

Abstract
The transforming growth factor (TGF)-β family is a group of structurally related, multifunctional growth factors, or ligands that are crucially involved in the development, regulation, and maintenance of animal tissues. In humans, the family counts over 33 members. These secreted ligands typically form multimeric complexes with two type I and two type II receptors to activate one of two distinct signal transduction branches. A striking feature of the family is its promiscuity, i.e., many ligands bind the same receptors and compete with each other for binding to these receptors. Although several explanations for this feature have been considered, its functional significance has remained puzzling. However, several recent reports have promoted the idea that ligand-receptor binding promiscuity and competition are critical features of the TGF-β family that provide an essential regulating function. Namely, they allow a cell to read and process multi-ligand inputs. This capability may be necessary for producing subtle, distinctive, or adaptive responses and, possibly, for facilitating developmental plasticity. Here, we review the molecular basis for ligand competition, with emphasis on molecular structures and binding affinities. We give an overview of methods that were used to establish experimentally ligand competition. Finally, we discuss how the concept of ligand competition may be fundamentally tied to human physiology, disease, and therapy.
Graphical abstract
Introduction
Transforming growth factor (TGF)-β family ligands are a group of structurally related, multifunctional cytokines that are found in all animals. They orchestrate many fundamental embryonic and adult developmental processes, including gastrulation and neurulation in the developing embryo, as well as expansion, homeostasis, and regeneration of adult tissues. Their central importance for animal cell physiology is underscored by their essentiality in both vertebrates and invertebrates 1.
In mammals, the family counts 33 different ligand genes, including TGF-β1-3, after which the family is named, bone morphogenetic proteins (BMP), growth differentiation factors (GDF), activins, nodal, anti-Mullerian hormone (AMH), inhibins and leftys 2. Their extraordinary degree of molecular conservation and functional preservation across species indicates that there is significant selective pressure to maintain a diversity of ligands and suggest that each ligand has unique and critical biological functions 3.
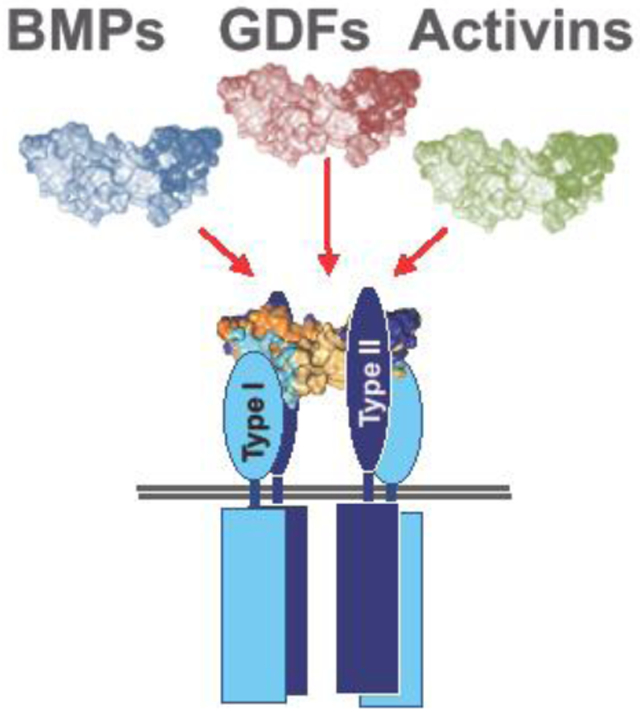
In general, TGF-β family ligands exert their function by forming a hexameric signaling complex that comprises two type I and two type II TGF-β family receptors. Formation of such a complex results in the activation of SMAD signal transduction pathways 1 (Figure 1). In this framework, type II receptors activate type I receptors by phosphorylating a glycine/serine-rich “GS domain” near their cytoplasmic kinase domain, which leads to recruitment and phosphorylation of the receptor-activated SMAD transcription factors (R-SMAD) by the type I receptor. Phosphorylated R-SMADs subsequently hetero-oligomerize with SMAD4 and translocate to the nucleus to regulate gene expression. R-SMADs belong to one of two signaling branches: the SMAD1/5/8 branch or the SMAD2/3 branch (Figure 1A). While SMAD pathways are the principal (canonical) mode by which TGF-β family ligand signals are transduced, activation of non-SMAD pathways by TGF-β family ligands is also well established. As this subject has recently been reviewed 4, we will only discuss SMAD mediated TGF-β family signaling here.
Figure 1. The TGF-β signaling pathway.

A. Schematic representation of TGF-β family pathway activation and action. Dimeric ligand binding induces assembly of two type I- and two type II-receptors into a hexameric signaling complex, allowing the type II receptors to activate the type I receptors by phosphorylation. The active type I receptors in turn phosphorylate R-SMADs, which then form trimeric complexes with SMAD4, translocate to the nucleus and regulate gene expression. R-SMADs are divided in two functionally different branches, the SMAD1/5/8 branch (a.k.a. “the BMP-pathway”), and the SMAD2/3 branch (a.k.a. “the Activin/TGF-β-pathway”). Ligands on the left side are colored blue, on the right side they are colored green to represent their pathway selectivity. Downstream gene regulation, therefore, depends on which type I receptor forms part of the active signaling complexes. B. The TGF-β/activin and the BMP subfamilies each share a subset of type I receptors and downstream effector SMADs. Notably, the family has over 33 ligand genes in humans but only five type II and seven type I receptors, indicating that there is substantial overlap in receptor utilization.
Although the mechanisms of pathway activation and signal transduction appear to be straightforward and direct, real-life implementation and interpretation of TGF-β family signaling is very complex and subject to many regulatory checkpoints and competing inputs. Thus, whether the SMAD1/5/8 or the SMAD2/3 branch is activated in a cell can depend on many factors (collectively defined as ‘context’), including receptor expression and ligand accessibility. For example, when receptor availability is limited, all present ligands that can bind the expressed receptors must compete to form a signaling complex. The binding affinity of ligands to receptors, therefore, will help determine in a complex environment which ligand will signal and, thus, which SMAD branch will be activated.
The large number of ligands stands in stark contrast to the relatively limited number of receptors and an even smaller number of SMAD pathways that mediate their signals in cells. In humans, over 40 ligands (including homodimers and heterodimers) interact with combinations of 7 type I and 5 type II receptors to activate one of two signaling branches mediated by the transcription factors SMAD2/3 or SMAD1/5/8 (Figure 1B). The difference between ligand number (input) and SMAD transcription factors (output) has long presented a conundrum as to why so many ligands are needed. One hypothesis is that ligands compete for receptor binding and thereby regulate the activity of each other, thus enabling cells to interpret complex information that is perceived as combinations of ligands rather than a single input. This line of thinking is not new but has gained interest through several recently published studies 5, 6, 7, 8, 9, 10. Here, we summarize what is known about ligand binding and signaling through specific receptors and we review the current literature on competition between ligands as one out of several ways to regulate physiological and pathological signaling in the TGF-β family. We outline the molecular basis for ligand competition. We review how ligand competition was determined experimentally. Finally, we discuss the relevance of ligand competition to human physiology, disease, and therapy.
Ligand structure
TGF-β family ligands are synthesized as precursors. They consist of a signal peptide, a prodomain, and a C-terminal mature domain 11. Furin-like proteases separate the prodomain from the mature domain through proteolytic cleavage 12. Most mature domains form disulfide-linked dimers. However, GDF-3, GDF-9, BMP-15, lefty1, and lefty2 likely form non-covalent dimers as they lack the cysteine involved in the intermolecular disulfide bond 1, 13. The mature domains of some ligands remain non-covalently associated with their prodomains 14 and may be kept in a latent form until activated, while the mature domains of other ligands remain associated with but are not inhibited by their prodomains 15, 16.
The dimeric ligands resemble two hands that form a butterfly-like shape (Figure 2A). This architecture is conserved within the family, even as structural details vary. In the hand analogy, the N-terminus is considered the ‘thumb’, the second loop forming the alpha helix is the ‘palm’, the first and third loops, which form the beta-sheet are the ‘fingers’, and the ‘knuckle’ epitope is the region between the fingertips and the palm (Figure 2B). The loop that precedes the alpha helix or palm is called the ‘prehelix loop’ (Figure 2B) and is part of the ‘wrist’ epitope. Ligands interact with type I receptors via their ‘wrist’ epitope, and BMPs, GDFs, and activins interact with type II receptors via the ‘knuckle’ epitope (Figure 2C, D).1
Figure 2. Ligand architecture exemplified by BMP-9.

A. The butterfly-like structure of mature dimeric BMP-9 is shown. For clarity, each protomer in the mature, dimeric BMP-9 is colored in a different shade of blue. B. The secondary structure of a ligand protomer is frequently described as a left hand. Structural features have been given names that match the left-hand analogy, including knuckle and palm regions (grey highlight), as well as the wrist helix, prehelix loop, thumb, and fingers (blue highlight). C. Mature dimeric BMP-9 is shown rotated by 90 degrees relative to A with a schematic representation of the type I receptor (grey line) and type II receptor (orange line) binding site located, respectively, near the palm and knuckle regions. D. Surface model showing the BMP-9-ALK1-ActRIIB complex. The orientation is the same as C. The palm region in BMP-9 interacts with the type I receptor (ALK1, grey), the knuckle epitope interacts with the type II receptor (ActRIIB, orange). Images were generated using models of BMP-9 (1ZKZ, 153) and BMP-9 in complex with receptor ectodomains (ECD) (BMP-9/ALK1-ECD/ActRIIB-ECD, 4FAO, 184). Figures were generated using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.)E-G. Schematic representation of the three proposed modes of ligand-receptor assembly (inspired by 17, 18). The figure illustrates important differences in the contact surface between ligands (monomers, blue and light blue), the type I receptors (grey), and type II receptors (orange) for the three paradigms of ligand-receptor assembly. White stars indicate high-affinity binding surfaces between ligand and receptor. E. BMPs bind receptors via a lock-and-key mechanism where the shapes of the binding interfaces in receptors and ligands fit well. The BMPs bind the type II receptors via the knuckle epitope, whereas binding to the type I receptors happens at the concave dimer interface of the ligand and is largely dependent on the ligand pre-helix loop. F. In contrast to BMPs, activins bind type II receptors with high affinity but still position the type II receptors like BMPs at the ligand knuckle epitope. Also, in this case, the type I receptor binds at the concave dimer interface of the ligand. G. TGF-β utilizes a cooperative mode of ligand-receptor assembly, whereby the type II receptor is bound at the ligand fingertip, enabling binding of the type I receptor, ALK5.
Ligand-receptor interaction
Ligands can be divided into three groups based on how they associate with their type I and type II receptors 1, 17, 18. Most ligands bind one receptor with high affinity, leading to the widely accepted model of sequential signaling complex assembly. Generally, TGF-βs, activins, and some GDFs bind their type II receptors with high affinity and their type I receptors with low affinity. These ligands are therefore believed to form a complex with their type II receptors first, then engage type I receptors. Members of the BMP/GDF family, including BMP-2 and BMP-4, bind their type I receptors with high affinity and their type II receptors with lower affinity. These ligands therefore presumably interact with their type I receptors first, then engage their type II receptors 19. Some ligands, including BMP-9 and BMP-10, bind both type I and type II receptors with high affinity 20, 21. These ligands could recruit both types of receptors at the same time. Notably, type I receptors interact with a limited group of ligands, whereas the three type II receptors ActRIIA, ActRIIB, and BMPRII are shared by many ligands of the activin, BMP and GDF families (Table 1).
