

Abstract
Injury induces retinal Müller glia of certain cold-blooded vertebrates, but not those of mammals, to regenerate neurons. To identify gene regulatory networks that reprogram Müller glia into progenitor cells, we profiled changes in gene expression and chromatin accessibility in Müller glia from zebrafish, chick, and mice in response to different stimuli. We identified evolutionarily conserved and species-specific gene networks controlling glial quiescence, reactivity, and neurogenesis. In zebrafish and chick, the transition from quiescence to reactivity is essential for retinal regeneration, whereas in mice, a dedicated network suppresses neurogenic competence and restores quiescence. Disruption of nuclear factor I transcription factors, which maintain and restore quiescence, induces Müller glia to proliferate and generate neurons in adult mice after injury. These findings may aid in designing therapies to restore retinal neurons lost to degenerative diseases.
Graphical Abstract
INTRODUCTION:
The ability to regenerate retinal neurons after injury varies drastically among vertebrate species. Teleost fish such as zebrafish can regenerate all major retinal cell types after injury by reprogramming Müller glia to a progenitor-like state. In the post-hatch chick, Müller glia can generate small numbers of neurons after injury but lose regenerative ability later in life. In contrast, mammalian Müller glia do not spontaneously regenerate lost retinal neurons. Although some genes that promote retinal regeneration have been identified, the core gene regulatory networks controlling Müller glia reprogramming remain largely unknown but can be identified through cross-species transcriptomic and epigenomic analysis.
RATIONALE:
To identify injury-induced changes in Müller glia, we performed bulk RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) to separately profile gene expression and chromatin accessibility in both mouse and zebrafish. These assays included multiple time points following N-methyl-d-aspartate (NMDA) and light treatments, which damage inner retinal neurons and photoreceptors, respectively. We also conducted single-cell RNA-seq (scRNA-seq) to identify changes in gene expression after NMDA treatment and light damage, as well as after treatment with exogenous factors that induced injury-independent reprogramming, in mouse, zebrafish, and chick retinas. We then developed a computational tool, which we call integrated regulatory network analysis (IReNA), to integrate gene expression and chromatin accessibility in order to reconstruct regulatory networks of Müller glia in response to diverse stimuli. Finally, using loss-of-function approaches, we validated functions of candidate factors controlling Müller glia reprogramming.
RESULTS:
We generated 100 RNA-seq and 40 ATAC-seq samples from zebrafish and mice, and obtained 105,666, 85,051, and 77,924 single retinal cells from zebrafish, chick, and mice, respectively. In all three species, Müller glia acquired a reactive state after treatments. In chick and zebrafish, Müller glia passed through this reactive state before becoming proliferative and neurogenic. In mice, however, Müller glia reverted to a resting state after injury. By integrating these datasets, we identified changes in gene expression and chromatin accessibility after each treatment. Cross-species analysis identified evolutionarily conserved and species-specific gene regulatory networks that control the transition of the quiescent, reactive, and proliferative Müller glia after stimulation. In mice, a dedicated network restored Müller glia to a quiescent state. In contrast, in zebrafish and chick, genes selectively expressed in reactive Müller glia promoted the transition to a proliferative and neurogenic progenitor state. Loss of function of genes selectively expressed in reactive Müller glia, such as hmga1 and yap1, inhibited Müller glia reprogramming in zebrafish. In chick, pharmacological disruption of fatty acid–binding protein 5, 7, and 8 (FABP5/7/8) activity inhibited injury-induced transition from quiescence to neurogenic competence. Finally, deletion of nuclear factor I factors a, b, and x (Nfia/b/x), which maintain and restore a glial quiescent state, resulted in Müller glia reprogramming into retinal bipolar and amacrine interneurons in adult mice after injury.
