

Abstract
The contribution of altered mitochondrial Ca2+ handling to metabolic and functional defects in type 2 diabetic (T2D) mouse hearts is not well understood. In this study, we show that the T2D heart is metabolically inflexible and almost exclusively dependent on mitochondrial fatty acid oxidation as a consequence of mitochondrial calcium uniporter complex (MCUC) inhibitory subunit MCUb overexpression. Using a recombinant endonuclease-deficient Cas9-based gene promoter pulldown approach coupled with mass spectrometry, we found that MCUb is upregulated in the T2D heart due to loss of glucose homeostasis regulator nuclear receptor corepressor 2 repression, and chromatin immunoprecipitation assays identified peroxisome proliferator–activated receptor α as a mediator of MCUb gene expression in T2D cardiomyocytes. Upregulation of MCUb limits mitochondrial matrix Ca2+ uptake and impairs mitochondrial energy production via glucose oxidation by depressing pyruvate dehydrogenase complex activity. Gene therapy displacement of endogenous MCUb with a dominant-negative MCUb transgene (MCUbW246R/V251E) in vivo rescued T2D cardiomyocytes from metabolic inflexibility and stimulated cardiac contractile function and adrenergic responsiveness by enhancing phospholamban phosphorylation via protein kinase A. We conclude that MCUb represents one newly discovered molecular effector at the interface of metabolism and cardiac function, and its repression improves the outcome of the chronically stressed diabetic heart.
Introduction
Diabetes is a complex metabolic syndrome afflicting a growing number of people globally (1). Although associated with a variety of pathologies, cardiovascular complications are the major cause of mortality and morbidity in patients with diabetes (2,3). Diabetes results in a cardiomyopathy characterized by decreased cardiomyocyte function, increased myocardial fatty acid utilization, decreased glucose utilization, increased myocardial oxygen consumption, and decreased cardiac efficiency (4,5), primarily as the result of changes in metabolism and Ca2+ handling within the myocyte (3,6,7). Furthermore, diabetic cardiomyopathies are characterized by depressed sympathoadrenergic signaling, which regulates chronotropic, inotropic, and lusitropic cardiac function to meet changing tissue perfusion needs (8–10).
In cardiomyocytes, the cAMP-dependent protein kinase A (PKA) mediates sympathoadrenergic signaling and influences cardiac function by phosphorylating many targets responsible for regulating Ca2+ handling and homeostasis (10). PKA-dependent phosphorylation of the l-type calcium channel complex located in the plasmalemma results in increased Ca2+ influx into myocytes; phosphorylation of phospholamban (PLN) on the longitudinal sarcoplasmic reticulum (SR) results in increased SR Ca2+ ATPase (SERCA2a) activity, which in return stimulates SR Ca2+ loading, while ryanodine receptor 2 phosphorylation at the junctional SR enhances SR Ca2+ release (10). Collectively, these posttranslational modifications increase cardiac contractility (positive inotropy) and occur in subcellular microdomains where PKA is anchored by A-kinase anchoring proteins and where the levels of ATP and cAMP are also shaped by adenyl cyclases (ACs) and phosphodiesterases (10).
Evidence increasingly supports the hypothesis that mitochondria modulate Ca2+ influx/efflux and alter the amplitude and the spatiotemporal distribution of intracellular Ca2+ levels (7). It is therefore becoming more widely accepted that mitochondrial Ca2+ (mCa2+) plays an important role in both stimulating mitochondrial respiratory function and contributing to whole-cell Ca2+ homeostasis.
Ca2+ passively diffuses through the outer mitochondrial membrane and is transported into the mitochondrial matrix by the mCa2+ uniporter complex (MCUC). The MCUC consists of pore-forming, scaffold, and regulatory subunits. MCU is the main pore-forming subunit (11–15); the MCU paralog MCUb is the other pore-forming subunit. Specific, conserved differences in two amino acids (W246 and V251) make MCUb an inhibitory subunit (12).
While the contribution of MCUb to MCUC regulation has been recently characterized in the context of acute cardiac stress (16,17), no information related to chronic cardiac stress has yet been published. In this study, we examined the effects related to excessive MCUb expression in type 2 diabetic (T2D) cardiomyocytes. We identified the etiology of MCUb upregulation and studied the mitochondrial and extramitochondrial mechanisms that are impacted by its overexpression. To ameliorate the negative effects from MCUb overexpression, we generated a dominant-negative MCUb (dnMCUb) mutant (MCUbW246R/V251E) and used this to shed light on the complex relationships between mitochondrial metabolism, cardiac function, and responsiveness to sympathoadrenergic stimulation.
Research Design and Methods
All investigations conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication number 85–23, revised 1996) and were conducted in accordance with guidelines established by the University of California, San Diego Institutional Animal Care and Use Committee. T2D was induced in 12-week-old C57BL/6 mice with a high-fat diet (HFD; 60% calories from fat) (Envigo) and a single streptozotocin (STZ) injection of 75 mg/kg, as previously reported (18). T2D progression was monitored over the course of 6 months (Supplementary Table 1). In vivo cardiac-enhanced and liver-detargeted AAV-9.45 transgene delivery was performed by direct jugular vein injection, as described (6). MCUb was cloned from mouse heart, and the dnMCUb was generated by introducing two point mutations (CTG>CAG and TGT>AGT), resulting in MCUbW246R/V251E. Empty AAV (AAV-ctr), AAV encoding C-terminal FLAG-tagged murine MCUbW246R/V251E (AAV-dnMCUb), or mitochondria-targeted MityCAM (AAV-MityCAM) were injected 18–20 weeks after STZ injection (3 × 1011 genome copies in 100 μL). Experiments were carried out 6 months after STZ/HFD treatment and 4 weeks after AAV injection. AAV-ctr was delivered in both nondiabetic control (CTR) and T2D mice, and no significant effects were reported (6) (Supplementary Fig. 1). Loss-of-function studies were carried out in 12-week-old mice receiving AAV-MCUb 4 weeks postinjection. On average, ∼4 × 105 transgene copies/µg guide DNA were detected in experimental hearts receiving gene therapy.
Ca2+-tolerant cardiomyocytes were isolated from ventricular tissue as described (6), plated in DMEM (4.5 g/L glucose) supplemented with blebbistatin (5 μg/mL) on laminin and adenovirus or siRNA/Lipofectamine RNAiMAX (Invitrogen) precoated surfaces, and cultured for 24 h to allow protein manipulation to occur. Pharmacological inhibition studies or experiments requiring cell fixation were initiated 24 h after plating.
The MCUb gene promoter pulldown approach was adapted from Tsui et al. (19). Using an MCUb promoter-specific guide RNA (sgRNA) and a recombinant 7XHis–endonuclease-deficient Cas9 (dCas9)–3XFLAG expressed and purified from Escherichia coli BL21 Rosetta (DE3), cardiomyocytes isolated from six control and six T2D mouse hearts were pooled, fixed, lysed, and sheared, and equal amounts of total protein were combined with preformed sgRNA-dCas9 ribonucleoprotein complexes. FLAG-tagged dCas9 and coprecipitated genomic DNA fragments and proteins were purified using anti-FLAG M2-conjugated agarose beads. Quantitative RT-PCR (RT-qPCR) was used for MCUb promoter analysis, and liquid chromatography coupled with tandem mass spectroscopy (LC-MS/MS) analysis of precipitated proteins was conducted.
dCas9-coprecipitated proteins and lysed cardiomyocyte preparations were trypsin-digested and analyzed by ultra-high-pressure LC-MS/MS as described (20). Phosphopeptides were enriched with TiO2 and metal-based resins (IMAC). Those peptides not retained by TiO2 and IMAC were used to describe the cardiomyocyte proteome, and peptides from all experiments were analyzed. A complete list of proteins identified in proteomics, phosphoproteomics, and dCas9 coprecipitation is reported in Supplementary Spreadsheet 1.
Chromatin immunoprecipitation (ChIP) was performed using equal amounts of pooled, sheared chromatin from fixed/lysed cardiomyocytes and anti–nuclear receptor corepressor 2 (Ncor2), –peroxisome proliferator–activated receptor (PPAR) γ, -PPARα, or normal anti-rabbit/mouse IgG control (5 μg each). Genomic DNA fragments isolated from pulldowns were analyzed by qPCR analysis for MCUb promoter fragments at positions −150 and −1,000. Relative quantification of the MCUb gene promoter fragments was calculated using the ΔΔ threshold cycle method, referring to the amount of MCUb gene promoter pulled down by a nonspecific sgRNA-dCas9 ribonucleoprotein complex (dCas9 plus human MCU sgRNA).
Strand-specific total RNA-sequencing (RNA-seq) libraries were prepared from rRNA-depleted RNA from mouse hearts and sequenced on an Illumina NextSeq 500. Functional gene enrichment and pathway analysis were performed using Metascape (21). Supplementary Spreadsheet 1 lists all genes with transcripts showing significant differences (adjusted P value ≤0.05) between groups.
Glucose and fatty acid metabolism were measured in isolated working hearts using [5-3H]glucose and [9,10-3H]palmitate as described (6). Mitochondrial ATP production was inferred by monitoring the linear increase of d-luciferin bioluminescence when luciferase was supplied with ATP from mitochondria.