Table 1. Interactions between ligands and receptors in the TGF-β family.
| Ligand (protein/gene) | Type II receptors | Type I receptors | References |
|---|---|---|---|
| Inhibina/INHA | ActRIIA, ActRIIB | No type 1 receptor | 48 |
| Activin Ab/INHBA | BMPRII, ActRIIA, ActRIIB | ALK2, ALK4, ALK5, ALK7 | 5, 59, 128, 129, 130, 131, 132, 133, 134 |
| Activin Bb/INHBB | BMPRII, ActRIIA, ActRIIB | ALK2, ALK4, ALK5, ALK7 | 5, 122, 131, 132, 133, 135, 136 |
| Activin Cb/INHBC | ActRIIA, ActRIIB | No type 1 receptor | 49 |
| Activin Eb/INHBE | N.D. | N.D. | |
| TGF-β1/TGFB1 | TGFβRII | ALK5, ALK1 | 14, 59, 129, 137, 138, 139, 140, 141 |
| TGF-β2/TGFB2 | TGFβRII | ALK5 | 14, 139, 142 |
| TGF-β3/TGFB3 | TGFβRII | ALK5, ALK1 | 14, 139, 140, 141 |
| BMP-2/BMP2 | BMPRII, ActRIIA, ActRIIB | ALK2, ALK3, ALK6 | 134, 143, 144, 145, 146 |
| BMP-3/BMP3 | ActRIIB | No type 1 receptor | 51, 147 |
| BMP-4/BMP4 | BMPRII, ActRIIB | ALK3, ALK6 | 5, 146, 148, 149 |
| BMP-5/BMP5 | N.D. | ALK2, ALK3, ALK6 | 120, 150, 151 |
| BMP-6 (VGR1)/BMP6 | BMPRII, ActRIIA, ActRIIB | ALK2, ALK3, ALK6 | 5, 143, 147 |
| BMP-7 (OP-1)/BMP7 | BMPRII, ActRIIA, ActRIIB | ALK2, ALK3, ALK6 | 5, 134, 144, 145, 148, 149, 152 |
| BMP-8A (OP-2)/BMP8A | N.D. | N.D. | |
| BMP-8B (OP-3)/BMP8B | N.D. | N.D. | |
| BMP-9/GDF2 | BMPRII, ActRIIA, ActRIIB | ALK1, ALK2 | 5, 6, 20, 21, 121, 153, 154, 155 |
| BMP-10/BMP10 | BMPRII, ActRIIA, ActRIIB | ALK1-3, ALK6 | 5, 21, 154, 155, 156, 157 |
| GDF-1/GDF1 | ActRIIA, ActRIIB | ALK4, ALK7 | 158, 159, 160 |
| GDF-3 (VGR2)/GDF3 | ActRIIA, ActRIIB | ALK4, ALK7 | 158, 161, 162 |
| GDF-5 (BMP-14)/GDF5 | BMPRII, ActRIIA | ALK3, ALK6 | 134, 163 |
| GDF-6 (BMP-13)/GDF6 | BMPRII, ActRIIA | ALK3, ALK6 | 157, 163 |
| GDF-7 (BMP12)/GDF7 | BMPRII, ActRIIA | ALK3, ALK6 | 157 |
| GDF-8 (Myostatin)/MSTN | ActRIIA, ActRIIB | ALK4, ALK5, ALK7 | 5, 10, 131, 133, 164 |
| GDF-9/GDF9 | BMPRII | ALK5, ALK6 | 165, 166, 167 |
| GDF-9B/BMP15 | BMPRII | ALK3, ALK6 | 168, 169, 170 |
| GDF-10 (BMP-3b)/GDF10 | TGFβRII, ActRIIA | ALK4, ALK5 | 171, 172 |
| GDF-11 (BMP-11)/GDF11 | ActRIIA, ActRIIB | ALK4, ALK5, ALK7 | 5, 131, 133, 173, 174 |
| GDF-15c/GDF15 | No type II receptord | No type I receptord | 126, 127 |
| MIS/AMH | AMHRII | ALK2, ALK3, ALK6 | 175 |
| Nodal/NODAL | BMPRII, ActRIIA, ActRIIB | ALK4 | 5, 52, 176 |
| Lefty1/LEFTY1 | ActRIIA, ActRIIB | No type 1 receptor | 52 |
| Lefty2/LEFTY2 | ActRIIA, ActRIIB | No type 1 receptor | 52 |
N.D.: not determined
Inhibin A and inhibin B are heterodimers of the inhibin α (INHA) monomer and an inhibin β monomer, βA (INHBA) or βB (INHBB), respectively.
The activins are homodimers of inhibin β monomers, but heterodimers of the inhibin β monomers also exist.
In the abovementioned butterfly structure model, BMP and activin ligands interact with ActRIIA and ActRIIB via the ‘knuckle’ epitope (Figure 2E and 2F). Two neighboring positions within the knuckle epitope, which contain mostly hydrophobic amino acids, are critical for binding. Although structures of BMPRII-ligand complexes are not yet available, it is suggested that BMPRII binds ligands using a geometry that is similar to ActRIIA and ActRIIB 22. In contrast to BMPs and activins, TGF-β 1-3 have a specific type II receptor, TGFBRII. They do not bind ActRIIA, ActRIIB, or BMPRII as they have large, polar amino acids in key knuckle epitope positions. Instead, TGF-βs bind TGFBRII via their fingertips, which contain a unique structure that is not found in other ligands (Figure 2G) 23. The fingertip-binding geometry likely allows TGFBRII and the type I receptor ALK5 to interact cooperatively 23.
Pathway activation and intracellular signal transduction
SMAD proteins relay TGF-β family signals inside the cell 24, 25, 26. They are highly conserved among vertebrates and can be divided into three functionally distinct groups: receptor SMADs (R-SMADs: SMAD1/5/8 and SMAD2/3), a common SMAD (SMAD4 in vertebrates), and inhibitory SMADs (I-SMADs: SMAD6 and SMAD7). R-SMADs and SMAD4 have two MAD-homology domains (MH1 and MH2), whereas I-SMADs only have one (MH2) 27. As SMADs contact DNA with their MH1 domain 28, only R-SMADs and SMAD4 are directly involved in regulating gene expression. However, I-SMADs modulate TGF-β family signaling by, e.g., blocking the R-SMAD interaction with type I receptors 29.
Binding of a ligand to two type I and type II receptors initiates TGF-β family signaling 30, 31. Specifically, formation of a ligand-receptor signaling complex triggers activation of the type I receptors, as type II receptors phosphorylate a regulatory glycine/serine-rich “GS domain” on type I receptors 32. This alters the binding specificity of the GS domain, reducing its affinity for the inhibitory protein FKBP12, and enhancing recruitment of R-SMADs, which in turn leads to their carboxy-terminal phosphorylation 33, 34, 35. Once C-terminally phosphorylated, R-SMADs dissociate from the receptor complex and associate with SMAD4. The heteromeric R-SMAD-SMAD4 complex then translocates to the nucleus to regulate gene expression 36.
A structure in type I receptors (specifically, the ‘L45’ loop) likely determines which R-SMADs are recruited to an active signaling complex and, thus, are activated by a particular ligand (Figure 3) 37, 38, 39. Based on this specificity, R-SMADs are grouped into two branches: the SMAD1/5/8 branch (a.k.a. “the BMP-pathway”), which is activated by the type I receptors activin receptor-like kinase (ALK)1, ALK2, ALK3, and ALK6, and the SMAD2/3 branch (a.k.a. “the Activin/TGF-β-pathway”), which is activated by the type I receptors ALK4, ALK5, or ALK7. Significantly, SMAD2/3 and SMAD1/5/8 regulate different target gene sets 24, 40. Which SMAD branch is activated therefore has fundamental consequences for a cell. For example, in myeloma cells, activation of SMAD1/5/8 inhibits cell proliferation and, in many cases lead to cell death. By contrast, activation of SMAD2/3 has little or no effect on myeloma cell survival 6, 41, 42. Similarly, SMAD1/5/8 primes 3T3-L1 cells to become adipocytes, whereas SMAD2/3 signaling suppresses 3T3-L1 adipogenesis, possibly by reprogramming these cells toward a different fate 43.
Figure 3. Activation of type I receptor kinase and R-SMADs.

Type I receptor activation is inhibited by the protein FKBP12 (left panel). FKBP12 binds the GS domain of type I receptors and prevents its phosphorylation and activation. FKBP12 is released upon ligand binding, or by the macrolide antibiotic FK506. FKBP12 release makes the GS domain available for phosphorylation by type II receptors (middle panel). The phosphorylated GS domain interacts with a basic patch near the L3 loop in the R-SMAD (right panel). The interaction between the L45 loop in the type I receptor and the L3 loop in R-SMAD mediates specificity. The C-terminal S-X-S motif in the R-SMAD is then phosphorylated by the activated type I receptor kinase.
Ligand competition
TGF-β family receptors are promiscuous, i.e., they bind two or more different ligands with varying affinities using the same recognition site (Figure 4A) 1, 5, 14, 19, 44, 45. Among type II receptors, ActRIIA and ActRIIB bind the greatest number of ligands, with BMPRII following closely behind (reported ligand-receptor interactions are summarized in Table 1) 5. As the number of available receptors per cell is likely limited 46, 47, ligands that engage the same receptors have to compete with each other for binding to these receptors. One consequence of this condition is that high-affinity ligands can block low-affinity ligands from interacting with a particular receptor and from inducing signaling. Established ligand-receptor-competition examples are listed in Table 2.
Figure 4. TGF-β family receptor promiscuity and functional outcomes of ligand competition.

The promiscuous nature of TGF-β ligand-receptor interactions can be visualized by superimposing ligand-receptor complexes. Specifically, the knuckle region of BMP-9 (4FAO 184) was used as template and other ligand-ActRIIA/B complexes were superimposed, including GDF11 (6MAC, beige, 18), BMP-2 ( 2H62, bright orange, 44), Activin A (1NYU, yellow, 18), and BMP7 (1LX5, olive, 185). A. The BMP-9 complex is shown as surface model with BMP-9 colored blue, ALK1 colored grey, and ActRIIB colored orange. The superimposed type II receptor ECDs including ActRIIB in the BMP-9 complex are shown for one protomer as Cα-traces/sticks. The figure to the right represents an orthogonal view of the left image. Ligand competition in the TGF-β family and various possible consequences for R-SMAD-mediated signaling are shown. B. Schematic of canonical BMP/GDF signaling. BMP/GDF (blue) bind type II and type I receptors (light and dark blue, respectively) and activate the SMAD1/5/8 pathway. C. An antagonistic ligand, such as inhibin (red), competes with BMP/GDF for binding to type II receptors but does not recruit type I receptors or activate R-SMAD signaling. Inhibin is an antagonist, as it suppresses BMP/GDF mediated SMAD1/5/8. D. A high affinity ligand, such as activin or myostatin (green), competes for binding to type II receptors and recruits a different type I receptor. Activin/myostatin suppress BMP/GDF mediated SMAD1/5/8 signaling and activate the SMAD2/3 branch causing a switch in signaling pathway use. E. A BMP/GDF ligand (grey) competes with another BMP/GDF (blue) for type II receptor binding and activates SMAD1/5/8 signaling. Different combinations of BMP/GDFs will activate SMAD1/5/8 signaling with distinctive potencies that can be additive, subtractive, or other. F. A high affinity ligand (green), such as activin A, forms a non-signaling complex (NSC) with type II receptors (ActRIIA/B) and the type I receptor ALK2 to inhibit BMP/GDF. Activin A blunts BMP/GDF mediated SMAD1/5/8 signaling in this context.
Table 2. Competition for receptor binding between TGF-β family ligands.
| BMP-2 | BMP-4 | BMP-5 | BMP-6 | BMP-7 | BMP-9 | BMP-10 | Activin A | Activin B | Nodal | GDF-5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Activin A | ActRII5, 57 | ActRII6 | ActRII5, 9 | ActRII5, 6 | ActRII5 | ||||||
| Activin B | ActRII5 | ActRII5 | |||||||||
| Activin C | ActRII49, 55 | ActRII49 | |||||||||
| GDF-8 | ActRII5 | ActRIIB10 | |||||||||
| GDF-11 | ActRII5 | ND8 | ND8 | ND8 | ND8 | ||||||
| GDF-3 | ND177, 178 | ||||||||||
| GDF-5 | ALK3179, ND8 | ND8 | ND8 | ||||||||
| GDF-6 | ND8 | ND8 | ND8 | ||||||||
| GDF-7 | ND8 | ||||||||||
| BMP-2 | ND180, ActRIIB56 | ActRII5 | ActRII5, ND8 | ActRII5, 57 | ALK3179, ND8 | ||||||
| BMP-3 | ND8, 181 | ActRIIB51, 56, ND8 | ND8 | ND8 | ND8 | ND8 | ActRIIB51 | ||||
| BMP-3b | ND172, 181 | ND172 | ND172 | ND172 | |||||||
| BMP-7 | ActRIIA9 | ||||||||||
| BMP-8a | ND8 | ND8 | |||||||||
| BMP-9 | ActRII5 | ActRII5 | |||||||||
| BMP-10 | ActRII5, ND8 | ND8 | ActRII5 | ActRII5 | |||||||
| Inhibin | ActRII, BMPRII54 | ActRII54 | ActRII54 | ActRII48, 53, 182, 183 | ActRII54 | ||||||
| Lefty1 | ActRII52 | ||||||||||
| Lefty2 | ActRII52 |
The table provides an overview of reported competition between TGF-β family ligands, including which receptor(s) the ligands supposedly compete for in cases where this has been investigated. The ligands listed in the first row indicate agonist ligands, whereas the ligands listed in the first column indicate ligands that compete with the agonist ligands by one of our proposed modes of competition illustrated in Figure 4. In the cells, we have indicated the receptor for which the ligands compete. ActRII: indicates ActRIIA and ActRIIB, ND: Not Determined, indicates competition for an unknown receptor. The table is likely not complete since no single literature search will cover this information.
