

Abstract
Rugged analytical methods for the screening and identity confirmation of anthocyanins require a dedicated sample preparation, chromatographic setup, and the reliable generation of multiple identification points to confirm identity against the wide range of phenolic compounds typically present in food, beverage, and plant material samples. To this end, combinations of spectroscopic and mass spectrometric detection are frequently employed for this application to provide higher confidence in the absence of authentic standards. In the present work, low‐field drift tube ion mobility (DTIM) separation is evaluated for this task using a LC–DAD–DTIM–QTOFMS method. DTIM‐MS allows accurate determination of collision cross sections (DTCCS) for all analysed compounds as well as a precise alignment tool for reconciling fragment and precursor ions in data independent acquisition mode. The presented approach thereby allows for an anthocyanin screening method taking true advantage of all dimensions of the analytical platform: relative retention (RPLC), UV/VIS absorption spectrum, accurate mass, DTCCSN2, and confirmed high‐resolution fragment ions. From the analysis of authentic standards and several berry samples primarily from the Vaccinium genus, Level 1 confirmation data for six anthocyanins from the cyanidin family, and Level 2 confirmation for a further 29 anthocyanins confirmed in berry samples is provided. The method and accompanying dataset provided as part of this work provides a means to develop anthocyanin screening methods using the ion mobility dimension as an additional alignment and filtering parameter in data independent analysis acquisition across any LC–IM–MS platform.
Keywords: Anthocyanins, Berries, Confirmation, Identity, Ion mobility‐mass spectrometry, Natural products
Abbreviations
- DTIM‐MS
drift tube ion mobility‐mass spectrometry
- MF
molecular feature
- QTOFMS
quadrupole time‐of‐flight mass spectrometry
1. Introduction
Anthocyanins are one of the most important subclasses of flavonoids and are widely present in leaves, flowers, and fruits, mainly in outer cell layers of some fruits such as red grapes, black currants, strawberries, cranberries, blueberries and blackberries, among others [1]. They are responsible for red, blue, purple, and violet colours [2]. Anthocyanins are the most stable glycosides of anthocyanidins and the large structural diversity of this subclass of flavonoids is associated with variations in the aglycone and sugar structures as well as in the number, position, and acylation of sugar attached to each aglycone [3]. The most common anthocyanidins in vascular plants are pelargonidin, cyanidin, peonidin, delphinidin, petunidin, and malvidin [4]. Anthocyanins are postulated to provide numerous health benefits associated with their consumption such as those from their antioxidant properties, cardiovascular protection [5], and antidiabetes [6], anti‐inflammatory [7], and antiobesity [8, 9] effects. They also present anticarcinogen properties [10], promising chemopreventive ability against cholangiocarcinoma by Opisthorchis viverrini infection [11] and anti‐invasive effects on human colon cancer cells [12]. These health benefits render anthocyanins an attractive choice as compounds for possible use as nutritional supplements, constituents of functional food formulas and medicines, among others, in addition to interest in anthocyanins as natural food colorants [13]. Owing to these health benefits, the determination of their structures plays a very important role in many areas of food science.
For suitable analysis, the extraction of anthocyanins requires consideration of several parameters such as extractant and acid agent. The latter enhances the formation and stability of the anthocyanins in its flavylium cationic form without degradation. The most used extractants are mainly organic solvents, such as acetonitrile (ACN), ethanol, and methanol, and the most used acid agents are phosphoric, formic, and citric acids [14]. Generally, anthocyanin extraction methods are assisted by soaking, stirring, enzymes, or ultrasound (US). This latter option provides some advantages such as lower extraction time, reduced amount of extractant, and a higher extraction yield [15]. Regarding chromatographic separation, the most used technique for anthocyanins is RPLC, and the most used column and mobile phases are C18 columns [14] and highly acidic mobile phases (e.g., 4–10% formic acid (FA), or 0.1–0.6% trifluoroacetic acid) [16, 17, 18, 19]. Highly acidic mobile phases (pH < 2) are required to ensure that the equilibrium is strongly shifted toward the formation of the flavylium cationic form in solution and provides a marked improvement in the chromatographic efficiency [13, 17, 20, 21, 22]. Currently, the official method of anthocyanins analysis described by the AOAC International is the spectrophotometric pH differential method, a fast and direct quantitative method (AOAC, 2005) [23] that is not convenient for probing the exact identities of individual species. However, the most common detection techniques for anthocyanins are DAD and MS or tandem mass spectrometry (MS/MS). DAD can tentatively distinguish between the main flavonoids as anthocyanins exhibit UV/vis absorption spectra characterized by maximum absorption in the visible range around 520 nm. DAD also offers some information of the aglycone structure, and glycosylation and acylation patterns [24, 25, 26]. However, it does not provide enough structural information to allow identification of individual anthocyanins within complex samples, which is an important limitation because the limited commercial availability of reference standards. In contrast, mass spectrometric detection provides higher sensitivity, decreased LOD, and detailed structural information compared to LC–DAD. Several MS instruments have been used for anthocyanins analysis such as simple quadrupole, triple quadrupole, and quadrupole‐time‐of flight (Q‐TOF), among others. Among them, Q‐TOF with ESI has been the most used [14]. Low resolution approaches using MS/MS are mainly appropriate for structure elucidation and compound identity confirmation since information concerning to the aglycone structure, number and type of sugars, and other substituents, such as acyl groups, can be acquired [27]. However, conclusive compound identification remains challenging because some non‐anthocyanin phenolic compounds, such as glycosylated flavanols, show similar mass spectral characteristics [28] and, moreover, use of high‐resolution fragment spectra does not allow discrimination among glycosidic isomers. For these reasons, chromatographic separation of anthocyanins is still necessary. In the last two decades, high‐performance RPLC coupled with DAD and MS (HPLC–DAD–MS) has proven to be the most suitable method for identification of anthocyanins in plant extracts [29, 30, 31, 32] and fruit juices [33, 34]. Compound identity confirmation is based on LC elution order, UV/vis spectra as well as mass spectral information (accurate mass and fragmentation spectra [27]).
