

Summary
CD8+ T cell recognition of peptide epitopes plays a central role in immune responses against pathogens and tumors. However, the rules that govern which peptides are truly recognized by existing T cell receptors (TCRs) remain poorly understood, precluding accurate predictions of neo-epitopes for cancer immunotherapy. Here, we capitalize on recent (neo-)epitope data to train a predictor of immunogenic epitopes (PRIME), which captures molecular properties of both antigen presentation and TCR recognition. PRIME not only improves prioritization of neo-epitopes but also correlates with T cell potency and unravels biophysical determinants of TCR recognition that we experimentally validate. Analysis of cancer genomics data reveals that recurrent mutations tend to be less frequent in patients where they are predicted to be immunogenic, providing further evidence for immunoediting in human cancer. PRIME will facilitate identification of pathogen epitopes in infectious diseases and neo-epitopes in cancer immunotherapy.
Keywords: tumor immunology, immunogenicity, TCR recognition, neo-epitope predictions, immunoediting
Graphical Abstract
Highlights
Development of a predictor of immunogenic CD8 T-cell epitopes (PRIME)
PRIME shows improved prediction accuracy on neo-epitopes
PRIME reveals molecular determinants of TCR recognition
PRIME supports immunoediting of recurrent cancer mutations
Schmidt et al. develop a predictor of immunogenicity (PRIME) for CD8 T cell epitopes that captures antigen presentation on HLA molecules and TCR recognition. Their results reveal molecular determinants of TCR recognition and support immunoediting acting on recurrent cancer mutations.
Introduction
The CD8+ T cell immune response is key to recognize and kill infected and malignant cells. Over the last decade, the success of cancer immunotherapy demonstrated that harnessing and boosting CD8+ T-cell-mediated control and elimination of cancer cells is clinically relevant.1, 2, 3, 4 A remaining central question is why some peptides specifically expressed by infected or cancer cells and displayed on class I human leukocyte antigen (HLA-I) molecules are recognized by CD8+ T cells and elicit specific immune responses while others do not, as the former provide ideal targets for vaccines1,3,5 and T-cell-based therapies.2 Unfortunately, even the latest machine learning epitope prediction tools display low accuracy, with a precision typically around or lower than 5% when applied to neo-epitope predictions from mutations identified by exome sequencing in cancer.5, 6, 7, 8, 9
At the molecular level, CD8+ T cell recognition of peptide epitopes is based on a series of specific events.10 First, the peptide is cleaved from the source protein by the proteasome, is transported into the endoplasmic reticulum (ER) and binds to HLA-I molecules. HLA-I molecules bind 8- to 12-mer peptides, are mainly encoded by three genes (HLA-A/B/C), and are highly polymorphic across the human population. Upon stable binding, the peptide-HLA-I (pHLA) complex is presented on the surface of cells. Second, the T cell receptor (TCR) can bind to the pHLA complex, which initiates the formation of the immune synapse and ultimately can lead to the killing of infected or malignant cells.
Much work has been done to characterize the binding (either affinity or stability) and presentation of peptides on HLA-I molecules, and several HLA-I ligand predictors are publicly available.11, 12, 13, 14, 15, 16 These tools primarily focus on binding to HLA-I, which is the most selective and arguably the best understood step in antigen presentation. Several approaches demonstrated further improvement in predictions by integrating signals from cleavage and antigen transport, presentation hotspots, gene expression of the source protein, or clonality of the mutations for cancer neo-epitopes.16, 17, 18, 19, 20, 21 However, these additional features are not always experimentally available in typical studies of pathogen or cancer epitopes, and some can be confounded by tumor purity or sequencing protocols. Importantly, information about whether the pHLA complexes are recognized by CD8+ T cells is typically not included in the training of these tools.
Less is known about the rules determining whether a pHLA complex can be recognized by CD8+ T cells. In cancer neo-epitope analyses, some studies observed that agretopicity, defined as the ratio between the affinity for HLA-I of the mutated and the wild-type (WT) peptide, has predictive power for immunogenicity,8,22 especially when the mutations fall on anchor positions.23 Similarity to existing epitopes (also referred to as foreignness) was also proposed as a way to improve selection of neo-epitope candidates.8,19,24 Other studies considered dissimilarity-to-self based on the hypothesis that mutated neo-epitopes similar to unmodified peptides are less likely to elicit T cell recognition due to central tolerance.25,26 However, it can be challenging to properly define DisToSelf in the context of TCR recognition.27 Studies focusing mainly on pathogens and cancer testis antigens suggested that peptide hydrophobicity plays a role in TCR recognition,28 although these results are not fully consistent with those of other studies.29 Moreover, these two approaches are not widely used in today’s class I epitope prediction pipelines.
Improved predictions of immunogenicity, defined here as evidence of recognition by some CD8+ T cells, will have several applications. From a clinical point of view, it will facilitate the development of peptide-based T cell vaccines in infectious diseases or personalized cancer immunotherapy.1,3,30 From a more fundamental point of view, it can guide mechanistic understanding of the molecular and biophysical determinants of TCR recognition. Moreover, it could provide insight into immune pressure acting on cancer mutations in human, a process often referred to as immunoediting,31 by taking advantage of large cancer genomics datasets available from thousands of patients.32 Unfortunately, both the limited accuracy of existing epitope predictors and the difficulty of building in silico models fully capturing cancer mutagenesis processes in the absence of immune selection make it challenging to use cancer genomics studies for analyzing immunoediting in human tumors.33
Here, we capitalize on recent (neo-)epitope data to train a predictor of immunogenic epitopes (PRIME). Unlike studies mainly focusing on HLA-I binding or antigen presentation,13,15, 16, 17,34,35 our approach is able to disentangle HLA-I binding from molecular TCR recognition propensity. This is accomplished by a careful annotation and analysis of epitope residues with minimal impact on binding to HLA-I molecules. Our results show that biophysical properties of TCR-pHLA interactions can be learned in this way, improve prediction of neo-epitopes, and lead to a better molecular understanding of the molecular determinants of TCR recognition. Looking at cancer genomics data, we observe that recurrent mutations tend to be less frequent in patients where they are predicted to be immunogenic, demonstrating that PRIME predictions are consistent with immunoediting in human cancer.
Results
Immunogenicity predictions beyond binding and presentation on HLA-I molecules
To develop accurate immunogenicity prediction tools that capture biophysical aspects of TCR recognition, we compiled and curated a dataset of 4,958 peptides derived from pathogens or cancer testis antigens (1,153 immunogenic and 476 non-immunogenic) as well as cancer mutations (129 immunogenic and 3,200 non-immunogenic) that were experimentally tested for immunogenicity in multiple studies in human (Figure 1A; STAR methods; Table S1).47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57
Figure 1.

Immunogenicity predictions beyond binding and presentation on HLA-I molecules
(A) Summary of the data used to benchmark existing tools and develop our new immunogenicity predictor.
(B) Allelic coverage in the immunogenic and non-immunogenic mutated peptides tested in cancer neo-epitope studies for the 15 most frequent HLA-I alleles. “Other (n)” indicates the cumulative frequency of the n remaining HLA-I alleles.
(C) Ability of existing predictors to distinguish between immunogenic and non-immunogenic mutated peptides in cancer neo-epitope studies. The x axis shows the p values of the Wilcoxon test between immunogenic and non-immunogenic peptides for the scores of each predictor (see Figure S1C).
(D) Graphical representation of the immunogenicity predictor combining affinity to HLA-I and TCR binding signals. PΩ stands for the position of last epitope residue. Residues at MIA positions (P4 to PΩ−1 in this example with HLA-B∗44:03) are shown in yellow, and other epitope residues are shown in blue. The HLA is shown in gray and the TCR in light blue and light green.
(E) AUC and PRAUC values of the 10-fold cross-validation (left), the leave-one-study-out cross-validation (middle), and the leave-one-allele-out cross-validation (right).
(F) AUC and PRAUC values obtained based on mouse neo-epitopes from Capietto et al.23
Among mutated peptides tested in cancer neo-epitope studies, we observed similar HLA-I allele coverage in the immunogenic and non-immunogenic peptides (Figure 1B). As expected, the over-representation of HLA-A∗02:01 epitopes was stronger when including data from pathogens and cancer testis antigens (Figure S1A). We also observed slightly more 9-mers among immunogenic peptides (Figure S1B). This may be due to the fact that longer peptides bulging out of the HLA-I binding site could interfere with TCR binding, although we cannot exclude that some studies reporting mainly immunogenic epitopes were restricted to 9-mers.
We then examined how existing predictors could distinguish immunogenic from non-immunogenic mutated peptides in cancer neo-epitope studies. As expected, our results show that neo-epitopes were enriched in peptides with better binding to HLA-I predicted by either MixMHCpred,12 NetMHCpan15 with eluted ligands (NetMHCpanEL) or binding affinity (NetMHCpanBA) predictions, MHCflurry,13 HLAthena,16 or NetMHCstabpan14 (Figures 1C and S1C). This is the case even if most peptides used in this benchmark, including the non-immunogenic ones, had been preselected based on binding affinity predictions. We next explored predictors related to antigen processing, TCR recognition, or immune tolerance. The predicted cleavage (NetChop),47 the predicted antigen transport (TAP),48 and the predicted immunogenicity (IEDB.imm)29 had limited power to predict immunogenicity (Figure 1C). The ratio of binding affinity between the mutant and the WT peptides did not reach statistical significance either, and the trend was mainly driven by mutations observed at anchor positions, where the ratio correlates most with the actual binding affinity (Figures S1D and S1E). The dissimilarity-to-self (DisToSelf) (STAR methods) demonstrated some statistical power to distinguish immunogenic from non-immunogenic cancer mutated peptides (Figure 1F). The recent method of Wells et al.8 (TESLA; STAR methods) that combines antigen presentation with agretopicity and peptide foreignness displayed similar enrichment as HLA-I ligand predictors (Figure 1C) and similar precision (Figure S1F).
To explore other peptide-intrinsic properties that may be important for TCR binding and recognition and avoid confounding factors related to affinity to HLA-I, we annotated for each HLA-I allele the positions in ligand sequences with minimal impact on HLA-I affinity (referred to as MIA positions) and that can potentially interact with the TCR (Figure 1D). To this end, we capitalized on our set of curated HLA-I motifs.12 For every allele, we selected specific positions between the fourth (P4) and the second to last (PΩ-1) that displayed low specificity (STAR methods; Table S2). The amino acid preferences at these positions are much less influenced by the binding to HLA-I, thereby preventing an important confounding factor when analyzing TCR binding and recognition. These amino acid frequencies were minimally correlated with predicted affinity to HLA-I (Figure S1G). We then combined the predicted binding to HLA-I (encoded as MixMHCpred −log(%rank)) with the frequency of each amino acid at these positions (i.e., 1+20 dimensional vector) to train a predictor of immunogenic epitope (PRIME) using a logistic regression (STAR methods; Figure 1D). Peptides with a predicted binding to HLA-I higher than a threshold T (T = 5% on %rank) were further given a score of 0 in the logistic regression, because they are very unlikely to bind to HLA. The same thresholding was applied to the other predictors. 10-fold cross-validation (Figure S2A) for all mutated peptides in neo-epitope studies demonstrated improvement with PRIME compared to other predictors both in terms of area under the receiver operative curve (AUC) and area under the precision-recall curve (PRAUC) (Figure 1E). Standard cross-validation results can be artificially boosted by batch effects because different peptides from the same study are found in both the training and testing sets. To overcome this issue, we selected the seven neo-epitope studies with at least five experimentally validated positives and negatives and performed a leave-one-study-out cross-validation (Figure S2A). Our results demonstrated improved prediction accuracy with PRIME (Figure 1E; Table S3). To exclude biases due to higher frequency of specific HLA-I alleles, we performed a leave-one-allele-out cross-validation across the nine alleles with at least 5 experimentally validated positives and negatives in our neo-epitope dataset (Figure S2A). Here again, PRIME performed better than other tools (Figure 1E; Table S3).