Ca2+ uptake was measured in isolated mitochondria by monitoring Calcium Green-5N (Thermo Fisher Scientific) fluorescence with 10 μmol/L CaCl2 pulses. The MCU inhibitor Ru360 was used to ensure MCUC-dependent mCa2+ uptake specificity. Cytosolic Ca2+ (cCa2+) transients were recorded in isolated cardiomyocytes with Indo-1 AM (22); mCa2+ transients were recorded with MityCAM (6). Transients were recorded upon electric stimulation at 0.3 Hz to preserve and extend contractile performance in vitro.
Echocardiography was performed on lightly anesthetized mice using the VisualSonics Vevo 2100 ultrasound system (18). Cardiomyocyte contractility was assessed by edge detection of cultured cardiomyocytes (6).
Left ventricular developed pressure (LVP) was assessed ex vivo as previously described (23). Hearts were paced at 400 bpm, and resulting pressure waves were analyzed for pressure development in basal or stimulated (1 μmol/L isoproterenol) conditions as reported (24).
Cardiac creatine kinase (CK), SERCA2a and PKA activity, and cardiac cAMP levels were measured in flash-frozen and pulverized cardiac tissues. Total CK activity was determined following the described method (25). SERCA2a SR Ca2+ uptake was assessed as reported by Kranias and Hajjar (26). PKA activity was measured similarly as described for cGMP-dependent protein kinase (PKG) activity and described in Schwaerzer et al. (27) by substituting cGMP with cAMP and DT-2 for PKG activity inhibition. cAMP levels were measured using a commercially available cAMP ELISA kit (ab65355; Abcam) following the manufacturer’s instructions.
A total of 2–8 mg of pure cardiac mitochondrial protein lysates was fractionated by gel filtration using Sephadex G-200 (Sigma-Aldrich), and presence of FLAG-tagged proteins was investigated by Western blot in fractions corresponding to those in which protein standards (Bio-Rad Laboratories) from 670 to 50 kDa were collected.
Results are presented as mean ± SD. One-way ANOVA followed by appropriate post hoc multiple-comparisons test or unpaired Student t test was used for comparison between two or more groups, respectively. Gaussian distribution was always assumed. Adjusted P value ≤0.05 was considered to be statistically significant.
Data and Resource Availability
All data generated or analyzed for this study will be made fully available to researchers upon any reasonable request. No applicable resources were generated or analyzed during the current study.
Results
Hearts from T2D mice were studied 6 months after administering a single STZ injection and beginning HFD feeding, allowing sufficient time for diabetes-induced decreased cardiac function to develop (18). T2D mice used in these studies were glucose intolerant, mildly hyperglycemic and insulin intolerant, severely hyperlipidemic, and characterized by hyperinsulinemia (Supplementary Table 1 and Supplementary Fig. 2), in line with prior reports (18).
MCUb Overexpression in T2D Cardiomyocytes Results From Metabolic Maladaptations
We initially characterized the transcriptome and proteome of the T2D heart model in order to identify altered pathways. RNA-seq and LC-MS/MS analysis were performed to quantify levels of gene transcripts extracted from whole hearts and protein peptides from isolated cardiomyocytes. Volcano plots and gene ontology analysis are shown for RNA-seq and LC-MS/MS (Fig. 1A and B). Both analyses converged in identifying the T2D heart as primarily dependent on fatty acid metabolism. Moreover, analysis of transcripts and proteins that were less abundant in T2D versus CTR mice highlighted that glucose and Ca2+ homeostasis were highly impacted by the onset and development of T2D. In both analyses, the pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4) was more abundant in T2D. PDK4 is known to inhibit the pyruvate dehydrogenase complex (PDC) by phosphorylating the E1α subunit, diminishing the conversion of pyruvate to acetyl-CoA, and thereby contributing to glucose metabolism regulation (28,29). We confirmed the RNA-seq and proteomics results by measuring rates of mitochondrial glucose and palmitic acid oxidation in isolated working hearts from CTR and T2D mice (Fig. 1C). Significantly lower glucose oxidation and significantly higher palmitic acid oxidation rates in T2D versus CTR hearts suggest a metabolic switch. Furthermore, and in line with the omics findings, we observed significantly decreased PDC activity (Fig. 1D) and increased PDC E1α S293 phosphorylation (Fig. 1E).
Figure 1.
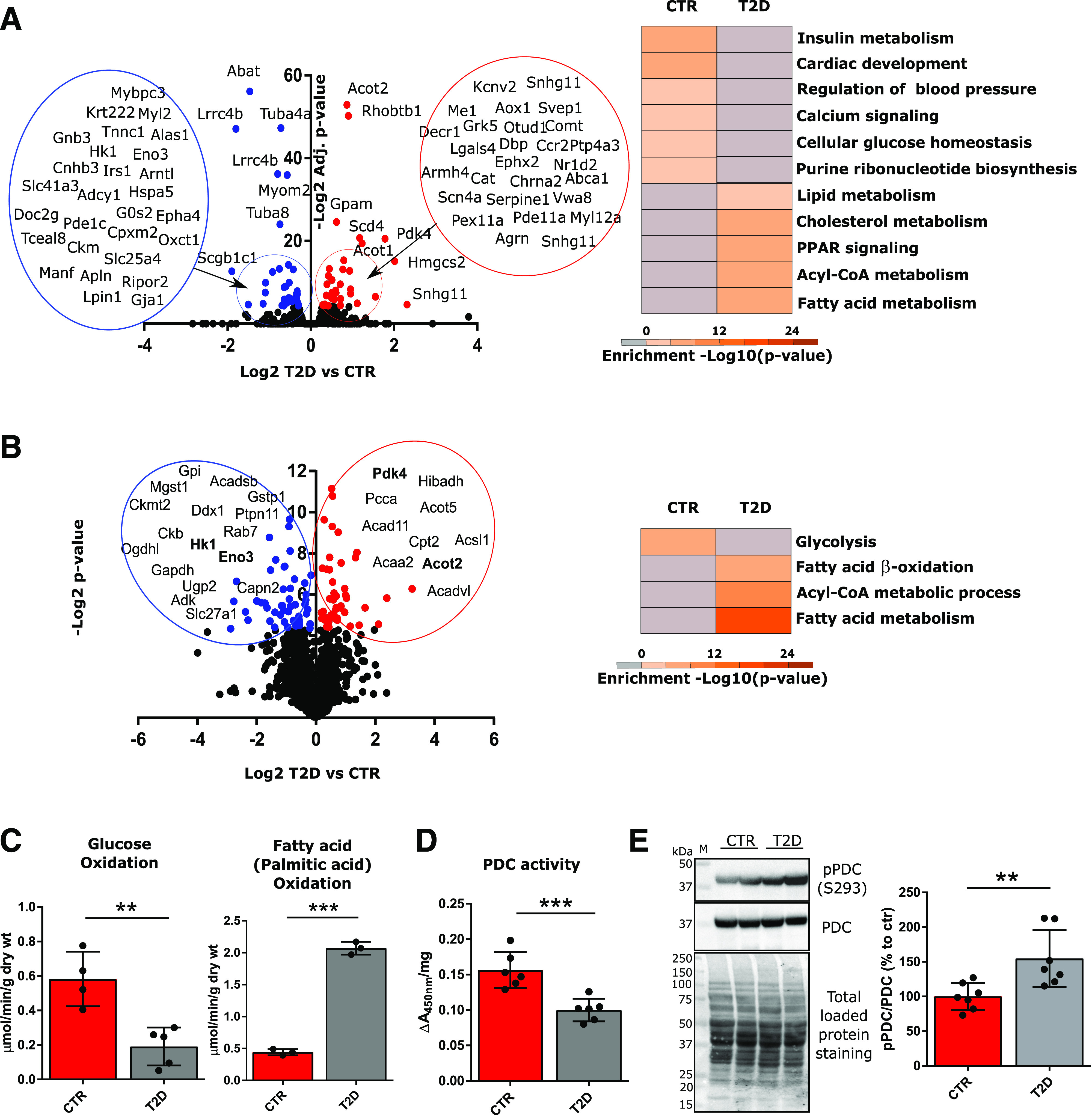
Metabolic inflexibility of the T2D heart. Transcriptomics analysis (A) was conducted on protein-coding RNAs extracted from two whole hearts per group, and proteomics analysis (B) was conducted on the phosphopeptide-depleted peptides (taken from the flow through following TiO2 and IMAC-based phosphopeptide enrichment) from isolated cardiomyocytes from two hearts per group. Volcano plots display Log2 differences between T2D vs. CTR, plotted vs. −Log2-adjusted (adjusted t test P values [RNA-seq] or −Log2 t test P values [proteomics]). Red dots correspond to significantly upregulated genes and proteins; blue dots correspond to significantly downregulated genes/proteins in T2D vs. CTR. Gene ontology analysis was performed with Metascape on genes with P values ≤0.05, and significantly enriched pathways are shown (B, right). Significantly up/downregulated proteins for which mRNAs were also significantly up/downregulated (from RNA-seq analysis) are highlighted in bold (B, left). C: Glucose and fatty acid (palmitate) oxidation in working heart preparations, reported as micromoles per minute per gram dry weight (wt). Oxidation rates were measured in four hearts per group. D: PDC activity was measured in mitochondrial homogenates using a microplate assay kit following the reduction of NAD+, coupled to the reduction of a reporter dye (450 nm). Data were normalized per milligram of mitochondrial protein content. Enzyme activities are representative of mitochondrial preparations from six hearts per group. E: Western blot analysis of phosphorylated PDC (E1α subunit, S293) in mitochondrial lysates. Total PDC levels and total protein staining of PVDF membranes, as well as summarized densitometric band analysis, are shown. Western blots are representative of seven mice. Data are presented as mean ± SD. Unpaired Student t test was used for statistical analysis. **P < 0.01; ***P < 0.001. M, molecular weight marker.
