

Abstract
Protein chemical modifications are important tools for elucidating chemical and biological functions of proteins. Several strategies have been developed to implement these modifications, including enzymatic tailoring reactions, unnatural amino acid incorporation using the expanded genetic codes, and recognition-driven transformations. These technologies have been applied in metalloenzyme studies, specifically in dissecting their mechanisms, improving their enzymatic activities, and creating artificial enzymes with non-natural activities. Herein, we summarize some of the recent efforts in these areas with an emphasis on a few metalloenzyme case studies.
Keywords: Metalloenzymes, Biorthogonal reactions, Protein engineering, Non-canonical amino acids, Biocatalysis
Proteins have a remarkable range of catalytic, structural, and regulatory functions using the 20 canonical amino acids (cAAs) as their building blocks. The functions of proteins are further diversified to a level well beyond those possessed by the 20 cAAs through enzymatic-driven post-translational modifications (PTMs), such as methylations, sulfations, phosphorylations, and glycosylations. Moreover, chemical modifications of a protein of interest (POI) with special probes enable functional characterizations of the POI, including dynamics, localization, and protein-protein interactions [1,2]. In pharmaceutical settings, the modification of protein therapeutics with polyethylene glycol (PEG) enhances their stability and circulation half-life [3]. Due to these reasons, a wide variety of protein modification methodologies have been explored to acquire exquisite control of these macromolecules. In this review, we discuss some of the recent strategies for protein modifications as well as several metalloenzymes case studies focusing on mechanistic investigation and enzyme engineering.
1. Chemical modifications of proteins
Protein chemical modification approaches can be roughly classified into three categories: 1) modifications via the reactivities of cAAs; 2) ribosomal-mediated incorporation of noncanonical amino acids (ncAAs); 3) modifications via affinity-driven ligand-directed reactions. This section presents the recent progress in each of these approaches. The reactions discussed in this section are summarized in Table 1.
Table 1.
Chemical modifications of cAAs.
| Lys | Lys is a convenient nucleophilic handle for many reactions. Site-selective Lys modification is challenging, yet, may be achieved by harnessing the pKa differences among various Lys residues. |
| Cys | The thiol of Cys can be modified using many different reactions, e.g., alkylation and thiol-ene chemistry. However, some of the resulting adducts may not be stable. New thiol-targeting modification reagents are being developed to obtain stable conjugates. |
| Tyr | The Tyr sidechain may exist in a phenol or a phenolate form. This allows selective modification by controlling the pH of the reaction. Common reactions include diazonium couplings and alkylation via π-allylpalladium complexes. |
| Trp | The Trp indole moiety offers an opportunity for selective modification via metal-mediated C–H functionalization reaction. |
1.1. Chemical modifications of canonical amino acids (cAAs)
Chemical modifications of amino acids offer straightforward approaches to modify proteins. Both the reactivity and relative abundance of these residues play crucial roles in determining the selectivity of the chemical modifications. Reactions targeting highly abundant residues such as Lys often result in a mixture of modified proteins; nevertheless, significant progress has been made to obtain site-selective Lys modifications [1,2]. On the other hand, low abundance residues such as Cys and Tyr might offer an opportunity for site-selective protein modifications. Many of these reactions can be performed within living cells as part of routine experiments. Currently, methods applicable for studies in multicellular organisms are of great interest in this field.
1.1.1. Lysine modifications
Among the 20 cAAs, Lys is an attractive choice for modifications due to both its high natural abundance and the number of biocompatible chemical reactions with its nucleophilic primary amine. Lys can be modified using electrophiles, such as squaric acids, isothiocyanates, sulfonyl chlorides, activated esters (including N-hydroxysuccinimide-based [NHS]), and 2-amino-2-methoxyethyl (Fig. 1a–e) [1,4,5]. Because of its high natural abundance, Lys modifications are often useful in cases where site-selectivity is not crucial or where multiple modification sites are desired. For instance, the NHS-based ortho-nitrobenzyl esters and aldehydes are used to modify vitronectin, an extracellular matrix glycoprotein, in photopatternable hydrogels. This system provides simplified environments for studying cellular responses to physiological cues [6]. By reversibly patterning the presentation of vitronectin on the hydrogel, spatial and temporal control over osteogenic stem cell differentiation is achieved.
Fig. 1.

Strategies for Lys chemical modifications. (a) The squaric acid ester reactions, (b) formation of thiourea using isothiocyanate, (c) the sulfonyl chloride reactions, (d) production of amides using the NHS-ester, and (e) the 2-imino-2-methoxyethyl reactions.
Due to its high natural abundance, site-specific modifications of Lys residues are challenging. In recent years, methods for achieving site-selectivity have emerged. In one case, by taking advantage of the different reactivities of Lys residues in various local microenvironments, Matos et al. computationally designed sulfonyl acrylates which afforded chemo- and regio-selective modifications [7]. Using the designed sulfonyl acrylates, a single Lys residue in several proteins, including the therapeutic antibody Trastuzumab, was modified without disturbing the structure and function of the POIs. With this method, an improved site-selectivity is achieved because the reaction favors Lys residues with the lowest pKa in the POIs (Lys33 of lysozyme with a pKa of 9.5, Lys300 of annexin V with a pKa of 10.3, and Lys100 of C2Am with a pKa of 10.1). However, this strategy might not be applicable in some POIs where the Lys with the lowest pKa is inaccessible. In another case, to prepare an antibody-drug conjugate (ADC), Rader and co-workers site-specifically conjugated β-lactam derivatives to a Lys residue (pKa ~6) in the h38C2 antibody via aldol and retro-aldol reactions [8]. This approach results in homogenous ADC, which might be useful for further therapeutic development.
1.1.2. N-terminal protein modifications
In addition to Lys residues, the N-terminal primary amine of a protein also displays unique reactivity. This site-selective modification is commonly used in peptide synthesis and has been applied in native chemical ligation (NCL) reactions (Fig. 2a) [9]. In NCL reactions, the thiolate of the N-terminal Cys residue of one peptide reacts with the C-terminal thioester of another peptide. The resulting thioester intermediate undergoes an acyl shift to form a native peptide bond (Fig. 2a). An alternative method for N-terminal modifications is the expressed protein ligation (EPL) method, where a recombinant protein can be conjugated to a synthetic peptide containing the desired modification (Fig. 2b) [[10], [11], [12]]. This approach enables site-selective modification when the desired site is close to the N- or C-terminus of a POI. For example, the N-terminal ultrafast split intein was genetically fused to the C-terminal of histone H2B, which then reacted with a synthetic C-terminal intein fragment bearing a pre-ubiquitinated Lys120. The reaction between the two intein fragments results in a ubiquitinated H2B (H2B–K120Ub) [13]. The semisynthetic H2B–K120Ub is operative and can induce chromatin signaling in isolated nuclei. Additionally, the N-terminus has been used to generate reactive ketones through a pyridoxal-5′-phosphate (PLP) catalyzed transamination reaction; however, this reaction is not compatible with some N-terminal residues, such as Lys and Gln due to side reactions [[14], [15], [16]]. The resulting ketone or aldehyde can be used for the oxime/hydrazine ligation. For instance, the N-terminus of streptavidin was modified to an α-ketoamide allowing its immobilization onto micropatterned surfaces through oxime ligation [17]. This approach immobilizes the proteins in a specific orientation, which is critical for maintaining their bioactivity. Moreover, strategies have been developed to modify the N-termini with small molecules, such as a reductive alkylation using an aldehyde-containing 2-pyridinecarboxaldehyde (2-PCA) [18]. This N-alkylation reaction has been applied to install a secondary amine on human insulin. The resulting secondary amine preserves the positive charge on the N-terminus of human insulin, and the bioactivity of the modified insulin is comparable to that of unmodified insulin. Despite these advances, there remain many challenges. For example, the N-termini of many proteins may not be accessible for modifications or might be essential for their biological/catalytic activities. In addition, a careful control over the reaction conditions is required for N-terminal modifications.
Fig. 2.

Strategies for modifying a protein's N-terminus. (a) The NCL approach, using one peptide with an N-terminal Cys and a second peptide with a C-terminal thioester. (b) A semisynthetic protein is generated using EPL. In this method, the N-terminal fragment of the POI is fused with an intein and the fusion protein is produced by overexpression. A synthetic peptide fragment containing the desired modifications and with an N-terminal Cys is then used to replace the ultrafast split intein, creating the target protein with the desired modifications at the sites of interest. The red stars represent protein modifications. This figure is adapted from Ref. [12].
1.1.3. Cysteine modifications
Cys is another residue of choice for chemical modifications due to the high nucleophilicity of its thiol and its low natural abundance in proteins, both of which favor single-site modification in POIs [1,5,19]. Under well-controlled pH conditions, selective modification of a Cys residue over other nucleophilic residues such as Lys and His can be obtained [20]. The Cys thiol can be modified using electrophilic α-halocarbonyls (iodoacetyl, bromoacetyl, or chloroacetyl) [21,22], maleimides [23,24], methanesulfonate, or phenylthiosulfonate (Fig. 3a) [25,26]. Maleimides have been widely used to synthesize ADCs, including the FDA-approved ADCs, e.g., brentuximab vedotin, trastuzumab emtansine, and certolizumab pegol [2]. However, the thioether of maleimide conjugates can undergo cleavage via the thiol-exchange or hydrolysis-driven reactions. The resulting products from thiol exchange lead to premature loss of the drug's efficacy and increased toxicity in vivo [27]. These issues can be addressed by minimizing the thiol exchange reaction or by converting the maleimide thio-adducts to stable ring-open conjugates. The latter has been accomplished through the development of new self-hydrolyzing maleimides, which exhibit superior pharmacokinetic properties [28].
Fig. 3.

Methods for Cys residue modifications. (a) Chemical modifications of Cys with commonly used reagents such as halocarbonyls, maleimides, sulfones. (b) Conjugation of antibodies to dyes via the amine-to-thiol coupling reagent, CBTF. (c) ADC construction via disulfide bridging using dibromomaleimide. All blue spheres represent other inert functional groups in the probe and the red stars represent modifications to be incorporated.
In recent efforts, new generations of thiol-targeting modification reagents have been developed, including electron-deficient alkynes [29], 3-arylpropiolonitrile [30,31], allenamides [32], the thiol-yne reactions [33], and carbonylacrylic reagents [34,35]. For example, Wagner et al. reported an amine-to-thiol coupling reagent, sodium 4-((4-(cyanoethynyl)benzoyl)oxy)-2,3,5,6-tetrafluorobenzenesulfonate (CBTF), which contains an arylpropionitrile functional group instead of a maleimide (Fig. 3b). The resulting conjugates exhibit superior stability in plasma compared to that of maleimide conjugates [31]. In a recent study, Bernardes et al. rationally designed carbonylacrylic reagents, which undergo thiol-Michael addition using the Cys residues from of the POIs [34]. Multiple proteins, including antibodies, were modified using this approach. The modified antibodies were not only homogenous, but also resistant to degradation in plasma. Additional approaches for the Cys-selective protein modification via vinyl/alkyl pyridine, azanorbornadiene bromovinyl sulfone, and diazo reagents, have been reported by the Bernardes Group [[35], [36], [37]].
In cases where the POIs lack a thiol functional group, disulfide modification serves as an alternative target. Multiple reagents including bissulfones, allyl sulfones, alkynes, and 3,4-disubstituted maleimides have been developed to site-selectively modify the disulfides of proteins as summarized in recent reviews [5,38]. For instance, using 2,3-dibromo maleimide with a C-2 (glycine derived) linker, Doxorubicin (DOX)-antibody conjugates were produced through a bis-alkylation reaction (Fig. 3c). This approach results in homogenous ADCs with enhanced pharmacological properties [39]. Oxetane, an oxygen (ether) containing four-membered ring, has also be used to modify protein disulfides via a site-selective bis-alkylation reaction [40,41]. In one of the reports, oxetane was installed onto a genetically detoxified diphtheria toxin (CRM197 protein) and the resulting modified protein exhibits increased immunogenicity in vivo [40].
1.1.4. Aromatic residue modifications
In addition to Cys, the relatively low natural abundance of aromatic residues, including His, Tyr, Trp, and Phe, offer alternative targets for site-specific modifications. However, obtaining a site-specific modification for one aromatic residue over another remains challenging. The reactivity of the ionizable side chain of Tyr is dependent on its protonation state, which allows the reactivity of Tyr to be modulated by controlling the pH of the reactions. Under acidic conditions, the aromatic ε-carbons adjacent to the hydroxyl group may undergo diazonium couplings (Fig. 4a) [42]. In a recent study, salmon's calcitonin was conjugated to linear monomethoxy PEG using this approach. The resulting conjugates maintain the ability to reduce the concentration of calcium ions in the plasma. In conditions where the pH approaches the pKa of the phenol side chain of the Tyr residue (pKa of ~10), alkylation or acylation reactions of the oxygen can occur [[43], [44], [45]]. Another approach for site-selective Tyr modification is to use the π-allylpalladium complexes (Fig. 4b) [43]. Using this method, a novel polarity-sensitive fluorescent probe was installed onto Tyr108 of a bovine Cu/Zn superoxide dismutase (SOD) which allowed the conformational changes of the Tyr-containing domain to be monitored. This example might serve as a general approach for studying protein conformational changes if the domain of interest has a Tyr residue.
Fig. 4.