As a recent commercial development, ion mobility‐MS (IM‐MS) has been shown to be a novel analytical platform especially useful for the differentiation and characterization of isomers and isobars [35]. IM‐MS allows separation of gaseous ions according to size, shape, and charge [36] and is nested between the chromatographic separation and mass analyzer(s) [37]. A Hadamard transform drift tube ion mobility mass spectrometry (HT‐DTIM‐MS) instrument was previously used to obtain the anthocyanins profile from blueberry, raspberry, blackberry, strawberry, and pomegranate [37]. Since this study was performed, new commercial developments in DTIM‐MS have demonstrated excellent interlaboratory agreement of IM separations and particularly the derived collision cross‐section (DTCCS) values [38], which is an essential requirement for utilizing IM information on a routine basis. As DTIM‐MS is the only commercially available IM analyzer that retains a direct link to the fundamental zero‐field relationships derived for relating IM arrival time to DTCCS values, experimental investigations on this instrument class are not only valuable for the elaboration of a new analytical workflow, but also providing reference CCS values that can support similar applications on other classes of IM‐MS instrumentation. Therefore, the objective of the present research was to develop a LC‐IM‐QTOFMS method for the identity confirmation of anthocyanins in a range of common berries (primarily Vaccinium sp.) using RPLC in combination with DAD and DTIM‐QTOFMS. This approach allows collection of a wide range of experimental identification points of each compound, supporting the differentiation of anthocyanins from other subclasses of flavonoids with the same m/z, as well as retaining the possibility to subsequently evaluate other phenolic classes in the same dataset. Optimized conditions for the chromatographic separation of anthocyanin extracts were employed and both positive and negative mode were assessed to provide as much complementary information as possible for the standards and samples evaluated.
2. Materials and Methods
2.1. Reagents and chemicals
Full details can be found in the Supporting Information.
2.2. Instruments and apparatus
A US water bath (44 kHz, model XUBA1) from Grant Instruments (Cambridge, United Kingdom) and a vortex shaker from IKA (Wilmington, NC) were used for standards and samples preparation. A Mikro 200 R centrifuge from Hettich Zentrifugen (2424B rotor) was also used for sample preparation.
The instrument platform comprised of an autosampler robot (Dual Rail MPS 2 Gerstel [Mülheim an der Ruhr, Germany], an Agilent Technologies 1290 Infinity II LC System (binary pump and a thermostated column compartment), an Agilent 1200 DAD module, and an Agilent 6560 DTIM‐QTOFMS equipped with a dual Jetstream (AJS) ESI source. An Agilent 1200 Series Nano Pump was used for simultaneous spraying of mass reference for online reference mass calibration. This analytical instrumentation was controlled through the Mass Hunter Workstation Acquisition Software (TOF/QTOF) (version B.09.00) from Agilent Technologies (Santa Clara, CA). The injection sequence was controlled using the Gerstel Maestro Configuration Maestro 1.49.3 software.
2.3. Sample collection
Five different berry samples were purchased in a local supermarket (Vienna, Austria) in October, 2018: lingonberries, red currants, cranberries, blueberries, and blackberries, Supporting Information Fig. S1. Red currants were labeled as originating in Austria, blackberries from Italy, blueberries from Peru, cranberries from Poland, and lingonberries from Sweden. At least 125 g of each were purchased. Fruits were ripe, fresh, with a firm consistency and a pronounced color.
2.4. Anthocyanin extraction and sample preparation
Anthocyanins were extracted from fruit samples using a modification of the protocol previously optimized by Canuto et al. [39]. Full details of the sample preparation can be found in the Supporting Information.
2.5. LC–DAD–IM‐QTOFMS analysis
After optimization of sample preparation and chromatographic separation steps, the diluted standards were analyzed by different acquisition modes: LC‐DAD–TOFMS, LC‐DAD–IM‐TOFMS, and LC‐DAD–QTOFMS, in positive and negative ionization modes. Samples were analyzed by LC‐DAD–TOFMS and LC‐DAD–IM‐TOFMS in both ionization modes, and by LC‐DAD–QTOFMS and LC‐DAD–IM‐QTOFMS using only the positive ionization mode. Chromatographic separation was performed using a Zorbax SB column (50 × 2.1 mm i.d., 1.8 μm dp) from Agilent using a temperature of 50°C. The mobile phases were 7.5% v/v FA in water (phase A) and 7.5% v/v FA in ACN (phase B). The injection volume was 5 μL and the injector needle was washed between injections with methanol and water. Full LC gradient details are contained in the Supporting Information.
For LC‐IM‐QTOFMS measurements, an established secondary single‐field calibration approach was used with reference DTCCS values and methodology determined in [38]. A collision energy of 40 V was used to obtain the fragmentation (high energy) spectral data. Full details of the applied acquisition methods are provided in the Supporting Information.
2.6. Data processing
The more complex datasets generated in this study required the use of vendor software and Skyline [40]. Full details of the data processing are contained in the Supporting information. A complete transition list compatible with Skyline including retention times (RT), fragments, and accurate DTCCSN2 values of precursors is also provided in the Supporting information.
3. Results and Discussion
3.1. Assessment of standards
Commercial standards of six anthocyanins (see Section 2.1) of the same aglycone, cyanidin (the most common anthocyanidin in fruits [41]), were selected and analyzed by LC–DAD–TOFMS and LC–DAD–IM‐TOFMS methods. While the use of high‐resolution MS and addition of DTCCSN2 are proposed to be the most conclusive for correct identity confirmation of anthocyanins, we note that annotation of anthocyanins is known to be misleading without DAD because other flavonoids can be easily misannotated as anthocyanins using conventional MS data processing tools designed for metabolomics‐type workflows [42]. Therefore, one aim of this work was to critically assess the value of DTCCSN2 for identity confirmation of anthocyanins using highest level identity confirmation as a starting point.
