

Abstract
Therapy resistance to a selective B-Raf inhibitor (BRAFi) poses a challenge in treating patients with BRAF-mutant melanomas. Here, we report that RNA interference of mortalin (HSPA9/GRP75), a mitochondrial molecular chaperone often upregulated and mislocalized in melanoma, can effectively induce death of vemurafenib-resistant progenies of human B-RafV600E melanoma cell lines, A375 and Colo-829. Mortalin depletion induced death of vemurafenib-resistant cells at similar efficacy as observed in vemurafenib-naïve parental cells. This lethality was correlated with perturbed mitochondrial permeability and was attenuated by knockdown of adenine nucleotide translocase (ANT) and cyclophilin D (CypD), the key regulators of mitochondrial permeability. Chemical inhibition of MEK1/2 and ERK1/2 also suppressed mortalin depletion-induced death and mitochondrial permeability in these cells. These data suggest that mortalin and MEK/ERK regulate an ANT/CypD-associated mitochondrial death mechanism(s) in B-RafV600E melanoma cells and that this regulation is conserved even after these cells develop BRAFi resistance. We also show that doxycycline-induced mortalin depletion can effectively suppress the xenografts of vemurafenib-resistant A375 progeny in athymic nude mice. These findings suggest that mortalin has potential as a candidate therapeutic target for BRAFi-resistant BRAF-mutant tumors.
Keywords: BRAF inhibitor, mortalin, ERK, mitochondrial permeability, drug resistance
1. INTRODUCTION
Activating mutations affecting B-Raf kinase domain, e.g., B-RafV600E,K,D, are the most frequently detected oncogenic alterations in cutaneous melanomas, making B-Raf and its effector pathway mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) key therapeutic targets in these tumors [1]. Recently, the Food and Drug Administration has approved different precision medicine therapies targeting B-Raf/MEK/ERK pathway, including B-Raf inhibitor (BRAFi) monotherapies using vemurafenib (trade name Zelboraf) and dabrafenib (trade name Tafinlar), and the combination therapy using dabrafenib and the MEK1/2 inhibitor, trametinib (trade name Mekinist). Although these drugs have advanced the therapy of BRAF-mutant melanomas (reviewed in [2–4]), many tumors initially responsive to these drugs developed resistance, eventually causing patient casualty. Therefore, strategies to treat therapy-resistant BRAF-mutant tumors are required.
While functioning as the energy and building block supplier to support cell proliferation and survival, mitochondria can cause cell death upon permeabilization of their outer or inner membranes [5, 6]. These lethal events are mainly mediated by the release of various death signals through different mitochondrial channels and regulators, including adenine nucleotide translocase (ANT), cyclophilin D (CypD), voltage-dependent anion channel, and mitochondrial calcium uniporter [5, 6]. In response to various metabolic stresses, these proteins participate, either directly or indirectly, in the formation of a mitochondrial membrane-spanning permeability transition pore which can cause metabolic catastrophe in cells [7–11]. There have been growing interests in exploiting mitochondrial death mechanisms for tumor therapy because metabolic reprogramming often occurs in tumor cells and increases the chance for various mitochondrial stresses [6, 12].
Mortalin (HSPA9/GRP75/PBP74) is a mitochondrial chaperone that is often upregulated and mislocalized in tumor cells, including melanoma, pancreatic, colon, liver, brain, and breast cancers [13–17]. Mortalin promotes tumorigenesis by facilitating tumor cell proliferation and survival, stemness, epithelial-mesenchymal transition, and angiogenesis [18–22]. We previously reported that deregulated MEK/ERK activity in BRAF-mutant melanoma cells can increase mitochondrial permeability but mortalin counteracts this increase by limiting a physical interaction between ANT3 and CypD. As such, mortalin depletion in these cells results in perturbed mitochondrial permeability and, consequently, tumor cell death [23]. Based upon these data, we have proposed that mortalin is a potential therapeutic target for BRAF-mutant melanoma and have begun addressing whether mortalin depletion can suppress BRAFi-resistant melanoma cells via a similar mechanism. In this study, we demonstrate that mortalin depletion can also suppress vemurafenib-resistant B-RafV600E melanoma cells by causing perturbed mitochondrial permeability in a MEK/ERK-dependent manner.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
Vemurafenib-resistant progenies of A375 and Colo-829 (A375R and Colo-829R, respectively) and their parental cells were gifts from Dr. Richard Marais at Univ. of Manchester [24]. A375 and A375R were maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (Invitrogen). Colo-829 and Colo-829R were maintained in RPMI 1640 (Invitrogen) supplemented with 10% FBS, 1% sodium pyruvate, and 1% nonessential amino acids. A375-dox-shMort, A375R-dox-shMort, Colo-829-dox-shMort, and Colo-829R-dox-shMort were generated by stably infecting A375, A375R, Colo-829, and Colo829R with lentiviral pTRIPZ doxycycline-inducible microRNA-adapted shRNA targeting human mortalin (pTRIPZ-dox-shMort, Open Biosystems, V3THS_362249), respectively. Doxycycline and cisplatin were purchased from Sigma-Aldrich (St. Louis, MO). AZD6244 and SCH772984 were purchased from Selleck Chemicals (Houston, TX) and ChemieTek (Indianapolis, IN), respectively.
2.2. Plasmids, RNA interference, and recombinant viruses
pLKO.1-shANT3 lentiviral constructs (TRCN0000045014, TRCN0000045015) and pLKO.1-shCypD lentiviral constructs (TRCN0000232681, TRCN0000232682) were purchased from Sigma-Aldrich. Lentivirus was generated and used as previously described [25].
2.3. Immunoblotting
For immunoblotting, equal amount of proteins in cell lysates in 62.5 mM Tris (pH 6.8)/2% SDS containing the protease and phosphatase inhibitor cocktails were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA), and blocked with 5% nonfat dry milk or BSA in 0.1 M Tris (pH 7.5)/ 0.9% NaCl/0.05% Tween 20 prior to blotting. Antibodies and their dilutions are as follows: phospho-MEK1/2 (Ser217/221 for MEK1 and Ser222/226 for MEK2, #9121), 1: 2500; MEK1/2 (#9122), 1: 2500; ERK1/2 (#9102), 1: 2500; cleaved lamin A (#2035), 1: 2500; PARP (#9542), 1: 5000 (Cell Signaling Technology, Boston, MA, USA); phospho-ERK1/2 (Thr202/Tyr204, sc-16982), 1: 2500; mortalin (sc-133137), 1: 5000; p21CIP1 (sc-56335), 1: 2500; p27KIP1 (sc-528), 1: 2500; CypD (sc-376061), 1: 2500 (Santa Cruz Bio- technology, Santa Cruz, CA, USA); β-actin (A2228), 1: 10000 (Sigma-Aldrich); ANT3 (AP12188), 1: 2500 (Abgent, San Diego, CA); E2F1 (MS-879), 1: 2500 (Thermo Fisher Scientific, Waltham, MA). Chemiluminescence signals of immunoblots were visualized by SuperSignal West Pico and Femto chemiluminescence kits (Thermo Fisher Scientific), captured by ChemiDoc XRS+ (Bio-Rad, Hercules, CA), and analyzed by Image Lab software (Bio-Rad) for densitometry.
