

Abstract
Background and Aims
The composition of the human gut microbiota is linked to health and disease, and knowledge of the impact of therapeutics on the microbiota is essential to decipher their biological roles and to gain new mechanistic insights. Here we report the effect of aldafermin, an analog of the gut hormone FGF19, versus placebo on the gut microbiota in a prospective, phase 2 study in patients with NASH.
Approach and Results
A total of 176 patients with biopsy‐confirmed nonalcoholic steatohepatitis (NASH) (nonalcoholic fatty liver disease activity score ≥ 4), fibrosis (F1‐F3 by NASH Clinical Research Network criteria), and elevated liver fat content (≥ 8% by magnetic resonance imaging–proton density fat fraction) received 0.3 mg (n = 23), 1 mg (n = 49), 3 mg (n = 49), and 6 mg (n = 28) aldafermin or placebo (n = 27) for 12 weeks. Stool samples were collected on day 1 and week 12 and profiled using 16S ribosomal RNA gene sequencing; 122 patients had paired stool microbiome profiles at both day 1 and week 12. Overall, the state of the gut microbial community was distinctly stable in patients treated with aldafermin, with all major phyla and genera unaltered during therapy. Patients treated with aldafermin showed a significant, dose‐dependent enrichment in the rare genus Veillonella, a commensal microbe known to have lactate‐degrading and performance‐enhancing properties, which correlated with changes in serum bile acid profile.
Conclusions
Veillonella may be a bile acid–sensitive bacteria whose enrichment is enabled by aldafermin‐mediated suppression of bile acid synthesis and, in particular, decreases in toxic bile acids. This study provides an integrated analysis of gut microbiome, serum bile acid metabolome, imaging, and histological measurements in clinical trials testing aldafermin for NASH. Our results provide a better understanding of the intricacies of microbiome–host interactions (clinicaltrials.gov trial No. NCT02443116).
Abbreviations
- DCA
deoxycholic acid
- FDR
false discovery rate
- FGF19
fibroblast growth factor 19
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GC‐MS
gas chromatography–mass spectrometry
- GDCA
glycodeoxycholic acid
- IQR
interquartile range
- LFC
liver fat content
- MRI
magnetic resonance imaging
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- OTU
operational taxonomic unit
- PDFF
proton density fat fraction
- rRNA
ribosomal RNA
- TDCA
taurodeoxycholic acid
The human gut microbiome has a pivotal role in the maintenance of health.( 1 ) Microbiome provides the host with enhanced metabolic capability, protection against pathogens, education of the immune system, and modulation of gastrointestinal development.( 2 , 3 , 4 ) The microbiome can be perturbed by conditions such as nonalcoholic steatohepatitis (NASH), a serious progressive form of nonalcoholic fatty liver disease (NAFLD) affecting more than 5% of the US adult population.( 5 , 6 )
The functional significance of the gut microbiome in the progression of liver disease in NASH and other chronic liver diseases is not well characterized. Dysbiotic microbiome is observed among individuals with NAFLD.( 7 , 8 ) Multiple human studies, mostly cross‐sectional, have evaluated the association between gut dysbiosis and the spectrum of NAFLD.( 9 , 10 , 11 , 12 , 13 , 14 ) Studies comparing the bacterial taxonomic composition between patients with NAFL or NASH and control subjects have yielded variable and often contradictory findings.( 9 , 10 , 11 , 12 , 13 , 14 ) Due to the cross‐sectional nature of these published human studies, most clinical evidence supports an association between dysbiosis and NAFLD, but mechanistic links have not been established.
NASH is characterized by complex host–microbiome interactions, but little is known about systemic alterations during therapies, their effects on biological processes, or potential impact on microbiome. Key questions on microbiome‐linked disease states and the underpinnings of these links remain unexplored. How dynamic is the microbiome during drug treatment? Which changes in the microbiome represent causes rather than effects of change? Which elements of a microbiome might be responsible for health outcomes, and how do they integrate with therapies? Why do therapies work in some individuals but not others? It is essential to create a global and simultaneous profile of both host and microbial factors in individuals on therapy over time, to fully understand the molecular pathways that are affected, and how the therapy affects biological responses in patients.
There is an intimate relationship between microbiome and the host factor bile acids. The gut microbiota deconjugate bile acids and convert primary bile acids to secondary bile acids. Bile acids are important molecules that activate several host receptors, including farnesoid X receptor and TGR5, influencing the host metabolism and immunity.( 15 , 16 ) Bile acids prevent intestinal bacterial overgrowth, both directly (via membrane damaging effects) and indirectly (via production of antimicrobial proteins). Fibroblast growth factor receptor 19 (FGF19) is a gut hormone that plays a central role in regulating bile acid metabolism.( 17 , 18 ) Through the FGFR4‐KLB receptor, FGF19 potently suppresses cholesterol 7alpha‐hydroxylase (CYP7A1), which encodes the first and rate‐liming enzyme in de novo synthesis of bile acids. Through the FGFR1c‐KLB receptor, FGF19 improves insulin sensitivity and energy homeostasis. Aldafermin (also known as NGM282 or M70), an engineered FGF19 analogue, has demonstrated robust activity in animal models of NASH.( 19 , 20 ) Importantly, a 12‐week treatment with aldafermin inhibited bile acid synthesis, reduced steatosis as measured by magnetic resonance imaging (MRI) proton density fat fraction (PDFF), and improved hepatic inflammation and fibrosis in patients with NASH.( 21 , 22 )
To gain a holistic view of host–microbe interactions over time during aldafermin therapy, we prospectively collected specimens, including serum, plasma, liver biopsy and stool, as well as MRI‐PDFF images, in a phase 2 clinical trial of aldafermin in patients with NASH. Using multiple complementary approaches to assess the mechanisms of human and microbial activity longitudinally, we followed the dynamics of NASH disease during aldafermin therapy and had measurements of clinically relevant parameters from 176 patients. These data provided valuable information to help identify associations between changes in the microbiome and conditions, as well as mechanisms that are critical in host–microbiome interplay.
Experimental Procedures
Patients
The study was conducted in compliance with International Conference on Harmonization, E6 Good Clinical Practice, and Declaration of Helsinki, and all patients provided written, informed consent. The study protocol was approved by the local ethics committees before study initiation. Patients were eligible if they met the following inclusion criteria: 18 to 75 years of age at the time of screening; biopsy‐confirmed NASH diagnosis as defined by the NASH Clinical Research Network (CRN) Histologic Scoring System, with a minimum NAFLD Activity Score (NAS) of 4 (≥1 point in each component of steatosis, lobular inflammation, and hepatocellular ballooning); stage 1, 2, or 3 fibrosis; liver fat content (LFC) ≥ 8% as assessed by MRI‐PDFF; and elevated alanine aminotransferase (ALT) (≥19 IU/L in females; ≥30 IU/L in males). Exclusion criteria included clinically significant acute or chronic liver disease unrelated to NASH, evidence of drug‐induced steatohepatitis, history or presence of compensated or decompensated cirrhosis, liver transplantation, any cardiovascular event or evidence of active cardiovascular disease within 6 months of screening, and type 1 diabetes. Patients taking medications for diabetes were required to be on a stable regimen for at least 3 months before day 1 and maintain a stable regimen during the study period. Initiation of any medications or products for diabetes or weight loss was prohibited from screening to day 1 until the end of the study.
Microbiome profiling was conducted in three separate cohorts of patients in this phase 2 study. Cohort 1 was a placebo‐controlled, double‐blind study comparing aldafermin 3 mg (n = 27) and 6 mg (n = 28) versus placebo (n = 27) for 12 weeks; cohort 2 was a dose‐expansion study evaluating aldafermin 0.3 mg (n = 23), 1 mg (n = 21), and 3 mg (n = 22) for 12 weeks; cohort 3 further expanded the assessment of aldafermin 1 mg in an additional 28 patients for 12 weeks. The primary endpoint was change from baseline to week 12 in LFC as measured by MRI‐PDFF. Patients were stratified by diabetes status. Patients were required to maintain their normal level of diet, physical activity, and lifestyle throughout the entire study. Aldafermin‐associated low‐density lipoprotein–cholesterol change was managed with rosuvastatin in cohorts 2 and 3. Overall, a total of 176 patients with NASH received 0.3 mg (n = 23), 1 mg (n = 49), 3 mg (n = 49), 6 mg (n = 28) aldafermin or placebo (n = 27) for 12 weeks. At each study visit, blood samples were collected for clinical laboratory tests, including liver enzymes, lipid panels, complete blood counts, bile acids, and biomarkers. Stool samples were collected at day 1 and week 12. Details on procedures and treatment in cohorts 1‐3 were previously reported.( 21 , 22 , 23 ) The clinicaltrials.gov trial number is NCT02443116.