Measurements of RT, UV/vis spectrum, absorbance at 280 and 510 nm (wavelengths for selective detection and accurate quantitation of anthocyanins) [43], high‐resolution MS and DTCCSN2 data of analytical standards were obtained to establish reference values for a range of common anthocyanins especially to later provide Level 1 identity confirmation in fruits. Table 1 summarizes determined RT, m/z, DTCCSN2 values and mass error of selected standards. The primary ions of interest ([M]+ and [M‐2H]–) were determined with experimental mass error less than 2.5 ppm in all cases. Two of the standards investigated were isomeric (cyanidin‐3‐O‐sophoroside and cyanidin‐3,5‐di‐O‐glucoside) and could be discriminated with a difference of 2% in their DTCCSN2 values. Experimental DTCCSN2 values of [M]+ ion and [M‐2H]– ions were found to be very similar for each anthocyanin, which is not unexpected given the partially rigid structure of the aglycones and the likely absence of charge migration. For example, DTCCSN2 values of cyanidin‐3,5‐di‐O‐glucoside were 240.8 and 240.7 Å2, for the [M]+ and [M‐2H]– species, respectively. However, DTCCSN2 (‐) values of postulated [M‐2H+H2O]– adducts [42] could not be determined for any of the anthocyanins as signals were not observed using the experimental IM‐TOFMS conditions employed. A hypothesis of faster interconversion of carbinol and quinidol species and a dependence on the mobile phase pH for the detection of this species has been proposed [43]. Our previous work [44] using DTIM‐MS allowed detection of these species in IM‐TOFMS analysis of wine samples using a less acidic mobile phase (0.1% v/v FA), but current results obtained using 7.5% v/v FA supports the mobile phase pH dependency hypothesis. Thus, while negative mode might be used for confirmation screening for the [M‐2H]– form, the additional postulated species [M‐2H+H2O]– is suggested here to not be amenable for workflows optimized for anthocyanin determination.
Table 1.
Accurate mass and DTCCSN2 data of anthocyanins standards determined in both positive and negative ionization modes
| [M]+ | [M‐2H]– | |||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Formula | RT (min) | Exp. mass | Mass error (ppm) | DTCCSN2 (Å2) | Exp. Mass | Mass error (ppm) | DTCCSN2 (Å2) |
| Cyanidin | C15H11O6 | 20.83 | 287.0552 | 0.7 | 166.2 | 285.0398 | −2.5 | 162.9 |
| Cyanidin‐3‐O‐arabinoside | C20H19O10 | 15.51 | 419.0966 | −1.7 | 198.2 | 417.0827 | <0.1 | 198.6 |
| Cyanidin‐3‐O‐galactoside | C21H21O11 | 13.18 | 449.1072 | −1.3 | 202.7 | 447.0932 | −0.2 | 198.5 |
| Cyanidin‐3‐O‐rutinoside | C27H31O15 | 16.37 | 595.1650 | −1.2 | 234.6 | 593.1512 | <0.1 | 234.1 |
| Cyanidin‐3‐O‐sophoroside | C27H31O16 | 13.18 | 611.1600 | −1.1 | 236.0 | 609.1460 | −0.2 | 230.7 |
| Cyanidin‐3,5‐di‐O‐glucoside | C27H31O16 | 11.86 | 611.1596 | −1.8 | 240.8 | 609.1461 | <0.1 | 240.7 |
Determined DTCCSN2 values for anthocyanins are shown in Fig. 1A. In which, the anthocyanidin (highlighted in white) exhibited an tA of 21.68 ms while cyanidin‐3,5‐di‐O‐glucoside (red) had the largest tA with 32.17 ms. The same order of tA obtained for anthocyanin standards was also obtained for anthocyanins in fruits in a previous study [37]. RTs obtained were between 11.86 (cyanidin‐3,5‐di‐O‐glucoside) and 20.83 min (cyanidin). However, the order of RT of standards is different to the tA order, which arises from the chemical selectivity offered by the RPLC separation that is not available with DTIM separation. The determined DTCCSN2 values versus m/z of each anthocyanin revealed a characteristic trend line [45] for anthocyanins, for both positive and negative ions (Fig. 1B). These characteristic trends of anthocyanins in the mobility‐m/z space can be used to help support identity confirmation workflows in the absence of authentic standards. A wavelength of 510 nm was selected to determine the anthocyanins in fruits as recommended in literature [43, 46, 47]. Finally, an innovative data processing approach was performed to reveal all the anthocyanins of a same aglycone, in this case cyanidin, in a lingonberry extract sample. Following analysis of the sample and standards, the IM‐MS file was filtered by the accurate m/z (287.0556) yielding a three‐dimensional extracted ion chromatogram revealing all possible cyanidin‐related species (Supporting Information Fig. S3). The combination of RT and tA of these individual peaks can be used to confirm if the species is an in‐source fragment, intact aglycone or post‐IM fragment of a glycosylated anthocyanin. Of particular interest here is the accurate match of the tA of cyanidin from in‐source fragmentation that allows confirmation with comparison to the DTCCSN2value of the cyanidin standard.
Figure 1.
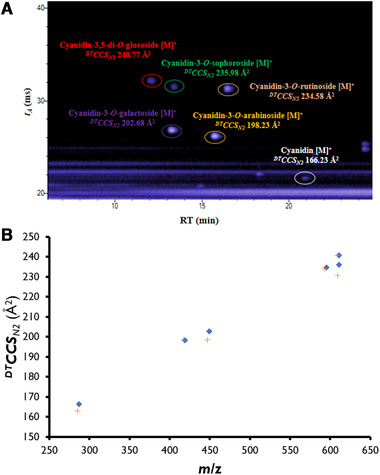
(A) IM–TOFMS TIC of anthocyanins standards illustrating RT (min) and tA (ms) for each standard. Anthocyanins are highlighted by different colors, red (cyanidin‐3,5‐di‐O‐glucoside), green (cyanidin‐3‐O‐sophoroside), purple (cyanidin‐3‐O‐galactoside), orange (cyanidin‐3‐O‐arabinoside), pink (cyanidin‐3‐O‐rutinoside), and white (cyanidin). (B) Representation of determined DTCCSN2 value versus m/z of each Level 1 anthocyanin for the primary positive and negative ions. [M]+ ions are shown as diamonds and [M‐2H]– ions as crosses.
3.2. Distribution of anthocyanins in berry samples
Different fruit samples from the Vaccinium family (blueberry, lingonberry, cranberry), as well as blackberry and red currant were prepared in accordance with the optimized method and analyzed by LC–DAD–TOFMS and LC–DAD–IM–TOFMS. Fig. 2 shows the DAD chromatograms obtained at 510 nm for each one of the samples. Among them, blueberry and cranberry presented a higher number of peaks corresponding with anthocyanins in comparison with the rest of the samples (lingonberry, blackberry, and red currant).
