

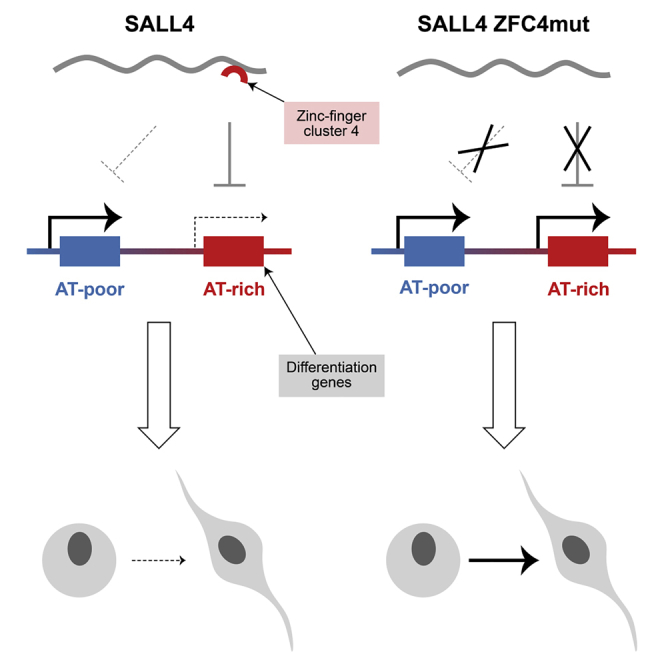
Summary
Mammalian genomes contain long domains with distinct average compositions of A/T versus G/C base pairs. In a screen for proteins that might interpret base composition by binding to AT-rich motifs, we identified the stem cell factor SALL4, which contains multiple zinc fingers. Mutation of the domain responsible for AT binding drastically reduced SALL4 genome occupancy and prematurely upregulated genes in proportion to their AT content. Inactivation of this single AT-binding zinc-finger cluster mimicked defects seen in Sall4 null cells, including precocious differentiation of embryonic stem cells (ESCs) and embryonic lethality in mice. In contrast, deletion of two other zinc-finger clusters was phenotypically neutral. Our data indicate that loss of pluripotency is triggered by downregulation of SALL4, leading to de-repression of a set of AT-rich genes that promotes neuronal differentiation. We conclude that base composition is not merely a passive byproduct of genome evolution and constitutes a signal that aids control of cell fate.
Keywords: DNA base composition, SALL4, gene regulation, differentiation, embryonic stem cells, pluripotency
Graphical Abstract
Highlights
-
•
SALL4 binds to short AT-rich motifs via its C-terminal zinc-finger cluster, ZFC4
-
•
SALL4 ZFC4 represses early differentiation genes in proportion to their AT content
-
•
Mutating ZFC4 leads to abnormal neuronal differentiation and embryonic lethality
-
•
The other zinc-finger clusters ZFC1 and ZFC2 seem dispensable in embryonic stem cells
The mammalian genome comprises domains with varying ratios of A/T to G/C base pairs. The significance of mosaic base composition was unknown, but we discovered that the stem cell protein SALL4 reads A/T content to inhibit the expression of many genes, thereby stabilizing the pluripotent state.
Introduction
A/T and G/C base pairs are nonrandomly distributed within mammalian genomes, forming large and relatively homogeneous AT-rich or GC-rich regions that usually encompass several genes together with their intergenic sequences. Base compositional domains are often evolutionarily conserved (Bernardi et al., 1985; Holmquist, 1989; Bickmore and Sumner, 1989; Costantini et al., 2009) and coincide with other genomic features (Bickmore and van Steensel, 2013), including early/late-replicating regions (Hiratani et al., 2009; Holmquist et al., 1982), lamina-associated domains (Meuleman et al., 2013), and topologically associating domains (Jabbari and Bernardi, 2017). Despite these interesting correlations, it is unclear whether conserved AT-rich and GC-rich domains are passive byproducts of evolution or DNA base composition can play an active biological role (Eyre-Walker and Hurst, 2001; Duret et al., 2006; Arhondakis et al., 2011). Exemplifying this second hypothesis, CpG islands represent conserved GC-rich domains (Bird, 1986) that are specifically bound by proteins recognizing unmethylated “CG” dinucleotides (Lee et al., 2001; Voo et al., 2000) to modulate chromatin structure and regulate gene expression (Thomson et al., 2010; Blackledge et al., 2010; Farcas et al., 2012; Wu et al., 2013; He et al., 2013).
Here, we tested the hypothesis that AT-rich DNA can be interpreted by specific proteins that recognize short AT-rich motifs whose frequency tracks fluctuations in base composition across the genome (Quante and Bird, 2016). To identify novel AT-binding proteins, we utilized a DNA pull-down mass spectrometry screen in mouse embryonic stem cells (ESCs), which are pluripotent and can be differentiated in culture. As a top hit, we identified SALL4, which is a multi-zinc-finger protein that restrains differentiation of ESCs (Yuri et al., 2009; Miller et al., 2016) and participates in several physiological processes, including neuronal development (Böhm et al., 2008; Sakaki-Yumoto et al., 2006; Tahara et al., 2019), limb formation (Akiyama et al., 2015; Koshiba-Takeuchi et al., 2006), and gametogenesis (Chan et al., 2017; Hobbs et al., 2012; Xu et al., 2017; Yamaguchi et al., 2015). Deletion of the Sall4 gene leads to embryonic lethality shortly after implantation (Sakaki-Yumoto et al., 2006; Elling et al., 2006; Warren et al., 2007). In humans, failure of SALL4 function is the cause of two severe developmental disorders: the recessive genetic disorder Okihiro syndrome (Al-Baradie et al., 2002; Kohlhase et al., 2002) and embryopathies due to treatment during pregnancy with the drug thalidomide (Donovan et al., 2018; Matyskiela et al., 2018). Despite its biological and biomedical importance, the molecular functions of SALL4 are incompletely understood. The extreme N terminus interacts specifically with the nucleosome remodeling and deacetylase (NuRD) co-repressor complex and can account for the transcriptional repression caused by SALL4 recruitment to reporter genes (Lauberth and Rauchman, 2006; Lu et al., 2009). In addition, there is evidence that the zinc-finger clusters bind to DNA (Sakaki-Yumoto et al., 2006; Xiong et al., 2016) or protein partners (Hobbs et al., 2012; Tanimura et al., 2013), though their precise developmental roles are unclear. The present work demonstrates that many of the defects seen in Sall4 null ESCs, including precocious differentiation, are mimicked by inactivation of a single zinc-finger cluster that interacts with AT-rich motifs. We go on to show that the ability of SALL4 to sense DNA base composition is essential to restrain transcription of genes that promote differentiation.
Results
A screen for AT-binding proteins in ESCs identifies SALL4
To identify proteins able to sense base composition, we used a DNA affinity purification approach coupled with stable isotope labeling with amino acids in cell culture (SILAC) quantitative mass spectrometry (Spruijt et al., 2013a, 2013b). Mouse ESC protein extracts were mixed with double-stranded DNA probes carrying variable runs of 5 base pairs composed only of A or T nucleotides (AT-1 and AT-2). As a negative control, matched probes with AT runs interrupted by G or C nucleotides were used as bait (Ctrl-1 and Ctrl-2). To robustly assess DNA-binding specificity, experiments were performed both in the “forward” (heavy-labeled AT probe versus light-labeled Ctrl-probe) and “reverse” (heavy-labeled Ctrl-probe versus light-labeled AT probe) orientations, which were considered as biological replicates. Proteins that bind specifically to AT runs show a high ratio in the forward experiments (Figure 1A, x axes) and a low ratio in the reverse experiments (Figure 1A, y axes). Mass spectrometry identified a consistent set of AT-binding proteins that largely overlapped between replicate experiments (Figure S1A) and between unrelated AT-rich probes (Figure S1B). High-confidence hits included proteins with well-characterized AT-binding domains such as AT hooks (Aravind and Landsman, 1998; Filarsky et al., 2015) (HMGA1, HMGA2, PRR12, and BAZ2A) and “AT-rich interaction domains” (Patsialou et al., 2005) (ARID3A, ARID3B, and ARID5B), thereby validating the screen (Figure 1A; Table S1). Three Spalt-like (SALL) family proteins (Sweetman and Münsterberg, 2006) (SALL1, SALL3, and SALL4) and most components of the NuRD complex (Tong et al., 1998; Wade et al., 1998; Xue et al., 1998; Zhang et al., 1998) were also recovered (Figure 1A). The most consistently enriched protein in our mass spectrometry screen was SALL4, whose AT binding we confirmed by western blot analysis using a variety of probes with one (AT-3) or more AT runs (Figure S1C). Considering their reported interaction with NuRD (Lauberth and Rauchman, 2006), we suspected that SALL proteins might be responsible for recruitment of this co-repressor complex to AT-rich DNA. To test this, we used extracts from mouse ESCs in which the Sall4 gene is disrupted (Sall4 knockout [S4KO] ESCs) (Sakaki-Yumoto et al., 2006; Miller et al., 2016). As predicted, recovery of NuRD components by AT-rich DNA was greatly reduced compared to wild type (WT) in the absence of SALL4 (Figure 1B).
Figure 1.

Identification of novel AT-binding proteins in ESCs by DNA pull-down mass spectrometry
(A) Scatterplots representing SILAC-based DNA affinity purifications, comparing AT-rich DNA probes (AT-1 and AT-2) with control probes having interrupted AT runs (Ctrl-1 and Ctrl-2). The ratio of the quantified proteins in the forward experiment (Heavy-AT/Light-Ctrl) is plotted on the x axis, and the ratio for the same proteins in the reverse experiment (Heavy-Ctrl/Light-AT) is plotted on the y axis. Proteins were considered to be specific interactors when showing at least a 2-fold ratio in both the forward and reverse experiments. These proteins cluster in the bottom right quadrant.
(B) DNA pull-down with AT-rich (AT-2) or control (Ctrl-2) probes followed by western blot analysis for SALL4 and NuRD components using wild-type (WT) or Sall4 knockout (S4KO) ESC protein extracts.
(C) Protein alignment of mouse SALL family members indicating conserved protein domains, including C2H2 zinc-finger clusters (ZFC1–ZFC4).
(D) Diagram showing the mutations or deletion introduced within SALL4 ZFC4 by CRISPR-Cas9.
(E) DNA pull-down with AT-rich (AT-3) or control (Ctrl-3) probe followed by western blot analysis for SALL4 using WT or Sall4 ZFC4mut/Δ ESC protein extracts. SALL4 levels were quantified and normalized to input. Data points indicate independent replicate experiments and error bars standard deviation.
(F) SALL4 immunofluorescence in the indicated ESC lines. DNA was stained with DAPI, showing dense clusters of AT-rich pericentric chromatin. Scale bars, 3 μm.
SALL4 binds to short AT-rich motifs via C2H2 zinc-finger cluster 4 (ZFC4)
Mammalian genomes encode four SALL family proteins (SALL1–SALL4), which each contain several clusters of C2H2 zinc fingers. Based on similarities in amino acid sequence between family members, the clusters are classified as ZFC1–ZFC4 (Figure 1C). SALL1, SALL3, and SALL4 all possess ZFC4 (Figure S1D), but SALL2 lacks this domain and was not recovered in our screen for AT-binding proteins. ZFC4 of both SALL1 and SALL4 was previously shown to interact with AT-rich heterochromatin in transfection assays (Sakaki-Yumoto et al., 2006; Yamashita et al., 2007), suggesting that it might be responsible for AT binding. To further characterize this domain, we used CRISPR-Cas9 to either delete ZFC4 (ZFC4Δ) or mutate two residues (T919D and N922A; mutated residues are shown in red) that we predicted would be involved in DNA binding (ZFC4mut) (Figure 1D). Homozygous mouse ESC lines expressing both mutated SALL4 proteins were obtained (Figure S1E), both of which retained the ability to interact with NuRD components by co-immunoprecipitation (Figure S1F). The interaction of SALL4 ZFC4mut or ZFC4Δ proteins with AT-rich sequences was drastically reduced (>10-fold) by inactivation of ZFC4, as assessed by the DNA pull-down assay (Figure 1E). This strongly suggests that the ZFC4 domain of SALL4 is primarily responsible for pull-down by AT-rich DNA. We next explored the in vivo DNA-binding properties of SALL4 ZFC4 in our mutant ESC lines. Heterochromatic foci, identified by DAPI staining in mouse cells, contain a high concentration of AT-rich satellite DNA (Matsuda and Chapman, 1991; Cerda et al., 1999; Guenatri et al., 2004) and therefore provide a low-resolution cellular assay for preferential AT binding. Immunostaining with a SALL4 antibody recognizing a preserved epitope in the two mutant proteins revealed a striking loss of ZFC4mut and ZFC4Δ protein localization at DAPI-dense foci (Figure 1F), further confirming that this zinc-finger cluster is necessary for AT targeting.