At the same time, the competing ligand may activate its specific R-SMAD branch. For example, activin A suppressed SMAD1/5/8 signaling by BMP-2 and BMP-7 while it activated SMAD2/3 signaling, causing a gradual, concentration dependent switch in signal transduction pathway utilization 5. However, if competing ligands activate the same SMAD branch, they can produce a spectrum of responses that depend on specific ligand groupings, their relative concentrations, and receptor expression levels of a particular cell. For example, BMP4 exhibited both antagonistic and synergistic interactions with BMP-family ligands. The precise effect depended on the specific ligand with which BMP-4 was paired 8. This observation led the authors to propose that the ability to form many competing ligand-receptor complexes with distinct affinities and activities likely enables a cell to process complex information encoded in ligand combinations 8. We further speculate that the ability to respond to multiple ligands simultaneously or in sequence enables a cell to exhibit input-output plasticity in a fluctuating environment.
Antagonistic ligand competition
While ligand binding competition or cross-inhibition is an inherent molecular property of the TGF-β family, the outcomes of this competition vary depending on the specific ligands involved. E.g., some TGF-β family ligands bind receptors but do not activate SMADs. They include inhibin, activin C, BMP-3, lefty1 and lefty2 48, 49, 50, 51, 52. These ligands may be regarded as true antagonists, as they regulate SMAD signaling either by forming heterodimers with agonistic ligands to produce growth factors that are devoid of signaling activity 50, or by competing with agonistic ligands for receptor binding to block SMAD pathway activation (Figure 4B, C).
For example, inhibin α and inhibin β are true antagonists. They consist of one inhibin α subunit (encoded by the INHA gene) and one inhibin βA, or βB subunit (encoded by the INHBA and INHBB gene, respectively). Inhibin A, like activin A, binds ActRIIA and ActRIIB. They, therefore, compete for binding to these receptors. But in contrast to activin A, inhibin A does not recruit the type I receptor ALK4 into the ligand-receptor complex, thus acting as a true antagonist of activin A signaling 53. The antagonistic relationship between inhibins and activins is essential to the control of pituitary follicle-stimulating hormone release and for normal gonadal function 48. Inhibin A also suppressed signaling by several BMPs/GDFs. This inhibition was associated with its interaction with ActRIIA, ActRIIB, or BMPRII 54.
Activin C is another antagonistic ligand as it blunted the growth inhibitory effect of activin A in LNCaP prostate cancer cells 55, and suppressed the growth-promoting effect of both activin A and activin B in COV434 granulosa cells. Its antagonistic effect was proposed to be dependent on type 2 receptor binding 49. Supporting this conclusion, SMAD2 activity decreased when activin C was overexpressed in vivo 55.
BMP-3 is also considered to be an antagonistic ligand, as it has no known type I receptor but can compete with activin and BMP-4 for binding to ActRIIB 51, 56. BMP-3 may also act as a general BMP antagonist as it can inhibit signaling by many BMPs 8.
Lefty1 and lefty2 are unusual ligands in that they share a uniquely extended C-terminal sequence, and do not form covalent dimers as they lack the cysteine residue required for dimerization. Nevertheless, they competed with nodal for binding to ActRIIA and ActRIIB and inhibited Nodal signaling 52.
A special example of antagonistic competition may occur when a cell lacks the cognate type I receptor for an agonistic ligand. For example, BMP-10 competes with BMP-2 and BMP-7 for binding to shared type II receptors 5. However, BMP-10 would fail to activate SMAD1/5/8 signaling in cells that lack its cognate receptor, ALK1. As many cells have restricted receptor expression, this special example of antagonism could be a prevalent mode of regulation.
Agonistic ligand competition
Agonistic ligands, i.e. ligands that can activate SMAD pathways, may also compete with other agonistic ligands. E.g., a SMAD2/3-activating ligand could antagonize a SMAD1/5/8-activating ligand (Figure 4D) to inhibit SMAD1/5/8 signaling. Two examples are GDF-11 and GDF-8 (myostatin), which activate SMAD2/3 via the type I receptors ALK4, ALK5, or ALK7. Both antagonized the low-affinity ligands BMP-2 and BMP-7 by competing for ActRIIA and ActRIIB and inhibited BMP-2 and BMP-7 mediated SMAD1/5/8 signaling in HepG2 liver carcinoma cells 5. Likewise, activin A inhibited signaling of several BMPs while activating its canonical SMAD2/3 pathway 5, 6, 9, 57. Agonistic ligands that bind receptors with high affinity can, therefore, broadly act as antagonists of low-affinity ligands and inhibit their signaling.
In a more subtle scenario, two agonistic ligands may bind the same receptors and activate the same pathway but bind receptors with different affinities and activate signaling with different potencies (Figure 4E). In that case, ligands’ pairings could result in stronger or weaker signaling than expected given the responses of the individual ligands. For example, BMP7 and BMP4 exhibited synergistic or antagonistic signaling depending on the ligand with which they were paired 8. Another example is seen in multiple myeloma cells where activin A weakly activates SMAD1/5/8 via ALK2, but at the same time antagonizes a strong BMP-mediated SMAD1/5/8 signaling due to its stronger binding affinity for type II receptors 6, 58.
Finally, agonistic ligands could form ‘non-signaling’ complexes (NSCs) with a particular receptor (Figure 4F). For example, activin A activates the SMAD2/3 branch via the type I receptor ALK4 59. However, activin A may also form an inhibitory NSC with ALK2, as it inhibited BMP-6 and BMP-7 signaling in cells where ALK2 is the main type I receptor 60, 61. Competition in this case involved the ligand interaction with ALK2 and type II receptors. The authors of this work suggested that the NSC could restrict ALK2 and type II receptor accessibility or prevent engagement of receptors with BMPs to inhibit ALK2-mediated BMP signaling.
In conclusion, the cross-inhibition or receptor binding-competition paradigm offers a unifying molecular mechanism that explains the broadly inhibitory or regulatory action of both agonistic and antagonistic ligands, as well as their role in signaling and non-signaling complexes.
Experimental determination of ligand competition
Several approaches have been used to study competition between ligands for receptor binding and downstream signaling. Direct approaches, such as surface plasmon resonance (SPR) or enzyme-linked immunosorbent assay (ELISA), can detect physical interactions between ligands and receptors, providing a molecular readout. Indirect approaches, such as cell-based assays, can be used to examine the biological effects of ligand combinations on signaling, offering a functional readout. Here we discuss how different approaches were used to establish competition between TGF-β family ligands.
ELISA can show ligand binding competition directly. In a typical ELISA format, ligands are captured on a plate coated with a ligand-specific antibody. After washing away any unbound substances, ligand binding is detected with an enzyme-conjugated secondary antibody that recognizes a different epitope on the ligand. To investigate receptor binding competition, this format is adjusted so that a ligand is captured with a recombinant Fc-fusion receptor instead of an antibody (Figure 5A). Importantly, the secondary antibody must recognize an epitope on the ligand that is different from the receptor-binding site. As ligand capture levels can be estimated from the intensity of the substrate signal, the changing intensities of the substrate signal can be used to determine the degree of competition between two ligands or molecules. Of note, this approach is limited to ligands that bind the recombinant Fc-fusion receptor with sufficient affinity to remain bound after washing of the plates. Thus, BMP-9 was captured on an ELISA plate coated with the Fc-fusion receptors ActRIIA-Fc and ActRIIB-Fc. Activin A prevented BMP-9 binding to these receptors in a dose-dependent manner. Notably, this inhibition could be blunted by adding the activin A trap Follistatin. Together, these findings showed that activin A competes with BMP-9 for binding to its type II receptors ActRIIA and ActRIIB 6.
Figure 5. Methods to measure ligand competition.

A. ELISA measures levels of ligand captured using Fc-fusion receptors (ActRIIA-Fc, ActRIIB-Fc). Binding of ligand (here BMP-9) is detected with a target-ligand specific, enzyme-conjugated antibody (here anti-BMP-9). A competing ligand (here activin A) is not detected by the that antibody. Competition between activin A and BMP-9 therefore presents as an activin A concentration dependent reduction in the target-specific antibody signal. B. Surface plasmon resonance (SPR) can directly measure competition between ligands for binding to a receptor. Here, a BMP-7 antibody that targets the type I receptor binding site is immobilized and BMP-7 is captured by this antibody. ActRIIA-Fc binds to the captured BMP-7. ActRIIA-Fc is incubated with varying concentrations of activin A. Competition between BMP-7 and activin A presents as an activin A concentration dependent decrease in ActRIIA-Fc binding to captured BMP-7. C. Cell-based assays measure the effect of competition on SMAD signaling via SMAD responsive luciferase reporters. Luciferase activity correlates with SMAD activity. Here, competition between BMP-7 and activin A presents as an activin A concentration dependent decrease in the BMP-7 mediated SMAD1/5/8 signal. D. Antibodies and inhibitors that specifically block the type II or type I receptor binding sites help elucidate the molecular basis for ligand competition.
SPR can be used to demonstrate the ligand competition directly using several different experimental formats (Figure 5B). At the most basic level, a receptor is cross-linked to a sensor chip or captured using a receptor-specific antibody that leaves the ligand-binding surface accessible. Saturating amounts of one ligand are injected followed by injection of a second ligand. Displacement of the first ligand by the second ligand would indicate that the two ligands compete for the same binding epitope on a receptor. Although this observation would support the binding-competition model, this format does not discriminate between the displacement of one ligand by another and other circumstances that result in a reduced SPR signal. A more definitive SPR format was used to demonstrate that activin A inhibits binding of BMP-7 to its type II receptor ActRIIA. In this experiment, BMP-7 was captured on the sensor chip using a BMP-7-specific monoclonal antibody. ActRIIA-Fc bound the captured BMP-7 in the absence of Activin A. However, ActRIIA-Fc binding to BMP-7 decreased with increasing concentrations of activin A, and was completely suppressed by excess activin A. This experiment directly showed that activin A blocks BMP-7 binding to its type II receptor ActRIIA 5. Variations of this experimental format could be used to investigate competitive inhibition for an expanded range of examples 62.
Like ELISA and SPR, cell-based assays can also be used to measure competition directly, as ligands associate with receptors on the surface of cells. Such measurements could be achieved using radiolabeled ligands along with immunoprecipitation of the receptor 10, or with ligand-specific antibodies and flow cytometry 63. For example, COS-1 cells transfected with BMP-7 receptors were radiolabeled with [125I]BMP-7. Increasing concentrations of myostatin (GDF-8) reduced [125I]BMP-7 binding to these cells 10, indicating that competition can be measured in a whole-cell context. However, this example probes competition for binding to the cell surface but cannot identify the specific cell-surface molecule for which BMP-7 and GDF-8 competed.
While cell-based assays can directly probe competition, they are best suited to evaluate ligand competition indirectly by detecting SMAD activation, or by analyzing downstream effects that are based on transcriptional responses, including SMAD target gene transcription, reporter gene expression assays (e.g., BRE-Luc for SMAD1/5/8 64 or CAGA-Luc for SMAD2/3 65, Figure 5C), cell viability, proliferation, or differentiation. These approaches support a direct comparison of signal or biological response intensities, enabling examination of the balance in signaling between the two main SMAD branches, a key factor controlling cell fate decisions. Notably, when measuring effects on transcriptional responses, the read-out could be affected by several independent factors, such as expression levels of the co-transcription factor SMAD4, the inhibitors SMAD6 and SMAD7, or R-SMADs 66.