Human Fecal Sample Collection and Extraction
Each patient provided fresh stool samples that were delivered immediately to the laboratory in an ice bag using insulating polystyrene foam containers. In the laboratory it was aliquoted and immediately stored at −80°C and shipped on dry ice. A frozen aliquot of each fecal sample was processed, and genomic DNA was isolated from stool samples using the MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA) according to the manufacturer’s instructions. DNA concentration was measured by NanoDrop (Thermo Fisher Scientific, Waltham, MA).
Sequencing of 16S Ribosomal RNA Gene Amplicon
16S ribosomal RNA (rRNA) gene‐sequencing methods were adapted from the methods developed for the National Institutes of Health Human Microbiome Project. The 16S ribosomal DNA V4 region was amplified using the Eppendorf thermocycler (Enfied, CT) under the following conditions: initial denaturation at 95°C for 2 minutes followed by 33 amplification cycles of 20 seconds at 95°C, 45 seconds at 50°C, and 90 seconds at 72°C, followed by final extension at 72°C for 10 minutes and sequenced on the MiSeq platform (Illumina, San Diego, CA) using the 2 × 250 base pair paired‐end protocol, yielding pair‐end reads that overlap almost completely. The primers (515F:5′ AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA 3′ and 806R:5′ CAAGCAGAAGACGGCATACGAGATTCCCTTGTCTCCAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT‘3) used for amplification contain adapters for MiSeq sequencing and single‐end barcodes, allowing pooling and direct sequencing of PCR products. The read pairs were demultiplexed based on the unique molecular barcodes, and reads were merged using USEARCH v7.0.1090, allowing zero mismatches and a minimum overlap of 50 bases. Merged reads are trimmed at first base with Q5. In addition, a quality filter was applied to the resulting merged reads, and reads containing above 0.05 expected errors were discarded. 16S rRNA gene sequences were clustered into operational taxonomic units (OTUs) at 97% similarity using the UPARSE algorithm. OTUs were mapped to an optimized version of the SILVA database containing only the 16S v4 region to determine taxonomies. Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A custom script constructed a rarefied OTU table from the output files generated in the previous two steps for downstream analyses of alpha diversity, beta diversity, and phylogenetic trends.
Richness and Diversity Index
For richness estimation, related to the number of OTUs, we use the observed OTU index. Biodiversity on how uniformly the sequences are spread into the different observed OTUs was estimated with the nonparametric Shannon formula.
Liver Biopsy
During the screening period and before randomization, patients underwent a liver biopsy or provided a liver biopsy tissue specimen obtained within the previous 6 months. Tissue samples were prepared and read by qualified local pathologists to verify NASH according to the NASH CRN histologic scoring system. The NAS, with a score of 8 representing the highest disease severity, is the sum of scores of the three components of the histological scoring system (steatosis, ballooning, and inflammation). Steatosis in hepatocytes was scored as 0, 1, 2, or 3 if there were less than 5%, 5%‐33%, 33%‐66%, or greater than 66% hepatocytes with fat, respectively. Ballooning degeneration in hepatocytes was scored as 0, 1, or 2 if there were none, few ballooned cells, or many ballooned cells, respectively. Lobular inflammation was scored as 0, 1, 2, or 3 if there were no foci, <2 foci, 2‐4 foci, or >4 foci per ×200 field, respectively. The Kleiner scoring system of NAFLD fibrosis (0‐4 for stages F0‐F4) was used to evaluate the stage of fibrosis in each specimen, with higher scores representing more severe fibrosis.
Liver Imaging by MRI Assessment
Patients underwent MRI for assessment of liver‐fat content by MRI‐PDFF at baseline and week 12 (end of treatment). Scans were performed on qualified and standardized instruments at 1.5 T or 3 T field strength. The local MRI facilities completed a qualification process before performing study MRI examinations, and ongoing quality assurance was conducted during the study. The MRI‐PDFF acquisition protocols included a six‐echo 2D gradient‐recalled echo sequence, and image data were transferred to the Center for Advanced Magnetic Resonance Development at Duke University for central calculation and measurement of MRI‐PDFF using an established technique.
Bile Acid Metabolome
Serum bile acid metabolome was quantified using gas chromatography–mass spectrometry (GC‐MS) methods. An Ultimate 3000 RSLC system (Dionex; Thermo Fisher Scientific) coupled to a 4000 QTRAP mass spectrometer via a TurboIonSpray electrospray ionization (SCIEX, Toronto, Canada) was operated in the negative ion mode. A BEH C18 (2.1 × 150 mm, 1.7 μm) UHPLC column (Waters Corp., Milford, MA) was used for the gradient elution, with 0.01% formic acid in water (solvent A) and 0.01% formic acid in acetonitrile (solvent B) as the mobile phase.
Statistics
Differences between populations were analyzed using Kruskal‐Wallis or Mann‐Whitney U tests (two‐sided). To account for multiple comparisons, the Benjamini‐Hochberg false discovery rate (FDR) adjustments were applied to the P values. Spearman’s correlation coefficient was computed using taxa abundances. Categorical parameters were compared using chi‐square or Fisher’s exact tests. A post hoc analysis was performed to evaluate whether patients with ≥ 70% reduction in LFC had specific changes in their gut microbiome. Analyses were performed in SAS version 9.4 and R, incorporating both public and proprietary packages.
Results
To better characterize the mechanisms of host–microbiome interaction during aldafermin therapy, we prospectively followed 176 patients with NASH longitudinally for 12 weeks in a phase 2 aldafermin trial (Fig. 1A). Overall, the trial population was 69% female, 6% non‐white, with a mean (SD) age of 51.2 (10.6) years; 49% had type 2 diabetes. The baseline mean (SD) MRI‐PDFF was 18.6 (6.8)%. Stages 1, 2, and 3 fibrosis was present in 35%, 32%, and 32% of patients, respectively. Baseline patient demographic and characteristics are provided in Table 1.
Fig. 1.
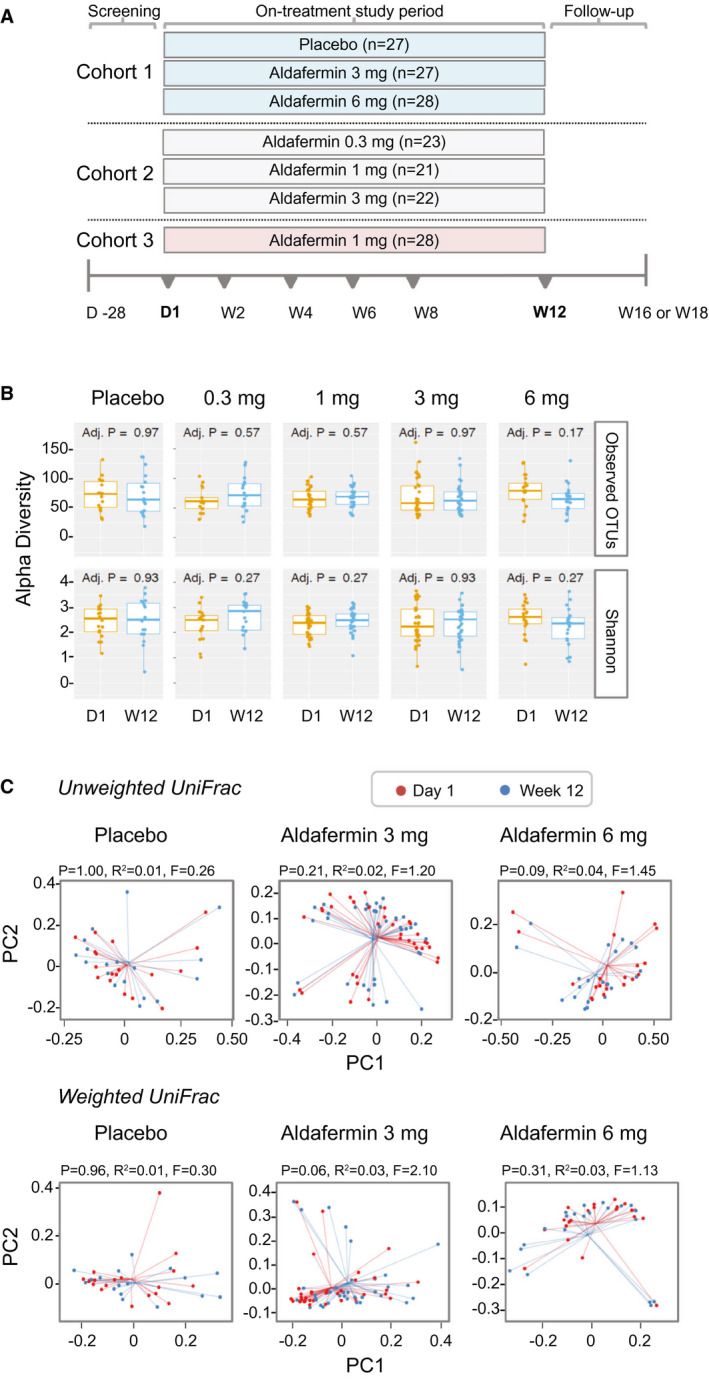
Stable gut microbiome with aldafermin therapy in patients with NASH. (A) Study design. Cohort 1 was a placebo‐controlled, double‐blind study comparing aldafermin 3 mg and 6 mg versus placebo for 12 weeks; cohort 2 was a dose‐expansion study evaluating aldafermin 0.3 mg, 1 mg, and 3 mg for 12 weeks; and cohort 3 further expanded the assessment of aldafermin 1 mg in additional patients for 12 weeks. Overall, a total of 176 patients with NASH received 0.3 mg (n = 23), 1 mg (n = 49), 3 mg (n = 49), or 6 mg (n = 28) aldafermin or placebo (n = 27) for 12 weeks in this phase 2 trial of aldafermin. The 16S rRNA sequencing of fecal samples collected at baseline (day 1) and week 12 (end of treatment) was performed. Patients had liver biopsy at baseline and underwent MRI and laboratory tests at baseline and week 12. (B) Gut microbial richness and evenness measured by alpha diversity (P values by Mann‐Whitney U test with FDR corrections using Benjamini‐Hochberg method). (C) Gut microbial beta biodiversity. Beta diversity was evaluated using UniFrac‐based analysis. In the principal coordinates analysis, no clustering was observed at the PC1 versus PC2 plot, indicating stable phylogenetic composition of the samples. All patients with paired stool samples at both day 1 and week 12 were included in the analysis: placebo (n = 18), aldafermin 0.3 mg (n = 17), 1 mg (n = 30), 3 mg (n = 36), and 6 mg (n = 21). Abbreviations: D1, day 1; W12, week 12.