To define the sequence preference of the purified ZFC4 domain, we performed systematic evolution of ligands by exponential enrichment (SELEX) coupled with high-throughput sequencing (HT-SELEX), whereby a library of initially random DNA sequences immobilized on beads was subjected to repeated cycles of binding and amplification (Jolma et al., 2010; Nitta et al., 2015). After 0, 1, 3, or 6 cycles, DNA was analyzed by high-throughput sequencing to detect enriched motifs. For comparison, we performed HT-SELEX on other SALL4 zinc-finger clusters (ZFC1 and ZFC2) (Figure 2A) and also included a sample without added proteins to control for PCR bias. Strikingly, with increasing cycles of ZFC4 binding, the base composition of the whole library gradually shifted toward higher AT content, but this effect was not seen with ZFC1, ZFC2, or the negative control (Figure 2B). Progressive A/T motif enrichment was also apparent for ZFC4 alone (Figure 2C). To determine the minimum number of A/T base pairs required for enrichment, we separated the oligomers (k-mers) into different groups (Figure 2C). Enrichment was only detectable in k-mers containing 4 or more A/T base pairs, suggesting that this is the minimum target sequence. After 6 cycles, the most enriched SELEX motif was “ATATT” (Figure S2A), which also corresponds to the preferred sequence identified by DNA pull-down using all possible combinations of AT 5-mers (Figure S2B). However, this is one of several target sequences, as multiple other AT-rich sequences had similar enrichment scores (Figures 2B and S2A). All 16 possible A/T 5-mers are enriched more than G/C containing 5-mers, with k-mers containing TATA among the least favored AT-rich motifs for ZFC4 binding. We conclude that ZFC4 targets a broad range of short motifs that are composed only of A and T, whereas ZFC1 and ZFC2 showed no apparent DNA sequence specificity.
Figure 2.

Characterization of SALL4 C2H2 zinc-finger cluster 4 (ZFC4) DNA binding in vitro and in vivo
(A) SALL4 ZFC1, ZFC2, and ZFC4 protein fragments used for in vitro HT-SELEX experiments. A sample without added protein was used as a negative control.
(B) Base composition of HT-SELEX libraries following 1, 3, and 6 cycles with ZFC1 (dark gray), ZFC2 (light gray) and ZFC4 purified proteins (blue).
(C) Relative enrichment of 5-mer motifs categorized by AT content at cycles 0, 1, 3, and 6 of HT-SELEX with SALL4 ZFC1 (dark gray), ZFC2 (light gray), ZFC4 (blue), and negative control (black) samples. All 16 possible A/T 5-mer motifs are ordered according to their enrichment at cycle 6 of ZFC4 HT-SELEX.
(D) Venn diagram showing the overlap of SALL4 ChIP-seq peaks between WT and ZFC4mut ESCs.
(E) Profile plot and heatmap showing SALL4 ChIP-seq signal at SALL4 WT ChIP-seq peaks in the indicated cell lines.
(F) Venn diagram showing the overlap of SALL4 ChIP-seq peaks detected in WT ESCs with ATAC-seq peaks (accessible chromatin) and CpG islands.
(G) Results from de novo motif analysis at SALL4 WT ChIP-seq peaks (summit ±250 bp) showing the relative frequency of each DNA motif and its associated E-value. ATAC-seq peaks were used as a control for regions of accessible chromatin.
(H) Analysis of the DNA base composition surrounding SALL4 ChIP-seq peaks (summit ±250 bp) in WT (blue) and ZFC4mut (red) ESCs. CpG islands and ATAC-seq peaks coincide with regions of accessible chromatin and are shown for comparison.
See also Figure S2.
ZFC4 mutation drastically reduces SALL4 chromatin binding in vivo
To assess the influence of ZFC4 on SALL4 chromatin occupancy in vivo, we performed chromatin immunoprecipitation sequencing (ChIP-seq) using two anti-SALL4 antibodies (one monoclonal and one polyclonal) recognizing a C-terminal epitope that is distant from C2H2 zinc-finger clusters. We first determined antibody specificity (Kidder et al., 2011; Landt et al., 2012; Uhlen et al., 2016) by assessing SALL4 ChIP signal in S4KO ESCs as a negative control. Over 15,000 nonspecific ChIP-seq peaks were observed with the polyclonal anti-SALL4 antibody compared with only 280 peaks for the monoclonal antibody (Figures S2C and S2D). We therefore analyzed exclusively data obtained with the anti-SALL4 monoclonal antibody, considering only ChIP-seq peaks that were consistent between independent replicate experiments in WT or ZFC4mut ESCs (Figure S2E). In agreement with its reported localization at enhancers (Miller et al., 2016; Xiong et al., 2016), we observed that SALL4 ChIP-seq peaks in WT cells were enriched in the histone marks H3K27ac and H3K4me1 (Chronis et al., 2017), which typically mark these genomic sites (Figure S2F). Strikingly, ZFC4mut cells lost ∼95% of ChIP-seq peaks compared to WT (Figure 2D). Heatmaps confirmed the depletion of SALL4 peaks, although we noted a small amount of bound ZFC4mut at a subset of WT binding sites (Figure 2E).
We compared WT SALL4-binding sites as a whole with regions of open chromatin identified by assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq), which detects accessible DNA, including enhancers and promoters. SALL4 peaks largely coincide with a subset of ATAC-seq peaks, while avoiding CpG island promoters (Figure 2F). The AT-binding specificity of SALL4 suggests that this protein might preferentially associate with open chromatin sites that are more AT rich than average. The complete absence of SALL4 at ATAC-seq peaks within CpG islands (Figure 2F), within which runs of As and Ts are rare, is compatible with this notion. To quantify this effect, we used de novo motif analysis to determine whether SALL4 peaks were consistent with a bias toward AT-rich motifs. First, by seeking recurrent motifs (<8 base pairs) coincident with SALL4 peaks, we identified short AT-rich motifs that were highly enriched at the majority (∼90%) of SALL4-binding sites compared with lower levels of enrichment (∼60%) in open chromatin generally (Figure 2G). As a second approach, we determined the base composition at SALL4-bound regions by analyzing the DNA sequences surrounding SALL4 ChIP-seq peak summits (±250 bp). SALL4 binding sites are relatively AT rich (50%–70% AT) (Figure 2H) compared with ATAC-seq peaks as a whole (40%–60% AT) (Figure 2H). Taken together, the data suggest that AT-motif binding is responsible for the presence of SALL4 at a subset of open chromatin sites.
SALL4 ZFC4 represses the expression of early differentiation genes in a base-composition-dependent manner
To determine whether SALL4 binding to AT-rich DNA causes gene expression changes that correlate with base composition, we performed RNA sequencing (RNA-seq) in WT, ZFC4mut, ZFC4Δ, and S4KO ESCs. Sall4 gene knockout resulted in the dysregulation of several thousand genes (Figure 3A). Both ZFC4 mutations caused the dysregulation of fewer genes, many of which overlapped with those affected in S4KO cells (Figure 3A). To test the relationship between AT composition and gene expression, genes differentially regulated in both ZFC4mut and ZFC4Δ ESCs (Figure 3A, red filling) were divided into five equal categories according to AT content across the entire transcription unit (Figure 3B), and the level and direction of transcriptional change was compared between them. In agreement with our hypothesis, genes differentially regulated in ZFC4mut/Δ cells showed progressively increased upregulation with rising AT content (Figure 3C). To quantify the strength of the relationship between AT content and gene expression, we fitted a linear regression model and calculated coefficient estimates. This independent approach, which reveals the variation in gene expression that can be attributed to base composition, confirmed that the positive relationship between AT content and upregulation in the ZFC4 mutants is significant (false discovery rate [FDR] < 0.01; see STAR methods; Table S2; Figure S3A). In contrast, genes differentially regulated in S4KO, but not in either of the ZFC4 mutant ESCs (Figure 3A, gray filling), showed a nonsignificant correlation (FDR > 0.01) and an effect size close to zero (Figures 3D, S3B, and S3C). The results show that the subset of SALL4-regulated genes that is dependent on ZFC4 is repressed in pluripotent cells according to the AT richness of their genomic setting.
Figure 3.

SALL4-mediated transcriptional regulation in relation to DNA base composition
(A) Venn diagram showing the overlap of differentially expressed genes detected by RNA-seq among S4KO, ZFC4mut, and ZFC4Δ ESCs. ZFC4-regulated genes are indicated in red and ZFC4-independent genes in gray.
(B) Profile plot showing the density of A/T nucleotides around the transcription unit of ZFC4-regulated genes divided into five equal categories according to AT content. TSS, transcription start site; TES, transcription end site.
(C and D) Correlation between gene mis-regulation (log2 fold change versus WT) and DNA base composition in Sall4 mutant ESCs. ZFC4-regulated (C) and ZFC4-independent (D) genes were divided into five equal categories depending on their AT content.
(E) Diagram representing S4KO ESC lines carrying SALL4 or EGFP (control) expression constructs under control of a doxycycline-inducible promoter.
(F) Venn diagram showing the overlap of differentially expressed genes detected by RNA-seq following a 48-h doxycycline induction in the ESC lines presented in (E). SALL4-responsive genes are indicated in blue and EGFP-responsive genes in green.
(G and H) Correlation between SALL4-induced gene expression changes and DNA base composition. SALL4-responsive (G) and ZFC4-regulated (H) genes were divided into five equal categories depending on their AT content, and their relative expression levels were analyzed in the indicated ESC lines.
(I) Diagram showing the protocol used to characterize early differentiation of WT ESCs.
(J) Venn diagram showing the overlap between genes changing during early differentiation of WT cells (day 0 versus day 2) with genes de-regulated in Sall4 mutant ESCs. Genes were divided into three categories: SALL4-independent genes (light blue), SALL4-dependent genes controlled by ZFC4 (red), and SALL4-dependent genes not controlled by ZFC4 (gray).
(K) Correlation between gene expression changes occurring during early differentiation and DNA base composition in WT cells. SALL4-independent genes (light blue), SALL4-dependent genes controlled by ZFC4 (red), and SALL4-dependent genes not controlled by ZFC4 (gray) were divided into five equal categories depending on their AT content, and their relative expression levels were analyzed at day 2 of differentiation.
To further test the hypothesis that AT binding by ZFC4 mediates repression according to base composition, we examined the reverse situation of SALL4 overexpression on transcription. This was performed by expressing SALL4, or as a negative control enhanced green fluorescent protein (EGFP), from a doxycycline-inducible promoter following random integration of expression constructs in S4KO ESCs (Figure 3E). After 48 h of induction, SALL4 was robustly overexpressed in these cells (Figures S3D and S3E). To characterize the effect of SALL4 reexpression on transcription, we performed RNA-seq on induced (+Dox) and noninduced (−Dox) cell lines (Figure 3F). As expected, gene expression changes in cells overexpressing SALL4 were anticorrelated with expression changes seen in S4KO cells (Figure S3F). Separation of differentially expressed genes into categories according to their AT content as before revealed that SALL4 expression caused transcriptional repression that was strikingly proportional to the base composition of the affected genes (Figures 3G and S3G). A similar relationship was observed when looking at genes differentially regulated in ZFC4mut/Δ ESCs (Figure 3H). Linear regression analysis again confirmed the significance of these relationships (Figures S3H and S3I). As a control, we applied the same analysis to the minority of genes whose expression was altered in response to EGFP induction (Figure 3F, green filling). In this case, there was no apparent relationship between fold change and base composition (Figures S3J and S3K), as confirmed by quantitative analysis (Figure S3L). Together, our results strongly suggest that SALL4 directly regulates gene expression in response to base composition via its zinc-finger cluster, ZFC4.