At the most basic level, indirect cell-based assays can show how the signaling response of one ligand is affected by another ligand. As most activins, BMPs and GDFs use the same type II receptors and frequently share type I receptors for signaling, high-affinity ligands like activin A may be expected to inhibit signaling by ligands that interact with the same receptors with lower affinity. Thus, activin A antagonized BMP-2- and BMP-7 dependent SMAD1/5/8 signaling in HepG2 cells carrying the SMAD1/5/8 responsive BRE reporter 5. Critically, activin A also activated its canonical SMAD2/3 signaling pathway, revealing the profound biological consequences of the ligand competition paradigm, which can result in both SMAD signaling antagonism and SMAD pathway switching. In another example, activin A blunted BMP-9 induced apoptosis in INA-6 and IH-1 multiple myeloma cells and inhibited BMP-9-induced phosphorylation of SMAD1/5/8 6, showing both biological and molecular consequences of ligand competition. Notably, activin A also inhibited BMP-6 dependent SMAD1/5/8 phosphorylation, highlighting the broad applicability of ligand competition. A subsequent analysis using reporter cells delineated cellular response profiles to combinations of BMP ligands. This work showed how specific ligand combinations produced distinct, cell-dependent outcomes, thus supporting the conclusion that promiscuous BMP receptor-ligand interactions enable cells to process multi-ligand inputs to produce complex responses that include additive, subtractive, and multiplicative signaling outputs 8. Notably, receptor expression levels shaped responses to ligand combinations, indicating how ligands could have distinct roles in different cell types.
A powerful tool to study ligand competition at the cellular level uses receptor or growth factor specific monoclonal antibodies or other inhibitors (Figure 5D). One example of this approach exploited critical functionalities of the three activin A inhibitors ActRIIA-Fc, Follistatin, and SB-431542 5. ActRIIA-Fc and Follistatin trap activin A and block its binding to receptors. By contrast, SB-431542 is a small molecule kinase inhibitor that blocks activin A signaling but does not prevent activin A from binding its receptors 67. Both ActRIIA-Fc and Follistatin rescued BMP-2 and/or BMP-7 signaling in the presence of activin A whereas SB-431542 did not. Similarly, Follistatin blunted activin A antagonism of BMP-6 and BMP-9 6. Collectively, these results support the conclusion that activin A antagonism is a direct consequence of competition for type II (and possibly also type I) receptor binding.
A particularly striking and therapeutically important example of competition and its blockade with monoclonal antibodies was reported in a group of studies that discovered activin A as a key driver in the pathology of Fibrodysplasia Ossificans Progressiva (FOP) 60, 61, 68. FOP is a genetic disorder characterized by episodic formation and growth of heterotopic bone in connective tissue. It arises from missense mutations in the kinase domain of the type I receptor ALK2 (which is encoded by ACVR1). In cells that harbor wild-type receptors, activin A antagonizes BMP-6, BMP-7, and BMP-9 signaling by forming an inhibitory, non-signaling complex with the type I receptor ALK2 and the type II receptors ActRIIA and ActRIIB. However, FOP-causing ALK2 variants perceive activin A as an agonist that aberrantly activates the SMAD1/5/8 pathway 60. To elucidate the molecular basis of activin A antagonism in the context of ALK2 non-signaling complexes, these studies exploited monoclonal antibodies that target the receptor-interacting epitopes of activin A, monoclonal antibodies that target the ligand-binding epitopes of receptors, and the small molecule kinase inhibitor SD208 6, 60, 61. Both REGN2476, an anti-activin A monoclonal antibody that blocks its type II receptor binding site, and H4H10442, an anti-activin A antibody that blocks its type I receptor binding site, blunted activin A antagonism of BMP-7 as evidenced by the rescue of BMP-7 signaling in cells co-treated with activin A and either antibody. By contrast, the anti-ALK4 antibody MAB222 and the small molecule ALK4/ALK5 inhibitor SD208 inhibited activin A-mediated SMAD2/3 signaling as expected but did not blunt activin A antagonism of BMP-6. Together, these results indicated that extracellular competition for ligand-receptor binding was the key step in activin A dependent antagonism of BMP-6/BMP-7 61. Notably, REGN2476 inhibited activin dependent SMAD1/5/8 signaling mediated by ALK2 variants found in FOP and suppressed heterotopic ossification in an animal model of FOP. These insights were effectively translated into the clinic, as data from a phase II trial by Regeneron Pharmaceuticals (NCT03188666) indicates that the anti-activin A antibody REGN2477, which has similar properties to H4H10442, could markedly reduce new abnormal bone formation and flare-ups in FOP patients.
In conclusion, combinations of monoclonal antibodies, extracellular antagonists, and small molecule kinase inhibitors have helped elucidate the molecular logic and biological function of the competition paradigm, firmly establishing cross-inhibition or competition as a common mechanism for modulating TGF-β family signaling activation and regulation (Figure 5D). These critical insights helped drive the development of a novel, disease-modifying therapy for FOP patients.
Physiological and pathological roles of ligand competition
In addition to the now well-established roles for activin A as competitive BMP-6/BMP-7 antagonist in the context of ActRIIA/ActRIIB-ALK2 dependent signaling and its gain of function in ALK2 variants that cause FOP 69, competition between TGF-β family ligands may also have critical physiologic and clinical relevance for bone and muscle development, osteoporosis, sarcopenia, cachexia, pulmonary arterial hypertension, tissue fibrosis, and cancers (recently reviewed 70) (Figure 6).
Figure 6. Proposed pathological roles of ligand competition.

A. Fibrodysplasia Ossificans Progressiva (FOP). In wild-type cells, activin A antagonizes BMP signaling by forming an inhibitory, non-signaling complex with ALK2 and type II receptors. In FOP, disease-causing ALK2 variants perceive activin A as an agonist that aberrantly activates the SMAD1/5/8 pathway, and, thus, induces osteogenic differentiation of fibroadipogenic progenitors to form ectopic bone. B. Wasting Syndrome/Cachexia. BMP signaling is vital for muscle and adipose tissue formation and maintenance. Elevated activin/GDF levels in patients with pancreatic cancers may antagonize BMP-SMAD1/5/8 activity in these tissues, contributing to cancer cachexia. C. Pulmonary Arterial Hypertension (PAH). Deficient BMP-SMAD1/5/8 and increased activin-SMAD2/3 signaling may contribute to endothelial arterial cell proliferation in PAH patients, and, thus, to the narrowing of pulmonary arteries. Increased activin/GDF-11 levels could promote PAH by shifting the balance from SMAD1/5/8 to SMAD2/3 signaling. D. Multiple Myeloma (MM). BMP signaling helps maintain MM cells in a slow-proliferating state. Activin A expression is induced in the bone marrow of MM patients and may contribute to tumor cell expansion by antagonizing BMP activity. Also, activin A may alter bone homeostasis and contribute to MM bone disease by suppressing osteoblastic bone formation and inducing osteoclastic bone destruction.
For instance, BMPRII knock-out in bone-forming cells resulted in a high bone mass phenotype 7. Strikingly, BMPRII ablation blunted activin dependent SMAD2/3 signaling but did not affect BMP dependent SMAD1/5/8 signaling. This observation led the authors to propose that BMPs outcompete activins for binding to the remaining type II receptors ActRIIA and ActRIIB to suppress activin A signaling and increase bone mass in this genetic background. Similarly, several studies showed that BMP signaling controls muscle mass, as BMP-mediated SMAD1/5/8 activation induced muscle growth, whereas its inhibition resulted in muscle atrophy 71, 72. Extracellular myostatin and activin inhibitors both induced BMP dependent SMAD1/5/8 signaling in skeletal muscle and increased muscle mass, supporting the conclusion that myostatin and activin antagonize BMPs in muscles by competing for the same receptors to negatively regulate BMP signaling and muscle mass 73, 74. Collectively, these findings indicate that cross-regulation/crosstalk of BMP and activin/myostatin pathways is fundamentally important for bone and muscle homeostasis and that proper re-balancing of these signals using extracellular antagonists could be exploited to regulate bone and muscle mass.
In addition to these physiologically relevant examples, three examples stand out where TGF-β family ligand binding competition may be a pathology driving factor and/or a critical point for therapeutic intervention, including cachexia, pulmonary arterial hypertension, and multiple myeloma. Cachexia is a wasting syndrome characterized by the progressive loss of adipose and skeletal muscle tissues that affects most advanced cancer patients, and that may be responsible for approximately 20% of all cancer deaths 75. Recent findings have implicated activin A in its development and progression, as serum levels correlate with the levels of muscle wasting and mortality in patients with pancreatic ductal adenocarcinoma 76, malignant pleural mesothelioma 77, colorectal, and lung cancers 78. Also, many well-established animal models of cachexia present elevated activin A levels, and activin A or myostatin overexpression leads to muscle wasting in mice 79, 80, 81. These findings have indicated that activin/myostatin antagonism could have therapeutic potential for treating cancer cachexia and other types of muscle wasting. Indeed, extracellular activin/myostatin inhibitors, such as the ligand traps ActRIIA-Fc and ActRIIB-Fc, the antagonist Follistatin, and the ActRIIB neutralizing antibody Bimagrumab, reversed the cachectic phenotype in several animal models, including inhibin-α knockout mice, which develop gonadal tumors, and mice bearing C26 colon carcinoma, G361 melanoma, Lewis lung carcinoma, or TOV21G ovarian carcinoma xenografts 74, 82, 83, 84, 85, 86.
Although these results firmly establish that extracellular activin/myostatin antagonism prevents cachexia-induced muscle wasting, thus implicating SMAD2/3 signaling as a key effector of muscle atrophy, recent findings indicate that the mechanism of cachexia pathogenesis and therapeutic action may critically include reduced SMAD1/5/8 signaling and ligand binding competition 87. For example, studies found that SMAD1/5/8 activation is a fundamental hypertrophic signal in mouse muscles 71, 72, that BMP signaling antagonism with the kinase inhibitor LDN-193189 caused significant atrophy of tibialis anterior myofibers 72 and injured shoulder muscles 88, and that overexpression of the extracellular BMP antagonist noggin led to severe muscle wasting in both hyper-muscular Mstn−/− and wild-type mice 72. These findings reveal the importance of BMP-SMAD1/5/8 signaling in muscle mass maintenance 87. Notably, extracellular activin/myostatin antagonism with Follistatin inhibited SMAD2/3 signaling, as expected, but also activated the SMAD1/5/8 pathway in muscles of C57B1/6 mice 73, 89. Also, activin/myostatin antagonism with their pro-domains both inhibited SMAD2/3 and activated SMAD1/5/8 signaling in muscles of C26 tumor-bearing BALB/c mice, leading the authors of this work to speculate that multiple TGF-β family ligands may cooperate to regulate myostatin activity in muscle-wasting pathologies 73, 74. Together, these findings support a model where extracellular ligand competition could be a key principle driving the cachectic phenotype, as elevated levels of the high-affinity ligands activin or myostatin could both activate their canonical SMAD2/3 signaling pathway and suppress SMAD1/5/8 signaling by blocking the binding of BMPs to their cognate receptors and, thus, promote muscle atrophy. Notably, at least one example showed that extracellular inhibition of these high-affinity ligands rebalanced SMAD pathway activation in mice and reversed the cachectic phenotype 74, an observation that was recapitulated using Follistatin in healthy animals and culture 5, 73. By contrast, the intracellular activin signaling inhibitor SB431542 did not prevent muscle loss in C26 cachectic animals 90, or rebalance SMAD pathway signaling in vitro 5, highlighting a fundamental functional difference between intracellular and extracellular TGF-β family antagonism.
Like cachexia, Pulmonary Arterial Hypertension (PAH) is also driven by a perturbation in SMAD signaling that could be attributed to ligand-binding competition. PAH is a progressive disorder characterized by elevated blood pressure in the lung arteries 91. It is caused by the abnormal remodeling of the small peripheral lung vasculature through proliferation, migration, and survival of vascular cells within the pulmonary arterial wall. The resulting vasoconstriction and small lung artery obstruction lead to increased pulmonary arterial pressure, progressive right heart functional decline, and, eventually, right heart failure 92.