Table 1.
Patient Demographics and Baseline Characteristics
| Parameters | Patients (n = 176) |
|---|---|
| Age (years) | 51.2 (10.6) |
| Weight (kg) | 100.2 (21.5) |
| BMI (kg/m2) | 36.4 (7.1) |
| Sex | |
| Female | 121 (69%) |
| Male | 55 (31%) |
| Race | |
| Asian | 3 (2%) |
| Black | 2 (1%) |
| White | 166 (94%) |
| Other | 5 (3%) |
| Ethnicity | |
| Hispanic/Latino | 82 (47%) |
| Non‐Hispanic/Latino | 94 (53%) |
| Type 2 diabetes | |
| Yes | 87 (49%) |
| No | 89 (51%) |
| Histology | |
| Fibrosis stage | |
| 1 | 62 (35%) |
| 2 | 56 (32%) |
| 3 | 56 (32%) |
| 4 | 2 (1%) |
| Total NAS score | 5.3 (1.2) |
| Ballooning | |
| 0 (none) | 6 (3%) |
| 1 (few ballooned cells) | 85 (48%) |
| 2 (many ballooned cells) | 84 (48%) |
| Steatosis | |
| 0 (<5%) | 0 |
| 1 (5%‐33%) | 40 (23%) |
| 2 (34%‐66%) | 76 (43%) |
| 3 (>66%) | 59 (34%) |
| Inflammation | |
| 0 (none) | 0 |
| 1 (<2) | 59 (34%) |
| 2 (2‐4) | 100 (57%) |
| 3 (>4) | 16 (9%) |
| LFC by MRI‐PDFF | |
| LFC (%) | 18.6 (6.8) |
Note: Values are presented as mean (SD) or n (%). Percentages may not total 100 because of rounding.
Abbreviations: BMI, body mass index; NAS, nonalcoholic fatty liver disease activity score.
Integrated longitudinal molecular profiles of microbial and host activity were generated by analyzing 288 stool samples, 176 liver biopsies, 343 liver MRI‐PDFF images, and more than 800 blood samples. Multiple molecular profiles were generated from the same visit, allowing concurrent changes to be observed in host and microbial molecular and clinical activity over time.
Patients With NASH Treated With Aldafermin Had Stable Microbiome
Gut microbiome composition of the patients was determined by 16S rRNA gene sequencing from stool‐extracted DNA. The V4 hypervariable region of the 16S rRNA gene was amplified using the Illumina MiSeq technology. An average of 17,404 sequences per sample were generated after filtering. The 16S rRNA gene sequences were clustered into OTUs at a similarity cutoff value of 97% using the UPARSE algorithm. A total of 84.9% of reads produced were mapped to the SILVA(v4) database.
A total of 122 patients had paired stool microbiome profiles at both day 1 and week 12, and were included in the microbiome analysis. We compared the microbial richness (observed OTUs) and the Shannon diversity index for the placebo and aldafermin groups. We found no significant differences in both alpha diversity indices between placebo and aldafermin samples at either baseline or week 12 (end of treatment) (Fig. 1B). We evaluated between‐sample diversity (beta diversity) within each group by UniFrac analysis. Both weighted and unweighted uniFrac‐based principal coordinates analysis showed no clustering between the two time points in all groups, indicating stable phylogenetic composition of the samples over time (Fig. 1C).
At the phylum level, Bacteroidetes and Firmicutes dominated the fecal microbial communities of all groups, followed by Proteobacteria. Compared with placebo controls, patients treated with aldafermin had similar levels of Bacteroidetes, Firmicutes, Proteobacteria, Verrucomicrobia, Fusobacteria, Actinobacteria, Tenericutes, Cyanobacteria, Lentisphaerae, Synergistetes, Euryarchaeota, and Spirochaetae (Fig. 2A,B). No significant difference was observed between groups at each time point as well as between visits within each treatment group.
Fig. 2.
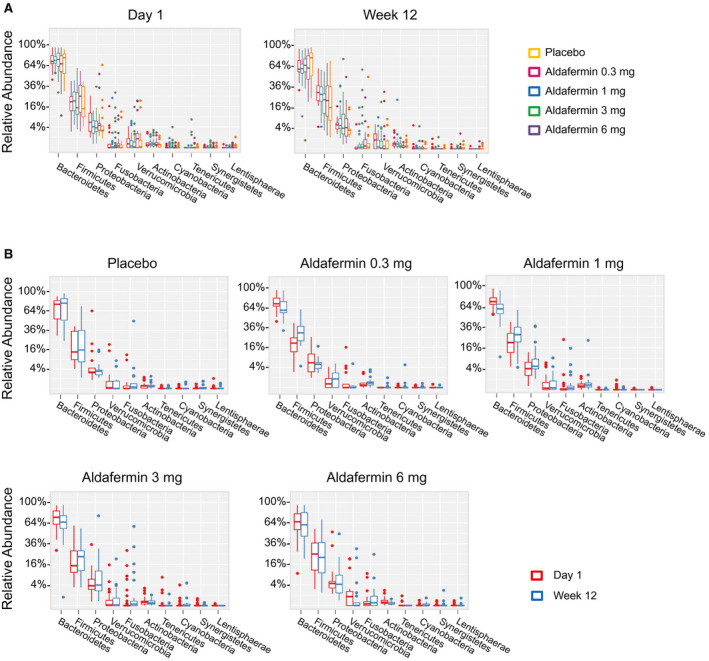
Phylogenetic abundance at the phylum level. (A) Relative abundance of top phyla by visits in patients treated with placebo or aldafermin. Taxonomic compositions of the most prevalent phyla are displayed, with ranks ordered from left to right by their decreasing abundance. Only the top 10 phylotypes are shown for clarity (P > 0.05 for all comparisons by Kruskal‐Wallis test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). (B) Relative abundance of top phyla by treatment groups (P > 0.05 for all comparisons by Mann‐Whitney U test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). The boxes represent the interquartile range (IQR), from the first and third quartiles, and the inside line represents the median. The whiskers denote the lowest and highest values within 1.5 IQR from the first and third quartiles. The circles represent outliers beyond the whiskers. All patients with paired stool samples at both day 1 and week 12 were included in the analysis: placebo (n = 18), aldafermin 0.3 mg (n = 17), 1 mg (n = 30), 3 mg (n = 36), and 6 mg (n = 21).