Methods for Tyr modification. (a) Chemical modification of Tyr via diazonium couplings. (b) Conjugation of Tyr in SOD to a fluorescent probe using π-allylpalladium complexes. The blue spheres represent the remaining functional group.
In the case of Trp, modifications of its side chain via metal-catalyzed reactions have been developed (Fig. 5), including alkynylations [46], C–H arylations [47], and photoinduced cycloadditions of tetrazoles [48]. However, these approaches often involve non-biocompatible reaction conditions such as the use of organic solvents and at high temperatures. An alternative strategy for selective modification of Trp consists of an N-oxy radical, 9-azabicyclo[3.3.1]nonane-3-one-N-oxy, coupled with NaNO2 under ambient conditions (Fig. 5) [49]. Using this approach, Kanai and co-workers achieved Trp-selective modifications on myoglobin, lysozyme, BSA, and the β2-microglobulin antibody. This study opens new avenues for Trp modifications without using toxic metals under biocompatible conditions, further paving the way for functional modulation of therapeutic targets such as antibodies. In addition to the aforementioned cases, efforts for site-specific modifications of other proteinogenic residues are well summarized in recent reviews [[50], [51], [52]].
Fig. 5.

Metal-free chemical modifications of Trp residues using the N-oxy radical. The pink spheres represent other functional groups in the probes.
1.2. Protein modifications using ncAAs
Protein modifications using chemical reactions are powerful approaches to selectively install the desired chemical handles on the POIs. Manipulating the protein translation machinery for direct incorporation of ncAAs is another desirable approach, which enables the introduction of novel functionalities and reactivities beyond those offered by the 20 cAAs. Incorporating ncAAs into proteins has been widely applied in biocatalysis to enhance the activity and selectivity of enzymes, and even allows researchers to obtain novel catalytic reactions that are naturally unavailable [[53], [54], [55]]. This section presents some of the recent examples of using the genetic code expansion method to incorporate ncAAs into the POIs.
1.2.1. Genetic code expansion
The natural genetic code is comprised of 64 codons encoded by four nucleobases. Among these, three codons serve as stop codons, while the remaining 61 are recognized by transfer RNAs (tRNAs), and are charged by their cognate tRNA synthetases (aaRSs) with one of the cAAs. All organisms encode the same 20 amino acids (AAs) with the exceptions of pyrrolysine [56] and selenocysteine [57]. The ability to employ and modify the existing protein translation machinery to incorporate ncAAs into proteins has opened new avenues for protein modifications. One of the strategies relies on the promiscuity of some tRNAs, which can be charged with ncAAs that are structurally analogous to their cognate amino acids (Fig. 6a) [58]. In this method, an auxotrophic bacterial host cannot produce the cAA and relies on the exogenously supplied ncAA. This approach results in a global incorporation of the ncAA to the proteome. One of the prominent examples is the substitution of Met with Se-Met to incorporate heavy atoms into the POIs for crystallographic phasing [59]. This strategy is straightforward and does not require genetic manipulation of the protein translation components. Instead, this method relies on the polyspecificity of the aaRS-tRNA pair, meaning that only ncAAs with structural similarities to the cognate amino acids can be incorporated, limiting the number of modifications. Additionally, global incorporation of ncAAs might alter protein folding, which could result in a perturbation of the activity and/or stability of the POIs.
Fig. 6.

Strategies for incorporating ncAAs in vivo. (a) Residue-specific incorporation of ncAAs using the endogenous aaRS-tRNA pair of an auxotrophic host. (b) Site-specific incorporation of ncAAs via the amber codon suppression method using an orthogonal aaRS-tRNA pair.
A powerful alternative approach to site-specifically incorporate ncAAs into the POIs is via genetic code expansion (Fig. 6b). In this method, the amber stop codon (UAG) is used to encode the ncAA of interest because of its low usage in Escherichia coli (~7–8%). The amber codon is recognized by an engineered aaRS-tRNA pair for the ncAA of interest. The aaRS-tRNA pair must also be orthogonal, i.e., not interfering with the endogenous translation system (Fig. 6b). For example, the tyrosyl-tRNA synthetase TyrRS-tRNACUA pair from Methanocaldococcus jannaschii is orthogonal in E. coli and other bacteria; the TyrRS-tRNACUA and LeuRS-tRNACUA pairs from E. coli are orthogonal in eukaryotic cells; the pyrrolysyl-tRNA synthetase PylRS-tRNACUA pairs from Methanosarcina barkeri and Methanosarcina mazei are orthogonal in both bacteria and eukaryotic cells [54,60,61]. Site-specifically modified POIs can be obtained, but the production yield is normally limited by the expression level of the exogenous aaRS-tRNA pairs and the presence of release factor 1 (RF-1), which recognizes the UAG triplet and terminates translation. Recently, an E. coli host has been engineered by removing RF-1 from the E. coli genome. Additionally, 95 out of the 273 amber stop codons were replaced with other more frequently used stop codons. After this engineering, the growth defects of the E. coli host were minimized when it was used to overexpress ncAA-containing proteins [62,63]. Most importantly, the ncAA incorporation efficiency is >98% in this engineered host strain, allowing a scalable production of the target ncAA-containing protein.
1.2.2. Next-generation genetic code expansion
To date, more than 200 ncAAs have been incorporated into POIs using the amber suppression method, thereby expanding the chemical functionalities and reactivities of proteins [54,64]. Thus far, the vast majority of studies employing this technology are restricted to the incorporation of single ncAAs into the POIs. The ability to incorporate multiple ncAAs into a protein might offer new opportunities for advanced biophysical studies and the synthesis of enhanced protein-based therapeutics. To achieve such goals, the enhanced specificity and orthogonality of aaRS-tRNA pairs is essential. Orthogonal aaRS-tRNA pairs can charge multiple ncAAs during the translation process. Thus, to site-selectively incorporate multiple ncAAs, the orthogonal aaRS-tRNA pairs must be highly selective. Several engineering approaches have been developed to address these challenges. Liu et al. employed the phage-assisted continuous evolution (PACE) technology to evolve a PylRS variant with 45-fold higher catalytic efficiency compared to that of the wild-type [65]. Through this technology, a TyrRS variant with improved selectivity toward p-iodo-l-phenylalanine was also obtained. Engineering of the aaRS-tRNA pairs has been proven useful in incorporating ncAAs at multiple sites. In a recent study, Chatterjee and co-workers simultaneously incorporated three distinct bioconjugation handles onto the GFP protein by employing three orthogonal pairs to decode three different codons. These pairs include the E. coli-derived tryptophanyl pair, the archaeal tyrosyl, and pyrrolysyl pairs. This work allows facile multiple-site labeling of proteins without an exogenous chemical catalyst. However, the incorporation efficiency is only 2% of the wild-type yield [66,67].
Besides improving the specificity of the aaRS-tRNA pair, evolving other translation machinery components has also been important for the efficient incorporation of ncAAs into multiple sites of the POIs. To facilitate the incorporation of ncAAs with bulky side-chains or altered backbones, ribosome engineering has been attempted. The small subunit of the ribosome contains 16S rRNA and binds mRNA, while the large subunit contains 23S rRNA. Both subunits are involved in coordinating the translation process. There are two major challenges in ribosome engineering. First, the activity and fidelity of ribosomes are essential for cellular survival. Second, the 23S rRNA subunit can freely exchange between the native and the orthogonal 16S rRNAs in the cell.
To overcome the former issue, orthogonal (O-)ribosome-mRNA pairs were evolved [68,69]. In these pairs, an O-ribosome containing O–16S rRNA and mutations in the anti-Shine-Dalgarno (ASD) sequence was introduced in E. coli (Fig. 7a). This O-ribosome can selectively translate an orthogonal mRNA (O-mRNA) containing the O-ribosome-binding site, but not native mRNA transcripts. The O-ribosome was further engineered to incorporate different ncAAs into proteins [68,70]. The endogenous and orthogonal ribosomes share the 23S rRNA. Therefore, further efforts on 23S rRNA engineering have also been attempted. These efforts amplified the contribution of new 23S rRNA to O-mRNA translation and minimized the lethal effect of new 23S rRNA in endogenous translation processes. To achieve this goal, Chin et al. reported an orthogonal ribosome with the two subunits linked through an optimized orthogonal RNA staple [71]. This engineered stapled ribosome can maintain activity comparable to that of the orthogonal parent ribosome with minimized association with the endogenous 16S or 23S subunit (Fig. 7a). This technology opens avenues for further improvement of the orthogonal ribosomes to incorporate more ncAAs [71].
Fig. 7.