Figure 2.
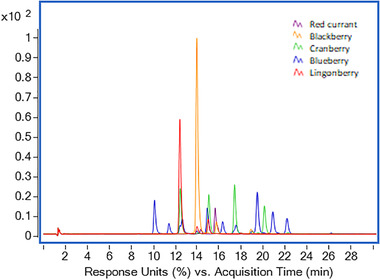
DAD chromatograms obtained at 510 nm for different fruits samples assessed in this study: red currant (purple), blackberry (orange), cranberry (green), blueberry (blue), and lingonberry (red).
In total, 38 anthocyanins were identity confirmed across the five berry types. All of the species determined were anthocyanidin‐glycosides. Identity confirmed anthocyanins include cyanidin‐, delphinidin‐, pelargonidin‐, peonidin‐, malvidin‐, and petunidin‐glycosides, which are the most common species in plants, fruits, and vegetables [41]. The distribution of these anthocyanidins in fruits is reported to be 50, 12, 12, 12, 7, and 7% by weight, respectively [3]. However, this distribution depends on fruit type. For example, the distribution of these anthocyanidins in cranberry has been determined to be 40, 15, 5, 10, 10, and 15%, respectively. None of the fruit samples contained all 38 anthocyanins. A total of 25 of them were confirmed in blueberry, 18 in cranberry, 13 in lingonberry, 11 in blackberry, and only 7 in red currant. The full list of identity confirmed metabolites and the abbreviations used in the following text are shown in Table S2 of the Supporting Information including supporting literature.
Blueberry is classified as multiple anthocyanin fruit [54] and was found to contain a large diversity of anthocyanins. Blueberry contained mainly delphinidin, malvidin, and petunidin glycosides (Dp‐3‐O‐ara, Dp‐3‐O‐xyl, Dp‐3‐O‐gal, Dp‐3‐O‐glc, Mv‐3‐O‐ara, Mv‐3‐O‐xyl, Mv‐3‐O‐gal, Mv‐3‐O‐glc, Pt‐3‐O‐ara, Pt‐3‐O‐gal and Pt‐3‐O‐glc), one acetylated malvidin glycoside (Mv‐3‐acetylglc), two malonylated malvidin glycosides [Mv‐3‐malonylhex (I) and Mv‐3‐malonylhex (II)], and two coumaroylated anthocyanins (Mv‐3‐p‐coumhex and Pt‐3‐p‐coumhex). Several cyanidin and peonidin glycosides (Cy‐3‐O‐ara, Cy‐pent (II), Cy‐3‐O‐gal, Cy‐3‐O‐glc, Cy‐3‐sam, Pn‐3‐O‐ara, Pn‐3‐O‐gal, and Pn‐3‐O‐glc), one pelargonidin glycoside (Pl‐3‐O‐gal) were also found.
Regarding cranberry, lingonberry, blackberry, and red currant, these have been previously classified as cyanidin‐ and peonidin‐rich fruits [54]. Cranberry was found to contain six cyanidin glycosides, one acylated cyanidin glycoside, and two peonidin glycosides. Cyanidin glycosides were Cy‐3‐O‐ara, Cy‐pent (I), Cy‐3‐O‐gal, Cy‐3‐O‐glc, and Cy‐dipent, an acylated cyanidin glycoside (Cy‐3‐cinnamoylhex) and catechin‐Cy‐3‐hex. Confirmed peonidin glycosides were Pn‐3‐O‐ara and Pn‐3‐O‐gal. Moreover, other anthocyanins, including Dp‐3‐O‐ara, Dp‐3‐O‐gal, Dp‐3‐O‐glc, Pt‐3‐O‐ara, Pt‐3‐O‐gal, Pl‐3‐O‐gal, Mv‐3‐O‐ara, Mv‐3‐O‐gal, were also confirmed.
Lingonberry was found to contain seven cyanidin glycosides, one acylated cyanidin glycosides, and one peonidin glycoside. The cyanidin glycosides were Cy‐3‐O‐ara, Cy‐pent (I), Cy‐3‐O‐gal, Cy‐3‐O‐glc, Cy‐dipent, Cy‐3‐sam, and catechin‐Cy‐3‐hex. The acylated cyanidin glycoside was Cy‐3‐cinnamoylhex. One peonidin glycoside was identity confirmed as Pn‐3‐O‐gal. Other confirmed anthocyanins were Dp‐3‐O‐ara, Dp‐3‐O‐gal, Mv‐3‐O‐gal, and Pl‐3‐O‐glc.
For blackberry, seven cyanidin glycosides were identity confirmed, which were Cy‐3‐O‐ara, Cy‐pent (II), Cy‐3‐O‐glc, Cy‐3‐sam, Cy‐3,5‐di‐O‐glc, Cy‐di‐hex, and Cy‐3‐O‐rut, and two acylated cyanidin glycosides including one acylated anthocyanin (Cy‐3‐cinnamoylhex) and one malonylated anthocyanin (Cy‐3‐malonylglc). Pl‐3‐O‐gal and Pn‐3‐O‐rut were also confirmed.
Finally, red currant was found to contain the lowest number of confirmed anthocyanins, which were Cy‐3‐O‐glc, Cy‐3‐O‐rut, Cy‐3‐sam, Cy‐xylosylrut, Cy‐di‐hex, Cy‐sambubiosyl‐rha, and Dp‐3‐O‐glc.
Even though all identity confirmed anthocyanins in berries were anthocyanidin glycosides, different sugar linkages and isomers could be confirmed in many cases. Fig. S4 of the Supporting Information shows a profile of confirmed anthocyanins contained in each berry sample. Cranberry, lingonberry, and blackberry contained mainly cyanidin monoglycosides (including acetylated) species, while blueberry contained mostly delphinidin, malvidin, and cyanidin monoglycosides (including acylated) species. In general, the berry samples contained more anthocyanidin monoglycosides than di‐ or triglycosides. Blackberry presented the highest number of anthocyanidin diglycosides (cyanidin diglycosides). It is also noteworthy that the presence of cyanidin triglycosides, such as Cy‐sambubiosyl‐rha and Cy‐3‐O‐(2″‐xyl) rut in red currant could also be confirmed.