To gain a clear image of the taxonomic changes in gut microbiomes in these patients, abundant genera were examined. At the genus level, Bacteroides was the dominant phylotype in all groups. The abundance of the top 30 genera, including Bacteroides, Prevotellaceae, Alistipes, Lachnospiraceae, Faecalibacterium, Parabacteroides, Escherichia, Lachnoclostridium, Akkermansia, Fusobacterium and Parasutterella, were similar between the placebo and aldafermin groups (Fig. 3A,B).
Fig. 3.
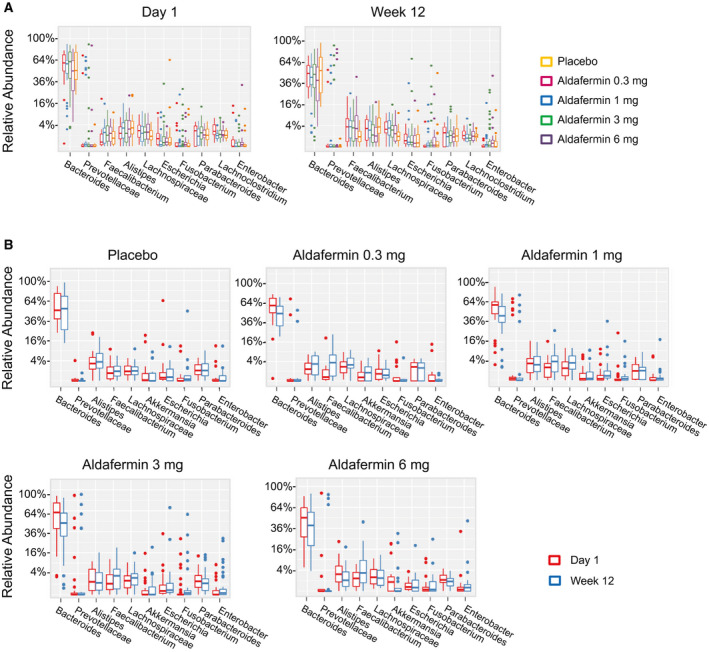
Phylogenetic abundance at the genus level. (A) Relative abundance of top genera by visits in patients treated with placebo or aldafermin. The phylotypes with median relative abundances greater than 0.01% of total abundance in either the placebo or aldafermin groups were included for analysis. Only the 10 most abundant genera in each group are shown for clarity (P > 0.05 for all comparisons by Kruskal‐Wallis test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). (B) Relative abundance of top genera by treatment groups (P > 0.05 for all comparisons by Mann‐Whitney U test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). The boxes represent the IQR, from the first and third quartiles, and the inside line represents the median. The whiskers denote the lowest and highest values within 1.5 IQR from the first and third quartiles. The circles represent outliers beyond the whiskers. All patients with paired stool samples at both day 1 and week 12 were included in the analysis: placebo (n = 18), aldafermin 0.3 mg (n = 17), 1 mg (n = 30), 3 mg (n = 36), and 6 mg (n = 21).
Overall, both the richness and the diversity of gut microorganisms were stable during aldafermin treatment.
Aldafermin Treatment Enriches the Rare Genus Veillonella
Strikingly, of all genera, the rare genus Veillonella was the only phylotype that was significantly altered with aldafermin treatment. Mean relative abundance of Veillonella increased by 4‐fold, 5‐fold, 33‐fold, and 30‐fold from baseline to week 12 (end of treatment) in patients receiving aldafermin 0.3 mg, 1 mg, 3 mg, and 6 mg, respectively, whereas no changes were observed in the placebo group (Fig. 4A). At week 12, 82%, 83%, 75%, and 86% of patients receiving aldafermin 0.3 mg, 1 mg, 3 mg, and 6 mg, respectively, had detectable 16S Veillonella signal present in their fecal samples, compared with 33% in the placebo group (Fig. 4B).
Fig. 4.
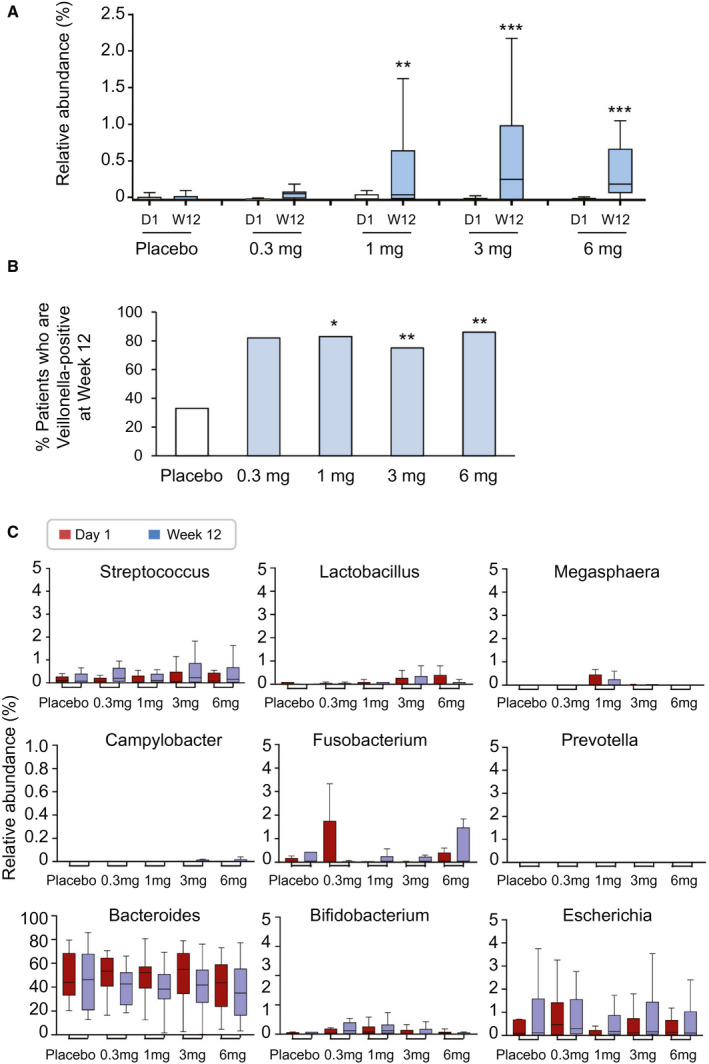
Aldafermin enriches the rare genus Veillonella. (A) Box and whisker plot of relative abundance of Veillonella. Stool samples from patients treated with placebo, aldafermin 0.3 mg, aldafermin 1 mg, aldafermin 3 mg, and aldafermin 6 mg were assessed by 16S rRNA gene sequencing. Dose‐dependent increases in Veillonella abundance were observed in the aldafermin groups, but not in the placebo group (***P < 0.001, **P < 0.01 vs. placebo by Kruskal‐Wallis test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). (B) Proportion of subjects with fecal samples that were positive for Veillonella at week 12. Presence of Veillonella in stool samples was considered positive (**P < 0.01, *P < 0.05 vs. placebo by Fisher’s exact test). (C) Aldafermin did not affect other microbes that are of oral origin or typically associated with Veillonella, nor did it affect microbes that ferment other substrates or have an ethanol‐producing property. Shown are box and whisker plots of relative abundance in placebo and aldafermin groups. All patients with paired stool samples at both day 1 and week 12 were included in the analysis: placebo (n = 18), aldafermin 0.3 mg (n = 17), 1 mg (n = 30), 3 mg (n = 36), and 6 mg (n = 21). Abbreviations: D1, day 1; W12, week 12.
Veillonella is a lactate‐fermenting bacteria that generally reside in the oral cavity. In addition to Veillonella, several other microbial genera, including Streptococcus, Lactobacillus and Megasphaera, also originate mostly from the mouth. No significant difference in abundance was observed in these bacteria (Fig. 4C). No enrichment was seen in taxonomically unrelated bacteria, which tend to be associated with Veillonella, such as Campylobacter and Fusobacterium (Fig. 4C). Microbial genera that can ferment xylan and cellulose to produce short‐chain fatty acids, such as Prevotella, were not affected by aldafermin (Fig. 4C). Elevated representation of ethanol‐producing bacteria, including Bacteroides, Bifidobacterium and Escherichia, was reported in the NASH microbiome.( 24 ) We saw no changes in the microbes which are capable of producing alcohol following aldafermin treatment (Fig. 4C).
In summary, the rare genus Veillonella was the only taxa exhibiting a significant difference with aldafermin treatment compared with placebo. Aldafermin appeared to enrich Veillonella in a dose‐dependent manner.
Correlation Between Veillonella and Bile Acid Species
As FGF19 blocks bile acid synthesis by inhibition of CYP7A1, we hypothesized that these changes in Veillonella might be the consequences of the inhibition of bile acid synthesis. Therefore, we examined a correlation between these changes in microbiota and bile acids.