Interestingly, Gene Ontology (GO) analysis of ZFC4-regulated genes identified GO terms associated with neuronal differentiation, morphogenesis, gonad development, and kidney development (Table S3), all of which are adversely affected in S4KO mice and embryos (Sakaki-Yumoto et al., 2006; Böhm et al., 2008; Tahara et al., 2019; Koshiba-Takeuchi et al., 2006; Akiyama et al., 2015; Hobbs et al., 2012; Yamaguchi et al., 2015; Xu et al., 2017; Chan et al., 2017; Warren et al., 2007). This suggests the possibility that SALL4 plays an essential role in the transition between self-renewing ESCs and the differentiated state by preferentially suppressing the expression of AT-rich developmental genes, thus preventing premature loss of pluripotency. If so, AT-rich genes that are aberrantly upregulated in the absence of ZFC4 should be activated during the normal differentiation program of WT cells (Rao et al., 2010). To test this, we performed RNA-seq on WT ESCs following 2 days of monolayer differentiation (Figure 3I) (Aubert et al., 2002). Although they represent a small fraction of all transcriptional changes taking place at these stages, SALL4-regulated genes overlapped significantly with genes whose expression changes naturally between day 0 (ESCs) and day 2 of differentiation (Figure 3J). Importantly, ZFC4-regulated genes, but not other categories of genes, are upregulated at this early stage in proportion to AT richness (Figures 3K, S3M, and S3N). Thus, AT-rich genes that are repressed by SALL4 in ESCs are activated soon after the exit from pluripotency.
SALL4 ZFC4 is critical for neuronal differentiation and embryonic development
Previous work demonstrated that disruption of the Sall4 gene leads to increased stem cell differentiation (Yuri et al., 2009; Miller et al., 2016). To test whether disrupting ZFC4 alone leads to phenotypic defects, we compared ZFC4mut and S4KO ESCs. Consistent with previous evidence showing that SALL4 is dispensable for the maintenance of pluripotency (Sakaki-Yumoto et al., 2006; Yuri et al., 2009; Tsubooka et al., 2009), both S4KO and ZFC4mut ESCs expressed normal levels of OCT4 (Figure S4A) and showed efficient self-renewal, with a modest decrease observed in S4KO ESCs (Figure S4B). Next, we used a monolayer differentiation assay, as described above, to assess the propensity of these cell lines to acquire a neuronal fate. After 5 days in N2B27 medium, ESCs lacking SALL4 or expressing a ZFC4 mutant protein generated many more TUJ1-positive cells compared to WT cells (Figure 4A). Further confirming increased neuronal differentiation, qRT-PCR analyses identified increased transcription of Tuj1 (4- to 12-fold), Ascl1 (3- to 6-fold), and Nestin (∼2-fold) in S4KO, ZFC4mut, and ZFC4Δ ESCs at day 5 of differentiation (Figure 4B). By this assay, inactivation of ZFC4 phenocopies complete loss of SALL4 protein.
Figure 4.

Phenotypic effects of SALL4 ZFC4 mutation on neuronal differentiation
(A) TUJ1 immunofluorescence in the indicated ESC lines cultured in serum/leukemia inhibitory factor (LIF) medium, and following differentiation for 5 days in N2B27 medium. DNA was stained with DAPI. Scale bars, 100 μm.
(B) qRT-PCR analysis of the neuronal markers Tuj1, Ascl1, and Nestin in the indicated cell lines following differentiation for 5 days in N2B27 medium. Transcript levels were normalized to TATA-binding protein (TBP) and expressed relative to WT. Data points indicate independent replicate experiments and error bars standard deviation.
See also Figure S4.
In order to observe the effects of ZFC4 mutation on embryonic development, we generated a ZFC4mut mouse line by blastocyst injection of heterozygous Sall4ZFC4mut/WT ESCs. F1 mice were crossed and their progeny analyzed at different stages of development. While ZFC4mut homozygous embryos were obtained at Mendelian ratios during early development, none survived until birth (Figures 5A and 5B). By embryonic day 10.5 (E10.5), homozygous embryos presented gross morphological abnormalities, which were not observed in controls (Figure 5C). Importantly, the ZFC4mut protein was expressed at levels similar to those seen in WT embryos (Figures S5A and S5B). Early embryonic mortality of ZFC4 mutant mice is reminiscent of the phenotype observed in S4KO mice, although the latter die earlier in development, shortly after implantation (by E5.5–E6.5) (Elling et al., 2006; Sakaki-Yumoto et al., 2006). Taken together, our in vitro and in vivo experiments indicate that mutation of ZFC4 alone phenocopies important aspects of the S4KO phenotypes seen in both ESCs and embryos. It follows that this DNA-binding domain is a key contributor to SALL4 biological function.
Figure 5.

Mutation of SALL4 ZFC4 causes embryonic lethality
(A) Table showing the number of live pups and embryos at different stages of development and their associated genotype. Animals were crossed to obtain ZFC4mut heterozygous (Het), homozygous (Hom), or WT progeny.
(B) Diagram showing the results from crossing ZFC4mut heterozygote mice. ZFC4mut homozygous animals die during embryonic development.
(C) Representative images of WT, ZFC4mut heterozygous (Het), and homozygous (Hom) embryos at E10.5, taken at the same magnification.
See also Figure S5.
C2H2 zinc-finger clusters ZFC1 and ZFC2 are dispensable for SALL4 function in ESCs
SALL4 contains two C2H2 zinc-finger clusters, ZFC1 and ZFC2, in addition to ZFC4. To determine their contribution to SALL4 function, we used CRISPR-Cas9 to delete the central segment of endogenous SALL4 protein, which contains the zinc-finger clusters ZFC1 and ZFC2, while leaving ZFC4 and the N-terminal domain intact (Figure 6A). ESCs homozygous for this ZFC1-2Δ knockin allele lack full-length SALL4, but, as expected, ZFC1-2Δ protein retained the ability to interact with SALL1 and NuRD components (Figure S6A). Immunostaining showed that ZFC1-2Δ resembled WT SALL4 by being enriched at heterochromatic foci, indicating that ZFC4 binding to this AT-rich DNA in vivo is unaffected by the internal deletion (Figure 6B). To characterize ZFC1-2Δ chromatin binding in more detail, we performed ChIP-seq (Figure S6B), as described above. In contrast to the dramatic effect of ZFC4 inactivation on SALL4 ChIP-seq peaks, ZFC1-2Δ occupancy of the genome closely resembled that of WT SALL4 (Figure S6C). In addition, both the average ChIP-seq signal (Figure 6C) and AT-rich profile (Figure 6D) of WT SALL4 peaks were preserved in ZFC1-2Δ cells. We conclude that ZFC1 and ZFC2 contribute minimally to the genome binding profile of SALL4, further supporting the view that ZFC4 is the primary determinant of DNA binding. Comparative RNA-seq analysis between WT, ZFC4mut, and ZFC1-2Δ ESCs revealed that SALL4 ZFC1-2Δ and ZFC4mut affect largely nonoverlapping sets of genes (Figure 6E). The effects of ZFC1-2Δ on transcription were independent of base composition, whereas ZFC4 regulated genes in proportion to their AT richness (Figure 6F, S6D, S6E and S6F). Finally, we examined the phenotypic consequences of ZFC1-2 deletion by assaying monolayer neuronal differentiation of our mutant ESCs. Unlike S4KO and ZFC4mut ESCs, ZFC1-2Δ cells did not show evidence of increased differentiation as assessed by TUJ1 immunofluorescence (Figure 6G) and RT-qPCR analysis of neuronal markers at day 5 of differentiation (Figure S6G).
Figure 6.

Effects of SALL4 ZFC1 and ZFC2 deletion in ESCs on chromatin binding, gene expression, and differentiation
(A) Diagram showing the in-frame deletion within the Sall4 coding sequence, generated by CRISPR-Cas9.
(B) SALL4 ZFC1-2Δ localization determined by immunofluorescence in the indicated ESC lines. DNA was stained with DAPI, showing dense clusters of AT-rich pericentric chromatin. Scale bars, 3 μm.
(C) Heatmap and profile plot showing SALL4 ChIP-seq signal at SALL4 WT ChIP-seq peaks in the indicated cell lines.
(D) Analysis of the DNA base composition surrounding SALL4 ChIP-seq peaks (summit ±250 bp) in WT (blue) and ZFC1-2Δ (purple) ESCs.
(E) Venn diagram showing the overlap of differentially expressed genes detected by RNA-seq between ZFC4mut and ZFC1-2Δ ESCs. ZFC4-regulated genes are indicated in red and ZFC1/2-regulated genes in purple.
(F) Correlation between gene mis-regulation (log2 fold change versus WT) and DNA base composition in Sall4 mutant ESCs. ZFC4-regulated (red) and ZFC1/2-regulated (purple) genes were divided into five equal categories depending on their AT content.
(G) TUJ1 immunofluorescence in the indicated ESC lines cultured in serum/LIF medium and following differentiation for 5 days in N2B27 medium. DNA was stained with DAPI. Scale bars, 100 μm.
To further characterize the differentiation defects of Sall4 mutant ESCs, we performed an RNA-seq time-course experiment with WT, S4KO, ZFC4mut, and ZFC1-2Δ cell lines at days 0 (ESCs), 2, and 5 of the differentiation protocol (Figure 7A). In agreement with our previous base-composition analyses, absence of SALL4 or inactivation of ZFC4 weakened repression, leading to premature activation of ZFC4-regulated AT-rich genes at all differentiation time points (Figure S7A). In contrast, ZFC1/2-regulated genes showed no preferential upregulation during differentiation and no correlation with base composition in any of the cell lines (Figure S7B). Moreover, principal-component analysis (PCA) showed that WT and ZFC1-2Δ samples clustered together at all time points, while S4KO and ZFC4mut formed an independent cluster at days 2 and 5 (Figure S7C). Accordingly, differential expression analysis across our time series revealed few differences between WT and ZFC1-2Δ, while the transcriptomes of S4KO and ZFC4mut were significantly disturbed (Figure 7B). Emphasizing the similarity of S4KO and ZFC4mut, genes differentially regulated in these cell lines were highly correlated both at day 2 and day 5 of differentiation (Figures 7C and S7D). Also, genes associated with neuronal differentiation were upregulated in both cell lines, whereas expression of these genes in ZFC1-2Δ cells was unaffected (Figure 7D). We conclude that the characteristic premature differentiation phenotype associated with SALL4 deficiency is mimicked by inactivation of ZFC4, but not by a large deletion of the central domain that includes ZFC1 and ZFC2.
Figure 7.

Transcriptional effects of SALL4 zinc-finger cluster mutations during neuronal differentiation
(A) Diagram of the RNA-seq time-course experiment comparing the differentiation potential of WT and Sall4 mutant ESCs.
(B) Differential gene expression analysis between WT and Sall4 mutant cell lines during neuronal differentiation at days 2 and 5 (adjusted p value < 0.05). Additional WT replicates were used as a control (WT versus WT).
(C) Scatterplot showing the relative expression levels of genes deregulated in differentiating S4KO cells (see B, gray bars) correlating with their expression in ZFC4mut cells at days 2 and 5 of differentiation.
(D) Relative expression levels (log2 fold change versus WT) of genes associated with the GO term “positive regulation of neuron differentiation” (GO:0045666) in Sall4 mutant cell lines at days 2 and 5 of differentiation. Additional WT replicates were used as a control (WT versus WT). Stars indicate statistical significance calculated from bootstrapped two-sided t tests (p value < 0.05).