2.4. Cell viability, death and apoptosis assays
Cell viability was determined by trypan blue exclusion assay (Invitrogen) or flow cytometry of cells stained with the fluorescent DNA intercalator TO-PRO 3 (Invitrogen). Apoptosis was determined by staining with Annexin V-FITC conjugate (Invitrogen) and TO-PRO 3 using the Guava EasyCyte flow cytometry system (MilliporeSigma, Billerica, MA, USA), as previously described. Data were analyzed by FCS EXPRESS software (De Novo Software, Los Angeles, CA, USA).
2.5. Calcein retention assay
Mitochondrial membrane permeability was analyzed using MitoProbe Transition Pore Assay kit (Thermo Fisher Scientific), according to the manufacturer’s instruction. Calcein fluorescence retaining in cells after cobalt quenching was determined by the Guava EasyCyte flow cytometry system (MilliporeSigma) and FCS EXPRESS software (De Novo Software).
2.6. In vivo xenograft experiments
Two million cells in 200 μL Hank’s balanced salt solution were subcutaneously inoculated into the rear flanks of 5-week-old female athymic nude (nu/nu) mice (The Jackson laboratory, Bar Harbor, ME, USA). Once palpable, tumors were measured using Vernier calipers every other day. Tumor volumes were calculated using the formula: length × width × height × 0.5236. When tumor volumes reached about 50 mm3, mice bearing each cell line were sorted into four groups to achieve equal distribution of tumor size in all the treatment groups. Doxycycline (2 mg/mL) was administered via drinking water for two weeks. At the end of the experiment, animals were euthanized by CO2 asphyxiation. Investigators were not blinded. All animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committee at Medical College of Wisconsin, Milwaukee, WI, USA.
2.7. Quantification and statistical analysis
All graphs represent the mean ± the standard error of the mean (SEM) of technical replicates (n) in a representative experiment, unless specified otherwise. All experiments, excluding animal work, were repeated at least three times to confirm data consistency. Statistical significance was determined by two-tailed paired student’s t test, one-way ANOVA with Dunnett posttests or two-way ANOVA with Bonferroni posttests using Prism (GraphPad Software, La Jolla, CA, USA).
3. RESULTS
3.1. Mortalin depletion induces death of vemurafenib-resistant B-RafV600E melanoma cells
To determine the effects of mortalin depletion in vemurafenib-naïve and -resistant B-RafV600E tumor cells in comparison, we generated A375R-dox-shMort model by infecting A375R cells, a vemurafenib-resistant progeny of the human melanoma line A375 [24], with the lentiviral doxycycline-inducible small hairpin RNA (shRNA)-mediated mortalin knockdown system, pTRIPZ-dox-shMort (Fig. 1A). We previously confirmed the specificity of pTRIPZ-dox-shMort in A375 cells by conducting a rescue experiment using exogenous mortalin constructs that are non shRNA-targetable [23]. Doxycycline treatment induced substantial mortalin depletion in A375R-dox-shMort cells within 5 days at a similar degree as observed in A375-dox-shMort cells (Fig. 1B). Similarly as in A375-dox-shMort cells, doxycycline increased phosphorylation of MEK1/2 and ERK1/2, an effect of mortalin depletion due to decreased access of protein phosphatase 1α to MEK1/2 [26], and lamin A cleavage in A375R-dox-shMort cells (Fig. 1B). The cleavage of lamin A and poly-(ADP-ribose)-polymerase (PARP) are key surrogate markers for caspase-dependent apoptosis [27]. PARP cleavage was not noticeably detected in A375R-dox-shMort cells although it was mildly increased in A375-dox-shMort cells upon doxycycline treatment (Fig. 1B). Also similarly as in A375-dox-shMort cells, doxycycline treatment decreased cellular levels of the S-phase transcription factor E2F1 and increased the levels of the cyclin-dependent kinase inhibitor p21CIP1 in A375R-dox-shMort cells, albeit at much lower degrees (Fig. 1B). However, doxycycline did not notably affect p27KIP1 levels in both A375-dox-shMort and A375R-dox-shMort cells (Fig. 1B). In contrast to A375-dox-shMort and A375R-dox-shMort cells, the control A375-pTRIPZ and A375R-pTRIPZ cells did not exhibit any similar effects in response to doxycycline (Fig. 1A and 1B)