We previously reported that the metabolic inflexibility characterizing the type 1 diabetic heart was rescued by restoring MCU protein levels, thus stimulating mCa2+ trafficking and mitochondrial glucose oxidation, as a probable consequence of decreased PDC E1α S293 phosphorylation and increased PDC activity (6). Building on these findings in the type 1 diabetic heart and guided by Metascape (21) analysis of RNA-seq showing disrupted Ca2+ homeostasis in the T2D heart, we measured MCU protein levels in isolated T2D cardiomyocytes but did not observe a significant change (Fig. 2A). However, we did detect a significant increase in protein and mRNA levels of the MCU paralog MCUb (Fig. 2A and B). MCUb is reported to inhibit MCUC-based mCa2+ import (12), and its relevance to cardiac pathophysiology in the context of acute stress has been recently investigated (18,19). In T2D cardiomyocytes, we measured diminished mitochondrial matrix Ca2+ import rates (Fig. 2C) and consequently lower matrix mCa2+ levels (Fig. 2D), which were associated with diminished mitochondrial ATP production rates (Fig. 2E) and impaired cardiomyocyte and whole heart contractility (Fig. 2F and G). To establish a cause–effect relationship between elevated MCUb levels and mitochondrial metabolic inflexibility in T2D hearts, we further performed loss-of-function studies in normal, nondiabetic mice by overexpressing MCUb via AAV. Twelve-week-old mice underwent in vivo gene delivery of AAV-MCUb, and 4 weeks after injection, the presence of MCUb in high-molecular-weight mitochondrial complexes was determined by fractioning mitochondrial lysates with Sephadex G-200 (Sigma-Aldrich) gel filtration. Western blot analysis of FLAG-tagged MCUb in fractions relative to molecular weight from 50 to >670 kDa is reported in Fig. 3A and shows that MCUb was detected in the void volume fractions (>200 kDa), thus suggesting MCUb incorporation in high-molecular-weight complexes. mCa2+ uptake rates were then measured in freshly isolated mitochondria with Calcium Green (Fig. 3B). Overexpression of MCUb in CTR hearts reduced Ca2+ uptake into mitochondria, led to significantly increased phosphorylated PDC levels (Fig. 3C), and decreased mitochondrial glucose oxidation (Fig. 3D), thus impairing energy production (Fig. 3E), as occurred in T2D hearts.
Figure 2.
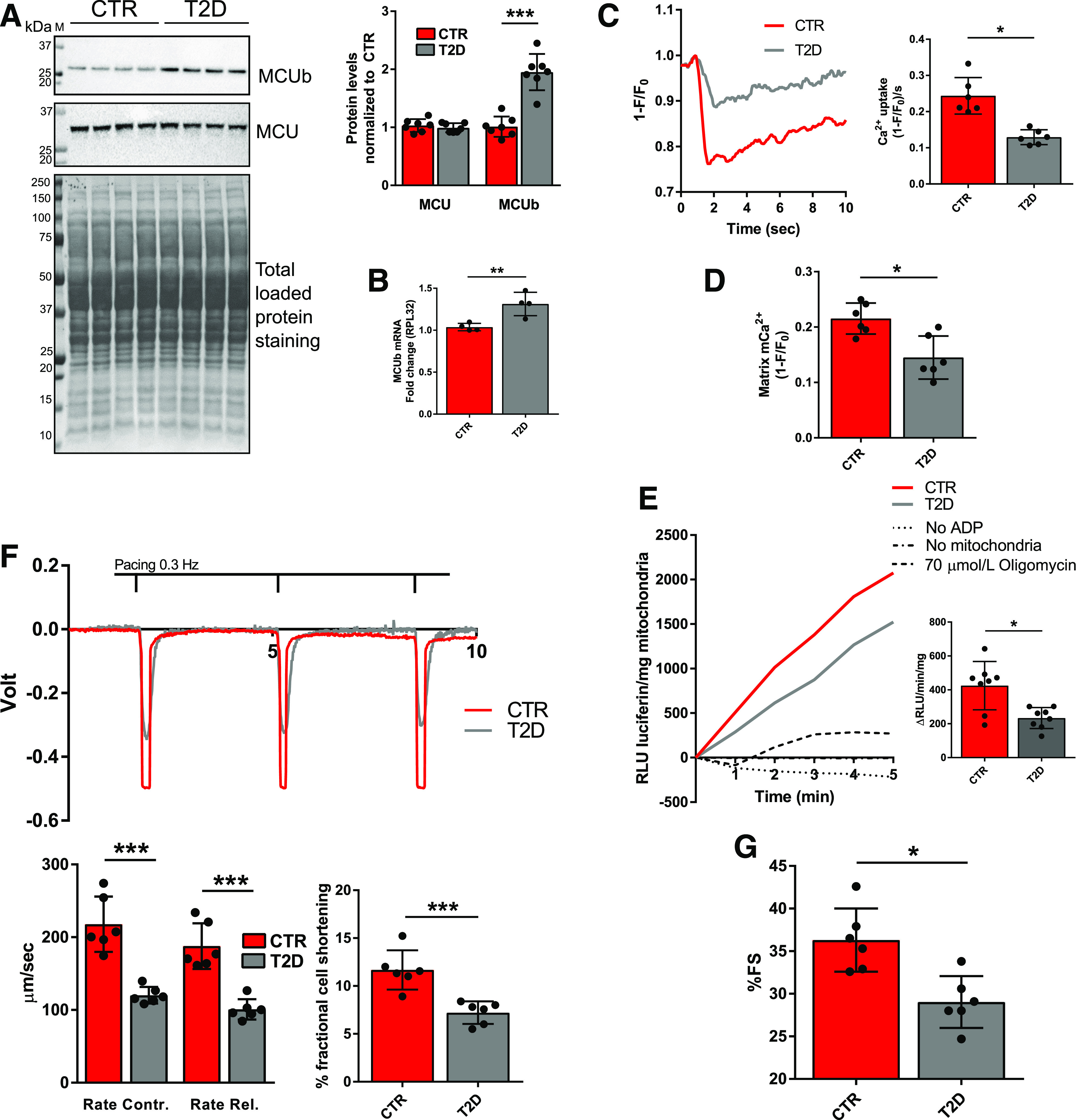
MCUb overexpression in the T2D heart is associated with decreased mCa2+ import rate and level, decreased mitochondrial ATP production, and decreased cardiac function. A: Western blot analysis of MCUb and MCU in isolated cardiomyocyte homogenates. Total protein staining of polyvinylidene difluoride membranes was used as loading control. Summarized densitometric band analysis is shown. Western blots are representative of cardiomyocyte preparations obtained from seven mice per group. B: RT-qPCR analysis of MCU mRNA levels in CTR and T2D mouse hearts (four per group). 60S ribosomal protein L32 (RPL32) was used as the housekeeping gene for normalization. C: Representative mCa2+ transients measured in isolated cardiomyocytes from CTR and T2D. mCa2+ transients were assessed in paced-contracting cardiomyocytes (0.3 Hz) using MityCAM, which was delivered in vivo by injection of AAV-MityCAM. mCa2+ uptake rates were quantified and are reported in the bar graph as averaged maximal drop of MityCAM fluorescence (F) induced by electrical stimulation over normalized stable MityCAM fluorescence (F0 = 1) at time zero prior to initiation of electrical stimulation per second ([1 − F/F0]/s). Data are representative of recordings from cells isolated from six mice per group. D: mCa2+ levels reported as averaged maximal drop of MityCAM fluorescence (F) induced by electrical stimulation over stable MityCAM fluorescence (F0 = 1) at time zero prior to initiation of electrical stimulation (1 − F/F0). Data are representative of recordings from cells isolated from six mice per group. E: ATP production, expressed as the rate of linear increase of luciferin luminescence (RLU luciferin) per milligram of mitochondria. Rates were analyzed in mitochondrial preparations from eight mice per group in the presence of 5 mmol/L ADP, 0.15 mg/mL d-luciferin, and 1 μg/mL recombinant firefly luciferase. As negative controls, the assays were conducted in absence of either mitochondria or ADP or in the presence of 70 μmol/L of the ATP synthase inhibitor oligomycin. F: Cardiomyocyte contractility was assessed in vitro by edge detection and analyzed with Felix32 software. Rate of contraction (Contr.) (+dL/dt) and rate of relaxation (Rel.) (−dL/dt) are reported in micrometers per second upon conversion of volts (V) into millimeters (mm). Percentage fractional cell shortening was calculated as (ΔV[Vmax − V0]/cell size [mm]) ∗ 100. Data are representative of recordings from paced-contracting (0.3 Hz) isolated cells from six mice per group. G: In vivo heart function was assessed by M-mode echocardiography; percentage of fractional shortening (%FS) is shown. Data are representative of six mice per group. Data are presented as mean ± SD. Unpaired Student t test was used for statistical analysis of A and B. One-way ANOVA followed by Dunnett multiple-comparisons test was used for statistical analysis in C–G. *Adjusted P < 0.05; **P < 0.01; ***P < 0.001. M, molecular weight marker.