Discussion
SALL4 targets a broad range of AT-rich motifs via the zinc-finger cluster ZFC4. While the ZFC4 domain has previously been implicated in binding to AT-rich repetitive DNA found in mouse major satellite (Yamashita et al., 2007), its biological significance was unknown. Our study demonstrates that ZFC4 is a key domain mediating SALL4 biological function in ESCs. Its inactivation drastically reduces peaks of SALL4 binding to the genome, suggesting that this domain plays a critical role in SALL4 targeting to chromatin. Disruption of the genomic binding pattern is accompanied by mis-regulation of a subset of all SALL4-regulated genes, many of which are implicated in neuronal differentiation, which is the preferred fate of ESCs in culture. Accordingly, ESCs expressing SALL4 lacking a functional ZFC4 domain phenocopy S4KO ESCs by displaying precocious differentiation toward the neuronal lineage. Importantly, we found that this gene set is normally activated as WT ESCs commence differentiation in culture to give neurons, a process that coincides with downregulation of SALL4 protein (Rao et al., 2010). The importance of ZFC4 is further demonstrated by the embryonic lethal phenotype of ZFC4mut homozygotes.
Levels of ZFC4 mutant proteins are somewhat reduced in the mutant ESCs (50%–75% of WT), but we consider it unlikely that this contributes to the biased effect on expression of AT-rich genes. The changes in genome occupancy revealed by ChIP and immunostaining are much greater than 2-fold and are unlikely to be due to the relatively modest reduction in protein levels. In addition, the early embryonic lethality of the ZFC4 mutation is much more severe than that seen in mice heterozygous for the Sall4 KO allele, which can be viable and fertile despite having 50% less protein (Sakaki-Yumoto et al., 2006). Notably, the reduction in ZFC4mut protein seen in ESCs is not reproduced in mouse embryos, as mutant and WT proteins are present at indistinguishable levels, yet the phenotype is nevertheless extremely severe. On the other hand, loss of the only AT-binding domain in the protein offers an attractive explanation for this phenomenon. Human genetics provides further support for the central importance of ZFC4. Mutations in the SALL4 gene cause Okihiro syndrome (Al-Baradie et al., 2002; Kohlhase et al., 2002; Terhal et al., 2006), with most patients carrying frameshift or nonsense mutations leading to deletion or severe truncation of the protein. The only reported disease-causing missense mutation (H888R) affects a zinc-coordinating histidine that is expected to specifically inactivate ZFC4 (Miertus et al., 2006), although this has not been tested experimentally.
Evidence regarding the functional significance of two other zinc-finger clusters, ZFC1 and ZFC2, is limited, although an affinity of ZFC1 for hydroxymethylcytosine has been reported (Xiong et al., 2016). Importantly, simultaneous deletion of ZFC1 and ZFC2 has a minimal effect on genome occupancy, gene expression, and propensity to differentiate of ESCs. Thus, the well-known role of SALL4 in stabilization of the pluripotent state appears to be largely attributable to the DNA-binding specificity of ZFC4. Our observations agree with previous studies using transfection assays that indicated that the naturally occurring isoform SALL4B, which closely resembles ZFC1-2Δ in lacking ZFC1 and ZFC2 and is expressed at much lower levels than the full-length SALL4A form, retains biological activity in pluripotent cells (Rao et al., 2010; Miller et al., 2016). Although these results suggest that these two C2H2 zinc-finger clusters are dispensable for SALL4 function in ESCs, we note that their sequence is highly conserved between fruit flies and humans. It is therefore likely that ZFC1 and ZFC2 are functional in other developmental contexts, such as limb development and/or gametogenesis.
At first sight, the correlation with base composition across the extended transcription unit contrasts with the relatively sharp peaks of SALL4 binding observed by ChIP-seq. In fact, it remains to be determined whether SALL4 acts at distance from AT-rich motifs in discrete regulatory elements or by binding broadly to AT-rich sequences dispersed through gene bodies. The latter would be challenging to detect by ChIP due to the high abundance of AT-rich motifs throughout the genome (potentially in excess of 10 million target sites) in contrast with the low abundance of SALL4 protein in ESCs (2,000–3,000 copies per cell) (Zhang et al., 2017). As a result of this discrepancy, percent occupancy of any one target site is likely to be extremely low. Further work is required to distinguish the effects on gene expression of dispersed versus focal SALL4 binding. An obvious potential mediator of repression by SALL4 is the NuRD co-repressor complex, which has long been known to associate with the N terminus of SALL4 (Lauberth and Rauchman, 2006). The role of NuRD recruitment for SALL4 function has been questioned, however (Miller et al., 2016). Another poorly understood aspect of SALL4 biochemistry is its interaction with other members of the SALL family (Kiefer et al., 2003; Sweetman et al., 2003). Notably, our screen for AT-binding proteins also identified SALL1 and SALL3, which both interact with SALL4 and might contribute to sensing AT content via their closely similar ZFC4 domains. Given the importance of SALL4 in development and disease, these issues deserve further investigation.
Our results demonstrate that cell-type-specific genes residing within AT-rich domains are susceptible to repression by the transcriptional repressor, SALL4, thereby preventing differentiation. Vertebrate genomes are on average relatively AT rich (60% A/T), and therefore, the short A/T motifs to which it binds occur throughout the genome with frequencies that vary probabilistically according to local base composition. As base composition is a constant feature of the genome, regulation is achieved by varying the availability of the base composition reader itself. Accordingly, as cells enter differentiation, expression of SALL4 drops (Rao et al., 2010), raising the possibility that differentiation is triggered by loss of SALL4-mediated inhibition of key developmental genes. Global regulation of this kind confers the ability to modulate expression of multi-gene blocks using relatively few base composition readers and is potentially more economical than controlling each gene by a separate mechanism. Our finding that this relatively simple system may underlie large-scale switching of gene expression programs indicates that base compositional domains are not merely a biologically irrelevant byproduct of genome evolution but constitute a signal that is advantageous to the organism.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SALL4 | Abcam | ab29112; RRID: AB_777810 |
| SALL4 | Santa Cruz Biotechnology | sc-101147; RRID: AB_1129262 |
| CHD4 | Abcam | ab70469; RRID: AB_2229454 |
| MTA1 | Santa Cruz Biotechnology | sc-9446; RRID:AB_649545 |
| MTA1 | Santa Cruz Biotechnology | sc-373765; RRID: AB_10917039 |
| SALL1 | Abcam | ab41974; RRID: AB_777807 |
| GATAD2A | Abcam | ab87663; RRID: AB_1952305 |
| MBD3 | Bethyl | A302-528A; RRID: AB_1998980 |
| HDAC1 | Santa Cruz Biotechnology | sc-6298; RRID: AB_2279712 |
| HDAC1 | Santa Cruz Biotechnology | sc-81598; RRID: AB_2118083 |
| OCT4 | Abcam | ab19857; RRID: AB_445175 |
| TUJ1 | BioLegend | 801201; RRID: AB_2313773 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant ZFC4 (SALL4 residues 859-1028) | This paper | N/A |
| Recombinant ZFC1 (SALL4 residues 320-486) | This paper | N/A |
| Recombinant ZFC2 (SALL4 residues 551-662) | This paper | N/A |
| Critical commercial assays | ||
| KAPA RNA Hyperprep Kit with RiboErase (RNA-seq library preparation) | Roche | 08098131702 |
| KAPA Hyperprep Kit (ChIP-seq library preparation) | Roche | 07962347001 |
| Deposited data | ||
| Raw and processed data used to generate the Figures | This paper | Mendeley Data: https://doi.org/10.17632/rwzttj9pn2.1 |
| RNA-seq of WT, S4KO, ZFC4mut and ZFC4Δ ESCs | This paper | Array Express: E-MTAB-7343 |
| RNA-seq of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs | This paper | Array Express: E-MTAB-7655 |
| ChIP-seq of anti-SALL4 in WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs | This paper | Array Express: E-MTAB-9197 |
| Time-course (day 0, 2 and 5) RNA-seq of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs | This paper | Array Express: E-MTAB-9198 |
| RNA-seq of S4KO cells integrated with Sall4 cDNA or EGFP cDNA with a doxycycline inducible promoter | This paper | Array Express: E-MTAB-9202 |
| HT-SELEX of recombinant C2H2 zinc finger domains of SALL4 | This paper | Array Express: E-MTAB-9236 |
| ATAC-seq in WT ESCs | This paper | Array Express: E-MTAB-9245 |
| ChIP-seq of H3K4me1 and H3K27ac in WT ESCs | Chronis et al., 2017 | Gene Expression Omnibus: GSE90893 |
| Experimental models: cell lines | ||
| Mouse: Wild-type embryonic stem cell line; E14Ju09 | Hooper et al., 1987 | N/A |
| Mouse: Sall4 knockout embryonic stem cell line | Provided by Brian Hendrich laboratory (Miller et al., 2016) | N/A |
| Mouse: Sall4 ZFC4mut embryonic stem cell line | This paper | N/A |
| Mouse: Sall4 ZFC4Δ embryonic stem cell line | This paper | N/A |
| Mouse: Sall4 ZFC1-2Δ embryonic stem cell line | This paper | N/A |
| Mouse: Sall4 knockout embryonic stem cell line with doxycycline-inducible Sall4 construct | This paper | N/A |
| Mouse: Sall4 knockout embryonic stem cell line with doxycycline-inducible EGFP construct | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: Sall4 ZFC4mut | This paper | N/A |
| Oligonucleotides | ||
| DNA pulldown oligonucleotides | This paper; see Table S1 | N/A |
| CRISPR/Cas9 oligonucleotides and genotyping primers | This paper; see Table S1 | N/A |
| RT-qPCR primers | This paper; see Table S1 | N/A |
| HT-SELEX oligonucleotides | This paper; see Table S1 | N/A |
| Recombinant DNA | ||
| Cas9/gRNA expression plasmid | Ran et al., 2013 | Addgene PX330 |
| PiggyBac vector with doxycycline-inducible construct and Hygromycin resistance cassette | This paper | N/A |
| PiggyBac vector with constitutively expressed Tet-On 3G and Zeocin resistance cassette | This paper | N/A |
| Hyperactive PiggyBac transposase expression plasmid | Yusa et al., 2011 | N/A |
| Software and Algorithms | ||
| Logolas | Dey et al., 2018 | N/A |
| sailfish v0.9.2 | Patro et al., 2014 | N/A |
| DESeq2 v1.28.0 | Love et al., 2014 | N/A |
| bedtools nuc | Quinlan and Hall, 2010 | N/A |
| deepTools | Ramírez et al., 2014 | N/A |
| clusterProfiler | Yu et al., 2012 | N/A |
| Trimmomatic v0.33 | Bolger et al., 2014 | N/A |
| bwa-mem v0.7.17 | Li, 2013) | N/A |
| Picard toolkit | http://broadinstitute.github.io/picard/ | N/A |
| computeGCBias | Benjamini and Speed, 2012 | N/A |
| MACS v2.1.2 | Zhang et al., 2008 | N/A |
| MEME-ChIP v5.1.0 | Machanick and Bailey, 2011 | N/A |
| Bioinformatics analysis – command line arguments | This paper (Methods S1) | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Adrian Bird (a.bird@ed.ac.uk).
Materials availability
Reagents generated in this study are available upon request from the Lead Contact.
Data and code availability
Raw and processed high-throughput sequencing data was deposited on Array Express, as described in the Table below. Python scripts and source code used for bioinformatic analyses, raw western blot and microscopy images, as well as other types of unprocessed and processed data used to generate the figures are available on Mendeley Data (https://doi.org/10.17632/rwzttj9pn2.1).
