

Abstract
Background and Aims
Hepatic ischemia‐reperfusion (I/R) injury, which mainly involves inflammatory responses and apoptosis, is a common cause of organ dysfunction in liver transplantation (LT). As a critical mediator of inflammation and apoptosis in various cell types, the role of tripartite motif‐containing (TRIM) 27 in hepatic I/R injury remains worthy of study.
Approach and Results
This study systemically evaluated the putative role of TRIM27/transforming growth factor β–activated kinase 1 (TAK1)/JNK (c‐Jun N‐terminal kinase)/p38 signaling in hepatic I/R injury. TRIM27 expression was significantly down‐regulated in liver tissue from LT patients, mice subjected to hepatic I/R surgery, and hepatocytes challenged by hypoxia/reoxygenation (H/R) treatment. Subsequently, using global Trim27 knockout mice (Trim27‐KO mice) and hepatocyte‐specific Trim27 transgenic mice (Trim27‐HTG mice), TRIM27 functions to ameliorate liver damage, reduce the inflammatory response, and prevent cell apoptosis. In parallel in vitro studies, activating TRIM27 also prevented H/R‐induced hepatocyte inflammation and apoptosis. Mechanistically, TRIM27 constitutively interacted with the critical components, TAK1 and TAK1 binding protein 2/3 (TAB2/3), and promoted the degradation of TAB2/3, leading to inactivation of TAK1 and the subsequent suppression of downstream JNK/p38 signaling.
Conclusions
TRIM27 is a key regulator of hepatic I/R injury by mediating the degradation of TAB2/3 and suppression of downstream TAK1‐JNK/p38 signaling. TRIM27 may be a promising approach to protect the liver against I/R‐mediated hepatocellular damage in transplant recipients.
Abbreviations
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- ASK1
apoptosis signal‐regulating kinase‐1
- AST
aspartate aminotransferase
- BAD
BCL2‐associated agonist of cell death
- BAX
Bcl2‐associated x
- BCL2
B‐cell lymphoma 2
- Ccl2
C‐C motif chemokine ligand 2
- CQ
chloroquine
- Cxcl2
chemokine (C‐X‐C motif) ligand 2
- ERK
extracellular regulated protein kinase
- GSEA
gene set enrichment analysis
- GST
glutathione S‐transferase
- HA
hemagglutinin
- H&E
hematoxylin and eosin
- H/R
hypoxia/reoxygenation
- HTG
hepatocyte‐specific transgenic
- IF
immunofluorescence
- IHC
immunohistochemistry
- IKK
IκB kinase
- Il1b
interleukin‐1β
- Il6
interleukin‐6
- IP
immunoprecipitation
- I/R
ischemia/reperfusion
- JNK
c‐Jun N‐terminal kinase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knockout
- LT
liver transplantation
- MAPK
mitogen‐activated protein kinase
- NF‐κB
nuclear factor kappa B
- NTG
nontransgenic
- PCA
principal component analysis
- p‐JNK
phosphorylated JNK
- p‐p38
phosphorylated p38
- p‐p65
phosphorylated p65
- p‐TAK1
phosphorylated TAK1
- RFP
ret finger protein
- RING
really interesting new gene
- RNA‐Seq
RNA sequencing
- ROS
reactive oxygen species
- TAB2/3
TAK1 binding protein 2/3
- TAK1
transforming growth factor β–activated kinase 1
- Tnfα
tumor necrosis factor‐α
- TRIM
tripartite motif‐containing
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
wild type
Liver ischemia‐reperfusion (I/R) injury is an important cause of liver damage occurring during liver transplantation (LT), complex liver resection, hemorrhagic shock, and severe liver trauma surgery attributed to long ischemic time.( 1 ) Hepatic I/R injury has been recognized as the most challenging factor for delayed early‐graft dysfunction and acute and chronic rejection as well as a key obstacle to expanding the donor organ pool in LT.( 2 ) However, despite the obvious clinical importance, mechanisms that account for liver I/R injury are only partially understood and no effective intervention is available to prevent or treat this condition in humans. Thus, therapeutic concepts to combat hepatic I/R injury are urgently needed to improve the outcomes of LT.
Hepatic I/R injury is a dynamic process involving cell damage directly caused by ischemia and more‐severe liver cell damage caused by the inflammation associated with reperfusion.( 3 ) The mechanisms underlying hepatic I/R injury are complex. However, it is now becoming clear that the immune response plays a central role during I/R injury, and that this response is characterized by the recruitment and activation of immune cells associated with the innate and adaptive immune system.( 4 ) During hepatic I/R injury, reactive oxygen species (ROS) activate redox‐regulated transcription factors, such as nuclear factor kappa B (NF‐κB), which trigger the secretion of proinflammatory mediators, including cytokines, chemokines, and adhesion molecules, leading to cell apoptosis and tissue injury.( 5 ) Accordingly, the identification of pivotal regulators targeting of these signaling molecules may attenuate I/R injury.
The tripartite motif‐containing (TRIM) protein family has been reported to be involved in many biological processes, such as cell proliferation and apoptosis, innate immunity, and inflammatory response, among others.( 6 ) Previous study reported that TRIM47 is a critical mediator of cerebral I/R injury through regulating inflammation.( 7 ) Moreover, TRIM8 deficiency attenuated hepatic I/R injury by mediating inflammation responses and apoptosis.( 8 ) As a member of the TRIM family, which contains similar domains of a RING (really interesting new gene) finger, B‐box zinc finger, and coiled‐coil domain, TRIM27 was first reported as a fusion protein of the RET proto‐oncogene in 1988.( 9 , 10 ) TRIM27 has been shown to negatively regulate antiviral and inflammatory responses through inhibition NF‐κB and interferon activation mediated by IκB kinase (IKK)α/β/ε.( 11 ) Moreover, TRIM27 knockdown induced cell apoptosis in ovarian cancer cells by up‐regulating phosphorylated p38 (p‐p38).( 12 ) However, whether and how TRIM27 plays a role in hepatic I/R injury is worthy of study.
In the present study, hepatic I/R treatment significantly down‐regulated TRIM27 expression. The in vivo and in vitro studies demonstrated that TRIM27 alleviates liver damage by suppressing apoptosis and inhibiting inflammatory response during hepatic I/R injury. Mechanistically, TRIM27 was identified as a negative regulator of transforming growth factor β–activated kinase 1 (TAK1) by mediating the degradation of TAK1 binding protein 2/3 (TAB2/3) and suppression of downstream TAK1/JNK (c‐Jun N‐terminal kinase)/p38 signaling. Together, these findings identified a previously undiscovered mechanism for the regulation of hepatic I/R injury and a promising approach to protect the liver against I/R‐related insult.
Materials and Methods
Clinical LT Samples
Nine adult LT recipients were recruited at The First Affiliated Hospital of Zhengzhou University (Zhengzhou, China). Liver biopsies were obtained from the left lobe before ischemia (pre: before organ perfusion) and at approximately 2 hours after portal reperfusion (post: before the abdominal closure). Informed consent forms were signed by all subjects. None of the donor organs were obtained from executed prisoners or other institutionalized persons. The study was conducted in accordance with the principles outlined in the 1975 Declaration of Helsinki, and all samples are for experimental purposes only. All procedures involving human samples were approved by The First Affiliated Hospital of Zhengzhou University.
Animals
Male mice (8‐10 weeks of age, 25 ± 2 g) were housed in polycarbonate cages under a 12:12‐hour light‐dark photo cycle, and they had access to food and water ad libitum throughout the study period. Humane care was given to animals in adherence with the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH; publication 86‐23 revised in 1985). All animal procedures were approved by the Ethics Committee of The First Affiliated Hospital of Zhengzhou University. Trim27 knockout (Trim27‐KO) mice and hepatocyte‐specific Trim27 transgene (Trim27‐HTG) mice in the C57BL/6J background were generated with a detailed description in the Supporting Information.
Mouse Liver I/R Injury Model
We used an established 70% warm hepatic I/R injury model as described.( 3 ) In general, after mice were anesthetized, a midline laparotomy was performed to expose the liver. Next, the left and middle portal vein branches in the liver were clamped with an atraumatic microvascular clamp to interrupt the blood supply. After ischemia for 1 hour, the clamp was released for reperfusion. At each indicated time point after reperfusion, mice were anesthetized to collect liver samples and serum for further analysis. Mice that underwent the same surgical procedure without vasculature occlusion served as sham controls.
Examination of Liver Function
Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by the ADVIA 2400 Chemistry System (Siemens, Tarrytown, NY), according to the manufacturer’s protocols, to assess extent of liver damage in animals.
Histological Staining
Liver samples were fixed with formalin and embedded with paraffin and were sectioned serially at 4‐ to 5‐µm thickness. After deparaffinization and rehydration, standard, hematoxylin and eosin (H&E) staining was carried out to visualize the pattern in necrotic areas of the liver. Images were observed and captured using a light microscope (Olympus, Tokyo, Japan).
Immunohistochemistry and Immunofluorescence Staining
Immunohistochemistry (IHC) and immunofluorescence (IF) staining are described in detail in the Supporting Information.