Figure 3.
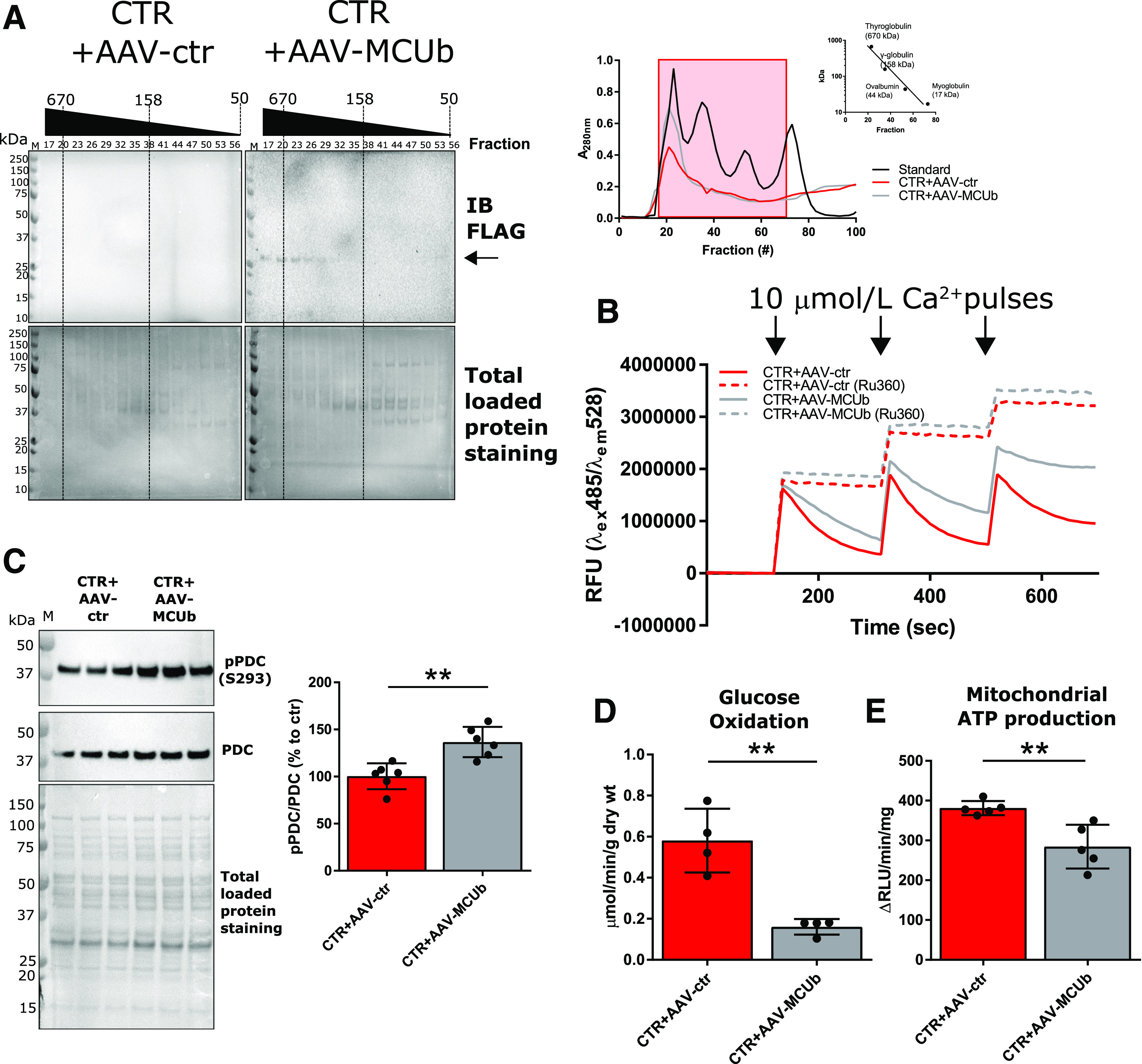
Overexpression of MCUb in normal CTR mouse hearts impacts mitochondrial metabolism, recapitulating the T2D-like phenotype. A: Western blot analysis of FLAG-MCUb in mitochondrial homogenates prefractioned with Sephadex G-200 (left). Absorbance at 280 nm was used to monitor retention time of mitochondrial homogenates and known molecular standards (right; Log10 kDa vs. retention time reported in inset graph). Fractions (from 17 to 56) corresponding to standards ranging from ∼670 to ∼100 kDa (red-boxed at right) were analyzed. Total loaded protein stained on polyvinylidene difluoride membranes was used as loading control. B: Representative Calcium Green-5N traces for mCa2+ import assays. A total of 100 µg of isolated mitochondria was incubated in respiratory buffer, and 10 μmol/L Ca2+ were pulsed (arrows). As control, mitochondria were incubated with 100 μmol/L of the MCU inhibitor Ru360. Data are representative of six mice per group. C: Western blot analysis of pPDC (E1α subunit, S293) in mitochondrial lysates (N = 6). Total PDC levels and total protein staining of polyvinylidene difluoride membranes, as well as summarized densitometric band analysis, are shown. D: Glucose oxidation in working heart preparations, reported as micromoles per minute per gram dry weight (wt). Oxidation rates were measured in four hearts per group. E: Mitochondrial ATP production rates was measured using mitochondria isolated from five hearts per group. Unpaired Student t test was used for statistical analysis. **P < 0.01. em, emission; ex, excitation; IB, immunoblot; M, molecular weight marker.
To investigate the causes of MCUb overexpression in the T2D heart, and given increased MCUb mRNA levels in T2D, we probed for regulators of MCUb gene expression. We adapted the locus pulldown approach published by Tsui et al. (19), performing MCUb promoter pulldown with an endonuclease-deficient clustered regularly interspaced short palindromic repeats (CRISPR)–associated protein 9 (dCas9). The MCUb gene promoter was pulled down from sheared chromatin extracted from fixed cardiomyocytes using a ribonucleoprotein complex formed from a recombinant dCas9 and an sgRNA (Supplementary Fig. 3). Proteins coprecipitated with dCas9 were analyzed by LC-MS/MS and sorted for subcellular localization and gene ontology categories (Fig. 4). Nuclear proteins responsible for gene expression regulation are highlighted, and several proteins were differentially associated with the MCUb promoter in CTR and T2D cardiomyocytes. Nuclear receptor corepressor 2 (Ncor2, also known as SMRT) and coactivator of the CREBP SRCAP were associated with the MCUb promoter in CTR, but not in T2D cardiomyocytes. In contrast, RNA polymerase III (POL3B) and histone-lysine N-methyltransferase enzyme ASH1L (aka KMT2H) were associated with the MCUb promoter in T2D, but not CTR cardiomyocytes.
Figure 4.