Engineering of other translational machinery components. (a) Aggregated ribosome engineering efforts enable the incorporation of a wide variety of ncAAs. (b) Engineering of the ribosome to efficiently decode quadruplet codons.
Other exciting technologies for genetic code expansion extend beyond the canonical 64 codons. The first glimpse of quadruplet codon usage was reported from Salmonella typhimurium, in which a tRNAGly containing the frameshifted CCCC anticodon was observed [72]. However, the efficiency of natural ribosomes for decoding quadruplet codons is extremely low. To overcome this challenge, Chin and co-workers engineered translation components that make use of quadruplet codons, instead of the traditional triplet ones (Fig. 7b) [70]. In this work, Chin and co-workers evolved an orthogonal ribosome (ribo-Q1) and a variant Seryl-tRNA synthetase/tRNA pair that can incorporate ncAAs at the quadruplet codon. This system was then used to incorporate azide and alkyne-containing amino acids to a calcium-binding messenger protein, calmodulin.
Another important piece of work is a semisynthetic bacteria with an expanded genetic code [73]. In this study, Romesberg et al. introduced an unnatural base pair (UBP) comprised of two deoxynucleoside triphosphates (dNaM and dTPT3). Theoretically, the addition of these two UBPs enables the incorporation of 152 new unnatural codons. In this study, the authors identified nine unnatural codons that can be used to efficiently incorporate ncAAs. Among these, three codons are orthogonal and can be used for three distinct ncAAs, which results in the first 67-codon semisynthetic organism.
1.2.3. Chemical modifications of the incorporated ncAAs
Genetic code expansion allows for the site-specific incorporation of a variety of ncAAs into a POI, providing useful chemical handles for biorthogonal reactions [1,74]. Several functional groups including azides, alkynes, alkenes, and tetrazines, can be incorporated through the amber codon suppression method, which are subsequently modified by appropriate reagents. For instance, the azides or alkynes of incorporated ncAAs can undergo a copper-catalyzed azide-alkyne cycloaddition (CuAAC), which is termed the “CuAAC click chemistry reaction” (Fig. 8a) [75,76]. Due to its high specificity and having a reasonable reaction rate, the click chemistry has been applied in the analysis of the cellular proteome via biorthogonal non-canonical amino acid tagging (BONCAT) [77,78]. By replacing Met in the cell culture media with an azide- or alkyne-containing ncAA such as azidohomoalanine (AHA) or homopropargylglycine (HPG), the newly synthesized proteins are marked with azide or alkyne functionality and are distinguishable from the pool of preexisting proteins (Fig. 8b and c). The labeling time with AHA should be adjusted according to the protein synthesis rate of the target cell type. Subsequently, the AHA- or HPG-containing proteins are covalently attached to an affinity tag such as disulfide biotin alkyne tag (DST-alkyne) via click chemistry. The newly synthesized and affinity-tagged proteins can be analyzed by conventional biochemical studies or using high-resolution mass spectrometry (Fig. 8c). In a recent study, an alkyne Nε-(propargyloxycarbonyl)-l-lysine (AlkK) was incorporated into the proteome of murine tissue slices and the brains of live mice expressing an orthogonal pyrrolysyl pair introduced by viral injection [78]. The alkyne-bearing proteins are covalently linked to streptavidin beads via diazobenzene linkers, which are then cleaved to release the labeled proteome for analysis by tandem mass spectrometry. This method has been applied to characterize newly synthesized proteins after pharmacological treatments for a particular cell type [79,80].
Fig. 8.

Modifications of ncAAs using click chemistry. (a) Copper-catalyzed conjugation of azide and alkyne. (b) Examples of azide- and alkyne-bearing ncAAs used for click chemistry. (c) Use of click chemistry to label newly synthesized proteins (shown in yellow) using BONCAT technology. (d) Proteins with strained alkyne-containing amino acids can be further modified via SPAAC. The red and blue spheres represent other functional groups in the probes.
The CuAAC reaction involves the use of toxic metal catalysts and exogenous ligands. As an alternative, the strain-promoted azide-alkyne cycloaddition (SPAAC) has emerged as a powerful copper-free click chemistry for the modification of the POIs (Fig. 8d) [1,81]. Several strained alkynes have been developed, including difluorinated cyclooctyne, dibenzoccyclooctynol, and biarylazacyclooctynone. Some of these functional groups can be incorporated into the POIs via the amber codon suppression system [[82], [83], [84], [85]]. The copper-free click chemistry has been implemented in a variety of applications, which are well summarized in recent reviews [80,86,87]. Another emerging strategy for ncAA modifications is the photo-click chemistry, which involves a photo-inducible dipolar [3 + 2]-cycloaddition reaction between an alkenyl-ncAA and a nitrile imine [88,89]. This reaction results in the formation of fluorescent adducts, allowing not only for the in vitro modification of isolated proteins, but also the visualization of proteins in living cells [[89], [90], [91]]. These examples illuminate the general applicability of the biorthogonal reactions in the site-specific modifications of the POIs under complex biological environments, including living cells and multi-cellular organisms with minimal disruption of the biological functions and activities of the POIs.
1.3. Ligand-directed modifications of proteins
In some cases, proteins possess high binding affinities for their respective binding partners. The interactions between proteins and their binders allow for affinity-driven site-specific modifications. A variety of recognition-driven chemical modification strategies have been developed, including ligand-directed (LD) modifications [1,2,92]. The LD chemical modifications of proteins have been developed for endogenous protein labeling by harnessing the specific interactions between the POIs and small molecules. In this approach, the labeling reagent contains an affinity ligand, a reporter tag, and a reactive moiety (Fig. 9a). The binding of the ligand to the POI places the reactive unit of the labeling reagent in proximity to residues close to the ligand binding site, which enhances the selective modification. The LD approach has been applied in the labeling of carbonic anhydrase (CA) [93]. In this study, the specific ligand, benzenesulfonamide, is conjugated to a synthetic probe, which includes a fluorophore via the phenylsulfonate linkage (Fig. 9a). Upon binding to the target protein, a nucleophilic side chain in the POI reacts with the electrophilic phenylsulfonate ester group. This reaction leads to the labeling of the POI, while at the same time, the ligand may dissociate. Therefore, the functions of the POI are not disrupted after the chemical modification. Additionally, the LD approach used in the labeling of CA could be applied to both in vitro and in vivo studies [93].
Fig. 9.

Affinity-driven protein chemical modifications. (a) The LD-based protein labeling approach. (b) The LDNASA-based covalent inhibitor developed through LD chemical modification. Red and yellow star each represents a probe. Blue and pink spheres each represent a functional group.
Non-covalent and reversible ligand binders are often useful in many applications. However, in some cases, the ability to form a stable covalent adduct might be important, such as in the development of inhibitors. In a recent study, Hamachi et al. reported a new ligand-directed N-acyl-N-alkyl sulfonamide (LDNASA)-based covalent inhibitor, which targets chaperone Hsp90 (Fig. 9b) [94,95]. LDNASA enabled the site-specific modification of Lys58, which is a residue near the ligand's binding site in Hsp90. This reaction leads to a covalent adduct between Hsp90 and its inhibitor (Fig. 9b). Treatment of cancer cells with an LDNASA-linked inhibitor results in irreversible covalent modification of Hsp90 at Lys58 by the inhibitor and reduces Hsp90's molecular chaperone activity in cancer cells.
2. Utilizing protein modifications in the studies of metalloenzymes
Chemical modifications and the expansion of the genetic code allow the incorporation of structurally and chemically diverse ncAAs into the POIs, enabling the study of the biological functions and regulation of proteins including metalloenzymes. In addition to these approaches, protein modifications can also be achieved by installing an abiotic metallo cofactor to a protein scaffold, resulting in an artificial metalloenzyme [[96], [97], [98]]. These technologies have opened new avenues for investigating enzymatic reaction mechanisms and engineering of metalloenzymes with new activities. Among these metalloenzymes, non-heme iron (NHFe) and heme enzymes catalyze a remarkable range of chemical transformations, including hydroxylation, endoperoxidation, and desaturation at the expense of molecular oxygen as the final electron acceptor [[99], [100], [101], [102]]. Recent efforts in utilizing these strategies in metalloenzyme engineering and their biochemical applications are summarized in several recent reviews [97,[103], [104], [105], [106]]. This section presents selected case studies, from recent years, of NHFe, heme, and Cu-containing enzymes where ncAAs are used as novel tools. We summarize the discussed cases in Table 2.
Table 2.
Applications of protein modification in metalloenzyme studies.
| Enzyme | Applications |
|---|---|
| Cysteine dioxygenase (CDO) |
|
| Sulfoxide synthase OvoA |
|
| Myoblogin and its variants |
|
| Heme copper oxidase (HCO) |
|
| |
| |
| Cytochrome c P450 |
|
| Azurin (Az) |
|
2.1. Probing the role of residues in NHFe enzymes’ active sites
Post-translational modifications (PTMs) are required for proper protein functionalities, including sub-cellular localization, protein-protein interactions, and catalysis. For instance, a protein-derived thioether Cys-Tyr crosslink enhances the cysteine oxidation activity of the NHFe cysteine dioxygenase enzyme (CDO, Fig. 10a and b) [107]. In CDO, the octahedral ferrous iron is coordinated by a 3-His motif (Fig. 10b). The Cys-Tyr crosslink biogenesis in CDO is an autocatalytic oxidation reaction, in which the oxygen-activated iron center oxidizes the residues at the active site (Cys and Tyr) rather than the substrate, L-Cys [108]. Despite the important role that the Cys-Tyr crosslink plays in the catalytic activity, the mechanistic details of the biogenesis of this crosslink remain unclear, partly due to the challenges of obtaining a homogeneous population of either cross- or uncross-linked CDO. In a recent study, Liu and co-workers employed the amber codon suppression method to replace Tyr157 with a halogen-substituted Tyr in human CDO [109]. The fluorine- and chlorine-containing CDO variants remained active, albeit with a lower activity (2–10% of the wild-type activity). Intriguingly, the Cys93-Tyr157 crosslink is also observed among these variants. Given the high bond dissociation energy, C–F bond cleavage was unexpected in the Cys-Tyr crosslink biogenesis (Fig. 10c). The oxidative C–F bond cleavage might take place during the oxidation of Tyr157 by the iron center or the Cys93 radical. This study represents the first reported case of an oxidative C–F bond cleavage mediated by an NHFe enzyme [109].
Fig. 10.

Using ncAAs in the study of the Cys-Tyr cofactor biogenesis in CDO. (a) Cysteine oxidation mediated by CDO. (b) The 3-His motif and the Cys-Tyr crosslink in wild-type CDO (shown in green) in complex with the L-Cys substrate (shown in purple). The iron center is shown in a yellow sphere (PDB ID: 6BGF). (c) C–F bond cleavage in CDO-catalysis. (d) The structure of F2-Tyr CDO•L-Cys without crosslink (shown in blue, PDB ID: 6BPS) overlays with the structure of mature F2-Tyr CDO•L-Cys (shown in pink, PDB ID: 6BPV), revealing two conformations of Cys93. The fluorine atoms are shown in light blue. (e) A ternary complex of F2-Tyr CDO•L-Cys•NO (shown in magenta) reveals that one conformation of Cys93 is 3.1 Å from the NO and primed to be oxidized by the metallocenter (PDB ID: 6BPR).
The uncross-linked structure of F2-Tyr containing CDO reveals that Cys93 assumes two conformations prior to the Cys-Tyr cofactor formation (Fig. 10d) [109]. During the crosslink biogenesis, the sulfur atom of Cys93 might rotate toward the iron center, which is supported by a recently reported structure of the uncross-linked F2-TyrCDO• L-Cys•NO ternary complex (Fig. 10e) [110]. This structure revealed an interaction between NO and one conformer of Cys93, suggesting that cysteine oxidation may occur prior to the crosslink formation. The oxidation of Cys93 by the iron center is further supported by a computational study, which suggests that an iron-bound oxygen species oxidizes Cys93 as the first step of the crosslink biogenesis, instead of the Tyr157 oxidation, as previously proposed [108,111]. The studies of the CDO cofactor biogenesis highlight the general applicability of site-specific modifications of POIs in mechanistic studies. Due to the important roles of Tyr in enzymatic catalysis, the ability to control the structural features and chemical properties (reduction potential and the pKa) of active Tyr residue has been sought after in several cases [55,[112], [113], [114], [115], [116]].
Ergothioneine is a potent antioxidant and has been proposed to be a longevity vitamin [117,118]. Due to its potential benefits, ergothioneine biosynthesis has received considerable interest. In the mycobacterial ergothioneine biosynthetic pathway, a NHFe sulfoxide synthase, EgtB, mediates the oxidative coupling between hercynine and γ-glutamyl-cysteine (γ-Glu-Cys, Fig. 11b) [119]. Recently, the fungal ergothioneine biosynthetic pathway was reported, in which a sulfoxide synthase, Egt1, catalyzes the C–S bond formation between hercynine and L-Cys (Fig. 11b) [120,121]. A similar reaction has also been observed in the biosynthesis of another potent antioxidant, ovothiol A. In the ovothiol biosynthesis, a sulfoxide synthase, OvoA, catalyzes the oxidative coupling between L-His and L-Cys (Fig. 11b) [122,123]. These ergothioneine and ovothiol sulfoxide synthase reactions differ in terms of their substrate selectivity and their products' C–S bond regioselectivity. In EgtB- and Egt1-catalysis, the C–S bond is formed at the ε-carbon of hercynine's imidazole ring, while OvoA-catalysis incorporates the C–S bond at the δ-carbon of L-His’ imidazole ring (Fig. 11b). The structure of Mycobacterium thermoresistible's EgtB reveals a 3-His iron coordination site similar to that of CDO (Fig. 12a) [124]. Mutating the active site Tyr377 to Phe suppressed the sulfoxide synthase activity and the oxidation of γ-Glu-Cys is the dominant activity in the mutant. Recently, the crystal structure of a sulfoxide synthase from Candidatus Chloracidobacterium thermophilum has also been reported and its iron center also has a 3-His iron coordination ligand environment, too [125,126]. Based on the EgtB crystal structure, three different mechanistic models have been proposed based on calculations using the density functional theory (DFT) or the quantum mechanics/molecular mechanics (QM/MM) methods (Fig. 12c) [[127], [128], [129]]. The key questions are whether the reaction involves a sulfenic acid intermediate and what role the active site Tyr377 plays in EgtB-catalysis. In Fig. 12c, two independent theoretical studies reported by Tian and Wei suggested that the sulfur's oxidation to sulfenic acid is the first half of the reaction (Path IA-IC), while another study by Faponle et al. proposed that the oxidative C–S bond formation is the first half of EgtB-catalysis (Path II) [[127], [128], [129]]. In these two mechanistic models (pathway I vs pathway II), different functions have been proposed for the active site Tyr377. In the Tian model, Tyr377 functions as a Lewis acid/base (Path I, Fig. 12c). In the Faponle model, Tyr377 plays a key role in a proton-coupled electron transfer (PCET) process (Path II, Fig. 12c). Due to the similarities of these reactions, all of the mechanistic discussions regarding EgtB-catalysis are extended to OvoA-studies [112].
Fig. 11.