We performed targeted metabolomic profiling of serum bile acids using GC‐MS and correlated the abundance of Veillonella with concentrations of bile acid species. There were marked reductions in the levels of bile acid species—especially the more toxic, glycine‐conjugated, hydrophobic bile acids, among them glycocholic acid (GCA), glycochenodeoxycholic acid (GCDCA), and glycodeoxycholic acid (GDCA)—in patients treated with aldafermin (Fig. 5A). At week 12, decreases of 27%, 52%, 73%, and 51% in GCA, decreases of 21%, 33%, 56%, and 26% in GCDCA, and decreases of 18%, 67%, 80%, and 75% in GDCA were seen in patients receiving 0.3 mg, 1 mg, 3 mg, or 6 mg aldafermin, respectively. In contrast, no significant changes were seen in placebo‐treated patients. The hydrophobic, secondary bile acid deoxycholic acid (DCA) that was previously implicated in bile acid–associated carcinogenesis in animal models( 25 ) was significantly and dose‐dependently reduced with aldafermin treatment (decreases of 47%, 66%, 84%, and 92% in DCA were seen in patients receiving 0.3 mg, 1 mg, 3 mg, or 6 mg aldafermin, respectively, compared with a 22% increase in patients receiving placebo).
Fig. 5.
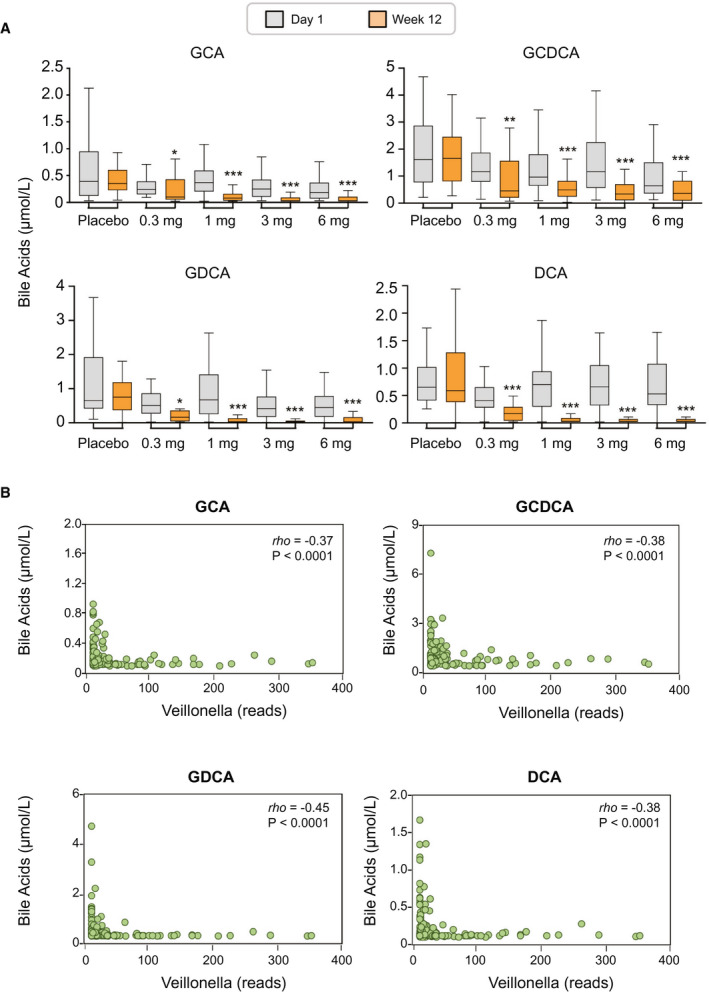
Enrichment of Veillonella inversely correlates with toxic, hydrophobic bile acids. (A) Box and whisker plots of serum bile acids from placebo and aldafermin‐treated patients by GC‐MS. Shown are concentrations of GCA, GCDCA, GDCA, and DCA at baseline (day 1) and end‐of‐treatment (week 12) (***P < 0.001, **P < 0.01, *P < 0.05 vs. placebo): placebo (n = 27), aldafermin 0.3 mg (n = 23), 1 mg (n = 49), 3 mg (n = 49), and 6 mg (n = 28). (B) Correlation between Veillonella OTU reads and concentrations of GCA, GCDCA, GDCA, and DCA at week 12. Spearman’s correlation coefficients were calculated; rho and P values are shown on the graphs.
A correlation analysis was conducted between Veillonella and bile acid metabolome, including GCA, taurocholic acid, GCDCA, taurochenodeoxycholic acid, GDCA, taurodeoxycholic acid (TDCA), glycolithocholic acid, taurolithocholic acid, glycoursodeoxycholic acid, tauroursodeoxycholic acid, glycohyodeoxycholic acid, taurohyodeoxycholic acid, cholic acid, chenodeoxycholic acid, DCA, lithocholic acid, and ursodeoxycholic acid (Table 2). At week 12, abundance of Veillonella significantly and inversely correlated with concentrations of the more toxic, hydrophobic bile acids. The Spearman’s correlation coefficients were rho = −0.45, P < 0.0001 for GDCA; rho = −0.38, P < 0.0001 for GCDCA; rho = −0.38, P < 0.0001 for DCA; rho = −0.37, P < 0.0001 for GCA; and rho = −0.36, P < 0.0001 for TDCA (Table 2 and Fig. 5B).
Table 2.
Correlation Between the Relative Abundance of Veillonella and Bile Acid Metabolome at Week 12
| Bile Acid Metabolome | rho | P Value |
|---|---|---|
| Conjugated primary bile acids | ||
| GCA | −0.37 | <0.0001 |
| TCA | −0.24 | 0.01 |
| GCDCA | −0.38 | <0.0001 |
| TCDCA | −0.11 | 0.25 |
| Conjugated secondary bile acids | ||
| GDCA | −0.45 | <0.0001 |
| TDCA | −0.36 | <0.0001 |
| GLCA | −0.30 | 0.001 |
| TLCA | −0.15 | 0.11 |
| GUDCA | −0.28 | 0.002 |
| TUDCA | −0.13 | 0.16 |
| GHDCA | −0.24 | 0.009 |
| THDCA | −0.03 | 0.73 |
| Unconjugated primary bile acids | ||
| CA | −0.20 | 0.03 |
| CDCA | −0.17 | 0.07 |
| Unconjugated secondary bile acids | ||
| DCA | −0.38 | <0.0001 |
| LCA | −0.27 | 0.003 |
| UDCA | −0.10 | 0.29 |
Note: Values are presented as Spearman’s correlation coefficients and P values. Significant correlation with rho > 0.30 are shown in bold.
Abbreviations: CA, cholic acid; CDCA, chenodeoxycholic acid; GHDCA, glycohyodeoxycholic acid; GLCA, glycolithocholic acid; GUDCA, glycoursodeoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; THDCA, taurohyodeoxycholic acid; TLCA, taurolithocholic acid; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid
This correlation analysis led us to hypothesize that Veillonella could be sensitive to bile acids, and the suppression of bile acids by aldafermin allows them to survive and colonize the human gut.
Correlation Between Gut Microbiome and Liver Fat
Previous studies have shown that several phyla were significantly associated with liver steatosis,( 10 ) including Proteobacteria, Actinobacteria and Verrucomicrobia, which were positively correlated with liver steatosis, and Firmicutes and Euryarchaeota, which were negatively correlated. We evaluated the relationship between fecal microbiota and LFC using data from the pooled cohort of patients in our studies.
We compared the microbiome composition in patients with the top quartile versus the bottom quartile of LFC at baseline. Although no differences were observed between quartile 1 (Q1) and quartile 4 (Q4) subjects in the Shannon diversity index, differences were seen in observed OTUs (Fig. 6A). Due to the differences in the number of observed OTUs, we saw a trend in clustering between Q1 and Q4 in unweighted UniFrac analysis, while no clustering was observed in weighted UniFrac (Fig. 6B). A comparison of Q1 and Q4 subjects revealed no significant differences at the phylum and genus levels (Fig. 6C). No significant differences in top phyla or genera were noted either by LFC quantile or by steatosis grade at baseline (Supporting Tables S1‐S4).
Fig. 6.
