butan-2-yl)carbamate (1).
To a flask were added carbamate derivative 18 (48 mg, 0.1 mmol) and THF (1 mL). To the mixture were added triethylamine (24 μL, 0.17 mmol), succinimidyl carbonate 16 (26 mg, 0.1 mmol), and then 10% Pd/C (10 mg). The mixture was placed under a hydrogen balloon. The flask containing the mixture was evacuated under vacuum briefly and then flushed with hydrogen. This was repeated two more times, and then the mixture was left to stir under a balloon of hydrogen at 23 °C for 17 h. The mixture was then filtered through Celite with ethyl acetate, concentrated under reduced pressure, and purified by flash chromatography on SiO2 using 50% EtOAc/CH2Cl2 as the eluent to yield compound 1 (Darunavir) (25 mg, 53%) as an amorphous solid. Rf = 0.5 (75% EtOAc/hexanes, SiO2 plate). 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 7.8 Hz, 2H), 7.23 (m, 5H), 6.77 (d, J = 8.3 Hz, 2H), 5.64 (d, J = 5.1 Hz, 1H), 5.04 (dd, J = 20.8, 8.5 Hz, 2H), 3.87 (td, J = 20.4, 16.5, 8.6 Hz, 5H), 3.78–3.60 (m, 3H), 3.22–2.75 (m, 8H), 1.85–1.74 (m, 1H), 1.62 (t, J = 10.7 Hz, 1H), 1.45 (d, J = 13.4 Hz, 1H), 0.92 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3) δ 155.6, 149.8, 137.8, 129.6, 129.5, 128.6, 127.1, 126.7, 115.0, 109.5, 73.5, 72.9, 71.1, 69.8, 58.9, 55.3, 53.8, 45.6, 35.8, 27.4, 25.9, 20.3, 20.1. HRMS (APCI) m/z; [M + H]+ calcd C27H38N3O7S, 548.2425; found 548.2430.
Supplementary Material
ACKNOWLEDGMENTS
Financial support of this work was provided by the National Institutes of Health (Grant AI150466). The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c02396.
1H and 13C NMR spectra for all new compounds; HPLC data for compound 11 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c02396
The authors declare no competing financial interest.
Contributor Information
Arun K. Ghosh, Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana 47907, United States.
Shivaji B. Markad, Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana 47907, United States
William L. Robinson, Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana 47907, United States
REFERENCES
- (1).Hernán MA The Effect of Combined Antiretroviral Therapy On the Overall Mortality of HIV-Infected Individuals. AIDS 2010, 24, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).The Antiretroviral Therapy in Lower Income Countries (ART-LINC) Collaboration. Mortality of HIV-1-Infected Patients in the First Year of Antiretroviral Therapy: Comparison Between Low-Income and High-Income Countries. Lancet 2006, 367, 817–824. [DOI] [PubMed] [Google Scholar]
- (3).Ghosh AK; Osswald HL; Prato G Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Protease Inhibitors in AIDS Therapy; Ogden R; Flexner C, Eds.; Marcel Dekker: New York, 2001; pp 1–310. [Google Scholar]
- (5).FDA approves Darunavir on June 23, 2006: FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.html.