Although not a focus of the present work, some interpretation of relative anthocyanin content can be interpreted from the DAD chromatogram peak areas following confirmation of peak identities. Blueberry presented a higher content of Dp‐3‐O‐gal, Mv‐3‐O‐gal, Pt‐3‐O‐gal, Mv‐3‐O‐glc, Pt‐3‐O‐glc, Mv‐3‐O‐ara, Dp‐3‐O‐glc, Dp‐3‐O‐ara, Pt‐3‐O‐ara, and Cy‐3‐O‐gal compared to other anthocyanins. These anthocyanins were determined to be the most abundant in a previous study focused on the quantitation of anthocyanins [54], in which Dp‐3‐O‐gal, Mv‐3‐O‐gal, Pt‐3‐O‐gal, Mv‐3‐O‐glc were found at concentrations between 0.47 and 1.10 mg/g of blueberry extract.
In the case of cranberry, the most abundant anthocyanins were Pn‐3‐O‐gal, Pn‐3‐O‐ara, Cy‐3‐O‐gal, and Cy‐3‐O‐ara. These anthocyanins have been determined as the most abundant in cranberry in a previous study [48] using HPLC–ESI–MS revealing concentrations between 13.2 and 49.6 mg/100 g of cranberry pomace.
Regarding lingonberry, the most abundant anthocyanins were Cy‐3‐O‐gal, Cy‐3‐O‐ara, and Cy‐3‐O‐glc, which was also previously reported by Lee et al. [49]. In fact, Cy‐3‐gal is reported to be the predominant anthocyanin contained in lingonberry [54].
Cy‐3‐O‐glc, Cy‐3‐O‐rut, and Cy‐3‐O‐xyl were the most dominant anthocyanins in blackberry, and Cy‐3‐O‐(2″‐xyl)rut, Cy‐3‐O‐rut, and Cy‐3‐sam in red currant. Even though these fruits do not have one predominant anthocyanin, both contain one major anthocyanin, which is Cy‐3‐O‐glc in the case of blackberry and Cy‐3‐O‐(2″‐xyl)rut in the case of red currant [37, 47].
3.3. Identity confirmation potential of LC–DAD–IM‐TOFMS for anthocyanins
The analyses of five different fruits extracts by LC–DAD–TOFMS and LC–DAD–IM‐TOFMS allowed the identity confirmation of 38 anthocyanins in berry samples. Among them, four could be confirmed as Level 1 by matching with chemical standards used in this study (Cy‐3‐O‐gal, Cy‐3‐O‐rut, Cy‐3,5‐di‐O‐glc, and Cy‐3‐O‐ara). Due to a general lack of commercial anthocyanin standards, the identity confirmation of other anthocyanins was based on multiple identification points such as comparison of relative elution order with literature, UV/vis spectral information, feasibility of DTCCSN2 values, accurate mass, isotopologue distribution, and characteristic fragment ions. As can be seen in Table S2 of the Supporting Information, anthocyanins formed a dominant [M]+ ion in all cases in positive ionization mode, while approximately half of these were found to form a [M‐2H]– ion and none of them formed a previously postulated [M‐2H+H2O]– adduct when using highly acidic mobile phase conditions. Anthocyanins were identity confirmed with an average absolute mass error of 2.1 ppm. Comparison of the IM and m/z data for the identity confirmed anthocyanins in fruit revealed characteristic trend lines as highlighted in Section 3.1 (Fig. 1A) for standards, which provides a means to constrain the identity confirmation in ambiguous cases [50]. Fig. 3A shows the trends lines obtained after representation of determined DTCCSN2values versus m/z for each anthocyanin in positive and negative ionization modes. Finally, the correlation between determined DTCCSN2 values measured for the primary positive and negative mode species for each anthocyanin (Fig. 3B) was very close to 1 (0.9505), which was consistent with the results observed for the Level 1 standards.
Figure 3.
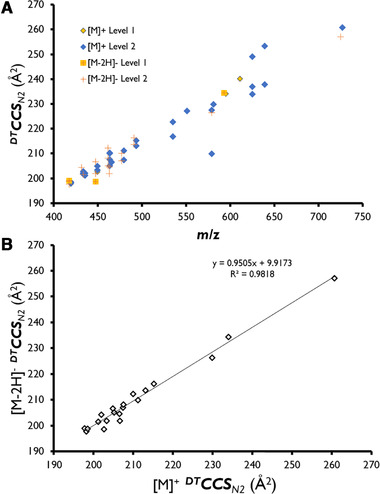
(A) Representation of determined DTCCSN2 value versus m/z of all identity‐confirmed anthocyanins for positive and negative ions. [M]+ ions are shown as diamonds and [M‐2H]– ions as crosses. (B) Representation of determined DTCCSN2 values for negative ion versus DTCCSN2 values for positive ion for all identity‐confirmed anthocyanins.
Regarding order of elution of anthocyanins with the applied chromatographic conditions, this depended on anthocyanidin identity, linked sugar, and the acylation of the linked sugar. Therefore, it is emphasized that a reliable LC method and knowledge of relative retention characteristics remain as a critical part of the identity confirmation workflow for this application. This is further emphasized upon close examination of some results. For example, compounds 22 and 26 are confirmed as cyanidin pentosides with almost identical DTCCSN2 values. Assessment of previous literature did not allow unambiguous confirmation of the glycoside and linkage in each case according to the sample type. Similarly, compounds 33 and 35 (Cy‐malonylhex (I) and (II)) were both found in blueberry and confirmed using fragment data, but could not be conclusively confirmed in the absence of standards. In this example, these compounds might differ in the position of the malonyl group with the 6“ position being most common, but occurrence of an isomer with the malonyl group in the 3” position has also been postulated by some authors [51, 52]. Alternatively, the sugar may be linked via a different position (e.g., 3 versus 5 position) with the malonyl at 6" position. Finally, it is also theoretically possible that the only the sugar identity differs (i.e., ‐galactoside in addition to ‐glucoside), but the large DTCCSN2 difference (227.6 versus 209.9 Å2) for the isomers indicates that one of the aforementioned positional differences is more likely than a simple variation of the hexoside identity. Finally, two coumarylhexoside compounds were detected in blueberry (compounds 37 and 38), which both yielded two apparent conformers separated in the DTIM cell for the [M]+ species and associated fragment ions. There are several possibilities to consider in explaining the results for these two compounds. First, structural isomerism, such as cis/trans geometry of the coumaryl group, ortho‐ or meta‐ coumaric acid ester attachment to the sugar, or variation of the hexoside identity, but none of these options are considered likely as some degree of RPLC separation can be expected in such cases [53]. Second, a different acylation position or sugar position (as considered most likely for compounds 33 and 35) may be possible, but no literature data are available to support this and RPLC separation should again resolve such a difference. Finally, different stable conformers of the same ion species only detectable via the DTIM separation remains as a possibility. This assessment is supported by the coelution and consistent fragmentation results for the conformers of both examples, but requires further theoretical investigation and assessment of analytical standards to provide further confirmation.