To further investigate the importance of both affinity to HLA and amino acids at MIA positions, we trained PRIME only with the frequencies of amino acids at MIA positions. The performance was much lower (Figure S2B). We then attempted to randomize the amino acids at MIA positions and retrained PRIME on such data (STAR methods). Our results show that predictions became less accurate (Figure S2B).
We next investigated the effect of combining NetChop, TAP, IEDB.imm, or DisToSelf with affinity to HLA-I (MixMHCpred). In general, the combinations were better than the single predictors but less accurate than PRIME with only one exception (AUC for MixMHCpred combined with DisToSelf in the leave-one-allele-out cross-validation; Figure S2C). When combining these four different predictors with PRIME, we observed mainly similar or lower performance, with again the exception of combining PRIME with DisToSelf in the leave-one-allele-out cross-validation (Figure S2D). Using the threshold on binding affinity or not had limited impact on the performance of PRIME and HLA-I ligand predictors but more impact on other predictors, as expected (Figure S2E).
We then asked whether PRIME could also be used for predicting mouse neo-epitopes in the study of Capietto et al.,23 where 409 mutated peptides identified by exome sequencing have been tested for immunogenicity. In terms of AUC, PRIME outperformed existing tools that can be used with mouse major histocompatibility complex (MHC) alleles. The difference with NetMHCpanBA was modest, and NetMHCpanBA displayed better PRAUC (Figure 1F), although this comparison may be biased by the fact that the peptides experimentally tested in Capietto et al. had been preselected based on binding affinity predictions with NetMHCpan. MixMHCpred and HLAthena were not included because mouse MHC alleles are not available with these tools. Half-lives predicted by NetMHCstabpan were always lower than the 1.4h threshold derived in Wells et al.,8 preventing us to compute AUC and PRAUC values for the TESLA method.
In terms of computational efficiency, PRIME runs 10 times faster than MHCflurry and from 10 to 1,000 times faster than NetMHCpan and HLAthena (Figure 2). This makes it especially appropriate to score large datasets of potential epitopes.
Figure 2.
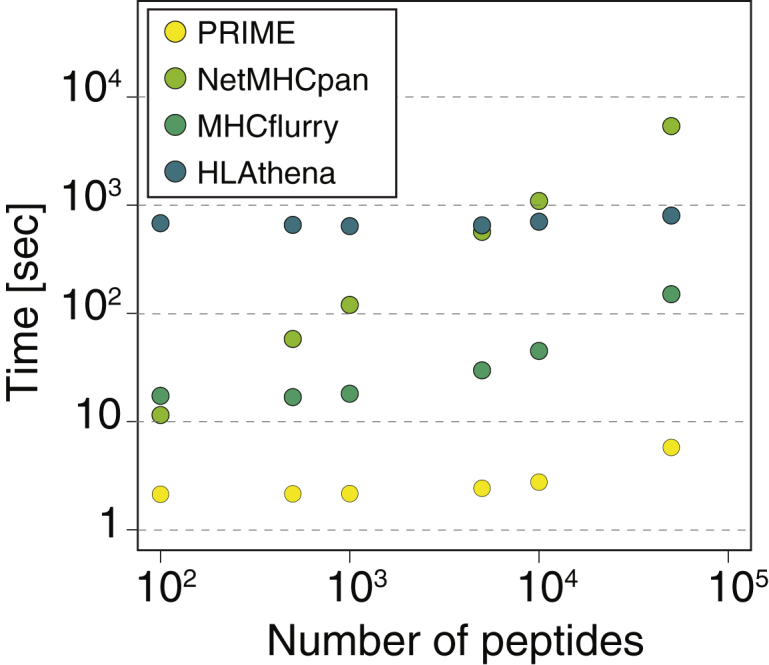
PRIME runs faster than other HLA-I ligand predictors
Computational efficiency of PRIME and different HLA-I ligand predictors for predictions with 6 HLA-I alleles.
PRIME correlates with structural avidity
Given that PRIME is capturing determinants of TCR recognition propensity, we hypothesized that it could also correlate with structural avidity of CD8+ T cells, which is a more quantitative measure of peptide immunogenicity.49 To this end, we used proprietary fluorescence-activated cell sorting (FACS)-based NTAmer technology to determine the monomeric pHLA-TCR dissociation rate (half-life [t1/2]), a physical parameter correlated with T-cell potency.50,51 We analyzed multiple CD8+ T cells with distinct TCRs recognizing thirteen known epitopes, including viral, tumor associated, and neo-epitopes (Figure S3A; Table S4A; STAR methods). PRIME predictions were significantly correlated with half-lives, while no significant correlation could be observed with affinity, stability, cleavage, TAP, or immunogenicity predictors (Figure 3). Significant correlations could be seen with DisToSelf, although this analysis may be influenced by the mixing of immunodominant pathogen epitopes with cancer epitopes, and the correlation mainly reflects the separation between pathogen and cancer epitopes. Of note, the ratio and the TESLA predictions were not included because they can only be applied to neo-epitopes. The version of PRIME trained without predicted affinity resulted in lower performance (Figure S3B). We further measured the cytolytic activity of T cell clones, recognizing eleven of the different epitopes by in vitro killing assay (EC50; STAR methods; Table S4B). In this case, PRIME displayed higher correlation than other predictors, although no tool reached statistical significance (Figure S3C). Overall, our results indicate that PRIME can be used to prioritize epitopes of high structural avidity, which are most promising for vaccine design and personalized cancer immunotherapy.
Figure 3.

PRIME correlates with structural avidity
Correlation between structural avidity (i.e., monomeric pHLA-TCR dissociation kinetics; t1/2) and the predictions of the different tools. Pearson correlation coefficients and p values are shown above each plot. Each point corresponds to the average log(t1/2) of different clones (Table S4A).
PRIME reveals determinants of TCR recognition
To gain insights into the molecular mechanisms underlying the improved predictions of immunogenicity with PRIME, we plotted the coefficients of the logistic regression corresponding to amino acids at MIA positions. We observed a striking correspondence with biophysical properties of amino acids. In particular, the three aromatic amino acids (W, F, and Y) showed the highest values, followed by the three other most hydrophobic ones (V, L, and I), whereas charged amino acids (especially R, K, and E) as well as Q, which are characterized by long and charged/polar sidechains, showed the opposite trend (Figure 4A). The same pattern was observed when training PRIME with different affinity predictors and a similar trend was seen when excluding peptides restricted to HLA-A∗02:01 (Figure S4A). A similar trend was also observed when considering the frequency of amino acids at MIA positions in neo-epitopes, normalized either by the one in human proteins or the one in non-immunogenic cancer mutant peptides with the same distribution of predicted affinity to HLA-I, the same HLA-I allele distribution and the same peptide length distribution as the neo-epitopes (Figure S4B).
Figure 4.

PRIME reveals molecular determinants of TCR recognition
(A) Ranking of the coefficients in PRIME corresponding to amino acids at MIA positions.
(B) IFNγ-ELISpot signals obtained after stimulating naive CD8+ T cells from three healthy donors (d1, d2, and d3) with all P5 variants of the HIV (ALIRILQQL) and CMV (NLVPMVATV) epitopes (two technical replicates).
(C) Spearman correlation coefficients between immunogenicity predictions and IFNγ ELISpot signals for P5 variants of the HIV and CMV epitopes from each donor. Stars indicate Spearman correlation p values smaller than 0.05.
(D) Functional avidity of CD8+ T cell responses (EC50) measured for five variants of the CMV epitopes used to vaccinate HLA-A∗02:01 transgenic mice (4–7 biological replicates). Data were renormalized for each epitope based on the signal at 103 nM. No response was detected for the peptide with K at P5 (values of 0 are shown).
To investigate whether PRIME truly captures properties related to TCR binding and recognition and not merely provides a better model of binding to HLA, we computed the amino acid frequency at MIA positions in naturally presented HLA-I ligands with the same allele/length distribution, normalized by the expected amino acid frequency in their source proteins (STAR methods). A distinct ranking of amino acids was observed with no enrichment in aromatic or hydrophobic residues (Figure S4C). We further attempted to predict HLA-I ligands coming from 10 samples not used in the training of MixMHCpred. MixMHCpred performed better than PRIME in all samples (Figure S4D), further indicating that the improvement in immunogenicity predictions with PRIME is likely not due to better predictions of binding to HLA-I.
The results of Figure 4A recapitulate some observations from previous studies but also highlight important discrepancies. For instance, in Chowell et al.,28 W and F were only slightly correlated with immunogenicity and Y was negatively correlated with immunogenicity, whereas C was the most over-represented amino acid among immunogenic peptides. In Calis et al.,29 charged amino acids, such as E or R, contributed positively to immunogenicity, whereas L and Y had a negative contribution. The correlation between the immunogenicity propensity determined in Calis et al.29 and the data in Figure 4A did not reach statistical significance (ρ = 0.38; p = 0.101; Figure S4E).
To experimentally validate these results, we selected two viral HLA-A∗02:01-restricted epitopes (ALIRILQQL from HIV and NLVPMVATV from CMV) and replaced the peptide residue at position 5 with each other amino acid. Position 5 is known to have limited impact on binding affinity to HLA-A∗02:01, thereby providing a well-suited test case for our immunogenicity predictions (Figures S4F and S4G). To assess the relative immunogenicity of the different peptide variants, naive CD8+ T cells were isolated from three HLA-A∗02:01-positive healthy donors and stimulated in vitro with each peptide separately (STAR methods; Figure S4H; Table S4C). In two out of three donors, variants showing high immunogenicity, as determined by interferon gamma (IFNγ) ELISpot, were enriched in residues predicted to confer high immunogenicity, with the only exception of a weak response for the proline variant of the CMV epitope in donor 2 (Figure 4B). When correlating the immunogenicity of the distinct variants with different predictors, we observed significant correlations in three cases with PRIME, MHCflurry and NetMHCpanBA (Figure 4C). The other predictors were less successful at capturing the immunogenicity signal present in these data. The good performance of MHCflurry and NetMHCpanBA may be explained by the fact that binding affinity data used to train these algorithms are enriched in verified CD8+ T cell epitopes, unlike HLA peptidomics data, which only capture presentation on HLA-I molecules and are the main source of training data for MixMHCpred, NetMHCpanEL, and HLAthena. Similar improvements in predictions with PRIME could be observed when analyzing immunogenicity data for HLA-A∗02:01 restricted peptide analogs tested in Tangri et al. (Figure S4I; Table S4D).52
To see whether the higher immunogenicity of aromatic residues could also be observed in vivo in mouse, we vaccinated HLA-A∗02:01 transgenic mice separately with five variants at P5 of the CMV epitope (NLVPMVATV) and measured the functional avidity of T cell responses 14 days post-immunization (STAR methods). Our results confirm that aromatic sidechains (W and F) triggered the highest avidity, whereas the positively charged sidechain (K) resulted in a total lack of recognition for this peptide (Figure 4D). In this case, E and M displayed similar T cell recognition. It is important to realize that the observations of Figure 4A reflect a general propensity of molecular TCR recognition but do not exclude recognition of less immunogenic residues in specific contexts. In particular, the vast potential diversity of the TCR repertoire can result in recognition of sub-optimal residues, although with lower probability.