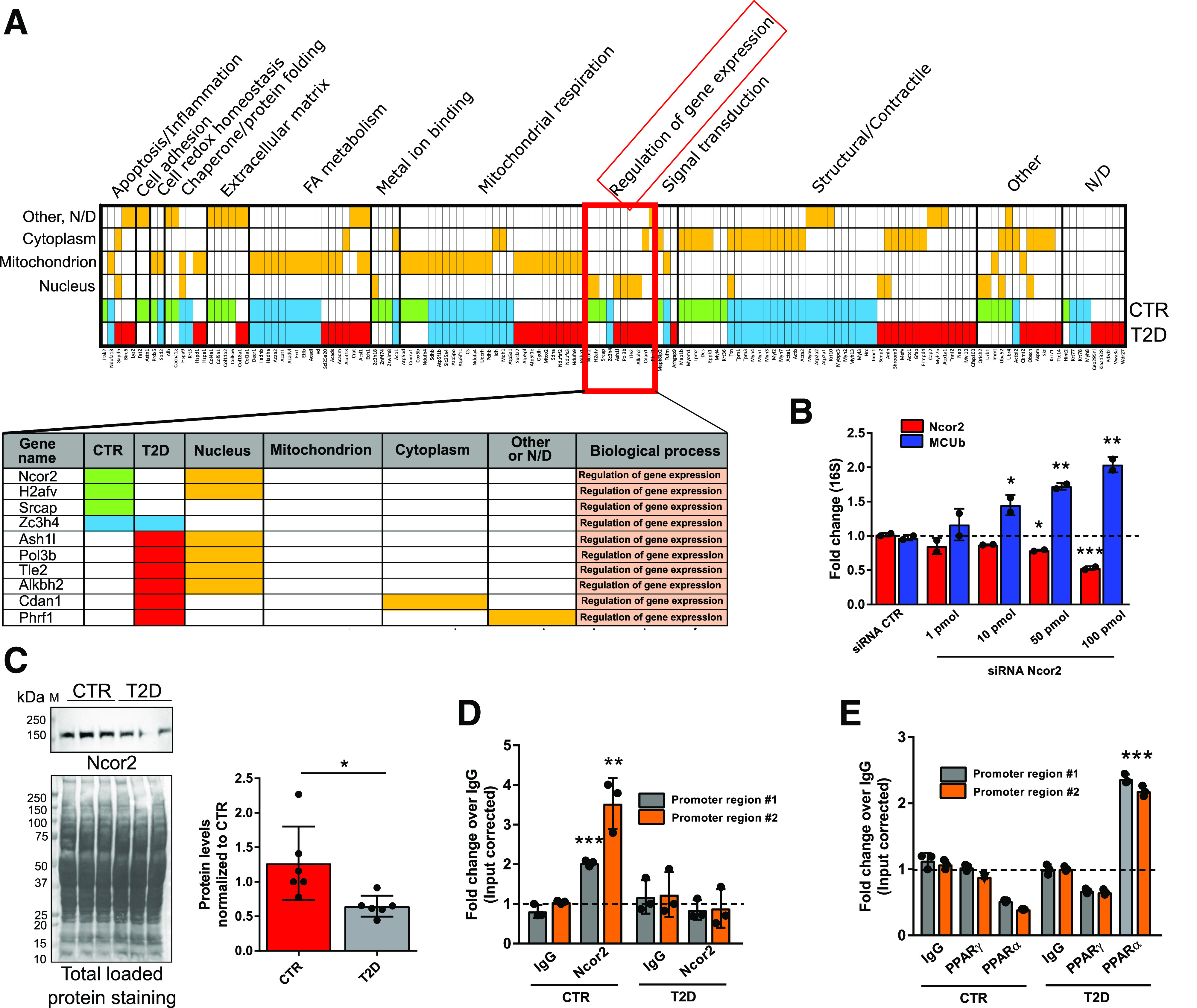
The Ncor2 modulates MCUb gene expression. The MCUb promoter was pulled down from lysed cardiomyocytes isolated from six pooled CTR and six pooled T2D hearts (description in research design and methods and in Supplementary Fig. 3). A: LC-MS/MS analysis of MCUb promoter-guided dCas9-coprecipitated proteins. Proteins detected only in CTR (green), only in T2D (red), or in both (blue) were sorted per subcellular localization and biological process (orange). Proteins related to the biological process “Regulation of gene expression” are highlighted. B: In vitro siRNA-mediated Ncor2 silencing. Cardiomyocytes from CTR hearts were transfected with Ncor2-specific or scrambled siRNA and cultured for 24 h. RT-qPCR analysis of MCUb and Ncor2 mRNA levels is reported. 16S rRNA (16S) was used as the housekeeping gene for normalization. The experiment was replicated two times. C: Ncor2 protein levels were investigated in total heart lysates from six mice. One representative Western blot and summarized densitometric band analysis from all blots are shown. Ncor2 band density in T2D samples was normalized to total protein membrane staining in the corresponding region of the blot. Ncor2 (D) and PPARγ/PPARα (E) ChIPs were performed using isolated cardiomyocytes from three CTR and T2D hearts. Myocytes were prepared as detailed in the Supplementary Material and pooled together before ChIP assays were conducted. As control, chromatin was immunoprecipitated using normal anti-mouse or anti-rabbit IgG. Presence of MCUb promoter fragments was investigated with two sets of primers annealing to ∼150-bp regions at the interface of promoter region #1 and ∼1 kb upstream from the first MCUb gene exon (promoter region #2), respectively. Data are presented as mean ± SD. One-way ANOVA followed by Dunnett multiple-comparisons test was used for statistical analysis in B. Unpaired Student t test was used for statistical analysis of C–E. *Adjusted P (or P) < 0.05; **adjusted P (or P) < 0.01; ***adjusted P (or P) < 0.001. FA, fatty acid; M, molecular weight marker; N/D, not determined.
Ncor2 functions as a critical metabolic modulator by mediating nuclear receptor signaling repression during high-fat feeding, thus avoiding aberrant lipid accumulation (30). The presence of Ncor2 on the MCUb promoter in cardiomyocytes from CTR, and not T2D, suggests that Ncor2-mediated MCUb transcription repression may be absent in T2D. It also suggests that MCUb transcription could be mediated by the PPARs. PPAR activity is intimately connected with nuclear receptor corepressors (31), and RNA-seq found upregulated PPAR signaling in T2D hearts (Fig. 1A).
To confirm the LC-MS/MS data obtained from MCUb promoter pulldown experiments and to test these hypotheses, we knocked down Ncor2 with siRNA in cultured cardiomyocytes from CTR hearts. RT-qPCR analysis showed a dose-dependent upregulation of MCUb with increasing Ncor2 siRNA (Fig. 4B). Furthermore, the levels of Ncor2 were found decreased in T2D hearts versus CTR (Fig. 4C), and ChIP studies showed Ncor2 binding to the MCUb promoter in CTR, but not T2D, cardiomyocytes (Fig. 4D), while PPARα, but not PPARγ, interacts with the same MCUb promoter regions in T2D cardiomyocytes, but not CTR (Fig. 3E).
A dnMCUb Mutant Rescues the T2D Heart
Arginine 251 (R251) and glutamic acid 256 (E256) in MCU were previously shown to confer Ca2+ influx inhibitory properties to the MCUC when mutated to the amino acids found at homologous positions in MCUb (tryptophan and valine, respectively) (12). Inverting this strategy, we substituted the highly conserved, hydrophobic residues tryptophan 246 (W246) and valine 251 (V251) in MCUb with the hydrophilic R and E. These amino acids are present at homologous positions in the MCU protein sequence. The resulting mutant MCUbW246R/V251E was FLAG-tagged and expressed in neonatal cardiomyocytes by adenoviral transduction, and mitochondrial matrix Ca2+ import rates were measured (Supplementary Fig. 4A and B). As previously reported, overexpression of a FLAG-tagged MCUb reduced mCa2+ uptake velocity (12,16,17). In contrast, MCUbW246R/V251E behaved as a dnMCUb that significantly increased mCa2+ uptake velocity, similar to that measured in cells expressing a FLAG-tagged MCU. Further characterization studies aimed to evaluate incorporation of dnMCUb into MCUC were carried out in isolated T2D cardiomyocytes transduced with Adv-dnMCUb. Interactions between dnMCUb and MCUC proteins were visualized in situ by proximity ligation assay (PLA) (Supplementary Fig. 4C). PLA demonstrated interaction between dnMCUb and MCU, EMRE, MICU1, and, to a lesser extent, MICU2, thus confirming that dnMCUb incorporates into the MCUC supercomplex. Similar studies were conducted in cardiomyocytes from T2D hearts transduced with either Adv-MCU or Adv-MCUb (Supplementary Fig. 4C, right panel). Interestingly, whereas exogenous MCU was found to interact with MCU, EMRE, MICU1, and MICU2, exogenous MCUb did not interact with MICU1. Levels of FLAG-tagged and endogenous proteins in isolated cardiomyocyte preparations undergoing PLA were determined by Western blot to assure presence of both targets used in the PLA (Supplementary Fig. 4D). Subsequently, we generated AAV-dnMCUb viral vectors and delivered them in vivo. AAV-dnMCUb or AAV-ctr (empty) was administered via jugular vein injection to T2D mice (3 × 1011 viral particles), 5 months post-STZ/HFD, as reported (6). One month after AAV administration, dnMCUb transgene expression was assessed in cardiomyocytes. Immunofluorescence staining of the FLAG-tagged dnMCUb shows efficient cardiomyocyte transduction (Supplementary Fig. 4E), and MCUb immunoprecipitates from fixed cardiomyocyte lysates revealed increased interaction between dnMCUb and MICU1/MICU2 in T2D plus AAV-dnMCUb hearts, but not T2D plus AAV-ctr (Supplementary Fig. 4F), thus confirming PLA results in Supplementary Fig. 4C. Consequently, dnMCUb was found incorporated into high-molecular-weight complexes by Sephadex G-200 fractionation of mitochondrial lysates (void volume fractions >200 kDa) (Fig. 5A).
Figure 5.