- (6).Llibre JM; Imaz A; Clotet B From TMC114 to Darunavir: Five Years of Data on Efficacy. AIDS Rev. 2013, 15, 112–121. [PubMed] [Google Scholar]
- (7).Ghosh AK; Sridhar PR; Kumaragurubaran N; Koh Y; Weber IT; Mitsuya H A Privileged Ligand for Darunavir and a New Generation of HIV-Protease Inhibitors That Combat Drug-Resistance. ChemMedChem 2006, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- (8).Ghosh AK; Chapsal BD; Weber IT; Mitsuya H Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- (9).On October 21, 2008, FDA granted traditional approval to Prezista (darunavir), coadministered with ritonavir and with other antiretroviral agents, for the treatment of HIV-1 infection in treatment-experienced adult patients. In addition, a new dosing regimen for treatment-naïve adult patients was subsequently approved. [Google Scholar]
- (10).Ghosh AK; Anderson DD; Weber IT; Mitsuya H Enhancing Protein Backbone Binding - A Fruitful Concept for Combating Drug-Resistant HIV. Angew. Chem., Int. Ed 2012, 51, 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Miller JF; Andrews CW; Brieger M; Furfine ES; Hale MR; Hanlon MH; Hazen RJ; Kaldor I; McLean EW; Reynolds D; Sammond DM; Spaltenstein A; Tung R; Turner EM; Xu RX; Sherrill RG Ultra-potent P1Modified Arylsulfonamide HIV Protease Inhibitors: The Discovery of GW0385. Bioorg. Med. Chem. Lett 2006, 16, 1788–1794. [DOI] [PubMed] [Google Scholar]
- (12).Cihlar T; He G-X; Liu X; Chen J; Hatada M; Swaminathan S; McDermott MJ; Yang Z-Y; Mulato AS; Chen X; Leavitt SA; Stray KM; Lee WA Suppression of HIV-1 Protease Inhibitor Resistance by Phosphonate-mediated Solvent Anchoring. J. Mol. Biol 2006, 363, 635–647. [DOI] [PubMed] [Google Scholar]
- (13).Dierynck I; Van Marck H; Van Ginderen M; Jonckers THM; Nalam MNL; Schiffer CA; Raoof A; Kraus G; Picchio G TMC310911, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor, Shows In Vitro an Improved Resistance Profile and Higher Genetic Barrier to Resistance Compared with Current Protease Inhibitors. Antimicrob. Agents Chemother 2011, 55, 5723–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ghosh AK; Martyr CD Darunavir (Prezista): A HIV-1 Protease Inhibitor for Treatment of Multidrug-Resistant HIV In Modern Drug Synthesis; Li JJ, Johnson DS, Eds.; Wiley: NJ, 2010; pp 29–44. [Google Scholar]
- (15).Ghosh AK; Kincaid JF; Walters DE; Chen Y; Chaudhuri NC; Thompson WJ; Culberson C; Fitzgerald PMD; Lee HY; McKee SP; Munson PM; Duong TT; Darke PL; Zugay JA; Schleif WA; Axel MG; Lin J; Huff JR Nonpeptidal P2-Ligands for HIV Protease Inhibitors: Structure-Based Design, Synthesis and Biological Evaluations. J. Med. Chem 1996, 39, 3278–3290. [DOI] [PubMed] [Google Scholar]
- (16).Ghosh AK; Chen Y Synthesis and Optical Resolution of High Affinity P2-Ligands for HIV-1 Protease Inhibitors. Tetrahedron Lett. 1995, 36, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ghosh AK; Leshchenko S; Noetzel M Stereoselective Photochemical 1,3-Dioxalene Addition to 5-Alkoxymethy1-2(5H>)-furanone: Synthesis of Bis-tetrahydrofuranyl Ligand for HIV Protease Inhibitor UIC-94–017 (TMC-114). J. Org. Chem 2004, 69, 7822–7829. [DOI] [PubMed] [Google Scholar]
- (18).Quaedflieg PJLM; Kesteleyn BRR; Wigerinck PBTP; Goyvaerts NMF; Vijn R; Liebregts CSM; Kooistra JHMH; Cusan C Stereoselective and Efficient Synthesis of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol. Org. Lett 2005, 7, 5917–5920. [DOI] [PubMed] [Google Scholar]