Chemical properties of Tyr analogs and the biosynthesis of ergothioneine and ovothiol A. (a) The non-canonical Tyrs that can be incorporated into the POIs using the amber codon suppression method possess different reduction potentials and pKas. (b) The oxidative C–S bond formation reactions mediated by sulfoxide synthases (EgtB/Egt1/OvoA) in the biosyntheses of ergothioneine and ovothiol A.
Fig. 12.

Structures and proposed mechanisms of sulfoxide synthases. (a) Crystal structure of EgtB with the metallocenter and the active site Tyr377. The iron center is shown as a yellow sphere and the water molecules are shown as red spheres. (b) The computational model of OvoA from Erwinia tasmaniensis with the proposed metallocenter and the active site Tyr417. (c) Proposed EgtB mechanisms.
To unravel the mechanism of OvoA-catalysis, Liu and co-workers harnessed the unique chemical properties of the non-canonical Tyrs [112]. Notably, the biochemical characterizations also revealed the cysteine oxidation activity in OvoA-catalysis [130]. The computational studies and biochemical results suggest that the sulfoxidation and cysteine oxidation activities might be two pathways branching out from a common intermediate in OvoA-catalysis. Due to this reason, the isotopically sensitive branching method was used to measure the kinetic isotope effect (KIE) of the OvoA reaction [112,131]. Tyr417 in OvoA is the EgtB's Tyr377 counterpart (Fig. 12a and b). The Tyr417 of OvoA was replaced with 2-amino-3-(4-hydroxy-3-(methylthio)-phenyl)propanoic acid (MtTyr) using the amber codon suppression system (Fig. 11a) [112]. The substrate KIE of the OvoAY417MtTyr variant is close to unity, which is comparable to that of the wild-type. This experimental result contradicts the model suggested by Wei et al., in which they predicted a primary substrate deuterium KIE as high as 5.7 [128]. To further investigate the role of Tyr417, Liu and co-workers replaced Tyr417 with 3-methoxytyrosine (MeOTyr) [132], which possesses a similar pKa but a much lower reduction potential than that of canonical Tyr [131]. The OvoAY417MeOTyr variant exhibited an inverse KIE, while the wild-type has a KIE close to unity. These results imply that Tyr417 might be part of redox-chemistries in OvoA-catalysis.
The model proposed by Faponle et al. [127] is more consistent with the experimental results described above. In this computational model, an inverse deuterium KIE was predicated if the δ-hydrogen of the imidazole ring was replaced with deuterium, which was indeed observed in the OvoAY417MeOTyr variant. Given the similarities between OvoA- and EgtB-catalysis, the OvoA mechanism will be discussed in the context of EgtB mechanistic models (Fig. 12c). In the model proposed by Faponle and co-worker, the oxygen binding and activation results in an FeIII-superoxo species (f). Then, the Tyr377 in EgtB participates in the PCET process through the active site's water network, resulting in the FeIII-hydroperoxo species (g) along with a Tyr377-based radical species (Fig. 12c). Subsequent nucleophilic or radical attack by the sulfur atom of γ-Glu-Cys on the imidazole's sidechain of hercynine affords a thioether product. In this step, the hydrogen atom from the hydroperoxo species relays back to the Tyr377-based radical, regenerating Tyr377 and producing an FeII-superoxo species (h). The FeII-superoxo species then abstracts a hydrogen atom from the imidazole ring to regenerate the aromaticity and results in an FeII-hydroperoxo intermediate (i), which undergoes oxidation to afford the sulfoxide product (Fig. 12c). Our MeOTyr-containing OvoA studies are consistent with the EgtB model suggested by Faponle et al. [127,131]. However, the OvoA structure is not available and it is possible that OvoA and EgtB might follow different mechanistic models. Additional kinetic and spectroscopic studies are needed to uncover the mechanistic details of these sulfoxide synthases.
There exists another enzymatic system where the role of Tyr in catalysis is under a heated debate. In the biosynthesis of a tremorgenic mycotoxin verruculogen (2), an α-ketoglutarate-dependent (αKG) NHFe enzyme, FtmOx1, mediates the endoperoxidation of fumitremorgin B (1) (Fig. 13a) [133,134]. Biochemical characterization under single-turnover reaction conditions in the absence of extra reductants (e.g., ascorbate) revealed that each cycle of FtmOx1-catalysis consumes two molecules of molecular oxygen and involves two distinct reactions: the endoperoxidation reaction to afford verruculogen (2) and the oxidation of verruculogen's C13-hydroxyl group to form a keto-product (3) (Fig. 13a) [113]. Compound 3 is the dominant product under single-turnover conditions. In the reported crystal structure of the FtmOx1•αKG binary complex, αKG coordinates to the metallocenter through a distal-type configuration, and Tyr224 is adjacent to the putative oxygen binding site (Fig. 13b) [113]. Structural, biochemical, and spectroscopic characterizations led to a mechanistic model involving the Tyr224 radical as one of the key species in FtmOx1-catalysis (Fig. 13c).
Fig. 13.

The endoperoxidation reaction mediated by FtmOx1. (a) The products of the FtmOx1 reactions under single-turnover conditions in the absence of extra reductants (e.g., ascorbate). (b) The distal-type αKG binding configuration in the FtmOx1•αKG binary complex (PDB ID: 4Y5S). αKG is shown in purple and the iron center is shown as a yellow sphere. Water is presented as a red sphere. (c) A proposed FtmOx1 mechanistic model involving the Tyr224 radical as one of the key species (shaded in green) under single-turnover conditions. The Tyr224 radical is also responsible for initiating the oxidation of the C13-hydroxyl group to a keto-product (13). Therefore, there are two reaction cycles in FtmOx1-catalysis under single-turnover conditions.
However, in a recent report, Bollinger and co-workers proposed that an alternative tyrosine residue (Tyr68) might be the tyrosyl radical site (Fig. 14) [135]. The authors reported that in the reaction catalyzed by the FtmOx1Y68F variant, a new major, uncharacterized product was formed. In FtmOx1-catalysis, under single-turnover conditions and in the absence of other reductants, the keto-product (3) is the dominant product. In this Tyr68 radical model (Fig. 14), Tyr68 is on the protein surface and fully solvent exposed, and the Fe-center is far from the substrate's C13–OH group. It will be challenging to explain formation of the keto-product (3) as the dominant product using this Tyr68 radical model [135]. In addition, it will be more convincing to discuss this Tyr68 radical mechanistic model after the structures of the reaction products were characterized [135]. Therefore, Tyr224 remains the best candidate for the proposed tyrosyl radical in FtmOx1-catalysis. To provide further evidence to differentiate between the two proposed mechanistic models, the use of non-canonical Tyr will be beneficial in characterizing the catalytic roles of the two Tyr residues in FtmOx1-catalysis, which are on-going efforts in our laboratory.
Fig. 14.

A proposed FtmOx1 mechanistic model involving a Tyr68-based radical (shaded in green).
2.2. Heme enzymes engineering
Another common type of iron-dependent enzyme is the heme-containing enzymes, in which the iron center is coordinated to a porphyrin as a tetradentate ligand. In addition to heme cofactors, these enzymes often possess additional metals and they catalyze a diverse range of reactions, e.g., copper (CuB) in heme copper oxidase (HCO) and iron (FeB) in nitric oxide reductase (NOR) [[136], [137], [138], [139], [140]]. Designing artificial heme enzymes is a powerful tool not only for facilitating in-depth mechanistic studies, but also for developing novel biocatalysts. Both rational enzyme design and directed evolution methods have been applied for heme-enzyme engineering where metal-ligands and the secondary coordination shell residues are modulated. The readers are referred to several recent reviews on artificial metalloenzyme engineering [103,104,[141], [142], [143], [144]]. The cytochrome c oxidase (CcO) is a membrane-bound protein from the HCO family. It catalyzes the reduction of O2 to water without the generation of toxic reactive oxygen species (ROS). In this reaction, the catalytic functions of the additional CuB site remain unclear [145]. Due to the difficulty of obtaining large amounts of membrane-bound CcO, an artificial model for this enzyme has been generated through engineering the heme-containing oxygen carrier protein, myoglobin from sperm whale Physeter macrocephalus (swMb, Fig. 15a). Based on the structural comparison between swMb and the native bovine CcO, residues His64, Leu29, and Phe34 from the swMb active site were selected to create the CuB binding site [145]. Both Leu29 and Phe34 were mutated to His to create the swMb L29H F43H variant, which is referred to as CuBMb. This engineered CuBMb variant exhibited biophysical properties similar to the heme-copper center from the HCO family. In addition, spectroscopic studies of the engineered CuBMb indicated that the CuB center enhances the molecular oxygen binding affinity to the heme center in the HCO reaction.
Fig. 15.