Fig. 1. Mortalin depletion effectively suppresses vemurafenib-resistant B-RafV600E melanoma cells.
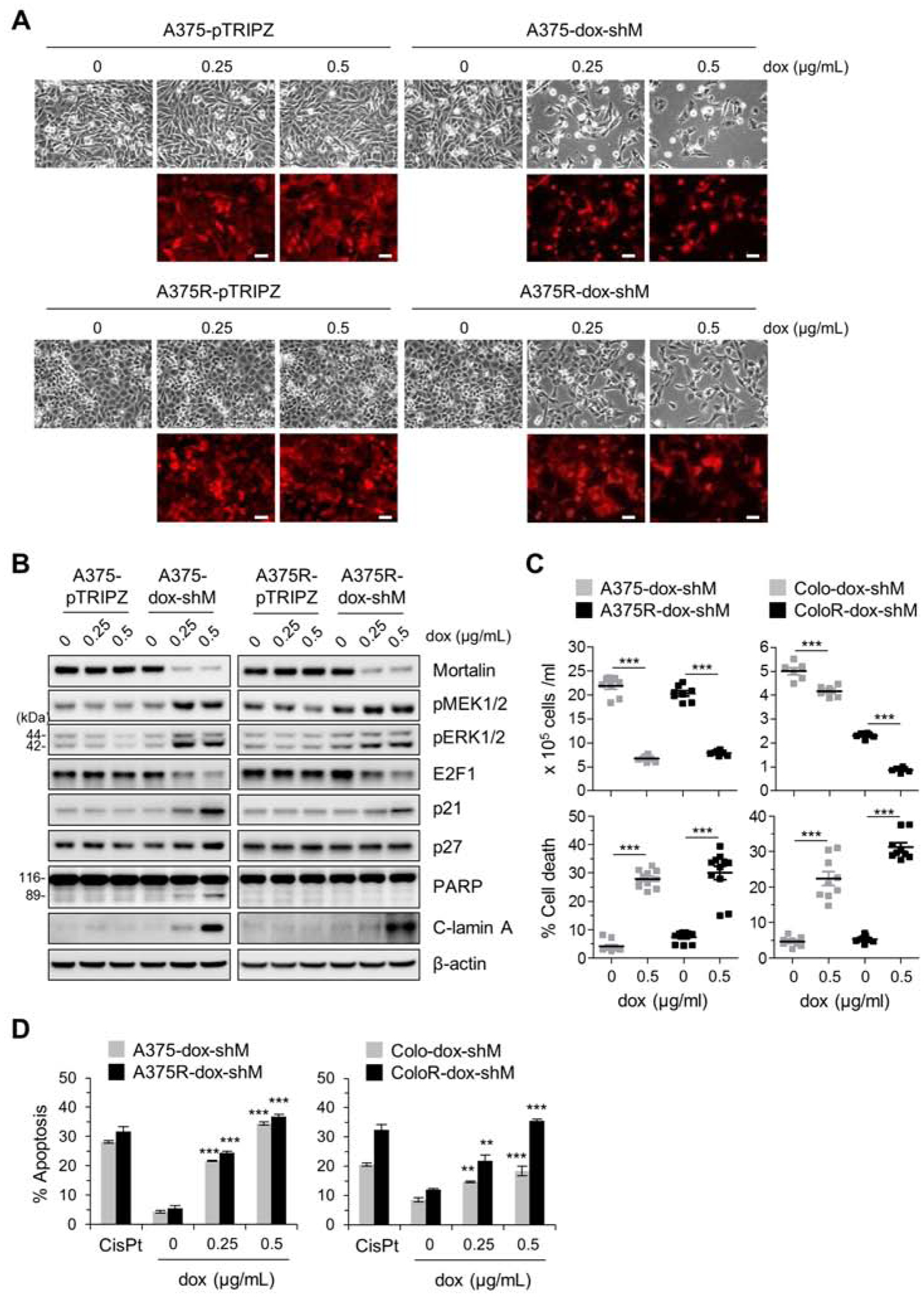
(A) Microscopic images of A375 and its vemurafenib-resistant progeny (A375R) stably expressing pTRIPZ-dox-shMort (A375-dox-shM and A375R-dox-shM, respectively) or the control pTRIPZ (A375-pTRIPZ and A375R-pTRIPZ, respectively). Cells were treated with doxycycline (dox) at indicated concentrations for 5 days. Red fluorescence indicates doxycycline-induced RFP expression and thus infection efficiency. Scale bar = 100 μm. (B) Western blot analyses of total lysates of cells treated as described in (A). C-lamin A indicates cleaved lamin A. β-actin is the control for equal protein loading. Densitometry of Western blots is shown in supplemental figure S1. (C) Cell viability determined by trypan blue exclusion assay after 5 day doxycycline treatment. Data are mean ± SEM from at least 6 independent experiments. (D) Apoptosis determined by Annexin V-FITC/TO-PRO 3 assay after 5 day doxycycline treatment. Colo-dox-shM and ColoR-dox-shM indicate Colo-829 and Colo829R stably expressing pTRIPZ-dox-shMort, respectively. Cells treated with 25 μM cisplatin (CisPt) for 24 hours are the positive control for apoptosis. Data are mean ± SEM, n = 3. Representative FACS histograms are shown in supplemental figure S2. P values are relative to untreated control. ** P < 0.01, and *** P < 0.001 by two-tailed paired student’s t test in (C) or by one-way ANOVA with Dunnett post-tests in (D).
Consistent with doxycycline-induced surrogate marker changes, A375R-dox-shMort cells exhibited substantial loss of cell viability associated with apoptotic cell death within 5 days of doxycycline treatment, as determined by trypan blue exclusion assays (Fig. 1C, left) and annexin V/TO-PRO-3 co-staining (Fig. 1D, left). This viability loss was quite comparable to viability loss of A375-dox-shMort cells under equal doxycycline treatment condition. Moreover, consistent with this observation in A375R model, pTRIPZ-dox-shMort-mediated mortalin depletion also induced substantial apoptotic cell death in Colo-829R cells, a vemurafenib-resistant progeny of Colo-829 melanoma line, similarly as it did in parental Colo-829 cells (Fig. 1C and 1D, right). These data demonstrate that mortalin depletion can effectively suppress B-RafV600E tumor cells, not only BRAFi-naïve but also BRAFi-resistant.
3.2. ANT3 and CypD mediate mortalin depletion-induced death of vemurafenib-resistant B-RafV600E melanoma cells by increasing mitochondrial permeability
We previously reported that the physical interaction between ANT and CypD, and subsequent increases in mitochondrial permeability underlie the lethal effects of mortalin depletion in BRAFi-naïve B-RafV600E tumor cells [23]. To determine whether mortalin depletion induces death of BRAFi-resistant B-RafV600E tumor cells via a similar mechanism, we determined whether knockdown of ANT and CypD can rescue A375R-dox-shMort and Colo-829R-dox-shMort cells from the lethal effects of mortalin depletion. Knockdown of ANT3, using two different shRNA constructs, substantially attenuated mortalin depletion-induced cell death and lamin A cleavage in these cells (Fig. 2A and 2B). Similarly, knockdown of CypD also rescued these cells from mortalin knockdown (Fig. 2C and 2D). These data demonstrate that ANT3 and CypD are required for mortalin depletion to induce cell death in B-RafV600E tumor cells, not only BRAFi-naïve but also BRAFi-resistant. Of note, although knockdown of ANT3 and CypD substantially rescued these cells from mortalin depletion-induced death, it did not effectively attenuate mortalin depletion-induced decreases in cell proliferation rates (Fig. 2A and 2C) and increases in p21CIP1 expression (Fig. 2C and 2D), which suggests that mortalin can regulate p21CIP1 expression and cell proliferation independently of ANT3 and CypD.