CONCLUSION:
We found that transition from quiescence through the reactive state is essential for Müller glia reprogramming in regeneration-competent species such as zebrafish and chick. Furthermore, proliferative and neurogenic competence are both suppressed by a dedicated gene regulatory network in mouse Müller glia. Cross-species RNA-seq and ATAC-seq data provide a comprehensive resource to study cellular responses to injury and Müller glia reprogramming. Our findings indicate that treatments targeting gene regulatory networks that repress neurogenic competence may help to facilitate reprogramming of mammalian Müller glia to neurons.
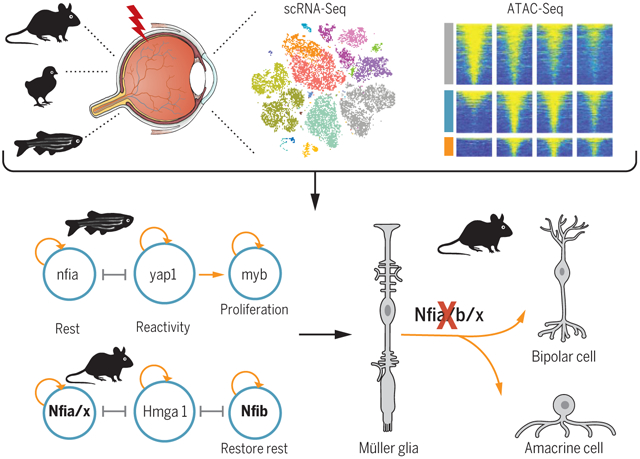
Control of retinal Müller glia reprogramming. RNA-seq and ATAC-seq were performed on Müller glia from zebrafish, chick, and mouse after different treatments. Integrative transcriptomic and epigenomic analysis revealed core regulatory networks controlling retinal regeneration. Loss of function of zebrafish genes expressed in reactive Müller glia blocked reprogramming, while in mouse Müller glia, disruption of Nfia/b/x, which maintain and restore quiescence, resulted in acquisition of neurogenic competence.
Loss of retinal neurons is a leading cause of blindness. Whereas mammals cannot regenerate lost retinal neurons, adult zebrafish regenerate all retinal neurons through reprograming Müller glia to a neurogenic progenitor-like state. In contrast, post-hatch chick Müller glia proliferate in response to injury, retain limited neurogenic competence, and produce small numbers of inner retinal neurons (1). In zebrafish, several injury-induced Müller glia genes that regulate neurogenic competence, such as ascl1a (2), promote Müller glia reprogramming. In mice, forced expression of Ascl1 in Müller glia can reprogram these cells into functional inner retinal neurons (3). Conversely, inhibiting the Hippo pathway stimulates Müller glia proliferation in mice after injury but does not induce neurogenic competence (4, 5).
To identify the genetic and epigenetic barriers to Müller glia proliferation and neurogenesis, we compared the molecular responses to retinal injury in multiple species with different regenerative abilities. We reconstructed evolutionarily conserved and species-specific gene regulatory networks controlling glial quiescence, reactivity, proliferation, and neurogenic competence. Our findings identify similarities and differences in the responses of Müller glia to injury in these species and reveal mechanisms that suppress neurogenic competence in mammalian Müller glia.
RNA-seq analysis of injury-regulated genes in Müller glia
To identify core gene regulatory networks that control Müller glia reprogramming in mouse and zebrafish, we examined two injury paradigms: light damage (outer retinal injury) and N-methyl-d-aspartate (NMDA) treatment (inner retinal injury) (1). We generated 100 bulk RNA sequencing (RNA-seq) libraries and 40 assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) libraries from fluorescence-activated cell sorting (FACS)–isolated Müller glia at multiple time points after injury (fig. S1, A to C, and table S1). Known Müller glia–specific genes were enriched in green fluorescent protein–positive (GFP+) samples, while GFP− samples were enriched for genes specific to other retinal cell types (fig. S1D and table S2). Principal component analysis of RNA-seq data revealed species-specific patterns of transcriptional changes after NMDA treatment and light damage. Transcriptional changes in mouse Müller glia occurred rapidly, within 3 hours after injury, but then reverted to a rest-like state (Fig. 1A). However, both NMDA treatment and light damage in zebrafish induced Müller glia to progressively diverge from the resting state (Fig. 1B).