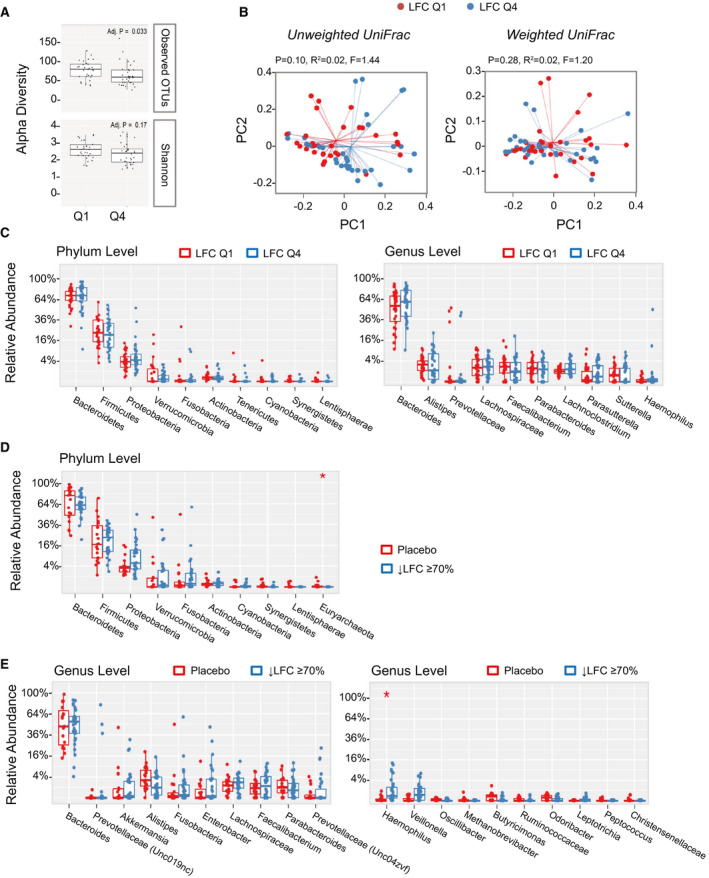
Correlation between gut microbiome and LFC measured by MRI‐PDFF. (A) Patients were grouped into the top quartile (Q4) versus the bottom quartile (Q1) according to baseline LFC. Differences in alpha diversity were observed between Q1 and Q4 subjects using the observed OTUs but not the Shannon index. (B) Weighted UniFrac analysis showed no separation between low liver fat (Q1) and high liver fat (Q4) populations at baseline, whereas unweighted UniFrac showed a trend of clustering. (C) No significant difference at the phylum and genus level between low liver fat (Q1) and high liver fat (Q4) populations. (D) Relative abundance of top phyla in subjects with ≥ 70% reduction in LFC (i.e., super‐responders). At the phylum level, Euryarchaeota significantly differed between aldafermin‐treated subjects who had ≥ 70% reduction in LFC and the placebo group (*P < 0.05 by Mann‐Whitney U test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). (E) Relative abundance of top genera in subjects with ≥ 70% reduction in LFC (i.e., super‐responders). Genera are displayed with ranks ordered from left to right by their decreasing abundance (left panel) or significance (right panel). Only the top 10 phylotypes are shown for clarity. At the genus level, although the top 10 genera are similar between groups, the minor genus Haemophilus is more abundant in subjects with ≥ 70% reduction in LFC than placebo‐treated subjects (*P < 0.05 by Mann‐Whitney U test with Benjamini‐Hochberg FDR‐adjusted multiple test correction). The boxes represent the IQR, from the first and third quartiles, and the inside line represents the median. The whiskers denote the lowest and highest values within 1.5 IQR from the first and third quartiles. The circles represent outliers beyond the whiskers. All patients with paired stool samples at both day 1 and week 12 were included in the analysis: placebo (n = 18), aldafermin 0.3 mg (n = 17), 1 mg (n = 30), 3 mg (n = 36), and 6 mg (n = 21).
Next, we compared the microbiome of patients who achieved ≥ 70% reduction in liver fat on aldafermin (“super‐responders”) versus placebo‐treated patients. Thirty patients (out of 176 patients) were identified to have achieved a 70% or greater reduction in liver fat after 12 weeks of treatment with aldafermin and had paired stool samples, versus none in the placebo group. At the phylum level, Euryarchaeota were reduced in the treated group with 70% or greater reduction in liver fat compared with the placebo group (P < 0.05 by Mann‐Whitney U test with FDR corrections using the Benjamini‐Hochberg method) (Fig. 6D). At the genus level, Methanobrevibacter, the major genus in Euryarchaeota trended toward reduction in subjects with ≥ 70% reduction in liver fat. Haemophilus was significantly enriched in subjects with ≥ 70% liver fat reduction after FDR correction (Fig. 6E). The super‐responders had significant reductions in ALT and aspartate aminotransferase (Supporting Fig. S1A); however, these changes did not appear to correlate with microbiome (Supporting Fig. S1B).
There were no significant differences in the gut microbiome seen between the various stages of fibrosis among these patients with NASH‐related fibrosis (Supporting Tables S5‐S6 and Supporting Fig. S2). Overall, the results remained consistent with Veillonella enrichment across the aldafermin treatment arms independent of baseline LFC or stage of fibrosis.
Therefore, by 16S rRNA sequencing, we found that several microbial taxa in gut microbiome were modulated in patients with ≥ 70% reduction in LFC.
Discussion
We performed integrated analyses of the gut microbiome using 16S rRNA sequencing, targeted metabolomics on serum bile acids, and performed histological and imaging measurements in patients with NASH enrolled in clinical trials of aldafermin. This well‐designed study in patients suggests a potentially causal role of the gut hormone FGF19 and specific changes in the gut microbiome, and that a certain microbiome signature may be used as a noninvasive pharmacodynamics biomarker of response to aldafermin versus placebo in patients with NASH. Therapeutic doses of aldafermin lead to an increase (ranging from 4‐fold to 33‐fold) in the lactate‐degrading microbe Veillonella, which was found to be enriched in the gut microbiome of elite athletes and associated with performance enhancement via lactate metabolism.( 26 ) This is the largest report to date on gut microbiome profiling in response to a therapeutic agent in patients with NASH. Our data provide a holistic view of the dynamic change of gut microbiome over time in response to therapy and reveal host–microbe interactions.
Much of our knowledge of the human microbiome comes from association studies that use either a cross‐sectional or case‐control design. Most studies are conducted at a single time point in a population with the disease, and consequently, these studies can only identify microorganisms that differentiate individuals with the disease and the control population. While providing valuable information, these studies are nearly impossible to separate associations from secondary effects, and therefore cannot establish causality. Well‐designed, longitudinal, prospective studies with multi‐omic profiling technique and integrated analysis are needed to decipher the complex microbiome–host interactions.
There are several innovative aspects in this study, including the incorporation of gut microbiome collection in a multicenter phase 2 trial, taking advantage of a unique opportunity for interrogating microbiome functionality in patients with NASH over a 12‐week period. Combined with liver biopsy, imaging, and laboratory data collection, this proof‐of‐concept multimodality approach can provide a wealth of information and insights about not only microbial dynamics, but also associated human host responses and microbial interrelationships. The study’s longitudinal design with integrated within‐subject profiles further allowed us to characterize the stability and dynamics of host–microbiome interactions during therapy. The presence of a placebo arm adds to the rigor of the study design. Importantly, our results demonstrate that the gut microbial community state is distinctly stable in patients treated with aldafermin. Ecological diversities (alpha and beta diversity) were similar between placebo and aldafermin‐treated patients, indicating a stable microbiome with aldafermin therapy. There was no significant difference in the abundance of all 12 phyla. Of the top 30 most abundant genera, no significant differences were observed in any of the phylotypes.
Although the gut microbiome consists of a stable core, a dynamic component exists that can be influenced by aldafermin. Importantly, we showed that a rare genus, Veillonella, was the only genus within the entire bacteria domain exhibiting a significant difference between the placebo and aldafermin groups. The increase in Veillonella showed a clear dose response to aldafermin, with relative abundance of Veillonella increased by 4‐fold, 5‐fold, 33‐fold, and 30‐fold from baseline to week 12 (end of treatment) in patients receiving aldafermin 0.3 mg, 1 mg, 3 mg and 6 mg, respectively, whereas no changes were observed in the placebo group. Furthermore, aldafermin‐associated enrichment of Veillonella inversely correlated with changes in bile acids, and especially the more toxic, hydrophobic bile acids. Bile acids have been long known for their major effects on the microbiome and the intestinal barrier function.( 15 , 27 ) Bile acids and the gut microbiota closely interact and modulate each other. Due to their detergent property, bile acids exert bactericidal effects: The more hydrophobic a bile acid is, the higher the bactericidal activity. Conversely, gut microbes can deconjugate and convert primary bile acids to secondary bile acids. Our results reveal the adaptive potential of the gut microbiota to bile acids, indicating a correlation between Veillonella and aldafermin therapy that can potentially influence the fitness of the host.