TCR-pHLA structures provide molecular interpretations for the predictions of PRIME
To structurally interpret the prevalent role of aromatic residues in TCR binding and recognition, we first considered a well-known HLA-A∗02:01 restricted tumor-associated antigen (ITDQVPFSV; PMEL209–217) that displayed an aromatic sidechain (F) at position 7. The peptide, along with the T2M variant, is recognized well by several TCRs,53 including the PMEL209–217-specific TCR SILv44. The latter was studied recently for its therapeutic potential.54 Consistent with the predictions of PRIME, substitution of phenylalanine at position 7 (P7) with alanine in the PMEL209(2M)–217 epitope did not impact binding to HLA-A∗02:01 (Figure 5A) but eliminated recognition by the SILv44 TCR (Figure 5B). To gain insights into the structural basis of this observation, we solved the X-ray structure of the SILv44-PMEL-HLA-A∗02:01 ternary complex (Table S5A; Figures S5A and S5B). Within the interface, the F7 sidechain inserts into a cleft on the TCR surface formed by sidechains and backbones of residues of CDR3β and CDR2β (Figure 5C). It forms a CH-π hydrogen bond with I94 of CDR3β and 17 van der Waals contacts with the TCR, burying 69 Å2 of solvent accessible surface area.
Figure 5.

TCR-pHLA structures provide molecular interpretations for the predictions of PRIME
(A) HLA-A∗02:01 binding of the PMEL209(2M)–217 epitope (IMDQVPFSV) and the F7A substitution (thermal stability measured by differential scanning fluorimetry). Solid lines represent bi-Gaussian fits to the data. The measured Tm values are 61.9°C ± 0.2°C for the epitope and 61.7°C ± 0.2°C for the F7A-modified epitope (fitted values with standard fitting errors).
(B) Recognition of the PMEL209(2M)–217 WT and F7A mutant by the SILv44 TCR. Solid line for the WT represents a 1:1 fit to the data, yielding a KD of 140 ± 20 μM (average and standard deviation of three independent measurements). No detectable binding was observed for the F7A modified epitope.
(C) Crystal structure of the SILv44 TCR in complex with PMEL209(2M)–217 presented by HLA-A∗02:01.
(D) Crystal structure of a TCR in complex with the HHAT68–76 L75F neo-epitope (KQWLVWLFL; PDB: 6UK4).27
(E) Frequencies of 9-mer epitope residues with sidechains directly contacting TCR residues in TCR-pHLA X-ray structures, normalized by the amino acid frequencies in the source proteins.
As a second example, we considered an ovarian cancer neo-epitope enriched in aromatic and hydrophobic residues (KQWLVWLFL; HHAT68–76; L75F mutant)5 in complex with a cognate TCR, for which we have recently solved the crystal structure.27 Our structure indicated a prevalent role of the sidechain of W at P6 in the interaction with the TCR, including hydrophobic packing with sidechains of CDR3α and CDR3β and a NH-π hydrogen bond with Y100 of CDR3α (Figure 5D). The mutated phenylalanine (F8) plays a less important role in the binding to the TCR compared to W6, mainly burying surface and making some hydrophobic contacts. These results indicate that T cell recognition of neo-epitopes is not restricted to mutations that specifically introduce new amino acids with much higher immunogenicity or stronger binding to HLA-I but also includes the situation where immunogenic amino acids are already present (W6 in this example) and mutations fall elsewhere on these epitopes.
We then surveyed publicly available X-ray structures of TCRs interacting with pHLA complexes (Table S5B). Consistent with the predictions of PRIME, we observed a clear enrichment of aromatic sidechains and a depletion of charged sidechains among epitope residues directly interacting with the TCR (Figure 5E; STAR methods). The correlation between the ranking of amino acids of Figure 4A and the one in Figure 5E was highly significant (ρ = 0.81; p = 1.03 × 10−5), despite the limited number of epitopes with available X-ray structures used to estimate amino acid frequencies in Figure 5E. In many of the TCR structures, the peptide aromatic residues penetrate between the TCR loops, forming a variety of interactions, as demonstrated by the large buried solvent-accessible surface areas (Figures S5C and S5D; Table S5C).
PRIME provides insight into immunoediting in human cancer
One of the hallmarks of cancer is the ability to escape immune recognition. This can be achieved by several mechanisms, such as physical barriers preventing T cell infiltration, establishment of an immuno-suppressive microenvironment, downregulation or alteration of the antigen presentation machinery,55 or negative selection of mutations giving rise to neo-epitopes—the so-called immunoediting.31,56
Here, we hypothesized that the ability of PRIME to integrate presentation on HLA-I and TCR recognition could be useful to investigate immunoediting in immunotherapy-naive tumors in human. Practically, we took advantage of the diversity of HLA-I alleles in the human population and reasoned that cancer mutations should undergo higher immune selective pressure (and therefore show lower frequency) in patients where they have higher probability to give rise to immunogenic neo-epitopes (Figure 6A).57 To explore this hypothesis, we collected non-synonymous mutations found in The Cancer Genome Atlas (TCGA) and predicted the PRIME score with all HLA-I alleles of each patient (STAR methods). For each mutation, we compared its frequency among patients where it would give rise to neo-epitopes () with its frequency among patients where it would not (; Figures 6A and 6B; STAR methods). For rare mutations with a low number (N) of occurrences in the TCGA cohort, we observed no differences between and (Figure 6C). However, as the number of occurrences of the mutations in the TCGA cohort increased, the mutations started to display lower frequency in patients where they would give rise to neo-epitopes (Figure 6C). These results indicate that recurrent mutations, which are often oncogenic, tend to be less frequent among patients where they would give rise to neo-epitopes. Consistent with previous studies,57,58 the trend is especially strong in colorectal cancer, suggesting that this tumor type may be especially prone to immunoediting (Figures 6D and S6A). To further test the robustness of our model, we reasoned that the trend should be sensitive to the actual HLA-I typing of the patients.57 The red circles and error bars in Figures 6C and 6D show the results obtained after randomly shuffling the HLA-I alleles in TCGA patients (STAR methods). As expected, the trend was much weaker after shuffling HLA-I alleles.
Figure 6.

PRIME provides insight into immunoediting in human cancer
(A) Proposed framework to study immunoediting acting on cancer mutations. For each mutation, patients are stratified based on whether the mutation would be immunogenic (, yellow rectangle) or would not (, red rectangle). The actual frequency of the mutation is compared between these two groups ( and , where stands for the subset of patients where the mutation is observed). These frequencies will be further compared to those obtained after shuffling the HLA-I alleles between patients (i.e., and ).
(B) Frequency of KRAS G12D mutation in patients where it would give rise to neo-epitopes () and where it would not (), together with the example of HLA-I alleles of a patient with the mutation in (upper row, with the two predicted epitopes) and one in (lower row).
(C) Top: average value of for mutations observed N times ( in the TCGA cohort. The color scale shows the number of distinct mutations for all values of the mutation occurrence (N > 0). Bottom: average value of for mutations observed at least Nmin times in the TCGA cohort is shown. The red circles and error bars correspond to randomized HLA-I alleles (mean and standard deviation of ).
(D) Top: average value of for mutations observed N times in colorectal tumors of TCGA. Bottom: average value of for mutations observed at least Nmin times in colorectal tumors of TCGA is shown.
The insets in (C) and (D) show the boxplots of the frequencies and for all mutations observed at least Nmin times in our TCGA samples (Nmin = 35 in C and Nmin = 10 in D). r and p values shown in the top plots in (C) and (D) correspond to the Pearson correlation coefficient (STAR methods). p values shown in the insets of the bottom plots in (C) and (D) correspond to paired Wilcoxon tests. All frequencies are shown in percentages (%).
We further attempted to investigate the potential effect of gene expression, clonality, and deleterious alterations in the antigen processing machinery (STAR methods). To this end, we restricted the analysis to patients for which expression data were available. For a given mutation, patients for which the source gene of the mutation was poorly expressed, for which the mutation was predicted to be sub-clonal, or with deleterious alterations in antigen processing machinery were removed from the set of patients with the mutation (M in Figure 6A) and frequencies were recomputed. An even slightly stronger trend could be observed, demonstrating the robustness of our observations (Figure S6B). Reversely, the trend was no longer observed when considering for each mutation only patients with low expression of the source gene of the mutation, where the mutations were predicted to be sub-clonal or with deleterious alterations in antigen presentation genes (Figure S6C).
Taken together, our results indicate that recurrent mutations are on average less frequent in patients where they are predicted to give rise to neo-epitopes, and this association depends on the patient’s HLA-I typing. For less common mutations, similar mutational frequencies are observed, and the values obtained with the actual HLA-I alleles of the patients overlap with those obtained after shuffling these alleles. Of note, the trend was weaker when using predictions only based on binding to HLA-I molecules, highlighting PRIME’s ability to better highlight immunoediting acting on recurrent mutations (Figure S6D).
Discussion
Accurate prediction of neo-epitope immunogenicity is a cornerstone for rational vaccine design and personalized cancer immunotherapy. While most recent studies focused on improving prediction of HLA-I binding and antigen presentation, we capitalized on recent (neo-)epitope data to train and validate a method that goes beyond binding to HLA-I molecules and captures molecular TCR recognition properties. PRIME not only improved prioritization of neo-epitopes but also correlated with structural avidity (Figure 3), which is promising to select optimal epitopes that could induce strong responses upon stimulation or vaccination.
Unlike previous approaches,29 PRIME combines presentation on HLA-I molecules with peptide-intrinsic TCR recognition propensity, which resulted in improved prediction accuracy. Our work also helps rationalize why some peptides with low HLA-I affinity can be well recognized by CD8+ T cells, as weak pHLA affinity can be offset by strong TCR binding enabled by particularly immunogenic amino acids. Consistent with previous studies,8,19,24 adding features linked to peptide foreignness or DisToSelf, which is an additional aspect of T cell recognition related to central tolerance and not considered in PRIME, may further improve predictions.
Our results reveal a convergence between immunogenicity and biophysical properties of amino acids. In particular, aromatic sidechains (i.e., W, F, and Y) showed the highest immunogenicity. These observations are attributable to the multifunctional properties of these aromatic side chains: while hydrophobic, they can also serve as hydrogen bond acceptors, pair with cations, engage in various stacking interactions, and possess limited rotational degrees of freedom. The corresponding bias against charged amino acids is explained similarly: burying charges within a protein interface is associated with energetically expensive desolvation penalties and necessitates precise alignments between opposing charges in the TCR.59 This also explains the low immunogenicity of histidine, which can easily be protonated due to its pKa of 6.0, close to physiological pH.