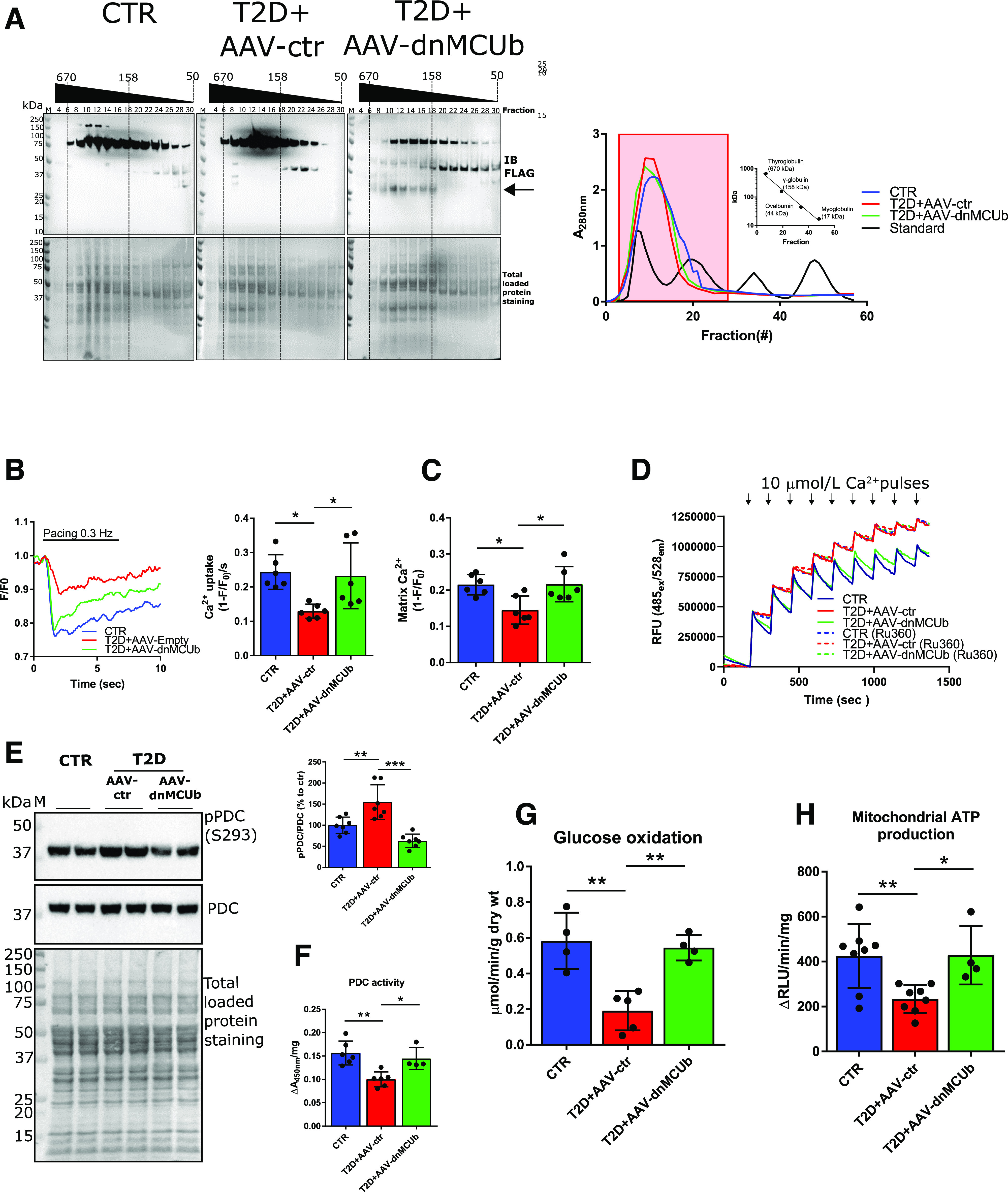
dnMCUb gene therapy rectifies mCa2+ handling, mitochondrial oxidative metabolism, and energy production. A: Western blot analysis of FLAG-dnMCUb in mitochondrial homogenates prefractioned with Sephadex G-200 (left). Absorbance at 280 nm was used to monitor retention time of mitochondrial homogenates and known molecular standards (A, right; Log10 kDa vs. retention time reported in inset graph). Fractions (from 4 to 30) corresponding to standards ranging from ∼670 to ∼100 kDa (red-boxed at right) were analyzed. Total loaded protein stained on PVDF membranes was used as loading control. Data are representative of mitochondria isolated from three pooled hearts per group. B: Representative mCa2 transients measured in cardiomyocytes isolated from CTR, T2D + AAV-ctr, and T2D + AAV-dnMCUb (N = 6). C: mCa2+ uptake rates and mCa2+ levels were quantified and are shown in the bar graphs. D: Representative Calcium Green-5N traces for mCa2+ import assays. A total of 300 µg of isolated mitochondria was incubated in respiratory buffer, and 10 μmol/L Ca2+ was pulsed (arrows). As control, mitochondria were incubated with 100 μmol/L of the MCU inhibitor Ru360. Data are representative of four mice per group. E: Western blot analysis of pPDC (E1α subunit, S293) in mitochondrial heart lysates (N = 6). Total PDC levels and total protein staining of polyvinylidene difluoride membranes as well as densitometric analysis are shown. F: PDC activity in mitochondrial homogenates (CTR and T2D + AAV-ctr, N = 6; T2D + AAV-dnMCUb, N = 4). G: Glucose oxidation in working heart preparations (N = 4/group). H: Mitochondrial ATP production rates (CTR and T2D + AAV-ctr, N = 8; T2D + AAV-dnMCUb, N = 4). Data are presented as mean ± SD. One-way ANOVA followed by Dunnett multiple-comparisons test was used for statistical analysis. *Adjusted P < 0.05; **adjusted P < 0.01; ***adjusted P < 0.001. IB, immunoblot; M, molecular weight marker; RFU, relative fluorescence units; RLU, relative luminescence unit; wt, weight.
After verifying dnMCUb incorporation into mitochondrial complexes and interaction with other MCUC members, we investigated the dnMCUb-driven effects on mitochondrial metabolism. AAV-dnMCUb brought T2D cardiomyocyte matrix Ca2+ import rates and mCa2+ levels toward CTR levels, measured with MityCAM (Fig. 5B and C) and Calcium Green (Fig. 5D). Furthermore, Western blot and kinetic studies displayed that dnMCUb treatment resulted in diminished PDC E1α phosphorylation (Fig. 5E) and rectified PDC activity (Fig. 5F). Mitochondria from T2D plus AAV-dnMCUb hearts also displayed signs of metabolic flexibility, as glucose oxidation rates were normalized, in spite of T2D (Fig. 5G), and displayed significantly higher mitochondrial energy production rates (Fig. 5H).
As Ca2+ is known to modulate kinase and phosphatase activity and thus indirectly control many downstream effects, we further characterized and compared the phosphoproteomes of cardiomyocytes from CTR, T2D, and T2D plus AAV-dnMCUb hearts. Phosphoproteomics profiling findings suggest a tendency toward restored protein phosphorylation in T2D plus AAV-dnMCUb cardiomyocytes, toward levels in CTR (Fig. 6A). Furthermore, gene ontology analysis of proteins with phosphopeptides twofold or more increased or decreased in T2D plus AAV-dnMCUb versus T2D plus AAV-ctr cardiomyocytes revealed phosphomodifications on proteins relevant for cardiac contraction by Ca2+ signaling, mitochondrial metabolism, mitochondrial respiration, and glucose and energy homeostasis. Phosphoproteomic profiling confirmed that dnMCUb treatment resulted in diminished PDC E1α phosphorylation in T2D cardiomyocytes at many sites (S15, S232, S293, S300, and S331), including the site recognized in Western blot analysis (Fig. 5E; S293). Furthermore, the analysis revealed significantly increased phosphorylation of PLN (S/T 16/17), as well as both cytosolic (M-type; T166) and mitochondrial (S-type; S319) CKs, the former being confirmed in Western blot analysis (Fig. 6B) and found associated with stimulated SERCA2a-dependent cCa2+ uptake into SR (Fig. 6C: SR speed of Ca2+ uptake measured with Indo-1) (Fig. 6D: SERCA2a activity), while the latter associated with increased global cardiac CK activity (Fig. 6E). In line with improved cCa2+ handling and cellular energy homeostasis, T2D plus AAV-dnMCUb cardiomyocytes also showed significantly boosted contractile function (Fig. 6F), and echocardiographic studies indicated significantly increased percentage of fractional shortening, despite absence of morphometric changes (Table 1), suggesting that the beneficial dnMCUb-driven effects were not caused by cardiac remodeling. In line with this evidence, T2D cardiomyocytes did not show significant upregulation of cardiac remodeling–associated genes or proteins (Supplementary Fig. 5).
Figure 6.