Fig. 1. RNA-seq analysis of mouse and zebrafish Müller glia.
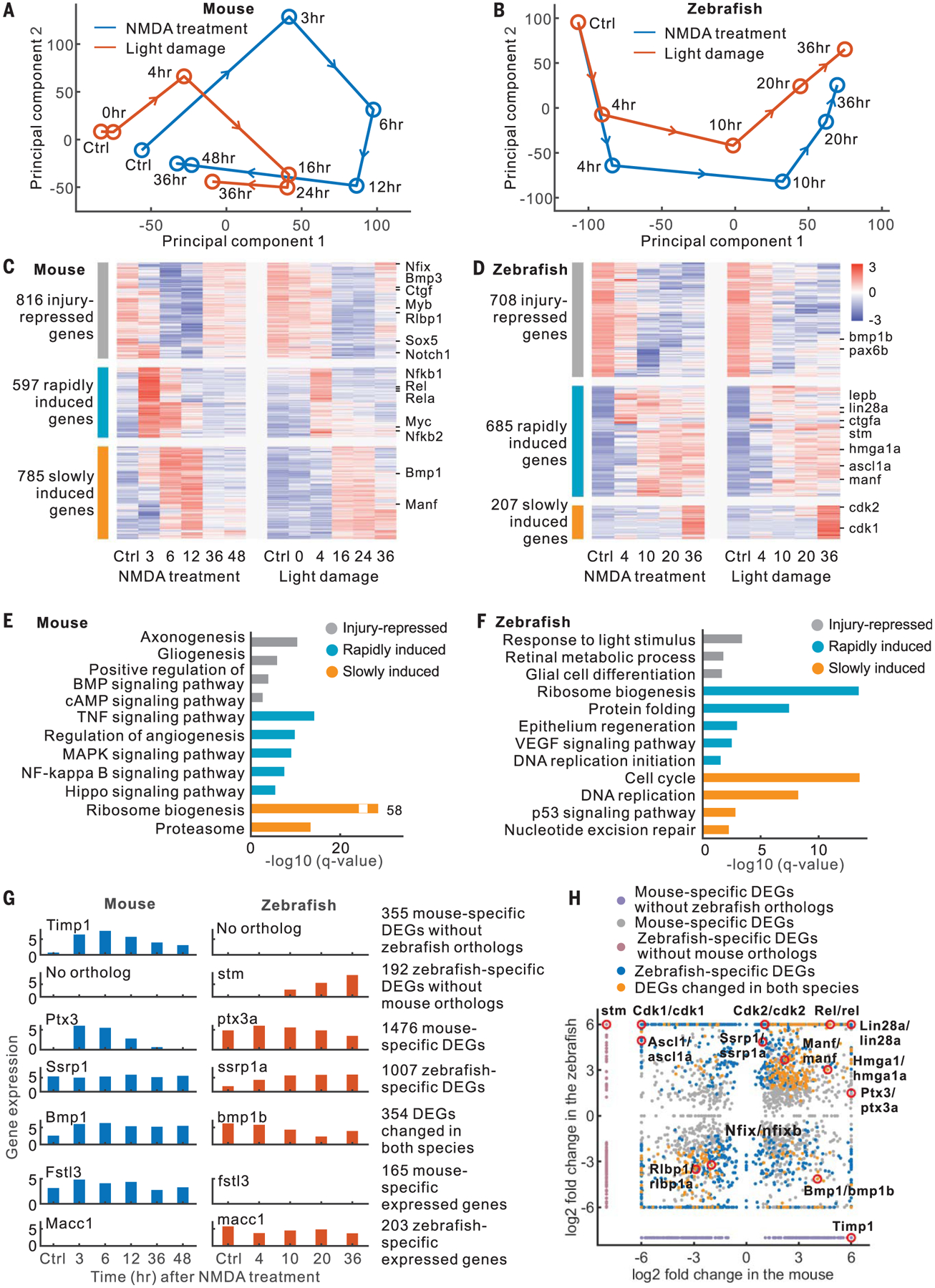
(A and B) Principal component analysis of RNA-seq data. (C and D) Expression profiles of DEGs. (E and F) Enriched functions of DEGs. BMP, bone morphogenetic protein; cAMP, cyclic adenosine monophosphate; VEGF, vascular endothelial growth factor. (G) Examples of DEGs and species-specific expressed genes. (H) Comparison of DEGs between mouse and zebrafish.