Fig. 2. Knockdown of ANT3 or CypD attenuates mortalin depletion-induced lethality in vemurafenib-resistant B-RafV600E melanoma cells.
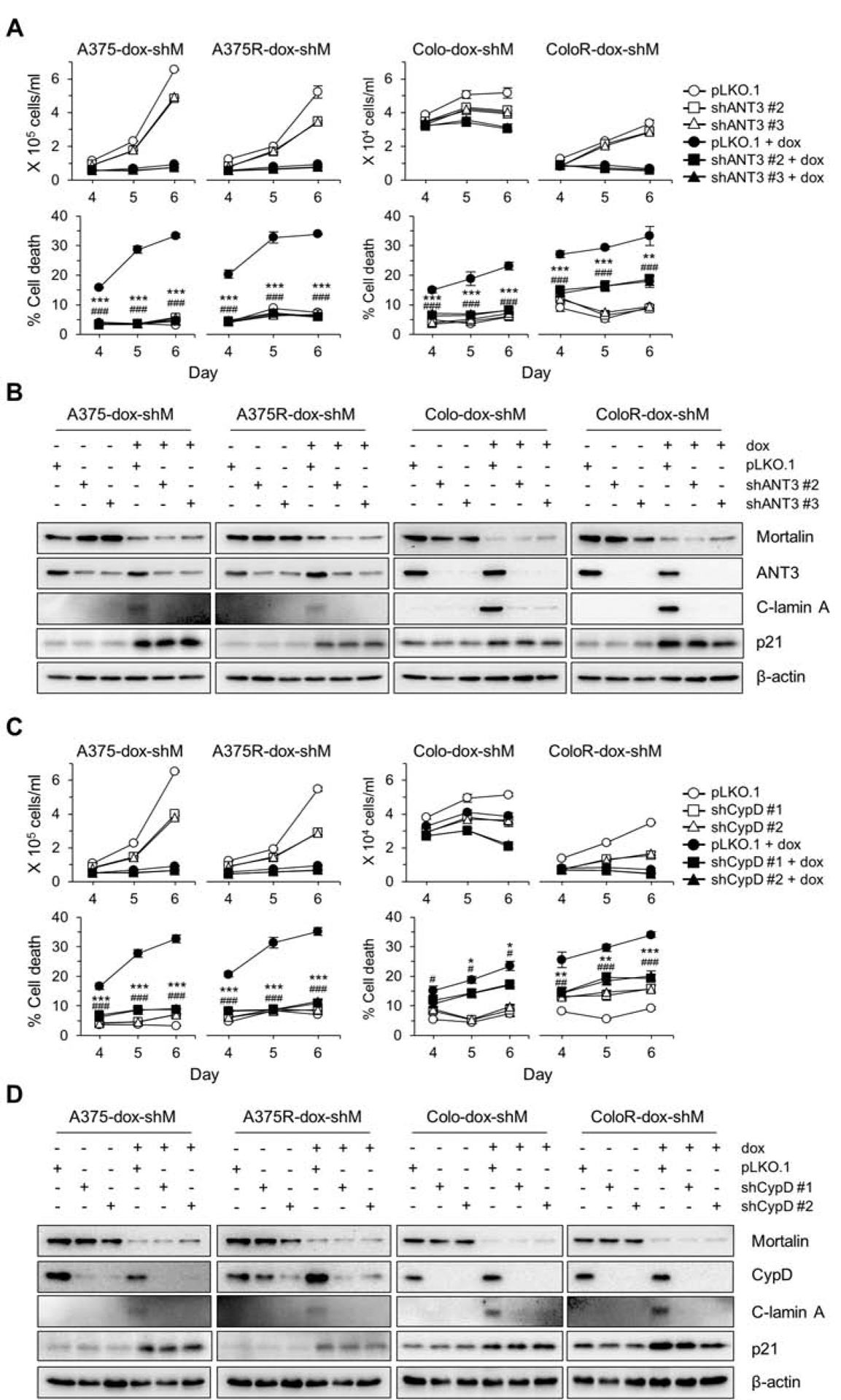
(A) Cell viability determined by TO-PRO 3 assay. Cells stably expressing pTRIPZ-dox-shMort (dox-shM) were infected with lentiviral pLKO.1-shANT3 or the control pLKO.1 for 24 hours prior to 5 day doxycycline (0.5 μg/mL) treatment. (B) Western blot analyses of total lysates of cells treated as described in (A). C-lamin A indicates cleaved lamin A. β-actin is the control for equal protein loading. Densitometry shown in supplemental figure S3A. (C) Cell viability determined by TO-PRO 3 assay. Cells stably expressing pTRIPZ-dox-shMort (dox-shM) were infected with pLKO.1-shCypD or the control pLKO.1 for 1 day prior to 5 day doxycycline (0.5 μg/mL) treatment. (D) Western blot analyses of total lysates of cells treated as described in (C). Densitometry shown in supplemental figure S3B. Blots are representative, and data (A and C) are mean ± SEM, n = 3. P values are relative to pLKO.1. * P < 0.05, ** P < 0.01, and *** P < 0.001; # P < 0.05, ## P < 0.01, and ### P < 0.001 by two-way ANOVA with Bonferroni post-tests. The asterisk and hash symbols for P values correspond to the first and the second RNA interference constructs, respectively.
Next, we determined the effects of mortalin knockdown on mitochondrial permeability in these cells using the calcein-acetyoxymethyl release assay [28]. In our time-course analyses, mortalin knockdown markedly increased mitochondrial permeability in A375R and Colo-829R cells in a time-dependent manner, which occurred at similar degrees as detected in vemurafenib-naïve parental cells (Fig. 3A). Moreover, knockdown of ANT3 or CypD substantially inhibited mortalin depletion-induced mitochondrial permeability in these cells at similar efficiency regardless of vemurafenib resistance (Fig. 3B and 3C). These data suggest that mortalin depletion induces lethality in vemurafenib-resistant B-RafV600E melanoma cells by causing perturbed mitochondrial permeability similarly as it does in vemurafenib-naïve B-RafV600E melanoma cells.
Fig. 3. Mortalin depletion perturbs mitochondrial permeability via ANT3 and CypD in vemurafenib-resistant B-RafV600E tumor cells.
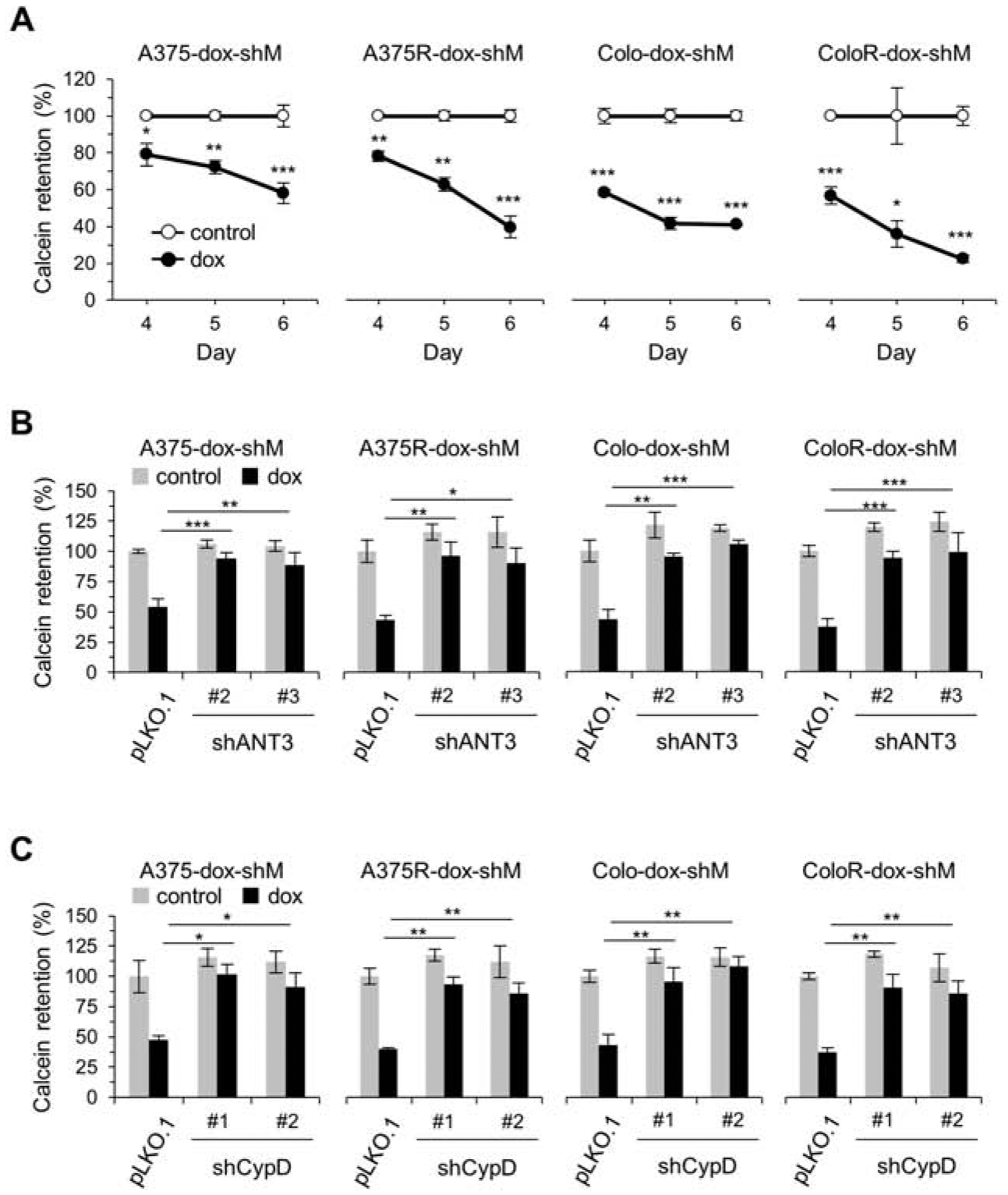
(A) Mitochondrial permeability determined by calcein retention assay. Cells stably expressing pTRIPZ-dox-shMort were treated with doxycycline (0.5 μg/mL) for the indicated time. (B) Calcein retention assay. Cells stably expressing pTRIPZ-dox-shMort (dox-shM) were infected with pLKO.1-shANT3 or the control pLKO.1 for 1 day prior to 5 day doxycycline (0.5 μg/mL) treatment. (C) Calcein retention assay. Cells stably expressing pTRIPZ-dox-shMort (dox-shM) were infected with pLKO.1-shCypD or the control pLKO.1 for 1 day prior to 5 day doxycycline (0.5 μg/mL). Representative FACS histograms for A to C are shown in supplemental figures S4A, S4B, and S4C, respectively. Data are means ± SEM, n =3. P values are relative to untreated controls at different time-points (A, two-tailed paired student’s t-test) or to the dox-treated pLKO.1 (B and C, two-way ANOVA with Bonferroni post-tests). * P < 0.05, ** P < 0.01, and *** P < 0.001.