RNA Sequencing Analysis
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). Single‐end libraries were prepared according to the manufacturer’s instructions. Sequencing was carried out using BGISEQ 500 with a read length of 50 base pairs. Reads were mapped to reference genome sequences (mm10) using HISAT2 software (version 2.1.0). Fragments per kilobase of exon model per million reads mapped values were calculated by StringTie software (version 1.3.3b), using default parameters. Differential gene expression was calculated utilizing DESeq2 software (version 1.2.10). RNA sequencing (RNA‐Seq) and transcriptomics data have been submitted to the Sequence Read Archive with the database identifiers PRJNA616148.
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) was performed on the Java GSEA (version 3.0) platform. The gene sets used in GSEA analysis were known Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and involved genes from the KEGG database. Only gene sets with a P value <0.05 and a false discovery rate value <0.25 were considered statistically significant.
KEGG Pathway Enrichment Analysis
KEGG pathway enrichment analysis was performed using Fisher’s exact test. Annotations of all genes in the selected genome were downloaded from the KEGG database. Only pathways with a P value <0.05 were considered significantly enriched.
Cell Culture and Treatment
Hepatocyte L02 cell lines were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). A hepatocyte hypoxia‐reoxygenation (H/R) experiment was performed as described.( 13 )
Quantitative RT‐PCR
Quantitative RT‐PCR analyses were performed with ChamQ SYBR qPCR Master Mix (Catalog No.: Q311‐02; Vazme, Nanjing, China) for quantification of mRNA expression as described.( 3 ) Results were normalized against β‐actin expression. Primer sequences of the target genes used in this study are listed in Supporting Table S1.
Western Blotting
Protein levels were detected using western blotting analysis, as reported. A ChemiDoc MP Imaging System (Bio‐Rad, Hercules, CA) was used to detect signals. Protein levels were quantified using ImageJ software (NIH, Bethesda, MD). All the antibodies used for western blotting analysis in this study are shown in Supporting Table S2.
Plasmid Construction
Plasmid construction is detailed in the Supporting Information.
Immunoprecipitation and Glutathione S‐Transferase Pull‐Down Assays
Immunoprecipitation (IP) and glutathione S‐transferase (GST) pull‐down assays were performed as described to identify interactions of TRIM27 with TAK1 and are detailed in the Supporting Information.( 3 )
Confocal Microscopy
L02 hepatocytes plated on glass coverslips in six‐well plates were cotransfected with Flag‐tagged TAK1 and hemagglutinin (HA)‐tagged TRIM27 plasmids for 24 hours. Cells were then labeled with tag antibody and viewed using a confocal laser microscope (LCS‐SP8‐STED; Leica).
Ubiquitination Assays
The in vivo ubiquitination assays were performed as described.( 14 ) Briefly, transfected cells were lysed in sodium dodecyl sulfate (SDS) lysis buffer (20 mM of Tris‐HCl [pH 7.4], 150 mM of NaCl, 1 mM of ethylenediaminetetraacetic acid, and 1% SDS) and heated at 95°C for 5 minutes to denature. Supernatants were subsequently diluted 10‐fold with IP buffer and subjected to IP with the indicated antibodies. Then, immunoblotting (IB) was performed with corresponding antibodies.
Statistical Analysis
SPSS software (version 21.0; IBM, Armonk, NY) was used for all statistical analyses in this study. All data are expressed as the mean ± SD. Statistical analyses used a two‐tailed Student t test for comparisons between two groups and one‐way analysis of variance (ANOVA) for comparisons between multiple groups. P < 0.05 was considered to be statistically significant.
Additional Methods
Details of these procedures are presented in the Supporting Information.
Results
Decreased TRIM27 Expression Levels Are Associated With Hepatic I/R Injury
We detected TRIM27 expression in liver biopsies from patients who underwent LT to explore the potential role of TRIM27 in liver I/R injury. Notably, TRIM27 protein and mRNA levels were significantly decreased in patient samples posttransplantation compared with the sample pretransplantation (Fig. 1A,B). We next assessed TRIM27 expression in liver lobes in wild‐type (WT) mice subjected to partial hepatic I/R injury, and a gradual down‐regulation of TRIM27 expression was observed from 1 to 24 hours post‐I/R surgery at both the mRNA and protein levels (Fig. 1C,D). Moreover, IHC staining further confirmed the significant down‐regulation of TRIM27 expression in the liver after reperfusion from both the clinical and mouse samples (Fig. 1E,F). In accordance with the in vivo results, protein expression TRIM27 was also significantly down‐regulated in hepatocytes subjected to H/R insult (Fig. 1G). In conclusion, these results suggest that TRIM27 plays an important role in liver I/R injury.
FIG. 1.
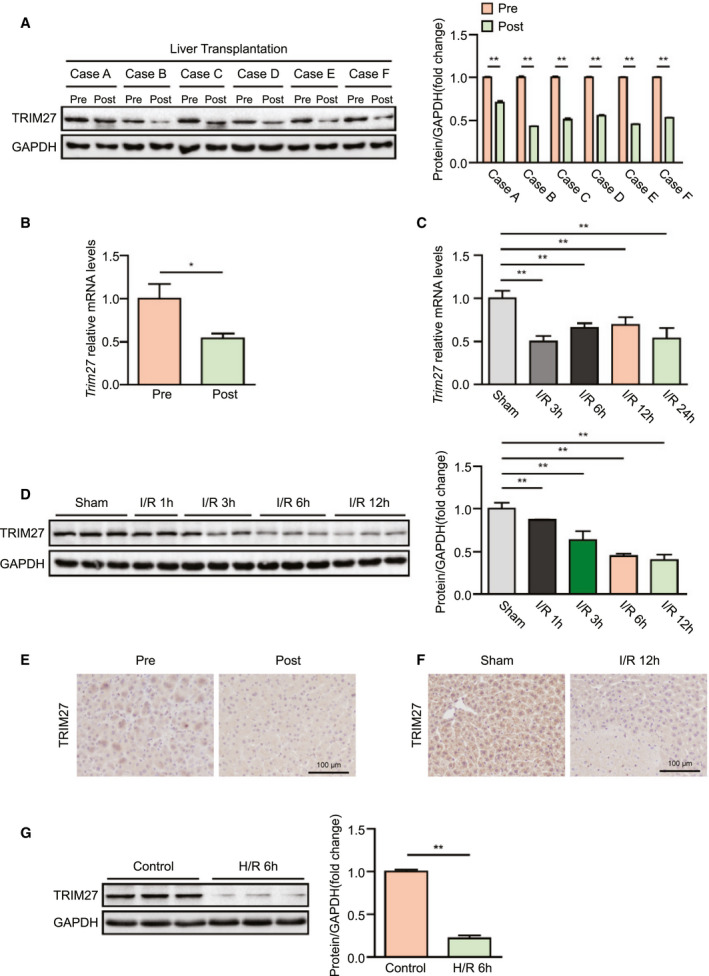
Decreased TRIM27 expression levels are associated with hepatic I/R injury. (A) TRIM27 protein expression in liver of LT patients. GAPDH served as the loading control (n = 6). (B) TRIM27 mRNA levels in liver of LT patients (n = 9). (C) Trim27 mRNA levels in liver from mice subjected to sham or ischemia treatment for 1 hour, followed by reperfusion for the indicated times (n = 6‐7 per time point). (D) TRIM27 protein expression in liver from mice subjected to sham or ischemia treatment for 1 hour, followed by reperfusion for the indicated times (n = 3 per time point). GAPDH served as the loading control. (E) IHC staining of TRIM27 in liver from LT patients (n = 4 per group). Scale bar, 100 μm. (F) IHC staining of TRIM27 in liver lobes from WT mice subjected to hepatic I/R surgery (n = 5 per group). Scale bar, 100 μm. (G) TRIM27 protein expression in cultured L02 hepatocytes challenged by H/R treatment. GAPDH served as the loading control. Representative of three independent experiments. All data are presented as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviation: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
TRIM27 Deletion Exacerbates LIVER Damage and the Inflammatory Response During Hepatic I/R Injury
On the basis of the expression level of TRIM27 being significantly decreased during hepatic I/R injury, we hypothesized a functional relevance of TRIM27 in hepatic I/R injury. We generated global Trim27‐KO mice to further investigate the role of TRIM27 in hepatic I/R injury, and TRIM27 deletion in the liver was confirmed by western blotting analyses (Fig. 2A,B). Importantly, TRIM27 deficiency significantly enhanced the elevated levels of ALT and AST in serum at 3 and 6 hours after liver I/R surgery compared to levels in WT control mice (Fig. 2C). Consistent with these findings, considerably greater necrotic area was observed in liver sections of Trim27‐KO mice, compared to WT controls, as assessed by H&E staining (Fig. 2D). These findings show that TRIM27 deficiency promotes I/R‐mediated liver insult.
FIG. 2.
