

SUMMARY
Compromised protein homeostasis underlies accumulation of plaques and tangles in Alzheimer’s disease (AD); however, little is known about the early mechanisms that contribute to this process. To objectively assess protein turnover at early stages of amyloid beta (Aβ) proteotoxicity, we used dynamic 15N metabolic labeling followed by proteomic analysis of amyloid precursor protein knock in mouse brains. At initial stages of Aβ accumulation, the turnover of proteins associated with presynaptic terminals is selectively impaired. Presynaptic proteins with impaired turnover, particularly synaptic vesicle (SV) associated proteins, have elevated levels, misfold in both a plaque dependent and independent manner, and interact with APP and Aβ. Concurrent with elevated levels of SV associated proteins, we found an enlargement of the SV pool as well as enhancement of presynaptic potentiation. Together, our findings reveal that the presynaptic terminal is particularly vulnerable and represents a critical site for manifestation of initial AD etiology.
A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
INTRODUCTION
Alzheimer’s disease (AD) is a common irreversible neurodegenerative disorder that gradually erodes cognition and memory with age. AD pathology is characterized by the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs), made of misfolded and aggregated amyloid beta peptides (Aβ) and hyperphosphorylated tau, respectively (Long and Holtzman, 2019). The presymptomatic phase of AD lasts for several years during which time Aβ42 peptides oligomerize, accumulate, and form fibrils years before NFTs appear and cognitive impairments manifest (De Strooper and Karran, 2016). Aβ peptides accumulate in brain regions with high levels of synaptic activity, and amyloid plaques are required for clinical progression of AD, but alone are not sufficient for AD (Brody et al., 2008; Selkoe and Hardy, 2016). Amyloid precursor protein (APP), a transmembrane protein that localizes to endosomal and presynaptic plasma membranes, is cleaved to form Aβ peptides (O’Brien and Wong, 2011). Amyloidogenic processing of APP during familial and late onset AD involves secretory trafficking and clathrin-mediated endocytosis. During this process, APP is proteolytically processed by the beta-site APP cleaving enzyme 1 (BACE1), and then by the γ-secretase complex (De Strooper et al., 1999; Vassar et al., 1999). Aβ is generated at multiple intracellular sites including the endoplasmic reticulum, the trans-golgi network, and at synapses (Cirrito et al., 2005; Greenfield et al., 1999). The majority of Aβ is released from intracellular stores into the extracellular space in an activity-dependent manner through the synaptic vesicle cycle (SVC) (Cirrito et al., 2008; Cirrito et al., 2005; Kamenetz et al., 2003).
Age is the primary risk factor for AD. Cellular quality control measures and proteostasis network efficiency decline during aging (Balch et al., 2008; Morimoto and Cuervo, 2014). Neurons are particularly vulnerable to age-associated deterioration since they are long-lived, post-mitotic cells that cannot dilute misfolded or damaged proteins through cellular division (Toyama and Hetzer, 2013). Aβ accumulates in the AD brain due to an imbalance in the rate of synthesis, folding, and degradation; ultimately leading to aggregation and formation of plaques (Shankar and Walsh, 2009). The requirement of amyloid plaques for the clinical manifestation of AD strongly supports the hypothesis that hampered protein turnover contributes to the pathogenesis. Clearance of Aβ involves proteasomal and lysosomal protein degradation pathways. However, accumulated oligomeric Aβ is a poor substrate for the proteasome, impairs protein degradation machinery, and consequently accelerates the accumulation of Aβ (Bustamante et al., 2018). Furthermore, Aβ interacts with many proteins, some of which are present in insoluble plaques, suggesting that additional proteins become misfolded, trapped, and functionally impaired (Liao et al., 2004; Xiong et al., 2019). While the importance of impaired proteostasis in AD is clear, a detailed understanding of the origins of this process has remained elusive (Bai et al., 2020). Identifying proteins with compromised turnover during early stages of AD pathogenesis could elucidate critical mechanisms of AD etiology and provide targets for therapeutic intervention.
We set out to advance our understanding of AD pathology by identifying proteins with impaired turnover during Aβ accumulation in vivo. Our analysis platform consisted of the three recently developed App knock in (App KI) mouse lines that express humanized Aβ using the endogenous promoter, thus maintaining physiological expression and processing of APP (Saito et al., 2014). App KI mice express combinations of App mutations found in early onset familial AD patients and represent a new opportunity to study Aβ pathology in vivo without the caveats that accompany traditional APP overexpression in transgenic models (Sasaguri et al., 2017). As a control, we used the App KI mice expressing the Swedish KM670/671NL (AppNL/NL) mutation. These mice do not have amyloid plaque deposits, nor display any cognitive defects even up to 24-months of age (Salas et al., 2018), thus these mice serve as a control that lacks amyloid pathology while maintaining production of Aβ, albeit mostly in the less toxic Aβ40 form. To model the effects of accumulating Aβ42, a particularly toxic form of Aβ, we used App KI mice expressing the Beyreuther/Iberian (I716F) and Swedish (AppNL-F/NL-F) mutations; and to model accelerated amyloid formation we used App KI mice expressing the Arctic (E693G), Iberian, and Swedish (AppNL-G-F/NL-G-F) mutations. We assessed protein turnover proteome-wide using dynamic in vivo 15N metabolic labeling of App KI mice with discovery-based proteomic analysis, as we have done in the past to identify extremely long lived proteins (Savas et al., 2016; Savas et al., 2012; Toyama et al., 2013).
We monitored protein turnover in multiple brain regions across different stages of Aβ proteotoxicity, with a focus on the early stages of pathology. We found that the axon terminal represents the predominant cellular compartment with hampered proteostasis, as presynaptic proteins have impaired turnover just as Aβ accumulation becomes detectable. Along with hampered turnover, these proteins were also found to have elevated levels early that was not due to compensatory increases in mRNA abundance, before a later reduction in protein levels. Many axon terminal proteins aggregated in both an amyloid plaque-dependent and -independent manner. Additionally, we found that Aβ and APP interact with SVC proteins, and Aβ impedes synaptic vesicle (SV) fusion. Finally, the SV pool is enlarged, and short-term potentiation is affected during the early stages of Aβ proteotoxicity before synaptic transmission becomes impaired. Altogether, these results reveal that the presynapse is among the earliest and most vulnerable cellular compartments affected by AD-like pathology and may play a critical role in the early development of synaptotoxicity and cognitive impairment in AD.
RESULTS
Development of a screen to identify proteins with diminished turnover during Aβ accumulation in App KI brains
We set out to advance our understanding of AD by identifying protein networks with impaired degradation dynamics in App KI brains (Saito et al., 2014). To achieve this goal, we used pulse-chase 15N stable isotope metabolic labeling with liquid chromatography tandem-mass spectrometry (LC-MS/MS)-based proteomic analysis (Savas et al., 2012; Toyama et al., 2013). This unique and powerful approach allows us to specifically investigate how the degradation of proteins is altered at different stages of AD-like pathology. First, we determined the timeline of pathological onset in AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F brains from two to twelve months of age (Figure 1). Consistent with previous reports, we found that AppNL/NL brains do not accumulate detectable levels of Aβ42 peptides or amyloid plaques in the cortex, hippocampus, or cerebellum (Salas et al., 2018). AppNL-F/NL-F mice have relatively mild pathology and do not have significantly elevated Aβ42 levels before ten months of age based on standard, ultrasensitive, and oligomeric ELISA assays (Figure 1A–C). Furthermore, AppNL-F/NL-F mice display minimum thioflavin S positive amyloid plaques by six months of age (Figure 1D–G). Analysis of the SDS-insoluble fraction by dot blot using a highly sensitive anti-amyloid fibril antibody revealed elevated levels at six months, suggesting that there is a small pool of Aβ fibrils accumulates in AppNL-F/NL-F brains (Figure 1H–I). AppNL-G-F/NL-G-F mice exhibit more aggressive pathology, displaying elevated Aβ42 levels as early as two months of age and an amyloid plaque load that is already near its peak by six months of age (Figure 1). In addition to Aβ42, we found that APP C-terminal fragments, α-CTF and β-CTF, were elevated in six-month old AppNL-F/NL-F and AppNL-G-F/NL-G-F cortical homogenates and synaptosome fractions relative to AppNL/NL (Figure S1). β-CTF, an APP cleavage product and direct precursor of Aβ, can independently affect neurons deleteriously (Jiang et al., 2019; Lauritzen et al., 2016). Notably, none of the App KI mice exhibit NFTs or neuronal loss and model very early preclinical amyloid pathology (Sasaguri et al., 2017).
Figure 1: Confirmation of Aβ42 levels and amyloid pathology in AppNL-F/NL-F, and AppNL-G-F/NL-G-F mouse brains.

(A) Aβ42 levels in cortical homogenates (GuHCl soluble) from AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice at 2, 4, 6, 8, 10, and 12 months of age as measured by sandwich ELISA with analytical sensitivity range of 15.6 – 1,000 pg / mL. (B) Aβ42 levels in cortical homogenates (GuHCl soluble) from AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice at 4, 6, and 8 months of age as measured by an ultrasensitive sandwich ELISA with analytical sensitivity range of 1.56 – 100 pg / mL. (C) Oligomeric Aβ42 levels in TBST soluble cortical extracts from AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice at 4, 6, and 8 months of age as measured by a sandwich ELISA using the oligomeric preferential MOAB-2 antibody. (D) Amyloid pathology in App KI cortex and hippocampus. Representative thioflavin S stained sagittal brain sections from 6- and 12-month-old AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice. Scale bar = 500 μm. (E) Quantification of amyloid plaque area from panel (D). Data represents average thioflavin S positive stained area relative to the total cortical or hippocampal area per section. (F) Amyloid pathology in App KI cerebellum. Representative brain sections from 6- and 12-month-old AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice stained with thioflavin S. Scale bar = 500 μm. (G) Quantification of amyloid plaque area from panel (F) Data represents average thioflavin S positive stained area relative to the total cerebellar area per section. (H) Dot blot analysis of Aβ (82E1), amyloid fibrils (LOC), and actin using aggregated protein fractions from cortical extracts of AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice at 6 months of age. (I) Quantification of Aβ (82E1) and amyloid fibrils (LOC) levels from panel (H). (A) N = 3 – 8 mice of mixed gender, (B-C) N = 4 mice of mixed gender, (E, G, I) N = 3 mice of mixed gender, per group. Each datapoint was an average of 2 – 3 30 μm sections per mouse. All data represent mean ± SD, analyzed by one-way ANOVA followed by Fisher’s LSD. Circles represent individual biological replicates. * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001.
A small cohort of female AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice (F0) were fed specialized chow, highly enriched with the stable nitrogen isotope 15N. As the mice consume the chow, newly synthesized proteins become increasingly enriched with 15N atoms. These F0 mice were exclusively fed the chow for six months starting after weaning. Then, these App KI mice were bred with males of the same genotype and the homozygous progeny (F1) continued to be metabolically labeled with 15N until weaning. After this period, the 15N labeled F1 mice were chased with a standard 14N chow for six months (Figure 2A). This two-generation experimental design generates mirrored “label swapped” datasets from which we can measure the 14N labeled peptides in the F0 generation and the 15N labeled peptides in the F1 generation. Since the labeling timeframe in the F0 and the chase timeframe in the F1 are identical (6 months each), we can assess both of these as independent and confirmatory experiments, strengthening our analysis of turnover proteome-wide and for individual proteins in brain extracts. First, we confirmed efficient 15N labeling of the brain proteome. In the F0 App KI female mice, after six months of labeling, the brain proteome had an average labeling efficiency ≥ 98% ± 2.80 (Figure S2A). In the F1 brains at weaning, right before the chase period, the brain proteome was also highly enriched with 15N, with an average labeling efficiency ≥ 98% ± 3.20 (Figure S2B). Our labeling and chase period is six months in part because at relatively short chase periods, chimeric proteins composed of both 15N and 14N atoms dominate the brain proteome (Savas et al., 2016). Consequently, our ability to thoroughly monitor global protein lifetimes at short time points is severely hindered, as chimeric proteins cannot be reliably identified by MS / MS and MS1 isotopic envelops broaden. Indeed, in our previous in vivo 15N pulse-chase studies using wild type (WT) mouse brains, we found the number of measured proteins after one-month chase was three times fewer than at very short or long chase periods (Figure S2C).
Figure 2: Development of an in vivo screen to identify proteins with impaired turnover in App KI brains.
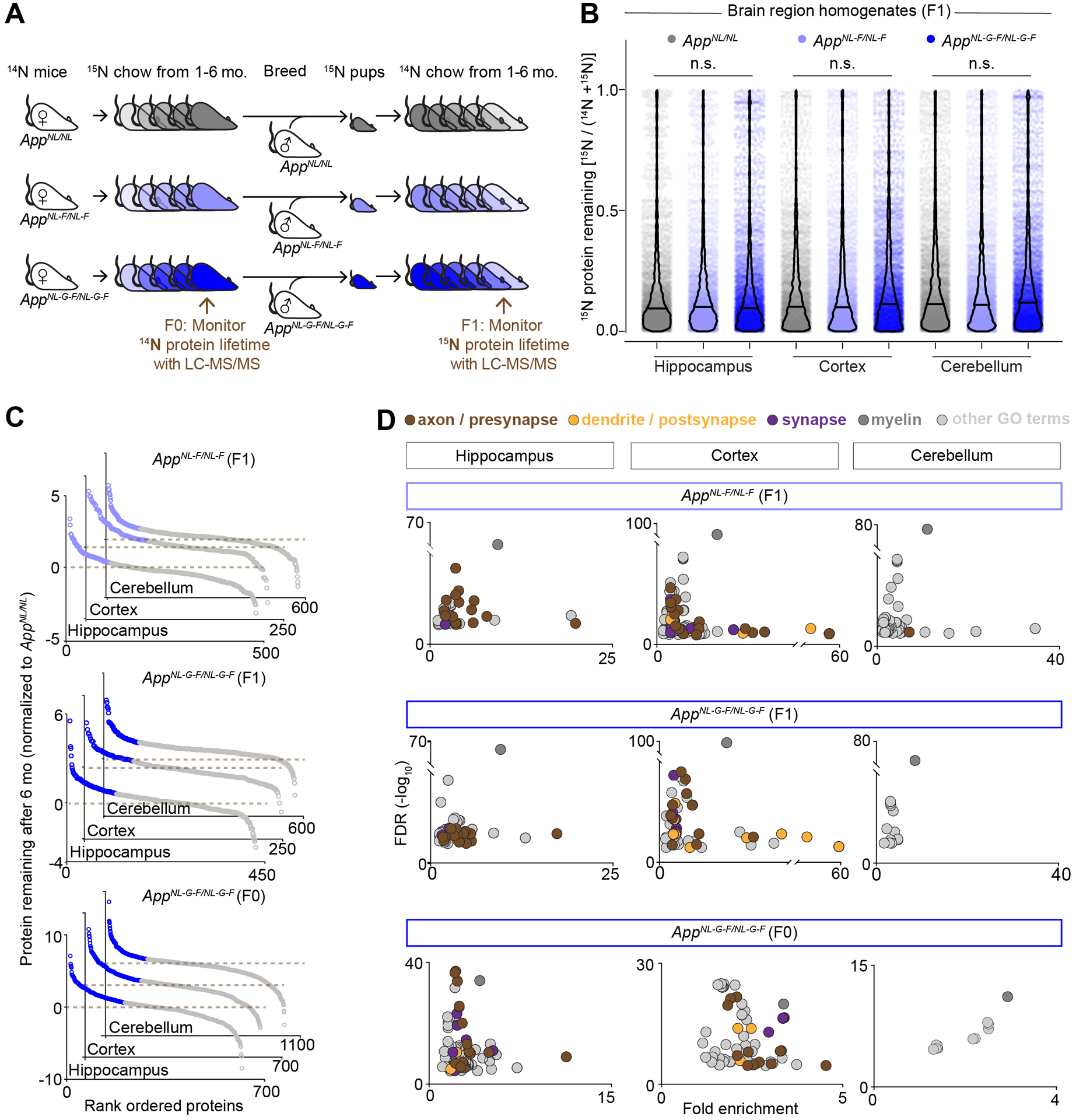
(A) Dynamic 15N labeling of App KI mice. Schematic depicting the metabolic labeling procedure of AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F dames (F0) with an 15N diet for six to seven months. Second generation 15N-labeled mice (F1) are further labeled with 15N during nursing and switched to an 14N diet at one month of age. Hippocampal, cortical, and cerebellar extracts from F0 and F1 mice at 7 months of age were analyzed by MS. Proteins remaining fully unlabeled (14N) in the F0 generation and heavy labeled (15N) in the F1 generation were monitored. (B) Global protein turnover [15N protein remaining = 15N / (15N+14N)] in F1 App KI mice was not significantly different between the App KI genotypes after six-month chase. N = 3565 – 4000 proteins from 4 mice per genotype. Bar represents median, analyzed by one-way ANOVA. (C) Protein turnover in AppNL-F/NL-F and AppNL-G-F/NL-G-F relative to AppNL/NL. (Top) F1 AppNL-F/NL-F and (Middle) F1 AppNL-G-F/NL-G-F protein turnover datasets of hippocampal, cortical, or cerebellar extracts relative to AppNL/NL based on 15N protein remaining. N = 3 – 4 mice per genotype. (Bottom) F0 AppNL-G-F/NL-G-F protein turnover datasets of hippocampal, cortical, or cerebellar extracts relative to AppNL/NL based on 14N protein remaining. N = 2 – 3 mice per genotype. (D) Gene ontology (GO) cell component enrichment analysis of proteins with ≥ 33% impaired turnover in AppNL-F/NL-F and AppNL-G-F/NL-G-F relative to AppNL/NL (blue circles from Figure 2C). Plots depict fold enrichment versus FDR (-log10), analyzed by Fisher’s exact test with FDR estimation. Brown represents GO terms related to axon / presynapse, yellow represents GO terms related to dendrite / postsynapse, purple represents GO terms related to entire synapse, dark grey is the GO term myelin, and grey represents all other GO terms.