Phosphoproteomic analysis reveals dnMCUb gene therapy significantly improves cardiac contractility by modulating PLN phosphorylation via PKA. A: Scatter plot shows Log2 differences in phosphopeptide abundance in cardiomyocytes isolated from CTR, T2D + AAV-ctr, and T2D + AAV-dnMCUb (N = 2 mice/group). The dashed red line indicates a trend toward restoration in T2D + AAV-dnMCUb toward CTR levels (left). Log2 differences between T2D + AAV-ctr vs. T2D + AAV-dnMCUb were plotted over −Log2 t test P values (volcano plots at center). Gene ontology analysis between groups (Log 2 < −0.5 or >0.5; right) was conducted with Metascape. Key phosphorylation sites on PDC, cardiac PLN, and CK (S-type is mitochondrial, and M-type is cytosolic) are highlighted. B: One representative Western blot of phosphorylated PLN (S16/T17) and SERCA2a in homogenates from cardiomyocytes isolated from four mice per group is shown. Total loaded protein stained on polyvinylidene difluoride membranes was used as loading control to normalize pPLN densitometric band analysis. C: cCa2+ transients and time of Indo-1 fluorescence decay (t decay; N = 5/group). D: SERCA2a activity (CTR and T2D + AAV-ctr, N = 6; T2D + AAV-dnMCUb, N = 4). E: CK activity measured in whole cardiac lysates (CTR, N = 6; T2D + AAV-ctr, N = 4; and T2D + AAV-dnMCUb, N = 4). F: Cardiomyocyte contractility assessed by edge detection. Representative profiles of paced-contracting (0.3 Hz) cardiomyocytes isolated from CTR (N = 6), T2D + AAV-ctr (N = 6), and T2D + AAV-dnMCUb (N = 3) (left). Rates of contraction and relaxation as well as percentage fractional cell shortening are reported (F, right). G: A graphical representation of the most recurrent serine (S)–centered phosphorylation consensus motifs is generated based on cardiomyocytes isolated from T2D + AAV-dnMCUb vs. T2D + AAV-ctr. H: Western blots show the levels of SERCA2a and pPLN in cardiomyocytes isolated from T2D and CTR mice and transduced ex vivo with either Adv-ctr or Adv-dnMCUb for 24 h. I: Western blots from cardiomyocytes treated as in H after subsequent pharmacological inhibition of PKA (H89), PKG (RP8-PET), or CAMKII (KN-93) was induced for 1 h with 100 μmol/L inhibitor. Western blots are representative of at least three independent experiments. J: PKA activity was measured in whole heart lysates (CTR N = 5, T2D + AAV-ctr N = 7, T2D + AAV-dnMCUb N = 6). Activity was measured in presence or absence of cAMP to determine basal and total activity, respectively. K: Cardiac cAMP levels (N = 5/group). L: LVP was measured in Langendorff preparations under basal physiological and adrenergic-stimulating (1 μmol/L isoproterenol [Iso]) conditions. Data are representative of six mice per group. Data are presented as mean ± SD. One-way ANOVA followed by Dunnett multiple-comparisons test was used for statistical analysis in B, C, E, J, and K. Two-way ANOVA followed by Sidak and Tukey multiple comparisons was used in I and L. *Adjusted P < 0.05; **adjusted P < 0.01; ***adjusted P < 0.001. M, molecular weight marker; rel., relative.
Table 1.
Echocardiography
| CTR | T2D + AAV-ctr | T2D + AAV-dnMCUb | Adjusted P values | ||
|---|---|---|---|---|---|
| T2D + AAV-ctr vs. CTR | T2D + AAV-ctr vs. T2D + AAV-dnMCUb | ||||
| BW (g) | 36 ± 3.6 | 55.17 ± 4.7 | 51.67 ± 2.6 | <0.0001*** | 0.2655 |
| HR M-mode (bpm) | 648.67 ± 50 | 596.17 ± 55 | 629.33 ± 20 | 0.1477 | 0.4155 |
| LVIDd (mm) | 4.05 ± 0.32 | 3.14 ± 0.24 | 3.25 ± 0.38 | 0.0008*** | 0.8104 |
| LVIDs (mm) | 2.59 ± 0.32 | 2.22 ± 0.08 | 2.00 ± 0.23 | 0.0425* | 0.2701 |
| %FS | 36.3 ± 3.4 | 29.03 ± 2.8 | 38.03 ± 4.7 | 0.014* | 0.0031** |
| IVSd (mm) | 0.75 ± 0.04 | 1.1 ± 0.07 | 0.99 ± 0.09 | <0.0001*** | 0.0569 |
| LVPWd (mm) | 0.71 ± 0.03 | 1.01 ± 0.11 | 0.94 ± 0.09 | <0.0001*** | 0.3847 |
| LVIDd/LVPWd | 5.75 ± 0.58 | 3.16 ± 0.65 | 3.48 ± 0.63 | <0.0001*** | 0.6519 |
| LVDd/BW | 0.114 ± 0.017 | 0.057 ± 0.006 | 0.06 ± 0.008 | <0.0001*** | 0.6411 |
| LVM(d) (mg) | 108.8 ± 13 | 120.43 ± 7.8 | 111.87 ± 17.7 | 0.3178 | 0.5135 |
| LVM/BW (mg/g) | 3.08 ± 0.61 | 2.19 ± 0.09 | 2.17 ± 0.35 | 0.0073** | 0.9957 |
Echocardiographic parameters were measured in six mice per group. Data are presented as mean ± SD. One-way ANOVA followed by Dunnett multiple-comparisons test was used for statistical analysis.
BW, body weight; %FS, percentage fractional shortening; HR, heart rate; IVSd, interventricular septal end diastole; LVDd, diastolic left ventricular dimension; LVIDd, left ventricular internal diameter in diastole; LVIDs, left ventricular internal diameter in systole; LVM(d), left ventricular mass in diastole; LVPWd, left ventricular posterior wall thickness in diastole.
Adjusted P < 0.05; **adjusted P < 0.01; ***adjusted P < 0.001.
In an attempt to explain how dnMCUb-driven restoration of mCa2+ levels modulates PLN phosphorylation, thus contributing to improved SR-dependent cCa2+ handling, we further analyzed the phosphopeptides detected in cardiomyocytes from T2D plus AAV-dnMCUb hearts. A graphical representation of the predominant motifs is presented in Fig. 6G. The motif “R..S” is a consensus sequence for three major kinases: PKA, PKG, and the Ca2+/calmodulin-dependent protein kinase II (CAMKII) (32,33). To discern which kinase was responsible for PLN phosphorylation following dnMCUb treatment, we manipulated PLN phosphorylation status in cardiomyocytes from T2D hearts transduced with Adv-dnMCUb and treated with pharmacological inhibitors of PKA (H89) (34), PKG (RP8-PET) (35), or CAMKII (KN-93) (24). In line with phosphoproteomics and Western blot analyses, phosphorylated PLN (pPLN) was lower in T2D versus CTR cardiomyocytes and restored following Adv-dnMCUb transduction (Fig. 6H). Interestingly, only the PKA inhibitor was capable of reversing the dnMCUb effect on pPLN, thus strongly suggesting that dnMCUb modulates PLN phosphorylation via PKA (Fig. 6I). This was further indicated by analysis of PKA activity (Fig. 6J) and levels of the PKA activator cAMP (Fig. 6K) in which both were significantly higher in lysates from T2D hearts when AAV-dnMCUb was administered. Finally, and in line with the idea that our treatment had beneficial effects on PKA activity and signaling, in Langendorff preparations, T2D plus AAV-dnMCUb hearts showed significantly increased LVP upon adrenergic stimulation, while T2D plus AAV-ctr hearts were unresponsive to this stimulation (Fig. 6L).
Discussion
Changes in cardiac metabolism and mitochondrial function precede cardiac dysfunction, indicating that metabolic remodeling is an early event in the progression of diabetes (36,37). The adult myocardium uses fatty acids as the primary source of energy (38); however, ATP production processes such as glucose oxidation that are more oxygen efficient are nonetheless important, especially upon increased tissue perfusion needs (10,39). The switch from one energy substrate to another is regulated by metabolic signals that act as allosteric regulators of key mitochondrial and cytosolic enzymes (Randle cycle; [28]). Dephosphorylation and activation of the PDC is one of the key control points of metabolic flexibility (28,37), and mCa2+ has been shown to directly and indirectly activate the PDC (6,28,29,40), thus stimulating mitochondrial glucose oxidation (6,28,29) and mitochondrial ATP production (41).
In this study, we found that MCUb levels were increased in T2D likely as a consequence of lower Ncor2 levels, diminished Ncor2 transcriptional repression, and increased PPARα transcriptional activity, as identified with our dCas9-driven MCUb promoter pulldown approach and further suggested by Ncor2 knockdown and ChIP experiments. In muscle, Ncor2 represses genes important for fatty acid catabolism (31) and associates with nuclear receptors, such as the PPARs, in the absence of their ligands to repress transcriptional activity (42). Studies indicate that diabetic hearts are associated with increased PPARα expression due to high circulating fatty acid levels and that cardiac PPARα activation increases expression of genes involved in fatty acid β-oxidation and suppresses glucose utilization by promoting PDC inhibition via PDK4-mediated phosphorylation (43,44). We propose that Ncor2 downregulation and the concomitant activation of PPARα is a mechanism by which the T2D heart metabolically adapts to glucose intolerance and hyperlipidemia via MCUb upregulation, thereby limiting mitochondrial matrix Ca2+ influx and favoring fatty acids over glucose for energy production (Fig. 7). We substantiated this hypothesis in loss-of-function studies (MCUb overexpressed in CTR hearts) and by expressing the dnMCUb transgene to counteract effects induced by endogenous MCUb overexpression. Overexpression of MCUb in CTR hearts recapitulated the T2D-like mitochondrial metabolic phenotype (decreased glucose oxidation and energy production via increased phosphorylated PDC [pPDC]). dnMCUb restored Ca2+ flux into the matrix and reverted the T2D heart metabolic inflexibility by reducing phosphorylation of the PDC E1α subunit, thus stimulating its activity, resulting in increased mitochondrial ATP production.