Veillonella is a lactate‐degrading microbe that uses lactate as their sole carbon source. In anaerobic conditions, Veillonella ferments lactate to produce the short‐chain fatty acids propionate. Veillonella has been shown to enrich in the gut microbiome of elite athletes and is associated with performance‐enhancing activity.( 26 ) An increase in Veillonella relative abundance was observed in marathon runners following a marathon; and inoculation of Veillonella into mice significantly increased exhaustive treadmill run time.( 26 ) Qin et al. have reported an enrichment of taxa of buccal origin, including Veillonella, in the gut microbiome of patients with cirrhosis, and concluded that an invasion of the lower gastrointestinal tract by oral bacteria occurs in liver cirrhosis.( 28 ) Chen et al. and more recently, Oh et al., also found an overrepresentation of genera, including Veillonella, in the gut microbiome of patients with cirrhosis.( 29 , 30 ) Liver cirrhosis is characterized by deficient levels of luminal bile acids in the gut.( 31 ) We postulate that an altered bile production in cirrhosis renders the gut more permissible and/or accessible to “foreign” bacteria, as bile resistance is required for survival in the human gut.( 32 ) Our results lead us to hypothesize that Veillonella may be sensitive to bile acids, and that its enrichment is an adaptive response to luminal bile acid deficiency, as in cirrhosis and cholestasis.( 28 , 33 ) This microbiota adaptation appears to be favorable, as fermenting lactate to propionate benefits the microbe by producing additional energy and benefits the host with the advantageous effect of eliminating the toxic lactate, whose accumulation has been associated with mortality in patients with cirrhosis.( 34 )
We have found that the abundance of the phylum Euryarchaeota was significantly reduced in patients who had achieved ≥ 70% liver fat reduction (super‐responders). Euryarchaeota uses H2 and CO2 to produce methane in the human gut and has received renewed interest in recent years.( 35 ) The reductions in Euryarchaeota were accompanied by corresponding changes in the genus Methanobrevibacter, the dominant methanogen in Euryarchaeota. Higher methane levels are reported to slow gastrointestinal transit and correlate with Methanobrevibacter in patients with constipation‐predominant irritable bowel syndrome.( 36 ) Interestingly, aldafermin treatment has been shown to accelerate gastrointestinal transit and improve symptoms in patients with functional constipation.( 37 ) A significant increase in the genus Haemophilus, which normally resides in the upper gastrointestinal tract and the airway, was observed in the super‐responders, echoing the enrichment of Veillonella.
Stool and serum biomarkers can have diagnostic and prognostic potential, as well as serve as biomarkers for monitoring response to treatment.( 6 , 38 , 39 , 40 ) Liver biopsy remains the gold standard for characterizing liver histology in patients with NASH; however, it is an invasive technique and subject to sampling errors and significant intra‐observer and interobserver variability. Hence, the need for noninvasive approaches to evaluate disease progression is particularly acute in NASH.( 41 ) Using the well‐characterized cohorts in this report, we assessed gut microbiome‐derived bacterial biomarkers for the detection of steatosis and fibrosis in NASH. We did not find significant differences in phyla and genera, previously reported as associated with steatosis or fibrosis.( 9 , 10 , 11 , 13 ) In a recent study designed to test the reproducibility of published results, Demir et al. compared 13 PubMed‐listed studies and found that most of the published differences could not be reproduced, neither in their own nor in other NAFLD cohorts.( 42 ) Differences in patient characteristics, duration of studies, sample sizes, dietary habits, medications, genetic and environmental factors, and the lack of standardized methods could contribute to the discrepant results. The difference among the studies highlights the possibility of chance findings and spurious results in small trials and the importance of larger trials with longer follow‐up. Moreover, although observational and cross‐sectional studies are relevant in providing compositional evidence on microbiome, randomized controlled trials and mechanistic studies are necessary to gain deeper understanding about the relationships between microbiome and disease pathogenesis or progression.
This study has several strengths. The prospective design and the detailed phenotyping of the biopsy‐confirmed NASH population allowed the identification of differences in the microbiome and host response during the course of aldafermin treatment. We further phenotyped these patients using advanced MRI and extensive laboratory measures at both baseline and end of treatment. Moreover, we integrated the microbiome data with the bile acid metabolomics, and revealed unexpected findings. This clinical trial evaluates a medical therapy in NASH that integrates microbiome profiling into the trial operation. The study’s longitudinal, complementary molecular measurements enabled the construction of a network of specific microbial taxa to bile acids metabolome as well as liver imaging and biopsy.
This study has the following limitations. First, the 16S‐based sequencing approach lacks the resolution required to identify bacteria on a species or strain level, and even different strains of the same bacterial species can exert different effects on the human host. Furthermore, taxonomic composition of the microbiome alone is often not a good correlate with host phenotype; this tended to be better predicted by microbial molecular function and gene expression. Second, in the past few years, key factors that affect microbiota composition have been identified, including diet.( 43 ) Although patients in this trial were required to maintain their normal level of diet, detailed diet information was not recorded. Moreover, this study was carried out within a geographically and genetically diverse population, and regional differences in life events or infectious disease exposure may change how microbiome dynamics contribute to the disease and response to therapy. Third, the current study focuses exclusively on bacterial communities, not the coexisting fungal and viral communities. Although bacteria dominate the gut microbiota, fungal and viral communities are increasingly recognized as integral members of the community, and trans‐kingdom interactions are likely to be in part responsible for ecological balance. Fourth, we acknowledge that this study may be underpowered, despite the fact that it is one of the largest stool microbiome profiling in patients with biopsy‐proven NASH. Microbiota analysis was designed to collect samples only at baseline (day 1) and week 12 (end of treatment). While blood samples and MRI scan were invariably collected/performed at clinic visits, some patients were unable to provide stool samples. Given this practical limitation in sample collection, missing stool samples at either time point were unavoidable, leading to fewer patients with paired microbiome data. Further studies with larger patient population and more frequent sampling are needed to better characterize the time course (onset) of aldafermin treatment on Veillonella, and to validate these findings. Finally, this is a proof‐of‐concept association study in patients who were followed longitudinally in a clinical trial testing a NASH therapy versus placebo; the precise underlying mechanisms were not identified. We acknowledge that this study does not provide evidence of causality. Future work is needed to examine whether the associations identified in the current study can be validated in studies with different therapeutic agents, and whether a direct cause‐and‐effect relationship can be established by in vitro and in vivo mechanistic research.
We are slowly advancing human microbiome research from cross‐sectional snapshot studies to trial‐setting research to microbiome‐based therapeutic modalities such as fecal microbiota transplant and probiotic interventions. However, effectively translating and applying findings accrued through catalog profiling requires well‐designed, large‐scale clinical trials with detailed and integrated characterization. As the role of the microbiota in NASH disease development, progression, and treatment is increasingly recognized, the need for focused, microbiome‐aware efforts to efficiently integrate disease characteristics and microbiome profiling is urgent. Our study showed that scientifically and ethically sound clinical research in microbiome can be conducted through seamless integration in clinical trials for therapeutics and can help inform treatment response. Such a study can also reveal mechanistic insights into the reciprocal interactions among hormones, metabolites and the human microbiome, representing the future of microbiome research.
Author Contributions
R.L., L.L., A.M.D., and S.A.H. were responsible for the study design. R.L. and S.A.H. were responsible for the data collection. R.L., L.L., and D.M.D. were responsible for the data analysis. R.L., L.L., D.M.D., A.M.D., H.D.L., S.A.H., and A.J.S. were responsible for the data interpretation. All authors were responsible for the manuscript review and writing. R.L., L.L., and D.M.D. were responsible for preparation of the tables and figures.
Supporting information
Supplementary Material
Acknowledgments
The authors thank Dr. Jessica Ferreyra for the insightful discussion on microbiome. They would also like to thank all of the patients who participated in this study, and the investigators, study coordinators, and staff of all of the participating clinical centers for their support and assistance.
Supported by NGM Biopharmaceuticals, National Institute of Environmental Health Sciences (5P42ES010337), and National Institute of Diabetes and Digestive and Kidney Diseases (P30DK120515).