We analyzed global protein turnover by measuring the levels of each protein’s 15N remaining [15N / (15N + 14N)], also termed fractional abundance, in App KI hippocampal, cortical, and cerebellar homogenates. In order to assess how protein turnover is affected at different stages of pathology, we analyzed a cohort of F1 App KI mice (n = 4 per genotype) that were chased with 14N for six months. Seven months of age represents a potentially informative time-point when AppNL-F/NL-F mice are just beginning to accumulate aggregated Aβ42 peptides; while AppNL-G-F/NL-G-F mice already show robust AD-like amyloid pathology with significantly elevated levels of Aβ42 peptides and amyloidosis (Figure 1). We observed that about 1,000 proteins in each dataset retained 15N with detectable levels after the six-month chase period (Figure S2D). On a proteome-wide level, there was not a systematic shift in turnover at seven months of age, as there was no significant difference in the average protein turnover in F1 AppNL-F/NL-F or AppNL-G-F/NL-G-F hippocampal, cortical, or cerebellar homogenates when compared to region matched extracts from AppNL/NL controls (Figure 2B). To strengthen our results with an independent cohort of mice, we also analyzed the age-matched, and label-swapped F0 generation (AppNL-G-F/NL-G-F and AppNL/NL dames) by measuring the 14N remaining [14N / (14N + 15N)]. Again, we found no evidence of globally altered protein turnover in AppNL-G-F/NL-G-F cortex or hippocampus compared to AppNL/NL (Figure S2E). For each dataset (e.g. F1 AppNL-G-F/NL-G-F cortex), we identified between 4,300 – 5,800 proteins, using a protein false discovery rate of ≤ 1% for each individual proteomic analysis, and > 63% of proteins overlapped in all biological replicates per brain region (Figure S2F).
Presynaptic terminal proteins have impaired turnover in cortical and hippocampal but not cerebellar AppNL-F/NL-F and AppNL-G-F/NL-G-F extracts
We next assessed each individual protein’s turnover by measuring the average 15N or 14N remaining among biological replicates, then comparing pathogenic genotypes (AppNL-F/NL-F and AppNL-G-F/NL-G-F) to the control genotype (AppNL/NL). Ratios > 1 for 15N remaining in the F1 or > 1 for 14N remaining in the F0 brains indicate proteins with impaired turnover presumably due to AD-like pathology. To home in on the most confident protein candidates, we confined our comparative analysis to the proteins quantified in at least 3 of 4 mice of each F1 genotype, or quantified in 2 of 2 AppNL-G-F/NL-G-F and 3 of 3 AppNL/NL mice for the F0 generation. We analyzed three experimental groups: AppNL-F/NL-F / AppNL/NL (F1), AppNL-G-F/NL-G-F / AppNL/NL (F1), and AppNL-G-F/NL-G-F / AppNL/NL (F0) for each cortical, hippocampal, and cerebellar analyses, generating nine datasets in total. Overall distributions of protein turnover ratios were similar among the datasets, and we identified a similar number of proteins with impaired turnover in all three-brain regions (Figure 2C and Table S1). To investigate if proteins with impaired turnover are associated with specific cellular compartments, a GO overrepresentation analysis was performed. We found that the most significantly enriched GO cell component terms from the cortical and hippocampal datasets were related to axons or the presynaptic compartment (Wilhelm et al., 2014). We found far more GO terms associated with the pre-compared to the post synapse (Figure 2D). In contrast, proteins with impaired turnover in the cerebellum, a brain region that typically exhibits pathology later in disease progression (Xu et al., 2019), were not associated with GO terms related to axon terminals (Table S2). Since the axon terminal was the prevailing compartment with impaired degradation, we investigated synapse density in six and twelve-month-old App KI mice. We found that synaptic density remains unaltered independent of plaques in the CA1 at both timepoints (Figure S3). These data are in line with a recent finding in 18-month old AppNL-F/NL-F animals using very sensitive SEQUIN analysis (Sauerbeck et al., 2020).
To further assess individual proteins with stunted degradation in the pathogenic genotypes, we focused on proteins associated with the GO term presynapse (GO: 0098793), as that term was overrepresented in all cortical and hippocampal datasets. These datasets comprised nearly the entire set of soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) including, Syntaxin 1B (Stx1b), Synaptobrevin 1 and 2 (Vamp1 and Vamp2), synaptosomal nerve-associated protein 25 (Snap25), and the calcium sensor Synaptotagmin 1 (Syt1) (Figure 3A and S4A–C). In addition, several key synaptic vesicle (SV) endocytosis factors, some of which are genetically associated to sporadic AD (Seshadri et al., 2010), had hampered turnover in the cortex and hippocampus of AppNL-F/NL-F and AppNL-G-F/NL-G-F mice. These proteins included clathrin coat assembly protein AP180 (Snap91), myc box-dependent interacting protein 1 (Bin1), and amphiphysin (Amph). Presynaptic proteins with impaired turnover, especially SV associated proteins, were much less prominent in the cerebellum datasets (Figure 3A and S4A–C). Many of the SV cycle proteins had slower turnover in the hippocampus and cortex of AppNL-F/NL-F mice, despite these mice just beginning to exhibit increases in misfolded Aβ peptides at this age (Figure 3A). Rather than solely rely on ratios, we also assessed whether any of these proteins had degradation impairments at the peptide level in AppNL-F/NL-F or AppNL-G-F/NL-G-F compared to AppNL/NL controls. Many presynaptic proteins identified by ratios, including Amph, Pip5k1c, Snca, Snap25, Sh3gl1, and Syn1 had significantly impaired turnover (Figure 3B–C and Table S1). Since proteins with impaired degradation may be preferentially ubiquitinated, we purified ubiquitinated proteins from six-month-old brain homogenates using TUBE-1 (Tandem Ubiquitin Binding Entity) capture resin. We found that ubiquitination levels of select presynaptic proteins were unchanged across App KI genotypes (Figure S4D–E). Unchanged ubiquitination levels signify a lack of evidence that these proteins are being preferentially tagged for trafficking or degradation. Based on these results, we focused further analysis on the SV associated proteins: t-SNAREs (Snap25 and Stx1b), v-Snares (Vamp1 and Vamp2), and the calcium sensor Syt1; as well as Snap91, Pip5k1c, Calm1, and Snca, because these proteins had the most robustly impaired turnover in AppNL-F/NL-F and AppNL-G-F/NL-G-F hippocampus and cortex, or had impaired turnover in all three hippocampal datasets.
Figure 3: Presynaptic proteins have hampered turnover in AppNL-F/NL-F and AppNL-G-F/NL-G-F cortex and hippocampus.
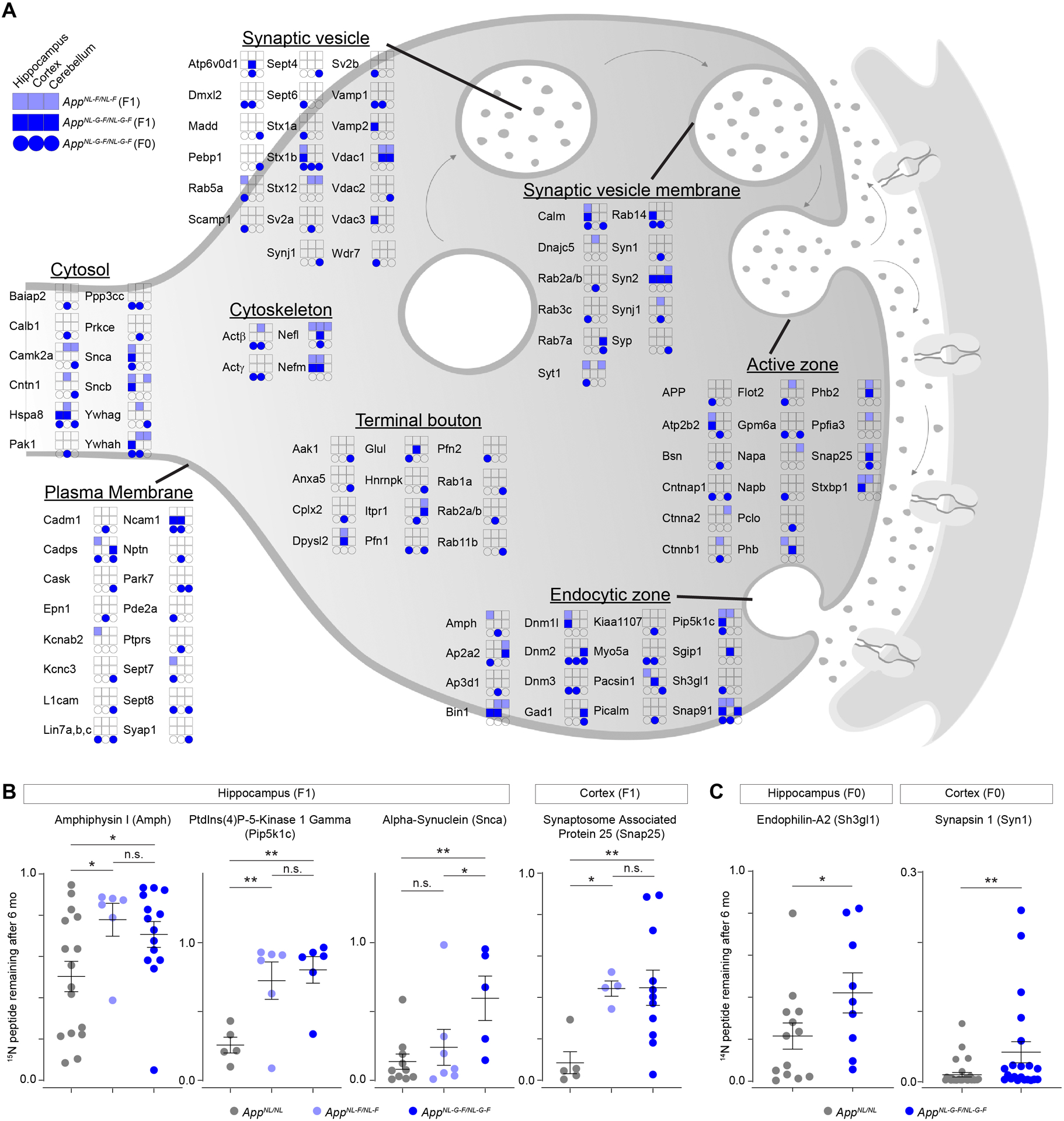
(A) Summary of presynaptic proteins (GO: 0098793) with impaired turnover in AppNL-F/NL-F and AppNL-G-F/NL-G-F compared to AppNL/NL. Proteins are grouped by sub compartments within the presynapse based on GO terms. Colored boxes and circles represent brain region and genotype where the presynaptic proteins had ≥ 33% reduced turnover after six months. (B) Turnover for the indicated presynaptic proteins based on 15N remaining analysis in the F1 mice. (C) Turnover for the indicated presynaptic proteins based on 14N remaining analysis in the F0 mice. (B-C) Circles represent individual peptide measurements. (B) N = 3 – 4 mice per genotype. Data represents mean ± SEM, analyzed with one-way ANOVA followed by Fisher’s LSD. (C) N = 2 AppNL-G-F/NL-G-F and 3 AppNL/NL mice. Data represents mean ± SEM, analyzed with unpaired Student’s t-test. * = p-value < 0.05 and ** = p-value < 0.01.
Steady state protein levels of the SV machinery are elevated at six months but slightly reduced at twelve months of age in AppNL-F/NL-F and AppNL-G-F/NL-G-F cortex
Dynamic 15N labeling analysis allowed us to assess how proteins’ turnover is affected by Aβ42 peptides and amyloid; however, it does not provide any information regarding potential alterations in protein steady state abundance. In order to investigate synaptic proteins’ abundance, we utilized fully 15N labeled WT mouse brains as an internal standard, as we have done in the past (Butko et al., 2013; Savas et al., 2015; Savas et al., 2017). We mixed WT 15N whole brain homogenates 1:1 with unlabeled 14N cortical or hippocampal homogenates from six- or twelve-month-old AppNL/NL, AppNL-F/NL-F, or AppNL-G-F/NL-G-F mice (Figure 4A). Next, we prepared crude synaptic fractions from the mixed 14N and 15N samples. Mixing the samples allows us to limit potential technical variation that may occur during the isolation of synaptosomes. We analyzed four biological replicates of each genotype with LC-MS/MS-based proteomics to obtain 14N / 15N peptide and protein ratios and finally generated ratios relative to the control (AppNL-F/NL-F / AppNL/NL or AppNL-G-F/NL-G-F / AppNL/NL). In both cortical and hippocampal tissue, we measured about 1,400 – 2,000 proteins in each dataset and found that at six months of age, most proteins significantly altered in AppNL-F/NL-F or AppNL-G-F/NL-G-F had generally increased levels compared to age matched AppNL/NL mice (Figure 4B–C, S5A, and Table S3). In contrast, at twelve months of age, most proteins with significantly altered levels in AppNL-F/NL-F or AppNL-G-F/NL-G-F cortical extracts had decreased abundance compared to AppNL/NL controls (Figure 4D–E and Table S3). Additionally, about 20% of these significantly altered proteins are associated with the GO term: presynapse (Figure 4B–E, insert).
Figure 4: Steady state level of the SV machinery is elevated at six months but slightly reduced at twelve months in AppNL-F/NL-F and AppNL-G-F/NL-G-F cortical synaptosome extracts.