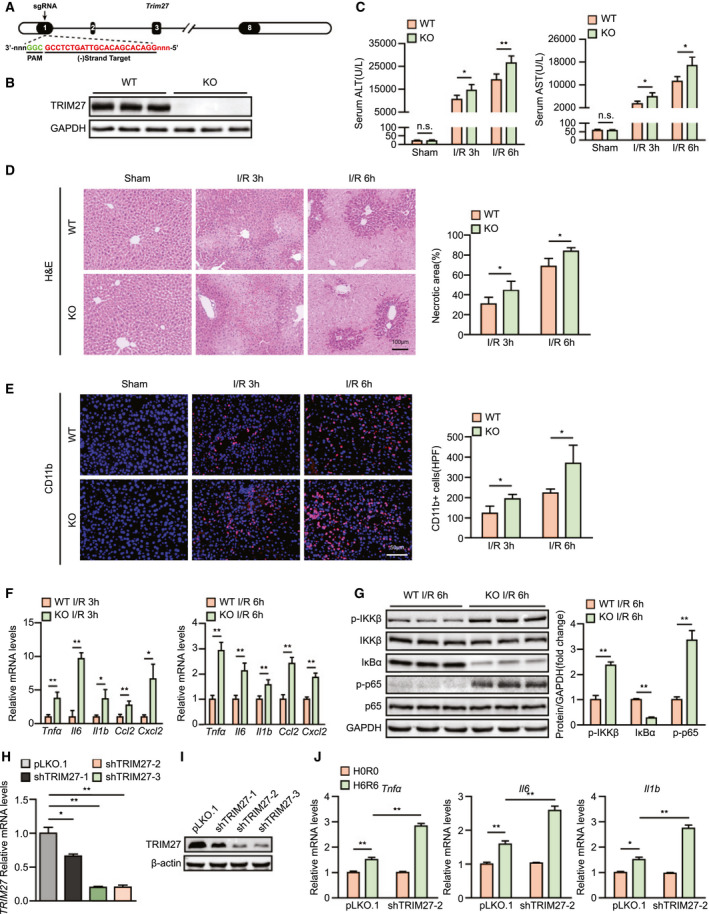
TRIM27 deletion exacerbates liver damage and inflammatory response during hepatic I/R injury. (A) Diagram of Trim27‐KO strategy. (B) TRIM27 protein expression in liver of WT and Trim27‐KO mice. GAPDH served as the loading control (n = 3 per group). (C) Serum ALT/AST activities in WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 7‐9 per group). (D) Representative histological H&E‐stained images and statistics showing necrotic areas in liver tissue from WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 6 per group). Scale bar, 100 μm. (E) Representative CD11b IF staining in liver lobes of WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (F) mRNA levels of proinflammatory factors (Tnfα, Il6, Il1b, Ccl2, and Cxcl10) in liver of WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). (G) Protein levels of NF‐κB signaling pathway molecules in liver of WT and Trim27‐KO mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (H) mRNA levels of TRIM27 in L02‐scramble and L02 TRIM27‐knockdown hepatocytes. Representative of three independent experiments. (I) TRIM27 protein expression in L02‐scramble and L02 TRIM27‐knockdown hepatocytes. β‐actin served as the loading control. Representative of three independent experiments. (J) mRNA levels of proinflammatory factors (Tnfα, Il6, and Il1b) in L02 TRIM27‐knockdown hepatocytes challenged by H/R treatment. Representative of three independent experiments. All data are presented as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviations: Cxcl10, chemokine (C‐X‐C motif) ligand 10; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HPF, high‐power field; IκBα, inhibitory κB α; n.s., not statistically significant; PAM, protospacer adjacent motif; p‐IKKβ, phosphorylated IκB kinase β; sgRNA, single‐guide RNA.
During hepatic I/R injury, the inflammatory response is initiated during the ischemic phase and persists throughout the entire process and is an essential factor in hepatic I/R injury.( 15 ) Therefore, we aimed to investigate whether TRIM27 could affect the inflammatory response after hepatic I/R injury in mice. At 3 and 6 hours after liver I/R surgery, IF staining indicated that the number of CD11b‐positive inflammatory cells in Trim27‐KO mouse liver was significantly increased compared to that in WT mouse liver (Fig. 2E). Moreover, mRNA levels of proinflammatory cytokines/chemokines, such as tumor necrosis factor alpha (Tnfα), interleukin‐6 (Il6), interleukin‐1β (Il1b), C‐C motif chemokine ligand 2 (Ccl2), and chemokine (C‐X‐C motif) ligand 2 (Cxcl2), in liver tissue were significantly increased in the Trim27‐KO group compared to those in WT controls (Fig. 2F). NF‐κB belongs to innate immune signaling and plays a critical role in regulating inflammatory responses during hepatic I/R injury.( 16 ) Interestingly, increased activation of NF‐κB signaling was observed in Trim27‐KO mouse liver subjected to hepatic I/R treatment compared to that in controls (Fig. 2G). To further examine whether TRIM27 directly influences inflammation in hepatocytes, we constructed a TRIM27‐knockdown L02 hepatocyte cell line by lentivirus infection for subsequent studies (Fig. 2H,I). As evidenced by RT‐PCR, hepatocytes with TRIM27 knockdown exhibited higher mRNA levels of inflammation factors than scramble controls after H/R challenge (Fig. 2J). Therefore, these findings indicate that TRIM27 regulates inflammation during hepatic I/R injury and that hepatocytes may serve as the target cell type.
TRIM27 Deficiency Aggravates Apoptosis During Hepatic I/R Injury
The important role of TRIM27 in regulating apoptosis has been reported.( 12 ) Thus, we examined whether TRIM27 directly regulates apoptosis and facilitates its protective effects on hepatic I/R injury. Compared to the WT I/R group, the number of apoptotic cells was significantly increased in livers of Trim27‐KO mice, as demonstrated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and c‐CASPASE3 IHC (Fig. 3A,B). Moreover, levels of the antiapoptotic gene, B‐cell lymphoma 2 (Bcl2), were significantly decreased and levels of the proapoptotic genes, BCL2‐associated agonist of cell death (Bad) and Bcl2‐associated x (Bax), were increased in liver from the Trim27‐KO group at 3 and 6 hours after hepatic I/R surgery compared to WT mice (Fig. 3C). Furthermore, expression of apoptosis‐related proteins BAD, BAX, BCL2, and c‐CASPASE‐3 exhibited a similar trend to mRNA levels in Trim27‐KO mouse liver compared to expression in WT mice during I/R injury (Fig. 3D). Consistent with the in vivo results, the proapoptotic effect of TRIM27 knockdown was also demonstrated in hepatocytes challenged by H/R insult (Fig. 3E). It can be deduced that TRIM27 deficiency promotes apoptosis during hepatic I/R injury.
FIG. 3.
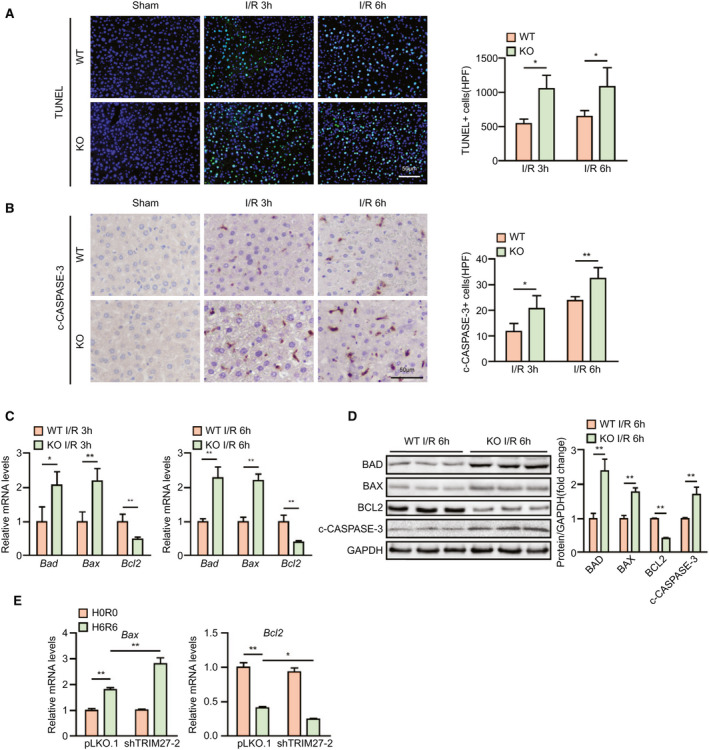
TRIM27 deficiency aggravates apoptosis during hepatic I/R injury. (A) TUNEL staining in liver sections from WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (B) IHC staining of c‐CASPASE‐3 in liver sections from WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (C) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in liver samples of WT and Trim27‐KO mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). (D) Western blotting analysis of apoptosis‐related molecules (BAD, BAX, BCL2, and c‐CASPASE‐3) protein levels in liver of WT and Trim27‐KO mice at 6 hours after hepatic I/R surgery. GAPDH served as a loading control (n = 3 per group). (E) mRNA levels of apoptosis‐related genes (Bax and Bcl2) in L02 TRIM27‐knockdown hepatocytes challenged by H/R treatment. Representative of three independent experiments. All data are shown as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviations: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HPF, high‐power field.
Hepatocyte‐Specific TRIM27 Overexpression Ameliorates Liver Damage and Inflammation Induced by Hepatic I/R Insult
Given that TRIM27 deficiency in the liver aggravates hepatic I/R injury, we tested whether TRIM27 overexpression protects the liver from I/R‐induced insult. We constructed Trim27‐HTG mice, and western blotting confirmed that TRIM27 was overexpressed in livers of the Trim27‐HTG mouse (Fig. 4A,B). As expected, Trim27‐HTG mice exhibited lower ALT and AST activities than those of nontransgenic (NTG) mice at 3 and 6 hours after liver I/R surgery (Fig. 4C). Histological analysis of livers also demonstrated reduced necrotic area in Trim27‐HTG mice subjected to hepatic I/R surgery compared to that of NTG mice (Fig. 4D). Furthermore, sustained TRIM27 expression weakened I/R‐induced inflammation. Trim27‐HTG mice showed decreased levels of proinflammatory cytokines/chemokines and reduced infiltration of immune cells into livers, and also suppressed NF‐κB signaling during hepatic I/R surgery compared to those of NTG mice (Fig. 4E,G). To further examine whether TRIM27 directly influences inflammation in hepatocytes, we generated a TRIM27‐overexpressed L02 hepatocyte cell line by lentivirus infection (Fig. 4H). Overexpression of TRIM27 inhibited the H/R‐triggered inflammatory response in hepatocytes (Fig. 4I). Together, these results demonstrate that TRIM27 in hepatocytes ameliorates liver damage and inflammation induced by hepatic I/R treatment.