3.3. MEK/ERK activity is required for mortalin depletion to induce death of vemurafenib-resistant B-RafV600E melanoma cells
We previously reported that mortalin negatively regulates MEK/ERK activity by facilitating the physical interaction between MEK1/2 and protein phosphatase 1α and that MEK/ERK activity is necessary for mortalin depletion to induce death of B-RafV600E-expressing cells [23, 26]. Because mortalin depletion increased MEK/ERK activity in vemurafenib-resistant A375R and Colo-829R cells, as determined by phosphorylation levels of MEK1/2 and ERK1/2, at similar efficiency in their parental cells (Fig. 1B and 4A), we determined whether MEK/ERK activity is also necessary for mortalin depletion to induce lethality in these vemurafenib-resistant cells. Indeed, when pretreated with selumetinib (AZD6244, MEK1/2 inhibitor) or SCH772984 (ERK1/2 inhibitor), A375R and Colo-829R cells exhibited substantially attenuated viability loss (Fig. 4B) and lamin A cleavage (Fig. 4A) in the face of mortalin depletion. Moreover, these MEK/ERK inhibitor pretreatments also attenuated mitochondrial permeability induced by mortalin depletion in these cells (Fig. 4C). The degree of rescue effects mediated by these MEK/ERK inhibitors was not notably different between vemurafenib-resistant cells and their parental cells (Fig. 4A to 4C). These data demonstrate that MEK/ERK activity is also necessary for mortalin depletion to induce lethality in vemurafenib-resistant B-RafV600E melanoma cells.
Fig. 4. Inhibition of MEK/ERK attenuates mortalin depletion-induced lethality and mitochondrial permeability in vemurafenib-resistant B-RafV600E cells.
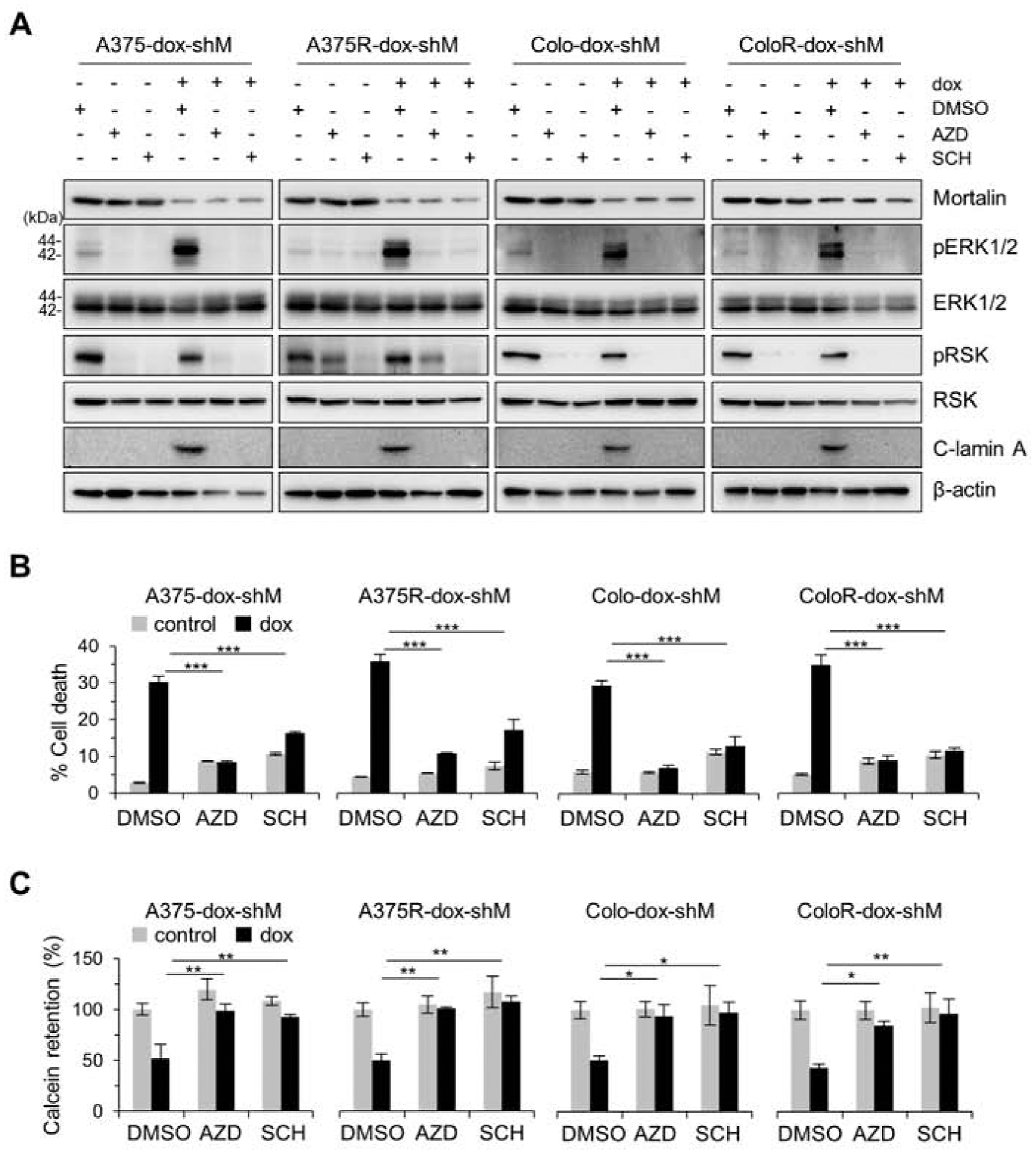
(A) Western blot analyses of total cell lysates. Cells stably expressing pTRIPZ-dox-shMort (dox-shM) were treated with doxycycline (0.5 μg/mL) for 3 days, and then co-treated with dox and 100 nM AZD6244 (AZD), 1 μM SCH772984 (SCH), or the vehicle control DMSO for 2 days before harvest. C-lamin A indicates cleaved lamin A. β-actin is the control for equal protein loading. Densitometry shown in supplemental figure S5. (B) TO-PRO 3 assays of cells treated as described in (A). (C) Calcein assays of cells treated as described in (A). Representative FACS histograms are shown in supplemental figure S6. Data are means ± SEM, n = 3. P values are relative to dox-treated DMSO control. * P < 0.05, ** P < 0.01, and *** P < 0.001 by two-way ANOVA with Bonferroni post-tests.