The most common causes of hereditary PAH (HPAH) are loss-of-function mutations in the type II receptor BMPRII (BMPR2), the type I receptor ALK1 (ACVRL1), the co-receptor endoglin (ENG), the ligand BMP-9 (GDF2), and their intracellular effectors SMAD4 and SMAD8 93. The genetics of HPAH, therefore, implicate the BMP-9 signaling axis and deficient SMAD1/5/8 signaling in the pathogenesis of HPAH 94. However, the penetrance of pathogenic BMPRII variants is low 95, indicating that additional factors are necessary for HPAH onset and progression.
Many studies have also found that inflammation plays a key role in the pathobiology of both HPAH and idiopathic PAH (IPAH) 96, 97, potentially linking cytokines secreted by immune cells such as activin A with these conditions 98. Although activins (and related ligands such as GDF-8 and GDF-11) share type II receptors with BMP-9, they do not intersect directly with the pathway and, thus, have not been considered until recently as drivers of, or targets for treating PAH. Nevertheless, reports showed that activin A levels were increased in the circulation of PAH patients and the lungs of mice with hypoxia-induced pulmonary hypertension (PH) 99. Also, activin A promoted proliferation of pulmonary artery smooth muscle cells (PASMCs) and its antagonism with a neutralizing antibody inhibited PASMC growth in vitro 100.
A recent study further showed that activin A, GDF-8, and GDF-11 levels were elevated in the distal pulmonary arterioles of patients with HPAH and IPAH, as well as in vessels of rat PH models. Also, activin A and GDF11 stimulated proliferation in HPAH-patient-derived pulmonary microvascular endothelial cells (PMVECs), and GDF-11 blunted BMP-9 mediated SMAD1/5/8 signaling in these cells 101. Collectively, these findings indicated that activins and the related GDFs could compete with BMP-9 for binding to surface receptors, causing a signaling imbalance that leads to vascular cell proliferation in PAH 101. To establish the functional significance of the binding-competition model in PAH, the authors of this work turned to ActRIIA-Fc, an extracellular antagonist that blocks binding of the proposed PAH ligands activin A, GDF-11, and GDF-8 to their type II receptors 101. ActRIIA-Fc both rescued BMP-9 signaling in the presence of GDF-11, and inhibited activin/GDF-8/-11-mediated proliferation of smooth muscle cells in vitro 101, recapitulating a previous finding, which showed that inhibition of BMP-2 and BMP-7 signaling by activin A can be suppressed by trapping activin A with ActRIIA-Fc 5. Significantly, ActRIIA-Fc blunted pathophysiologic remodeling of the pulmonary vasculature in several experimental models of PH and abrogated the ability of activins, GDF-8 and GDF-11 to compete with BMP-9 for receptor binding to restore BMP-9 signaling in vivo 101. These findings led the authors to propose that activins and/or GDFs are key drivers of PAH, and that receptor binding competition between these ligands and BMP-9 could be a key mechanism underlying PAH pathogenesis 101. Consistent with a critical role for binding competition, exogenous BMP-9 rescued, while the BMP-9 trap ALK1-Fc exacerbated PAH in animal models 102, 103, 104. Importantly, these new insights have provided a preclinical rationale for the clinical development of ActRIIA-Fc as therapeutic for treating PAH patients. Recent phase II clinical trial results by Acceleron Pharma using ActRIIA-Fc (a.k.a. Sotatercept) now indicate that rebalancing BMPRII signaling by inhibiting activins and GDFs could restore vascular homeostasis in PAH patients (NCT03496207 and NCT03496207), underscoring the potential impact of the competition paradigm in human diseases and their therapies.
Ligand binding competition also provides a molecular rationale for the role of activin A in multiple myeloma (MM) progression 57. MM is a malignant disease of the bone marrow that is characterized both by a pathological increase in antibody-producing plasma cells and osteolytic bone destruction 105. Although a definitive role for activin A in MM has not been firmly established, several groups have reported that elevated activin A blood plasma levels associate with advanced-stage disease, as indicated both by the degrees of plasma cell infiltration of the bone marrow and the levels of osteolysis in patients 106, 107. Mechanistically it is suggested that activin A enhances osteolytic bone destruction in MM patients by magnifying its natural biological role in bone remodeling, which includes activating bone-resorbing osteoclasts and inhibiting differentiation of bone-forming osteoblasts 108. Notably, ActRIIA-Fc increased bone formation and osteoblast number and reduced osteolytic lesions and the number of tumor cells in two distinct MM mouse models, providing direct evidence that activin antagonism has significant therapeutic potential for treating MM 106, 109.
Most MM cells express TGF-β family receptors and the ligand BMP-6 63, 110, 111. Although these cells do not produce activin A, they induce its secretion from bone marrow stromal cells (BMSC) via adhesion mediated JNK kinase activation 106, 112, 113. BMSCs are therefore the likely source of excess activin A in MM patients. Critically, several lines of evidence link activin A and its antagonism of BMP signaling with bone destruction. For example, activin A inhibited osteoblast differentiation in vitro as it both activated SMAD2/3 and inhibited SMAD1/5/8 signaling. Also, exposure of BMSCs to MM cell lines or primary cells increased SMAD2/3 phosphorylation and decreased osteoblast differentiation. By contrast, simultaneous treatment with ActRIIA-Fc decreased SMAD2/3 phosphorylation and rescued osteoblast differentiation 106. Collectively, these findings indicate that activin A could activate its canonical signaling pathway and inhibit BMP mediated SMAD1/5/8 signaling to drive bone destruction in MM patients. Although the role of activin A in MM bone destruction is well-established, a recent study showed that inhibition of the BMP pathway could also help restore bone mass 114.
In addition to its role in bone remodeling, activin A may also antagonize BMP signaling in MM cells to promote their proliferation. BMPs are potent inhibitors of the B cell lineage. They regulate B-cell growth and differentiation at multiple stages 115, 116, 117 and induce apoptosis via SMAD1/5/8-dependent repression of MYC 42. Notably, BMP-6 is expressed by many MM cells, and high expression levels predicted improved survival in untreated MM patients 63. BMP-6 also inhibited the proliferation of MM cell lines and survival of primary MM cells, suggesting that it (and possibly other BMPs) could help keep myeloma cells in a slow or non-proliferative state 63. Indeed, BMP-4 gene therapy blunted myeloma tumor growth in a humanized myeloma mouse model 118, and several BMPs induced growth arrest and apoptosis in MM cells in vitro 42, 119, 120, 121, 122, 123, underscoring the potential protective role of BMP ligands in MM. As MM cells, BMPs, and activin A are present in the bone marrow, and as activin A is elevated in the bone marrow of MM patients, it was proposed that the growth-inhibitory effect of BMPs on MM cells could be blunted by activin A to promote MM progression. Indeed, activin A antagonized BMP-6 and BMP-9-mediated apoptosis and signaling in INA-6 and IH-1 MM cells and prevented binding of these two ligands to their type II receptors 6. Similarly, activin A dampened the anti-proliferative activity of wild-type BMP-2 in KMS-12-BM cells, but not of BMP-2 variants that bind type II receptors with activin A-like high affinity 57. Consequently, these findings indicated that activin A likely promotes MM cell proliferation by competing with BMPs for binding to an overlapping set of cell surface receptors and inhibiting BMP dependent SMAD1/5/8 signaling 6, 57.
Thus, activin A could play a dual role in MM progression. On one hand, it may alter bone homeostasis in MM patients by suppressing osteoblastic bone formation and promoting osteoclastic bone destruction. On the other hand, it may promote MM cell proliferation by blunting the anti-proliferative activities of BMPs. Significantly, both roles are likely rooted in the receptor-binding competition paradigm, as activin A prevents binding of BMPs to their cognate receptors and inhibits activation of BMP mediated SMAD1/5/8 signaling.
Concluding remarks
A striking feature of TGF-β signaling pathways is the large number of ligands that can interact promiscuously with a much smaller group of receptors to activate one of two intracellular signal transduction pathways. The biological reason underlying what seems to be a contradictory principle (namely, ligand-receptor binding promiscuity along with signal transduction pathway convergence) has long puzzled. Several models have aimed to explain its purpose. For example, the redundant use of ligands and receptors was suggested to “offer robustness to genetic variation”, to permit “subtle forms of control and more flexibility during evolution”, or to provide signal processing capabilities that allow a cell to “perceive information encoded in combinations of ligands” 8, 124, 125. In addition to these concepts, recent work has demonstrated that ligand-receptor binding promiscuity, which manifests at the molecular level as competition of ligands for receptor binding, could provide a distinct mode of regulation that includes signaling antagonism or potentiation, and signal transduction pathway switching (see Figure 4) 5, 6. The ligand-receptor binding competition (or cross-inhibition) paradigm we discussed here provides a unifying molecular and functional logic that explains the previous models, and the diverse and pleiotropic effects attributed to many ligands in the family. Significantly, the biological impact of competition between ligands is fundamental. To illustrate the potential breadth of this paradigm, we presented several physiological and pathological examples where competition likely takes place, including physiological bone and muscle development, as well as pathological Cancer Cachexia, Pulmonary Arterial Hypertension, Fibrodysplasia Ossificans Progressiva, and Multiple Myeloma. In each one of these examples, the competition paradigm could explain how different ligands balance the activity of each other to produce, modulate, or shift a biological outcome. Critically, receptor-binding competition has been embraced as a possible mechanism underlying FOP and PAH, and its modulation (or rebalancing) has been successfully translated into the clinic with several Phase II trials showing considerable therapeutic potential.
Highlights.
Ligand-receptor binding-competition is a key feature of the TGF-β family.
- It enables:
- Ligands to function as agonists or antagonists depending on the cellular context.
- Cells to read and process complex information encoded in ligand combinations.
- Cells to respond to multiple ligands simultaneously or sequentially.
- Cells to exhibit input-output plasticity in a fluctuating environment.
The functional significance of Ligand-receptor binding-competition is emphasized by its roles in musculoskeletal development and in the pathology of several human diseases.
Acknowledgments:
Figures 1, 2E-G, 3, 4B-F, 5, and 6 were created with BioRender.com. Figures 2A-D and 4A were created with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.)
Funding: This research was funded by NIH R01 GM121499 (E.M.-H.), the Liaison Committee for education, research and innovation in Central Norway (T.H.), and the Joint Research Committee between St. Olav’s Hospital and Faculty of Medicine and Health Science, NTNU (T.H.).
Biography

Erik Martinez-Hackert is an Associate Professor of Biochemistry and Molecular Biology at Michigan State University. He became interested in TGF-β family signaling and regulation while working at Acceleron Pharma (Cambridge, MA, USA) on biologic therapeutics that target this family. He now works to determine how TGF-β family signaling growth factors interact with their receptors, how they activate their intracellular signaling pathways, and how they are regulated by co-receptors to elicit physiological and pathological responses in the musculoskeletal system.

Anders Sundan is a Professor of Cell Biology at the Norwegian University of Science and Technology (NTNU) in Trondheim, Norway. He has a long-standing interest in innate immunity as well as in oncogenesis and growth control in malignant plasma cells. He works to understand how myeloma cells may escape immune surveillance, and how the immune system may be reactivated in the treatment of multiple myeloma.

Toril Holien is a researcher and group leader with a PhD in Molecular Medicine from the Norwegian University of Science and Technology (NTNU) in Trondheim, Norway. She has a background as a Biomedical Laboratory Scientist and worked in clinical diagnostic labs as well as research labs before she did her PhD. Holien’s group at the Department of Clinical and Molecular Medicine, NTNU, is part of a larger translational research environment focusing on multiple myeloma and their lab is located in St. Olavs’s University Hospital. Her main research interest is how cell signaling pathways affect cell growth and survival, and how this can be targeted clinically for the benefit of patients. The group led by Holien studies TGF-β/BMP-signaling, or more specifically, mechanisms for ligand-receptor interactions and how these regulate downstream signaling.