FIG. 4.
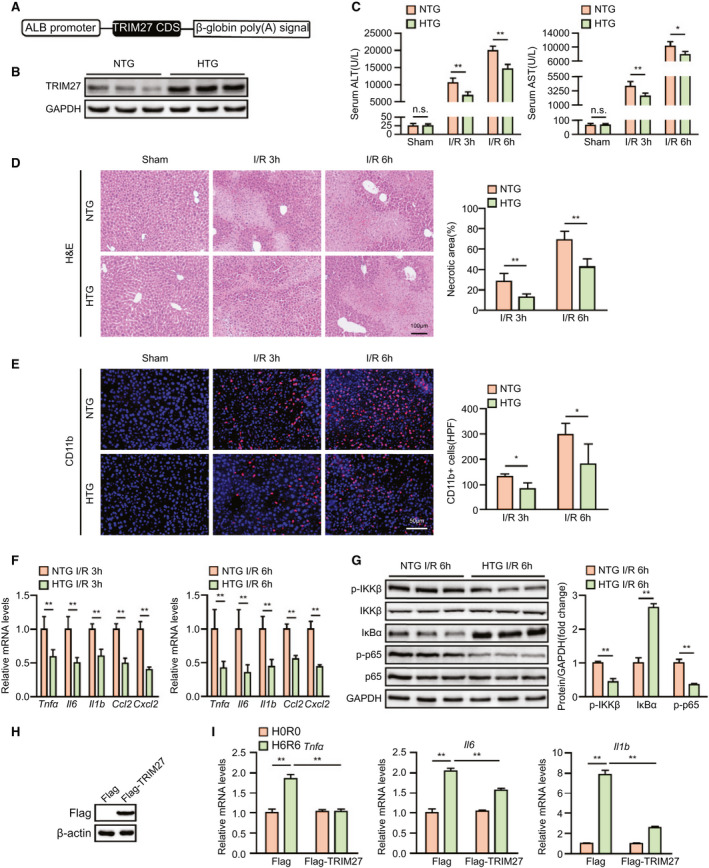
Hepatocyte‐specific TRIM27 overexpression ameliorates liver damage and inflammation induced by hepatic I/R insult. (A) Diagram of Trim27‐HTG strategy. (B) TRIM27 protein expression in liver of NTG and Trim27‐HTG mice. GAPDH served as the loading control (n = 3 per group). (C) Serum ALT/AST activities in NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 8 per group). (D) Representative histological H&E‐stained images and statistics showing necrotic areas in liver tissue from NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 6 per group). Scale bar, 100 μm. (E) Representative CD11b IF staining in liver lobes of NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (F) mRNA levels of proinflammatory factors (Tnfα, Il6, Il1b, Ccl2, and Cxcl10) in liver of NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). (G) Protein levels of NF‐κB signaling pathway molecules in liver of NTG and Trim27‐HTG mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (H) TRIM27 protein expression in L02‐scramble and L02 TRIM27‐overexpressed hepatocytes. β‐actin served as the loading control. Representative of three independent experiments. (I) mRNA levels of proinflammatory factors (Tnfα, Il6, and Il1b) in TRIM27‐overexpressed L02 hepatocytes challenged by H/R treatment. Representative of three independent experiments. All data are presented as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviations: ALB, albumin; CDS, coding sequence; Cxcl10, chemokine (C‐X‐C motif) ligand 10; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HPF, high‐power field; IκBα, inhibitory κB α; n.s., not statistically significant; p‐IKKβ, phosphorylated IκB kinase β.
TRIM27 Alleviates Apoptosis in Hepatic I/R Injury
Having determined that TRIM27 deficiency aggravated apoptosis in response to hepatic I/R injury, we next examined the effect of TRIM27 overexpression on apoptosis during liver I/R injury. TUNEL staining and IHC revealed that liver tissue obtained from Trim27‐HTG mice exhibited less cell apoptosis compared to tissue from counterpart NTG mice at 3 and 6 hours after reperfusion and was accompanied with decreased expression levels of the proapoptotic molecules, BAD, BAX, and c‐CASPASE‐3, and increased levels of the antiapoptotic molecule, BCL2 (Fig. 5A,D). Consistent with the decreased cell apoptosis observed in the in vivo studies, hepatocyte apoptosis was also alleviated in TRIM27‐overexpressed hepatocytes challenged by H/R treatment compared to controls (Fig. 5E). Collectively, these in vivo and in vitro results strongly demonstrate that TRIM27 in hepatocytes regulates apoptosis during hepatic I/R injury.
FIG. 5.
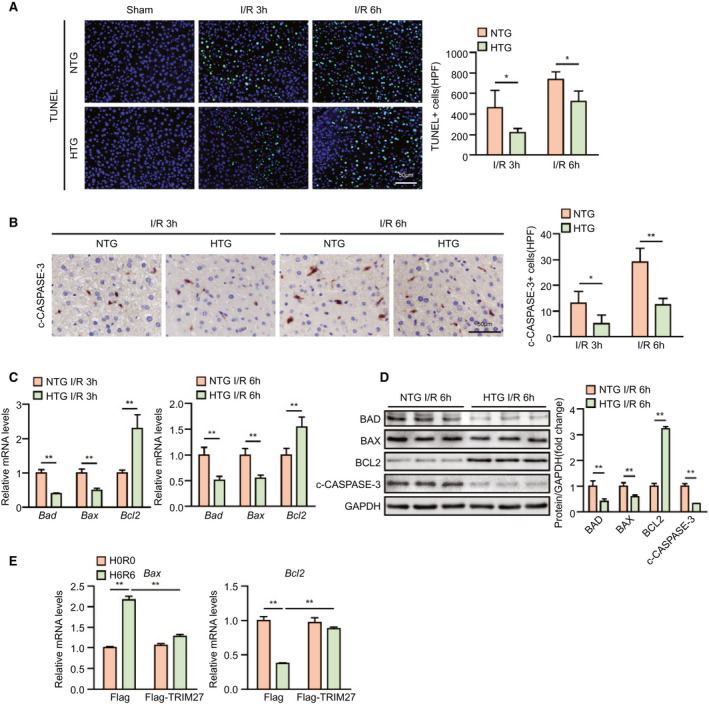
TRIM27 alleviates apoptosis in hepatic I/R injury. (A) TUNEL staining in liver sections from NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (B) IHC staining of c‐CASPASE‐3 in liver sections from NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). Scale bar, 50 μm. (C) mRNA levels of apoptosis‐related genes (Bad, Bax, and Bcl2) in liver samples of NTG and Trim27‐HTG mice at 3 and 6 hours after hepatic I/R surgery (n = 4 per group). (D) Western blotting analysis of apoptosis‐related molecules (BAD, BAX, BCL2, and c‐CASPASE‐3) protein levels in liver of NTG and Trim27‐HTG mice at 6 hours after hepatic I/R surgery. GAPDH served as a loading control (n = 3 per group). (E) mRNA levels of apoptosis‐related genes (Bax and Bcl2) in TRIM27‐overexpressed L02 hepatocytes challenged by H/R treatment. Representative of three independent experiments. All data are shown as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviations: GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HPF, high‐power field.
TRIM27 Inactivates JNK/p38 Signaling During the Hepatic I/R Process
To decipher the underlying mechanism of hepatic I/R injury driven by TRIM27 ablation, we performed RNA‐Seq with I/R‐challenged liver samples of WT and Trim27‐KO mice. Interestingly, principal component analysis (PCA) defined the primary determinants of differences between liver samples of WT and Trim27‐KO mice (PC1, 38.1%; PC2, 17.7%). All samples were clearly separated into their respective groups (Fig. 6A). Furthermore, GSEA indicated that genes most affected by Trim27‐KO were mainly involved in inflammation‐related pathways (Supporting Fig. S1). Genes in leading‐edge subsets of these pathways were collected and used for the KEGG pathway enrichment analysis. Results showed that the mitogen‐activated protein kinase (MAPK) signaling pathway, TNF signaling pathway, and chemokine signaling pathway were the top three significantly enriched signaling pathways contributing to TRIM27‐mediated hepatic I/R injury (Fig. 6B). Moreover, a heatmap of leading‐edge subsets of GSEA‐enriched pathways showed that Trim27 deletion mainly affected the expression trend of MAPK signaling‐related molecules (Fig. 6C). In line with the RNA‐seq results, Trim27 deficiency elevated JNK and p38 phosphorylation, whereas TRIM27 overexpression suppressed JNK and p38 phosphorylation at 6 hours after hepatic I/R injury (Fig. 6D,E). However, neither Trim27 deficiency nor overexpression affected extracellular regulated protein kinase (ERK) phosphorylation levels (Fig. 6D,E).
FIG. 6.