3.4. Mortalin depletion suppresses vemurafenib-resistant B-RafV600E melanoma in mice.
To further evaluate mortalin as a potential therapeutic target for vemurafenib-resistant B-RafV600E melanoma cells, we examined the effects of mortalin depletion on tumors derived from A375R-dox-shMort and the control A375R-pTRIPZ xenografts in mice. Orally administered doxycycline substantially suppressed the growth of A375R-dox-shMort xenografts (Fig. 5A and 5B) without causing marked loss of animal body weight (Fig. 5C). However, doxycycline treatment did not effectively suppress the growth of A375R-pTRIPZ xenografts while not notably affecting animal body weight (Fig. 5A to 5C). When these tumors were harvested at the end of treatment and analyzed, we found that doxycycline treatment notably increased lamin A cleavage while mildly increasing MEK1/2 phosphorylation in A375R-dox-shMort tumors, although it did not induce any similar effects in A375R-pTRIPZ tumors (Fig. 5D). Meanwhile, p21CIP1 levels were varied in these samples. These data are consistent with the effects of mortalin depletion in A375R cells in vitro, suggesting that mortalin targeting can effectively suppress vemurafenib-resistant B-RafV600E melanoma cells in vivo.
Fig. 5. Mortalin depletion suppresses vemurafenib-resistant B-RafV600E tumor xenografts in mice.
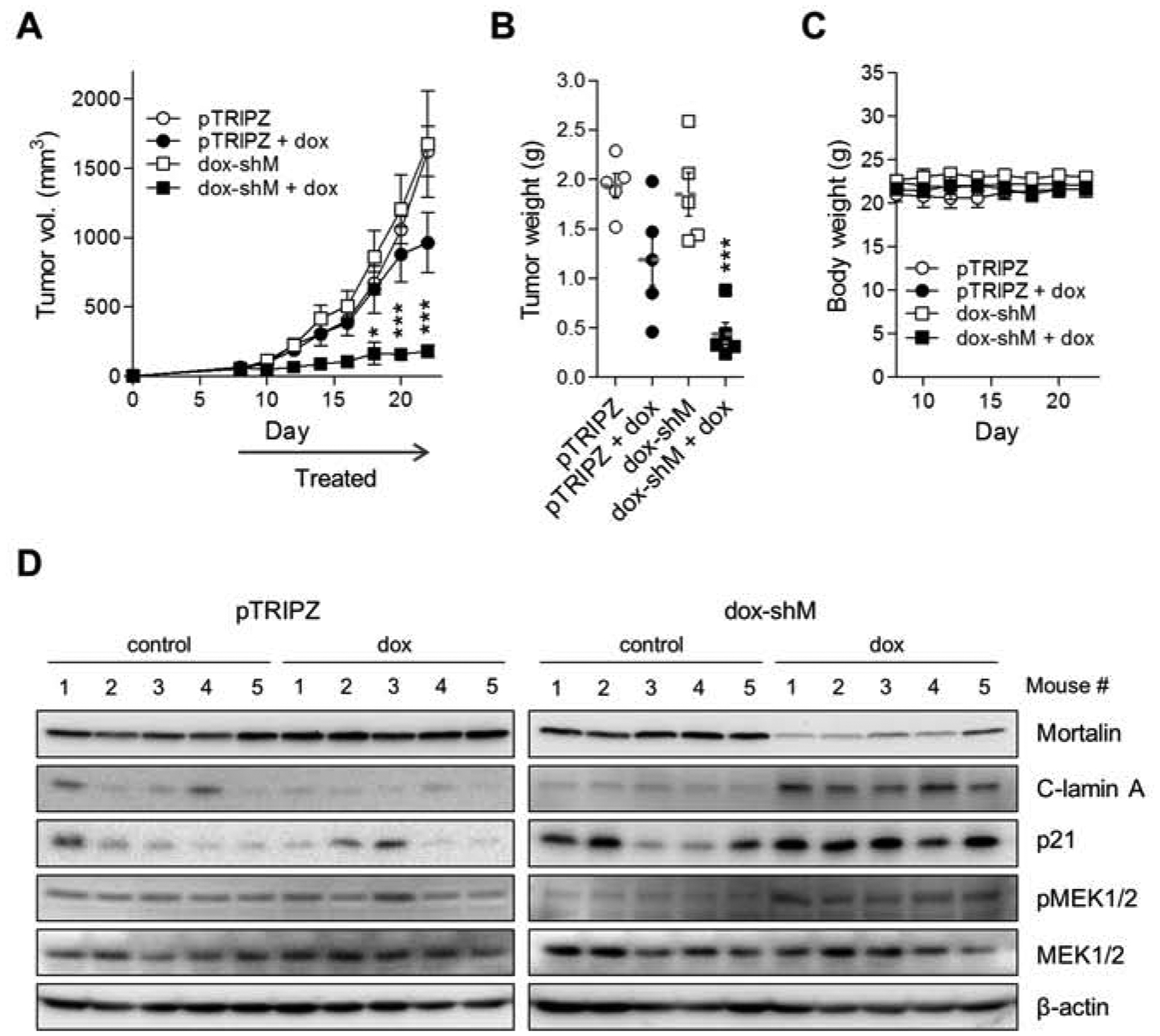
(A) Changes in tumor sizes in athymic mice bearing xenografts of A375R stably expressing pTRIPZ-dox-shMort (dox-shM) or the control pTRIPZ. Doxycycline was administered via drinking water (details in the methods section). (B) Weights of tumors collected at the end of the treatment in (A). Horizontal dotted line indicates mean. Each symbol represents one sample. (C) Body weight changes of animals during the treatment in (A). (D) Western blotting of the homogenates of tumors in (C). C-lamin A indicates cleaved lamin A. β-actin is the control for equal protein loading. Densitometry shown in supplemental figure S7. Data in (A to C) are mean ± SEM from a single cohort of five mice per treatment group. Each mouse developed one tumor. P values are relative to pTRIPZ. * P < 0.05, *** P < 0.001 (A, two-way ANOVA with Bonferroni post-tests; B, one-way ANOVA with Dunnett post-tests).
4. DISCUSSION
Our data demonstrate that mortalin depletion can effectively induce death of vemurafenib-resistant B-RafV600E melanoma cells by causing perturbed mitochondrial permeability. This lethal effect of mortalin depletion is consistent with its effects in BRAFi-naïve B-RafV600E melanoma cells, which we recently reported [23]. Noteworthy is that MEK/ERK activity is necessary for mortalin depletion to induce the lethality in B-RafV600E melanoma cells, whether BRAFi-naïve or BRAFi-resistant. Reactivation of MEK/ERK is a major mechanism underlying BRAFi therapy resistance of B-RafV600E tumor cells [29–33]. We thus speculate that MEK/ERK dependency of B-RafV600E melanoma cells might inevitably incur a mitochondrial weakness associated with mortalin, and this weakness is conserved in BRAFi-resistant B-RafV600E melanoma cells in which MEK/ERK is reactivated. Based upon these findings, we propose that mortalin has potential as a candidate therapeutic target for the treatment of BRAFi-resistant B-RafV600E melanomas, if they remain MEK/ERK-dependent (illustrated in Fig. 6).