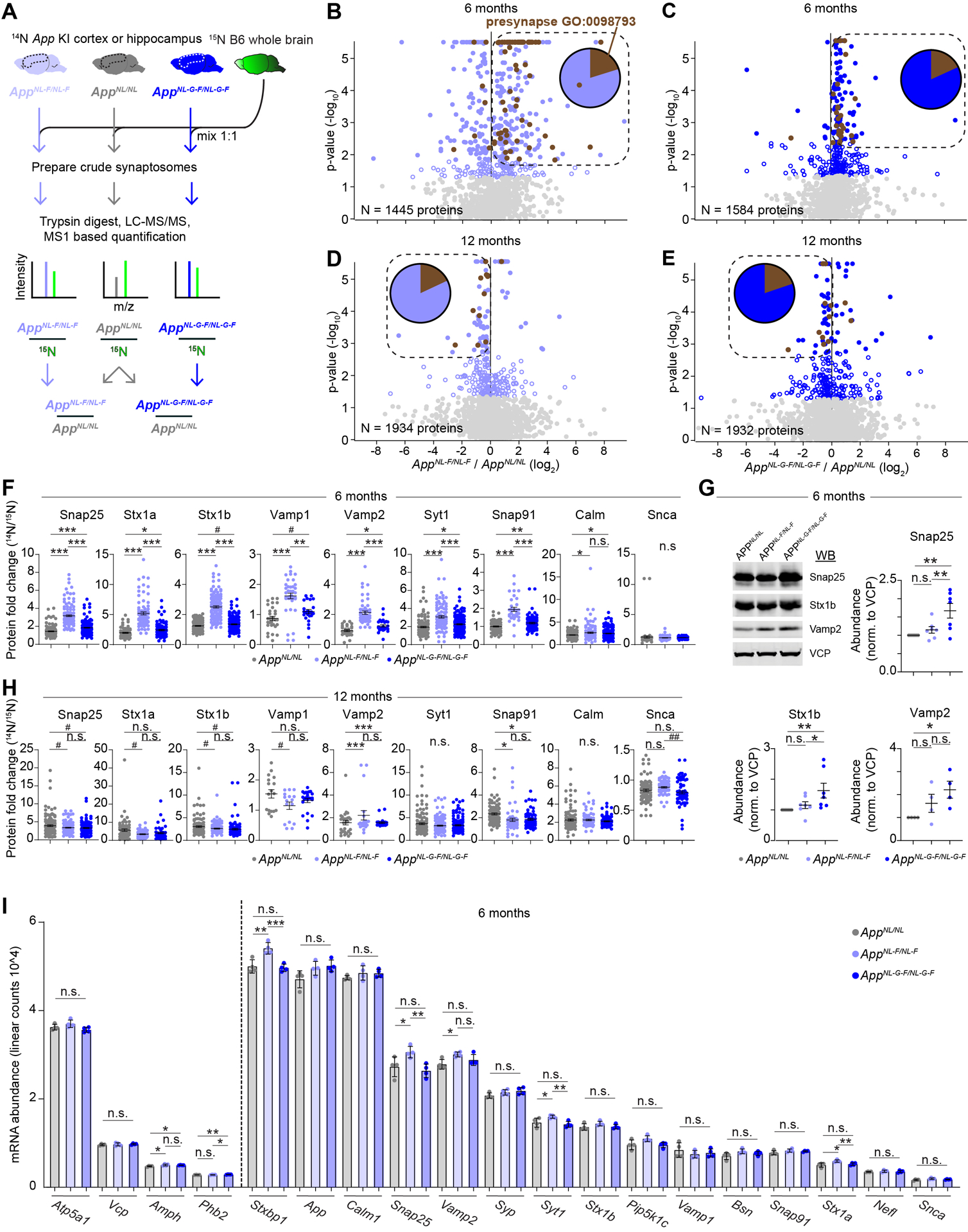
(A) Six- or twelve-month-old AppNL/NL, AppNL-F/NL-F, or AppNL-G-F/NL-G-F cortical or hippocampal homogenates were mixed 1:1 with 15N labeled WT whole brain homogenate, as an internal standard. Crude synaptosomes were prepared, 14N:15N ratio (relative abundance) for each protein was calculated, and a final comparison of AppNL-F/NL-F or AppNL-G-F/NL-G-F relative to AppNL/NL was made. (B-E) Volcano plots summarizing relative protein abundance in AppNL-F/NL-F or AppNL-G-F/NL-G-F compared to AppNL/NL. (B) Six-month-old AppNL-F/NL-F / AppNL/NL, (C) six-month-old AppNL-G-F/NL-G-F / AppNL/NL, (D) twelve-month-old AppNL-F/NL-F / AppNL/NL, (E) twelve-month-old AppNL-G-F/NL-G-F / AppNL/NL. Unfilled blue circles represent proteins with p-value < 0.05 by Student’s t-test, filled circles represent proteins with Benjamini-Hochberg (BH) adj. p-value < 0.05. Brown circles represent proteins from the GO term Presynapse: 0098793. Box encompasses BH significant proteins up or down regulated at six or twelve months respectively. Pie chart indicates presynaptic versus all other proteins in boxed region (F) Relative abundance for the indicated proteins with impaired turnover in six-month-old App KI mice. Circles represent individual peptides mapping to the respective protein. (G) Confirmation of elevated presynaptic proteins levels in AppNL-G-F/NL-G-F relative to AppNL/NL by WB analysis of synaptosome extracts. Circles represent individual biological replicates. (H) Relative abundance for the indicated proteins with impaired turnover in twelve-month-old App KI mice. Circles represent individual peptides mapping to the respective protein. (I) mRNA abundance from six-month-old App KI cortical homogenates of the indicated genes showing little to no change in gene expression. Circles represent individual biological replicates. Dotted line separates proteins, as determined by MS analysis in Figure 4B–C, with unchanged (left) or elevated protein levels (right) in AppNL-F/NL-F or AppNL-G-F/NL-G-F relative to AppNL/NL. (A-F, H, and I) N = 4 mice per genotype and age, (G) N = 4 – 7 per genotype. (F and H) Data represents mean ± SEM, analyzed by one-way ANOVA followed by FDR method of BH multiple comparison. (G) Data represents mean ± SEM, analyzed by one-way ANOVA followed by Fisher’s LSD. (I) Data represents mean ± SD, analyzed by one-way ANOVA followed by Fisher’s LSD. (G-I) * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. (F and H) # = p-value < 0.05, * = p-value < 0.05 with BH-adj, ** = p-value < 0.01 with BH-adj, *** = p-value < 0.001 with BH-adj.
When we specifically analyzed our proteins of interest, several of them, including Snap25, Vamp1, and Stx1b, that had impaired degradation in the cortex also had elevated levels in the cortex. Meanwhile proteins such as Calm, Vamp2, and Snca that had impaired degradation in the hippocampus also had elevated levels in the hippocampus (Figure 4F and S5B). We further confirmed elevated levels of a panel of SV cycle proteins by Western blotting (WB) (Figure 4G). The elevated synaptic protein levels seen at six months abated by twelve months and the trend shifted towards decreased synaptic protein fold change in AppNL-F/NL-F and AppNL-G-F/NL-G-F relative to AppNL/NL. This is consistent with the abundant evidence that synapses eventually degenerate in AD (Figure 4H) (Masliah et al., 1994b).
We then investigated whether the elevated abundance or impaired degradation of our proteins of interest was the result of increased gene expression by quantifying mRNA levels using Nanostring technologies. Most of these proteins were found to have unchanged mRNA levels across all genotypes at six-months (Figure 4I). Furthermore, the mRNA levels were unaffected at an earlier age, 3 months, but were decreased in the AppNL-G-F/NL-G-F at an older age, 12 months (Figure S5C–D). Taken together, our data show that presynaptic proteins have selectively impaired turnover and elevated steady state levels that are not due to compensatory changes in mRNA levels. However, we acknowledge the remote possibility that selective and enhanced protein translation could contribute to our findings. We also compared our findings on presynaptic proteins steady state levels with several recent proteomic studies on AD (Bai et al., 2020; Higginbotham et al., 2019; Savas et al., 2017; Seyfried et al., 2017) and found corroborating independent evidence that presynaptic proteins are increased early then decreased later in AD pathology (Table S4). For example, Syt1, Stx1a, Stx1b, and Stxbp1 were all found with elevated fold change early and reduced levels late in other mouse models of amyloid pathology and post mortem human AD brains.
SV machinery colocalizes with Aβ puncta in AppNL-G-F/NL-G-F hippocampus and coaggregates with Aβ and APP in AppNL-F/NL-F and AppNL-G-F/NL-G-F brain extracts
We assessed the localization pattern of these presynaptic proteins of interest relative to Aβ. Sagittal brain sections from six-month-old AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F mice were probed with an antibody for Aβ (which also detects β-CTF), and with antibodies for our panel of presynaptic proteins: Scna, Calm, Pip5k1c, Snap25, Snap91, Stx1b, Syt1, Vamp1, Vamp2, and Vglut1, as well as Vgat, which served as a negative control. In AppNL/NL and AppNL-F/NL-F brains, we detected no abnormal presynaptic protein staining patterns, nor obvious Aβ puncta in both hippocampal and cortical regions based on 10X magnification (Figure 5A–B, and S6A–B). In AppNL-G-F/NL-G-F brains, however, the presynaptic proteins with attenuated protein turnover were found aggregated in close proximity to Aβ puncta (Figure 5C and S6C). Conversely, Vgat, a marker of GABAergic terminals, had normal localization patterns despite abundant small and large Aβ puncta (Figure 5D). Quantification of the colocalization between presynaptic proteins and larger Aβ puncta (≥ 4-pixel radius) at 10X magnification showed robust colocalization in the AppNL-G-F/NL-G-F and nearly no colocalization in the AppNL/NL and AppNL-F/NL-F sections (Figure 5E and S6D). The very minor degree of colocalization in AppNL/NL and AppNL-F/NL-F likely represents nonspecific signals.
Figure 5: Altered presynaptic proteins colocalize with Aβ puncta in AppNL-G-F/NL-G-F brains and co-aggregate with Aβ in AppNL-F/NL-F and AppNL-G-F/NL-G-F brain extracts.

(A-C) Representative stitched 10X magnification IHC analysis of six-month-old App KI brains with Syt1 (green), Aβ (red), and Vglut (blue) antibodies. Localization patterns of synaptotagmin 1 (Syt1) are unaffected in (A) AppNL/NL and (B) AppNL-F/NL-F. (C) Syt1 colocalizes with Aβ puncta in AppNL-G-F/NL-G-F hippocampus and cortex. (D) Representative stitched 10X magnification IHC analysis with Syt1 (green), Aβ (red), and Vgat (blue) antibodies in AppNL-G-F/NL-G-F brains. Vgat does not colocalize with Aβ in AppNL-G-F/NL-G-F brain. (E) Quantification of 10X magnification images, quantifying the number of Aβ puncta ≥ 4 pixel radii colocalized with presynaptic proteins in the cortex. Circles represent individual biological replicates. Data represent average number of colocalized puncta ± SEM. (F) Representative 63X magnification IHC analysis of Aβ plaques in AppNL-G-F/NL-G-F brain sections immuno-stained with Snap91, Snca, Calm, Pip5k1c, Snap25, Vamp1, Stx1b, Syt1, and Vamp2 (green), Aβ (red), and Vglut (blue) or Vgat (blue). (G-H) Representative MIMS quantitative images from hippocampal sections of a F1 15N labeled (G) AppNL-G-F/NL-G-F or (H) AppNL/NL mouse that was chased with 14N for 6 months. Hue saturation intensity image represents 12C15N / 12C14N ratio as an intensity for each pixel, indicating areas where 15N remains in the CA1. Arrows indicate puncta with elevated 15N / 14N ratio. N indicates nuclei. (I) AppNL-G-F/NL-G-F hippocampal section that was adjacent to MIMS section (top), immunostained for Vamp1 (green) and Aβ (red). Arrows indicate puncta positive for 15N, Vamp1, and Aβ. (J) Coomassie stain of SDS gel analysis with detergent insoluble fractions from eight-month-old App KI cortices. (K-N) Immunoblots for (K) Aβ, (L) APP, (M) Stx1b, and (N) Syt1, with quantitation of the band intensity between the well and 250 kD normalized to the same area from the Coomassie stain. Data represent average band intensity ± SD relative to AppNL/NL except for Aβ. (F, K-N) N = 3 mice per genotype, analyzed with one-way ANOVA, followed by Fisher’s LSD, * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. Scale bar = 1 mm (A-D), 100 μm (F, I), 50 μm (G, H).
When we increased magnification to 63X, we found the Aβ-presynaptic protein colocalization patterns in AppNL-G-F/NL-G-F were frequently not direct overlaps, but rather a close proximity (Figure 5F). While all of the candidate proteins significantly colocalized with the Aβ puncta at low magnification, the extent and pattern of this colocalization greatly varied, suggesting multiple mechanisms may be contributing to impaired turnover of these proteins. Some presynaptic proteins appeared to accumulate extracellularly in plaques, others, such as Syt1, Vamp2, and Calm, accumulated predominantly in swollen axon terminals and dystrophic neurites surrounding the Aβ plaques (Figure 5F). Since the Aβ antibody also detects β-CTF, we cannot rule out β-CTF as a contributor to these findings.
To investigate the spatial distribution of the persisting 15N atoms in AppNL/NL and AppNL-G-F/NL-G-F brains, we performed multi-isotope imaging mass spectrometry (MIMS) in the CA1 region of the chased F1 generation (Steinhauser et al., 2012). MIMS measures the 15N / 14N ratios in tissue sections at high spatial resolution, allowing us to visualize and localize N-containing macromolecules, including proteins, that escape degradation (Figure 5G–H). We subsequently probed adjacent sections with antibodies for Aβ and Vamp1 to investigate if the 15N signal colocalized with plaques or hampered presynaptic proteins (Figure 5I). Indeed, we found that AppNL-G-F/NL-G-F sections harbor small punctate 15N signals measured by MIMS, some of which colocalized with Vamp1 and amyloid plaques, suggesting that Aβ aggregates can sequester these presynaptic proteins preventing their proper turnover. However, the vast majority of Aβ does not appear to persist for long periods.
To extend the imaging results, we assessed if these proteins were misfolded or insoluble similarly to Aβ. We isolated detergent insoluble fractions from App KI cortices and analyzed the proteins by SDS-PAGE followed by WB or gel band extraction followed by LC-MS/MS (geLC-MS/MS). For geLC-MS/MS, we excised the top of the well for in-gel digestion with trypsin then proteomic analysis, as the top of the gel should trap the misfolded insoluble proteins. As expected, LC-MS/MS nor WB revealed detectable Aβ in the AppNL/NL insoluble fractions, whereas aggregated Aβ was detected in both AppNL-F/NL-F and AppNL-G-F/NL-G-F brain extracts (Figure 5K and S6E). In addition, Scna, Vamp1, Syt1, Stx1a, Stx1b, Snap25, and Calm1 were found to have significantly increased levels in AppNL-F/NL-F and AppNL-G-F/NL-G-F insoluble fractions by MS (Figure S6E). Parallel APP, Stx1b, and Syt1 WB analyses revealed significantly elevated levels in AppNL-F/NL-F and AppNL-G-F/NL-G-F insoluble fractions (Figure 5L–N). To investigate the possibility that our panel of presynaptic proteins are trapped in the insoluble fraction during more advanced stages of pathology at 12 months of age, we isolated the SDS insoluble fraction and performed immuno-dot blot analysis. Indeed, every protein tested except Vgat and actin controls had elevated levels in both AppNL-F/NL-F and AppNL-G-F/NL-G-F compared to AppNL/NL cortical extracts, suggesting these proteins are commonly found in amyloid plaques in App KI brains (Figure S6F–G).
Aβ and APP interact with SV associated proteins and disrupt SNARE mediated vesicle fusion
Next, to investigate whether SVs or SV associated proteins physically interact or become trapped with Aβ in aggregates or plaques, we performed Aβ affinity purification on the aggregated protein fractions from App KI cortical extracts followed by LC-MS/MS. Notably, antibody used for Aβ affinity purification also recognizes β-CTF. As expected, Aβ was recovered in significantly higher amounts in 12-month-old AppNL-F/NL-F and AppNL-G-F/NL-G-F compared to AppNL/NL extracts (Figure 6A). We also found significantly more Syt1 and Stx1b co-purified with Aβ from AppNL-F/NL-F and AppNL-G-F/NL-G-F extracts. To test whether SV proteins also interact with full-length mutant APP in these App KI lines, we immuno-purified APP, using an antibody that specifically recognizes full-length APP, and analyzed the purified material with WB and LC-MS/MS. Consistent with previous findings (Del Prete et al., 2014; Gautam et al., 2015), we found that SV associated proteins robustly co-purified with APP (Figure S7), suggesting that both APP and Aβ interact at least indirectly with several SV proteins.
Figure 6: Aβ interacts with hampered presynaptic proteins and disrupts SNARE complex function.