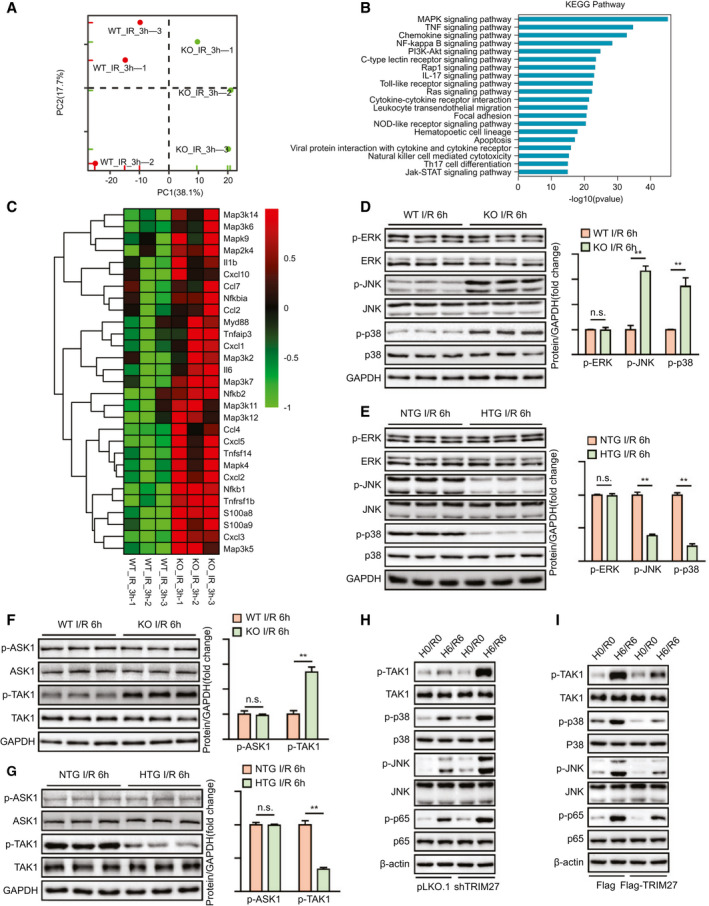
TRIM27 inactivates JNK/p38 signaling during the hepatic I/R process. (A) PCA images showing global sample distribution profiles analyzed by PCA. (B) KEGG pathway enrichment analysis of the major biological pathways contributing to TRIM27 function based on leading‐edge subsets of GSEA‐enriched, inflammation‐related pathways. The 20 most significantly enriched pathways are shown. The blue column represents ‐log10 P values for the enriched KEGG pathways. (C) Heatmap showing expression of inflammatory genes involved in GSEA‐enriched pathways. (D) Protein levels of the total and phosphorylated protein expression levels of classic MAPKs in liver of WT and Trim27‐KO mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (E) Protein levels of the total and phosphorylated protein expression levels of classic MAPKs in liver of NTG and Trim27‐HTG mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (F) Protein levels of the total and phosphorylated protein expression levels of classic MAP3Ks, including TAK1 and ASK1, in liver of WT and Trim27‐KO mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (G) Protein levels of the total and phosphorylated protein expression levels of classic MAP3Ks, including TAK1 and ASK1, in liver of NTG and Trim27‐HTG mice at 6 hours after hepatic I/R surgery. GAPDH served as the loading control (n = 3 per group). (H) Protein levels of the total and phosphorylated protein expression levels of TAK1, p38, JNK, and p65 in TRIM27 knockdown L02 hepatocytes challenged by H/R treatment. β‐actin served as a loading control. Representative of three independent experiments. (I) Protein levels of total and phosphorylated protein expression levels of TAK1, p38, JNK, and p65 in TRIM27‐overexpressed L02 hepatocytes challenged by H/R treatment. β‐actin served as a loading control. Representative of three independent experiments. All data are shown as the mean ± SD. Levels of statistical significance are indicated as: *P < 0.05; **P < 0.01. For statistical analysis, one‐way ANOVA with Bonferroni’s post‐hoc analysis or Tamhane’s T2 post‐hoc analysis and two‐tailed Student t test were used. Abbreviations: Ccl4, C‐C motif chemokine ligand 4; Ccl7, C‐C motif chemokine ligand 7; Cxcl1, chemokine (C‐X‐C motif) ligand 1; Cxcl2, chemokine (C‐X‐C motif) ligand 2; Cxcl3, chemokine (C‐X‐C motif) ligand 3; Cxcl5, chemokine (C‐X‐C motif) ligand 5; Cxcl10, chemokine (C‐X‐C motif) ligand 10; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; IL‐17, interleukin‐17; Jak, Janus kinase; Map2k4, mitogen‐activated protein kinase kinase 4; Map3k2, mitogen‐activated protein kinase kinase kinase 2; Map3k5, mitogen‐activated protein kinase kinase kinase 5; Map3k6, mitogen‐activated protein kinase kinase kinase 6; Map3k7, mitogen‐activated protein kinase kinase kinase 7; Map3k11, mitogen‐activated protein kinase kinase kinase 11; Map3k12, mitogen‐activated protein kinase kinase kinase 12; Map3k14, mitogen‐activated protein kinase kinase kinase 14; Mapk4, mitogen‐activated protein kinase 4; Mapk9, mitogen‐activated protein kinase 9; Myd88, MYD88 innate immune signal transduction adaptor; Nfkb1, nuclear factor kappa B subunit 1; Nfkb2, nuclear factor kappa B subunit 2; Nfkbia, nuclear factor kappa B inhibitor alpha; NOD, nucleotide‐binding and oligomerization domain; n.s., not statistically significant; p‐ASK1, phosphorylated ASK1; p‐ERK, phosphorylated ERK; Rap1, rapamycin 1; S100a8, S100 calcium‐binding protein A8; S100a9, S100 calcium‐binding protein A9; STAT, signal transducer and activator of transcription; Th17, T helper 17; Tnfaip3, TNF alpha‐induced protein 3; Tnfrsf1b, TNF receptor superfamily member 1B; Tnfsf14, TNF superfamily member 14.
We further explored the upstream kinase on which TRIM27 acts to modulate the JNK/p38 signaling pathways. Importantly, only TAK1, but not apoptosis signal‐regulating kinase 1 (ASK1), phosphorylation was dramatically increased by Trim27 KO in liver in response to hepatic I/R operation, whereas the phosphorylation of TAK1 was suppressed in liver from Trim27‐HTG mice without affecting ASK1 phosphorylation (Fig. 6F,G). We used TRIM27 knockdown and overexpressed hepatocytes to further detect whether TRIM27 regulated MAPK signaling during hepatocyte H/R insult. Again, TRIM27 inhibited the levels of phosphorylated TAK1 (p‐TAK1), phosphorylated JNK (p‐JNK), p‐p38, and phosphorylated p65 (p‐p65) in hepatocytes during H/R insult (Fig. 6H,I). These results suggest that TRIM27 regulates TAK1‐JNK/p38 signaling during hepatic I/R injury.
TRIM27 Interacts With TAK1 Through Its C‐Terminal Coiled Coil and Ret Finger Protein Domains
To determine how TRIM27 affects TAK1 signaling, we further examined the interaction between TRIM27 and TAK1. IF colocalization experiments indicated the colocalization of TRIM27 and TAK1 in the cytoplasm of L02 hepatocytes (Fig. 7A). Moreover, IP experiments demonstrated that TRIM27 immunoprecipitated with TAK1 (Fig. 7B,C). Furthermore, GST‐HA–tagged TAK1 protein efficiently pulled down Flag‐tagged TRIM27 and vice versa (Fig. 7D). The subsequent mapping experiment clarified that both the C‐terminal coiled coil and RFP (ret finger protein) domain of TRIM27 (amino acids 133‐513) were associated with TAK1, whereas the N‐terminal region segment of TAK (amino acids 1‐480) was responsible for its association with TRIM27 (Fig. 7E,F). Taken together, these results demonstrate that TRIM27 directly interacts with TAK1.
FIG. 7.