For H3K4me1 and H3K27ac ChIP-seq in ESCs, previously published data were obtained from GEO (accession number: GSE90893).
| Accession | Description |
|---|---|
| E-MTAB-7343 | RNA-seq of WT, S4KO, ZFC4mut and ZFC4Δ ESCs |
| E-MTAB-7655 | RNA-seq of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9197 | SALL4 ChIP-seq in WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9198 | Time course RNA-seq during differentiation (day 0, 2 and 5) of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9202 | RNA-seq of S4KO cells carrying Sall4 cDNA or EGFP cDNA under a doxycycline inducible promoter |
| E-MTAB-9236 | HT-SELEX of recombinant C2H2 zinc-finger domains of SALL4 |
| E-MTAB-9245 | ATAC-seq in WT ESCs |
Experimental model and subject details
In vivo animal studies (mouse)
The Sall4 ZFC4mut mouse line was generated by injection of CRISPR/Cas9-targeted heterozygous ESCs (see section above) into mouse blastocysts using standard methods. Resultant male and female chimeras were crossed with C57BL/6J wild-type animals at 7 weeks and coat color was used to identify germline offspring. Transmission of the targeted allele was confirmed by PCR (see primers) and Sanger sequencing. Heterozygotes identified from these crosses were inter-crossed to generate homozygotes. Animals were routinely genotyped by PCR combined with restriction fragment length polymorphism (RFLP) analysis using HaeII (restriction site introduced within ZFC4mut allele). Heterozygosity had no discernible phenotype in either sex at any age. The sex of homozygous blastocysts and embryos was not determined post-mortem.
All mice used in this study were bred and maintained at the University of Edinburgh animal facilities under standard conditions, and procedures were carried out by staff licensed by the UK Home Office and in accordance with the Animal and Scientific Procedures Act 1986 following initial approval by a local Animal Welfare and Ethical Review Board. All mice were housed within a SPF facility. They were maintained on a 12h light/dark cycle and given ad libitum access to food and water. They were housed in open top cages with wood chippings, tissue bedding and additional environmental enrichment in groups of up to ten animals. Mutant mice were caged with their wild-type littermates.
In vitro cell culture studies (mouse)
Cell culture conditions
E14Ju09, a clonal cell line derived from E14Tg2a ESCs (Hooper et al., 1987), was used as a wild-type cell line in this study. Sall4 ZFC4mut, ZFC4Δ, and ZFC1-2Δ ESC lines were derived from E14Ju09 ESCs using CRISPR/Cas9, as indicated below. Sall4 knockout ESCs were kindly provided by Brian Hendrich (Cambridge University) with agreement of Riuchi Nishinakamura (Kumamoto University) (Miller et al., 2016). SALL4 and EGFP doxycycline-inducible ESC lines were derived from Sall4 knockout ESCs using the PiggyBac (PB) transposon system, as indicated below.
All ESC lines were incubated at 37°C and 5% CO2 in gelatin-coated dishes containing Glasgow minimum essential medium (GMEM; GIBCO ref. 11710035) supplemented with 15% fetal bovine serum (batch tested), 1x L-glutamine (GIBCO ref. 25030024), 1x MEM non-essential amino acids (GIBCO ref.11140035), 1mM sodium pyruvate (GIBCO ref. 11360039), 0.1mM 2-mercaptoethanol (GIBCO ref. 31350010) and 100U/ml leukemia inhibitory factor (LIF, batch tested).
To differentiate ESCs into neurons, we performed monolayer neuronal differentiation (Aubert et al., 2002). ESCs were washed with PBS, dissociated using Accutase (StemPro ref. A1110501) and resuspended in N2B27 medium: 1:1 mix of Advanced DMEM/F-12 (GIBCO ref. 12634010) and Neurobasal (GIBCO ref. 21103049) supplemented with 1x L-Glutamine (GIBCO ref. 25030024), 1x MEM non-essential amino acids (GIBCO ref.11140035), 0.5x N-2 supplement (GIBCO ref. 17502048), 0.5x B-27 Supplement (GIBCO ref. 17504044) and 0.1mM 2-mercaptoethanol (GIBCO ref. 31350010). The appropriate number of cells (100,000 cells per well of a 6-well plate) was transferred into gelatin-coated plates containing N2B27 medium. The medium was changed every 2 days until analysis.
To assess self-renewal efficiency, ESCs were plated at clonal density (600 cells per well of a 6-well plate) in matrigel-coated (Corning ref. 354277) plates with N2B27 medium (see composition above) supplemented with “2i” inhibitors (Ying et al., 2008) (1μM PD0325901 (Axon ref. 1408) and 3μM CHIR99021 (Axon ref. 1386)) and 100U/ml LIF. Following 7 days of culture, cells were fixed and stained for alkaline phosphatase activity (AP) following manufacturer’s instructions (Sigma-Aldrich ref. 86R-1KT). AP-positive colonies were imaged using a brightfield microscope (Nikon Ti2) and automatically counted using the ImageJ software.
Genetic manipulation of ESCs
To mutate endogenous Sall4 genomic loci (ZFC4mut, ZFC4Δ and ZFC1-2Δ), E14Ju09 ESCs were modified by CRISPR/Cas9 (Ran et al., 2013). Guide RNAs were designed close to the desired mutation site (https://zlab.bio/guide-design-resources) and cloned into Cas9/gRNA co-expression plasmids (Addgene pX330, or derivative containing EGFP or a puromycin resistance cassette). Single-stranded repair DNA templates (ssDNAs) were ordered from Integrated DNA Technologies. ESCs (4x105 cells) were transfected with one (for point mutations) or two (for deletions) Cas9/gRNA plasmids and 10nmol of ssDNA template as appropriate. If a puromycin resistance cassette was used, cells were selected with puromycin and seeded at clonal density. If a fluorescent reporter was used, single cells were FACS-sorted and plated into wells of 96-well plates. ESC clones were expanded and their genomic DNA was extracted for genotyping by PCR (see primers) and Sanger sequencing.
To generate cell lines expressing a transgene of interest (Sall4 or EGFP cDNA) under a doxycycline-inducible promoter, Sall4 knockout ESCs were modified using the PiggyBac (PB) transposon system. 1x106 Sall4 knockout ESCs were transfected with two PiggyBac vectors (“PB-(TetO)8-Sall4-PGK-HygromycinR” or “PB-(TetO)8-EGFP-PGK-HygromycinR” + “PB-Tet-On 3G-IRES-ZeocinR”), together with a third plasmid expressing hyperactive PB transposase (Yusa et al., 2011) (pCMV-hyPBase). Approximately 48h post-transfection, ESCs were selected for 12 days with 200μg/ml hygromycin (doxycycline-inducible SALL4 or EGFP constructs) and 100μg/ml zeocin (Tet-On 3G transactivator construct). This experiment was repeated three times to obtain independent replicates for each cell line (SALL4 or EGFP). During selection, no doxycycline was added to the medium in order to prevent SALL4 or EGFP expression. To induce SALL4 or EGFP expression, cells were treated for 48h with 1μg/ml doxycycline (freshly prepared). For each replicate, SALL4 expression with and without doxycycline was controlled by RT-qPCR and immunofluorescence, as described below.
Method details
DNA pulldown and mass spectrometry
SILAC culture, preparation of heavy/light labeled nuclear protein extracts, DNA pulldowns and mass spectrometry were performed according to a previously published protocol (Spruijt et al., 2013b), with minor changes. Biotinylated bait (AT-run) and control (disrupted AT-run) DNA oligonucleotides (see Table S4) were purchased from Sigma-Aldrich and annealed as described. Poly(dI-dC) (Sigma-Aldrich ref. P4929) was used as competitor. Heavy- and light-labeled mouse ESC protein extracts were incubated with immobilized biotinylated DNA probes. After incubation and washes, beads from both pulldowns were combined after which bound proteins were digested with trypsin. Finally, the heavy/light ratio for the tryptic peptides was measured by LC-MS. Two replicate DNA pulldown/mass spectrometry experiments were performed with both bait/control pairs. The first experiment was done according to protocol using magnetic Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific ref. 65001) and in-gel digestion of samples after elution. In the second replicate experiment, agarose streptavidin beads (Thermo Fisher Scientific) were used and samples were digested on-beads prior to elution. Peptides were concentrated and desalted using StageTips (Rappsilber et al., 2003), before being analyzed on an EASY-nLC (Thermo Fisher Scientific) connected online to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Peptides were measured during a 120min acetonitrile gradient using CID fragmentation of the top 15 precursor ions, with a dynamic exclusion duration of 30sec. Raw data was analyzed using MaxQuant (Cox and Mann, 2008) version 1.3.0.5. Using Perseus (Tyanova et al., 2016), the data was filtered for contaminants, reverse hits and the number of (unique) peptides. A scatterplot of the filtered data was generated using R. Detailed results from mass spectrometry analyses are available in Table S1.
DNA pulldowns for subsequent western blot analysis (see below) required scaling down of oligonucleotides, beads, Poly(dI-dC) competitor and total buffer volumes for use with 100μg or 200μg of nuclear protein extract. After binding of DNA oligonucleotides and washes with DNA binding buffer, beads were washed twice with protein binding buffer containing 0.5% BSA and blocked for 15min at room temperature. After incubation with nuclear protein extract, beads were washed five times in protein binding buffer and bound proteins were eluted by incubating beads in 50μl of NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) for 15min at 70°C.
Immunoprecipitation
To prepare protein extracts for immunoprecipitation, ESCs were washed with PBS, trypsinised and collected in 15ml tubes. Following a centrifugation for 5min at 1,300rpm, the supernatant was removed and the cell pellet was resuspended in 1ml of lysis buffer (10mM NaCl, 1mM MgCl2, 20mM HEPES pH7.5, 0.1% (v/v) Triton X-100) freshly supplemented with 1x protease inhibitor cocktail (Roche ref. 11873580001) and 0.5mM DTT. After a 20min incubation on ice with occasional shaking, nuclei were pelleted by centrifugation at 4°C for 10min at 1,500rpm. Supernatant was removed and nuclei were resuspended in 1ml of lysis buffer freshly supplemented with 1x protease inhibitor cocktail and 0.5mM DTT. The material was transferred into 1.5ml LoBind tubes (Eppendorf) and supplemented with 250U of Benzonase nuclease (Sigma-Aldrich). After a 5min incubation at room temperature, samples were supplemented with NaCl to obtain a final concentration of 150mM NaCl. Samples were incubated on a rotating wheel for 30min at 4°C. Tubes were centrifuged at 4°C for 30min at 13,300rpm and supernatants (nuclear protein extracts) were transferred into new 1.5ml LoBind tubes. 50μl of nuclear protein extract was boiled for 5min at 90°C in 2x Laemmli buffer (Sigma-Aldrich ref. S3401) as input material. Nuclear extracts were used directly for immunoprecipitation or stored at −80°C.
For immunoprecipitations, 5μg of anti-SALL4 antibody (Abcam ref. ab29112, RRID:AB_777810) was added to each nuclear protein extract (Sall4 knockout protein extracts were used as negative control). Samples were incubated overnight at 4°C on a rotating wheel. 30μl of nProteinA Sepharose beads (GE Healthcare 4 Fast Flow), previously blocked with 0.5mg/ml BSA, were added to each nuclear extract and samples were incubated for 2h at 4°C on a rotating wheel. Samples were washed 5 times in lysis buffer freshly supplemented with 0.5mM DTT. Between each wash, samples were centrifuged at 4°C for 1min at 2,000rpm. After the final wash, beads were boiled for 5min at 90°C in 2x Laemmli buffer (Sigma-Aldrich ref. S3401) to elute the immunoprecipitated material.
Western blot
For western blot analysis, samples were loaded into 4%–15% Mini-PROTEAN TGX Precast gels (Bio-Rad), together with a fluorescent protein ladder (LI-COR ref. 928-60000). Proteins were separated by electrophoresis in SDS running buffer for ∼45min at 200V. Subsequently, proteins were transferred on a nitrocellulose membrane at 4°C overnight at 23V. The membrane was blocked for 1h at room temperature with PBS supplemented with 10% non-fat skimmed milk and 0.1% Tween. The membrane was then incubated for 90min at room temperature with primary antibodies (see Table S4) diluted at the appropriate concentration in PBS supplemented with 5% non-fat skimmed milk and 0.1% Tween. The membrane was washed 4 times with PBS supplemented with 0.1% Tween, and incubated for 2h at room temperature with fluorescently labeled (LI-COR IRDye) or HRP-conjugated (GE Healthcare) secondary antibodies diluted in PBS supplemented with 5% non-fat skimmed milk and 0.1% Tween. The membrane was finally washed 4 times with PBS supplemented with 0.1% Tween. Proteins were visualized using the LI-COR Odyssey CLx imaging system (fluorescence) or detected on film by chemiluminescence (PerkinElmer ECL kit). Western blot signal was quantified using the LI-COR Image Studio software by measuring the fluorescence intensity of appropriate protein bands.