CuBMb enzyme engineering. (a) The crystal structure of swMb (PDB ID: 1VXA). The residues Leu29, Phe43, His64, and Val68 were chosen to create a Cu binding site. (b) The crystal structure of bovine heart CcO (PDB ID: 1OCO). Tyr244 and His240 form a covalent crosslink, which was proposed to be essential for oxidase activity. Copper is shown as a blue sphere.
The creation of the CuBMb protein has enabled the subsequent efforts to improve its natural activities and even for creating non-natural activities. In CuBMb, ROS formation was inhibited, while H2O production did not change significantly relative to the wild-type swMb. HCOs contain a conserved Tyr-His crosslink in their active site and this crosslink has been proposed to be important for water production (Fig. 15b). Lu and co-workers replaced Gly65, a residue adjacent to the copper-binding site, with a Tyr residue in CuBMb. The water production rate of the CuBMbG65Y variant was dramatically enhanced compared to that of native CuBMb [146]. The importance of Tyr in CuBMbG65Y-catalysis motivated the authors to further investigate the contribution of a Tyr-His crosslink in HCO-catalysis using a ncAA incorporated by the amber-codon suppression method. In this study, Lu and co-workers chose (S)-2-Amino-3-(4-hydroxy-3-(1H-imidazole-1-yl)phenyl)propanoic acid (imiTyr) to mimic the Tyr-His crosslink [116]. The resulting imiTyr-CuBMb variant not only mediated the O2 reduction reaction with more than 1000 turnovers, but also minimized the ROS formation relative to the CuBMbG65Y variant. This study highlights the crucial role of the Tyr-His crosslink in HCO for selective H2O production with minimized ROS formation. The enhanced catalytic activity and selectivity of the engineered HCO reveals the value of protein modifications in developing robust biocatalysts.
NORs are similar to HCOs in that they both contain an additional iron-binding site near the heme-center. However, NORs catalyze the two-electron reduction of NO to N2O. Additionally, the creation of the NOR activity from a heme template (i.e. swMb) is challenging due to the lack of a NOR crystal structure. Based on their success in the CuBMb studies, Lu and co-workers created a swMb variant, FeBMb, with the NO reduction activity [140]. Starting from the CuBMb (swMb L29H F43H variant) scaffold, an additional Val68 to Glu mutation was introduced to mimic the Glu residue in NOR (Fig. 15a). The CuBMbV68E variant had the desired FeB site and also exhibited the NO reduction activity at a level comparable to that of the native NOR.
In these heme enzymes engineering efforts, besides mutating the metal-binding site, a heme analog can replace the native heme; a second heme cofactor can be added as well [147,148]. For instance, native heme b in an HCO has been replaced with monoformyl-, diformyl- and diacetyl hemes (Fig. 16a). By increasing the number of electron-withdrawing groups in these heme analogs, the reduction potential of the HCO increases proportionally [147]. The enhanced reduction potential resulted in an HCO with an increased dioxygen reduction activity. In another study, a manganese salen complex, Mn(salen), was introduced to swMb through a two-point covalent attachment strategy where Leu72 and Tyr103 were mutated to Cys. These two Cys residues of the swMbL72C_Y103C variant then reacted with the methane thiosulfate groups of the artificial cofactor to covalently attach Mn(salen) to the protein (Fig. 16a) [139]. The Mn(salen)-bearing swMb catalyzes the thioanisole sulfoxidation with a remarkably improved enantioselectivity (51%) relative to that of the apo swMb (<5%), (Fig. 16b).
Fig. 16.