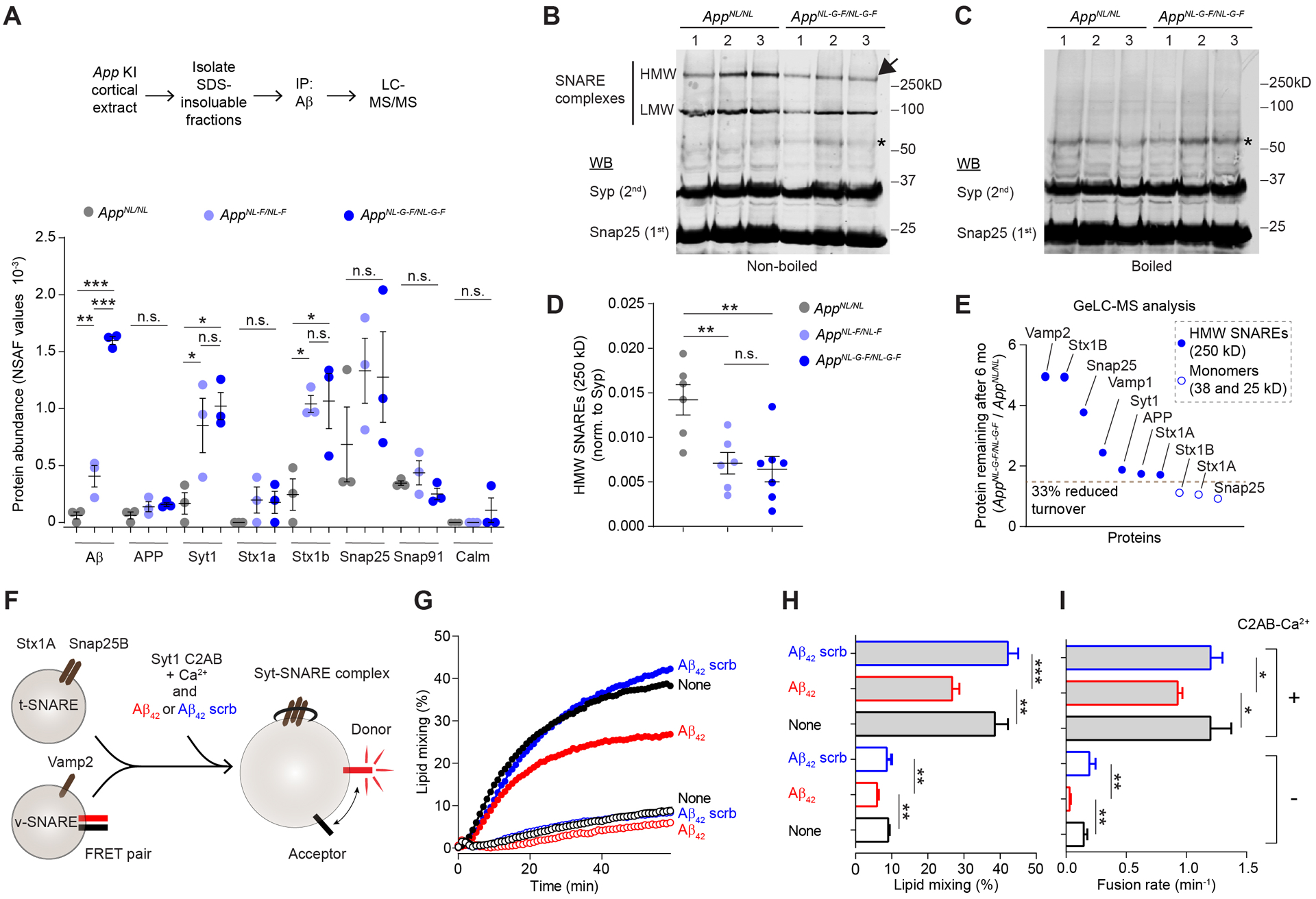
(A) Affinity purification of Aβ from aggregated protein fractions of twelve-month-old App KI cortex co-purifies Syt1 and Stx1b as analyzed by LC-MS/MS. Circles represent individual biological replicates. (B-C) Representative analysis of high molecular weight (HMW) and low molecular weight (LMW) SNARE complexes, nonboiled (B) and boiled (C). Samples from cortical extracts based on Snap25 immunoblotting shows reduced levels in AppNL-G-F/NL-G-F relative to AppNL/NL. (D) Quantification of HMW SNARE complex based on Snap25 relative to synaptophysin immunoblotting from non-boiled samples. Circles represent individual biological replicates. (E) Assessment of protein turnover in the context of the HMW SNARE complex relative to monomers. GeLC-MS/MS analysis of the 250, 38, and 25 kD gel slices from the F1 App KI cortical extracts from Figure 2. Plot shows 15N remaining in AppNL-G-F/NL-G-F relative to AppNL/NL is substantially higher for the indicated proteins in the HMW complex (filled circles) compared to the measures from monomeric Stx1A/B (38 kD) and Snap25 (25 kD) gel band slices (empty circles). Circles represent average 15N remaining. (F-I) Aβ42 inhibits SNARE-mediated membrane fusion in vitro using defined components. (F) Schematic of lipid mixing assays. Two proteoliposomes, one containing Stx1a and Snap25 and the other containing Vamp2 and a FRET pair were mixed in the presence or absence of Syt1•C2AB-Ca2+ and in the presence or absence of Aβ42 peptides or peptides with a scrambled Aβ42 sequence. (G) Time courses of lipid-mixing assays. Filled and empty circles indicate assays performed in the presence or absence of C2AB-Ca2+ respectively. Black circles indicate assays performed in the absence of additional peptides. Red and blue circles indicate assays performed in the presence of Aβ42 peptides or scrambled Aβ42 peptides, respectively. (H-I) Lipid mixing and fusion rate over sixty minutes. (A) N = 3 mice per genotype, (B-D) 6 mice per genotype, (E) 4 mice per genotype, and (F-I) N = 3 independent experiments. (A and D) mean ± SEM, analyzed with one-way ANOVA followed by Fisher’s LSD, (H-I) mean ± SD, analyzed with one-way ANOVA followed by Fisher’s LSD. * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001.
Taking advantage of the SDS resistant nature of the SNARE complex, we performed SDS-PAGE to measure the level of SNARE complexes in App KI cortical extracts (Figure 6B–C). We found reduced abundance of high molecular weight (HMW) SNARE complexes (approximately 250 kD) in AppNL-F/NL-F and AppNL-G-F/NL-G-F compared to AppNL/NL cortical extracts (Figure 6D). To begin to investigate whether the SNARE proteins that escape degradation are present in SNARE complexes, we performed GeLC-MS analysis on gel slices (250 kD) of F1 dynamically labelled AppNL-G-F/NL-G-F and AppNL/NL cortical extracts. Interestingly, we found that the fraction of old protein, based on residual 15N signal, in the HMW band that corresponds to the SNARE complex was substantially higher in AppNL-G-F/NL-G-F compared to AppNL/NL extracts. Meanwhile, there was no increase of old proteins in the bands at molecular weights corresponding to SNARE protein monomers (Figure 6E). This data suggests that the old SV associated proteins escaping degradation are present in HMW SNARE complexes.
To examine the possibility that Aβ42 peptides directly impair the function of trans-SNARE complexes, we assessed their impact on unregulated and Ca2+•Syt1 regulated membrane fusion utilizing a well-established in vitro lipid mixing assay (Figure 6F). We reconstituted two types of proteoliposomes: one containing Stx1a and Snap25, and the other containing Vamp2 and a lipidic FRET donor acceptor pair (Bao et al., 2016). The vesicles were incubated with and without the soluble C2AB domain of Syt1 and Ca2+, in the presence or absence of oligomerized Aβ42 or scrambled Aβ42 peptides. As expected, robust fusion was observed in the presence of C2AB and calcium. Scrambled Aβ42 peptides had no effect on unregulated or Ca2+•C2AB regulated membrane fusion. However, Aβ42 peptides significantly reduced both unregulated and Ca2+•C2AB regulated membrane fusion (Figure 6G–I). Taken together, Aβ and APP co-purify with SV associated proteins and Aβ42 can impair SNAREs in a purified reconstituted in vitro assay.
Enlarged SV pool in AppNL-F/NL-F and AppNL-G-F/NL-G-F brains
Seeing that we found increased levels of most SV associated proteins, due largely to compromised turnover (Figure 3 and 4), we next assessed presynaptic ultra-structure using electron tomography (ET). ET analysis of six-month-old App KI brains enabled us to measure SV size and density at the same age and brain regions that we observed elevated SV associated protein levels (Arthur et al., 2010). We focused on the hippocampal CA1 and cortical regions, in order to best correlate these EM-based measures with the proteomic data. (Figure 7A–C and S8A–C). By obtaining 100s of serial sections and reconstructing the three-dimensional volume we were able to generate 3D models of individual presynaptic sites suitable for quantification (Figure 7D–F, S8D–F, and Videos S1–3). Analysis of SV size revealed no difference in the average radius between the genotypes (Figure S8G). However, synaptic vesicle density was significantly increased in AppNL-F/NL-F and AppNL-G-F/NL-G-F CA1 and in AppNL-G-F/NL-G-F cortex compared to AppNL/NL (Figure 7G).
Figure 7: AppNL-G-F/NL-G-F hippocampus has an enlarged SV pool and enhanced post-tetanic potentiation.

(A-C) Representative electron tomography sections of individual synapses from App KI CA1 at six months of age. (D-F) Reconstructed 3-D models of the axon terminals from adjacent tomograms (100–150 planes per model). Plasma membrane is modeled in green; SVs are in blue; the PSD is in yellow, and large irregular structures are in purple. (G) Quantification of SV density (SV / nm3) in the hippocampal CA1 region (top) and cortex (bottom). AppNL-G-F/NL-G-F has greater SV density in CA1 and cortex, and AppNL-F/NL-F has greater density in CA1 compared to AppNL/NL. Each circle represents an individual synapse and the number of vesicles in that synapse. N = 3 mice and 3–7 synapses per mouse. Data represent mean ± SEM, analyzed with one-way ANOVA followed by Fisher’s LSD. (H) Representative traces of paired fEPSPs in AppNL-G-F/NL-G-F or AppNL/NL CA1 pyramidal cells, following stimulation of Schaffer collateral with increasing intensity. (I) Quantification showing no difference in field responses across stimulation intensities in AppNL-G-F/NL-G-F compared to AppNL/NL. (J) Representative traces from sequential stimulations across the indicated period. (K) Quantification showing no difference in paired stimulation ratio in AppNL-G-F/NL-G-F compared to AppNL/NL. (L) Plot representing cumulative fEPSPs (EPSP1 + EPSP2 + … + EPSP1200) in response to a train of stimulation (1200 at 20 Hz) in AppNL-G-F/NL-G-F and AppNL/NL slices. The average cumulative fEPSP is significantly elevated in AppNL-G-F/NL-G-F compared to AppNL/NL. (M) The number of functional SVs (N*q) is elevated in AppNL-G-F/NL-G-F CA1 compared to AppNL/NL. (N) mEPSC amplitude was not significantly different in AppNL-G-F/NL-G-F compared to AppNL/NL. (O) Potentiation following post theta burst stimulation (TBS) is significantly increased in AppNL-G-F/NL-G-F compared to AppNL/NL. (M-O) Data represents mean ± SEM, analyzed with Student’s t-test. * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. (A-F) Scale bar = 100 nm.
Finally, to determine if elevated levels of SV associated proteins and SV density influences synaptic transmission, we performed electrophysiological analyses in acute hippocampal sections. To assess synapses in the same region that we analyzed with ET, we stimulated Schaffer collaterals and recorded the postsynaptic field responses from the CA1 subregion of the hippocampus in six-month-old AppNL-G-F/NL-G-F and AppNL/NL. AppNL-G-F/NL-G-F electrophysiological recordings likely occur where there are plaques both in close and far proximity to the electrodes. Comparing field excitatory postsynaptic potential (fEPSP) across increasing stimulation intensities revealed no difference in the evoked synaptic response between slices from AppNL-G-F/NL-G-F and AppNL/NL mice, suggesting that there are no major differences in the strength or number of synaptic inputs to CA1 neurons (Figure 7H–I). Similarly, analysis of paired pulse facilitation of fEPSPs, which is a commonly used proxy of presynaptic release probability demonstrated no difference between slices from AppNL-G-F/NL-G-F and AppNL/NL mice (Figure 7J–K).
Using a functional approach to compare the readily releasable pool (RRP) in CA1 synapses, we analyzed the cumulative responses to long (1 min) trains of high frequency (20 Hz) stimuli. This stimulation method depletes the RRP of vesicles. The linear regression fit of the steady state region from the cumulative plots gave us a functional estimate of the RRP, the product of N (total number of releasable vesicles) and q (the quantal size) (Figure 7L) (Fernandes et al., 2015; Schneggenburger et al., 1999). We found that the measured N*q was significantly larger in the AppNL-G-F/NL-G-F compared AppNL/NL slices (Figure 7M). The quantal size (q) was directly measured by recording the amplitude of CA1 miniature EPSC (mEPSC) events in voltage clamp and was found to not be different in the two genotypes (Figure 7N), suggesting that the elevated N*q value in AppNL-G-F/NL-G-F is due to an increase in N (number of releasable vesicles). The increased number of releasable vesicles corresponds to the anatomical features of higher SV density in CA1 terminals found with EM analyses (Figure 7A–G). Finally, we analyzed another measure of presynaptic function by assessing the short-term potentiation observed after theta-burst stimulation (TBS) (Figure 7O). Consistent with the larger number of releasable vesicles in AppNL-G-F/NL-G-F mice, post TBS potentiation was significantly elevated in slice recordings from AppNL-G-F/NL-G-F compared to those from AppNL/NL controls (Andreoli et al., 1989; Lee et al., 2008). In summary, hippocampal CA1 synapses in AppNL-G-F/NL-G-F mice had normal basal synaptic characteristics but demonstrated an elevated functionally estimated RRP and elevated post-TBS potentiation, which was consistent with the anatomical findings that SV density is increased.
DISCUSSION
We investigated protein turnover in the recently developed App KI mouse models using dynamic 15N-labeling with discovery-based LC-MS/MS analysis (Saito et al., 2014; Savas et al., 2012). Our experiments revealed that glutamatergic presynaptic compartments in the hippocampus and cortex represent predominant sites where protein turnover is impaired in the early stages of pathology (Figure 3). Many presynaptic proteins, especially SV associated proteins with impaired turnover also have elevated levels early in pathology (Figure 4). Notably, our results in the AppNL-F/NL-F mice show that this proteostasis impairment occurs before widespread plaque formation and even before significantly increased Aβ42 levels are detectable by ultra-sensitive ELISA (Figure 1). Furthermore, presynaptic proteins had the highest fold change in six-month-old AppNL-F/NL-F samples. These results suggest that presynaptic protein abundance peaks very early during Aβ pathology and by the time amyloid plaques form, (i.e. in six-month-old AppNL-G-F/NL-G-F), steady state levels may already by trending back toward control levels before the eventual reduction that is typically associated with synaptic deterioration (Masliah et al., 1994b). Moreover, there is elevated APP and CTFs in synaptic fractions, which may propagate the production of toxic cleavage products causing a feed forward effect at presynaptic terminals. Since β-CTF is also detrimental at the synapse, especially in the lysosomal-autophagic pathway, its production and accumulation may contribute to the observed degradation perturbations (Kwart et al., 2019; Lauritzen et al., 2016). Altogether, presynaptic proteostasis likely represents a pioneering and under-appreciated synaptic defect in AD-like amyloid pathology.
The SVC is at the core of the presynaptic terminal and SNARE proteins are among the most impaired proteins in our datasets. The vast majority of previous evidence points to the postsynaptic membrane as the primary site of Aβ synaptic toxicity (DeBoer et al., 2014; Perdigao et al., 2020; Serrano-Pozo et al., 2011; Sheng et al., 2012). However, the localization and processing of APP mainly occurs at presynaptic terminals, and it is not without precedence to find that APP interacts with SVs (Masliah et al., 1994a; Oddo et al., 2003). APP and Aβ co-purify with SV associated proteins, including SNAREs (Figure 6A), possibly indicating APP and SVs are intrinsically linked and are functionally co-regulated (Del Prete et al., 2014; Fanutza et al., 2015; Gautam et al., 2015). Additionally, we confirmed HMW SNARE complexes have reduced levels in the context of amyloid pathology (Sharma et al., 2012; Yang et al., 2015). Aβ may compromise SNARE complex formation and hamper its turnover. Moreover, these data indicate SNARE complexes are maintained as a unit rather than being broken down and individually degraded. In similar regards, Aβ oligomers have been previously reported to block SNARE complex assembly through interaction with Stx1a, further supporting the connection between Aβ pathology and early presynaptic dysfunction (Yang et al., 2015). Together, our results suggest that disruption of SNARE function by Aβ in combination with hampered SV endocytosis likely represents a tipping point in presynaptic homeostasis, which may culminate in synaptic dysfunction and eventual synapse loss (Ovsepian et al., 2018).
In agreement with our findings that many SV associated proteins have elevated levels, SV density was also increased when measured by ET (Figure 7 and S8). Electrophysiological analysis revealed that these SVs are loaded with neurotransmitters and are capable of synaptic transmission. However, the readily releasable pool of vesicle was relatively larger in hippocampi with elevated Aβ. One possibility is that the enlarged SV pool arises in order to compensate for hampered SNAREs (Fornasiero et al., 2018; Kavalali, 2006). Our discovery that numerous SV proteins persist, presumably in old SVs in App KI brains suggests these SVs may have exocytic defects. This is consistent with prior studies showing that old SVs become gradually less capable of properly releasing their contents (Duncan et al., 2003). We also found hippocampal presynaptic short-term potentiation was altered (Figure 7). In the context of a behaving animal, we speculate that this abnormality may culminate as imbalanced neural circuits. This imbalance may eventually lead to neuronal hyperactivity and feedforward production, then subsequent accumulation of Aβ before, in parallel, or in concert with previously reported postsynaptic effects (Palop and Mucke, 2016; Perdigao et al., 2020; Sheng et al., 2012).