3.4. High‐resolution fragment spectra from data independent acquisition (LC—DAD–IM‐QTOFMS)
Generation of fragment‐level data using a data independent analysis (DIA) workflow is an attractive option for screening of anthocyanins in comparison to data dependent analysis (DDA) as the reconciliation of precursor and fragment ions can be addressed in data processing considering coelution (LC) and alignment in the DTIM dimension. The level of identity confirmation used for the annotated anthocyanins is indicated in Table S2 of the Supporting Information including literature. This table includes four metabolites identity‐confirmed at Level 1, 29 putatively annotated compounds (Level 2), and five putatively characterized compound classes (Level 3). The identity confirmation of the Level 2 compounds was additionally supported by confirmation of fragments, which were acquired by IM‐QTOFMS working at high and low energy alternating frames mode. Fragments allow clarification of ambiguous identity confirmations, for example, to discriminate between anthocyanins with the same formula, such as cyanidin‐O‐glucoside and delphinidin‐O‐rhamnoside [C21H21O11]+, with the additional benefit of IM (arrival time peak) alignment to confirm precursor‐fragment associations.
During the method development, it was observed that low collision energies (10–30 eV) led to insufficient fragmentation of anthocyanins, while at high energy (40–60 eV) the main fragment is the anthocyanidin (aglycone) moiety, with reductions in signal intensity observed at the upper end of this collision energy range. Using the optimized energy (40 eV), identity confirmation of some anthocyanins can nevertheless be supported as the accurate DTCCSN2 values allow a secondary confirmation of the coeluting fragments across low and high energy frames (i.e., via alignment of tA peaks). As can be seen in Table S2 of the Supporting Information, the majority of anthocyanins can be confirmed by detection of the primary aglycone fragment ion in the high energy frame. Other fragment ions (e.g., 213.0546 and 137.0234 for cyanidin) could be detected when high precursor abundances were present, but are much less reliable at low concentrations due to the unfavorably high fragment ion ratios (i.e., minor fragments can be readily expected to fall below LOD). An illustrative example from screening data processing in Skyline is shown in Fig. 4 whereby the aglycone fragments provide the most reliable means for identity confirmation with alignment by coelution and using arrival time peaks. Finally, a complete set of transitions for implementation in Skyline is included in the Supporting Information along with instructions for creation of an LC–IM–MS screening method for the Levels 1 and 2 confirmed anthocyanins reported in this study. This provided dataset can be used for setting up an anthocyanin screening method on any type of IM‐MS instrumentation.
Figure 4.
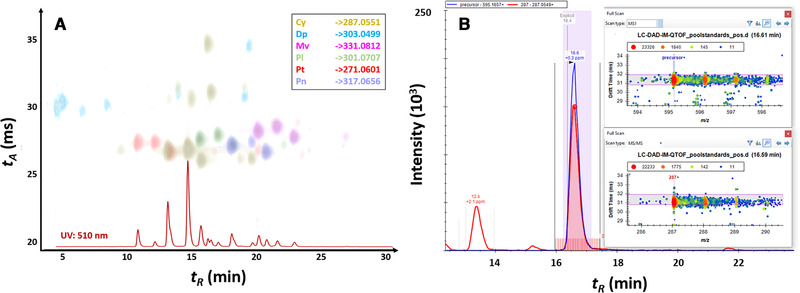
(A) LC–DAD–IM‐QTOFMS results for pooled sample containing five different berry types. Peaks shown correspond to high abundance aglycone fragments in high energy (CE 40 eV) frame with colours shown indicating the six major anthocyanin groups considered in this work. UV/Vis chromatogram (510 nm) is shown for reference. (B) Exemplary results from DIA‐based anthocyanin screening using RT, formula, DTCCSN2 and tA‐aligned precursor and fragment accurate mass results in Skyline (inset).
4. Concluding Remarks
An innovative LC–DAD–IM‐TOFMS method has been developed to identity confirmation of anthocyanins in fruits using a screening approach. Thirty‐eight anthocyanins were identity confirmed in five different berry types using this method. Among them, four were confirmed by standards at Level 1 while all others were supported by literature sources and their characteristic properties (i.e., trend and correlation of DTCCSN2 values, high‐resolution fragment spectra, retention order, and UV/Vis spectra). The berries assessed in this study exhibited different anthocyanin profiles in good correspondence with previous literature. The detailed characterization reported in this work includes multiple identification points for each species including (relative) RT, accurate mass, DTIM‐derived collision cross‐section, and fragment mass spectra. Particularly, the accurate DTCCSN2 data established in this work can be used as a training set to support further IM‐MS work focused on exploratory studies of less common anthocyanins in plant metabolism, as well as applications including determination of adulteration of food, wines, juices, and supplements. The DTCCSN2 dataset can also be valuable for future application of alternative chromatographic separations, such as HILIC and comprehensive two‐dimensional LC for anthocyanin analysis, where elution orders are currently less diagnostic due to limited literature studies performed.
M.M. Delgado‐Povedano thanks to the Ministerio de Educación, Cultura y Deporte (MECD) for an FPU scholarship (FPU14/03068) and the Ministerio de Ciencia Innovacion y Universidades for a mobility aid for a short stay in a foreign center (EST17/00363). Vienna Business Agency and EQ BOKU VIBT GmbH are acknowledged for providing mass spectrometry instrumentation.