When considering naturally presented HLA-I ligands identified by mass spectrometry (MS) (Figure S4C), we observed a different pattern compared to Figure 4A, supporting our conclusion that PRIME captures signals related to TCR recognition and not to HLA-I binding or antigen presentation. Figure S4C revealed a strong depletion of cysteine as well as a depletion of tryptophan. These amino acids can be chemically modified,60 which may explain why they are less well detected in peptides found by MS.17,34 As such, we cannot exclude that PRIME also partly corrects the bias against tryptophan that is likely present in HLA-I ligand predictors trained on MS data, like MixMHCpred.
For all tools considered in this work, we used an ad hoc threshold based on predictions of binding to HLA and presentation. The underlying motivation was to avoid cases of peptides with very low affinity to HLA-I but with a stretch of immunogenic residues at MIA positions, as these may be underrepresented in the training set of PRIME. The value of the threshold is a free parameter in PRIME and setting it to lower %rank will increase specificity while decreasing sensitivity.
In some of our analyses, naive CD8 T cells were used to probe T cell reactivity (Figure 4B). The main reason was to avoid biases due to previous exposure with specific epitopes. As such, these experiments capture general properties of epitopes to be recognized by some TCRs found in the TCR repertoire. This is sometimes referred to as antigenicity, although a consensus on this terminology has not been reached in cancer neo-epitope studies.
The idea of using cancer genomics data to investigate immunoediting mechanisms in human cancer has been explored in previous studies.32,57,42, 43, 44 For instance, mutated peptides generated by recurrent oncogenic mutations were shown to have lower predicted binding to HLA-I molecules compared to those from less frequent mutations.32 An important challenge in this type of analysis comes from the fact that non-synonymous cancer mutations display specific signatures reflecting a plethora of mechanisms, such as UV light, tobacco, DNA repair propensity, functional impact, codon degeneracy, etc.64 Accurate models of mutagenesis processes in cancer that would recapitulate all these different mechanisms, except for the immune selection, are difficult to design, and any bias in these models can result in wrong interpretations when using them to compare with actual data.33 The idea of stratifying patients based on the predicted immunogenicity of a mutation ensures that our findings do not rely on such theoretical models. This idea may therefore represent a powerful framework to explore immune pressure mechanisms in cancer, and our observations provide an independent validation of the results of Marty et al.32
Several reasons can explain the lack of signal detected for rare, mainly passenger, mutations. First, we cannot exclude that a certain fraction of these mutations are false-positives of mutation-calling algorithms. Second, immunoediting may be more difficult to statistically detect with our approach. Third, it is expected that many passenger mutations appear later in tumorigenesis and undergo lower immune selection.32 We also emphasize that negative selection of immunogenic mutations is only one of the multiple factors shaping the landscape of cancer mutations. This likely explains why we do not observe a complete absence of mutations in patients where they could give rise to immunogenic neo-epitopes (Figure 6C). This observation has important consequences for our understanding of tumor development and response to therapy. First, it confirms that several immunotherapy-naive tumors can afford having mutations giving rise to neo-epitopes, including driver mutations, such as KRAS G12D.65 Second, it may contribute to the success of cancer immunotherapy, because several immunogenic peptides will still be presented on cancer cells and could become clinically relevant upon boosting or engineering CD8+ T cells targeting them.
Investigation of the results obtained with each tumor separately (Figure S6A) demonstrated that a different trend was observed in melanoma (TCGA code: SKCM). Melanoma is among the tumors that are best recognized by T cells and show the best response rate to immunotherapy. Presentation of neo-antigens and cancer testis antigens is likely playing an important role in T cell recognition of tumors. As such, the lack of detectable immune selection of recurrent mutations in melanoma samples where they are predicted to be immunogenic is not inconsistent with the higher response rate observed in this tumor type.
The lack of signal observed for mutations poorly expressed, predicted to be sub-clonal, or found in patients with altered antigen presentation would be consistent with a lower immunoediting (Figure S6C). However, it is important to realize that recurrent mutations are underrepresented within this set of mutations. Moreover, the filters that we used have limitations (e.g., expression can be biased by the presence of non-malignant cells, clonality is challenging to estimate with single biopsies, and prediction of mutation functional effects, especially in HLA-I genes, is not fully reliable).
In vivo CD8+ T cell response against infected or cancer cells involves many other parameters beyond TCR binding (e.g., co-receptors, cytokines, T cell fitness, microenvironment, etc.). A holistic model of immunogenicity will ultimately need to integrate all of these aspects. However, many of these parameters are currently very difficult, if not impossible, to accurately measure experimentally in a clinical context, limiting the possibility to train and use such broad models for practical applications and quantitative predictions. By focusing on features that are readily available from peptide sequences, our immunogenicity predictor (http://prime.gfellerlab.org/) is suitable for large cohorts of patients in infectious diseases and cancer immunotherapy.
Limitations of study
One limitation of this study comes from the fact that immunogenic peptides were defined as peptides recognized by some T cells in T-cell assays (e.g., IFNγ-ELISpot). As such, our work uncovers properties of peptides that enhance their propensity to be physically recognized by TCRs (sometimes referred to as “antigenicity”) but does not robustly demonstrate that these peptides elicit a stronger response upon vaccination. This is likely one of the big, and currently unmet, challenges toward clinical applications of immunogenicity predictions in personalized cancer immunotherapy. A second limitation is that PRIME does not model the potential impact of central tolerance and therefore should preferentially be used on non-self peptides (e.g., mutated peptides in cancer or peptides from pathogens). Finally, although the set of verified neo-epitopes used in our benchmarks (i.e., 129 positives) is larger than in most recent neo-epitope prediction studies, it is still limited, and we anticipate that larger neo-epitope datasets will lead to additional refinements in immunogenicity predictions.
STAR★methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-hCD8 | Biolegend | 313904 |
| Anti-CCR7 | Biolegend | 353226 |
| Anti-CD45RA | Beckman Coulter | IM2711U |
| Biological samples | ||
| Healthy donors blood mononuclear cells and blood mononuclear cells and tumor infiltrating lymphocytes from cancer patients | Biobank from the Center of Experimental Therapeutics, Department of Oncology, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland. | Protocol 235/14 and 2016-02094, 2016-02166 and 2017-00490 |
| Chemicals, peptides, and recombinant proteins | ||
| CMV variant peptides | Peptide & Tetramer Core Facility, University of Lausanne | N/A |
| HIV variant peptides | Peptide & Tetramer Core Facility, University of Lausanne | N/A |
| Other peptides | Peptide & Tetramer Core Facility, University of Lausanne | N/A |
| pMHC NTAmers | Peptide & Tetramer Core Facility, University of Lausanne | N/A |
| RPMI | GIBCO | 61870-010 |
| MEM NEAA | GIBCO | 11140-035 |
| 2-Mercaptoethanol | GIBCO | 31350-010 |
| Sodium Pyruvat | GIBCO | 11360-033 |
| HEPES | Bio Concept | 5-31F00H |
| Pen/strep | Bio Concept | 4-01F00H |
| Human Serum | Biowest | S419H-100 |
| FBS | Biowest | S-1810-500 |
| IL2 | Novartis | Proleukina, PZN02238131 |
| DAPI | Sigma-Aldrich | 10236276001 |
| Incomplete Freund Adjuvant | Sigma-Aldrich | F5506-10ML |
| ODN1826 | Invivogen | Tlrl-1826-5 |
| Critical commercial assays | ||
| Human-Interferonγ release assay | Mabtech | 3420-2APT-10 |
| Human-CD8 isolation kit | Milteny | 130-045-201 |
| Murine-Interferonγ release assay | Mabtech | 3321-4APT-10 |
| Experimental models: cell lines | ||
| T2 cells | ATCC | CRL-1992 |
| CD4 blasts cells | In house | N/A |
| Experimental models: organisms/strains | ||
| HLA-A2.1/DR1 mice | Pajot et al.66 | N/A |
| Software and algorithms | ||
| NetMHCpan4.1 | Reynisson et al.15 | http://www.cbs.dtu.dk/services/NetMHCpan/ |
| MHCflurry1.6.1 | O’Donnell et al.13 | https://github.com/openvax/mhcflurry |
| HLAthena | Sarkizova et al., 201916 | http://hlathena.tools/ (executable shared by the authors, private communications) |
| NetChop3.0 | Nielsen et al.47; Stranzl and Lundegaard21 | http://www.cbs.dtu.dk/services/NetCTLpan/ |
| TAP | Peters et al.48; Stranzl and Lundegaard21 | http://www.cbs.dtu.dk/services/NetCTLpan/ |
| IEBD immunogenicity predictor | Calis et al.29 | http://tools.iedb.org/immunogenicity/ |
| NetMHCstabpan1.0 | Rasmussen et al.14 | http://www.cbs.dtu.dk/services/NetMHCstabpan/ |
| MixMHCpred2.1 | Gfeller et al.12 | https://github.com/GfellerLab/MixMHCpred |
| Prism version 8.0.0 | GraphPad Software, Inc | N/A |
| FlowJo 9.6.4 | FlowJo, LLC | N/A |
| Other | ||
| Chromium-51 radionuclide | Perkin Elmer | NEZ030S002MC |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Dr. David Gfeller (David.gfeller@unil.ch).
Materials availability
All unique materials and reagents generated in this study are available from the Lead Contact with a completed material transfer agreement.
Data and code availability
PRIME is freely available for academic researchers at the web interface http://prime.gfellerlab.org and as command-line tool https://github.com/GfellerLab/PRIME. All data used to train and validate PRIME are available in Supplementary Materials (Tables S1 and S4). The accession number for the X-ray structure reported in this paper is PDB: 6VM8.
Experimental model and subject details
Animal experiments
HLA-A2/DR1 mice (C57BL/6-Tg(HLA-A2.1)/Tg(HLA-DR1)/H-2class-I/class-II-knockout)66 were born and maintained in a conventional animal facility at the University of Lausanne, Switzerland. Experiments were conducted in accordance with the Cantonal Veterinary Office (License VD3321). Female and male of 6 to 10 weeks were randomly used for experiments.
Patients, Healthy donors and ethic statement
Peripheral blood samples were collected from HLA-A∗0201-positive healthy donors. Patients under study had stage III/IV metastatic melanoma, ovarian, non-small cell lung cancer and colorectal cancer and had received several lines of chemotherapy. Patients were enrolled under protocols approved by the respective institutional regulatory committees at the University of Pennsylvania, USA, and Lausanne University Hospital (CHUV), Switzerland (NCT00112242; https://www.clinicaltrials.gov). Patients and healthy donors’ recruitment, study procedures and blood withdrawal were approved by regulatory authorities and all signed written informed consents.
Cell lines and primary cells
HLA-A∗0201-positive TAP-deficient T2 cells (ATCC) and CD4 blasts were cultured in RPMI 1640 Glutamax media (GIBCO) supplemented with 10% FBS (Biowest) and penicillin/streptomycin (BioConcept). Primary CD8+ T cells were cultured in RPMI 1640 Glutamax media (GIBCO) supplemented with 8% Humsn serum (Biowest), non-essential amino acids (GIBCO), 2-mercaptoethanol (GIBCO), sodium pyruvate (GIBCO), HEPES (GIBCO), penicillin/streptomycin (BioConcept) and 150 U/ml of rhIL2 (Novartis). All cells were maintained at 37°C under 5% CO2 atmosphere.