Multiple lines of evidence from human AD support our discovery that the pioneering synaptic impairments in Aβ pathology manifest in presynaptic sites. First, BIN1 and PICALM represent major genetic risk factors and likely play key roles in AD etiology (Harold et al., 2009; Hu et al., 2011). Additionally, polymorphisms within DNM2, VAMP1 and SYNJ1 genes have been associated with increased risk for AD while polymorphisms in MUC6 gene influence AP2A2 expression, a gene encoding AP-2, which was previously implicated in AD pathogenesis (Aidaralieva et al., 2008; Katsumata et al., 2020; Miranda et al., 2018; Nelson et al., 2020; Sevlever et al., 2015). It is worth noting that these genetic alterations encompass both SV exo- and endocytosis factors suggesting that an imbalanced SVC may play a key role in AD. Second, ATP6V0D1, CALM1, SNAP91, SNCA, STXBP1, SNAP25, and VDAC1 among others, all have altered expression in postmortem AD brain (Akila Parvathy Dharshini et al., 2019; Canchi et al., 2019; Korolainen et al., 2010; Lanke et al., 2018; Manavalan et al., 2013; Yoshino et al., 2016). Third, and potentially most exciting, SNAP25, SYT1, SNCA, YWHAG, and NEFL represent prominent protein biomarkers with elevated levels in AD CSF or plasma liquid biopsies (Molinuevo et al., 2018; Sathe et al., 2019). One possibility is that these proteins are shuttled from the brain during AD due to misfolding and being poor substrates for degradation. Although there are limitations to using rodents in order to study neurodegeneration, these findings, in the context of many other compelling studies, support their utility for the discovery of important AD pathological mechanisms.
In summary, our discovery-based study identifies the presynaptic compartment and especially SV associated proteins as being the cardinal substrate of Aβ proteotoxicity. Our in vivo dynamic pulse-chase strategy represents a rare opportunity to uncover the earliest mechanisms underlying amyloid toxicity. These findings were hinted at in our previous studies with other AD mouse models (Savas et al., 2017), but were not as readily apparent as in these protein-turnover studies using second generation App KI mice. Due to numerous failed clinical AD trials focused on reducing Aβ levels, the importance of the amyloid cascade has been brought into question (Herrup, 2015). However, recent encouraging therapeutic results with Aducanumab and other antibodies against Aβ epitopes have reinvigorated the possibility that lowering Aβ plaques may represent a viable therapeutic strategy (Sevigny et al., 2016). In the context of our findings, Aβ lowering strategies may have to be administered extremely early in disease progression to limit presynaptic alterations and to obtain maximal therapeutic benefits.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey N Savas (jeffrey.savas@northwestern.edu).
Material Availability
This study did not generate new materials.
Data and Code Availability
Source data statement: The raw MS data have been deposited in MassIVE online database (MSV000085004) (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and upon acceptance will be available in the ProteomeXchange online database (http://www.proteomexchange.org/)
Code statement: This paper does not report original code
Scripts statement: Scripts were not used to generate the figures reported in this paper
Any addition information required to reproduce this word is available from the Lead Contact
Experimental Model and Subject Details
All experiments performed were approved by the Institutional Animal Care and Use Committee of Northwestern University (Protocols IS00004793 and IS00010858). The mice used were amyloid precursor protein knock-in mice (App KI), which were originally obtained from the research group led by Dr. Takaomi C. Saido at the Laboratory for Proteolytic Neuroscience, RIKEN Brain Science Institute, Saitama, Japan (Saito et al., 2014). All mice were genotyped by Transnetyx using real-time PCR. For euthanasia, mice were anesthetized with isoflurane followed by acute decapitation. Both male and female mice were used in these experiments.
Method Details
Pulse-Chase Metabolic Labeling in Mice
The general method for producing 15N-labeled mice was described previously (Savas et al., 2016; Savas et al., 2012). In brief, three female mice of each App KI genotype: AppNL/NL, AppNL-F/NL-F, and AppNL-G-F/NL-G-F (F0 generation), were fed a Spirulina-based chow enriched in 15N (Cambridge Isotopes Laboratories) for ~six months starting at P28. Males of the same genotypes were introduced around three-months-old and bred to produce 15N labeled pups (F1 generation). F1 mice stayed on the 15N chow until ~P35 and were then switched to the normal 14N chow for two to six months.
The 15N protein enrichment was calculated based on the shape of the peptide isotope envelope and reconstructed peak area (MacCoss et al., 2005). In the cortex, 15N protein enrichment was determined to be 90–95% for both F0 mice after six months of labeling and F1 mice at one month of age before they were switched to the 14N chow.
Preparation of Protein Extracts from App KI Mouse Brains
Brains were extracted and the hippocampus, frontal cortex, and cerebellum were immediately dissected in pH 7.4 phosphate buffered saline (PBS), then flash frozen in dry ice and ethanol. Brain regions were homogenized in homogenization buffer (4 mM HEPES, 0.32 M sucrose, 0.1 mM MgCl2) containing the following protease inhibitors: aprotinin, leupeptin, AEBSF, benzamidine, PMSF, and pepstatin A. A bead based Precellys 24 homogenizer was used. Volume of homogenization buffer per brain region was as follows: 300 μl for hippocampus, 300 μl for cerebellum, and 500 μl for frontal cortex. Protein concentration was then determined by BCA assay (Thermo Scientific, Cat# 23225) per manufacturer’s instructions, and optical density (OD) at 562 nm was read on a Synergy HTX multi-mode microplate reader (Biotek) and compared with the respective standard curve.
Preparation of Crude Synaptosome Fractions from Mouse Brain
Cortical homogenates were diluted with homogenization buffer and centrifuged at 1,000 × g for 15 minutes and the supernatant was collected. The collected supernatant was subsequently spun at 10,000 × g for 15 minutes, and the supernatant was discarded. The pellet (P2) was resuspended in 400 μl of homogenization buffer and the spin was repeated at 10,000 × g for 15 minutes, once again discarding the supernatant. The remaining pellet was resuspended in the appropriate volume and buffer for various assays.
Preparation of Amyloid Enriched, Aggregated Protein Fractions
Preparation were carried out following previous protocols with some modifications (Lu et al., 2013). Briefly, App KI cortices and hippocampi were minced and homogenized in equal volumes of “buffer C” containing 0.25 M sucrose, 3 mM EDTA, 0.1% sodium azide and protease inhibitor cocktail in 10 mM tris-HCl pH 7 for overnight mixing at 4°C. After raising the sucrose concentration of the mixture to 1.2 M, samples were centrifuged at 250,000 × g for 30 minutes at 4°C. Supernatant was removed and pellet was resuspended in “buffer C” with 1.9 M sucrose, followed by centrifugation at 125,000 × g for 30 minutes at 4°C. The top solid layer above the buffer was collected and washed in 50 mM Tris buffer, pH 8 and centrifuged at 8,000 × g for 15 minutes. Supernatant was removed and the pellet was resuspended in digestion buffer (2 mM CaCl2, 0.01 mg/ml DNase I in 50 mM Tris pH 8) and mixed overnight at 37°C. Next day, the samples were centrifuged at 8,000 × g. Supernatant was removed and the pellet was washed two times, then dispersed in SDS buffer (containing 1% SDS and 1.3 M sucrose in 50 mM Tris pH 8) followed by centrifugation at 200,000 × g for 1 hr. The pellet collected was highly enriched with amyloid fibrils and later used for further studies.
Mass Spectrometry (MS) Sample Preparation from Pulse Chase Metabolic Labeling in Mice
Proteins were precipitated using chloroform/methanol precipitation, denatured with 8 M urea and subsequently processed with ProteaseMAX following manufacturer’s instructions (Promega, Cat# V2072). The samples were reduced with 5 mM Tris(2-carboxyethyl)phosphine (TCEP) at room temperature (RT), alkylated in the dark with 10 mM iodoacetamide (IAA), then diluted with 50 mM ammonium bicarbonate (ABC) and quenched with 25 mM TCEP. Then the samples were digested with sequencing grade modified trypsin (Promega, Cat# V5280) overnight at 37°C. The reaction was subsequently stopped by acidification with 1% formic acid (FA) and desalted using HyperSep C18 Cartridges (Thermo Scientific, Cat# 60108–302) per manufacturer’s instructions then dried down with vacuum centrifugation. The desalted samples were fractionated using HyperSep Strong Cation Exchange (SCX) columns (Thermo Scientific, Cat# 60108–420) per manufacturer’s instruction. Fractions were eluted in 200 μl buffer at increasing ammonium acetate concentrations (20, 50, 100, 500, 2,000 mM ammonium acetate), then each fraction was desalted by Pierce C18 spin columns (Thermo Scientific, Cat# 89873) per manufacturer’s instructions and dried down again with vacuum centrifugation for future resuspension and MS analysis.
MS Sample Preparation for Immunoprecipitation MS (IP-MS)
50 μg of antibody was conjugated to 5 mg of beads (Dynabeads M-270 Epoxy, Invitrogen, Cat# 14301) rotating at RT overnight. Beads were washed in PBS, then incubated with cortical homogenates for APP IP experiments (homogenization buffer was supplemented with 1% Triton-X 100) or incubated with amyloid enriched fractions (see above) for Aβ IP experiments. Incubations occurred overnight, rotating at 4°C. Mixture was washed in PBS with 2 mM EDTA and 0.02% tween three times at 4°C. Finally, beads were eluted in 3 × SDS sample buffer with bromophenol blue while boiling. Eluted samples were precipitated using chloroform/methanol precipitation and digested as above. Trypsinized samples were desalted using Pierce C18 spin columns per manufacturer’s instructions and dried down again with vacuum centrifugation for future resuspension and MS analysis.
The following antibodies were used to conjugate beads:
Anti-Amyloid Precursor Protein Monoclonal Antibody (mAbP2–1) (specific for FL-APP) (Thermo Fisher Scientific Cat# OMA1–03132, RRID:AB_325526); Anti-beta-Amyloid (B-4) (also detects β-CTF) mouse monoclonal (Santa Cruz Biotechnology Cat# sc-28365, RRID:AB_626669); Control beads were conjugated to IgG from rabbit serum (Sigma-Aldrich, Cat#: 15006, RRID:AB_1163659).
Mass Spectrometry Sample Preparation for GeLC-MS
Cortical homogenates were mixed with 6X SDS sample buffer at a 5:1 ratio. The mixtures were sonicated for 5 minutes but not boiled, then separated by SDS-PAGE using a 10% Tris-Glycine gel (Thermo Scientific, Cat# XV00100PK20). We used SimplyBlue™ SafeStain kit (Thermo Fisher, Cat# LC6065) for gel staining per manufacturer’s instructions. Briefly, gels were fixed in 40% Methanol / 10% acetic acid with bromophenol blue for 45 minutes. Then the gel was destained in 8% acetic acid for 30 minutes and rinsed 3 times with ultrapure water for five minutes each. Appropriate areas of the gel were then cut into pieces for in-gel digestion, described below.
Gel pieces were covered in 10 mM TCEP and 50 mM ABC, and left incubating at 37°C for 1 hour. Liquid was removed and discarded then the gel piece was covered in 50 mM IAA in 50 mM ABC and left incubating at RT for 45 minutes in the dark. Liquid was removed and discarded, then the gel piece was covered in 50 mM TCEP and 50 mM ABC and left incubating at RT for 30 minutes. Liquid was removed and discarded, then the gel piece was washed in 50mM ABC three times. Next, the gel piece was put in 50 mM ABC with 1 μg sequencing grade modified trypsin and left overnight at 37°C shaking. The next day, the supernatant was collected into a new tube and the gel piece was incubated in 50% acetonitrile (ACN) and 5% FA at RT shaking for 20 minutes. Supernatant was collected and added to the previous supernatant. The 50% ACN and 5% FA incubation was repeated two more times. The combined supernatants were dried down with vacuum centrifugation. Finally, the sample was resuspended in 0.5% TFA and desalted with Pierce C18 spin column, and again dried down with vacuum centrifugation for future resuspension and MS analysis.
Mass Spectrometry Sample Preparation for Synaptic Protein Abundance
1 mg of AppNL/NL, AppNL-F/NL-F, or AppNL-G-F/NL-G-F cortical homogenate was mixed 1:1 with the same whole brain homogenate of a 15N labeled C57BL/6J mouse. Synaptosomes were then prepared from these mixed samples and digested for MS analysis as described above. Trypsinized samples were desalted using reverse phase IMCStips (IMCS, Cat# 04T-U1R05-1-20-96), then fractioned using HyperSep SCX columns using elutions with 50, 500, and 2,000 mM ammonia acetate. Fractions were finally desalted using Pierce C18 spin columns and dried down with vacuum centrifugation for future resuspension and MS analysis.
Mass Spectrometry Analysis
Dried samples were resuspended in 20 μl Buffer A (94.875% H2O with 5% ACN and 0.125% FA) and three micrograms, as determined by microBCA assay (Thermo Scientific, Cat# 23235) of each fraction or sample were loaded via auto-sampler with a Thermo EASY nLC 100 UPLC pump onto a vented Pepmap 100, 75um × 2 cm, nanoViper trap column coupled to a nanoViper analytical column (Thermo Scientific) with stainless steel emitter tip assembled on the Nanospray Flex Ion Source with a spray voltage of 2000 V. A coupled Orbitrap Fusion was used to generate MS data. Buffer A contained 94.785% H2O with 5% ACN and 0.125% FA, and buffer B contained 99.875 ACN with 0.125% FA.
For the SILAM experiments and synaptosome abundance experiments, the chromatographic run was 4.5 hours in total with the following profile of Buffer B: 2% for 7 mins, 2 – 7% for 1 min, 7 – 10% for 5 mins, 10 – 25% for 160 min, 25 – 33% for 40 min, 33 – 50% for 7 min, 50 – 95% for 5 min, 95% for 15 min, then back to 2% for the remaining 30 min.
For the IP-MS experiments and the GeLC/MS experiments, the chromatographic run was 2.5 hours in total with the following profile of Buffer B: 2 – 8% for 6 mins, 8 – 24% for 64 mins, 24 – 36% for 20 min, 36 – 55% for 10 min, 55 – 95% for 10 min, 95% for 10 min, then back to 2% for remaining 30 min.
Additional MS parameters include: Ion transfer tube temp = 300 °C, Easy-IC internal mass calibration, default charge state = 2 and cycle time = 3 s. Detector type set to Orbitrap, with 60 K resolution, with wide quad isolation, mass range = normal, scan range = 300 – 1500 m/z, max injection time = 50 ms, AGC target = 200,000, microscans = 1, S-lens RF level = 60, without source fragmentation, and datatype = positive and centroid. MIPS was set as on, included charge states = 2 – 6 (reject unassigned). Dynamic exclusion enabled with n = 1 for 30 s and 45 s exclusion duration at 10 ppm for high and low. Precursor selection decision = most intense, top 20, isolation window = 1.6, scan range = auto normal, first mass = 110, collision energy 30%, CID, Detector type = ion trap, OT resolution = 30 K, IT scan rate = rapid, max injection time = 75 ms, AGC target = 10,000, Q = 0.25, inject ions for all available parallelizable time.
Mass Spectrometry Data Analysis and Quantification
Protein identification, quantification, and analysis were performed with Integrated Proteomics Pipeline - IP2 (Integrated Proteomics Applications, Inc., San Diego, CA. http://www.integratedproteomics.com/) using ProLuCID (Eng et al., 1994; Xu et al., 2015), DTASelect2 (Cociorva et al., 2007; Tabb et al., 2002), Census (Park et al., 2008), and QuantCompare. Spectrum raw files were extracted into MS1, MS2 files using RawExtract 1.9.9 (http://fields.scripps.edu/downloads.php). The tandem mass spectra were searched against UniProt mouse protein database (downloaded on 03-25-2014), which included the App KI specific mutations (UniProt, 2015) and matched to sequences using the ProLuCID/SEQUEST algorithm (ProLuCID version 3.1) with 20 ppm peptide mass tolerance for precursor ions and 600 ppm for fragment ions. ProLuCID searches included all fully and half-tryptic peptide candidates that fell within the mass tolerance window and had with no-miscleavages. Carbamidomethylation (+57.02146 Da) of cysteine was considered as a static modification. Peptide/spectrum matches (PSMs) were assessed in DTASelect2 using the cross-correlation score (XCorr), and normalized difference in cross-correlation scores (DeltaCN). Peptide probabilities and false-discovery rates (FDR) were calculated based on a target/decoy database containing the reversed sequences of all the proteins appended to the target database (Elias and Gygi, 2007; Peng et al., 2003). For each brain region per biological replicates, proteins identified had a FDR of ≤1% at the protein level. Each protein identified was required to have a minimum of one peptide of minimal length of six amino acid residues.