The authors declare no conflict of interest.
Supporting information
Supporting information
Supporting information
Delgado‐Povedano MdM, de Villiers A, Hann S, Causon T. Identity confirmation of anthocyanins in berries by LC–DAD–IM‐QTOFMS. Electrophoresis 2020;00:1‐9. 10.1002/elps.202000274
Color online: See article online to view Figs. 1–4 in color.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
5 References
- 1. Panche, A. N. , Diwan, A. D. , Chandra, S.R. , J. Nutr. Sci. 2016, 5, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strack, D. , Wray, V. , in Harborne, J. B. (Ed.), The Flavonoids: Advances in Research Since 1986, Chapman and Hall, London: 1994, pp. 1–22. [Google Scholar]
- 3. Castañeda‐Ovando A., Pacheco‐Hernández Ma. de L., Páez‐Hernández Ma. E., Rodríguez J. A., Galán‐Vidal C. A., Food Chemistry 2009, 113, 859–871. [Google Scholar]
- 4. Willemse, C. M. , Stander, M. A. , de Villiers, A. , J. Chromatogr. A 2013, 1319, 127–140. [DOI] [PubMed] [Google Scholar]
- 5. He, J. , Giusti, M. M. , Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [DOI] [PubMed] [Google Scholar]
- 6. Rojo, L. E. , Ribnicky, D. , Logendra, S. , Poulev, A. , Rojas‐Silva, P. , Kuhn, P. , Dorn, R. , Grace, M. H. , Lila, M. A. , Raskin, I. , Food Chem. 2012, 131, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duarte Martino, H. S. , dos Santos Dias, M. M. , Noratto, G. , Talcott, S. , Mertens‐Talcotta, S. U. , J. Funct. Foods 2016, 23, 432–443. [Google Scholar]
- 8. Norberto, S. , Silva, S. , Meireles, M. , Faria, A. , Pintado, M. , Calhau, C. , J. Funct. Foods 2013, 5, 1518–1528. [Google Scholar]
- 9. Collins, B. , Hoffman, J. , Martinez, K. , Grace, M. , Lila, M. A. , Cockrell, C. , Nadimpalli, A. , Chang, E. , Chuang, C. C. , Zhong, W. , Mackert, J. , Shen, W. , Cooney, P. , Hopkins, R. , McIntosh, M. , J. Nutr. Biochem. 2016, 31, 150–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghasemzadeh, A. , Ghasemzadeh, N. , J. Med. Plants Res. 2011, 5, 6697–6703. [Google Scholar]
- 11. Intuyod, K. , Priprem, A. , Limphirat, W. , Charoensuk, L. , Pinlaor, P. , Pairojkul, C. , Lertrat, K. , Pinlaor, S. , Food Chem. Toxicol. 2014, 74, 206–215. [DOI] [PubMed] [Google Scholar]
- 12. Yun, J. W. , Lee, W. S. , Kim, M. J. , Lu, J. N. , Kang, M. H. , Kim, H. G. , Kim, D. C. , Choi, E. J. , Choi, J. Y. , Kim, H. G. , Lee, Y. K. , Ryu, C. H. , Kim, G. , Choi, Y. H. , Park, O. J. , Shin, S. C. , Food Chem. Toxicol. 2010, 48, 903–909. [DOI] [PubMed] [Google Scholar]
- 13. Valls, J. , Millán, S. , Martí, M. P. , Borràs, E. , Arola, L. , J. Chromatogr. A 2009, 1216, 7143–7172. [DOI] [PubMed] [Google Scholar]
- 14. Ongkowijoyo, P. , Luna‐Vital, D. A. , Gonzalez de Mejia, E. , Food Chem. 2018, 250, 113–126. [DOI] [PubMed] [Google Scholar]
- 15. Ince, A. E. , Sahin, S. , Sumnu, G. , J. Food Sci. Technol. 2014, 51, 2776–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopes‐da‐Silva, F. , de Pascual‐Teresa, S. , Rivas‐Gonzalo, J. C. , Santos‐Buelga, C. , Eur. Food Res. Technol. 2002, 214, 248–253. [Google Scholar]
- 17. de Villiers, A. , Cabooter, D. , Lynen, F. , Desmet, G. , Sandra, P. , J. Chromatogr. A 2009, 1216, 3270–3279. [DOI] [PubMed] [Google Scholar]
- 18. Xu, Y. , Simon, J. E. , Ferruzzi, M. G. , Ho, L. , Pasinetti, G. M. , Wu, Q. , J. Funct. Foods 2012, 4, 710–717. [Google Scholar]
- 19. Díaz‐García, M. C. , Obón, J. M. , Castellar, M. R. , Collado, J. , Alacid, M. , Food Chem. 2013, 138, 938–949. [DOI] [PubMed] [Google Scholar]
- 20. Marston, A. , Hostettmann, K. , in: Andersen, O. M. , Markham, K. R. (Eds.), Flavonoids: Chemistry, Biochemistry and Applications, CRC Press, Florida: 2005, pp. 1–36. [Google Scholar]
- 21. Santos‐Buelga, C. , García‐Viguera, C. , Tomas‐Barberan, F. A. , in: Santos‐Buelga, C. , Williamson, G. (Eds.), Methods in Polyphenol Analysis, The Royal Society of Chemistry, Cambridge: 2003, pp. 92–127. [Google Scholar]
- 22. Berente, B. , García, D. D. , Reichenbächer, M. , Danzer, K. , J. Chromatogr. A 2000, 871, 95–103. [DOI] [PubMed] [Google Scholar]
- 23. A.O.A.C 2005 , Official Methods of Analysis, AOAC Press, Philadelphia: 2005. [Google Scholar]
- 24. Hebrero, E. , Santos‐Buelga, C. , Rivas‐Gonzalo, J. C. , Am. J. Enol. Vitic. 1988, 39, 227–233. [Google Scholar]
- 25. Määttä‐Riihinen, K. R. , Kamal‐Eldin, A. , Mattila, P. H. , Gonzalez‐Paramas, A. M. , Torronen, A. R. , J. Agr. Food Chem. 2004, 52, 4477–4486. [DOI] [PubMed] [Google Scholar]