Method details
Collection of immunogenic and non-immunogenic peptides used for training PRIME
Immunogenic (n = 1,282) and non-immunogenic (n = 3,676) peptides were collected from many recent studies, and comprise both pathogen and cancer testis antigens as well as cancer neo-epitopes (Table S1 and Figure 1A). In these studies, immunogenicity was assessed using different T cell assays, such as INFγ ELISpot or pHLA tetramer staining. Only peptides with reported HLA-I restriction were considered. Non-immunogenic peptides were defined as peptides for which CD8+ T cell reactivity could not be observed in the experiments where they had been tested.
Existing epitope predictors used for benchmark
Benchmarking of the predictions was performed with several predictions. First, we used predictors of binding to HLA-I molecules and antigen presentation: MixMHCpred2.112; NetMHCpan4.1 considering both eluted ligand predictions (NetMHCpanEL) or binding affinity predictions (NetMHCpanBA)15; MHCflurry1.6.113; and HLAthena16. Percentile rank values (%rank) were used for all these predictors and are displayed as -log(%rank) in Figures. Cleavage predictions based on NetChop-3.047 and TAP transport efficiency48 were retrieved from the NetCTLpan website21, using the flanking regions consisting of the ten N- and ten C-terminal residues, when possible.
Next, we considered predictors that aim to capture signals related to immunogenicity beyond binding to HLA-I. The immunogenicity predictions with the method of Calis et al.29 were retrieved from the IEDB website (http://tools.iedb.org/immunogenicity/) and were referred to as “IEDB.imm” in the Figures. As %rank are more comparable between HLA-I alleles and recommended by several tools, the ratio of affinity between the mutated and the wt peptides (referred to as “Ratio”) was computed as log(%rank_wt) - log(%rank_mutant), based NetMHCpanEL %rank predictions. The dissimilarity-to-self (“DisToSelf”) was defined by computing the distance with all peptides of the same length in the human proteome based on the BLOSUM62 substitution matrix, and using the lowest value as the DisToSelf. Mathematically, for two peptides and , the distance was computed as , where stands for the BLOSUM62 entry for amino acids a and b. The method of Wells et al.8, referred to as TESLA in this manuscript, was implemented based on the proposed thresholds: predicted affinity < 34nM AND predicted stability > 1.4h AND (ratio_Kd < 0.1 OR Foreignness > 10−16). Affinity was predicted with NetMHCpan4.1 (Aff(nM)), stability was predicted with NetMHCstabpan1.0 (Thalf(h)), ratio_Kd was computed as the ratio between the predicted affinity (Aff(nM)) of the mutated and the wt peptide, foreignness was computed with the antigen.garnish package (https://github.com/andrewrech/antigen.garnish)26, as done in TESLA. Expression values could not be considered since they were not determined for the peptides analyzed in this study. The Ratio and the TESLA predictions were only computed for mutated peptides found in cancer, since they are not defined for pathogens or cancer testis antigens. The EDGE method35 and the method of Chowell at al.28 could not be included in this benchmark, as neither an executable nor a web interface is available.
Training of the immunogenicity predictor PRIME
To disentangle the influence of affinity to HLA-I molecules from other parameters, we first annotated positions with minimal impact on HLA-I affinity (MIA positions) and potentially interacting with the TCR for each HLA-I allele, using our set of HLA-I binding motifs derived from unbiased MS data. To this end, the information content (i.e., , where represents the frequency of amino acid i at position j in 9-mer ligands for a given allele) was computed for each position j = 4,…,8 and for each allele based on 9-mer ligands. Positions with information content lower than a threshold of 0.3 were defined as MIA positions. Position 4 in HLA-A∗02 alleles, position 5 in HLA-A∗68:02, position 6 in HLA-A∗25:02 and HLA-A∗26:01 and position 7 in HLA-A∗29:02 were further removed from the list, as they show residual specificity. This led to 6 different groups of alleles (g = 1,…,6) with specific MIA positions corresponding to: g = 1: P4 to P8 (e.g., HLA-A∗01:01); g = 2: P5 to P8 (e.g., HLA-A∗02:01); g = 3: P5, P7 and P8 (e.g., HLA-A∗02:03); g = 4: P4, P5, P7 and P8 (e.g., HLA-A∗25:01); g = 5: P4 to P6 and P8 (e.g., HLA-A∗29:02); g = 6: P4 and P6 to P8 (e.g., HLA-B∗08:01) (Table S2). For peptides of length other than 9, the MIA positions were defined for each group as follows: g = 1: P4 to PΩ-1; g = 2: P5 to PΩ-1; g = 3: P5 to PΩ-4 and PΩ-2 to PΩ-1; g = 4: P4, P5 and P7 to PΩ-1; g = 5: P4 to PΩ-3 and PΩ-1; g = 6: P4 and P6 to PΩ-1, where Ω stands for the length of the peptide.
We next trained a logistic regression (glmnet R package v2.0.16 (alpha = 0 and lambda = 1)) encoding each pHLA pair as a 21-dimensional vector consisting of the predicted affinity (-log(%rank)) of the peptide to the HLA-I molecule and the frequencies of the 20 amino acids at MIA positions29. Binding predictions were performed with MixMHCpred2.1. 2,800 (i.e., 50 peptides for 56 common HLA-I alleles used in our training set) additional 9-mer peptides randomly selected from the human proteome were added to the training set as negatives (i.e., non-immunogenic) to better match the real situation where non-immunogenic peptides are in excess compared to immunogenic ones. Peptides with low predicted binding to HLA-I (MixMHCpred %rank > T, with T = 5% in this work) were always predicted as non-immunogenic (i.e., given a score of 0 in the PRIME logistic regression). The same threshold was applied to the other tools used in all validation based on the predicted affinity (%rank) for each HLA-I affinity predictor and based on MixMHCpred %rank for the other tools (NetChop, TAP, IEDB.imm, Ratio, DisToSelf and TESLA) (i.e., giving a score of 1 minus the score of the peptide with the lowest score to compute the AUC). Logistic regressions were used when combining different predictors (i.e., NetChop, TAP, IEDB.imm or DisToSelf with MixMHCpred or PRIME, Figures S2C and S2D). Randomizing amino acids at MIA positions was performed by replacing the residues at these positions in each peptide with randomly selected residues from the human proteome.
In the upper panel of Figure S4B, amino acid frequencies at MIA positions were computed for all neo-epitopes and renormalized by those in human proteins, considering residues between the fifth and the second to last positions (this is to avoid including in those background frequencies residue that cannot be found at MIA positions by constructions, especially the starting Met). In the lower panel of Figure S4B, all neo-epitopes with at least one non-immunogenic cancer mutant peptide from the same allele, of the same length and with the same predicted affinity (based on 10 equal bins between the lowest and highest value of MixMHCpred log(%rank)) were considered to compute amino acid frequencies at MIA positions (115 out of 129 neo-epitopes). For renormalization, amino acid frequencies at MIA positions in non-immunogenic cancer mutated peptides were computed after weighting each of these peptides in order to have exactly the same distribution of alleles, peptide lengths and binding affinities as in the 115 neo-epitopes.
For each allele, percentile ranks (%rank) for PRIME were computed based on a dataset of 700,000 8- to 14-mer peptides (100,000 for each length) randomly selected from the human proteome, similarly to what is done for HLA-I ligand predictors such as MixMHCpred or NetMHCpan.
Cross-validation
Validation on neo-epitopes was first carried out with ten-fold cross-validation. To this end, the set of cancer mutated peptides was split into ten groups, and each group was iteratively used as testing set (Figure S2A). Similarly the other peptides (i.e., pathogens/cancer testis antigens + random negatives) were also split in ten groups and nine of them were iteratively used to train the algortithm. To ensure that our results are not biased by one specific study, we also used a leave-one-study-out cross-validation strategy (Figure S2A). Each neo-epitope study with at leasty five immunogenic and five non-immunogenic peptides (seven studies in total) were iteratively removed from the training of the predictor and used as testing set. The remaining peptides used only for training were also split into seven groups, and 6 of them were iteratively used for training (Figure S2A). Finally, we performed a leave-one-allele-out cross-validation over the set of HLA-I alleles with at least five immunogenic and five non-immunogenic peptides in the testing set. To avoid biases due to the similarity beteeen HLA-I alleles, peptides tested with alleles from the same super-type than the HLA-I allele used in the testing set were excluded from the training (Figure S2A). The area under the receiver operating curve (AUC) and the area under the Precision Recall curve (PRAUC) were used to assess the prediction accuracy.
Epitopes used for external validations
Data from Capietto et al.23 were retrieved from the original publication. For these epitopes, NetMHCpanBA4.1 was used to predict %rank used in PRIME, since MixMHCpred cannot run on mouse MHC alleles. Only peptides experimentally validated for immunogenicity were used in the benchmarking of Figure 1F (26 positives in total). Data from Tangri et al.52 for the analogs of the two epitopes (IMIGVLVGV from CEA and KVAELVHFL from MAGEA3) were manually retrieved from the Figures published in this study (Table S4D) and the ranking of the peptide analogs was used to compute Spearman correlation coefficients in Figure S4I. Only mutations at P5 were considered, since this position is the one with the lowest impact on affinity to HLA-A∗02:01 (Figures S4F and S4G).
Computational efficiency
In Figure 2, peptides of length 8 to 13 were used for PRIME, NetMHCpan and MHCflurry, and peptides of length 8 to 11 for HLAthena, since longer peptides are not supported. Predictions were performed with six common HLA-I alleles (HLA-A∗02:01, HLA-A∗03:01, HLA-B∗07:02, HLA-B∗08:01, HLA-C∗04:01, HLA-C∗07:02), on a single core (MacBookPro, 2.9 GHz Intel Core i7).
Measurements of structural avidity (half-lives) and cytolytic activity (EC50) of antigen-specific CD8+ T cells
CD8+ T cells directed against shared viral, tumor associated antigens and cancer neo-epitope of Figure 3 were isolated from patients as previously described5. Written informed consents were obtained from all patients and HDs. NTAmers were produced by the Peptide and Tetramer Core Facility of the University of Lausanne. NTAmers are dually labeled pHLA multimers built on NTA-Ni2+-His-tag interactions and were used for monomeric pHLA-TCR dissociation kinetics measurements as previously described51,67. Briefly, antigen-specific CD8+ T cells were stained for 45 min at 4°C in PBS, 0.2% BSA, 5 mM EDTA with cognate NTAmers. NTAmer staining was assessed at 4°C on a SORP-LSR II flow cytometer (BD Biosciences). Following 30 s of baseline acquisition, imidazole (100 mM) was added and Cy5 fluorescence was measured during the following 10 minutes. Data were analyzed using the kinetic module of FlowJo software (v.9.7.6, Treestar) and modeled (1-phase exponential decay) using Prism software (GraphPad) to determine the half-life (t1/2 = ln(2)/koff). The list of t1/2 for different TCRs tested with each epitope can be found in Table S4A, and the average of the logarithms of t1/2 for each epitope was used in Figure 3. The binding affinity ratio was not considered since it is not defined for tumor associated and viral antigens.
Cytolytic activity (Figure S3C) was measured by chromium release assay. 51Cr-labeled HLA-A∗0201-positive TAP-deficient T2 cells, or alternatively CD4 blasts cells, were pulsed with serial dilutions of peptides of interest, and incubated with antigen-specific CD8+ T cell clones at an effector/target ratio of 4:1 for 4 hours at 37°C. Percentages of specific lysis were calculated as 100 x (experimental – spontaneous release) / (total – spontaneous release). EC50 values were derived by dose-response curve analysis (log(agonist) versus response) using Prism software (GraphPad). The list of EC50 values for different TCRs tested with each epitope can be found in Table S4B, and the average of the logarithms of EC50 for each epitope was used in Figure S3C.