For the dynamic 15N experiments, each dataset was searched twice, once against light (14N) and then against heavy (15N) protein databases. After the results from ProLuCID were filtered using DTASelect2, ion chromatograms were generated using “Census” (MacCoss et al., 2003; Park et al., 2008). Census allows users to filter peptide ratio measurements based on a correlation threshold because the correlation coefficient (values between zero and one) represents the quality of the correlation between the unlabeled and labeled chromatograms and can be used to filter out poor quality measurements. This correlation coefficient also considers the retention time of the identified spectra. Peptide ratios measurements used for further analysis exceeded a profile score of 0.8. Fractional abundance was calculated from the 14N and 15N peptide chromatograms, and for each respective protein the 14N and 15N chromatograms for the respective peptides were averaged, then the protein remaining was calculated from those 14N and 15N averages to finally compare across genotypes
For the synaptosome abundance measurements, Census calculates peptide ion intensity ratios for each pair of extracted ion chromatograms. The core of the program is a linear least-squares correlation that is used to calculate the ratio (i.e., slope of the line) and closeness of fit [i.e., correlation coefficient (r)] between the data points of the unlabeled and labeled ion chromatograms. Census allows users to filter peptide ratio measurements based on a correlation threshold; the correlation coefficient (values range form 0–1) represents the quality of the correlation between the unlabeled and labeled chromatograms and can be used to filter out poor-quality measurements. In this study, only peptide ratios with the coefficient correlation values (r2) greater than 0.5 were used for further analysis. In addition, Census provides an automated method for detecting and removing statistical outliers. In brief, SDs are calculated for all proteins using their respective peptide ratio measurements. The Grubbs test (p value < 0.01) is then applied to remove outlier peptides. The outlier algorithm is used only when more than two peptides are found in the same protein, because the algorithm becomes unreliable for a small number of measurements. Final protein ratios were generated with QuantCompare, which uses Log two-fold change on the biological replicates. The statistical significance of the differential expression of all proteins was assessed using a two-tailed one-sample Student’s t-test on their corresponding peptide quantification ratios between both conditions. The obtained p values were FDR-adjusted for multiple hypothesis testing using the Benjamini–Hochberg correction (Benjamini and Hochberg, 1995). Proteins with FDR-adjusted p values < 0.05 and measured in at least two biological replicates in both conditions were considered for further analyses.
Online Databases
We performed GO (Cell Component) statistical overrepresentation analyses by using the Pantherdb (RRID:SCR_004869) (Mi et al., 2019). Specifically, we compared proteins with ≥ 0.33% reduced turnover in AppNL-F/NL-F or AppNL-G-F/NL-G-F relative to AppNL/NL (the query) to all proteins identified by LC-MS/MS based proteomic analysis in each brain region.
Immunohistochemistry and Thioflavin Staining
Mice were transcardially perfused with ice cold PBS (pH 7.4). Brains were sagittally hemisected and one half was post fixed in 4% paraformaldehyde overnight, cryoprotected in 30% sucrose for at least 24 hours, embedded in Tissue-Tek OCT Compound within cryomolds, and flash frozen on dry ice. Sagittal cryosections were prepared at 30 μm thickness and mounted onto gelatin-subbed slides (SouthernBiotech, Cat# SLD01-CS).
For thioflavin S staining, sections were prepared following standard procedures (Ly et al., 2011). Briefly sections were washed with 70% then 80% ethanol for 1 minute each then incubated in filtered Thioflavin S solution (1% in 80% of Ethanol) for 15 minutes in dark. Slides were subsequently washed with 70%, 80% ethanol, and distilled water for 1 minute each. Coverslips were mounted using Fluoromount-G (SouthernBiotech, Cat# 0100–01). Sections were imaged at the Northwestern University Center for Advanced Microscopy with a TissueGnostics system using a 10 × objective. Analysis was conducted using Fiji, and the Analyze puncta tool following thresholding. Cortical and hippocampal area size analyzed were kept consistent throughout each section.
For immunohistochemistry experiments, sections were washed first with PBS (3 × 10 minutes) to remove excess OCT, then washed with 0.01 M glycine (3 × 5 minutes) to remove excess formaldehyde, again with PBS (2 × 10 minutes), then washed with 0.2% Triton-X 100, PBS (1 hour), and once again with PBS (2 × 5 minutes). After washes, sections were blocked with 10% normal horse serum (HS) with 0.1% Triton X-100 for 2 hours at RT, then washed with PBS (2 × 5 minutes). Sections were incubated with the primary antibodies, diluted in 1% HS and 0.1% Triton-X overnight at 4°C. The next day, sections were washed with PBS (2 × 5 minutes), blocked with 3% HS and 0.1% Triton-X 100, again washed with PBS (2 × 5 minutes), then incubated with secondary antibodies diluted in 1% HS and 0.1% Triton-X 100 overnight at 4°C. The next day sections were washed with PBS (3 × 5 minutes), then incubated with DAPI solution for 10 minutes. After a final wash with PBS, coverslips were mounted with Fluoromount-G. Sections were imaged using a Nikon A1R confocal microscope at 10 and 63 × objectives. For colocalization analysis, merged images were imported to Fiji and the channels were spilt. After thresholding the images, the channel stained with Aβ antibody was used as a mask to overlay onto the other channels. The analyze puncta plugin was used, and a radius pixel size > 4 pixels was required to identify a colocalized puncta. For synapse density analysis, the Bassoon signal was used as a mask and overlayed onto PSD95 channel and the size criteria was > 1 pixel. Areas used for analysis were measured to ensure the same size area was used for each slice.
The following primary antibodies were used for immunohistochemistry:
Anti-Human Amyloid-beta (N) (82E1) (Also detects β-CTF), mouse monoclonal at 1:10,000 (Immuno-Biological Laboratories Cat# 10323, RRID:AB_10707424); Anti-AP180 (Snap91), rabbit polyclonal at 1:500 (Synaptic Systems Cat# 155 003, RRID:AB_887691); Anti-α Synuclein, rabbit polyclonal at 1:500 (Synaptic Systems Cat# 128 102, RRID:AB_887858); Anti-Bassoon, guinea pig polyclonal at 1:1,000 (Synaptic Systems Cat# 141 004, RRID:AB_2290619); Anti-Calmodulin, rabbit monoclonal at 1:500 (Thermo Fisher Scientific Cat# MA5–32074, RRID:AB_2809368); Anti-Pip5k1c, rabbit polyclonal at 1:500 (Novus Cat# NBP1–82986, RRID:AB_11029240); Anti-PSD-95, mouse monoclonal at 1:500 (Thermo Fisher Scientific Cat# MA1–046, RRID:AB_2092361); Anti-Snap25, rabbit polyclonal at 1:500 (Synaptic Systems Cat# 111 002, RRID:AB_887790); Anti-Synaptobrevin 2 (Vamp2), rabbit polyclonal at 1:500 (Synaptic Systems Cat# 104 202, RRID:AB_887810); Anti-Synaptotagmin 1/2 cytoplasmic tail, rabbit polyclonal at 1:500 (Synaptic Systems Cat# 105 003AF, RRID:AB_2744565); Anti-Syntaxin 1B, rabbit polyclonal at 1:500 (Synaptic Systems Cat# 110 402, RRID:AB_887901); Anti-Vamp1, rabbit polyclonal at 1:500 (Abcam Cat# ab41324, RRID:AB_1281203); Anti-Vesicular Glutamate Transporter 1 (VGLUT1), guinea pig polyclonal at 1:1,000 (Millipore Cat# AB5905, RRID:AB_2301751); and Anti-Vgat, guinea pig polyclonal at 1:200 (Synaptic Systems Cat# 131 004, RRID:AB_887873).
The following secondary antibodies were used for immunohistochemistry:
Anti-Rabbit IgG (H+L) Alexa Fluor 488, goat polyclonal at 1:5,000 (Thermo Fisher Scientific Cat# A-11034, RRID:AB_2576217); Anti-Mouse IgG (H+L) Alexa Fluor 568, goat polyclonal at 1:5,000 (Thermo Fisher Scientific Cat# A-11031, RRID:AB_144696); and Anti-Guinea pig IgG (H+L) Alexa Fluor 647, goat polyclonal at 1:5,000 (Abcam Cat# ab150187, RRID:AB 2827756).
ELISA Assays
Aβ42 ELISA assay: Levels of Aβ42were analyzed using two human Aβ42 ELISA kits (Thermo Scientific, Cat# KHB3441 and KHB3544) according to the manufacturer’s instructions. These kits had respective sensitivity/assay ranges of 15.6 – 1,000 pg / mL and 1.56 – 100 pg / mL. Briefly, App KI cortical homogenates (1 – 2 mg) were supplemented with 5M guanidine HCl and agitated for 1 hour at RT. Then, samples were diluted 1:10 in Standard Diluent Buffer supplied by the kits. 50 μl of sample was loaded into wells coated with Aβ42 antibody and incubated for 3 hours. After thoroughly washing, HRP-conjugated antibody was added to the sample wells for 30 minutes. After another wash step, samples were incubated with stabilized chromogen for 30 minutes, and the reaction was stopped with an acid-based Stop solution. Finally, OD was measured at 450 nm using a Synergy HTX multi-mode microplate reader (Biotek) and compared to a standard curve to determine the final concentration.
Oligomeric Aβ ELISA assay: Levels of oligomeric Aβ were analyzed using an Oligomeric amyloid-beta ELISA kit (Biosensis, Cat# BEK-2215–1P) according to manufacturer’s protocols. Briefly, 3 mg of App KI cortical homogenates was diluted with TBS buffer supplemented with protease inhibitors then centrifuged at 16,000 × g for 30 minutes at 4°C. The pellets were resuspended in TBS plus 1% Triton X-100 (TBST), sonicated for 5 minutes, and centrifuged at 16,000 × g for 30 minutes at 4°C. The TBST supernatant was collected, and protein concentration was measured by BCA. 100 μl of each sample was loaded into wells coated with the MOAB-2 monoclonal antibody that is preferential for oligomeric Aβ and incubated for 24 hours at 4°C. After thoroughly washing, detection antibody was added to the sample wells and incubated for 1 hour at RT followed by another wash. Samples were then incubated with streptavidin-HRP conjugate for 30 minutes at RT. After a final wash, TMB solution (3,3’,5,5’-tetramethylbenzidine) was incubated in each well for 8 minutes in the dark, quenched with a stop solution, and the OD was measured at 450 nm using a Synergy HTX multi-mode microplate reader (Biotek).
Western Blotting
For synaptic protein abundance experiments, synaptosome preparations were resuspended in a lysis buffer (150 mM NaCl, 50 mM Tris pH 8, 1 mM EDTA pH 8, 0.2% SDS, 0.5% Sodium Deoxychloate, 1% NP40 and protease inhibitors) and prepared as described below for gel loading.
For SNARE complex experiments, cortical homogenates were split into two samples with one half undergoing the standard preparation as described below and the other half bypassing the boiling step before gel loading.
For the ubiquitin blot experiments, cortical homogenates were incubated with TUBE1 Magnetic beads (Life Sensors, Cat# UM401M) for 3 hours rotating at 4°C, then washed with TBS-T, and eluted with 3X SDS buffer. Samples were boiled for 5 minutes before being ready to load onto gels. Our positive control for these experiments were HEK cells treated with the proteasome inhibitor MG-132. HEK cells were cultured in DMEM media with 10% FBS and maintained in a 37°C, 5% CO2 tissue culture incubator. HEK cells were treated with 10 μM MG-132 (EMD Millipore, Cat# 474790) for 12 hours before being lysed and prepared as described below for gel loading.
For detergent insoluble protein experiments, we followed the published methodology in (Gal et al., 2018) with some modifications. Briefly 1 mg of cortical homogenates was centrifuged at 20,000 × g for 30 minutes at 4°C. Pellets were solubilized in a low salt (LS) buffer (10 mM Tris-HCl, pH7.5, 5 mM EDTA, and 10% (w/v) sucrose) and centrifuged again at 20,000 × g for 30 minutes at 4°C. The supernatant was removed, and the pellet extracted with LS buffer supplemented with 1% Triton-X-100 and 0.5 M NaCl (TX buffer), then centrifuged at 180,000 × g for 30 minutes at 4°C. The pellets were reextracted with TX buffer and the 180,000 × g centrifugation was repeated for 30 minutes at 4°C. The supernatant was removed, and the pellets were extracted with myelin flotation buffer (TX buffer with 30% sucrose) and centrifuged at 180,000 × g for 30 minutes at 4°C. The supernatant was removed and the pellet was extracted with SARC buffer (LS buffer supplemented with 1% N-lauroylsarcosine and 0.5 M NaCl) and incubated for 2 hours 22°C on an end-over-end shaker, then centrifuged at 180,000 × g for 30 minutes at 22°C. The supernatant was removed, and the pellet was extracted with urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, and 30 mM Tris-HCl, pH 8.5) and centrifuged at 180,000 × g for 30 minutes at 4°C. This urea soluble supernatant was then prepared for Western blot analysis. All buffers were supplemented with protease inhibitors. All buffers except the urea buffer were also supplemented with 1 mM DL-dithiothreitol (DTT) immediately before centrifugation.
APP IP Western Blot
50 μg of antibody was conjugated to 5 mg of beads (Dynabeads M-270 Epoxy, Invitrogen 14301) rotating at RT overnight. Beads were washed in PBS, then incubated with cortical homogenates for 3 hours, rotating at 4°C. Mixture was then washed in PBS with 2 mM EDTA and 0.02% tween three times at 4°C. Finally, beads were eluted in 3 × SDS buffer while boiling then ready to load for Western blot.
Antibody for conjugating to beads was Amyloid Precursor Protein Monoclonal Antibody (mAbP2–1) (Thermo Fisher Scientific Cat# OMA1–03132, RRID:AB_325526).
Control beads were conjugated to IgG from mouse serum (Sigma-Aldrich, Cat#: 15381, RRID:AB_1163670).
Gel Loading
To prepare the samples for Western blot, each sample was mixed with 6 × SDS sample buffer at a 5:1 ratio. The mixtures were sonicated for 5 minutes, then boiled at 95°C for 5 minutes. 50 μg of sample was separated by SDS-PAGE using a 10% or 16% Tris-Glycine Gel (Thermo Scientific, Cat# XV00100PK20 and Cat# XP00162BOX). Gels were run at ~120 V for ~1.5 hours, then were wet transferred to a 0.2 μm nitrocellulose membrane. Membranes were subsequently washed with TBST and blocked with Odyssey Blocking Buffer (LI-COR, Cat# 92740003) in PBS for 1 hour then incubated overnight with primary antibody. The next day membranes were washed and incubated in secondary antibody for 1 hour at RT. Blots were imaged on an Odyssey CLx (Li-Cor).
Dot Blots
For detection of proteins in large aggregates, dot blot analysis was performed. Equal amount of proteins from amyloid enriched fractions (see preparation above) were loaded on the nitrocellulose membranes as dots followed by standard immunoblotting procedure (Chhangani et al., 2016).
The following primary antibodies were used for Western and dot blots:
Anti-Human Amyloid-beta (N) (82E1) (also detects β-CTF) mouse monoclonal at 1:1,000 (Immuno-Biological Laboratories Cat# 10323, RRID: AB_10707424); Anti-Amyloid beta precursor protein [Y188] rabbit monoclonal at 1:1,000 (Abcam Cat# ab32136, RRID: AB_2289606); Anti-AP180 (Snap91) rabbit polyclonal at 1:500 (Synaptic Systems Cat# 155 003, RRID: AB_887691); Anti-Gapdh (0411) mouse monoclonal at 1:2,000 (Santa Cruz Biotechnology Cat# sc-47724, RRID: AB_627678); Anti-Pip5k1c rabbit polyclonal at 1:500 (Novus Cat# NBP1–82986, RRID: AB_11029240); Anti-Snap25 rabbit polyclonal at 1:500 (Synaptic Systems Cat# 111 002, RRID: AB_887790); Anti-Synaptobrevin 2 (Vamp2) rabbit polyclonal at 1:500 (Synaptic Systems Cat# 104 202, RRID: AB_887810); Anti-Synaptophysin mouse monoclonal at 1:1,1000 (Sigma-Aldrich Cat# S5768, RRID: AB_477523); Anti-Synaptotagmin 1/2 cytoplasmic tail rabbit polyclonal at 1:500 (Synaptic Systems Cat# 105 003AF, RRID: AB_2744565); Anti-Syntaxin 1B rabbit polyclonal at 1:500 (Synaptic Systems Cat# 110 402, RRID: AB_887901); Anti-Vamp1 rabbit polyclonal at 1:500 (Abcam Cat# ab41324, RRID: AB_1281203); Anti-Ubiquitin P4D1 mouse monoclonal at 1:1,000 (Santa Cruz Biotechnology Cat# sc-8017, RRID: AB_628423); Anti-VCP mouse monoclonal at 1:2,000 (Abcam Cat# ab11433, RRID: AB_298039).
The following secondary antibodies were used for immunoblotting:
IRDye 800CW Donkey anti-Rabbit IgG antibody (LI-COR Biosciences Cat# 926–32213, RRID: AB_621848);
IRDye 680RD Donkey anti-Mouse IgG antibody (LI-COR Biosciences Cat# 925–68072, RRID: AB_2814912).