Immunofluorescence
For high resolution imaging, cells were plated on chambered coverslips (Ibidi ref. 80286). For lower magnification, cells were grown on standard tissue culture dishes. Cells were washed with PBS and fixed with 4% PFA for 10min at room temperature. After fixation, cells were washed with PBS and permeabilised for 10min at room temperature in PBS supplemented with 0.3% (v/v) Triton X-100. Samples were blocked for 1h30min at room temperature in blocking buffer: PBS supplemented with 0.1% (v/v) Triton X-100, 1% (w/v) BSA and 3% (v/v) serum of the same species as secondary antibodies were raised in (ordered from Sigma-Aldrich). Following blocking, samples were incubated overnight at 4°C with primary antibodies (see Table S4) diluted at the appropriate concentration in blocking buffer. After 4 washes in PBS supplemented with 0.1% (v/v) Triton X-100, samples were incubated for 2h at room temperature (in the dark) with fluorescently labeled secondary antibodies (Invitrogen Alexa Fluor Plus antibodies) diluted 1:1,000 in blocking buffer. Cells were washed 4 times with PBS supplemented with 0.1% (v/v) Triton X-100. DNA was stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5min at room temperature, and cells were submitted to a final wash with PBS. Samples were imaged by fluorescence microscopy (Nikon Ti2 or Zeiss LSM 880 with Airyscan). Images were analyzed and processed using the software Fiji.
RT-qPCR
Cells were directly lysed on the plate and total RNA was isolated using the RNeasy Plus Mini kit (QIAGEN ref. 74136), following manufacturer’s instructions. The quantity and purity of RNA samples were determined using a micro-volume spectrophotometer (Nanodrop ND-1000). RNA was reverse-transcribed with SuperScript IV and random hexamers (Invitrogen ref. 18091050), following manufacturer’s instructions. Triplicate qPCR reactions were set up in 384-well plates using the Takyon SYBR Mastermix (Eurogentec ref. UF-NSMT-B0701) and appropriate primer pairs (see Table S4). qPCR was performed and analyzed using the Roche LightCycler 480 machine. For each primer pair, a standard curve was performed to assess amplification efficiency and melting curves were analyzed to verify the production of single DNA species.
HT-Selex
SELEX coupled with high-throughput sequencing (HT-SELEX) was performed as previously described (Jolma et al., 2010; Nitta et al., 2015), in three independent replicate experiments. All oligonucleotides were ordered from Integrated DNA Technologies (see Table S4). SELEX libraries were generated by PCR and consisted of 20bp random sequences flanked by primer binding sites for amplification. Double-stranded DNA libraries were purified using the MinElute PCR Purification Kit (QIAGEN ref. 28004) and controlled on a 10% polyacrylamide gel.
For SELEX experiments, recombinant SALL4 ZFC1 (residues 320-486), ZFC2 (residues 551-662) or ZFC4 (residues 859-1028) with an N-terminal hexahistidine tag were expressed from a pET-based vector in E.coli BL21 (DE3) cells. Proteins were purified using a 5 mL Histrap FF column, followed by separation by ion exchange (6 mL ResS column) and size exclusion chromatography (Superdex 200 16/600, all columns from GE Healthcare). SELEX libraries (1.5μg for the first cycle, 200ng for subsequent cycles) were mixed with 1μg of hexahistidine-tagged SALL4 ZFC in 100μl of SELEX buffer (50mM NaCl, 1mM MgCl2, 0.5mM EDTA, 10mM Tris-HCl pH7.5, 4% glycerol) freshly supplemented with 5μg/ml Poly(dI-dC) and 0.5mM DTT. A negative control experiment (without addition of proteins) was also performed to control for technical bias during the SELEX protocol. Following a 10min incubation at room temperature on a rotating wheel, 50μl of Ni Sepharose 6 Fast Flow beads (GE Healthcare), previously equilibrated in SELEX buffer, were added to each sample and incubated for an additional 20min at room temperature on a rotating wheel. To remove non-specifically bound oligonucleotides, beads were washed 5 times with 1ml of SELEX buffer, freshly supplemented with 0.5mM DTT. Between each wash, samples were incubated for 5min at room temperature on a rotating wheel and centrifuged for 1min at 2,000 rpm. After the final wash, beads were resuspended in 100μl H2O and used directly for PCR using the high-fidelity Phusion DNA polymerase (NEB ref. M0530S). The minimum number of PCR cycles required to amplify each library was determined by running samples amplified with increasing PCR cycle numbers on 10% polyacrylamide gels. Amplified libraries were purified using the MinElute PCR Purification Kit and used for subsequent rounds of SELEX. To generate libraries for high-throughput sequencing at the desired number of SELEX cycles, libraries were amplified using primers containing Illumina adapters and unique barcodes. Double-stranded DNA libraries were purified using the QIAquick PCR Purification Kit (QIAGEN ref. 28104). Contaminating primers were eliminated by size exclusion using KAPA Pure beads (Roche ref. 07983271001) with a 3x beads-to-sample ratio. SELEX libraries with unique barcodes were pooled in equimolar amounts and sequenced using the Illumina MiSeq platform (EMBL GeneCore facility, Germany).
ChIP-seq
ChIP was performed as previously described (Stock et al., 2007), in two or three independent replicate experiments for each sample. For each ChIP, 25x106 ESCs were plated into 15cm dishes the day before the experiment. Cells were crosslinked at 37°C for 10min with 1% formaldehyde. Following quenching for 5min at room temperature with 125mM glycine, cells were washed 3 times with ice-cold PBS. Swelling buffer (10ml of 10mM KCl, 1.5mM MgCl2, 25mM HEPES pH7.9, 0.1% NP-40) freshly supplemented with 1x protease inhibitor cocktail (Roche ref. 11873580001) was added into each plate, followed by a 10min incubation at 4°C. Nuclei were collected by scraping and transferred into 15ml tubes. Samples were centrifuged at 4°C for 5min at 3,000rpm and the supernatant was removed. Crosslinked nuclei were quickly frozen on dry ice and stored at −80°C. Crosslinked nuclei were thawed on ice, resuspended in 2ml of sonication buffer (140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 50mM HEPES pH7.9) freshly supplemented with 1x protease inhibitor cocktail, and transferred into 1.5ml TPX tubes (Diagenode). Chromatin was sonicated by performing 20x sonication cycles (30sec on/ 30sec off) using the Bioruptor Twin instrument (Diagenode) with a 4°C water bath. Samples were centrifuged at 4°C for 30min at 13,000rpm to remove insoluble material. Supernatants (soluble chromatin fraction) were collected and transferred into 1.5ml LoBind tubes (Eppendorf). To evaluate the amount of chromatin in each sample, a 2μl aliquot was alkaline-lysed with 0.1M NaOH and measured using a micro-volume spectrophotometer (Nanodrop ND-1000).
For each immunoprecipitation, 700μg of chromatin was mixed with 5μg of anti-SALL4 antibody (Santa Cruz ref. sc-101147, RRID:AB_1129262 or Abcam ref. ab29112, RRID:AB_777810) in a total volume of 1ml of sonication buffer supplemented with 1x protease inhibitor cocktail. Sall4 knockout ESCs chromatin samples were used as a negative control. Samples were incubated overnight at 4°C on a rotating wheel. 50μl of either Protein A (ChIP with Abcam ref. ab29112) or Protein G (ChIP with Santa Cruz ref. sc-101147) magnetic beads (Invitrogen Dynabeads), previously equilibrated in sonication buffer, was added into each sample. Following a 3h incubation at 4°C on a rotating wheel, beads were extensively washed with 1ml of each of the following buffer: 1x with sonication buffer, 1x with wash buffer A (500mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 50mM HEPES pH7.9), 1x with wash buffer B (250mM LiCl, 1mM EDTA, 0.5% NP-40, 0.5% Na-deoxycholate, 20mM Tris pH8.0), 2x with TE buffer (Sigma-Aldrich ref. 93283). Between each wash, beads were incubated for 5min at room temperature on a rotating wheel. Finally, DNA was eluted by resuspending beads in 250μl of elution buffer (50mM Tris pH7.5, 1mM EDTA) freshly supplemented with 1% SDS, and by incubating samples at 65°C for 5 min. Samples were further incubated for 15min at room temperature on a rotating wheel and the supernatant (eluted chromatin) was collected into a new 1.5ml LoBind tube. The elution was repeated a second time to obtain 500μl of immunoprecipitated chromatin.
To extract DNA from immunoprecipitated chromatin or from the input material (50μl of soluble chromatin), crosslinking was reversed by incubating samples overnight at 65°C in a total volume of 500μl with 160mM NaCl and 20 μg/ml RNase A. Then, 5mM EDTA and 200 μg/ml Proteinase K were added to the samples, followed by a 2h incubation at 45°C. Finally, DNA was purified by phenol-chloroform extraction (Invitrogen ref. 15593031) followed by ethanol precipitation with 2x volumes of 100% ethanol, 0.1x volume of 3M sodium acetate, and 40μg of glycogen (Invitrogen ref. 10814010). Samples were incubated at −80°C for at least 1h and centrifuged at 4°C for 30min at 13,000rpm. The supernatant was removed and DNA pellets were washed with 70% EtOH. Following a final spin at 4°C for 15min at 13,000rpm, DNA pellets were air-dried and resuspended in 30-100μl TE buffer (Sigma-Aldrich ref. 93283) or H2O. DNA concentration was quantified using the Qubit dsDNA HS Assay Kit (Invitrogen ref. Q32854).
ChIP-seq libraries were prepared using the KAPA Hyperprep Kit (Roche ref. 07962347001) together with KAPA dual-indexed adapters (Roche ref. 08278555702), following manufacturer’s instruction. ChIP-seq libraries were quantified using the Qubit dsDNA HS Assay Kit (Invitrogen ref. Q32854) and fragment size was evaluated using the Agilent 2100 Bioanalyzer (Agilent High Sensitivity DNA Kit). ChIP-seq libraries with unique barcodes were pooled in equimolar amounts and sequenced using the Illumina HiSeq 4000 and NextSeq 500 platforms (EMBL GeneCore facility, Germany).
ATAC-seq
ATAC-seq was performed as previously described (Cholewa-Waclaw et al., 2019). ESC nuclei from three independent WT ESC replicates were isolated using hypotonic buffer (10mM Tris-HCl pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% Igepal CA-630). 50,000 nuclei were resuspended in 50μl of transposition reaction mix containing 2.5 μL Nextera Tn5 Transposase and 2x TD Nextera reaction buffer. The mix was incubated for 30 min at 37°C. DNA was purified and PCR amplified with the NEBNext High Fidelity reaction mix (NEB) to generate DNA libraries. Libraries were sequenced using the Illumina HiSeq 2500 platform with 75bp paired-end sequencing.
RNA-seq
For RNA-seq in ESCs, all cell lines were seeded at the same density in 6-well plates, in three or four independent replicate experiments for each sample. Following two days of culture, total RNA was extracted using the AllPrep DNA/RNA kit (QIAGEN) or the RNeasy Plus Mini kit (QIAGEN), following the manufacturer’s instructions and contaminating genomic DNA was removed by DNase I treatment. Before library preparation, equal amounts of either RNA sequins (Garvan Institute of Medical Research, Australia) or ERCC (Invitrogen) spike-in mix were added to each sample. Ribosomal RNA-depleted RNA-seq libraries were prepared using either the ScriptSeq Complete Gold Kit (Illumina) or the KAPA RNA Hyperprep Kit (Roche ref. 08098131702) together with indexed adapters, following the manufacturer’s instructions. RNA-seq libraries with unique barcodes were pooled in equimolar amounts and sequenced using the Illumina HiSeq 2500 (Wellcome Sanger Institute, UK), HiSeq X (Novogene Europe, UK) or NextSeq 500 (EMBL GeneCore facility, Germany) platforms.