- (19).Black DM; Davis R; Doan BD; Lovelace TC; Millar A; Toczko JF; Xie S Highly Diastereo- and Enantioselective Catalytic Synthesis of the bis-Tetrahydrofuran Alcohol of Brecanavir and Darunavir. Tetrahedron: Asymmetry 2008, 19, 2015–2019. [Google Scholar]
- (20).Yu RH; Polniaszek RP; Becker MW; Cook CM; Yu LHL Research and Development of an Efficient Synthesis of Hexahydrofuro[2,3-b]furan-3-ol Moiety–A Key Component of the HIV Protease Inhibitor Candidates. Org. Process Res. Dev 2007, 11, 972–980. [Google Scholar]
- (21).Canoy WL; Cooley BE; Corona JA; Lovelace TC; Millar A; Weber AM; Xie S; Zhang Y Efficient Synthesis of (3R,3aS,6aR)- Hexahydrofuro[2,3-b]furan-3-ol from Glycolaldehyde. Org. Lett 2008, 10, 1103–1106. [DOI] [PubMed] [Google Scholar]
- (22).Hanessian S Total Synthesis of Natural Products: The Chiron Approach; Pergamon Press Inc: Oxford, UK, 1983. [Google Scholar]
- (23).Hanessian S; Giroux S; Merner BL Design and Strategy in Organic Synthesis: From the Chiron Approach to Catalysis, 1st ed.; Wiley-VCH Verlag GmbH & Co.: 2013. [Google Scholar]
- (24).For a related allylsilane reaction, please see: García-Tellado F; de Armas P; Marrero-Tellado JJ Highly 1,2-trans Stereoselective Allylations of 1,2-O-Isopropylidene-Protected Glycofuranosides. Angew. Chem., Int. Ed 2000, 39, 2727–2729. [DOI] [PubMed] [Google Scholar]
- (25).de Armas P; García-Tellado F; Marrero-Tellado JJ Enantioselective Synthesis of Medium-Sized Ring-Bridged Oxabicycles by Ring-Closing Metathesis. Eur. J. Org. Chem 2001, 2001, 4423–4429. [Google Scholar]
- (26).Ghosh AK; Kulkarni S; Anderson DA; Hong L; Baldridge A; Wang Y-F; Chumanevich AA; Kovalevsky AY; Tojo Y; Amano M; Koh Y; Tang J; Weber IT; Mitsuya H Design, Synthesis, Protein-Ligand X-ray Structure, and Biological Evaluation of a Series of Novel Macrocyclic Human Immunodeficiency Virus-1 Protease Inhibitors to Combat Drug Resistance. J. Med. Chem 2009, 52, 7689–7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rosenthal A; Shudo S Branched-Chain Glycosyl -Amino Acids. I. Stereospecific Synthesis of 2-L-(3“Deoxy-1,2-O-isopropyli-dene-α-D-allofuranos-3-yl)glycine, an Analog of the Polyoxin Sugar Moiety. J. Org. Chem 1972, 37, 4391–4395. [DOI] [PubMed] [Google Scholar]
- (28).For a similar preparation of compound 12, see:; Himmelbauer M; Farcet J-B; Gagnepain J; Mulzer J An Approach to the Carbon Backbone of Bielschowskysin, Part 1: Photocyclization Strategy. Eur. J. Org. Chem 2013, 2013, 8214–8244. [Google Scholar]
- (29).Ghosh AK; Doung TT; McKee SP; Thompson WJ, N,N’-dissuccinimidyl carbonate: A Useful Reagent for Alkoxycarbonylation of Amines. Tetrahedron Lett. 1992, 33, 2781–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tamamura H; Koh Y; Ueda S; Sasaki S; Yamasaki T; Aoki M; Maeda K; Watai Y; Arikuni H; Otaka A; Mitsuya H; Fujii N Reduction of Peptide Character of HIV Protease Inhibitors That Exhibit Nanomolar Potency against Multidrug Resistant HIV-1 Strains. J. Med. Chem 2003, 46, 1764–1768. [DOI] [PubMed] [Google Scholar]
- (31).Ghosh AK; McKee SP; Duong TT; Thompson WJ An Efficient Synthesis of Functionalized Urethanes from Azides. J. Chem. Soc., Chem. Commun 1992, 1308–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.