Incorporation of heme analogs or artificial metal complexes to create heme-protein variants. (a) Non-native cofactors used in heme-protein variants. (b) The sulfoxidation reaction catalyzed by Mn(salen)-harboring swMb. (c) The incorporation of non-native cofactors using the heme transporter, ChuA, in vivo. (d) The cyclopropanation reactions catalyzed by P450s with Fe-DPIX.
In these studies, the heme analogs are introduced into the purified apo-enzymes by in vitro reconstitution [139,147]. This method often results in a low yield of the reconstituted holo-enzyme. To overcome this issue, an orthogonal enzyme/heme pair consisting of a heme-like cofactor and a complementary heme-binding protein has been developed (Fig. 16c) [148]. In this system, the native heme-transport channel, ChuA, is evolved to import the iron deuteroporphyrin IX (Fe-DPIX) cofactor, which binds selectively to an engineered cytochrome P450 scaffold from Bacillus megaterium. The non-natural cofactor containing variant does not catalyze the native P450 monooxygenation reactions. Instead, this artificial enzyme catalyzes cyclopropanation reactions (Fig. 16d) [148].
2.3. Copper enzymes engineering
Copper-containing metalloenzymes play essential roles in multiple biological processes, e.g., as electron transfer mediators due to the wide range of redox potentials of their metallo centers [[149], [150], [151]]. Of this family, Azurin (Az) from Pseudomonas aeruginosa is a type I copper protein which has been extensively investigated and re-engineered. The trigonal bipyramidal Cu2+ is bound by 2-His and 1-Cys in a plane with a Met121 and the carboxyl moiety of Gly45 binding at the axial positions (Fig. 17). To determine the role of the axial Met ligand in Az, Lu and co-workers replaced Met121 with multiple ncAAs through an expressed protein ligation method [152,153]. A linear correlation was observed between the hydrophobicity of the ncAAs and the redox potential of the copper center, with higher hydrophobicity resulting in higher reduction potentials. Other than the engineering of the copper-binding ligands, Az has also served as a protein scaffold for incorporating non-native cofactors because of the stability of the apo-protein. Ferrocene is an attractive organometallic catalyst, but its applications are limited because of its poor solubility in aqueous solutions. To resolve this issue, an Azurin-Ferrocene complex was generated via covalent attachment between Cys112 and 2-[(Methylsulfonyl)thio]ethylferrocene [154]. The reduction potential of the ferrocene complex-containing Az variant was increased compared to that of the wild-type. Engineering of the secondary coordination shell residues further enhanced the reduction potential of the Az variant. The tunability of Az's reduction potential makes it a good electron transfer mediator in biological systems and an excellent target for enzyme engineering.
Fig. 17.
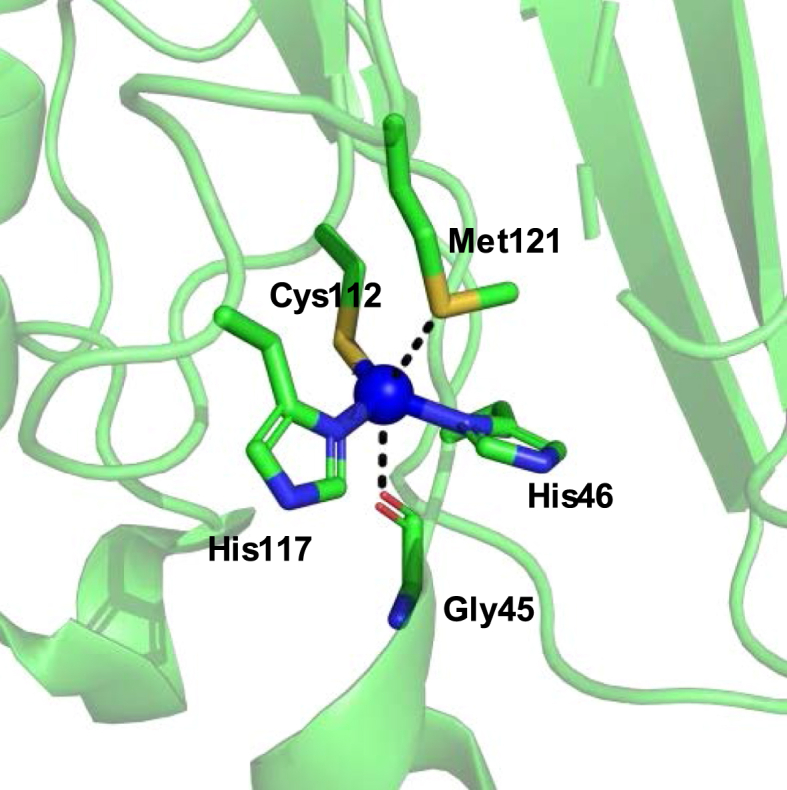
Metal-binding site of Azurin. The copper ion (shown in blue sphere) is coordinated by Gly45, His46, Cys112, His117, and Met121 at the active site of azurin (PDB ID: 4AZU).
3. Conclusions
In this review, we summarize some of the recent advances in protein modifications through both chemical and biological approaches, which are widely applied in biochemistry, chemical biology, cell biology, and drug discovery. In recent years, there are more cases of the application of these tools in enzyme mechanistic studies and functional engineering. We select some recent examples where introducing ncAAs or non-native cofactors either served as new mechanistic study tools or to introduce novel catalytic activities that do not exist in the wild-type enzymes.
Author contributions
Conceptualization, P.L.; writing of the original draft, N.N. and R.H.; review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by National Science Foundation (CHE-2004109 to P. L.)
Declaration of competing interest
The authors declare no conflict of interest.
Appendix A: Abbreviation List
- 2-PCA
2-pyridinecarbaldehyde
- AA
Amino acid
- aaRS
tRNA synthetase
- ADC
Antibody-drug conjugates
- AHA
Azidohomoalanine
- AlkK
Alkyne Nε-(propargyloxycarbonyl)-l-lysine
- BONCAT
Biorthogonal non-canonical amino acid tagging
- cAA
Canonical amino acid
- CBTF
4-(cyanoethynyl)benzoyl)oxy)-2,3,5,6-tetrafluorobenzenesulfonate
- CcO
Cytochrome c oxidase
- CDO
Cysteine dioxygenase
- CuAAC
Copper-catalyzed azide-alkyne cycloaddition
- CuB
Copper-binding site
- CuBMb
swMb L29H F43H variant
- DST-alkyne
Disulfide biotin alkyne tag
- EPL
Expressed protein ligation
- Fe-DPIX
Iron-deuteroporphyrin IX
- H2B–K20Ub
Ubiquitinated H2B
- HAT
Hydrogen-atom transfer
- HCO
Heme-copper oxidase
- HPG
Homopropargylglycine
- imiTyr
(S)-2-amino-3-(4-hydroxy-3-(1H-imidazol-1-yl)phenyl)propanoic acid
- KIE
Kinetic isotope effect
- LD
Ligand-directed
- LDNASA
Ligand-directed N-acyl-N-alkyl sulfonamide
- MeOTyr
3-methoxytyrosine
- Mn(salen)
Manganese-salen complex
- MtTyr
2-amino-3-(4-hydroxy-3-(methylthio)-phenyl)propanoic acid
- ncAA
Noncanonical amino acid
- NCL
Native chemical ligation
- NHFe
Non-heme iron enzyme
- NHS
N-hydroxysuccinimide
- NOR
Nitric oxide reductase
- PACE
Phage-assisted continuous evolution
- PCET
Proton-coupled electron transfer
- PCS
Primary coordination shell
- PEG
Polyethylene glycol
- PLP
Pyridoxal-5′-phosphate
- POI
Protein of interest
- PTC
Peptidyl transferase center
- PTM
Post-translational modification
- PylRS
Pyrrolysyl-tRNA synthetase
- QM/MM
Quantum mechanics/molecular mechanics
- RF-1
Release factor 1
- ROS
Reactive oxygen species
- SCS
Secondary coordination shell
- SD
Shine-Dalgarno
- SOD
Superoxide dismutase
- SPAAC
Strain-promoted azide-alkene cycloaddition
- swMb
Physeter microcephalus Myoglobin
- tRNA
Transfer RNA
- UBP
Unnatural base pair
- αKG
α-ketoglutarate
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
References
- 1.Sakamoto S., Hamachi I. Recent progress in chemical modification of proteins. Anal Sci. 2018;35(1) doi: 10.2116/analsci.18R003. [DOI] [PubMed] [Google Scholar]
- 2.Shadish J.A., DeForest C.A. Site-selective protein modification: from functionalized proteins to functional biomaterials. Matter. 2020;2(1):50–77. [Google Scholar]
- 3.Nischan N., Hackenberger C.P. Site-specific PEGylation of proteins: recent developments. J Org Chem. 2014;79(22):10727–10733. doi: 10.1021/jo502136n. [DOI] [PubMed] [Google Scholar]
- 4.Wurm F., Steinbach T., Klok H.A. One-pot squaric acid diester mediated aqueous protein conjugation. Chem Commun. 2013;49(71):7815–7817. doi: 10.1039/c3cc44039g. [DOI] [PubMed] [Google Scholar]
- 5.Spicer C.D., Davis B.G. Selective chemical protein modification. Nat Commun. 2014;5(1):4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- 6.DeForest C.A., Tirrell D.A. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat Mater. 2015;14(5):523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 7.Matos M.J., Oliveira B.L., Martinez-Saez N., Guerreiro A., Cal P., Bertoldo J. Chemo- and regioselective lysine modification on native proteins. J Am Chem Soc. 2018;140(11):4004–4017. doi: 10.1021/jacs.7b12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nanna A.R., Li X., Walseng E., Pedzisa L., Goydel R.S., Hymel D. Harnessing a catalytic lysine residue for the one-step preparation of homogeneous antibody-drug conjugates. Nat Commun. 2017;8(1):1112. doi: 10.1038/s41467-017-01257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson P.E., Muir T.W., Clark-Lewis I., Kent S. Synthesis of proteins by native chemical ligation. Science. 1994;266(5186):776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 10.Muir T.W. Semisynthesis of proteins by expressed protein ligation. Annu Rev Biochem. 2003;72(1):249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 11.Vila-Perelló M., Liu Z., Shah N.H., Willis J.A., Idoyaga J., Muir T.W. Streamlined expressed protein ligation using split inteins. J Am Chem Soc. 2013;135(1):286–292. doi: 10.1021/ja309126m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minnihan E.C., Yokoyama K., Stubbe J. Unnatural amino acids: better than the real things? F1000 Biol Rep. 2009;1:88. doi: 10.3410/B1-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.David Y., Vila-Perelló M., Verma S., Muir T.W. Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat Chem. 2015;7(5):394–402. doi: 10.1038/nchem.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilmore J.M., Scheck R.A., EsserKahn A.P., Joshi N.S., Francis M.B. N-terminal protein modification through a biomimetic transamination reaction. Angew Chem Int. 2006;45(32):5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 15.Scheck R.A., Francis M.B. Regioselective labeling of antibodies through N-terminal transamination. ACS Synth Biol. 2007;2(4):247–251. doi: 10.1021/cb6003959. [DOI] [PubMed] [Google Scholar]
- 16.Scheck R.A., Dedeo M.T., Iavarone A.T., Francis M.B. Optimization of a biomimetic transamination reaction. J Am Chem Soc. 2008;130(35):11762–11770. doi: 10.1021/ja802495w. [DOI] [PubMed] [Google Scholar]
- 17.Christman K.L., Broyer R.M., Tolstyka Z.P., Maynard H.D. Site-specific protein immobilization through N-terminal oxime linkages. J Mater Chem. 2007;17(19):2021–2027. [Google Scholar]
- 18.Jie W., Peng C. Development and applications of bioorthogonal cleavage reactions. Hua Hsueh Hsueh Pao. 2017;75(12):1173–1182. [Google Scholar]
- 19.Chalker J.M., Bernardes G.J., Davis B.G.A. “tag-and-modify” approach to site-selective protein modification. Acc Chem Res. 2011;44(9):730–741. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- 20.Crankshaw M.W., Grant G.A. Modification of cysteine. Curr Protein Pept Sci. 1996;3(1):15. 1. 1-.1. 8. [Google Scholar]
- 21.Goddard D.R., Michaelis L. Derivatives of keratin. J Biol Chem. 1935;112(1):361–371. [Google Scholar]
- 22.Lundell N., Schreitmüller T. Sample preparation for peptide mapping-a pharmaceutical quality-control perspective. Anal Biochem. 1999;266(1):31–47. doi: 10.1006/abio.1998.2919. [DOI] [PubMed] [Google Scholar]
- 23.Moore J., Ward W. Cross-linking of bovine plasma albumin and wool keratin. J Am Chem Soc. 1956;78(11):2414–2418. [Google Scholar]
- 24.Tsao T.-C., Bailey K. The extraction, purification and some chemical properties of actin. Biochim Biophys Acta. 1953;11:102–113. doi: 10.1016/0006-3002(53)90013-4. [DOI] [PubMed] [Google Scholar]
- 25.Gamblin D.P., Garnier P., Ward S.J., Oldham N.J., Fairbanks A.J., Davis B.G. Glycosyl phenylthiosulfonates (Glyco-PTS): novel reagents for glycoprotein synthesis. Org Biomol Chem. 2003;1(21):3642–3644. doi: 10.1039/b306990g. [DOI] [PubMed] [Google Scholar]
- 26.Bernardes G.J., Grayson E.J., Thompson S., Chalker J.M., Errey J.C., El Oualid F. From disulfide-to thioether-linked glycoproteins. Angew Chem Int. 2008;120(12):2276–2279. doi: 10.1002/anie.200704381. [DOI] [PubMed] [Google Scholar]
- 27.Fontaine S.D., Reid R., Robinson L., Ashley G.W., Santi D.V. Long-term stabilization of maleimide–thiol conjugates. Bioconjugate Chem. 2015;26(1):145–152. doi: 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- 28.Lyon R.P., Setter J.R., Bovee T.D., Doronina S.O., Hunter J.H., Anderson M.E. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat Biotechnol. 2014;32(10):1059–1062. doi: 10.1038/nbt.2968. [DOI] [PubMed] [Google Scholar]
- 29.Shiu H.-Y., Chong H.-C., Leung Y.-C., Zou T., Che C.-M. Phosphorescent proteins for bio-imaging and site selective bio-conjugation of peptides and proteins with luminescent cyclometalated iridium (III) complexes. Chem Commun. 2014;50(33):4375–4378. doi: 10.1039/c3cc48376b. [DOI] [PubMed] [Google Scholar]
- 30.Koniev O., Leriche G., Nothisen M., Remy J.-S., Strub J.-M., Schaeffer-Reiss C. Selective irreversible chemical tagging of cysteine with 3-arylpropiolonitriles. Bioconjugate Chem. 2014;25(2):202–206. doi: 10.1021/bc400469d. [DOI] [PubMed] [Google Scholar]
- 31.Kolodych S., Koniev O., Baatarkhuu Z., Bonnefoy J.-Y., Fo Debaene, Cianferani S. CBTF: new amine-to-thiol coupling reagent for preparation of antibody conjugates with increased plasma stability. Bioconjugate Chem. 2015;26(2):197–200. doi: 10.1021/bc500610g. [DOI] [PubMed] [Google Scholar]
- 32.Abbas A., Xing B., Loh T.P. Allenamides as orthogonal handles for selective modification of cysteine in peptides and proteins. Angew Chem Int. 2014;53(29):7491–7494. doi: 10.1002/anie.201403121. [DOI] [PubMed] [Google Scholar]
- 33.Conte M.L., Staderini S., Marra A., Sanchez-Navarro M., Davis B.G., Dondoni A. Multi-molecule reaction of serum albumin can occur through thiol-yne coupling. Chem Commun. 2011;47(39):11086–11088. doi: 10.1039/c1cc14402b. [DOI] [PubMed] [Google Scholar]
- 34.Bernardim B., Cal P.M., Matos M.J., Oliveira B.L., Martínez-Sáez N., Albuquerque I.S. Stoichiometric and irreversible cysteine-selective protein modification using carbonylacrylic reagents. Nat Commun. 2016;7(1):1–9. doi: 10.1038/ncomms13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernardim B., Matos M.J., Ferhati X., Compañón I., Guerreiro A., Akkapeddi P. Efficient and irreversible antibody–cysteine bioconjugation using carbonylacrylic reagents. Nat Protoc. 2019;14(1):86–99. doi: 10.1038/s41596-018-0083-9. [DOI] [PubMed] [Google Scholar]
- 36.Gil de Montes E., Istrate A., Navo C.D., Jiménez-Moreno E., Hoyt E.A., Corzana F. Stable pyrrole-linked bioconjugates through tetrazine-triggered azanorbornadiene fragmentation. Angew Chem Int. 2020;132(15):6255–6259. doi: 10.1002/anie.201914529. [DOI] [PubMed] [Google Scholar]