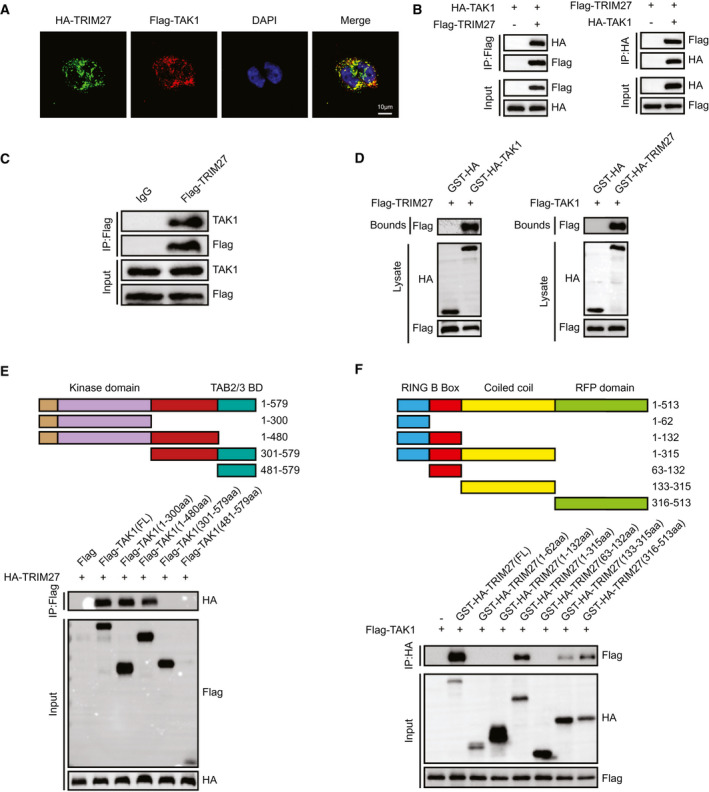
TRIM27 interacts with TAK1 through its C‐terminal coiled coil and RFP domain. (A) Representative confocal images show L02 cells transfected with HA‐tagged TRIM27 and Flagged TAK1. Proteins were visualized using anti‐Flag (red) and anti‐HA (green) antibodies, followed by fluorophore‐conjugated secondary antibodies after 24 hours of transfection. Nuclei were stained using DAPI (blue). n = 3 independent experiments per group with six images per group. Scale bar, 25 μm. (B) Flag‐tagged TRIM27 and HA‐tagged TAK1 plasmids were cotransfected into HEK293T cells. Anti‐Flag antibody (left panel) or anti‐HA antibody (right panel) were used for IP. Representative of three independent experiments. (C) Flag‐tagged TRIM27 was transfected into L02 hepatocytes. Anti‐Flag antibody was used for IP. Representative of three independent experiments. (D) Flag‐tagged TRIM27 and GST‐HA–tagged TAK1 plasmids or Flag‐tagged TAK1 and GST‐HA‐TRIM27 were cotransfected into HEK293T cells. Representative of three independent experiments. (E) Full‐length HA‐TRIM27 and various truncated forms of Flag‐TAK1 were cotransfected into HEK293T cells. An anti‐Flag antibody was used for IP. Representative of three independent experiments. (F) Full‐length Flag‐TAK1 and various truncated forms of GST‐HA‐TRIM27 were cotransfected into HEK293T cells. An anti‐HA antibody was used for IP. Representative of three independent experiments. Abbreviations: aa, amino acids; BD, binding domain; DAPI, 4′,6‐diamidino‐2‐phenylindole; IgG, immunoglobulin G.
TRIM27 Suppresses TAK1 Activity by Mediating the Degradation of TAB2/3
We further identified how TRIM27 mediated TAK1 inactivation. As an E3 ubiquitin ligase, TRIM27 was reported to mediate posttranslational modification of targeted protein through ubiquitin‐proteasome degradation.( 17 ) Moreover, given that K48‐ and K63‐linked polyubiquitination were essential for the activity of TAK1,( 18 , 19 , 20 ) we detected whether TRIM27 would promote the polyubiquitination of TAK1. Surprisingly, K48‐linked polyubiquitination of TAK1 was not changed, but the total ubiquitination and K63‐linked polyubiquitin state of TAK1 were dramatically decreased in the presence of TRIM27 (Fig. 8A). Furthermore, given that TRIM27 interacts with TAK1 independently of its RING domain (Fig. 7F), which confers E3‐ubiquitin ligase activity by specifically interacting with and promoting E2‐dependent ubiquitin conjugation,( 21 ) we speculated that TRIM27 suppressed the ubiquitination of TAK1 through an indirect manner. During TAK1 signal transduction, interactions with TAB2/3 are essential steps for TAK1 activation, and numerous studies reported that TAB2/3 binds to K63‐linked polyubiquitin chains to activate TAK1( 22 , 23 ); thus, we detected whether TRIM27 functioned at the level of the TAK1‐TAB2/3 complex. Interestingly, an IP assay showed an interaction between TRIM27 and TAB2/3 (Fig. 8B). Furthermore, overexpression of TRIM27 decreased TAB2/3 protein levels in a dose‐dependent manner (Fig. 8C). The ubiquitin‐proteasome and lysosomal pathways are two main strategies for protein degradation, which is one of the main strategies for blocking protein function in biological processes.( 24 ) Here, TRIM27‐mediated degradation of TAB2/3 was completely inhibited by chloroquine (CQ), but not MG132, which are inhibitors of the lysosome and proteasome pathways, respectively (Fig. 8D). In addition, TAB2/3 significantly potentiated K63‐linked polyubiquitin and phosphorylation of TAK1, which were largely abolished in the presence of TRIM27 (Fig. 8E,F). These results together indicate that TRIM27 suppresses TAK1 activity by mediating the degradation of TAB2/3.
FIG. 8.
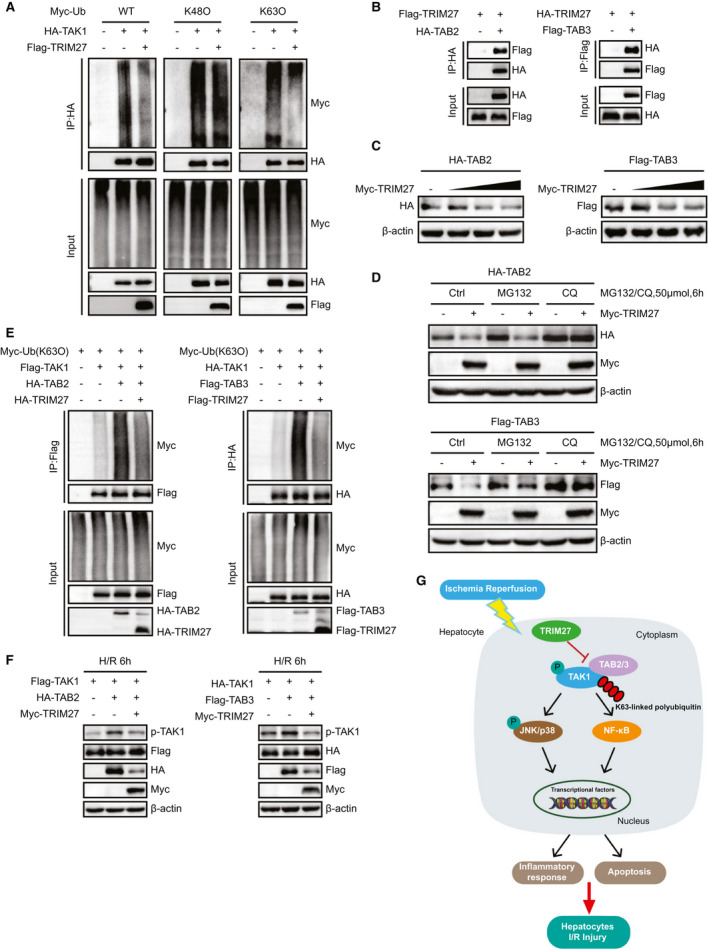
TRIM27 suppresses TAK1 activity by mediating the degradation of TAB2/3. (A) HA‐TAK1, Myc‐Ub, Myc‐Ub (K48O), and Myc‐Ub (K63O) plasmids were cotransfected into HEK293T in the presence or absence of TRIM27. Polyubiquitination of TAK1 was detected by IB with an anti‐Myc antibody after IP with anti‐HA agarose beads. Representative of three independent experiments. (B) Flag‐tagged TRIM27 and HA‐tagged TAB2 or HA‐tagged TRIM27 and Flag‐tagged TAB3 plasmids were cotransfected into HEK293T cells. Anti‐HA antibody (left panel) or anti‐Flag antibody (right panel) were used for IP. Representative of three independent experiments. (C) Protein level of HA‐TAB2 and Flag‐TAB3 in HEK293T cells transfected with increasing doses of Myc‐TRIM27 (0.5, 1, and 2 ug), HA‐TAB2, or Flag‐TAB3 plasmids. β‐actin served as a loading control. Representative of three independent experiments. (D) Protein level of HA‐TAB2 and Flag‐TAB3 in HEK293T cells cotransfected with Myc‐TRIM27, HA‐TAB2, or Flag‐TAB3 plasmids and treated with MG132, CQ, or DMSO. β‐actin served as a loading control. Representative of three independent experiments. (E) IP analyses of the K63O polyubiquitination of TAK1 from HEK293T cells expressing TAK1 and TAB2/3 in the presence or absence of TRIM27. Representative of three independent experiments. (F) Protein level of p‐TAK1 in L02 hepatocytes expressing TAK1 and TAB2/3 in the presence or absence of TRIM27 and challenged by H/R treatment. Representative of three independent experiments. (G) Schematic model showing the molecular mechanism underlying TRIM27‐mediated suppression of TAK1‐JNK/p38 signaling in hepatic I/R‐induced hepatocyte inflammation and apoptosis. Abbreviations: Ctrl, control; DMSO, dimethyl sulfoxide; Myc‐Ub, ubiquitin tagged with the myc epitope.
Discussion
The pathophysiology and mechanism of hepatic I/R injury have been comprehensively studied and reviewed in the past, and it has consequently been accompanied by a long list of potential therapeutic alternatives.( 25 ) Nevertheless, liver I/R injury still represents a serious problem in clinical practice, probably because very few basic (or translational) studies have successfully been applied at the bedside. In this study, the expression of TRIM27 in liver samples from transplantation patients was highly consistent with its changes in mouse liver subjected to hepatic I/R injury, indicating the close clinical correlation of TRIM27 with liver injury. The significant down‐regulation of TRIM27 may be regulated by upstream factors at the transcription level. For example, nicotine‐induced murine spermatozoa apoptosis by TRIM27 promoter hypomethylation and microRNA‐27a inhibits antiviral innate response by up‐regulating TRIM27.( 26 , 27 ) Through gain‐ and loss‐of‐function experiments with TRIM27, we found that TRIM27 negatively regulated inflammation and apoptosis during hepatic I/R injury. Furthermore, TRIM27 suppressed TAK1 activity by promoting TAB2/3 proteolysis, thus leading to suppression of the JNK/p38 and NF‐κB signaling pathways (Fig. 8G). These results together strongly suggest that TRIM27 is essential for the regulation of hepatic I/R injury and that TRIM27 may be a promising approach to protect the liver against I/R‐mediated hepatocellular damage in transplant recipients.