Analysis of naturally presented HLA-I ligands
A subset of naturally presented HLA-I ligands detected by MS from the compilation done in Gfeller et al.12 was selected to contain the same distribution of allele/length distribution as the neo-epitopes. These peptides were used to compute amino acid frequencies at MIA positions (Figure S4C). These frequencies were normalized by the amino acid frequencies between the fifth and the second-to-last positions of the set of source proteins detected in HLA-I peptidomics studies. This is done to prevent confounding factors mainly because the first (including the starting methionine) and last residues are never found at MIA positions, by construction.
HLA-I ligands from the ten HLA peptidomics samples measured in Gfeller et al.12 and not included in the training of MixMHCpred were used to benchmark predictions of HLA-I ligands with PRIME and MixMHCpred. AUC values (Figure S4D) were computed by taking as negatives for each sample 99-fold excess peptides randomly selected from the human proteome with length 8 to 14.
Analysis of antigen-specific CD8+ T cells from healthy donors and cancer patients
Peripheral blood mononuclear cells (PBMCs) were collected from HLA-A∗02:01 positive healthy donors (HDs). Fresh PBMCs were positively enriched using anti-CD8-coated magnetic beads (Miltenyi Biotec), stained in PBS, 0.2% BSA, 5 mM EDTA with anti-CCR7 and anti-CD45RA for 45 min at 4°C. After washing, cells were resuspended in PBS, 0.2% BSA, 5 mM EDTA containing DAPI and naive CD8+ T cells (CCR7+/CD45RA+) were directly sorted on a FACSAria flow cytometer (BD Biosciences). Purified naive CD8+ T cells were plated in 24-well plates (2x106/ml) and stimulated three times in vitro (every 10 days) with 1 μM of single CMV or HIV-derived peptides, irradiated autologous PBMC and 150 U/ml of rhIL-2. Ten days after the last stimulation the T cell cultures were tested for IFN-γ production by ELISpot following manufacturer’s instructions. Briefly, 100,000 CD8+ T cells were incubated for 16 h with 30,000 T2 cells priorly pulsed for 1 h with single CMV or HIV variant peptides (1 μM). A positive response was considered if the average number of spots in the peptide-exposed wells was ≥ 2-fold higher than the number of spots in the unstimulated wells, and there were ≥ 10 specific spots/100,000 T cells. For positive responses, the number of IFNγ-secreting cells reported in Table S4C correspond to the number of spots in the stimulated well minus the number of spots in the background (unstimulated well). The ELISpot assay was performed according to manufacturer’s instructions (Mabtech, Nacka Strand, Sweden). All measurements were performed in duplicates. As these experiments are based on pathogen derived peptides directly used to stimulate CD8 T cells, NetChop, TAP, Ratio and DisToSelf were not included in Figure 4C, as they are either not defined or less meaningful.
Peptides and pMHC NTAmers
Peptides and pMHC NTAmers were produced by the Peptide and Tetramer Core Facility (PTCF) of the University of Lausanne. Peptides were HPLC purified (≥90% pure), verified by MS and kept lyophilized at −80°C. NTAmers containing 5% glycerol, were aliquoted (5 ul), kept at −80°C and single used.
Mouse immunization
HLA-A∗02:01/DR1 transgenic, H-2−/− mice66 (n = 4-7 per condition) were immunized with peptides essentially as described68. In brief, single CMV-derived peptides and the DR1 restricted influenza HA306-318 peptide (10 μg each) were injected subcutaneously at the base of the tail in an emulsion containing PBS, IFA and ODN 1826 (InvivoGen, San Diego). Only one peptide (i.e., P5 variants of the CMV epitope NLVPMVATV) was injected in each mice. After two weeks mice were booster immunized and a fortnight later their spleens harvested, 100,000 splenocytes were incubated overnight with T2 cells previously pulsed with different concentrations (ranging from 10−3-103 nM) of single peptide at a 1:1 ratio. Production of IFNγ was assessed using a mouse ELISpot kit following the manufacturer’s instructions (Mabtech, Nacka Strand, Sweden). A positive response was considered positive if the number of spots in the peptide-exposed wells was ≥ 2-fold higher than the number of spots in the un-stimulated wells, and there were ≥ 10 specific spots/100,000 splenocytes. All measurements were performed in duplicates. The functional avidity of the different peptide-specific T cell responses was determined by calculating the peptide concentration able to mobilize 50% of the maximal number of spot forming unit (SFU).
Recombinant protein production and X-ray crystallography
Recombinant SILv44 TCR and HLA-A∗02:01 were generated from bacterially-produced inclusion bodies as previously described69. Peptides were synthesized commercially and obtained at > 95% purity. Proteins were refolded in vitro and purified using ion-exchange followed by size exclusion chromatography, concentrated, and concentrations determined using predicted extinction coefficients. Crystals of SILv44 bound to PMEL209(2M)-217 were grown in 20% PEG 3350 and 236 mM ammonium citrate dibasic at 6 mg/mL total protein concentration by vapor diffusion at 23°C. Diffraction data was collected on the 22ID beamline at the Advanced Photon Source at Argonne National Laboratories. Data was indexed, integrated, and scaled in HKL200070. The structure was solved by molecular replacement using the MoRDa pipeline within the CCP4 suite71. Following molecular replacement, the model was rebuilt using PHENIX Autobuild72. Multiple rounds of restrained refinement were performed using PHENIX Refine73. Evaluation of models and map fitting were performed using COOT74. Structures were evaluated by MolProbity75 during and after refinement. The fully refined complex was deposited to the Protein Data Bank and assigned accession code PDB: 6VM8 (see Table S5A). The assembled pHLA-TCR biological unit can be constructed by visualization of symmetry-related molecules.
Analysis of TCR binding and peptide/HLA-A∗02:01 thermal stability
Surface plasmon resonance (SPR) experiments were conducted on a Biacore T200 instrument in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% surfactant P-20 at 25°C as previously described69. TCRs were coupled to a Biacore CM5 sensor chip using amine coupling. Increasing concentrations of pHLA were flowed over immobilized SILv44 at a rate of 5 μL/min. Steady-state responses (RU) were determined by averaging the final 10 s of each injection and subtracting the response values from identical injections over a blank surface. Each concentration was injected in duplicate for each experiment. Binding affinity was determined by fitting a 1:1 binding curve to a plot of RU versus pHLA concentration. Both datasets were globally fit to enhance accuracy and precision of the determined KD76. The reported SILv44 KD value is the average and standard deviation of three independent measurements. Thermal stability experiments were measured by differential scanning fluorimetry (DSF) using SYPRO fluorescent dye as previously described77. RT-PCR excitation and emission wavelengths were set to 587 nm and 607 nm, respectively. Excess SYPRO orange was added to approximately 10 μM of protein. The temperature range spanned 20-95°C at a ramp rate of 1°C/min. Melting temperature values were determined by fitting the peak center of the first derivative of the melting curve fit to a Bigaussian function, with errors reported as standard fitting errors.
Analysis of known X-ray structures
TCR-pHLA X-ray structures were downloaded from the PDB. Structures with mutated HLA-I alleles, modified/non-peptidic epitopes or incomplete TCRs were not included. The final list of structures with 9-mer epitopes is available in Table S5B. For each structure, the contacts between the sidechains of the epitope and the TCR were computed, using a threshold of 4Å between heavy atoms. Epitope sidechains making direct contacts with the TCR are listed in Table S5B. Amino acid frequencies at these positions were computed and normalized by the average amino acid frequencies in the source proteins of the epitopes (for synthetic peptides, the amino acid frequency in the human proteome was used) (Figure 5E). As many structures contain redundant epitopes, each epitope, and the corresponding source protein, was weighted by the inverse of the number of epitopes that show at least 80% sequence identity, resulting in an effective number of sequences (i.e., sum of weights) equal to 22.44. Representative structures of each 9-mer epitope with aromatic sidechains interacting with the TCR are displayed in Figure S5C.
Analysis of TCGA data
TCGA mutations from BLCA, BRCA, CESC, COAD, GBM, HNSC, LGG, LIHC, LUAD, LUSC, OV, PAAD, PRAD, SKCM, STAD, THCA and UCEC tumor types were downloaded from the GDC data portal using the Aggregated Somatic Mutations files. Non-synonymous single nucleotide substitutions identified by MuSE, MuTect2, SomaticSniper and VarScan2 were considered. Mutations were mapped to the GRCh38 or GRCh37 proteome to retrieve all 9- to 11-mers encompassing each mutation. HLA-I typing was retrieved from The Cancer Immune Atlas (TCIA)78, and completed by data from Marty et al.32 for samples without HLA-I typing data in TCIA (8,317 tumor samples in total with available HLA-I typing, representing a total of 1,136,329 unique mutations, with 92.8% percent observed only once, 5.8% observed in two patients and 0.075% observed in more than five patients).
Expression values were used for all patients where they were available (7,736 patients) and a threshold of 1 FPKM was used to define genes that were poorly expressed. Clonality predictions were performed by comparing the mutant allelic fraction (average over all four variant callers) with the one expected after tumor purity correction. Tumor purity values were retrieved from Aran et al.79 or estimated with EPIC80 for samples absent from this study and with available expression data (8,185 samples in total). In first approximation, the expected mutant allelic fraction was defined as the coverage (based on the average of all four variant callers) multiplied by the purity and divided by 2. To account for variability in cancer genomic data and provide conservative predictions of sub-clonality, sub-clonal mutations were defined as those having a reported mutant allelic fraction lower than 50% of the expected one. Patients predicted to have altered antigen presentation machinery were defined as those harboring deleterious mutations (based on SIFT “deleterious” or “deleterious_low_confidence” predictions and PolyPhen “probably_damaging” or “possibly_damaging” predictions) in the HLA-A, HLA-B, HLA-C, B2M, TAP1, TAP2, TAPBP, TAPBPL, ERAP1 or ERAP2 genes. These filters removed 59.6% of actual mutation/patient pairs considered in Figure 6C.
Statistical model of immunoediting
To compute the frequency of a mutation among patients where it would give rise to neo-epitopes, and those where it would not, the PRIME %rank was computed for all 9- to 11-mers encompassing the mutation with each HLA-I allele of each patient. Patients with at least one peptide encompassing the mutated residue showing a PRIME %rank score lower or equal to 0.5% with at least one of the patient’s HLA-I alleles were assigned to the group where the mutation would be immunogenic (P+ in Figure 6A). Patients with all peptides showing a PRIME %rank larger or equal to 2% with all HLA-I alleles of the patient were assigned to the group where the mutation would not be immunogenic (). The frequency of the mutation was then computed within each group ( and ), where M indicates the set of patients that actually have the mutation and . Cases where P+ was equal to zero (< 2% of all mutations, including only PIK3CA_E542K among mutations observed more than 15 times) were not considered since the frequency could not be mathematically defined, although theses may represent another type of interesting immunoediting (i.e., mutations not predicted to be immunogenic in any patient). The mean value of the difference between and across mutations observed exactly N times or at least Nmin times are shown in Figures 6C and 6D. For mutations observed less than five times, a random subset of one thousand mutations was used.