- 37.Bernardim B., Dunsmore L., Li H., Hocking B., Nuñez-Franco R., Navo C.D. Precise installation of diazo-tagged side-chains on proteins to enable in vitro and in-cell site-specific labeling. Bioconjugate Chem. 2020;31(6):1604–1610. doi: 10.1021/acs.bioconjchem.0c00232. [DOI] [PubMed] [Google Scholar]
- 38.Kuan S.L., Wang T., Weil T. Site-selective disulfide modification of proteins: expanding diversity beyond the proteome. Chem Eur J. 2016;22(48):17112–17129. doi: 10.1002/chem.201602298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forte N., Chudasama V., Baker J.R. Homogeneous antibody-drug conjugates via site-selective disulfide bridging. Drug Discov Today Technol. 2018;30:11–20. doi: 10.1016/j.ddtec.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Martínez-Sáez N., Sun S., Oldrini D., Sormanni P., Boutureira O., Carboni F. Oxetane grafts installed site-selectively on native disulfides to enhance protein stability and activity in vivo. Angew Chem Int. 2017;56(47):14963–14967. doi: 10.1002/anie.201708847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee M.T., Maruani A., Baker J.R., Caddick S., Chudasama V. Next-generation disulfide stapling: reduction and functional re-bridging all in one. Chem Sci. 2016;7(1):799–802. doi: 10.1039/c5sc02666k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones M.W., Mantovani G., Blindauer C.A., Ryan S.M., Wang X., Brayden D.J. Direct peptide bioconjugation/PEGylation at tyrosine with linear and branched polymeric diazonium salts. J Am Chem Soc. 2012;134(17):7406–7413. doi: 10.1021/ja211855q. [DOI] [PubMed] [Google Scholar]
- 43.Chen S., Li X., Ma H. New approach for local structure analysis of the tyrosine domain in proteins by using a site-specific and polarity-sensitive fluorescent probe. Chembiochem. 2009;10(7):1200–1207. doi: 10.1002/cbic.200900003. [DOI] [PubMed] [Google Scholar]
- 44.Seim K.L., Obermeyer A.C., Francis M.B. Oxidative modification of native protein residues using cerium (IV) ammonium nitrate. J Am Chem Soc. 2011;133(42):16970–16976. doi: 10.1021/ja206324q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tilley S.D., Francis M.B. Tyrosine-selective protein alkylation using π-allylpalladium complexes. J Am Chem Soc. 2006;128(4):1080–1081. doi: 10.1021/ja057106k. [DOI] [PubMed] [Google Scholar]
- 46.Ruan Z., Sauermann N., Manoni E., Ackermann L. Manganese-catalyzed C-H alkynylation: expedient peptide synthesis and modification. Angew Chem Int. 2017;56(12):3172–3176. doi: 10.1002/anie.201611118. [DOI] [PubMed] [Google Scholar]
- 47.Schischko A., Ren H., Kaplaneris N., Ackermann L. Bioorthogonal diversification of peptides through selective ruthenium (II)-catalyzed C–H activation. Angew Chem Int. 2017;56(6):1576–1580. doi: 10.1002/anie.201609631. [DOI] [PubMed] [Google Scholar]
- 48.Siti W., Khan A.K., de Hoog H.-P.M., Liedberg B., Nallani M. Photo-induced conjugation of tetrazoles to modified and native proteins. Org Biomol Chem. 2015;13(11):3202–3206. doi: 10.1039/c4ob02025a. [DOI] [PubMed] [Google Scholar]
- 49.Seki Y., Ishiyama T., Sasaki D., Abe J., Sohma Y., Oisaki K. Transition metal-free tryptophan-selective bioconjugation of proteins. J Am Chem Soc. 2016;138(34):10798–10801. doi: 10.1021/jacs.6b06692. [DOI] [PubMed] [Google Scholar]
- 50.deGruyter J.N. Malins LR, Baran PS. Residue-specific peptide modification: a chemist's guide. Biochemistry. 2017;56(30):3863–3873. doi: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoyt E.A., Cal P.M., Oliveira B.L., Bernardes G.J. Contemporary approaches to site-selective protein modification. Nat Rev Chem. 2019;3(3):147–171. [Google Scholar]
- 52.Conibear A.C. Deciphering protein post-translational modifications using chemical biology tools. Nat Rev Chem. 2020:1–22. doi: 10.1038/s41570-020-00223-8. [DOI] [PubMed] [Google Scholar]
- 53.Drienovská I., Alonso-Cotchico L., Vidossich P., Lledós A., Maréchal J.-D., Roelfes G. Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem Sci. 2017;8(10):7228–7235. doi: 10.1039/c7sc03477f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drienovská I., Roelfes G. Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat Catal. 2020:1–10. [Google Scholar]
- 55.Won Y., Pagar A.D., Patil M.D., Dawson P.E., Yun H. Recent advances in enzyme engineering through incorporation of unnatural amino acids. Biotechnol Bioproc Eng. 2019;24(4):1–13. [Google Scholar]
- 56.Srinivasan G., James C.M., Krzycki J.A. Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science. 2002;296(5572):1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 57.Böck A., Forchhammer K., Heider J., Leinfelder W., Sawers G., Veprek B. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5(3):515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 58.Munier R., Cohen G. Incorporation of structural analogues of amino acids into bacterial proteins during their synthesis in vivo. Biochim Biophys Acta. 1959;31(2):378. doi: 10.1016/0006-3002(59)90011-3. [DOI] [PubMed] [Google Scholar]
- 59.Yang W., Hendrickson W.A., Crouch R.J., Satow Y. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249(4975):1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]
- 60.Liu C.C., Schultz P.G. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 61.Chin J.W. Expanding and reprogramming the genetic code. Nature. 2017;550(7674):53–60. doi: 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- 62.Lajoie M.J., Rovner A.J., Goodman D.B., Aerni H.-R., Haimovich A.D., Kuznetsov G. Genomically recoded organisms expand biological functions. Science. 2013;342(6156):357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mukai T., Hoshi H., Ohtake K., Takahashi M., Yamaguchi A., Hayashi A. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci Rep. 2015;5(1):1–9. doi: 10.1038/srep09699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dumas A., Lercher L., Spicer C.D., Davis B.G. Designing logical codon reassignment-Expanding the chemistry in biology. Chem Sci. 2015;6(1):50–69. doi: 10.1039/c4sc01534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bryson D.I., Fan C., Guo L.-T., Miller C., Söll D., Liu D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat Chem Biol. 2017;13(12):1253. doi: 10.1038/nchembio.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Italia J.S., Addy P.S., Wrobel C.J., Crawford L.A., Lajoie M.J., Zheng Y. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nat Chem Biol. 2017;13(4):446–450. doi: 10.1038/nchembio.2312. [DOI] [PubMed] [Google Scholar]
- 67.Italia J.S., Addy P.S., Erickson S.B., Peeler J.C., Weerapana E., Chatterjee A. Mutually orthogonal nonsense-suppression systems and conjugation chemistries for precise protein labeling at up to three distinct sites. J Am Chem Soc. 2019;141(15):6204–6212. doi: 10.1021/jacs.8b12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rackham O., Chin J.W. A network of orthogonal ribosome· mRNA pairs. Nat Chem Biol. 2005;1(3):159–166. doi: 10.1038/nchembio719. [DOI] [PubMed] [Google Scholar]
- 69.Hui A., De Boer H.A. Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc Natl Acad Sci Unit States Am. 1987;84(14):4762–4766. doi: 10.1073/pnas.84.14.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neumann H., Wang K., Davis L., Garcia-Alai M., Chin J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464(7287):441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 71.Schmied W.H., Tnimov Z., Uttamapinant C., Rae C.D., Fried S.D., Chin J.W. Controlling orthogonal ribosome subunit interactions enables evolution of new function. Nature. 2018;564(7736):444–448. doi: 10.1038/s41586-018-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Riddle D.L., Carbon J. Frameshift suppression: a nucleotide addition in the anticodon of a glycine transfer RNA. Nat New Biol. 1973;242(121):230–234. doi: 10.1038/newbio242230a0. [DOI] [PubMed] [Google Scholar]
- 73.Fischer E.C., Hashimoto K., Zhang Y., Feldman A.W., Dien V.T., Karadeema R.J. New codons for efficient production of unnatural proteins in a semisynthetic organism. Nat Chem Biol. 2020:1–7. doi: 10.1038/s41589-020-0507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lang K., Chin J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev. 2014;114(9):4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 75.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. A stepwise Huisgen cycloaddition process: copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int. 2002;41(14):2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 76.Tornøe C.W., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67(9):3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 77.Dieterich D.C., Link A.J., Graumann J., Tirrell D.A., Schuman E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci Unit States Am. 2006;103(25):9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krogager T.P., Ernst R.J., Elliott T.S., Calo L., Beránek V., Ciabatti E. Labeling and identifying cell-specific proteomes in the mouse brain. Nat Biotechnol. 2018;36(2):156–159. doi: 10.1038/nbt.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cañeque T., Müller S., Rodriguez R. Visualizing biologically active small molecules in cells using click chemistry. Nat Rev Chem. 2018;2(9):202–215. [Google Scholar]
- 80.Thirumurugan P., Matosiuk D., Jozwiak K. Click chemistry for drug development and diverse chemical–biology applications. Chem Rev. 2013;113(7):4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 81.Agard N.J., Prescher J.A., Bertozzi C.R. A strain-promoted [3 + 2] azide−alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126(46):15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 82.Codelli J.A., Baskin J.M., Agard N.J., Bertozzi C.R. Second-generation difluorinated cyclooctynes for copper-free click chemistry. J Am Chem Soc. 2008;130(34):11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ning X., Guo J., Wolfert M.A., Boons G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew Chem Int. 2008;47(12):2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jewett J.C., Sletten E.M., Bertozzi C.R. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J Am Chem Soc. 2010;132(11):3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nikić I., Estrada Girona G., Kang J.H., Paci G., Mikhaleva S., Koehler C. Debugging eukaryotic genetic code expansion for site-specific click-PAINT super-resolution microscopy. Angew Chem Int. 2016;55(52):16172–16176. doi: 10.1002/anie.201608284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim E., Koo H. Biomedical applications of copper-free click chemistry: in vitro, in vivo, and ex vivo. Chem Sci. 2019;10(34):7835–7851. doi: 10.1039/c9sc03368h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nguyen S.S., Prescher J.A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat Rev Chem. 2020:1–14. doi: 10.1038/s41570-020-0205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vrabel M., Carell T. 2016. Cycloadditions in bioorthogonal chemistry: Springer. [DOI] [PubMed] [Google Scholar]
- 89.Ramil C.P., Lin Q. Photoclick chemistry: a fluorogenic light-triggered in vivo ligation reaction. Curr Opin Chem Biol. 2014;21:89–95. doi: 10.1016/j.cbpa.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Song W., Wang Y., Qu J., Lin Q. Selective functionalization of a genetically encoded alkene-containing protein via “photoclick chemistry” in bacterial cells. J Am Chem Soc. 2008;130(30):9654–9655. doi: 10.1021/ja803598e. [DOI] [PubMed] [Google Scholar]
- 91.Yu Z., Pan Y., Wang Z., Wang J., Lin Q. Genetically encoded cyclopropene directs rapid, photoclick-chemistry-mediated protein labeling in mammalian cells. Angew Chem Int. 2012;51(42):10600–10604. doi: 10.1002/anie.201205352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baneyx F., Schwartz D.T. Selection and analysis of solid-binding peptides. Curr Opin Biotechnol. 2007;18(4):312–317. doi: 10.1016/j.copbio.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 93.Tsukiji S., Miyagawa M., Takaoka Y., Tamura T., Hamachi I. Ligand-directed tosyl chemistry for protein labeling in vivo. Nat Chem Biol. 2009;5(5):341. doi: 10.1038/nchembio.157. [DOI] [PubMed] [Google Scholar]
- 94.Tamura T., Hamachi I. Chemistry for covalent modification of endogenous/native proteins: from test tubes to complex biological systems. J Am Chem Soc. 2018;141(7):2782–2799. doi: 10.1021/jacs.8b11747. [DOI] [PubMed] [Google Scholar]
- 95.Tamura T., Ueda T., Goto T., Tsukidate T., Shapira Y., Nishikawa Y. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat Commun. 2018;9(1):1870. doi: 10.1038/s41467-018-04343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Akabori S., Sakurai S., Izumi Y., Fujii Y. An asymmetric catalyst. Nature. 1956;178(4528):323–324. [Google Scholar]