NanoString Gene Expression Analysis
RNA isolation was done using RNeasy Lipid Tissue Mini Kit (Quiagen, Cat# 74804) per manufacturer’s instructions. Briefly, frontal cortex was homogenized by Precellys in 1 mL of QIAzol Lysis Reagent, then incubated at RT for 10 minutes. 200 μl of chloroform was added to each sample, vortexed, incubated at RT for 3 minutes and centrifuged at 12,000 × g for 15 minutes at 4°C. The upper aqueous phase was then transferred to a new tube. One volume of 70% ethanol was added, and the mixture was vortexed well. Up to 700 μl of mixture was transferred to an RNeasy Mini Spin Column inside a 2 mL collection tube and centrifuged at RT for 15 seconds at 8,000 × g. Flow-through was discarded and process repeated for the remaining volume of mixture. Columns were washed with 700 μl of Buffer RW1 and centrifuged at 8,000 × g for 15 seconds at RT. Columns were then washed with 500 μl of Buffer RPE and centrifuged at 8,000 × g for 15 seconds at RT (X2). RNeasy columns were transferred into new collection tubes and eluted with 20 μl of RNase-free water by centrifuging for 1 minute at 8,000 × g (X2). RNA concentration was measured using a Take 3 micro volume plate (Biotek) read on a Synergy HTX multi-mode microplate reader (Biotek). Samples were sent to NanoString Technologies for mRNA abundance analysis using a custom CodeSet Design, which contained bar-coded hybridization probes against mRNAs for the following proteins: amphiphysin (Amph), apolipoprotein E (Apoe), amyloid precursor protein (App), ATP synthase F1 subunit alpha (Atp5a1), bassoon (Bsn), calmodulin-1 (Calm1), neurofilament light polypeptide (nefl), prohibitin-2 (Phb2), phosphatidylinositol-4-phosphate 5-kinase type-1 gamma (Pip5k1c), synaptosomal-associated protein 25 (Snap25), clathrin coat assembly protein AP180 (Snap91), alpha-synuclein (Snca), syntaxin-1A (Stx1a), syntaxin-1B (Stx1b), syntaxin-binding protein 1 (Stxbp1), synaptophysin (Syp), synaptotagmin-1 (Syt1), synaptobrevin-1 (Vamp1), synaptobrevin-2 (Vamp2), and valosin-containing protein (Vcp).
nSolver software was used for gene expression analysis, and, for each gene, transcript count was normalized to the geometric mean expression of 6 housekeeping genes: cytochrome c1 (Cyc1), 60S ribosomal protein L13 (Rpl13), 60S ribosomal protein L32 (Rpl32), TATA-binding protein (Tbp), ubiquitin-conjugating enzyme E2D 2A (Ube2d2a), ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 (Uqcrfs1). Background expression level in each sample was calculated as the mean plus standard deviation of raw counts for eight synthetic negative control RNA probes for that sample.
MIMS
Analyses were performed at the Brigham and Women’s Hospital Center for NanoImaging with the NanoSIMS 50L (Cameca) as described (Kim et al., 2014; Steinhauser et al., 2012). LR white embedded tissue samples were sectioned (0.5 μm), mounted on silicon wafers, and gold coated. The isotope ratio of 15N / 14N was measured as previously described by quantifying the 12C15N– / 12C14N–ratio. Samples were analyzed in automated chain analysis mode, with each tile acquired at 256 × 256 pixels, 50 μm × 50 μm field size. Images were visualized and analyzed using the freely available custom plugin to FIJI / ImageJ (National Institutes of Health) called OpenMIMS, version 3.0 (https://github.com/BWHCNI/OpenMIMS/wiki/Installation). For each field analyzed, images within the z-stack were first aligned using the “Autotrack” function in the “Stack Editing” tab. Data in successive images were then summed using the “Compress” function, also in the “Stack Editing” tab. HSI images representing the 12C15N / 12C14N ratios were generated using the “Display HSI” function in the “Process” tab. The lower end of the ratio range was set to 37, corresponding to the natural abundance for 12C15N of ~0.037%, and the upper end was adjusted to a multiple of 37 that placed average cytosol enrichment approximately within the middle of the range.
SNARE Fusion Assay
Lipid mixing assays were carried out as described (Bao et al., 2016; Bhalla et al., 2008; Tucker et al., 2004) in the absence or presence of 5 μM Aβ1–42 or a scrambled Aβ1–42 peptide (see below for preparation and sequences). Briefly, mixtures of v-SNARE containing nanodiscs (0.2 μM) and t-SNARE liposomes (0.5 μM) were incubated in the presence or absence of C2AB (1 μM) at 37°C for 10 minutes in reconstitution buffer plus 0.2 mM EGTA. After incubation Ca2+ (1 mM final concentration) was added. In this system, fusion between v-SNARE nanodiscs and t-SNARE liposomes resulted in the dilution of the nitro-2–1,3-benzoxadiazol-4-yl (NBD)-rhodamine FRET pair and consequent dequenching of the NBD fluorescence. The NBD signal was monitored for an additional 1 hour. After each run, 20 μl of 2.5% n-dodecyl β-D-maltoside (DDM) was added to each reaction to calculate the maximal fluorescence signal for an additional 25 minutes. Percentage of lipid mixing at each timepoint was normalized by subtracting the fluorescence signal at the beginning of the assay.
1 mM Aβ1–42 (Abcam Cat# ab120301) or scrambled Aβ1–42 (Bachem) were prepared with HFIP (Hexafluoroisopropanol) and incubated for 1 hour at RT, then aliquoted and dried overnight. Dried samples were further lyophilized by vacuum centrifugation for 1hr and stored at −20°C before use. Dried samples were resuspended in DMSO to 5 mM, vortexed, pulse spun and sonicated for 10 minutes. To prepare oligomers, cold buffer was added to bring to concentration to 100 μM and incubated at 4°C for 24 hours. Before use, oligomers were spun 12,000 × g for 10 minutes to remove potential fibril species.
The following peptides were used:
Aβ 1–42: (Abcam, Cat# ab120301) Sequence: DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA.
Aβ Scrambled: (Bachem, Cat# 4104168.1000) Sequence: YHAGVDKEVVFDEGAGAEHGLAQKIVRGFGVSDVSMIHINLF.
Electron Tomography
App KI mice were sacrificed by cervical dislocation in accordance to IACUC approved protocol. The brains were removed from the skull, washed in ice-cold PBS and cut into two halves along the longitudinal – sagittal axis. Brain hemispheres were placed into a soft plastic mold with the sagittal plane facing the bottom and completely covered with 2% low melting agarose in PSB (~40 – 42°C). The samples were incubated on ice for a couple of minutes, until the agarose block solidified. The agarose block was then fixed onto a specimen disc stage (sagittal plain on the top) by superglue and transferred to a buffer tray containing ice-cold PSB and submerged in a cooling bath filled with ice. A Leica VT1000 S Vibrating-blade Microtome (setting condition: knife travel speed 3.5 and frequency 8) was used to obtain 0.1 mm sagittal slices. The sagittal slices, corresponding to approximately sagittal section 14 (http://atlas.brain-map.org/atlas), were micro-dissected using 1 mm or 2 mm biopsy punches (Miltex, #33–31AA-P/25) to obtain 1 or 2 mm diameter punches of CA1 – hippocampal, CBX – cerebellum, and CTX – frontal cortex regions. Punches were briefly submerged in a cryoprotectant solution (2% sucrose, 150 mM Manitol in DMEM medium), and then rapidly frozen under high pressure in a Wohlwend Compact 02 High-Pressure Freezer (Engineering Office M. Wohlwend GmbH CH-9466 Sennwald/Switzerland). The frozen samples were then freeze substituted in 2% osmium tetroxide and 0.1% uranyl acetate in anhydrous acetone for three days on dry ice at −78.5°C. The sample temperature was then raised to −20°C, 4°C and then room temperature (with a hold of 12 hours, 4 hours, and 1 hour, respectively). After 1 hour at room temperature, the fixatives were washed out and samples were slowly infiltrated with EPON-Araldite over four days, followed by 48 hours of polymerization at 60°C (Ruhl et al., 2019). Serial, 300 nm thick sections of the embedded tissue were collected onto formvar-coated copper slot grids and post-stained using 2% uranyl acetate and Reynolds lead citrate. Imaging was performed using a Tecnai F30 (Thermo Scientific, Waltham MA) operated at 300 kV, using a OneView camera (Gatan, Inc. Pleasanton, CA) to record images at 1 – 2 nm pixel size. Serial, tilted views were collected from 60° to −60° at 1.5° increments along the dual axis using the SerialEM acquisition program (Mastronarde, 2005). Images were processed and tomographic reconstruction generated using the IMOD software package (Kremer et al., 1996; Mastronarde, 1997). Reconstructions were rendered by hand tracing synapse membranes of every other section, then meshing contours to give a precise rendering of the three-dimensional information. Vesicles were rendered by predicting the spherical nature of the vesicle surface and then contouring accordingly.
Electrophysiology
Animals were anesthetized using ketamine/xylazine (i.p.), and underwent rapid cardiac perfusion with ice-cold sucrose ACSF containing the following (in mM): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2, 4 MgCl2, 0.5 ascorbic acid, 0.1 kynurenic acid, and 0.01 dl-APV, equilibrated with 95% O2 and 5% CO2. The brain was rapidly removed under the same ice-cold sucrose ACSF, mounted on the vibratome stage (Leica), and horizontal hippocampal slices (350 μm) were prepared. Sections were placed in a recovery chamber and heated to 30°C for ~30 minutes in ACFS. The sucrose solution was gradually exchanged for an ACSF containing the following (in mM): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 1 CaCl2, 2 MgCl2, 0.1 kynurenic acid, and 0.01 dl-APV at room temperature until used for experiments. Individual slices were transferred to a recording chamber, perfused with a recording ACSF, containing the following (in mM): 125 NaCl, 2.4 KCl, 1.2 Na2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2 and visualized under DIC optics. Stimulation and recording electrodes were fabricated from borosilicate glass using a P-97 Micropipette Puller (Sutter Instrument). Stimulating electrodes were filled with the recording ACSF. Recording electrodes had a tip resistance of 3–5 MΩ when filled with internal solutions. For extracellular recordings, recording electrodes were filled with the same recording ACSF. fEPSPs were elicited in the CA1 region of hippocampi by stimulating electrodes placed in the Stratum Radiatum. The readily releasable pool (RRP) was measured by analyzing the cumulative amplitudes of the fEPSPs elicited by the long high frequency trains of stimuli (60 s at 20 Hz). The zero-time y-axis intersect of the linear regression fit obtained from the last 50 events of the cumulative plot was used to estimate the N*q, where N represents the total number of releasable synaptic vesicles and q represents the quantal size of synaptic responses. Theta-burst stimulation (TBS) consisting of 10 bursts of five stimuli at 100 Hz with an inter-burst interval of 200 ms (5 Hz) was used to measure post-high frequency potentiation. TBS potentiation was quantified as the ratio of the average of the 3 fEPSP slopes after TBS to the baseline (30 fEPSPs). For whole cell recordings, electrodes were filled with an internal solution containing the following (in mM): 95 CsF, 25 CsCl, 10 Cs-HEPES, 10 CS-EGTA, 2 NaCl, 2 Mg-ATP, 10 QX-314, 5 TEA-Cl, and 5 4-AP. Cells were voltage clamped at −70 mV for EPSC recordings isolated by applying the GABAA receptor antagonists picrotoxin (50 μM) and bicuculline (10 μM), and the Na+ channel blocker tetrodotoxin (TTX) (1 μM). All recordings were performed at 30°C. Data were collected and analyzed using pClamp 10 software (Axon Instruments) and Mini Analysis (Synaptosoft) software.
Quantification and Statistical Analysis
Statistical analyses were conducted using GraphPad Prism, version 8 (GraphPad Software, Inc.), IP2, pClamp10, Mini Analysis (Synaptosoft). Methods of quantification and statistical analysis, including the test used, the value and meaning of n, definition of center and dispersion are included in the figure legend and Method Details section of the corresponding experiment. In most cases, comparisons of two experimental groups were analyzed via unpaired t-tests and comparisons of multiple groups were analyzed via one-way ANOVA with a post hoc Fisher’s LSD test between each group. Brown-Forsythe test for SD differences was also performed when appropriate. Significance levels are defined in the figure legends but in most cases were considered significant when p ≤ 0.05 (one symbol represents p ≤ 0.05; two symbols represents p ≤ 0.01; three symbols represents p ≤ 0.001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APP (mAbP2–1) | Thermo Fisher Scientific | Cat# OMA1–031232; RRID:AB_ 325526 |
| Amyloid-Beta (B-4) | Santa Cruz Biotechnology | Cat# sc-28365; RRID:AB_626669 |
| IgG from Rabbit Serum | Sigma-Aldrich | Cat#: I5006, RRID:AB_1163659 |
| IgG from Mouse Serum | Sigma-Aldrich | Cat#: I5381 RRID:AB_1163670 |
| Amyloid-Beta (82E1) | Immuno-Biological Laboratories | Cat# 10323, RRID:AB_10707424 |
| AP180 (Snap91) | Synaptic Systems | Cat# 155 003, RRID:AB_887691 |
| α-Synuclein | Synaptic Systems | Cat# 128 102, RRID:AB_887858 |
| Bassoon | Synaptic Systems | Cat# 141 004, RRID:AB_2290619 |
| Calmodulin (Calm) | Thermo Fisher Scientific | Cat# MA5–32074, RRID:AB_2809368 |
| Pip5k1c | Novus | Cat# NBP1–82986, RRID:AB_11029240 |
| PSD-95 | Thermo Fisher Scientific | Cat# MA1–046, RRID:AB_2092361 |
| Snap25 | Synaptic Systems | Cat# 111 002, RRID:AB_887790 |
| Synaptobrevin 2 (Vamp2) | Synaptic Systems | Cat# 104 202, RRID:AB_887810 |
| Synaptotagmin 1/2 cytoplasmic tail | Synaptic Systems | Cat# 105 003AF, RRID:AB_2744565 |
| Syntaxin 1B | Synaptic Systems | Cat# 110 402, RRID:AB_887901 |
| Vamp1 | Abcam | Cat# ab41324, RRID:AB_1281203 |
| Vesicular Glutamate Transporter 1 (VGLUT1) | Millipore | Cat# AB5905, RRID:AB_2301751 |
| Vgat | Synaptic Systems | Cat# 131 004, RRID:AB_887873 |
| Anti-Rabbit IgG (H+L) Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| Anti-Mouse IgG (H+L) Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11031, RRID:AB_144696 |
| Anti-Guinea pig IgG (H+L) Alexa Fluor 647 | Abcam | Cat# ab150187 RRID:AB 2827756 |
| Amyloid beta precursor protein (Y188) | Abcam | Cat# ab32136, RRID: AB_2289606 |
| Gapdh | Santa Cruz Biotechnology | Cat# sc-47724, RRID: AB_627678 |
| Synaptophysin | Sigma-Aldrich | Cat# S5768, RRID: AB_477523 |
| Ubiquitin (P4D1) | Santa Cruz Biotechnology | Cat# sc-8017, RRID: AB_628423 |
| VCP | Abcam | Cat# ab11433, RRID: AB_298039 |
| IRDye 800CW Donkey anti Rabbit IgG | LI-COR Biosciences | Cat# 926–32213, RRID: AB_621848 |
| IRDye 680CW Donkey anti Mouse IgG | LI-COR Biosciences | Cat# 925–68072, RRID: AB_2814912 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse Express (15N, 98%) mouse feed prepared with Spirulina (U-15N, 98%+) | Cambridge Isotopes Laboratories | MF-SPIRULINA-A |
| ProteaseMax | Promega | Cat# V2071 |
| Sequencing grade modified Trypsin | Promega | Cat# V5280 |
| Proteasome Inhibitor MG-132 | EMD Millipore | Cat# 474790 |
| Beta-Amyloid Peptide (1–42) (human) Sequence: DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA | Abcam | Cat# ab120301 |
| Teplow’s Amyloid β-Protein (1–42) (scrambled II). Sequence: YHAGVDKEVVFDEGAGAEHGLAQKIVRGFGVSDVSMIHINLF | BACHEM | Cat# 4104168 |
| Critical Commercial Assays | ||
| 10% Tris-Glycine Gels | Thermo Fisher Scientific | Cat# XV00100PK20 |
| 16% Tris-Glycine Gelts | Thermo Fisher Scientific | Cat# XP00162BOX |
| SimplyBlue™ SafeStain Kit | Thermo Fisher Scientific | Cat# LC6065 |
| Odyssey Blocking Buffer | LI-COR | Cat# 92740003 |
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat# 23225 |
| Pierce microBCA Assay Kit | Thermo Fisher Scientific | Cat# 23235 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat# 74804 |
| Tandem Ubiquitin Binding Entities (TUBEs) | LifeSensors | Cat# UM401M |
| Amyloid Beta 42 Human ELISA Kit | Thermo Fisher Scientific | Cat# KHB3441 |
| Amyloid Beta 42 Human ELISA Kit, Ultrasensitive | Thermo Fisher Scientific | Cat# KHB3544 |
| Oligomeric Amyloid-beta ELISA Kit | Biosensis | Cat# BEK-2215–1P |
| Software and Algorithms | ||
| Graphpad | Graphpad | Version 8 |
| PANTHER | PANTHER | Version 15.0 |
| OpenMIMS FIJI Plugin | NIH | Version 3.0 |
| pClamp | Axon Instruments | Version 10 |
| Mini Analysis | Synaptosoft | Version 6 |
| Integrated Proteomics Pipeline(IP2) | Integrated Proteomics Applications, Inc | Version 5.0.1 |
| ProLuCID/SEQUEST algorithm | Yate Laboratory, The Scripps Research Institute | Version 3.1 |
| Census | Yate Laboratory, The Scripps Research Institute | Version 6.0 |
| RawExtract | Yate Laboratory, The Scripps Research Institute | Version 1.9.9 |
| Other | ||
| Gelatin-Subbed Microscope Slides | SouthernBiotech | Cat# SLD01-CS |
| Fluoromount-G | SouthernBiotech | Cat# 0100–01 |
| Jupiter C18 resin capillary | Phenomenex | N/A |
| Fusion Orbitrap mass spectrometer | Thermo Finnigan | N/A |
| Precellys 24 | Bertin Technology | N/A |
| nanoViper™ Analytical Column | Thermo Scientific | Cat# 164570 |
| HyperSep™ SCX Cartridges | ThermoFisher Scientific | Cat# 60108–420 |
| HyperSep™C18 Cartridges | Thermo Fisher Scientific | Cat# 60108–302 |
| Pierce C18 Spin Columns | Thermo Fisher Scientific | Cat# 89873 |
| RP IMCSTIPS® | IMCS | Cat# 04T-H6R05-1-20-96 |
| Dynabeads M-270 Epoxy | Invitrogen | Cat# 14301 |
| Synergy HTX multi-mode microplate reader | Biotek | N/A |
| Raw Proteomic Datasets | Massive | MSV000085004 |
ACKNOWLEDGMENTS
We thank Robert Vassar, Robert Kalb, Dimitri Krainc and members of the Savas laboratory for insightful discussions. We thank Hongkai Wang for contributing to the Thioflavin S imaging. Some of the imaging in this article was derived from the Northwestern University Center for Advanced Microscopy, which is generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.