Footnotes
Competing Interests: E.M.-H owns shares of Acceleron Pharma and is co-founder of Advertent Biotherapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yadin D, Knaus P, Mueller TD. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine & growth factor reviews 2016, 27: 13–34. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal 2019, 12(570). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harbor perspectives in biology 2016, 8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YE. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harbor perspectives in biology 2017, 9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aykul S, Martinez-Hackert E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J Biol Chem 2016, 291(20): 10792–10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, et al. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal 2015, 13(1): 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowery JW, Intini G, Gamer L, Lotinun S, Salazar VS, Ote S, et al. Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J Cell Sci 2015, 128(7): 1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antebi YE, Linton JM, Klumpe H, Bintu B, Gong M, Su C, et al. Combinatorial Signal Perception in the BMP Pathway. Cell 2017, 170(6): 1184–1196.e1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piek E, Afrakhte M, Sampath K, van Zoelen EJ, Heldin CH, ten Dijke P. Functional antagonism between activin and osteogenic protein-1 in human embryonal carcinoma cells. J Cell Physiol 1999, 180(2): 141–149. [DOI] [PubMed] [Google Scholar]
- 10.Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Molecular and cellular biology 2003, 23(20): 7230–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison CA, Al-Musawi SL, Walton KL. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth factors (Chur, Switzerland) 2011, 29(5): 174–186. [DOI] [PubMed] [Google Scholar]
- 12.Constam DB. Regulation of TGFbeta and related signals by precursor processing. Seminars in cell & developmental biology 2014, 32: 85–97. [DOI] [PubMed] [Google Scholar]
- 13.Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocrine reviews 2002, 23(6): 787–823. [DOI] [PubMed] [Google Scholar]
- 14.Hinck AP, Mueller TD, Springer TA. Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harbor perspectives in biology 2016, 8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mi LZ, Brown CT, Gao Y, Tian Y, Le VQ, Walz T, et al. Structure of bone morphogenetic protein 9 procomplex. Proceedings of the National Academy of Sciences of the United States of America 2015, 112(12): 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Fischer G, Hyvonen M. Structure and activation of pro-activin A. Nature communications 2016, 7: 12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin SJ, Lerch TF, Cook RW, Jardetzky TS, Woodruff TK. The structural basis of TGF-beta, bone morphogenetic protein, and activin ligand binding. Reproduction (Cambridge, England) 2006, 132(2): 179–190. [DOI] [PubMed] [Google Scholar]
- 18.Goebel EJ, Corpina RA, Hinck CS, Czepnik M, Castonguay R, Grenha R, et al. Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proceedings of the National Academy of Sciences of the United States of America 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nickel J, Sebald W, Groppe JC, Mueller TD. Intricacies of BMP receptor assembly. Cytokine & growth factor reviews 2009, 20(5-6): 367–377. [DOI] [PubMed] [Google Scholar]
- 20.Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, et al. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. Journal of cell science 2007, 120(Pt 6): 964–972. [DOI] [PubMed] [Google Scholar]
- 21.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109(5): 1953–1961. [DOI] [PubMed] [Google Scholar]
- 22.Kirsch T, Nickel J, Sebald W. BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. Embo j 2000, 19(13): 3314–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groppe J, Hinck CS, Samavarchi-Tehrani P, Zubieta C, Schuermann JP, Taylor AB, et al. Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Molecular cell 2008, 29(2): 157–168. [DOI] [PubMed] [Google Scholar]
- 24.Massagué J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. Embo j 2000, 19(8): 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113(6): 685–700. [DOI] [PubMed] [Google Scholar]
- 26.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425(6958): 577–584. [DOI] [PubMed] [Google Scholar]
- 27.Heldin CH, Moustakas A. Role of Smads in TGFbeta signaling. Cell and tissue research 2012, 347(1): 21–36. [DOI] [PubMed] [Google Scholar]
- 28.Tzavlaki K, Moustakas A. TGF-β Signaling. Biomolecules 2020, 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazawa K, Miyazono K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harbor perspectives in biology 2017, 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gipson GR, Goebel EJ, Hart KN, Kappes EC, Kattamuri C, McCoy JC, et al. Structural Perspective of BMP ligands and Signaling. Bone 2020: 115549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goebel EJ, Hart KN, McCoy JC, Thompson TB. Structural biology of the TGFβ family. Exp Biol Med (Maywood) 2019, 244(17): 1530–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370(6488): 341–347. [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 1996, 86(3): 435–444. [DOI] [PubMed] [Google Scholar]
- 34.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massague J. The TGF beta receptor activation process: an inhibitor-to substrate-binding switch. Molecular cell 2001, 8(3): 671–682. [DOI] [PubMed] [Google Scholar]
- 35.Chaikuad A, Bullock AN. Structural Basis of Intracellular TGF-beta Signaling: Receptors and Smads. Cold Spring Harbor perspectives in biology 2016, 8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. Journal of cell science 2001, 114(Pt 24): 4359–4369. [DOI] [PubMed] [Google Scholar]
- 37.Persson U, Izumi H, Souchelnytskyi S, Itoh S, Grimsby S, Engstrom U, et al. The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS letters 1998, 434(1-2): 83–87. [DOI] [PubMed] [Google Scholar]
- 38.Chen YG, Hata A, Lo RS, Wotton D, Shi Y, Pavletich N, et al. Determinants of specificity in TGF-beta signal transduction. Genes & development 1998, 12(14): 2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo RS, Chen YG, Shi Y, Pavletich NP, Massague J. The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-beta receptors. Embo j 1998, 17(4): 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill CS. Transcriptional Control by the SMADs. Cold Spring Harbor perspectives in biology 2016, 8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baughn LB, Di Liberto M, Niesvizky R, Cho HJ, Jayabalan D, Lane J, et al. CDK2 phosphorylation of Smad2 disrupts TGF-beta transcriptional regulation in resistant primary bone marrow myeloma cells. J Immunol 2009, 182(4): 1810–1817. [DOI] [PubMed] [Google Scholar]
- 42.Holien T, Vatsveen TK, Hella H, Rampa C, Brede G, Groseth LA, et al. Bone morphogenetic proteins induce apoptosis in multiple myeloma cells by Smad-dependent repression of MYC. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2012, 26(5): 1073–1080. [DOI] [PubMed] [Google Scholar]
- 43.Aykul S, Maust J, Floer M, Martinez-Hackert E. TGF-β Family Inhibitors Blunt Adipogenesis Via Non-Canonical Regulation Of SMAD Pathways. bioRxiv 2020. [Google Scholar]
- 44.Weber D, Kotzsch A, Nickel J, Harth S, Seher A, Mueller U, et al. A silent H-bond can be mutationally activated for high-affinity interaction of BMP-2 and activin type IIB receptor. BMC Struct Biol 2007, 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS letters 2012, 586(14): 1846–1859. [DOI] [PubMed] [Google Scholar]
- 46.Wakefield LM, Smith DM, Masui T, Harris CC, Sporn MB. Distribution and modulation of the cellular receptor for transforming growth factor-beta. The Journal of cell biology 1987, 105(2): 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondo S, Hashimoto M, Etoh Y, Murata M, Shibai H, Muramatsu M. Identification of the two types of specific receptor for activin/EDF expressed on Friend leukemia and embryonal carcinoma cells. Biochemical and biophysical research communications 1989, 161(3): 1267–1272. [DOI] [PubMed] [Google Scholar]
- 48.Lebrun JJ, Vale WW. Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Molecular and cellular biology 1997, 17(3): 1682–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marino FE, Risbridger G, Gold E. The inhibin/activin signalling pathway in human gonadal and adrenal cancers. Molecular human reproduction 2014, 20(12): 1223–1237. [DOI] [PubMed] [Google Scholar]
- 50.Mellor SL, Cranfield M, Ries R, Pedersen J, Cancilla B, de Kretser D, et al. Localization of activin beta(A)-, beta(B)-, and beta(C)-subunits in humanprostate and evidence for formation of new activin heterodimers of beta(C)-subunit. The Journal of clinical endocrinology and metabolism 2000, 85(12): 4851–4858. [DOI] [PubMed] [Google Scholar]
- 51.Gamer LW, Nove J, Levin M, Rosen V. BMP-3 is a novel inhibitor of both activin and BMP-4 signaling in Xenopus embryos. Developmental biology 2005, 285(1): 156–168. [DOI] [PubMed] [Google Scholar]
- 52.Sakuma R, Ohnishi Yi Y, Meno C, Fujii H, Juan H, Takeuchi J, et al. Inhibition of Nodal signalling by Lefty mediated through interaction with common receptors and efficient diffusion. Genes to cells : devoted to molecular & cellular mechanisms 2002, 7(4): 401–412. [DOI] [PubMed] [Google Scholar]
- 53.Xu J, McKeehan K, Matsuzaki K, McKeehan WL. Inhibin antagonizes inhibition of liver cell growth by activin by a dominant-negative mechanism. J Biol Chem 1995, 270(11): 6308–6313. [DOI] [PubMed] [Google Scholar]
- 54.Wiater E, Vale W. Inhibin is an antagonist of bone morphogenetic protein signaling. J Biol Chem 2003, 278(10): 7934–7941. [DOI] [PubMed] [Google Scholar]
- 55.Gold E, Jetly N, O'Bryan MK, Meachem S, Srinivasan D, Behuria S, et al. Activin C antagonizes activin A in vitro and overexpression leads to pathologies in vivo. The American journal of pathology 2009, 174(1): 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kokabu S, Gamer L, Cox K, Lowery J, Tsuji K, Raz R, et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol Endocrinol 2012, 26(1): 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seher A, Lagler C, Stuhmer T, Muller-Richter UDA, Kubler AC, Sebald W, et al. Utilizing BMP-2 muteins for treatment of multiple myeloma. PLoS One 2017, 12(5): e0174884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olsen OE, Hella H, Elsaadi S, Jacobi C, Martinez-Hackert E, Holien T. Activins as Dual Specificity TGF-beta Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules 2020, 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, et al. Characterization of type I receptors for transforming growth factor-beta and activin. Science (New York, NY) 1994, 264(5155): 101–104. [DOI] [PubMed] [Google Scholar]
- 60.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 2015, 7(303): 303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aykul S, Corpina RA, Goebel EJ, Cunanan CJ, Dimitriou A, Kim H, et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9: e54582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aykul S, Martinez-Hackert E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Analytical biochemistry 2016, 508: 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seckinger A, Meissner T, Moreaux J, Goldschmidt H, Fuhler GM, Benner A, et al. Bone morphogenic protein 6: a member of a novel class of prognostic factors expressed by normal and malignant plasma cells inhibiting proliferation and angiogenesis. Oncogene 2009, 28(44): 3866–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem 2002, 277(7): 4883–4891. [DOI] [PubMed] [Google Scholar]
- 65.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo j 1998, 17(11): 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gronroos E, Kingston IJ, Ramachandran A, Randall RA, Vizan P, Hill CS. Transforming growth factor beta inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Molecular and cellular biology 2012, 32(14): 2904–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Molecular pharmacology 2002, 62(1): 65–74. [DOI] [PubMed] [Google Scholar]
- 68.Alessi Wolken DM, Idone V, Hatsell SJ, Yu PB, Economides AN. The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 2018, 109: 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hwang C, Pagani CA, Das N, Marini S, Huber AK, Xie L, et al. Activin A does not drive post-traumatic heterotopic ossification. Bone 2020: 115473. [DOI] [PubMed] [Google Scholar]
- 70.Hudnall AM, Arthur JW, Lowery JW. Clinical Relevance and Mechanisms of Antagonism Between the BMP and Activin/TGF-beta Signaling Pathways. The Journal of the American Osteopathic Association 2016, 116(7): 452–461. [DOI] [PubMed] [Google Scholar]