For the RNA-seq time course experiment, cells were submitted to neuronal differentiation as previously described (see cell culture section), in two independent replicate experiments for each sample. At the appropriate time point, cells were directly lysed on the plate and total RNA was extracted using the RNeasy Plus Mini kit (QIAGEN), following the manufacturer’s instructions and contaminating genomic DNA was removed by DNase I treatment. Equal amounts of RNA sequins spike-in mix (Garvan Institute of Medical Research, Australia) were added to each sample and RNA-seq libraries were prepared by polyA-enrichment using the NEBNext Ultra II Library Prep Kit (NEB ref. E7645) together with indexed adapters. RNA-seq libraries with unique barcodes were pooled in equimolar amounts and sequenced using the Illumina NovaSeq platform (Novogene Europe, UK).
Quantification and statistical analysis
HT-SELEX analysis
All possible k-mers (width = 5) were searched individually in all SELEX libraries at different cycles. The fraction of reads containing the k-mer was considered as its abundance. Subsequently, top 3 abundant k-mers from ZFC4 SELEX library at cycle 6 were searched allowing one mismatch and a position frequency matrix (PFM) was generated for each. The PFM was used to visualize the motifs using Logolas (Dey et al., 2018).
RNA-seq analysis
Alignment-free quantification from RNA-seq data was performed using sailfish v0.9.2 (Patro et al., 2014). Annotation data was downloaded from Gencode and a transcriptome index was generated for assembly release M23. Differential gene expression analysis was performed using DESeq2 v1.28.0 (Love et al., 2014) and genes with adjusted p value < 0.05 were considered for further analyses. Genome wide base composition was calculated for 1 kilobase (kb) windows of the mouse genome using bedtools nuc (Quinlan and Hall, 2010) and the AT content BigWig track was generated. Base composition for multiple gene loci was calculated using deepTools computeMatrix (Ramírez et al., 2014). Gene ontology analysis for genes deregulated in ZFC4mut/Δ ESCs was performed using clusterProfiler Bioconductor package (Yu et al., 2012) and simplified GO terms from enrichGO function were used to identify enriched GO Terms with q-value < 0.01 as significance threshold (see Table S3). Bootstrapped two-sided t tests were used to associate statistical significance for comparisons of log2 fold changes in different Sall4 mutants compared to wild-type (WT) for neuronal differentiation genes. Command line arguments and source code for analysis is detailed in Methods S1.
ChIP-seq analysis
Sequencing reads were trimmed using Trimmomatic v0.33 (Bolger et al., 2014) and aligned to mm10 assembly using bwa-mem v0.7.17 (Li, 2013). PCR duplicate sequencing reads were removed using MarkDuplicates from Picard toolkit (http://broadinstitute.github.io/picard/). GC-bias was estimated for input chromatin samples using computeGCBias (Benjamini and Speed, 2012) from deepTools (Ramírez et al., 2014). Subsequently, both ChIP and input chromatin samples were corrected using the input chromatin estimated bias using correctGCBias. Peak calling on the GC-bias corrected BAM files was performed using MACS v2.1.2 (Zhang et al., 2008). BigWig tracks for ChIP over input chromatin were calculated using bamCompare. For meta-analyses of peak regions, ChIP signal scores per genome regions was calculated using computeMatrix. Motif discovery and motif enrichment analysis was performed using MEME-ChIP v5.1.0 (Machanick and Bailey, 2011) for ChIP-seq peaks with background sequences randomly sampled from accessible chromatin regions. Command line arguments and source code for analysis is detailed in Methods S1.
ATAC-seq analysis
Sequencing reads were trimmed using Trimmomatic v0.33 (Bolger et al., 2014) and aligned to mm10 assembly using bwa-mem v0.7.17 (Li, 2013). PCR duplicate sequencing reads were removed using MarkDuplicates from Picard toolkit (http://broadinstitute.github.io/picard/). Broad peak calling on the de-duplicated BAM files was performed using MACS v2.1.2 (Zhang et al., 2008).
Quantification of AT effect
Ordinary least-squares (OLS) linear regression was fitted by selecting RNA-seq log2 fold change as an endogenous variable and average AT content across the gene locus as an exogenous variable. For every model fit, the p value associated with the F-statistic and quantified AT effect with a confidence interval of 99% was used for further analysis. R2 values were estimated from a linear regression fit when log2 fold change is regressed against AT content across gene locus. p values obtained from all model fits were adjusted using the Benjamini/Hochberg multiple testing comparison and models with a false discovery rate (FDR) < 0.01 were deemed significant. Detailed results from statistical analyses are available in Table S2. Command line arguments and source code for analysis is detailed in Methods S1.
Acknowledgments
We thank Sara Giuliani for providing critical feedback on the manuscript, Dina De Sousa and Michal Prendecki for technical assistance, Shaun Webb for bioinformatics advice, David Kelly and the Centre Optical Instrumentation Laboratory facility for microscopy support, Martin Waterfall for support with flow cytometry, the Edinburgh Protein Production Facility for access to protein purification facilities, and Vladimir Benes (EMBL GeneCore facility, Germany) for support with high-throughput sequencing. We are grateful to Riuchi Nishinakamura (Kumamoto University) and Brian Hendrich (Cambridge University) for sharing S4KO ESCs and SALL4 expression plasmids. The Edinburgh Protein Production Facility and the Centre Optical Instrumentation Laboratory are funded by core grants (092076 and 203149) to the Wellcome Centre for Cell Biology at the University of Edinburgh. This work was primarily funded by a European Research Council advanced grant EC 694295 (Gen-Epix) and Wellcome Investigator Award 107930 to A.B., who is also a member of the Simons Initiative for the Developing Brain. K.S.S. was supported by a Sir Henry Wellcome Fellowship (grant 101489/Z/13/Z). T.Q. received an EU Marie Curie Fellowship. A.C. is supported by Wellcome Senior Fellowship 200898. The M.V. lab is part of the Oncode Institute, which is funded by the Dutch Cancer Society. C.G.S. is supported by NWO-VENI grant 722.016.003.
Author contributions
Conceptualization, A.B., T.Q., R.P., and K.C.; Methodology, R.P., K.C., T.Q., J.C.-W., C.G.S., M.V., and J.S.; Software, K.C.; Formal Analysis, K.C.; Investigation, R.P., K.C., T.Q., K.S.-S., G.A., B.A.-H., H.Y.L., A.C., J.C.-W., C.G.S., and J.S.; Writing – Original Draft, R.P., K.C., and A.B.; Writing – Review & Editing, R.P., K.C., and A.B.; Supervision, A.B.; Funding acquisition, A.B.
Declaration of interests
The authors declare no competing interests.
Published: January 5, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.046.
Supplemental information
References
- Akiyama R., Kawakami H., Wong J., Oishi I., Nishinakamura R., Kawakami Y. Sall4-Gli3 system in early limb progenitors is essential for the development of limb skeletal elements. Proc. Natl. Acad. Sci. USA. 2015;112:5075–5080. doi: 10.1073/pnas.1421949112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Baradie R., Yamada K., St Hilaire C., Chan W.-M., Andrews C., McIntosh N., Nakano M., Martonyi E.J., Raymond W.R., Okumura S. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002;71:1195–1199. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L., Landsman D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;26:4413–4421. doi: 10.1093/nar/26.19.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhondakis S., Auletta F., Bernardi G. Isochores and the regulation of gene expression in the human genome. Genome Biol. Evol. 2011;3:1080–1089. doi: 10.1093/gbe/evr017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert J., Dunstan H., Chambers I., Smith A. Functional gene screening in embryonic stem cells implicates Wnt antagonism in neural differentiation. Nat. Biotechnol. 2002;20:1240–1245. doi: 10.1038/nbt763. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Speed T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G., Olofsson B., Filipski J., Zerial M., Salinas J., Cuny G., Meunier-Rotival M., Rodier F. The mosaic genome of warm-blooded vertebrates. Science. 1985;228:953–958. doi: 10.1126/science.4001930. [DOI] [PubMed] [Google Scholar]
- Bickmore W.A., Sumner A.T. Mammalian chromosome banding: an expression of genome organization. Trends Genet. 1989;5:144–148. doi: 10.1016/0168-9525(89)90055-3. [DOI] [PubMed] [Google Scholar]
- Bickmore W.A., van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Bird A.P. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Blackledge N.P., Zhou J.C., Tolstorukov M.Y., Farcas A.M., Park P.J., Klose R.J. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell. 2010;38:179–190. doi: 10.1016/j.molcel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm J., Buck A., Borozdin W., Mannan A.U., Matysiak-Scholze U., Adham I., Schulz-Schaeffer W., Floss T., Wurst W., Kohlhase J., Barrionuevo F. Sall1, sall2, and sall4 are required for neural tube closure in mice. Am. J. Pathol. 2008;173:1455–1463. doi: 10.2353/ajpath.2008.071039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda M.C., Berríos S., Fernández-Donoso R., Garagna S., Redi C. Organisation of complex nuclear domains in somatic mouse cells. Biol. Cell. 1999;91:55–65. [PubMed] [Google Scholar]
- Chan A.-L., La H.M., Legrand J.M.D., Mäkelä J.-A., Eichenlaub M., De Seram M., Ramialison M., Hobbs R.M. Germline stem cell activity is sustained by SALL4-dependent silencing of distinct tumor suppressor genes. Stem Cell Reports. 2017;9:956–971. doi: 10.1016/j.stemcr.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J., Shah R., Webb S., Chhatbar K., Ramsahoye B., Pusch O., Yu M., Greulich P., Waclaw B., Bird A.P. Quantitative modelling predicts the impact of DNA methylation on RNA polymerase II traffic. Proc. Natl. Acad. Sci. USA. 2019;116:14995–15000. doi: 10.1073/pnas.1903549116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017;168:442–459.e20. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini M., Cammarano R., Bernardi G. The evolution of isochore patterns in vertebrate genomes. BMC Genomics. 2009;10:146. doi: 10.1186/1471-2164-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Dey K.K., Xie D., Stephens M. A new sequence logo plot to highlight enrichment and depletion. BMC Bioinformatics. 2018;19:473. doi: 10.1186/s12859-018-2489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan K.A., An J., Nowak R.P., Yuan J.C., Fink E.C., Berry B.C., Ebert B.L., Fischer E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane radial ray syndrome. eLife. 2018;7:e38430. doi: 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L., Eyre-Walker A., Galtier N. A new perspective on isochore evolution. Gene. 2006;385:71–74. doi: 10.1016/j.gene.2006.04.030. [DOI] [PubMed] [Google Scholar]
- Elling U., Klasen C., Eisenberger T., Anlag K., Treier M. Murine inner cell mass-derived lineages depend on Sall4 function. Proc. Natl. Acad. Sci. USA. 2006;103:16319–16324. doi: 10.1073/pnas.0607884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A., Hurst L.D. The evolution of isochores. Nat. Rev. Genet. 2001;2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- Farcas A.M., Blackledge N.P., Sudbery I., Long H.K., McGouran J.F., Rose N.R., Lee S., Sims D., Cerase A., Sheahan T.W. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. eLife. 2012;1:e00205. doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filarsky M., Zillner K., Araya I., Villar-Garea A., Merkl R., Längst G., Németh A. The extended AT-hook is a novel RNA binding motif. RNA Biol. 2015;12:864–876. doi: 10.1080/15476286.2015.1060394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenatri M., Bailly D., Maison C., Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Shen L., Wan M., Taranova O., Wu H., Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013;15:373–384. doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I., Takebayashi S., Lu J., Gilbert D.M. Replication timing and transcriptional control: beyond cause and effect--part II. Curr. Opin. Genet. Dev. 2009;19:142–149. doi: 10.1016/j.gde.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs R.M., Fagoonee S., Papa A., Webster K., Altruda F., Nishinakamura R., Chai L., Pandolfi P.P. Functional antagonism between Sall4 and Plzf defines germline progenitors. Cell Stem Cell. 2012;10:284–298. doi: 10.1016/j.stem.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist G.P. Evolution of chromosome bands: molecular ecology of noncoding DNA. J. Mol. Evol. 1989;28:469–486. doi: 10.1007/BF02602928. [DOI] [PubMed] [Google Scholar]