This work was supported by the NIH, Mechanisms of Aging and Dementia T32AG20506 and F31AG059364 to T.J.H.; R01AG061787, R01AG061865, and R21NS107761 to J.N.S.; R01MH099114 to AC; MH061876 to E.R.C., who is an Investigator of the Howard Hughes Medical Institute; AG061829 and the MCDB Neurodegenerative Disease Fund to M.H.B.S., as well as, the Cure Alzheimer’s Fund and a pilot award from the CNADC of Northwestern Medicine to J.N.S.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Nine supplemental figures, four supplemental tables, and three supplemental videos can be found in the supplemental information.
REFERENCES
- Aidaralieva NJ, Kamino K, Kimura R, Yamamoto M, Morihara T, Kazui H, Hashimoto R, Tanaka T, Kudo T, Kida T, et al. (2008). Dynamin 2 gene is a novel susceptibility gene for late-onset Alzheimer disease in non-APOE-epsilon4 carriers. J Hum Genet 53, 296–302. [DOI] [PubMed] [Google Scholar]
- Akila Parvathy Dharshini S, Taguchi YH, and Michael Gromiha M (2019). Exploring the selective vulnerability in Alzheimer disease using tissue specific variant analysis. Genomics 111, 936–949. [DOI] [PubMed] [Google Scholar]
- Andreoli A, Gognalons MV, Abensur J, Olivier C, Muhlebach AL, Tricot L, and Garrone G (1989). [Long-term clinical follow-up (2 years) of 78 patients hospitalized in a crisis situation]. Schweiz Arch Neurol Psychiatr (1985) 140, 439–458. [PubMed] [Google Scholar]
- Arthur CP, Dean C, Pagratis M, Chapman ER, and Stowell MH (2010). Loss of synaptotagmin IV results in a reduction in synaptic vesicles and a distortion of the Golgi structure in cultured hippocampal neurons. Neuroscience 167, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, et al. (2020). Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 105, 975–991 e977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, and Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Bao H, Goldschen-Ohm M, Jeggle P, Chanda B, Edwardson JM, and Chapman ER (2016). Exocytotic fusion pores are composed of both lipids and proteins. Nat Struct Mol Biol 23, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, and Chapman ER (2008). Analysis of the synaptotagmin family during reconstituted membrane fusion. Uncovering a class of inhibitory isoforms. J Biol Chem 283, 21799–21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, and Holtzman DM (2008). Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 321, 1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante HA, Gonzalez AE, Cerda-Troncoso C, Shaughnessy R, Otth C, Soza A, and Burgos PV (2018). Interplay Between the Autophagy-Lysosomal Pathway and the Ubiquitin-Proteasome System: A Target for Therapeutic Development in Alzheimer’s Disease. Front Cell Neurosci 12, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butko MT, Savas JN, Friedman B, Delahunty C, Ebner F, Yates JR 3rd, and Tsien RY (2013). In vivo quantitative proteomics of somatosensory cortical synapses shows which protein levels are modulated by sensory deprivation. Proc Natl Acad Sci U S A 110, E726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchi S, Raao B, Masliah D, Rosenthal SB, Sasik R, Fisch KM, De Jager PL, Bennett DA, and Rissman RA (2019). Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. Cell Rep 28, 1103–1116 e1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhangani D, Endo F, Amanullah A, Upadhyay A, Watanabe S, Mishra R, Yamanaka K, and Mishra A (2016). Mahogunin ring finger 1 confers cytoprotection against mutant SOD1 aggresomes and is defective in an ALS mouse model. Neurobiol Dis 86, 16–28. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, and Holtzman DM (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, and Holtzman DM (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Cociorva D, D LT, and Yates JR (2007). Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics Chapter 13, Unit 13 14. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- DeBoer SR, Dolios G, Wang R, and Sisodia SS (2014). Differential release of beta-amyloid from dendrite- versus axon-targeted APP. J Neurosci 34, 12313–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete D, Lombino F, Liu X, and D’Adamio L (2014). APP is cleaved by Bace1 in pre-synaptic vesicles and establishes a pre-synaptic interactome, via its intracellular domain, with molecular complexes that regulate pre-synaptic vesicles functions. PLoS One 9, e108576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Greaves J, Wiegand UK, Matskevich I, Bodammer G, Apps DK, Shipston MJ, and Chow RH (2003). Functional and spatial segregation of secretory vesicle pools according to vesicle age. Nature 422, 176–180. [DOI] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fanutza T, Del Prete D, Ford MJ, Castillo PE, and D’Adamio L (2015). APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. Elife 4, e09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, and Contractor A (2015). Epac2 Mediates cAMP-Dependent Potentiation of Neurotransmission in the Hippocampus. J Neurosci 35, 6544–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornasiero EF, Mandad S, Wildhagen H, Alevra M, Rammner B, Keihani S, Opazo F, Urban I, Ischebeck T, Sakib MS, et al. (2018). Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat Commun 9, 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Chen J, Katsumata Y, Fardo DW, Wang WX, Artiushin S, Price D, Anderson S, Patel E, Zhu H, et al. (2018). Detergent Insoluble Proteins and Inclusion Body-Like Structures Immunoreactive for PRKDC/DNA-PK/DNA-PKcs, FTL, NNT, and AIFM1 in the Amygdala of Cognitively Impaired Elderly Persons. J Neuropathol Exp Neurol 77, 21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam V, D’Avanzo C, Berezovska O, Tanzi RE, and Kovacs DM (2015). Synaptotagmins interact with APP and promote Abeta generation. Mol Neurodegener 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, and Xu H (1999). Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K (2015). The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18, 794–799. [DOI] [PubMed] [Google Scholar]
- Higginbotham L, Dammer EB, Duong DM, Modeste E, Montine TJ, Lah JJ, Levey AI, and Seyfried NT (2019). Network Analysis of a Membrane-Enriched Brain Proteome across Stages of Alzheimer’s Disease. Proteomes 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E, Dechairo B, John S, Van Eerdewegh P, Soares H, et al. (2011). Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One 6, e16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, et al. (2019). Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-betaCTF (C99). J Neurosci 39, 5255–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, and Malinow R (2003). APP processing and synaptic function. Neuron 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Katsumata Y, Fardo DW, Bachstetter AD, Artiushin SC, Wang WX, Wei A, Brzezinski LJ, Nelson BG, Huang Q, Abner EL, et al. (2020). Alzheimer Disease Pathology-Associated Polymorphism in a Complex Variable Number of Tandem Repeat Region Within the MUC6 Gene, Near the AP2A2 Gene. J Neuropathol Exp Neurol 79, 3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET (2006). Synaptic vesicle reuse and its implications. Neuroscientist 12, 57–66. [DOI] [PubMed] [Google Scholar]
- Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, and Steinhauser ML (2014). Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab 20, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolainen MA, Nyman TA, Aittokallio T, and Pirttila T (2010). An update on clinical proteomics in Alzheimer’s research. J Neurochem 112, 1386–1414. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, and Tessier-Lavigne M (2019). A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 104, 1022. [DOI] [PubMed] [Google Scholar]
- Lanke V, Moolamalla STR, Roy D, and Vinod PK (2018). Integrative Analysis of Hippocampus Gene Expression Profiles Identifies Network Alterations in Aging and Alzheimer’s Disease. Front Aging Neurosci 10, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, and Checler F (2016). Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol 132, 257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK, and Lee SH (2008). Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci 28, 7945–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI, and Peng J (2004). Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem 279, 37061–37068. [DOI] [PubMed] [Google Scholar]
- Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, and Tycko R (2013). Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly PT, Cai F, and Song W (2011). Detection of neuritic plaques in Alzheimer’s disease mouse model. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCoss MJ, Wu CC, Liu H, Sadygov R, and Yates JR 3rd (2003). A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem 75, 6912–6921. [DOI] [PubMed] [Google Scholar]
- MacCoss MJ, Wu CC, Matthews DE, and Yates JR 3rd (2005). Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal Chem 77, 7646–7653. [DOI] [PubMed] [Google Scholar]
- Manavalan A, Mishra M, Feng L, Sze SK, Akatsu H, and Heese K (2013). Brain site-specific proteome changes in aging-related dementia. Exp Mol Med 45, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Honer WG, Mallory M, Voigt M, Kushner P, Hansen L, and Terry R (1994a). Topographical distribution of synaptic-associated proteins in the neuritic plaques of Alzheimer’s disease hippocampus. Acta Neuropathol 87, 135–142. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, and Terry R (1994b). Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 174, 67–72. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (1997). Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120, 343–352. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda AM, Herman M, Cheng R, Nahmani E, Barrett G, Micevska E, Fontaine G, Potier MC, Head E, Schmitt FA, et al. (2018). Excess Synaptojanin 1 Contributes to Place Cell Dysfunction and Memory Deficits in the Aging Hippocampus in Three Types of Alzheimer’s Disease. Cell Rep 23, 2967–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, Fagan AM, Hampel H, Mielke MM, Mikulskis A, et al. (2018). Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol 136, 821–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI, and Cuervo AM (2014). Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci 69 Suppl 1, S33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Fardo DW, and Katsumata Y (2020). The MUC6/AP2A2 Locus and Its Relevance to Alzheimer’s Disease: A Review. J Neuropathol Exp Neurol 79, 568–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, and Wong PC (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34, 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, and LaFerla FM (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- Ovsepian SV, O’Leary VB, Zaborszky L, Ntziachristos V, and Dolly JO (2018). Synaptic vesicle cycle and amyloid beta: Biting the hand that feeds. Alzheimers Dement 14, 502–513. [DOI] [PubMed] [Google Scholar]
- Palop JJ, and Mucke L (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17, 777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, and Yates JR 3rd (2008). A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods 5, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, and Gygi SP (2003). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Perdigao C, Barata MA, Araujo MN, Mirfakhar FS, Castanheira J, and Guimas Almeida C (2020). Intracellular Trafficking Mechanisms of Synaptic Dysfunction in Alzheimer’s Disease. Front Cell Neurosci 14, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl DA, Bomba-Warczak E, Watson ET, Bradberry MM, Peterson TA, Basu T, Frelka A, Evans CS, Briguglio JS, Basta T, et al. (2019). Synaptotagmin 17 controls neurite outgrowth and synaptic physiology via distinct cellular pathways. Nat Commun 10, 3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, and Saido TC (2014). Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17, 661–663. [DOI] [PubMed] [Google Scholar]
- Salas IH, Callaerts-Vegh Z, D’Hooge R, Saido TC, Dotti CG, and De Strooper B (2018). Increased Insoluble Amyloid-beta Induces Negligible Cognitive Deficits in Old AppNL/NL Knock-In Mice. J Alzheimers Dis 66, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, and Saido TC (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36, 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathe G, Na CH, Renuse S, Madugundu AK, Albert M, Moghekar A, and Pandey A (2019). Quantitative Proteomic Profiling of Cerebrospinal Fluid to Identify Candidate Biomarkers for Alzheimer’s Disease. Proteomics Clin Appl 13, e1800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbeck AD, Gangolli M, Reitz SJ, Salyards MH, Kim SH, Hemingway C, Gratuze M, Makkapati T, Kerschensteiner M, Holtzman DM, et al. (2020). SEQUIN Multiscale Imaging of Mammalian Central Synapses Reveals Loss of Synaptic Connectivity Resulting from Diffuse Traumatic Brain Injury. Neuron 107, 257–273 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Park SK, and Yates JR 3rd (2016). Proteomic Analysis of Protein Turnover by Metabolic Whole Rodent Pulse-Chase Isotopic Labeling and Shotgun Mass Spectrometry Analysis. Methods Mol Biol 1410, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Ribeiro LF, Wierda KD, Wright R, DeNardo-Wilke LA, Rice HC, Chamma I, Wang YZ, Zemla R, Lavallee-Adam M, et al. (2015). The Sorting Receptor SorCS1 Regulates Trafficking of Neurexin and AMPA Receptors. Neuron 87, 764–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR 3rd, and Hetzer MW (2012). Extremely long-lived nuclear pore proteins in the rat brain. Science 335, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Wang YZ, DeNardo LA, Martinez-Bartolome S, McClatchy DB, Hark TJ, Shanks NF, Cozzolino KA, Lavallee-Adam M, Smukowski SN, et al. (2017). Amyloid Accumulation Drives Proteome-wide Alterations in Mouse Models of Alzheimer’s Disease-like Pathology. Cell Rep 21, 2614–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, and Neher E (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23, 399–409. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, and Hardy J (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, and Hyman BT (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, et al. (2010). Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. (2016). The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Sevlever D, Zou F, Ma L, Carrasquillo S, Crump MG, Culley OJ, Hunter TA, Bisceglio GD, Younkin L, Allen M, et al. (2015). Genetically-controlled Vesicle-Associated Membrane Protein 1 expression may contribute to Alzheimer’s pathophysiology and susceptibility. Mol Neurodegener 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. (2017). A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst 4, 60–72 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, and Walsh DM (2009). Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Burre J, and Sudhof TC (2012). Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci Transl Med 4, 147ra113. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, and Sudhof TC (2012). Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser ML, Bailey AP, Senyo SE, Guillermier C, Perlstein TS, Gould AP, Lee RT, and Lechene CP (2012). Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 481, 516–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR 3rd (2002). DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, and Hetzer MW (2013). Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker WC, Weber T, and Chapman ER (2004). Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438. [DOI] [PubMed] [Google Scholar]
- UniProt C (2015). UniProt: a hub for protein information. Nucleic Acids Res 43, D204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. [DOI] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, et al. (2014). Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. [DOI] [PubMed] [Google Scholar]
- Xiong F, Ge W, and Ma C (2019). Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimers Dement 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Xu J, Patassini S, Rustogi N, Riba-Garcia I, Hale BD, Phillips AM, Waldvogel H, Haines R, Bradbury P, Stevens A, et al. (2019). Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun Biol 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim J, Kim HY, Ryoo N, Lee S, Kim Y, Rhim H, and Shin YK (2015). Amyloid-beta Oligomers May Impair SNARE-Mediated Exocytosis by Direct Binding to Syntaxin 1a. Cell Rep 12, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino Y, Mori T, Yoshida T, Yamazaki K, Ozaki Y, Sao T, Funahashi Y, Iga JI, and Ueno SI (2016). Elevated mRNA Expression and Low Methylation of SNCA in Japanese Alzheimer’s Disease Subjects. J Alzheimers Dis 54, 1349–1357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data statement: The raw MS data have been deposited in MassIVE online database (MSV000085004) (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and upon acceptance will be available in the ProteomeXchange online database (http://www.proteomexchange.org/)
Code statement: This paper does not report original code
Scripts statement: Scripts were not used to generate the figures reported in this paper
Any addition information required to reproduce this word is available from the Lead Contact