- 26. Hong, V. , Wrolstad, R. E. , J. Agr. Food Chem. 1990, 38, 708–715. [Google Scholar]
- 27. Abad‐García, B. , Berrueta, L. A. , Garmón‐Lobato, S. , Gallo, B. , Vicente F., J. Chromatogr. A 2009, 1216, 5398–5415. [DOI] [PubMed] [Google Scholar]
- 28. Sun, J. , Lin, L. Z. , Chen, P. , Rapid Commun. Mass Spectrom. 2012, 26, 1123–1133. [DOI] [PubMed] [Google Scholar]
- 29. Ha, T. J. , Lee, M. H. , Park, C. H. , Pae, S. B. , Shim, K. B. , Ko, J. M. , Shin, S. O. , Baek, I. Y. , Park, K. Y. , J. Agr. Food Chem. 2010, 58, 2571–2576. [DOI] [PubMed] [Google Scholar]
- 30. de Rosso, V. V. , Mercadante, A. Z. , J. Agr. Food Chem. 2007, 55, 9135–9141. [DOI] [PubMed] [Google Scholar]
- 31. Chirinos, R. , Campos, D. , Betalleluz, I. , Giusti, M. M. , Schwartz, S. J. , Tian, Q. , Pedreschi, R. , Larondelle, Y. , J. Agr. Food Chem. 2006, 54, 7089–7097. [DOI] [PubMed] [Google Scholar]
- 32. Hong, H. T. , Netzel, M. E. , O'Hare, T. J. , Food Chem. 2020, 319, 126515. [DOI] [PubMed] [Google Scholar]
- 33. Oh, Y. S. , Lee, J. H. , Yoon, S. H. , Oh, C. H. , Choi, D. S. , Choe, E. , Jung, M. Y. , J. Food Sci. 2008, 73, C378–C389. [DOI] [PubMed] [Google Scholar]
- 34. Fanali, C. , Dugo, L. , D'Orazio, G. , Lirangi, M. , Dachà, M. , Dugo, P. , Mondello, L. , J. Sep. Sci. 2011, 34, 150–159. [DOI] [PubMed] [Google Scholar]
- 35. Inutan, E. D. , Wager‐Miller, J. , Narayan, S. B. , Mackie, K. , Trimpin, S. , Int. J. Ion Mobil. Spectrom. 2013, 16, 145–159. [Google Scholar]
- 36. Lapthorn, C. , Pullen, F. , Chowdhry, B. Z. , Mass Spectrom. Rev. 2013, 32, 43–71. [DOI] [PubMed] [Google Scholar]
- 37. Liu, W. , Zhang, X. , Siems, W. F. , Hill Jr., H. H. , Yin, D. , Food Chem. 2015, 177, 225–232. [DOI] [PubMed] [Google Scholar]
- 38. Stow S. M., Causon T. J., Zheng X., Kurulugama R. T., Mairinger T., May J. C., Rennie E. E., Baker E. S., Smith R. D., McLean J. A., Hann S., Fjeldsted J. C., Anal. Chem. 2017, 89, (17), 9048–9055.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Canuto, G. A. B. , Oliveira, D. R. , da Conceição, L. S. M. , Farah, J. P. S. , Tavares, M. F. M. , Food Chem. 2016, 192, 566–574. [DOI] [PubMed] [Google Scholar]
- 40. MacLean, B.X. , Pratt, B. S. , Egertson, J. D. , MacCoss, M. J. , Smith, R. D. , Baker, E.S. , J. Am. Soc. Mass Spectrom. 2018, 29, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khoo, H. E. , Azlan, A. , Tang, S. T. , Lim, S. M. , Food Nutr. Res. 2017, 61, 1361779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun J., Lin, L. , Chen, P. , Rapid Commun. Mass Spectrom. 2012, 26, 1123–1133. [DOI] [PubMed] [Google Scholar]
- 43. de Villiers, A. , Venter, P. , Pasch, H. , J. Chromatogr. A 2016, 1430, 16–78. [DOI] [PubMed] [Google Scholar]
- 44. Causon, T. , Ivanova‐Petropulos, V. , Petrusheva, D. , Bogeva, E. , Hann, S. , Anal. Chim. Acta 2019, 1052, 179–189. [DOI] [PubMed] [Google Scholar]
- 45. May, J. C. , Goodwin, C. R. , Lareau, N. M. , Leaptrot, K. L. , Morris, C. B. , Kurulugama, R. T. , Mordehai, A. , Klein, C. , Barry, W. , Darland, E. , Overney, G. , Imatani, K. , Stafford, G. C. , Fjeldsted, J. C. , McLean, J. A. , Anal. Chem. 2014, 86, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Merken, H. M. , Beecher, G. R. , J. Agric. Food Chem. 2000, 48, 577–599. [DOI] [PubMed] [Google Scholar]
- 47. Sánchez‐Ilárduya, M. B. , Sánchez‐Fernández, C. , Viloria‐Bernal M., López‐Márquez, D. M. , Berrueta, L. A. , Gallo, B. , Vicente, F. , Aust. J. Grape Wine Res. 2012, 18, 203–214. [Google Scholar]
- 48. White, B. L. , Howard, L. R. , Prior, R. L. , J. Agr. Food Chem. 2010, 58, 4030–4036. [DOI] [PubMed] [Google Scholar]
- 49. Lee, J. , Finn, C. E. , J. Funct. Foods 2012, 4, 213–218. [Google Scholar]
- 50. Nichols, C. M. , Dodds, J. N. , Rose, B. S. , Picache, J. A. , Morris, C. B. , Codreanu, S. G. , May, J. C. , Sherrod, S. D. , McLean, J. A. , Anal. Chem. 2018, 90, 14484–14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Donner, H. , Gao., L. , Mazza, G. , Food Res. Int. 1997, 30, 637–643. [Google Scholar]
- 52. Petersson, E.V. , Puerta, A. , Bergquist, J. , Turner, C. , Electrophoresis 2008, 29, 2723–2730. [DOI] [PubMed] [Google Scholar]
- 53. Sigurdson, G.T. , Tang, P. , Giuisti, M.M. , Molecules 2018, 23, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fang J., Nutrition 2015, 31, 1301–1306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.