To preserve existing linkage between HLA-I alleles in the analysis with randomized HLAs, all alleles of one patient were randomly swapped with those of another patient, further requesting that no patient gets the same HLA-I alleles as the original ones after randomizing. 20 different random seeds were used in this randomization of HLA-I alleles (mean and standard deviation shown with red circles and error bars in Figures 6C and 6D).
Quantification and statistical analysis
Enrichment analysis for neo-epitopes
Two-sided Wilcoxon tests were used to measure the enrichment of high scoring peptides among neo-epitopes in Figure 1C for each predictor.
Half-lives and EC50
For each epitope, average half-life was computed by taking the average of the logarithm of the half-lives measured for different clones (Table S4A). Similarly, the log of EC50 values for different clones (Table S4B) were averaged in Figure S3C. Pearson correlation coefficients used in Figures 3 and S3C and corresponding P values were computed with the cor.test function in R.
Correlation with ELISpot signals
Correlation between ELISpot signals (Figure 4C) or ranking (Figure S4I) and different predictors were measured with Spearman correlation and corresponding P values were computed with the cor.test function in R.
Analysis of TCGA data
Mutations were grouped based on their number of occurrences (N) in TCGA patients considered in this work. Pearson correlation between log(N) and < were computed in Figures 6C and 6D and statistical significance was assessed based on the cor.test function in R. Two-sided Wilcoxon test were used in the insets of Figures 6C and 6D.
Additional resources
The new immunogenicity predictor is available through the online interface http://prime.gfellerlab.org/ and code (including command-line executables) can be downloaded from: https://github.com/GfellerLab/PRIME.
Acknowledgments
We are thankful to Aymeric Auger for help with the FACS and Nathalie Rufer for sharing T cell clones. J.R.D. and B.M.B. acknowledge support from the National Institutes of Health (grants R35GM118166 andUL1TR002529). A.R.S. acknowledges support by a fellowship from the Walther Cancer Foundation Interdisciplinary Interface Training Project. J.R. and D.G. acknowledge support from the Swiss Cancer Research Foundation (KFS-4104-02-2017) and the University of Lausanne (Financement en Recherche Fondamentale). A.H. acknowledges support from the Swiss National Science Foundation (310030_182384). We are thankful to the authors of HLAthena for sharing a command line executable of their methods. We are thankful for Rachel Marty for sharing HLA-I typing information of the TCGA samples. The results here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
Author contributions
D.G. and A.H. designed the study; D.G. coordinated the project; D.G. performed the bioinformatics analyses; A.R.S., J.R.D., and B.M.B. performed the X-ray crystallography and TCR binding analyses; B.M.B., A.R.S., and D.G. analyzed protein structures; J.S., M.M., S.B., J.C., and A.H. performed the T cell assays; J.R., V.B., and D.G. developed the website; and J.R., S.J.C., D.E.S., G. Ciriello, M.B.-S., and G. Coukos provided reagents and feedback on the methods and manuscript. B.M.B. and D.G. wrote the manuscript.
Declaration of interests
The authors declare no competing interests. G. Ciriello is a member of the advisory board of Cell Reports Medicine.
Published: February 16, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2021.100194.
Contributor Information
Brian M. Baker, Email: brian-baker@nd.edu.
Alexandre Harari, Email: alexandre.harari@chuv.ch.
David Gfeller, Email: david.gfeller@unil.ch.
Supplemental information
List of immunogenic and non-immunogenic peptides used to train PRIME. References to studies (Column “StudyOrigin”) are: Bobisse5, Bassani-Sternberg55, Bentzen56, Cohen57, Strønen58, Kalaora59, McGranahan6, van Rooij60, Rajasagi61, Rizvi62, Robbins7, Snyder63, Wick64, Sahin3, Ott1, Dengue65, Calis29. Several neo-epitopes had been compiled in Bjerregaard et al.25. Pathogen/cancer testis in studies “Dengue” and “Calis,” as well as random peptides, were only used in the training of PRIME.
List of AUC and PRAUC values of each predictor used in Figures 1E and S2B–S2E for each study used iteratively as test set in the leave-one-study-out cross-validation and each HLA-I allele used iteratively in the leave-one-allele-out cross-validation.
(A) Structural avidity (i.e., half-lives; t1/2) measurements for different viral, tumor associated or cancer mutated epitopes. The different measurements correspond to distinct TCRs found in patients. (B) EC50 measurements for different viral, tumor associated or cancer mutated epitopes. The different measurements correspond to distinct TCRs found in patients. (C) Results of the IFNγ ELISpot assays for the P5 variants of the HIV (ALIRILQQL) and the CMV (NLVPMVATV) epitopes, after background corrections. 0 values indicate cases that could not be distinguished from the background. Standard deviations (SD) were computed based on two independent replicates, except for the CMV epitope with Donor 1 where the limited number of cells allowed for only one replicate. (D) Ranking of analogs of the CEA and MAGEA3 epitopes reported in Tangri et al.20 with variants at P5. The ranking was manually extracted from Figure 2 of this reference for all peptides that could be unambiguously identified there.
(A) X-ray data and refinement statistics for the SILv44-PMEL209(2M)-217/HLA-A∗02:01 structure. (B) List of TCR-pHLA X-ray structures used in Figure 5E. (C) List of structures of pHLA-TCR complexes used in Figure S5C with information about HLA-I restriction, peptide sequence, type of interactions mediated by aromatic sidechains and buried solvent accessible surface area (Å2).
References
- 1.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg S.A., Restifo N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 4.Zacharakis N., Chinnasamy H., Black M., Xu H., Lu Y.-C., Zheng Z., Pasetto A., Langhan M., Shelton T., Prickett T. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018;24:724–730. doi: 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobisse S., Genolet R., Roberti A., Tanyi J.L., Racle J., Stevenson B.J., Iseli C., Michel A., Le Bitoux M.-A., Guillaume P. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 2018;9:1092. doi: 10.1038/s41467-018-03301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robbins P.F., Lu Y.-C., El-Gamil M., Li Y.F., Gross C., Gartner J., Lin J.C., Teer J.K., Cliften P., Tycksen E. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells D.K., van Buuren M.M., Dang K.K., Hubbard-Lucey V.M., Sheehan K.C.F., Campbell K.M., Lamb A., Ward J.P., Sidney J., Blazquez A.B., Tumor Neoantigen Selection Alliance Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020;183:818–834.e13. doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadav M., Jhunjhunwala S., Phung Q.T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T.K., Fritsche J., Weinschenk T. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 10.Neefjes J., Jongsma M.L.M., Paul P., Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 11.Gfeller D., Bassani-Sternberg M. Predicting antigen presentation-what could we learn from a million peptides? Front. Immunol. 2018;9:1716. doi: 10.3389/fimmu.2018.01716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gfeller D., Guillaume P., Michaux J., Pak H.-S., Daniel R.T., Racle J., Coukos G., Bassani-Sternberg M. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 2018;201:3705–3716. doi: 10.4049/jimmunol.1800914. [DOI] [PubMed] [Google Scholar]
- 13.O’Donnell T.J., Rubinsteyn A., Laserson U. MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 2020;11:42–48.e7. doi: 10.1016/j.cels.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen M., Fenoy E., Harndahl M., Kristensen A.B., Nielsen I.K., Nielsen M., Buus S. Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J. Immunol. 2016;197:1517–1524. doi: 10.4049/jimmunol.1600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu T., Wang S., Xu L., Zhou Q., Singla N., Gao J., Manna S., Pop L., Xie Z., Chen M. Tumor neoantigenicity assessment with CSiN score incorporates clonality and immunogenicity to predict immunotherapy outcomes. Sci. Immunol. 2020;5:eaaz3199. doi: 10.1126/sciimmunol.aaz3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller M., Gfeller D., Coukos G., Bassani-Sternberg M. ‘Hotspots’ of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization. Front. Immunol. 2017;8:1367. doi: 10.3389/fimmu.2017.01367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stranzl T., Larsen M.V., Lundegaard C., Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–368. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan F., Duitama J., Al Seesi S., Ayres C.M., Corcelli S.A., Pawashe A.P., Blanchard T., McMahon D., Sidney J., Sette A. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014;211:2231–2248. doi: 10.1084/jem.20141308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capietto A.-H., Jhunjhunwala S., Pollock S.B., Lupardus P., Wong J., Hänsch L., Cevallos J., Chestnut Y., Fernandez A., Lounsbury N. Mutation position is an important determinant for predicting cancer neoantigens. J. Exp. Med. 2020;217:e20190179. doi: 10.1084/jem.20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balachandran V.P., Łuksza M., Zhao J.N., Makarov V., Moral J.A., Remark R., Herbst B., Askan G., Bhanot U., Senbabaoglu Y., Australian Pancreatic Cancer Genome Initiative. Garvan Institute of Medical Research. Prince of Wales Hospital. Royal North Shore Hospital. University of Glasgow. St Vincent’s Hospital. QIMR Berghofer Medical Research Institute. University of Melbourne, Centre for Cancer Research. University of Queensland, Institute for Molecular Bioscience. Bankstown Hospital. Liverpool Hospital. Royal Prince Alfred Hospital, Chris O’Brien Lifehouse. Westmead Hospital. Fremantle Hospital. St John of God Healthcare. Royal Adelaide Hospital. Flinders Medical Centre. Envoi Pathology. Princess Alexandria Hospital. Austin Hospital. Johns Hopkins Medical Institutes. ARC-Net Centre for Applied Research on Cancer Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bjerregaard A.-M., Nielsen M., Jurtz V., Barra C.M., Hadrup S.R., Szallasi Z., Eklund A.C. An analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol. 2017;8:1566. doi: 10.3389/fimmu.2017.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richman L.P., Vonderheide R.H., Rech A.J. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019;9:375–382.e4. doi: 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devlin J.R., Alonso J.A., Ayres C.M., Keller G.L.J., Bobisse S., Vander Kooi C.W., Coukos G., Gfeller D., Harari A., Baker B.M. Structural dissimilarity from self drives neoepitope escape from immune tolerance. Nat. Chem. Biol. 2020;16:1269–1276. doi: 10.1038/s41589-020-0610-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowell D., Krishna S., Becker P.D., Cocita C., Shu J., Tan X., Greenberg P.D., Klavinskis L.S., Blattman J.N., Anderson K.S. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. USA. 2015;112:E1754–E1762. doi: 10.1073/pnas.1500973112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calis J.J.A., Maybeno M., Greenbaum J.A., Weiskopf D., De Silva A.D., Sette A., Keşmir C., Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013;9:e1003266. doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahin U., Oehm P., Derhovanessian E., Jabulowsky R.A., Vormehr M., Gold M., Maurus D., Schwarck-Kokarakis D., Kuhn A.N., Omokoko T. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–112. doi: 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- 31.Dunn G.P., Old L.J., Schreiber R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 32.Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J., Carter H. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Eynden J., Jiménez-Sánchez A., Miller M.L., Larsson E. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet. 2019;51:1741–1748. doi: 10.1038/s41588-019-0532-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bassani-Sternberg M., Chong C., Guillaume P., Solleder M., Pak H., Gannon P.O., Kandalaft L.E., Coukos G., Gfeller D. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 2017;13:e1005725. doi: 10.1371/journal.pcbi.1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulik-Sullivan B., Busby J., Palmer C.D., Davis M.J., Murphy T., Clark A., Busby M., Duke F., Yang A., Young L. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018;37:55–63. doi: 10.1038/nbt.4313. [DOI] [PubMed] [Google Scholar]