- 71.Winbanks CE, Chen JL, Qian H, Liu Y, Bernardo BC, Beyer C, et al. The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. The Journal of cell biology 2013, 203(2): 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, et al. BMP signaling controls muscle mass. Nat Genet 2013, 45(11): 1309–1318. [DOI] [PubMed] [Google Scholar]
- 73.Davey JR, Watt KI, Parker BL, Chaudhuri R, Ryall JG, Cunningham L, et al. Integrated expression analysis of muscle hypertrophy identifies Asb2 as a negative regulator of muscle mass. JCI Insight 2016, 1(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen JL, Walton KL, Hagg A, Colgan TD, Johnson K, Qian H, et al. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proceedings of the National Academy of Sciences 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aoyagi T, Terracina KP, Raza A, Matsubara H, Takabe K. Cancer cachexia, mechanism and treatment. World J Gastrointest Oncol 2015, 7(4): 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhong X, Pons M, Poirier C, Jiang Y, Liu J, Sandusky GE, et al. The systemic activin response to pancreatic cancer: implications for effective cancer cachexia therapy. J Cachexia Sarcopenia Muscle 2019, 10(5): 1083–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paajanen J, Ilonen I, Lauri H, Jarvinen T, Sutinen E, Ollila H, et al. Elevated Circulating Activin A Levels in Patients With Malignant Pleural Mesothelioma Are Related to Cancer Cachexia and Reduced Response to Platinum-based Chemotherapy. Clin Lung Cancer 2020, 21(3): e142–e150. [DOI] [PubMed] [Google Scholar]
- 78.Loumaye A, de Barsy M, Nachit M, Lause P, van Maanen A, Trefois P, et al. Circulating Activin A predicts survival in cancer patients. J Cachexia Sarcopenia Muscle 2017, 8(5): 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walton KL, Chen JL, Arnold Q, Kelly E, La M, Lu L, et al. Activin A-Induced Cachectic Wasting Is Attenuated by Systemic Delivery of Its Cognate Propeptide in Male Mice. Endocrinology 2019, 160(10): 2417–2426. [DOI] [PubMed] [Google Scholar]
- 80.Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. Faseb j 2014, 28(4): 1711–1723. [DOI] [PubMed] [Google Scholar]
- 81.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science (New York, NY) 2002, 296(5572): 1486–1488. [DOI] [PubMed] [Google Scholar]
- 82.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142(4): 531–543. [DOI] [PubMed] [Google Scholar]
- 83.Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach-Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet Muscle 2016, 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012, 3(1): 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochemical and biophysical research communications 2010, 391(3): 1548–1554. [DOI] [PubMed] [Google Scholar]
- 86.Li Q, Kumar R, Underwood K, O'Connor AE, Loveland KL, Seehra JS, et al. Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Molecular human reproduction 2007, 13(9): 675–683. [DOI] [PubMed] [Google Scholar]
- 87.Sartori R, Gregorevic P, Sandri M. TGFbeta and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends in endocrinology and metabolism: TEM 2014, 25(9): 464–471. [DOI] [PubMed] [Google Scholar]
- 88.Liu X, Joshi S, Ravishankar B, Laron D, Kim HT, Feeley BT. Bone morphogenetic protein signaling in rotator cuff muscle atrophy and fatty infiltration. Muscles Ligaments Tendons J 2015, 5(2): 113–119. [PMC free article] [PubMed] [Google Scholar]
- 89.Winbanks CE, Weeks KL, Thomson RE, Sepulveda PV, Beyer C, Qian H, et al. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. The Journal of cell biology 2012, 197(7): 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Levolger S, Wiemer EAC, van Vugt JLA, Huisman SA, van Vledder MG, van Damme-van Engel S, et al. Inhibition of activin-like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Scientific reports 2019, 9(1): 9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ (Clinical research ed) 2018, 360: j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Woodcock CC, Chan SY. The Search for Disease-Modifying Therapies in Pulmonary Hypertension. J Cardiovasc Pharmacol Ther 2019, 24(4): 334–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garcia-Rivas G, Jeijes-Sánchez C, Rodriguez D, Garcia-Pelaez J, Trevino V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med Genet 2017, 18(1): 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Orriols M, Gomez-Puerto MC, Ten Dijke P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cellular and molecular life sciences : CMLS 2017, 74(16): 2979–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. American journal of respiratory and critical care medicine 2012, 186(9): 892–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. Journal of the American College of Cardiology 2009, 54(1 Suppl): S10–19. [DOI] [PubMed] [Google Scholar]
- 97.Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, et al. Inflammation in pulmonary arterial hypertension. Chest 2012, 141(1): 210–221. [DOI] [PubMed] [Google Scholar]
- 98.Phillips DJ, de Kretser DM, Hedger MP. Activin and related proteins in inflammation: not just interested bystanders. Cytokine & growth factor reviews 2009, 20(2): 153–164. [DOI] [PubMed] [Google Scholar]
- 99.Yndestad A, Larsen KO, Oie E, Ueland T, Smith C, Halvorsen B, et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. Journal of applied physiology (Bethesda, Md : 1985) 2009, 106(4): 1356–1364. [DOI] [PubMed] [Google Scholar]
- 100.Kudryashova TV, Shen Y, Pena A, Cronin E, Okorie E, Goncharov DA, et al. Inhibitory Antibodies against Activin A and TGF-β Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. Int J Mol Sci 2018, 19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yung LM, Yang P, Joshi S, Augur ZM, Kim SSJ, Bocobo GA, et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci Transl Med 2020, 12(543). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ormiston ML, Upton PD, Li W, Morrell NW. The promise of recombinant BMP ligands and other approaches targeting BMPR-II in the treatment of pulmonary arterial hypertension. Glob Cardiol Sci Pract 2015, 2015(4): 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Long L, Ormiston ML, Yang X, Southwood M, Gräf S, Machado RD, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nature medicine 2015, 21(7): 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nikolic I, Yung LM, Yang P, Malhotra R, Paskin-Flerlage SD, Dinter T, et al. Bone Morphogenetic Protein 9 Is a Mechanistic Biomarker of Portopulmonary Hypertension. American journal of respiratory and critical care medicine 2019, 199(7): 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Silbermann R, Roodman GD. Myeloma bone disease: Pathophysiology and management. J Bone Oncol 2013, 2(2): 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vallet S, Mukherjee S, Vaghela N, Hideshima T, Fulciniti M, Pozzi S, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(11): 5124–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Terpos E, Kastritis E, Christoulas D, Gkotzamanidou M, Eleutherakis-Papaiakovou E, Kanellias N, et al. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2012, 23(10): 2681–2686. [DOI] [PubMed] [Google Scholar]
- 108.Nicks KM, Perrien DS, Akel NS, Suva LJ, Gaddy D. Regulation of osteoblastogenesis and osteoclastogenesis by the other reproductive hormones, Activin and Inhibin. Molecular and cellular endocrinology 2009, 310(1-2): 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chantry AD, Heath D, Mulivor AW, Pearsall S, Baud'huin M, Coulton L, et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2010, 25(12): 2633–2646. [DOI] [PubMed] [Google Scholar]
- 110.Hose D, Moreaux J, Meissner T, Seckinger A, Goldschmidt H, Benner A, et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 2009, 114(1): 128–143. [DOI] [PubMed] [Google Scholar]
- 111.Grcević D, Kusec R, Kovacić N, Lukić A, Lukić IK, Ivcević S, et al. Bone morphogenetic proteins and receptors are over-expressed in bone-marrow cells of multiple myeloma patients and support myeloma cells by inducing ID genes. Leukemia research 2010, 34(6): 742–751. [DOI] [PubMed] [Google Scholar]
- 112.Silbermann R, Bolzoni M, Storti P, Guasco D, Bonomini S, Zhou D, et al. Bone marrow monocyte-/macrophage-derived activin A mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2014, 28(4): 951–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Palma BD, Guasco D, Pedrazzoni M, Bolzoni M, Accardi F, Costa F, et al. Osteolytic lesions, cytogenetic features and bone marrow levels of cytokines and chemokines in multiple myeloma patients: Role of chemokine (C-C motif) ligand 20. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 2016. 30(2): 409–416. [DOI] [PubMed] [Google Scholar]
- 114.Gooding S, Olechnowicz SWZ, Morris EV, Armitage AE, Arezes J, Frost J, et al. Transcriptomic profiling of the myeloma bone-lining niche reveals BMP signalling inhibition to improve bone disease. Nature communications 2019, 10(1): 4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kersten C, Sivertsen EA, Hystad ME, Forfang L, Smeland EB, Myklebust JH. BMP-6 inhibits growth of mature human B cells; induction of Smad phosphorylation and upregulation of Id1. BMC immunology 2005, 6: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huse K, Bakkebø M, Oksvold MP, Forfang L, Hilden VI, Stokke T, et al. Bone morphogenetic proteins inhibit CD40L/IL-21-induced Ig production in human B cells: differential effects of BMP-6 and BMP-7. European journal of immunology 2011, 41(11): 3135–3145. [DOI] [PubMed] [Google Scholar]
- 117.Bollum LK, Huse K, Oksvold MP, Bai B, Hilden VI, Forfang L, et al. BMP-7 induces apoptosis in human germinal center B cells and is influenced by TGF-β receptor type IALK5. PLoS One 2017, 12(5): e0177188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Westhrin M, Holien T, Zahoor M, Moen SH, Buene G, Stordal B, et al. Bone Morphogenetic Protein 4 Gene Therapy in Mice Inhibits Myeloma Tumor Growth, But Has a Negative Impact on Bone. JBMR plus 2020, 4(1): e10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hjertner O, Hjorth-Hansen H, Borset M, Seidel C, Waage A, Sundan A. Bone morphogenetic protein-4 inhibits proliferation and induces apoptosis of multiple myeloma cells. Blood 2001, 97(2): 516–522. [DOI] [PubMed] [Google Scholar]
- 120.Ro TB, Holt RU, Brenne AT, Hjorth-Hansen H, Waage A, Hjertner O, et al. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene 2004, 23(17): 3024–3032. [DOI] [PubMed] [Google Scholar]
- 121.Olsen OE, Wader KF, Misund K, Vatsveen TK, Ro TB, Mylin AK, et al. Bone morphogenetic protein-9 suppresses growth of myeloma cells by signaling through ALK2 but is inhibited by endoglin. Blood Cancer J 2014, 4: e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Olsen OE, Sankar M, Elsaadi S, Hella H, Buene G, Darvekar SR et al. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. Journal of cell science 2018, 131(11). [DOI] [PubMed] [Google Scholar]
- 123.Kawamura C, Kizaki M, Yamato K, Uchida H, Fukuchi Y, Hattori Y, et al. Bone morphogenetic protein-2 induces apoptosis in human myeloma cells with modulation of STAT3. Blood 2000, 96(6): 2005–2011. [PubMed] [Google Scholar]
- 124.Llimargas M, Lawrence PA. Seven Wnt homologues in Drosophila: a case study of the developing tracheae. Proceedings of the National Academy of Sciences of the United States of America 2001, 98(25): 14487–14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dudley AT, Robertson EJ. Overlapping expression domains of bone morphogenetic protein family members potentially account for limited tissue defects in BMP7 deficient embryos. Dev Dyn 1997, 208(3): 349–362. [DOI] [PubMed] [Google Scholar]
- 126.Breit SN, Tsai VW, Brown DA. Targeting Obesity and Cachexia: Identification of the GFRAL Receptor-MIC-1/GDF15 Pathway. Trends in molecular medicine 2017, 23(12): 1065–1067. [DOI] [PubMed] [Google Scholar]
- 127.Olsen OE, Skjaervik A, Stordal BF, Sundan A, Holien T. TGF-beta contamination of purified recombinant GDF15. PLoS One 2017, 12(11): e0187349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ebner R, Chen RH, Lawler S, Zioncheck T, Derynck R. Determination of type I receptor specificity by the type II receptors for TGF-beta or activin. Science (New York, NY) 1993, 262(5135): 900–902. [DOI] [PubMed] [Google Scholar]
- 129.Attisano L, Carcamo J, Ventura F, Weis FM, Massague J, Wrana JL. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 1993, 75(4): 671–680. [DOI] [PubMed] [Google Scholar]