- Holmquist G., Gray M., Porter T., Jordan J. Characterization of Giemsa dark- and light-band DNA. Cell. 1982;31:121–129. doi: 10.1016/0092-8674(82)90411-1. [DOI] [PubMed] [Google Scholar]
- Hooper M., Hardy K., Handyside A., Hunter S., Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- Jabbari K., Bernardi G. An isochore framework underlies chromatin architecture. PLoS ONE. 2017;12:e0168023. doi: 10.1371/journal.pone.0168023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder B.L., Hu G., Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat. Immunol. 2011;12:918–922. doi: 10.1038/ni.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer S.M., Ohlemiller K.K., Yang J., McDill B.W., Kohlhase J., Rauchman M. Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum. Mol. Genet. 2003;12:2221–2227. doi: 10.1093/hmg/ddg233. [DOI] [PubMed] [Google Scholar]
- Kohlhase J., Heinrich M., Schubert L., Liebers M., Kispert A., Laccone F., Turnpenny P., Winter R.M., Reardon W. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 2002;11:2979–2987. doi: 10.1093/hmg/11.23.2979. [DOI] [PubMed] [Google Scholar]
- Koshiba-Takeuchi K., Takeuchi J.K., Arruda E.P., Kathiriya I.S., Mo R., Hui C.C., Srivastava D., Bruneau B.G. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat. Genet. 2006;38:175–183. doi: 10.1038/ng1707. [DOI] [PubMed] [Google Scholar]
- Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth S.M., Rauchman M. A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J. Biol. Chem. 2006;281:23922–23931. doi: 10.1074/jbc.M513461200. [DOI] [PubMed] [Google Scholar]
- Lee J.-H., Voo K.S., Skalnik D.G. Identification and characterization of the DNA binding domain of CpG-binding protein. J. Biol. Chem. 2001;276:44669–44676. doi: 10.1074/jbc.M107179200. [DOI] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 https://arxiv.org/abs/1303.3997 1303.3997. [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Jeong H.W., Kong N., Yang Y., Carroll J., Luo H.R., Silberstein L.E., Yupoma, Chai L. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE. 2009;4:e5577. doi: 10.1371/journal.pone.0005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P., Bailey T.L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y., Chapman V.M. In situ analysis of centromeric satellite DNA segregating in Mus species crosses. Mamm. Genome. 1991;1:71–77. doi: 10.1007/BF02443781. [DOI] [PubMed] [Google Scholar]
- Matyskiela M.E., Couto S., Zheng X., Lu G., Hui J., Stamp K., Drew C., Ren Y., Wang M., Carpenter A. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018;14:981–987. doi: 10.1038/s41589-018-0129-x. [DOI] [PubMed] [Google Scholar]
- Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.-B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013;23:270–280. doi: 10.1101/gr.141028.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miertus J., Borozdin W., Frecer V., Tonini G., Bertok S., Amoroso A., Miertus S., Kohlhase J. A SALL4 zinc finger missense mutation predicted to result in increased DNA binding affinity is associated with cranial midline defects and mild features of Okihiro syndrome. Hum. Genet. 2006;119:154–161. doi: 10.1007/s00439-005-0124-7. [DOI] [PubMed] [Google Scholar]
- Miller A., Ralser M., Kloet S.L., Loos R., Nishinakamura R., Bertone P., Vermeulen M., Hendrich B. Sall4 controls differentiation of pluripotent cells independently of the nucleosome remodelling and deacetylation (NuRD) complex. Development. 2016;143:3074–3084. doi: 10.1242/dev.139113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta K.R., Jolma A., Yin Y., Morgunova E., Kivioja T., Akhtar J., Hens K., Toivonen J., Deplancke B., Furlong E.E.M., Taipale J. Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife. 2015;4:e04837. doi: 10.7554/eLife.04837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R., Mount S.M., Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsialou A., Wilsker D., Moran E. DNA-binding properties of ARID family proteins. Nucleic Acids Res. 2005;33:66–80. doi: 10.1093/nar/gki145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante T., Bird A. Do short, frequent DNA sequence motifs mould the epigenome? Nat. Rev. Mol. Cell Biol. 2016;17:257–262. doi: 10.1038/nrm.2015.31. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187-91. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Zhen S., Roumiantsev S., McDonald L.T., Yuan G.-C., Orkin S.H. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol. Cell. Biol. 2010;30:5364–5380. doi: 10.1128/MCB.00419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Sakaki-Yumoto M., Kobayashi C., Sato A., Fujimura S., Matsumoto Y., Takasato M., Kodama T., Aburatani H., Asashima M., Yoshida N., Nishinakamura R. The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development. 2006;133:3005–3013. doi: 10.1242/dev.02457. [DOI] [PubMed] [Google Scholar]
- Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W.T.C., Bauer C., Münzel M., Wagner M., Müller M., Khan F. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Spruijt C.G., Baymaz H.I., Vermeulen M. Identifying specific protein–dna interactions using SILAC-based quantitative proteomics. In: Bina M., editor. Gene Regulation: Methods and Protocols. Humana Press; 2013. pp. 137–157. [DOI] [PubMed] [Google Scholar]
- Stock J.K., Giadrossi S., Casanova M., Brookes E., Vidal M., Koseki H., Brockdorff N., Fisher A.G., Pombo A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007;9:1428–1435. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- Sweetman D., Münsterberg A. The vertebrate spalt genes in development and disease. Dev. Biol. 2006;293:285–293. doi: 10.1016/j.ydbio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Sweetman D., Smith T., Farrell E.R., Chantry A., Münsterberg A. The conserved glutamine-rich region of chick csal1 and csal3 mediates protein interactions with other spalt family members. Implications for Townes-Brocks syndrome. J. Biol. Chem. 2003;278:6560–6566. doi: 10.1074/jbc.M209066200. [DOI] [PubMed] [Google Scholar]
- Tahara N., Kawakami H., Chen K.Q., Anderson A., Yamashita Peterson M., Gong W., Shah P., Hayashi S., Nishinakamura R., Nakagawa Y. Sall4 regulates neuromesodermal progenitors and their descendants during body elongation in mouse embryos. Development. 2019;146:dev177659. doi: 10.1242/dev.177659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N., Saito M., Ebisuya M., Nishida E., Ishikawa F. Stemness-related factor Sall4 interacts with transcription factors Oct-3/4 and Sox2 and occupies Oct-Sox elements in mouse embryonic stem cells. J. Biol. Chem. 2013;288:5027–5038. doi: 10.1074/jbc.M112.411173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhal P., Rösler B., Kohlhase J. A family with features overlapping Okihiro syndrome, hemifacial microsomia and isolated Duane anomaly caused by a novel SALL4 mutation. Am. J. Med. Genet. A. 2006;140:222–226. doi: 10.1002/ajmg.a.31060. [DOI] [PubMed] [Google Scholar]
- Thomson J.P., Skene P.J., Selfridge J., Clouaire T., Guy J., Webb S., Kerr A.R.W., Deaton A., Andrews R., James K.D. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J.K., Hassig C.A., Schnitzler G.R., Kingston R.E., Schreiber S.L. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- Tsubooka N., Ichisaka T., Okita K., Takahashi K., Nakagawa M., Yamanaka S. Roles of Sall4 in the generation of pluripotent stem cells from blastocysts and fibroblasts. Genes Cells. 2009;14:683–694. doi: 10.1111/j.1365-2443.2009.01301.x. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., Rimm D.L., Rodriguez H., Hiltke T., Snyder M., Yamamoto T. A proposal for validation of antibodies. Nat. Methods. 2016;13:823–827. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voo K.S., Carlone D.L., Jacobsen B.M., Flodin A., Skalnik D.G. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell. Biol. 2000;20:2108–2121. doi: 10.1128/mcb.20.6.2108-2121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade P.A., Jones P.L., Vermaak D., Wolffe A.P. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr. Biol. 1998;8:843–846. doi: 10.1016/s0960-9822(98)70328-8. [DOI] [PubMed] [Google Scholar]
- Warren M., Wang W., Spiden S., Chen-Murchie D., Tannahill D., Steel K.P., Bradley A. A Sall4 mutant mouse model useful for studying the role of Sall4 in early embryonic development and organogenesis. Genesis. 2007;45:51–58. doi: 10.1002/dvg.20264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Johansen J.V., Helin K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell. 2013;49:1134–1146. doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Xiong J., Zhang Z., Chen J., Huang H., Xu Y., Ding X., Zheng Y., Nishinakamura R., Xu G.-L., Wang H. Cooperative action between SALL4A and TET proteins in stepwise oxidation of 5-methylcytosine. Mol. Cell. 2016;64:913–925. doi: 10.1016/j.molcel.2016.10.013. [DOI] [PubMed] [Google Scholar]
- Xu K., Chen X., Yang H., Xu Y., He Y., Wang C., Huang H., Liu B., Liu W., Li J. Maternal Sall4 is indispensable for epigenetic maturation of mouse oocytes. J. Biol. Chem. 2017;292:1798–1807. doi: 10.1074/jbc.M116.767061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Wong J., Moreno G.T., Young M.K., Côté J., Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.L., Tanaka S.S., Kumagai M., Fujimoto Y., Terabayashi T., Matsui Y., Nishinakamura R. Sall4 is essential for mouse primordial germ cell specification by suppressing somatic cell program genes. Stem Cells. 2015;33:289–300. doi: 10.1002/stem.1853. [DOI] [PubMed] [Google Scholar]
- Yamashita K., Sato A., Asashima M., Wang P.-C., Nishinakamura R. Mouse homolog of SALL1, a causative gene for Townes-Brocks syndrome, binds to A/T-rich sequences in pericentric heterochromatin via its C-terminal zinc finger domains. Genes Cells. 2007;12:171–182. doi: 10.1111/j.1365-2443.2007.01042.x. [DOI] [PubMed] [Google Scholar]
- Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuri S., Fujimura S., Nimura K., Takeda N., Toyooka Y., Fujimura Y., Aburatani H., Ura K., Koseki H., Niwa H., Nishinakamura R. Sall4 is essential for stabilization, but not for pluripotency, of embryonic stem cells by repressing aberrant trophectoderm gene expression. Stem Cells. 2009;27:796–805. doi: 10.1002/stem.14. [DOI] [PubMed] [Google Scholar]
- Yusa K., Zhou L., Li M.A., Bradley A., Craig N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., LeRoy G., Seelig H.-P., Lane W.S., Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95:279–289. doi: 10.1016/s0092-8674(00)81758-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Smits A.H., van Tilburg G.B.A., Jansen P.W.T.C., Makowski M.M., Ovaa H., Vermeulen M. An interaction landscape of ubiquitin signaling. Mol. Cell. 2017;65:941–955.e8. doi: 10.1016/j.molcel.2017.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed high-throughput sequencing data was deposited on Array Express, as described in the Table below. Python scripts and source code used for bioinformatic analyses, raw western blot and microscopy images, as well as other types of unprocessed and processed data used to generate the figures are available on Mendeley Data (https://doi.org/10.17632/rwzttj9pn2.1).
For H3K4me1 and H3K27ac ChIP-seq in ESCs, previously published data were obtained from GEO (accession number: GSE90893).
| Accession | Description |
|---|---|
| E-MTAB-7343 | RNA-seq of WT, S4KO, ZFC4mut and ZFC4Δ ESCs |
| E-MTAB-7655 | RNA-seq of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9197 | SALL4 ChIP-seq in WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9198 | Time course RNA-seq during differentiation (day 0, 2 and 5) of WT, S4KO, ZFC4mut and ZFC1-2Δ ESCs |
| E-MTAB-9202 | RNA-seq of S4KO cells carrying Sall4 cDNA or EGFP cDNA under a doxycycline inducible promoter |
| E-MTAB-9236 | HT-SELEX of recombinant C2H2 zinc-finger domains of SALL4 |
| E-MTAB-9245 | ATAC-seq in WT ESCs |