Fig. 6. Graphic summary of the study.
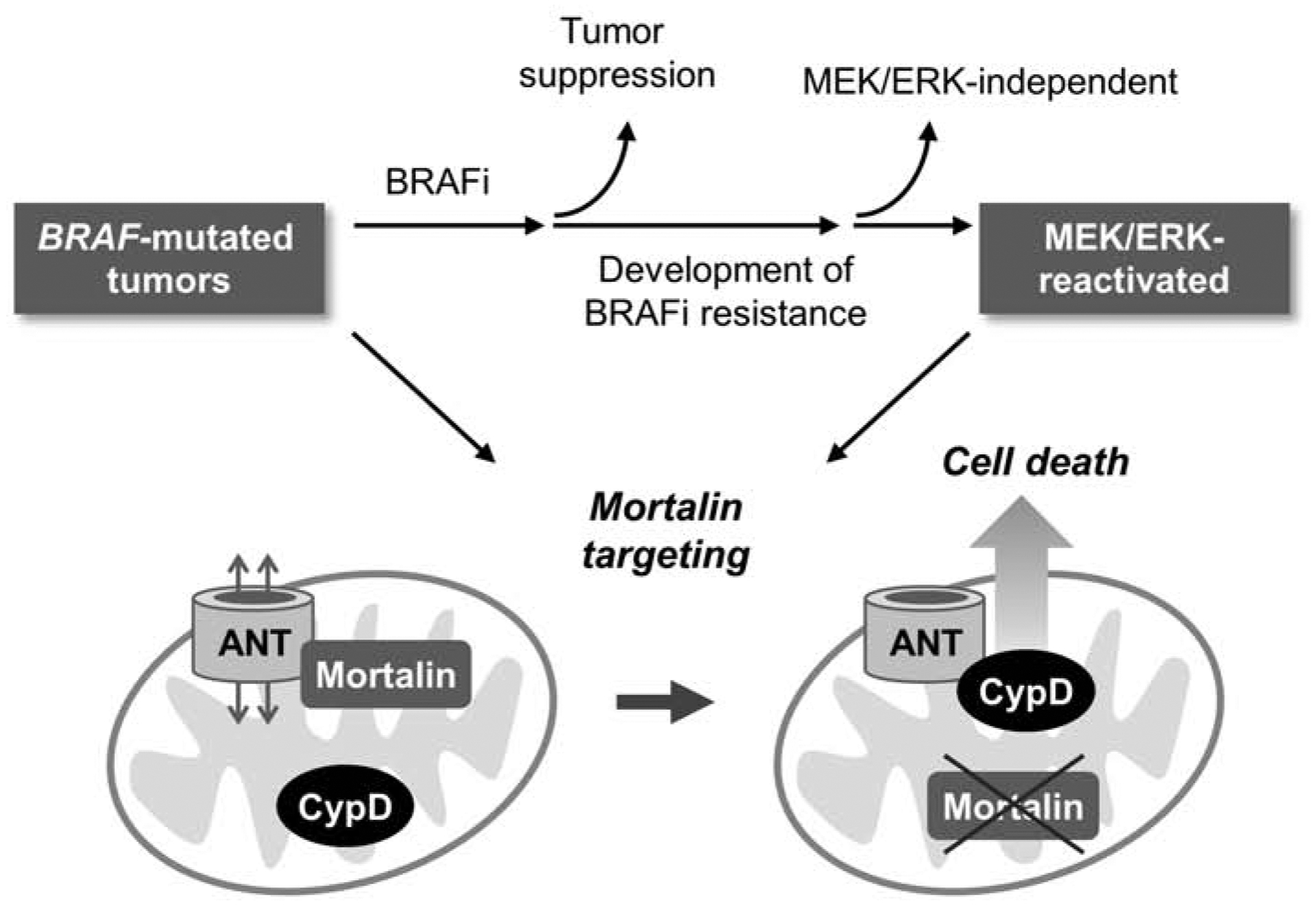
Most BRAF-mutated tumors develop resistance to BRAFi therapy, wherein MEK/ERK reactivation is a major mechanism of drug resistance. ANT is a mitochondrial channel critical for bioenergetics but it can also mediate cell death in collaboration with CypD, the gatekeeper of mitochondrial permeability transition pore. Deregulated MEK/ERK activity increases this lethal risk but mortalin counteracts it by inhibiting ANT-CypD interaction. Our study suggests that these processes are intact and exploitable in MEK/ERK-dependent BRAFi-resistant tumors.
Mortalin is a molecular chaperone upregulated in melanoma in correlation with poor patient survival in melanoma [23]. We recently identified ANT3 as a novel client for mortalin and demonstrated that mortalin and MEK/ERK mediate a highly coordinated regulation of ANT3 interaction with CypD to facilitate the survival of B-RafV600E melanoma cells [23]. Although ANTs regulate cellular bioenergetics through ATP/ADP exchange across the mitochondrial inner membrane [8], they can trigger death signals if complexed with CypD [34, 35]. We hypothesize that mortalin has a critical role to suppress this lethal potential of ANTs and CypD in BRAF-mutant melanoma cells while promoting ANT-mediated bioenergetics. This notion is in line with recent observations that mitochondrial metabolism is reprogramed to facilitate BRAF-mutant melanoma maintenance [36–38]. It is possible that BRAF-mutant melanoma cells are prone to a stress associated with metabolic reprogramming in mitochondria and mortalin is a key chaperone that prevents this stress. Targeting mitochondrial death mechanisms has been proposed as a therapeutic strategy for cancer treatment [5, 6], and mortalin might have potential as a candidate target to implement this strategy in melanoma.
It is noteworthy that although knockdown of ANT and CypD effectively rescued B-RafV600E melanoma cells from lethal effects of mortalin depletion, it did not effectively rescue these cells from cytostatic effects of mortalin depletion, which suggests that mortalin regulates cell proliferation and survival via separate mechanisms. This notion is consistent with increased p21CIP1 expression in these cells and our previous observations that mortalin depletion induces cell cycle arrest in B-RafV600E melanoma cells by facilitating MEK/ERK-regulated mechanisms, including transcription of CDKN1A (encoding p21CIP1) through the transcription factors TP53 and SP1 [17, 39]. These mechanisms may also possibly be conserved in BRAFi-resistant melanoma cells and remain to be determined. In conclusion, our findings suggest that mortalin is an Achilles heel in BRAF-mutated melanomas, not only BRAFi-naïve but also BRAFi-resistant, and provide a rationale to consider mortalin inhibition as a therapeutic strategy to overcome the drug resistance.
Supplementary Material
Highlights.
Mortalin depletion effectively induces death of vemurafenib-resistant B-RafV600E melanoma cells.
This lethality is correlated with perturbed mitochondrial permeability and is attenuated by knockdown of adenine nucleotide translocase (ANT) and cyclophilin D (CypD), the key regulators of mitochondrial permeability.
Chemical inhibition of MEK1/2 and ERK1/2 suppresses mortalin depletion-induced death and mitochondrial permeability in vemurafenib-resistant B-RafV600E melanoma cells.
ANT/CypD-associated mitochondrial death mechanism(s) is regulated by mortalin and MEK/ERK and is an Achilles heel in BRAF-mutated melanomas even after these cells develop B-Raf inhibitor resistance.