Figure 7.

Model for the onset of metabolic inflexibility and cardiac dysfunction in T2D hearts and proposed mechanism mediated by dnMCUb gene therapy. In response to diet alteration and glucose intolerance, a reduced level of Ncor2 induces PPARα-dependent MCUb overexpression, which alters mCa2+ handling and pushes the metabolic oxidative balance toward utilization of fatty acids as an almost unique source for energy production. Cardiac dysfunction and unresponsiveness to adrenergic stimulation are correlated to lower levels of ATP produced in mitochondria, with the concomitant reduced capability of the CK system to preserve the energy both inside (CK-S type) and outside (CK-M type) the mitochondrial compartment, possibly due to lower phosphorylation levels detected in both systems. Therapeutic intervention intended to inhibit the negative effects mediated by the MCUb-dependent reduced Ca2+ influx rates into the mitochondrial matrix, rectifies the global cardiac phosphoproteome toward normal levels, stimulates glucose oxidation by enhancing the activity of the PDC, and favors the conditions to produce and preserve more energy both inside and outside of mitochondria. Higher CK activity and AC-dependent cAMP levels are proposed as mechanisms that favor the conditions to stimulate the PKA-dependent phosphorylation of PLN, which translate into ameliorated cardiac function and adrenergic responsiveness.
Data presented in this study align with a developing hypothesis that mCa2+ handling coordinates ATP supply for cardiomyocyte contraction (6,39,45). Furthermore, our data demonstrate that restoring mCa2+ handling influences the phosphorylation status of many intra- and extramitochondrial proteins and are foundational for better understanding the mechanisms coupling mitochondrial energy production to cardiac function. In detail, we observed that improved mCa2+ import influences cCa2+ handling by enhancing SERCA2a activity via PKA-dependent modulation of PLN phosphorylation. Even though differences in SERCA2a and PKA activity as well as cAMP levels between T2D versus CTR were only trends, likely due to the disruption of the cAMP-PKA-PLN-SERCA microdomains in whole heart lysates, the dnMCUb treatment was accompanied by significantly increased SERCA2a and total PKA activity and cAMP levels and resulted in enhanced cardiac inotropic function at both basal and stimulated conditions. Interestingly, phosphoproteomic profiling identified significant phosphorylation of mitochondrial (S-type) and cytosolic (M-type) CK isoforms. CK enzyme activity is known to be regulated by phosphorylation (25), and CKs catalyze the reversible conversion of creatine plus ATP to phosphocreatine and ADP, functioning both as an ATP shuttle from mitochondria to cytosol and as primary cardiac energy reserve. Mice receiving the dnMCUb transgene showed rectified total CK activity. Hence, we propose that increased mitochondrial ATP levels as well as rectified CK shuttles favor conditions within cardiomyocytes to produce more cAMP, likely by activating ACs, and to sustain the activity of PKA, which in turn stimulates cCa2+ loading into the SR via PLN phosphorylation and contributes to rectify responsiveness to sympathoadrenergic stimulation (Fig. 7).
Taken together, our study identifies a new therapeutic target for amelioration of chronically stressed hearts characterized by metabolic inflexibility and impaired cardiac function.
Article Information
Funding. This work was supported by Veterans Health Administration Office of Research and Development Merit Review Award BX003429 (to W.H.D.), the P. Robert Majumder Charitable Foundation, and the University of California Institute for Mexico and the United States–Consejo Nacional de Ciencia y Tecnología grant CN 19-157 (to W.H.D.). Imaging studies were supported by National Institutes of Health grant NS047101 (to the University of California, San Diego Neuroscience Microscopy Imaging Core).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. F.C., B.T.S., and W.H.D. designed the study. F.C., J.S., D.E.C., S.H., A.D., T.D., J.A.S., C.W.B., and M.G. conducted experiments and analyzed data. F.C., B.T.S., and W.H.D. wrote the manuscript. W.H.D. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.13342229.
References
- 1.Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 2014;103:137–149 [DOI] [PubMed] [Google Scholar]
- 2.Belke DD, Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 2004;6:424–429 [DOI] [PubMed] [Google Scholar]
- 3.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014;57:660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus. Circ Res 2019;124:121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dillmann WH Diabetic cardiomyopathy. Circ Res 2019;124:1160–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suarez J, Cividini F, Scott BT, et al. . Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J Biol Chem 2018;293:8182–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang CH, Wei YH. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J Biomed Sci 2017;24:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West TM, Wang Q, Deng B, et al. . Phosphodiesterase 5 associates with β2 adrenergic receptor to modulate cardiac function in type 2 diabetic hearts. J Am Heart Assoc 2019;8:e012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q, Liu Y, Fu Q, et al. . Inhibiting insulin-mediated β2 adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation 2017;135:73–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Wang WE, Zhang X, et al. . Cardiomyocyte PKA ablation enhances basal contractility while eliminates cardiac β-adrenergic response without adverse effects on the heart. Circ Res 2019;124:1760–1777 [DOI] [PubMed] [Google Scholar]
- 11.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011;476:336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raffaello A, De Stefani D, Sabbadin D, et al. . The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 2013;32:2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancak Y, Markhard AL, Kitami T, et al. . EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013;342:1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vais H, Mallilankaraman K, Mak DD, et al. . EMRE is a matrix Ca(2+) sensor that governs gatekeeping of the mitochondrial Ca(2+) uniporter. Cell Rep 2016;14:403–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patron M, Checchetto V, Raffaello A, et al. . MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 2014;53:726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert JP, Luongo TS, Tomar D, et al. . MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 2019;140:1720–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huo J, Lu S, Kwong JQ, et al. . MCUb induction protects the heart from postischemic remodeling. Circ Res 2020;127:379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fricovsky ES, Suarez J, Ihm SH, et al. . Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol 2012;303:R689–R699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsui C, Inouye C, Levy M, et al. . dCas9-targeted locus-specific protein isolation method identifies histone gene regulators. Proc Natl Acad Sci U S A 2018;115:E2734–E2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guttman M, Betts GN, Barnes H, Ghassemian M, van der Geer P, Komives EA. Interactions of the NPXY microdomains of the low density lipoprotein receptor-related protein 1. Proteomics 2009;9:5016–5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Zhou B, Pache L, et al. . Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004;53:3201–3208 [DOI] [PubMed] [Google Scholar]
- 23.Belke DD, Swanson E, Suarez J, Scott BT, Stenbit AE, Dillmann WH. Increased expression of SERCA in the hearts of transgenic mice results in increased oxidation of glucose. Am J Physiol Heart Circ Physiol 2007;292:H1755–H1763 [DOI] [PubMed] [Google Scholar]
- 24.Grimm M, Ling H, Willeford A, et al. . CaMKIIδ mediates β-adrenergic effects on RYR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic β-adrenergic stimulation. J Mol Cell Cardiol 2015;85:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin G, Liu Y, MacLeod KM. Regulation of muscle creatine kinase by phosphorylation in normal and diabetic hearts. Cell Mol Life Sci 2009;66:135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 2012;110:1646–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwaerzer GK, Kalyanaraman H, Casteel DE, et al. . Aortic pathology from protein kinase G activation is prevented by an antioxidant vitamin B12 analog. Nat Commun 2019;10:3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Randle PJ Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 1998;14:263–283 [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) 2014;11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang S, Suh JM, Atkins AR, et al. . Corepressor SMRT promotes oxidative phosphorylation in adipose tissue and protects against diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A 2011;108:3412–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev 2013;27:819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem 1991;266:15555–15558 [PubMed] [Google Scholar]
- 33.Kemp BE, Pearson RB. Protein kinase recognition sequence motifs. Trends Biochem Sci 1990;15:342–346 [DOI] [PubMed] [Google Scholar]
- 34.Xin W, Li N, Cheng Q, Fernandes VS, Petkov GV. Constitutive PKA activity is essential for maintaining the excitability and contractility in guinea pig urinary bladder smooth muscle: role of the BK channel. Am J Physiol Cell Physiol 2014;307:C1142–C1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalyanaraman H, Schwaerzer G, Ramdani G, et al. . Protein kinase G activation reverses oxidative stress and restores osteoblast function and bone formation in male mice with type 1 diabetes. Diabetes 2018;67:607–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchanan J, Mazumder PK, Hu P, et al. . Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005;146:5341–5349 [DOI] [PubMed] [Google Scholar]
- 37.Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci 2004;1015:202–213 [DOI] [PubMed] [Google Scholar]
- 38.Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res 2017;113:411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luongo TS, Lambert JP, Yuan A, et al. . The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 2015;12:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denton RM Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 2009;1787:1309–1316 [DOI] [PubMed] [Google Scholar]
- 41.Wescott AP, Kao JPY, Lederer WJ, Boyman L. Voltage-energized calcium-sensitive ATP production by mitochondria. Nat Metab 2019;1:975–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol 2007;17:292–301 [DOI] [PubMed] [Google Scholar]
- 43.Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci 2017;24:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008;114:195–210 [DOI] [PubMed] [Google Scholar]
- 45.Rasmussen TP, Wu Y, Joiner ML, et al. . Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A 2015;112:9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]