Numerous studies have revealed strong evidence for the critical involvement of inflammation in hepatic I/R injury.( 28 ) During the ischemic phase, initial parenchymal cell damage occurs as a result of glycogen consumption and ATP depletion, leading to subsequent activation of resident Kupffer cells. During the reperfusion period, production of ROS further contributes to activation of innate immune signaling pathways and triggers production of cytokines and chemokines, such as Tnfα, Il1b, and Ccl2, which recruit CD11b inflammatory cells from the bone marrow, and subsequent infiltration into the liver.( 29 , 30 , 31 ) TRIM27, a well‐known E3 ubiquitin ligase, is in a unique position to down‐regulate immune response at multiple levels.( 32 ) For instance, TRIM27 negatively regulates CD4 T cells by ubiquitinating and inhibiting class II phosphoinositide 3‐kinase (PI3K)‐C2β.( 33 ) Consistently, here we demonstrated that TRIM27 deletion in mice enhanced the gene expression of Tnfa, Il1b, Il6, and Ccl2 after hepatic I/R injury. Moreover, TRIM27 deficiency enchanced CD11b‐positive inflammatory cell infiltration, as well as activation of the NF‐κB axis, whereas hepatocyte specificity TRIM27 overexpression had the opposite effect. Additionally, the in vitro experiments also indicated that TRIM27 inhibited the increased Tnfα, Il1b, and Il6 in hepatocytes after H/R treatment. These results demonstrated that TRIM27 in hepatocytes affected the recruitment of CD11b‐positive inflammatory cells by mediating the expression of inflammatory factors, thereby suppressing inflammatory response during liver I/R injury. However, given that we used TRIM27 to completely knock down mice, we cannot rule out whether TRIM27 plays a role in other cell types, like immune cells, during hepatic I/R injury; this needs further study.
Apoptosis, also known as programmed cell death, is a physiological cell suicide process that is essential in development and homeostasis.( 34 ) Recent evidence indicated that apoptosis is another essential step in the pathogenesis of hepatic I/R injury.( 35 ) Conflicting roles of TRIM27 in apoptosis signaling have been reported. Previous studies showed that TRIM27 deficiency protected dopaminergic neurons from apoptosis.( 36 ) Interestingly, other studies have revealed that TRIM27 knockdown induced cell apoptosis in ovarian cancer cells by down‐regulating the expression of phosphorylated protein kinase B (AKT),( 12 ) which is consistent with our results. Consequently, the distinct partners of TRIM27 and their different biological activities may be responsible for its dual roles. Our present study demonstrated that TRIM27 ameliorated hepatocyte apoptosis, as indicated by the increased level of the antiapoptotic molecule, BCL2, and reduced levels of proapoptotic factors, such as cleaved CASPASE‐3, BAX, and BAD, in liver of Trim27‐HTG mice after hepatic I/R injury compared to that of NTG control mice, whereas liver of Trim27‐KO mice exhibited the opposite effect. Thus, we identified TRIM27 as an efficient regulator of apoptosis in response to hepatic I/R.
It has been well documented that the TAK1‐JNK/p38 signaling pathway plays a crucial role during hepatic I/R injury.( 37 ) TAK1 has been implicated in the regulation of a diverse range of cellular processes, that include cellular death and inflammatory responses, and acts as a key regulator of hepatic I/R injury.( 3 , 38 ) In addition, recent studies have demonstrated that the JNK/p38 pathway is activated by stress‐related stimuli and is involved in I/R injury.( 39 ) Down‐regulation of TRIM27 expression was reported to inhibit the proliferation of ovarian cancer cells by up‐regulating the expression of p‐p38.( 12 ) However, how TRIM27 fine‐tunes the inhibition of TAK1‐JNK/p38 signaling remains worthy of study. In the current study, we observed significantly lower levels of phosphorylated TAK1‐JNK/p38 signaling in TRIM27‐overexpressed liver and hepatocytes after I/R or H/R insult, respectively, whereas TRIM27 KO or knock‐down liver and hepatocytes showed an opposite trend. Further experiments demonstrated that TRIM27 physically interacted with TAK1 by specific binding domains. These observations indicate that TAK1 is a target of TRIM27 in the protection against hepatic I/R injury. However, the role of TAK1 in TRIM27‐regulated hepatic I/R injury still needs further verification.
Previous studies have shown that the polyubiquitination of TAK1 is important for TAK1 activation.( 20 , 40 ) For instance, TRIM8 modulates NF‐κB activation by targeting TAK1 for K63‐linked polyubiquitination.( 41 ) Moreover, Itch and Cyld act together to deactivate TAK1 through lysine 48 (Lys48)‐linked ubiquitination.( 20 ) As an E3 ubiquitin ligase, TRIM27 mediates posttranslational modification of targeted protein, mainly through ubiquitin.( 17 ) However, in the present study, we identified TRIM27 as a negative regulator of TAK1 activation by suppressing the K63‐linked polyubiquitin state of TAK1. This molecular event seemed to make it difficult to explain its E3 ligase activity, suggesting that TRIM27 may regulate the activation of TAK1 indirectly. Upon TAK1 activation, strong interactions between unanchored K63 polyubiquitin chains and TAB2/3 induce conformational changes in TAK1 and result in autophosphorylation of TAK1.( 23 ) TAK1 further phosphorylates the MAPK kinases (e.g., mitogen‐activated protein kinase kinase kinase 3 and mitogen‐activated protein kinase kinase kinase 6) and IKKβ. Here, we found that TRIM27 interacted with TAB2/3 and catalyzed the degradation of TAB2/3. Moreover, TAB2/3‐potentiated K63‐linked polyubiquitination and phosphorylation of TAK1 were largely abolished in the presence of TRIM27. These findings suggest that targeting TAB2/3 may represent strategies for TAK1 inhibition and may yield hepatic I/R injury therapy.
Proteins are mainly degraded by the ubiquitin‐proteasome and autophagy‐lysosome pathways.( 42 ) It was reported that TRIM38 inhibits TNFα‐ and IL‐1β–triggered NF‐κB activation by mediating the lysosomal‐dependent degradation of TAB2/3, which resulted in decreased activity of TAK1.( 24 ) In the present study, we found that TRIM27‐mediated degradation of TAB2/3 was completely inhibited by an inhibitor of the lysosome (CQ), but not an inhibitor of the proteasome (MG132). These findings indicated that TRIM27 mediated the degradation of TAB2/3 through the lysosomal pathway. However, the potential mechanism is unclear. Recent evidence showed that ubiquitin‐specific peptidase 15 induced cleavage of Lys48‐linked ubiquitination of TAB2 and also inhibited neighbor of BRCA1 gene 1–mediated selective autophagic TAB3 degradation.( 43 ) Therefore, TRIM27 may also mediate the protein stability of TAB2/3 through ubiquitination or autophagy, which needs further study.
Overall, our results identify TRIM27 as a protective regulator in liver subjected to I/R surgery by mediating the inflammatory response and apoptosis. In addition, TRIM27 mediated the degradation of TAB2/3 and suppression of downstream TAK1‐JNK/p38 signaling. Our studies provide insight into a therapeutic target and a useful prognostic biomarker for hepatic I/R injury.
Author Contributions
S.‐Y.C., H.‐P.Z., J.L., and S.‐J.Z. participated in research design; S.‐Y.C., H.‐P.Z., J.L., J.‐H.S., H.‐W.T., Y.Z., P.‐H.W., J.‐K.Z., Y.‐T.H., Z.‐H.W., X.‐Y.S., H.Y., and B.‐W.H. conducted experiments; all authors performed data analysis and interpretation; S.‐Y.C., H.‐P.Z., and J.L. drafted the paper. S.‐J.Z. supervised the study, and all authors read and approved the final manuscript.
Supporting information
Supplementary Material
Supported by grants from the National Natural Science Foundation of China (nos.: U1604282, 81671958) and a supporting plan for scientific and technological innovation talents in colleges and universities of Henan Province (19HASTIT003).
Potential conflict of interest: Nothing to report.
Contributor Information
Wen‐Zhi Guo, Email: fccguowz@zzu.edu.cn.