- 36.Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016;7:13404. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bentzen A.K., Marquard A.M., Lyngaa R., Saini S.K., Ramskov S., Donia M., Such L., Furness A.J.S., McGranahan N., Rosenthal R. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016;34:1037–1045. doi: 10.1038/nbt.3662. [DOI] [PubMed] [Google Scholar]
- 38.Cohen C.J., Gartner J.J., Horovitz-Fried M., Shamalov K., Trebska-McGowan K., Bliskovsky V.V., Parkhurst M.R., Ankri C., Prickett T.D., Crystal J.S. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest. 2015;125:3981–3991. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strønen E., Toebes M., Kelderman S., van Buuren M.M., Yang W., van Rooij N., Donia M., Böschen M.-L., Lund-Johansen F., Olweus J., Schumacher T.N. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 40.Kalaora S., Barnea E., Merhavi-Shoham E., Qutob N., Teer J.K., Shimony N., Schachter J., Rosenberg S.A., Besser M.J., Admon A., Samuels Y. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget. 2016;7:5110–5117. doi: 10.18632/oncotarget.6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Rooij N., van Buuren M.M., Philips D., Velds A., Toebes M., Heemskerk B., van Dijk L.J.A., Behjati S., Hilkmann H., El Atmioui D. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013;31:e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajasagi M., Shukla S.A., Fritsch E.F., Keskin D.B., DeLuca D., Carmona E., Zhang W., Sougnez C., Cibulskis K., Sidney J. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124:453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wick D.A., Webb J.R., Nielsen J.S., Martin S.D., Kroeger D.R., Milne K., Castellarin M., Twumasi-Boateng K., Watson P.H., Holt R.A., Nelson B.H. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin. Cancer Res. 2014;20:1125–1134. doi: 10.1158/1078-0432.CCR-13-2147. [DOI] [PubMed] [Google Scholar]
- 46.Weiskopf D., Yauch L.E., Angelo M.A., John D.V., Greenbaum J.A., Sidney J., Kolla R.V., De Silva A.D., de Silva A.M., Grey H. Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. J. Immunol. 2011;187:4268–4279. doi: 10.4049/jimmunol.1101970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen M., Lundegaard C., Lund O., Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 48.Peters B., Bulik S., Tampe R., Van Endert P.M., Holzhütter H.-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 2003;171:1741–1749. doi: 10.4049/jimmunol.171.4.1741. [DOI] [PubMed] [Google Scholar]
- 49.Allard M., Couturaud B., Carretero-Iglesia L., Duong M.N., Schmidt J., Monnot G.C., Romero P., Speiser D.E., Hebeisen M., Rufer N. TCR-ligand dissociation rate is a robust and stable biomarker of CD8+ T cell potency. JCI Insight. 2017;2:e92570. doi: 10.1172/jci.insight.92570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hebeisen M., Allard M., Gannon P.O., Schmidt J., Speiser D.E., Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front. Immunol. 2015;6:582. doi: 10.3389/fimmu.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt J., Guillaume P., Irving M., Baumgaertner P., Speiser D., Luescher I.F. Reversible major histocompatibility complex I-peptide multimers containing Ni(2+)-nitrilotriacetic acid peptides and histidine tags improve analysis and sorting of CD8(+) T cells. J. Biol. Chem. 2011;286:41723–41735. doi: 10.1074/jbc.M111.283127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tangri S., Ishioka G.Y., Huang X., Sidney J., Southwood S., Fikes J., Sette A. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J. Exp. Med. 2001;194:833–846. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Z., Theoret M.R., Touloukian C.E., Surman D.R., Garman S.C., Feigenbaum L., Baxter T.K., Baker B.M., Restifo N.P. Poor immunogenicity of a self/tumor antigen derives from peptide-MHC-I instability and is independent of tolerance. J. Clin. Invest. 2004;114:551–559. doi: 10.1172/JCI21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eby J.M., Smith A.R., Riley T.P., Cosgrove C., Ankney C.M., Henning S.W., Paulos C.M., Garrett-Mayer E., Luiten R.M., Nishimura M.I. Molecular properties of gp100-reactive T-cell receptors drive the cytokine profile and antitumor efficacy of transgenic host T cells. Pigment Cell Melanoma Res. 2019;32:68–78. doi: 10.1111/pcmr.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaretsky J.M., Garcia-Diaz A., Shin D.S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D.Y., Abril-Rodriguez G., Sandoval S., Barthly L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DuPage M., Mazumdar C., Schmidt L.M., Cheung A.F., Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Angelova M., Mlecnik B., Vasaturo A., Bindea G., Fredriksen T., Lafontaine L., Buttard B., Morgand E., Bruni D., Jouret-Mourin A. Evolution of metastases in space and time under immune selection. Cell. 2018;175:751–765.e16. doi: 10.1016/j.cell.2018.09.018. [DOI] [PubMed] [Google Scholar]
- 59.Singh N.K., Riley T.P., Baker S.C.B., Borrman T., Weng Z., Baker B.M. Emerging concepts in TCR specificity: rationalizing and (maybe) predicting outcomes. J. Immunol. 2017;199:2203–2213. doi: 10.4049/jimmunol.1700744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perdivara I., Deterding L.J., Przybylski M., Tomer K.B. Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: chemical artifact or post-translational modification? J. Am. Soc. Mass Spectrom. 2010;21:1114–1117. doi: 10.1016/j.jasms.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castro A., Ozturk K., Pyke R.M., Xian S., Zanetti M., Carter H. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med. Genomics. 2019;12(Suppl 6):107. doi: 10.1186/s12920-019-0544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang F., Kim D.-K., Nakagawa H., Hayashi S., Imoto S., Stein L., Roth F.P. Quantifying immune-based counterselection of somatic mutations. PLoS Genet. 2019;15:e1008227. doi: 10.1371/journal.pgen.1008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zapata L., Pich O., Serrano L., Kondrashov F.A., Ossowski S., Schaefer M.H. Negative selection in tumor genome evolution acts on essential cellular functions and the immunopeptidome. Genome Biol. 2018;19:67. doi: 10.1186/s13059-018-1434-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., PCAWG Mutational Signatures Working Group. PCAWG Consortium The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran E., Ahmadzadeh M., Lu Y.-C., Gros A., Turcotte S., Robbins P.F., Gartner J.J., Zheng Z., Li Y.F., Ray S. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pajot A., Michel M.-L., Fazilleau N., Pancré V., Auriault C., Ojcius D.M., Lemonnier F.A., Lone Y.-C. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur. J. Immunol. 2004;34:3060–3069. doi: 10.1002/eji.200425463. [DOI] [PubMed] [Google Scholar]
- 67.Hebeisen M., Schmidt J., Guillaume P., Baumgaertner P., Speiser D.E., Luescher I., Rufer N. Identification of rare high-avidity, tumor-reactive CD8+ T cells by monomeric TCR-ligand off-rates measurements on living cells. Cancer Res. 2015;75:1983–1991. doi: 10.1158/0008-5472.CAN-14-3516. [DOI] [PubMed] [Google Scholar]
- 68.Boucherma R., Kridane-Miledi H., Bouziat R., Rasmussen M., Gatard T., Langa-Vives F., Lemercier B., Lim A., Bérard M., Benmohamed L. HLA-A∗01:03, HLA-A∗24:02, HLA-B∗08:01, HLA-B∗27:05, HLA-B∗35:01, HLA-B∗44:02, and HLA-C∗07:01 monochain transgenic/H-2 class I null mice: novel versatile preclinical models of human T cell responses. J. Immunol. 2013;191:583–593. doi: 10.4049/jimmunol.1300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis-Harrison R.L., Armstrong K.M., Baker B.M. Two different T cell receptors use different thermodynamic strategies to recognize the same peptide/MHC ligand. J. Mol. Biol. 2005;346:533–550. doi: 10.1016/j.jmb.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 70.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 71.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 72.Terwilliger T.C., Grosse-Kunstleve R.W., Afonine P.V., Moriarty N.W., Zwart P.H., Hung L.W., Read R.J., Adams P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blevins S.J., Baker B.M. Using global analysis to extend the accuracy and precision of binding measurements with T cell receptors and their peptide/MHC ligands. Front. Mol. Biosci. 2017;4:2. doi: 10.3389/fmolb.2017.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hellman L.M., Yin L., Wang Y., Blevins S.J., Riley T.P., Belden O.S., Spear T.T., Nishimura M.I., Stern L.J., Baker B.M. Differential scanning fluorimetry based assessments of the thermal and kinetic stability of peptide-MHC complexes. J. Immunol. Methods. 2016;432:95–101. doi: 10.1016/j.jim.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 79.Aran D., Sirota M., Butte A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Racle J., de Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife. 2017;6:1865. doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of immunogenic and non-immunogenic peptides used to train PRIME. References to studies (Column “StudyOrigin”) are: Bobisse5, Bassani-Sternberg55, Bentzen56, Cohen57, Strønen58, Kalaora59, McGranahan6, van Rooij60, Rajasagi61, Rizvi62, Robbins7, Snyder63, Wick64, Sahin3, Ott1, Dengue65, Calis29. Several neo-epitopes had been compiled in Bjerregaard et al.25. Pathogen/cancer testis in studies “Dengue” and “Calis,” as well as random peptides, were only used in the training of PRIME.
List of AUC and PRAUC values of each predictor used in Figures 1E and S2B–S2E for each study used iteratively as test set in the leave-one-study-out cross-validation and each HLA-I allele used iteratively in the leave-one-allele-out cross-validation.
(A) Structural avidity (i.e., half-lives; t1/2) measurements for different viral, tumor associated or cancer mutated epitopes. The different measurements correspond to distinct TCRs found in patients. (B) EC50 measurements for different viral, tumor associated or cancer mutated epitopes. The different measurements correspond to distinct TCRs found in patients. (C) Results of the IFNγ ELISpot assays for the P5 variants of the HIV (ALIRILQQL) and the CMV (NLVPMVATV) epitopes, after background corrections. 0 values indicate cases that could not be distinguished from the background. Standard deviations (SD) were computed based on two independent replicates, except for the CMV epitope with Donor 1 where the limited number of cells allowed for only one replicate. (D) Ranking of analogs of the CEA and MAGEA3 epitopes reported in Tangri et al.20 with variants at P5. The ranking was manually extracted from Figure 2 of this reference for all peptides that could be unambiguously identified there.
(A) X-ray data and refinement statistics for the SILv44-PMEL209(2M)-217/HLA-A∗02:01 structure. (B) List of TCR-pHLA X-ray structures used in Figure 5E. (C) List of structures of pHLA-TCR complexes used in Figure S5C with information about HLA-I restriction, peptide sequence, type of interactions mediated by aromatic sidechains and buried solvent accessible surface area (Å2).
Data Availability Statement
PRIME is freely available for academic researchers at the web interface http://prime.gfellerlab.org and as command-line tool https://github.com/GfellerLab/PRIME. All data used to train and validate PRIME are available in Supplementary Materials (Tables S1 and S4). The accession number for the X-ray structure reported in this paper is PDB: 6VM8.