- 130.Rejon CA, Hancock MA, Li YN, Thompson TB, Hebert TE, Bernard DJ. Activins bind and signal via bone morphogenetic protein receptor type II (BMPR2) in immortalized gonadotrope-like cells. Cell Signal 2013, 25(12): 2717–2726. [DOI] [PubMed] [Google Scholar]
- 131.Khalil AM, Dotimas H, Kahn J, Lamerdin JE, Hayes DB, Gupta P, et al. Differential Binding Activity of TGF-beta Family Proteins to Select TGF-beta Receptors. The Journal of pharmacology and experimental therapeutics 2016, 358(3): 423–430. [DOI] [PubMed] [Google Scholar]
- 132.Mathews LS, Vale WW. Characterization of type II activin receptors. Binding, processing, and phosphorylation. J Biol Chem 1993, 268(25): 19013–19018. [PubMed] [Google Scholar]
- 133.Walker RG, Czepnik M, Goebel EJ, McCoy JC, Vujic A, Cho M, et al. Structural basis for potency differences between GDF8 and GDF11. BMC biology 2017, 15(1): 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Heinecke K, Seher A, Schmitz W, Mueller TD, Sebald W, Nickel J. Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC biology 2009, 7: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tsuchida K, Nakatani M, Yamakawa N, Hashimoto O, Hasegawa Y, Sugino H. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Molecular and cellular endocrinology 2004, 220(1-2): 59–65. [DOI] [PubMed] [Google Scholar]
- 136.Canali S, Core AB, Zumbrennen-Bullough KB, Merkulova M, Wang CY, Schneyer AL, et al. Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice. Endocrinology 2016, 157(3): 1146–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Massague J, Czech MP, Iwata K, DeLarco JE, Todaro GJ. Affinity labeling of a transforming growth factor receptor that does not interact with epidermal growth factor. Proceedings of the National Academy of Sciences of the United States of America 1982, 79(22): 6822–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Massague J, Like B. Cellular receptors for type beta transforming growth factor. Ligand binding and affinity labeling in human and rodent cell lines. J Biol Chem 1985, 260(5): 2636–2645. [PubMed] [Google Scholar]
- 139.Lin HY, Moustakas A, Knaus P, Wells RG, Henis YI, Lodish HF. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-beta receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-beta ligands. J Biol Chem 1995, 270(6): 2747–2754. [DOI] [PubMed] [Google Scholar]
- 140.Lux A, Attisano L, Marchuk DA. Assignment of transforming growth factor beta1 and beta3 and a third new ligand to the type I receptor ALK-1. J Biol Chem 1999, 274(15): 9984–9992. [DOI] [PubMed] [Google Scholar]
- 141.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo j 2002, 21(7): 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rotzer D, Roth M, Lutz M, Lindemann D, Sebald W, Knaus P. Type III TGF-beta receptor-independent signalling of TGF-beta2 via TbetaRII-B, an alternatively spliced TGF-beta type II receptor. Embo j 2001, 20(3): 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ebisawa T, Tada K, Kitajima I, Tojo K, Sampath TK, Kawabata M, et al. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J Cell Sci 1999, 112 (Pt 20): 3519–3527. [DOI] [PubMed] [Google Scholar]
- 144.Liu F, Ventura F, Doody J, Massague J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Molecular and cellular biology 1995, 15(7): 3479–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sako D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J Biol Chem 2010, 285(27): 21037–21048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xia Y, Yu PB, Sidis Y, Beppu H, Bloch KD, Schneyer AL, et al. Repulsive guidance molecule RGMa alters utilization of bone morphogenetic protein (BMP) type II receptors by BMP2 and BMP4. J Biol Chem 2007, 282(25): 18129–18140. [DOI] [PubMed] [Google Scholar]
- 147.Allendorph GP, Isaacs MJ, Kawakami Y, Izpisua Belmonte JC, Choe S. BMP-3 and BMP-6 structures illuminate the nature of binding specificity with receptors. Biochemistry 2007, 46(43): 12238–12247. [DOI] [PubMed] [Google Scholar]
- 148.ten Dijke P, Yamashita H, Sampath TK, Reddi AH, Estevez M, Riddle DL, et al. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem 1994, 269(25): 16985–16988. [PubMed] [Google Scholar]
- 149.Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, et al. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proceedings of the National Academy of Sciences of the United States of America 1995, 92(17): 7632–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Beck HN, Drahushuk K, Jacoby DB, Higgins D, Lein PJ. Bone morphogenetic protein-5 (BMP-5) promotes dendritic growth in cultured sympathetic neurons. BMC Neurosci 2001, 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pang J, Zuo Y, Chen Y, Song L, Zhu Q, Yu J, et al. ACVR1-Fc suppresses BMP signaling and chondro-osseous differentiation in an in vitro model of Fibrodysplasia ossificans progressiva. Bone 2016, 92: 29–36. [DOI] [PubMed] [Google Scholar]
- 152.Yamashita H, ten Dijke P, Huylebroeck D, Sampath TK, Andries M, Smith JC, et al. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J Cell Biol 1995, 130(1): 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brown MA, Zhao Q, Baker KA, Naik C, Chen C, Pukac L, et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J Biol Chem 2005, 280(26): 25111–25118. [DOI] [PubMed] [Google Scholar]
- 154.Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, et al. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol 2008, 22(12): 2689–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mitrofan CG, Appleby SL, Nash GB, Mallat Z, Chilvers ER, Upton PD, et al. Bone morphogenetic protein 9 (BMP9) and BMP10 enhance tumor necrosis factor-alpha-induced monocyte recruitment to the vascular endothelium mainly via activin receptor-like kinase 2. J Biol Chem 2017, 292(33): 13714–13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mazerbourg S, Hsueh AJ. Genomic analyses facilitate identification of receptors and signalling pathways for growth differentiation factor 9 and related orphan bone morphogenetic protein/growth differentiation factor ligands. Hum Reprod Update 2006, 12(4): 373–383. [DOI] [PubMed] [Google Scholar]
- 157.Mazerbourg S, Sangkuhl K, Luo CW, Sudo S, Klein C, Hsueh AJ. Identification of receptors and signaling pathways for orphan bone morphogenetic protein/growth differentiation factor ligands based on genomic analyses. J Biol Chem 2005, 280(37): 32122–32132. [DOI] [PubMed] [Google Scholar]
- 158.Andersson O, Bertolino P, Ibanez CF. Distinct and cooperative roles of mammalian Vg1 homologs GDF1 and GDF3 during early embryonic development. Developmental biology 2007, 311(2): 500–511. [DOI] [PubMed] [Google Scholar]
- 159.Cheng SK, Olale F, Bennett JT, Brivanlou AH, Schier AF. EGF-CFC proteins are essential coreceptors for the TGF-beta signals Vg1 and GDF1. Genes & development 2003, 17(1): 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Andersson O, Reissmann E, Jornvall H, Ibanez CF. Synergistic interaction between Gdf1 and Nodal during anterior axis development. Developmental biology 2006, 293(2): 370–381. [DOI] [PubMed] [Google Scholar]
- 161.Chen C, Ware SM, Sato A, Houston-Hawkins DE, Habas R, Matzuk MM, et al. The Vg1-related protein Gdf3 acts in a Nodal signaling pathway in the pre-gastrulation mouse embryo. Development (Cambridge, England) 2006, 133(2): 319–329. [DOI] [PubMed] [Google Scholar]
- 162.Andersson O, Korach-Andre M, Reissmann E, Ibanez CF, Bertolino P. Growth/differentiation factor 3 signals through ALK7 and regulates accumulation of adipose tissue and diet-induced obesity. Proceedings of the National Academy of Sciences of the United States of America 2008, 105(20): 7252–7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Erlacher L, McCartney J, Piek E, ten Dijke P, Yanagishita M, Oppermann H, et al. Cartilage-derived morphogenetic proteins and osteogenic protein-1 differentially regulate osteogenesis. J Bone Miner Res 1998, 13(3): 383–392. [DOI] [PubMed] [Google Scholar]
- 164.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proceedings of the National Academy of Sciences of the United States of America 2001, 98(16): 9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vitt UA, Mazerbourg S, Klein C, Hsueh AJ. Bone morphogenetic protein receptor type II is a receptor for growth differentiation factor-9. Biol Reprod 2002, 67(2): 473–480. [DOI] [PubMed] [Google Scholar]
- 166.Mazerbourg S, Klein C, Roh J, Kaivo-Oja N, Mottershead DG, Korchynskyi O, et al. Growth differentiation factor-9 signaling is mediated by the type I receptor, activin receptor-like kinase 5. Mol Endocrinol 2004, 18(3): 653–665. [DOI] [PubMed] [Google Scholar]
- 167.Jia J, Jin J, Chen Q, Yuan Z, Li H, Bian J, et al. Eukaryotic expression, Co-IP and MS identify BMPR-1B protein-protein interaction network. Biol Res 2020, 53(1): 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Moore RK, Otsuka F, Shimasaki S. Molecular basis of bone morphogenetic protein-15 signaling in granulosa cells. J Biol Chem 2003, 278(1): 304–310. [DOI] [PubMed] [Google Scholar]
- 169.Pulkki MM, Mottershead DG, Pasternack AH, Muggalla P, Ludlow H, van Dinther M, et al. A covalently dimerized recombinant human bone morphogenetic protein-15 variant identifies bone morphogenetic protein receptor type 1B as a key cell surface receptor on ovarian granulosa cells. Endocrinology 2012, 153(3): 1509–1518. [DOI] [PubMed] [Google Scholar]
- 170.Chang HM, Cheng JC, Klausen C, Leung PC. BMP15 suppresses progesterone production by down-regulating StAR via ALK3 in human granulosa cells. Mol Endocrinol 2013, 27(12): 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Upadhyay G, Yin Y, Yuan H, Li X, Derynck R, Glazer RI. Stem cell antigen-1 enhances tumorigenicity by disruption of growth differentiation factor-10 (GDF10)-dependent TGF-beta signaling. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(19): 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Matsumoto Y, Otsuka F, Hino J, Miyoshi T, Takano M, Miyazato M, et al. Bone morphogenetic protein-3b (BMP-3b) inhibits osteoblast differentiation via Smad2/3 pathway by counteracting Smad1/5/8 signaling. Molecular and cellular endocrinology 2012, 350(1): 78–86. [DOI] [PubMed] [Google Scholar]
- 173.Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M, Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes & development 2002, 16(21): 2749–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Andersson O, Reissmann E, Ibanez CF. Growth differentiation factor 11 signals through the transforming growth factor-beta receptor ALK5 to regionalize the anterior-posterior axis. EMBO reports 2006, 7(8): 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Josso N, Clemente N. Transduction pathway of anti-Mullerian hormone, a sex-specific member of the TGF-beta family. Trends in endocrinology and metabolism: TEM 2003, 14(2): 91–97. [DOI] [PubMed] [Google Scholar]
- 176.Aykul S, Ni W, Mutatu W, Martinez-Hackert E. Human Cerberus prevents nodal-receptor binding, inhibits nodal signaling, and suppresses nodal-mediated phenotypes. PLoS One 2015, 10(1): e0114954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Levine AJ, Levine ZJ, Brivanlou AH. GDF3 is a BMP inhibitor that can activate Nodal signaling only at very high doses. Developmental biology 2009, 325(1): 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Levine AJ, Brivanlou AH. GDF3, a BMP inhibitor, regulates cell fate in stem cells and early embryos. Development (Cambridge, England) 2006, 133(2): 209–216. [DOI] [PubMed] [Google Scholar]
- 179.Klammert U, Mueller TD, Hellmann TV, Wuerzler KK, Kotzsch A, Schliermann A, et al. GDF-5 can act as a context-dependent BMP-2 antagonist. BMC biology 2015, 13: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jeffery TK, Upton PD, Trembath RC, Morrell NW. BMP4 inhibits proliferation and promotes myocyte differentiation of lung fibroblasts via Smad1 and JNK pathways. American journal of physiology Lung cellular and molecular physiology 2005, 288(2): L370–378. [DOI] [PubMed] [Google Scholar]
- 181.Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet 2001, 27(1): 84–88. [DOI] [PubMed] [Google Scholar]
- 182.Martens JW, de Winter JP, Timmerman MA, McLuskey A, van Schaik RH, Themmen AP, et al. Inhibin interferes with activin signaling at the level of the activin receptor complex in Chinese hamster ovary cells. Endocrinology 1997, 138(7): 2928–2936. [DOI] [PubMed] [Google Scholar]
- 183.Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000, 404(6776): 411–414. [DOI] [PubMed] [Google Scholar]
- 184.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem 2012, 287(33): 27313–27325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Greenwald J, Groppe J, Gray P, Wiater E, Kwiatkowski W, Vale W, et al. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Molecular cell 2003, 11(3): 605–617. [DOI] [PubMed] [Google Scholar]