Potential conflict of interest: Dr. Loomba consults, advises, and received grants from Boehringer‐Ingelheim, Bristol‐Myers Squibb, Cirius, Eli Lilly, Galmed, Gilead, Intercept, Janssen, Merck, NGM, Pfizer, Prometheus, and Siemens. He consults and advises Anylam/Regeneron, Arrowhead, AstraZeneca, Bird Rock, Celgene, CohBar, Conatus, Gemphire, Glympse, GNI, GRI, Inipharm, Ionis, Metacrine, Novartis, Novo Nordisk, Promethera, Sanofi, and Viking. He received grants from Allergan, Galectin, GE, Grail, Madrigal, NuSirt, and pH Pharma. Dr. Lieu is employed and owns stock in NGM. Dr. DePaoli is employed and owns stock in NGM. Dr. Ling is employed and owns stock in NGM. Dr. Harrison consults, advises, received grants, and owns stock in Akero, Cirius, Galectin, Genfit, Metacrine, and NGM. He consults, advises, and received grants from Axcella, CymaBay, Hepion, High Tide, Madrigal, North Sea, Novo Nordisk, Poxel, Sagimet, and Terns. He consults, advises, and owns stock in HistoIndex. He consults and advises Altimmune, Echosens, Foresite, Intercept, and Prometic. He consults and received grants from Civi, Enyo, Gilead, Novartis, and Viking. He consults for Canfite, Fortress, Kowa, and Medpace. He advises Arrowhead. He received grants from Ridgeline. Dr. Sanyal consults and received grants from Conatus, Gilead, Echosens‐Sandhill, Malinckrodt, Salix, Novartis, Galectin, and Sequana. He consults and owns stock in GenFit, Hemoshear, Durect, and Indalo. He consults for Immuron, Intercept, Pfizer, Boehringer Ingelheim, Nimbus, Lilly, Novo Nordisk, Fractyl, Allergan, Chemomab, Affimmune, Teva, Aredlyx, Terns, ENYO, Birdrock, Albireo, Sanofi, Takeda, Janssen, Zydus, BASF, AMRA, Perspectum, Owl, Poxel, Servier, Second Genome, General Electric, and 89 Bio. He received grants from Bristol‐Myers Squibb. He received royalties from Elsevier and Uptodate. He owns stock in Exhalenz, Akarna, and Tiziana.
The copyright line for this article was changed on 10 February 2021 after original online publication.
Contributor Information
Rohit Loomba, Email: roloomba@ucsd.edu.
Lei Ling, Email: lling@ngmbio.com.
References
Author names in bold designate shared co‐first authorship
- 1. Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host‐bacterial mutualism in the human intestine. Science 2005;307:1915‐1920. [DOI] [PubMed] [Google Scholar]
- 2. Sonnenburg JL, Backhed F. Diet‐microbiota interactions as moderators of human metabolism. Nature 2016;535:56‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature 2016;535:75‐84. [DOI] [PubMed] [Google Scholar]
- 4. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature 2016;535:65‐74. [DOI] [PubMed] [Google Scholar]
- 5. Younossi ZM, Tampi R, Priyadarshini M, Nader F, Younossi IM, Racila A. Burden of illness and economic model for patients with nonalcoholic steatohepatitis in the United States. Hepatology 2019;69:564‐572. [DOI] [PubMed] [Google Scholar]
- 6. Sharpton SR, Ajmera V, Loomba R. Emerging role of the gut microbiome in nonalcoholic fatty liver disease: from composition to function. Clin Gastroenterol Hepatol 2019;17:296‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caussy C, Loomba R. Gut microbiome, microbial metabolites and the development of NAFLD. Nat Rev Gastroenterol Hepatol 2018;15:719‐720. [DOI] [PubMed] [Google Scholar]
- 9. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab 2017;25:1054‐1062.e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoyles L, Fernandez‐Real JM, Federici M, Serino M, Abbott J, Charpentier J, et al. Molecular phenomics and metagenomics of hepatic steatosis in non‐diabetic obese women. Nat Med 2018;24:1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo‐Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016;63:764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2013;11:868‐875.e861‐863. [DOI] [PubMed] [Google Scholar]
- 13. Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—a longitudinal study. PLoS One 2013;8:e62885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang B, Jiang X, Cao M, Ge J, Bao Q, Tang L, et al. Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non‐alcoholic fatty liver disease. Sci Rep 2016;6:32002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 2006;103:3920‐3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284:1365‐1368. [DOI] [PubMed] [Google Scholar]
- 17. Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR‐FGF15/19 pathway. Dig Dis 2015;33:327‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov 2016;15:51‐69. [DOI] [PubMed] [Google Scholar]
- 19. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun 2017;1:1024‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DePaoli AM, Zhou M, Kaplan DD, Hunt SC, Adams TD, Learned RM, et al. FGF19 analog as a surgical factor mimetic that contributes to metabolic effects beyond glucose homeostasis. Diabetes 2019;68:1315‐1328. [DOI] [PubMed] [Google Scholar]
- 21. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non‐alcoholic steatohepatitis: a multicentre, randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet 2018;391:1174‐1185. [DOI] [PubMed] [Google Scholar]
- 22. Harrison SA, Rossi SJ, Paredes AH, Trotter JF, Bashir MR, Guy CD, et al. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology 2020;71:1198‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rinella ME, Trotter JF, Abdelmalek MF, Paredes AH, Connelly MA, Jaros MJ, et al. Rosuvastatin improves the FGF19 analogue NGM282‐associated lipid changes in patients with non‐alcoholic steatohepatitis. J Hepatol 2019;70:735‐744. [DOI] [PubMed] [Google Scholar]
- 24. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601‐609. [DOI] [PubMed] [Google Scholar]
- 25. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97‐101. [DOI] [PubMed] [Google Scholar]
- 26. Scheiman J, Luber JM, Chavkin TA, MacDonald T, Tung A, Pham LD, et al. Meta‐omics analysis of elite athletes identifies a performance‐enhancing microbe that functions via lactate metabolism. Nat Med 2019;25:1104‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sorribas M, Jakob MO, Yilmaz B, Li H, Stutz D, Noser Y, et al. FXR modulates the gut‐vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol 2019;71:1126‐1140. [DOI] [PubMed] [Google Scholar]
- 28. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014;513:59‐64. [DOI] [PubMed] [Google Scholar]
- 29. Chen Y, Ji F, Guo J, Shi D, Fang D, Li L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci Rep 2016;6:34055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S, Singh S, et al. A Universal Gut‐Microbiome‐Derived Signature Predicts Cirrhosis. Cell Metab 2020;32:901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwartz CC, Almond HR, Vlahcevic ZR, Swell L. Bile acid metabolism in cirrhosis. V. Determination of biliary lipid secretion rates in patients with advanced cirrhosis. Gastroenterology 1979;77:1177‐1182. [PubMed] [Google Scholar]
- 32. Merritt ME, Donaldson JR. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J Med Microbiol 2009;58:1533‐1541. [DOI] [PubMed] [Google Scholar]
- 33. Wang Y, Gao X, Zhang X, Xiao Y, Huang J, Yu D, Li X, et al. Gut microbiota dysbiosis is associated with altered bile acid metabolism in infantile cholestasis. mSystems 2019;4:e00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drolz A, Horvatits T, Rutter K, Landahl F, Roedl K, Meersseman P, et al. Lactate improves prediction of short‐term mortality in critically Ill patients with cirrhosis: a multinational study. Hepatology 2019;69:258‐269. [DOI] [PubMed] [Google Scholar]
- 35. Gaci N, Borrel G, Tottey W, O'Toole PW, Brugere JF. Archaea and the human gut: new beginning of an old story. World J Gastroenterol 2014;20:16062‐16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghoshal U, Shukla R, Srivastava D, Ghoshal UC. Irritable bowel syndrome, particularly the constipation‐predominant form, involves an increase in methanobrevibacter smithii, which is associated with higher methane production. Gut Liv 2016;10:932‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oduyebo I, Camilleri M, Nelson AD, Khemani D, Nord SL, Busciglio I, et al. Effects of NGM282, an FGF19 variant, on colonic transit and bowel function in functional constipation: a randomized phase 2 trial. Am J Gastroenterol 2018;113:725‐734. [DOI] [PubMed] [Google Scholar]
- 38. Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun 2019;10:1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Link between gut‐microbiome derived metabolite and shared gene‐effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018;68:918‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut‐liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Castera L, Friedrich‐Rust M, Loomba R. Noninvasive assessment of liver disease in patients with nonalcoholic fatty liver disease. Gastroenterology 2019;156:1264‐1281.e1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Demir M, Lang S, Martin A, Farowski F, Wisplinghoff H, Vehreschild M, et al. Phenotyping non‐alcoholic fatty liver disease by the gut microbiota: Ready for prime time? J Gastroenterol Hepatol 2020. Apr 8. 10.1111/jgh.15071. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 43. Falony G, Joossens M, Vieira‐Silva S, Wang J, Darzi Y, Faust K, et al. Population‐level analysis of gut microbiome variation. Science 2016;352:560‐564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material