- 97.Davis H.J., Ward T.R. Artificial metalloenzymes: challenges and opportunities. ACS Cent Sci. 2019;5(7):1120–1136. doi: 10.1021/acscentsci.9b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller K.R., Paretsky J.D., Follmer A.H., Heinisch T., Mittra K., Gul S. Artificial iron proteins: modeling the active sites in non-heme dioxygenases. Inorg Chem. 2020;59(9):6000–6009. doi: 10.1021/acs.inorgchem.9b03791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Solomon E.I., Decker A., Lehnert N. Non-heme iron enzymes: contrasts to heme catalysis. Proc Natl Acad Sci Unit States Am. 2003;100(7):3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gao S.-S., Naowarojna N., Cheng R., Liu X., Liu P. Recent examples of α-ketoglutarate-dependent mononuclear non-haem iron enzymes in natural product biosyntheses. Nat Prod Rep. 2018;35(8):792–837. doi: 10.1039/c7np00067g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Song H., Naowarojna N., Cheng R., Lopez J., Liu P. Adv Protein Chem Struct Biol: Elsevier; 2019. Non-heme iron enzyme-catalyzed complex transformations: endoperoxidation, cyclopropanation, orthoester, oxidative CC and CS bond formation reactions in natural product biosynthesis. pp. 1–61. [DOI] [PubMed] [Google Scholar]
- 102.Poulos T.L. Heme enzyme structure and function. Chem Rev. 2014;114(7):3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brandenberg O.F., Fasan R., Arnold F.H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr Opin Biotechnol. 2017;47:102–111. doi: 10.1016/j.copbio.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schwizer F., Okamoto Y., Heinisch T., Gu Y., Pellizzoni M.M., Lebrun V. Artificial metalloenzymes: reaction scope and optimization Strategies. Chem Rev. 2018;118(1):142–231. doi: 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- 105.Himiyama T., Okamoto Y. Artificial metalloenzymes: from selective chemical transformations to biochemical applications. Molecules. 2020;25(13):2989. doi: 10.3390/molecules25132989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liang A.D., Serrano-Plana J., Peterson R.L., Ward T.R. Artificial metalloenzymes based on the biotin–streptavidin technology: enzymatic cascades and directed evolution. Acc Chem Res. 2019;52(3):585–595. doi: 10.1021/acs.accounts.8b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siakkou E., Rutledge M.T., Wilbanks S.M., Jameson G.N. Correlating crosslink formation with enzymatic activity in cysteine dioxygenase. Biochim Biophys Acta Protein Proteonomics. 2011;1814(12):2003–2009. doi: 10.1016/j.bbapap.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 108.Dominy J.E., Hwang J., Guo S., Hirschberger L.L., Zhang S., Stipanuk M.H. Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J Biol Chem. 2008;283(18):12188–12201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li J., Griffith W.P., Davis I., Shin I., Wang J., Li F. Cleavage of a carbon–fluorine bond by an engineered cysteine dioxygenase. Nat Chem Biol. 2018;14(9):853–860. doi: 10.1038/s41589-018-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li J., Koto T., Davis I., Liu A. Probing the Cys-Tyr cofactor biogenesis in cysteine dioxygenase by the genetic incorporation of fluorotyrosine. Biochemistry. 2019;58(17):2218–2227. doi: 10.1021/acs.biochem.9b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Simmons C.R., Liu Q., Huang Q., Hao Q., Begley T.P., Karplus P.A. Crystal structure of mammalian cysteine dioxygenase a novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281(27):18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 112.Chen L., Naowarojna N., Song H., Wang S., Wang J., Deng Z. Use of a tyrosine analogue to modulate the two activities of a nonheme iron enzyme ovoA in ovothiol biosynthesis, cysteine oxidation versus oxidative C–S bond formation. J Am Chem Soc. 2018;140(13):4604–4612. doi: 10.1021/jacs.7b13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yan W., Song H., Song F., Guo Y., Wu C.-H., Sae Her A. Endoperoxide formation by an α-ketoglutarate-dependent mononuclear non-haem iron enzyme. Nature. 2015;527(7579):539–543. doi: 10.1038/nature15519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 114.Yu Y., Lv X., Li J., Zhou Q., Cui C., Hosseinzadeh P. Defining the role of tyrosine and rational tuning of oxidase activity by genetic incorporation of unnatural tyrosine analogs. J Am Chem Soc. 2015;137(14):4594–4597. doi: 10.1021/ja5109936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Q., Hu M., Zhang W., Jiang L., Perrett S., Zhou J. Probing the function of the Tyr-Cys cross-link in metalloenzymes by the genetic incorporation of 3-methylthiotyrosine. Angew Chem Int. 2013;52(4):1203–1207. doi: 10.1002/anie.201207229. [DOI] [PubMed] [Google Scholar]
- 116.Liu X., Yu Y., Hu C., Zhang W., Lu Y., Wang J. Significant increase of oxidase activity through the genetic incorporation of a tyrosine-histidine cross-link in a myoglobin model of heme-copper oxidase. Angew Chem Int. 2012;51(18):4312–4316. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cheah I.K., Halliwell B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2012;1822(5):784–793. doi: 10.1016/j.bbadis.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 118.Ames B.N. Prolonging healthy aging: longevity vitamins and proteins. Proc Natl Acad Sci Unit States Am. 2018;115(43):10836–10844. doi: 10.1073/pnas.1809045115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seebeck F.P. In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J Am Chem Soc. 2010;132(19):6632–6633. doi: 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- 120.Hu W., Song H., Sae Her A., Bak D.W., Naowarojna N., Elliott S.J. Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus Neurospora crassa ergothioneine biosynthetic pathway. Org Lett. 2014;16(20):5382–5385. doi: 10.1021/ol502596z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Irani S., Naowarojna N., Tang Y., Kathuria K.R., Wang S., Dhembi A. Snapshots of C-S cleavage in Egt2 reveals substrate specificity and reaction mechanism. Cell Chem Biol. 2018;25(5):519–529. doi: 10.1016/j.chembiol.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Braunshausen A., Seebeck F.P. Identification and characterization of the first ovothiol biosynthetic enzyme. J Am Chem Soc. 2011;133(6):1757–1759. doi: 10.1021/ja109378e. [DOI] [PubMed] [Google Scholar]
- 123.Naowarojna N., Cheng R., Chen L., Quill M., Xu M., Zhao C. Mini-review: ergothioneine and ovothiol biosyntheses, an unprecedented trans-sulfur strategy in natural product biosynthesis. Biochemistry. 2018;57(24):3309–3325. doi: 10.1021/acs.biochem.8b00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goncharenko K.V., Vit A., Blankenfeldt W., Seebeck F.P. Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway. Angew Chem Int. 2015;54(9):2821–2824. doi: 10.1002/anie.201410045. [DOI] [PubMed] [Google Scholar]
- 125.Naowarojna N., Irani S., Hu W., Cheng R., Zhang L., Li X. Crystal structure of the ergothioneine sulfoxide synthase from Candidatus Chloracidobacterium thermophilum and structure-guided engineering to modulate tts substrate selectivity. ACS Catal. 2019;9(8):6955–6961. doi: 10.1021/acscatal.9b02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stampfli A.R., Goncharenko K.V., Meury M., Dubey B.N., Schirmer T., Seebeck F.P. An alternative active site architecture for O2 activation in the ergothioneine biosynthetic EgtB from Chloracidobacterium thermophilum. J Am Chem Soc. 2019;141(13):5275–5285. doi: 10.1021/jacs.8b13023. [DOI] [PubMed] [Google Scholar]
- 127.Faponle A.S., Seebeck F.P., de Visser S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J Am Chem Soc. 2017;139(27):9259–9270. doi: 10.1021/jacs.7b04251. [DOI] [PubMed] [Google Scholar]
- 128.Wei W.J., Siegbahn P.E., Liao R.Z. Theoretical study of the mechanism of the non-heme iron enzyme EgtB. Inorg Chem. 2017;56(6):3589–3599. doi: 10.1021/acs.inorgchem.6b03177. [DOI] [PubMed] [Google Scholar]
- 129.Tian G., Su H., Liu Y. Mechanism of sulfoxidation and C–S bond formation involved in the biosynthesis of ergothioneine catalyzed by ergothioneine synthase (EgtB) ACS Catal. 2018;8(7):5875–5889. [Google Scholar]
- 130.Song H., Her A.S., Raso F., Zhen Z., Huo Y., Liu P. Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Org Lett. 2014;16(8):2122–2125. doi: 10.1021/ol5005438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen L., Naowarojna N., Chen B., Xu M., Quill M., Wang J. Mechanistic studies of a nonheme iron enzyme OvoA in ovothiol biosynthesis using a tyrosine analogue, 2-amino-3-(4-hydroxy-3-(methoxyl) phenyl) propanoic acid (MeOTyr) ACS Catal. 2019;9(1):253–258. [Google Scholar]
- 132.Yu Y., Zhou Q., Wang L., Liu X., Zhang W., Hu M. Significant improvement of oxidase activity through the genetic incorporation of a redox-active unnatural amino acid. Chem Sci. 2015;6(7):3881–3885. doi: 10.1039/c5sc01126d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kato N., Suzuki H., Takagi H., Uramoto M., Takahashi S., Osada H. Gene disruption and biochemical characterization of verruculogen synthase of Aspergillus fumigatus. Chembiochem. 2011;12(5):711–714. doi: 10.1002/cbic.201000562. [DOI] [PubMed] [Google Scholar]
- 134.Steffan N., Grundmann A., Afiyatullov S., Ruan H., Li S.M. FtmOx1, a non-heme Fe(II) and α-ketoglutarate-dependent dioxygenase, catalyses the endoperoxide formation of verruculogen in Aspergillus fumigatus. Org Biomol Chem. 2009;7(19):4082–4087. doi: 10.1039/b908392h. [DOI] [PubMed] [Google Scholar]
- 135.Dunham N.P., Del Río Pantoja J.M., Zhang B., Rajakovich L.J., Allen B.D., Krebs C. Hydrogen donation but not abstraction by a tyrosine (Y68) during endoperoxide installation by verruculogen synthase (FtmOx1) J Am Chem Soc. 2019;141(25):9964–9979. doi: 10.1021/jacs.9b03567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chan S.I., Li P.M. Cytochrome c oxidase: understanding nature's design of a proton pump. Biochemistry. 1990;29(1):1–12. doi: 10.1021/bi00453a001. [DOI] [PubMed] [Google Scholar]
- 137.Collman J.P., Devaraj N.K., Decréau R.A., Yang Y., Yan Y.-L., Ebina W. A cytochrome c oxidase model catalyzes oxygen to water reduction under rate-limiting electron flux. Science. 2007;315(5818):1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kaila V.R., Verkhovsky M.I. Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110(12):7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 139.Carey J.R., Ma S.K., Pfister T.D., Garner D.K., Kim H.K., Abramite J.A. A site-selective dual anchoring strategy for artificial metalloprotein design. J Am Chem Soc. 2004;126(35):10812–10813. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 140.Yeung N., Lin Y.-W., Gao Y.-G., Zhao X., Russell B.S., Lei L. Rational design of a structural and functional nitric oxide reductase. Nature. 2009;462(7276):1079–1082. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lin Y. Rational design of artificial metalloenzymes: case studies in myoglobin. Prog Chem. 2018;30(10):1464–1474. [Google Scholar]
- 142.Lin Y.-W. Rational design of metalloenzymes: from single to multiple active sites. Coord Chem Rev. 2017;336:1–27. [Google Scholar]
- 143.Nastri F., Chino M., Maglio O., Bhagi-Damodaran A., Lu Y., Lombardi A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem Soc Rev. 2016;45(18):5020–5054. doi: 10.1039/c5cs00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Reetz M.T. Directed evolution of artificial metalloenzymes: a universal means to tune the selectivity of transition metal catalysts? Acc Chem Res. 2019;52(2):336–344. doi: 10.1021/acs.accounts.8b00582. [DOI] [PubMed] [Google Scholar]
- 145.Sigman J.A., Kwok B.C., Lu Y. From myoglobin to heme-copper oxidase: design and engineering of a CuB center into sperm whale myoglobin. J Am Chem Soc. 2000;122(34):8192–8196. [Google Scholar]
- 146.Miner K.D., Mukherjee A., Gao Y.-G., Null E.L., Petrik I.D., Zhao X. A designed functional metalloenzyme that reduces O2 to H2O with over one thousand turnovers. Angew Chem Int. 2012;51(23):5589–5592. doi: 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bhagi-Damodaran A., Petrik I.D., Marshall N.M., Robinson H., Lu Y. Systematic tuning of heme redox potentials and its effects on O2 reduction rates in a designed oxidase in myoglobin. J Am Chem Soc. 2014;136(34):11882–11885. doi: 10.1021/ja5054863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reynolds E.W., McHenry M.W., Cannac F., Gober J.G., Snow C.D., Brustad E.M. An evolved orthogonal enzyme/cofactor pair. J Am Chem Soc. 2016;138(38):12451–12458. doi: 10.1021/jacs.6b05847. [DOI] [PubMed] [Google Scholar]
- 149.Liu J., Chakraborty S., Hosseinzadeh P., Yu Y., Tian S., Petrik I. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem Rev. 2014;114(8):4366–4469. doi: 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Festa R.A., Thiele D.J. Copper: an essential metal in biology. Curr Biol. 2011;21(21):R877–R883. doi: 10.1016/j.cub.2011.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Messerschmidt A. 2010. Copper metalloenzymes. [Google Scholar]
- 152.Berry S.M., Ralle M., Low D.W., Blackburn N.J., Lu Y. Probing the role of axial methionine in the blue copper center of azurin with unnatural amino acids. J Am Chem Soc. 2003;125(29):8760–8768. doi: 10.1021/ja029699u. [DOI] [PubMed] [Google Scholar]
- 153.Garner D.K., Vaughan M.D., Hwang H.J., Savelieff M.G., Berry S.M., Honek J.F. Reduction potential tuning of the blue copper center in Pseudomonas aeruginosa azurin by the axial methionine as probed by unnatural amino acids. J Am Chem Soc. 2006;128(49):15608–15617. doi: 10.1021/ja062732i. [DOI] [PubMed] [Google Scholar]
- 154.Hwang H.J., Carey J.R., Brower E.T., Gengenbach A.J., Abramite J.A., Lu Y. Blue ferrocenium azurin: an organometalloprotein with tunable redox properties. J Am Chem Soc. 2005;127(44):15356–15357. doi: 10.1021/ja054983h. [DOI] [PubMed] [Google Scholar]