Acknowledgements
We thank Dr. Richard Marais at Univ. of Manchester for vemurafenib-resistant melanoma cells. We also thank former Park lab members for technical supports. This study was supported by the NIH/National Cancer Institute grant R01CA138441 to J.-I.P.
Abbreviations:
- ANT
adenine nucleotide translocase
- BRAFi
B-Raf inhibitor
- c-lamin A
cleaved lamin A
- CypD
cyclophilin D
- ERK
extracellular signal-regulated kinase
- HSP
heat shock protein
- MEK
mitogen-activated protein kinase kinase
- PARP
Poly (ADP-ribose) polymerase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors have no competing interest to disclose.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Sullivan RJ, Flaherty K, MAP kinase signaling and inhibition in melanoma, Oncogene, 32 (2013) 2373–2379. [DOI] [PubMed] [Google Scholar]
- [2].Karoulia Z, Gavathiotis E, Poulikakos PI, New perspectives for targeting RAF kinase in human cancer, Nat Rev Cancer, 17 (2017) 676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kidger AM, Sipthorp J, Cook SJ, ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway, Pharmacol Ther, 187 (2018) 45–60. [DOI] [PubMed] [Google Scholar]
- [4].Roskoski R Jr., Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas, Pharmacol Res, 117 (2017) 20–31. [DOI] [PubMed] [Google Scholar]
- [5].Kroemer G, Galluzzi L, Brenner C, Mitochondrial membrane permeabilization in cell death, Physiol Rev, 87 (2007) 99–163. [DOI] [PubMed] [Google Scholar]
- [6].Leanza L, Zoratti M, Gulbins E, Szabo I, Mitochondrial ion channels as oncological targets, Oncogene, 33 (2014) 5569–5581. [DOI] [PubMed] [Google Scholar]
- [7].Bonora M, Pinton P, A New Current for the Mitochondrial Permeability Transition, Trends Biochem Sci, 44 (2019) 559–561. [DOI] [PubMed] [Google Scholar]
- [8].Chevrollier A, Loiseau D, Reynier P, Stepien G, Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism, Biochim Biophys Acta, 1807 (2011) 562–567. [DOI] [PubMed] [Google Scholar]
- [9].He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE, Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase, Proc Natl Acad Sci U S A, 114 (2017) 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hurst S, Baggett A, Csordas G, Sheu SS, SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca(2+) influx, and regulation of mitochondrial permeability transition pore opening, J Biol Chem, 294 (2019) 10807–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov EV, ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore, Cell Rep, 26 (2019) 11–17 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Green DR, Galluzzi L, Kroemer G, Cell biology. Metabolic control of cell death, Science, 345 (2014) 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Daugaard M, Rohde M, Jaattela M, The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions, FEBS Lett, 581 (2007) 3702–3710. [DOI] [PubMed] [Google Scholar]
- [14].Kaul SC, Deocaris CC, Wadhwa R, Three faces of mortalin: a housekeeper, guardian and killer, Exp Gerontol, 42 (2007) 263–274. [DOI] [PubMed] [Google Scholar]
- [15].Lee AS, Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential, Nat Rev Cancer, 14 (2014) 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu PK, Hong SK, Starenki D, Oshima K, Shao H, Gestwicki JE, Tsai S, Park JI, Mortalin/HSPA9 targeting selectively induces KRAS tumor cell death by perturbing mitochondrial membrane permeability, Oncogene, 39 (2020) 4257–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI, A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase, Mol Cell Biol, 33 (2013) 4051–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen J, Liu WB, Jia WD, Xu GL, Ma JL, Huang M, Deng YR, Li JS, Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition, Int J Oncol, 44 (2014) 247–255. [DOI] [PubMed] [Google Scholar]
- [19].Na Y, Kaul SC, Ryu J, Lee JS, Ahn HM, Kaul Z, Kalra RS, Li L, Widodo N, Yun CO, Wadhwa R, Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis, Cancer Res, 76 (2016) 2754–2765. [DOI] [PubMed] [Google Scholar]
- [20].Rozenberg P, Kocsis J, Saar M, Prohaszka Z, Fust G, Fishelson Z, Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer, Int J Cancer, 133 (2013) 514–518. [DOI] [PubMed] [Google Scholar]
- [21].Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, Kaul SC, Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis, Int J Cancer, 118 (2006) 2973–2980. [DOI] [PubMed] [Google Scholar]
- [22].Yun CO, Bhargava P, Na Y, Lee JS, Ryu J, Kaul SC, Wadhwa R, Relevance of mortalin to cancer cell stemness and cancer therapy, Sci Rep, 7 (2017) 42016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu PK, Hong SK, Chen W, Becker AE, Gundry RL, Lin CW, Shao H, Gestwicki JE, Park JI, Mortalin (HSPA9) facilitates BRAF-mutant tumor cell survival by suppressing ANT3-mediated mitochondrial membrane permeability, Sci Signal, 13 (2020) 13: pii: eaay1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, Lorigan P, Springer C, Larkin J, Jorgensen C, Marais R, Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma, Cancer Discov, 3 (2013) 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hong SK, Yoon S, Moelling C, Arthan D, Park JI, Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling, J Biol Chem, 284 (2009) 33006–33018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu PK, Hong SK, Park JI, Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells, Mol Cell Biol, 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rosen A, Casciola-Rosen L, Macromolecular substrates for the ICE-like proteases during apoptosis, J Cell Biochem, 64 (1997) 50–54. [DOI] [PubMed] [Google Scholar]
- [28].Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM, p53 opens the mitochondrial permeability transition pore to trigger necrosis, Cell, 149 (2012) 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song JY, Niessen HWM, Rozeman EA, Geukes Foppen MH, Blank CU, Peeper DS, Cancer drug addiction is relayed by an ERK2-dependent phenotype switch, Nature, 550 (2017) 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Konieczkowski DJ, Johannessen CM, Garraway LA, A Convergence-Based Framework for Cancer Drug Resistance, Cancer Cell, 33 (2018) 801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Maik-Rachline G, Seger R, The ERK cascade inhibitors: Towards overcoming resistance, Drug Resist Updat, 25 (2016) 1–12. [DOI] [PubMed] [Google Scholar]
- [32].Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, Ruchalski K, Seifert H, Larkin J, Dahlman KB, Johnson DB, Algazi A, Sosman JA, Ribas A, Lo RS, Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction, Cancer Cell, 27 (2015) 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sale MJ, Cook SJ, Intrinsic and acquired resistance to MEK1/2 inhibitors in cancer, Biochem Soc Trans, 42 (2014) 776–783. [DOI] [PubMed] [Google Scholar]
- [34].Halestrap AP, Brenner C, The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death, Curr Med Chem, 10 (2003) 1507–1525. [DOI] [PubMed] [Google Scholar]
- [35].Halestrap AP, Woodfield KY, Connern CP, Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase, J Biol Chem, 272 (1997) 3346–3354. [DOI] [PubMed] [Google Scholar]
- [36].Haq R, Fisher DE, Widlund HR, Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation, Clin Cancer Res, 20 (2014) 2257–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR, Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF, Cancer Cell, 23 (2013) 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P, PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress, Cancer Cell, 23 (2013) 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Karkhanis M, Park JI, Sp1 regulates Raf/MEK/ERK-induced p21(CIP1) transcription in TP53-mutated cancer cells, Cell Signal, 27 (2015) 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.