Shui‐Jun Zhang, Email: zhangshuijun@zzu.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Yang W, Chen J, Meng Y, Chen Z, Yang J. Novel targets for treating ischemia‐reperfusion injury in the liver. Int J Mol Sci 2018;19 pii: E1302. doi: 10.3390/ijms19051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu Y, Lu T, Zhang C, Xu J, Xue Z, Busuttil RW, et al. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia‐reperfusion injury. J Hepatol 2019;71:719‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guo WZ, Fang HB, Cao SL, Chen SY, Li J, Shi JH, et al. Six‐transmembrane epithelial antigen of the prostate 3 deficiency in hepatocytes protects the liver against ischemia‐reperfusion injury by suppressing transforming growth factor‐β‐activated kinase 1. Hepatology 2020;71:1037‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fan Q, Tao R, Zhang H, Xie H, Lu L, Wang T, et al. Dectin‐1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 2019;139:663‐678. [DOI] [PubMed] [Google Scholar]
- 5. Motiño O, Francés DE, Casanova N, Fuertes‐Agudo M, Cucarella C, Flores JM, et al. Protective role of hepatocyte cyclooxygenase‐2 expression against liver ischemia‐reperfusion injury in mice. Hepatology 2019;70:650‐655. [DOI] [PubMed] [Google Scholar]
- 6. Jaworska AM, Wlodarczyk NA, Mackiewicz A, Czerwinska P. The role of TRIM family proteins in the regulation of cancer stem cell self‐renewal: concise review. Stem Cells 2020;38:165‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hao MQ, Xie LJ, Leng W, Xue RW. Trim47 is a critical regulator of cerebral ischemia‐reperfusion injury through regulating apoptosis and inflammation. Biochem Biophys Res Comm 2019;515:651‐657. [DOI] [PubMed] [Google Scholar]
- 8. Tao Q, Tianyu W, Jiangqiao Z, Zhongbao C, Xiaoxiong M, Long Z, et al. Tripartite motif 8 deficiency relieves hepatic ischaemia/reperfusion injury via TAK1‐dependent signalling pathways. Int J Biol Sci 2019;15:1618‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang HX, Xu ZS, Lin H, Li M, Xia T, Cui K, et al. TRIM27 mediates STAT3 activation at retromer‐positive structures to promote colitis and colitis‐associated carcinogenesis. Nat Commun 2018;9:3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu J, Feng X, Tian Y, Wang K, Gao F, Yang L, et al. Knockdown of TRIM27 expression suppresses the dysfunction of mesangial cells in lupus nephritis by FoxO1 pathway. J Cell Physiol 2019;234:11555‐11566. [DOI] [PubMed] [Google Scholar]
- 11. Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci 2017;42:297‐311. [DOI] [PubMed] [Google Scholar]
- 12. Ma Y, Wei Z, Bast RC, Wang Z, Li Y, Gao M, et al. Downregulation of TRIM27 expression inhibits the proliferation of ovarian cancer cells in vitro and in vivo . Lab Invest 2016;96:37‐48. [DOI] [PubMed] [Google Scholar]
- 13. Yan ZZ, Huang YP, Wang X, Wang HP, Ren F, Tian RF, et al. Integrated omics reveals tollip as an regulator and therapeutic target for hepatic ischemia‐reperfusion injury in mice. Hepatology 2019;70:1750‐1769. [DOI] [PubMed] [Google Scholar]
- 14. Bai L, Chen MM, Chen ZD, Zhang P, Tian S, Zhang Y, et al. F‐box/WD repeat‐containing protein 5 mediates the ubiquitination of apoptosis signal‐regulating kinase 1 and exacerbates nonalcoholic steatohepatitis in mice. Hepatology 2019;70:1942‐1957. [DOI] [PubMed] [Google Scholar]
- 15. van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev 2012;23:69‐84. [DOI] [PubMed] [Google Scholar]
- 16. Napetschnig J, Wu H. Molecular basis of NF‐κB signaling. Ann Rev Biophys 2013;42:443‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng Q, Hou J, Zhou YE, Yang Y, Xie B, Cao X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res 2015;25:1121‐1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landström M. The TAK1‐TRAF6 signalling pathway. Int J Biochem Cell Biol 2010;42:585‐589. [DOI] [PubMed] [Google Scholar]
- 19. Aashaq S, Batool A, Andrabi KI. TAK1 mediates convergence of cellular signals for death and survival. Apoptosis 2019;24:3‐20. [DOI] [PubMed] [Google Scholar]
- 20. Ahmed N, Zeng M, Sinha I, Polin L, Wei WZ, Rathinam C, et al. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat Immunol 2011;12:1176‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajsbaum R, García‐Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3‐ubiquitin ligase family in innate antiviral immunity. J Mol Biol 2014;426:1265‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF‐κB signaling pathways. Nat Immunol 2011;12:695‐708. [DOI] [PubMed] [Google Scholar]
- 23. Ajibade AA, Wang HY, Wang RF. Cell type‐specific function of TAK1 in innate immune signaling. Trends Immunol 2013;34:307‐316. [DOI] [PubMed] [Google Scholar]
- 24. Hu MM, Yang Q, Zhang J, Liu SM, Zhang Y, Lin H, et al. TRIM38 inhibits TNFα‐ and IL‐1β‐triggered NF‐κB activation by mediating lysosome‐dependent degradation of TAB2/3. Proc Natl Acad Sci U S A 2014;111:1509‐1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gracia‐Sancho J, Casillas‐Ramírez A, Peralta C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: a 2015 update. Clin Sci (Lond) 2015;129:345‐362. [DOI] [PubMed] [Google Scholar]
- 26. Zheng Q, Hou J, Zhou Y, Yang Y, Cao X. Type I IFN‐inducible downregulation of microRNA‐27a feedback inhibits antiviral innate response by upregulating Siglec1/TRIM27. J Immunol 2016;196:1317‐1326. [DOI] [PubMed] [Google Scholar]
- 27. Nie D, Zhang D, Dai J, Zhang M, Zhao X, Xu W, et al. Nicotine induced murine spermatozoa apoptosis via up‐regulation of deubiquitinated RIP1 by Trim27 promoter hypomethylation. Biol Reprod 2016;94:31. [DOI] [PubMed] [Google Scholar]
- 28. Jiménez‐Castro MB, Cornide‐Petronio ME, Gracia‐Sancho J, Peralta C. Inflammasome‐mediated inflammation in liver ischemia‐reperfusion injury. Cells 2019;8:1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song P, Zhang J, Zhang Y, Shu Z, Xu P, He L, et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int J Mol Med 2018;41:935‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Xu P, Song P, Wang H, Zhang Y, Hu Q, et al. CCL2‐CCR2 signaling promotes hepatic ischemia/reperfusion injury. J Surg Res 2016;202:352‐362. [DOI] [PubMed] [Google Scholar]
- 31. Nakashima H, Nakashima M, Kinoshita M, Ikarashi M, Miyazaki H, Hanaka H, et al. Activation and increase of radio‐sensitive CD11b+ recruited Kupffer cells/macrophages in diet‐induced steatohepatitis in FGF5 deficient mice. Sci Rep 2016;6:34466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med 2011;3:513‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cai X, Srivastava S, Sun Y, Li Z, Wu H, Zuvela‐Jelaska L, et al. Tripartite motif containing protein 27 negatively regulates CD4 T cells by ubiquitinating and inhibiting the class II PI3K‐C2β. Proc Natl Acad Sci U S A 2011;108:20072‐20077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng CS, Wan S, Yim AP. Pulmonary ischaemia‐reperfusion injury: role of apoptosis. Euro Respir J 2005;25:356‐363. [DOI] [PubMed] [Google Scholar]
- 35. Zhang S, Jiang S, Wang H, Di W, Deng C, Jin Z, et al. SIRT6 protects against hepatic ischemia/reperfusion injury by inhibiting apoptosis and autophagy related cell death. Free Radic Biol Med 2018;115:18‐30. [DOI] [PubMed] [Google Scholar]
- 36. Liu Y, Zhu M, Lin L, Fan X, Piao Z, Jiang X. Deficiency of Trim27 protects dopaminergic neurons from apoptosis in the neurotoxin model of Parkinson's disease. Brain Res 2014;1588:17‐24. [DOI] [PubMed] [Google Scholar]
- 37. Wang X, Mao W, Fang C, Tian S, Zhu X, Yang L, et al. Dusp14 protects against hepatic ischaemia‐reperfusion injury via Tak1 suppression. J Hepatol 2017. Sep 6. 10.1016/j.jhep.2017.08.032. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 38. Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol 2014;49:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Wang Q, Zhou W, Zhou Z, Tang PY, Xu T, et al. GPCR kinase 2‐interacting protein‐1 protects against ischemia‐reperfusion injury of the spinal cord by modulating ASK1/JNK/p38 signaling. FASEB J 2018:fj201800548 2018 Jun 18. 10.1096/fj.201800548. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 40. Fan Y, Yu Y, Mao R, Zhang H, Yang J. TAK1 Lys‐158 but not Lys‐209 is required for IL‐1β‐induced Lys63‐linked TAK1 polyubiquitination and IKK/NF‐κB activation. Cell Signal 2011;23:660‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Q, Yan J, Mao AP, Li C, Ran Y, Shu HB, et al. Tripartite motif 8 (TRIM8) modulates TNFα‐ and IL‐1β‐triggered NF‐κB activation by targeting TAK1 for K63‐linked polyubiquitination. Proc Natl Acad Sci U S A 2011;108:19341‐19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Majumder P, Baumeister W. Proteasomes: unfoldase‐assisted protein degradation machines. Biol Chem 2019;401:183‐199. [DOI] [PubMed] [Google Scholar]
- 43. Zhou Q, Cheng C, Wei Y, Yang J, Zhou W, Song Q, et al. USP15 potentiates NF‐κB activation by differentially stabilizing TAB2 and TAB3. FEBS J 2020. Jan 5. 10.1111/febs.15202. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material