We found 2198 differentially expressed genes (DEGs) in mice and 1600 DEGs in zebrafish that were common between NMDA treatment and light damage (expression correlation > 0.5, fig. S1E). These DEGs showed three groups of changes in both zebrafish and mouse. The first group was highly expressed in resting Müller glia and repressed after injury (Fig. 1, C and D). The remaining two groups exhibited rapid and slow injury-dependent induction, respectively. Gene Ontology analysis showed species-specific differences in DEGs. For instance, rapidly induced mouse Müller glia genes included components of the tumor necrosis factor (TNF), mitogen-activated protein kinase (MAPK), nuclear factor κB (NFκB), and Hippo pathways (Fig. 1E), whereas in zebrafish, they were enriched for ribosome biogenesis (Fig. 1F). Slowly induced mouse genes were enriched for ribosome biogenesis, whereas in zebrafish, they were enriched for cell cycle-related functions, reflecting the fact that zebrafish Müller glia reenter the cell cycle at 25 hours after light damage (6).
To identify species-specific genes that changed in Müller glia, including candidate regulators of neurogenic competence, we compared zebrafish and mouse DEGs and classified them into three categories (Fig. 1, G and H, and table S3). These include 547 DEGs lacking orthologs in the other species, 2483 DEGs with orthologs in both species but with species-specific changes, and 354 DEGs with different patterns of injury-induced changes in two species. In addition, we identified 368 genes that are not differentially expressed after injury but show significant differences in baseline expression between mouse and zebrafish Müller glia (Fig. 1G and table S3).
ScRNA-seq analysis of zebrafish, chick, and mouse retinas
To comprehensively identify genes regulating Müller glia reprogramming, we conducted single-cell RNA sequencing (scRNA-seq) of mouse, zebrafish, and chick retinas after NMDA treatment or light damage. We also treated retinas with exogenous factors that induce reprogramming independent of injury, such as TNFα and a gamma-secretase inhibitor (T+R) in zebrafish (7), and FGF2 and insulin (growth factors) in chick (8). In mice, we tested FGF2 and insulin combined with NMDA treatment, which induces limited proliferation but not neurogenic competence (9). We profiled 77,924 cells in mice, 85,051 cells in chick, and 105,666 cells in zebrafish (fig. S2, A and B, and table S1).
Our scRNA-seq analysis separated cells into different clusters (Fig. 2, A to D, and figs. S2 and S3) that we annotated using known retinal cell type markers (table S4). We identified all major retinal cell types in each species, including a subset of zebrafish rod-restricted progenitor cells that expressed nr2e3 and neurod1 (Fig. 2D and fig. S2, F and G). Rod progenitors had 101 and 195 DEGs after NMDA treatment and light damage, respectively, 97 and 92% of which were also changed in Müller glia after injury (fig. S3D). We also identified resting and activated Müller glia in all three species after each treatment. Müller glia were overrepresented relative to their abundance in intact retina, accounting for 25.8, 38.2, and 16.7% of retinal cells in zebrafish, chick, and mouse, respectively, likely reflecting bias in cell survival and capture efficiency (10).
Fig. 2. ScRNA-seq analysis of mouse, chick, and zebrafish retinas.
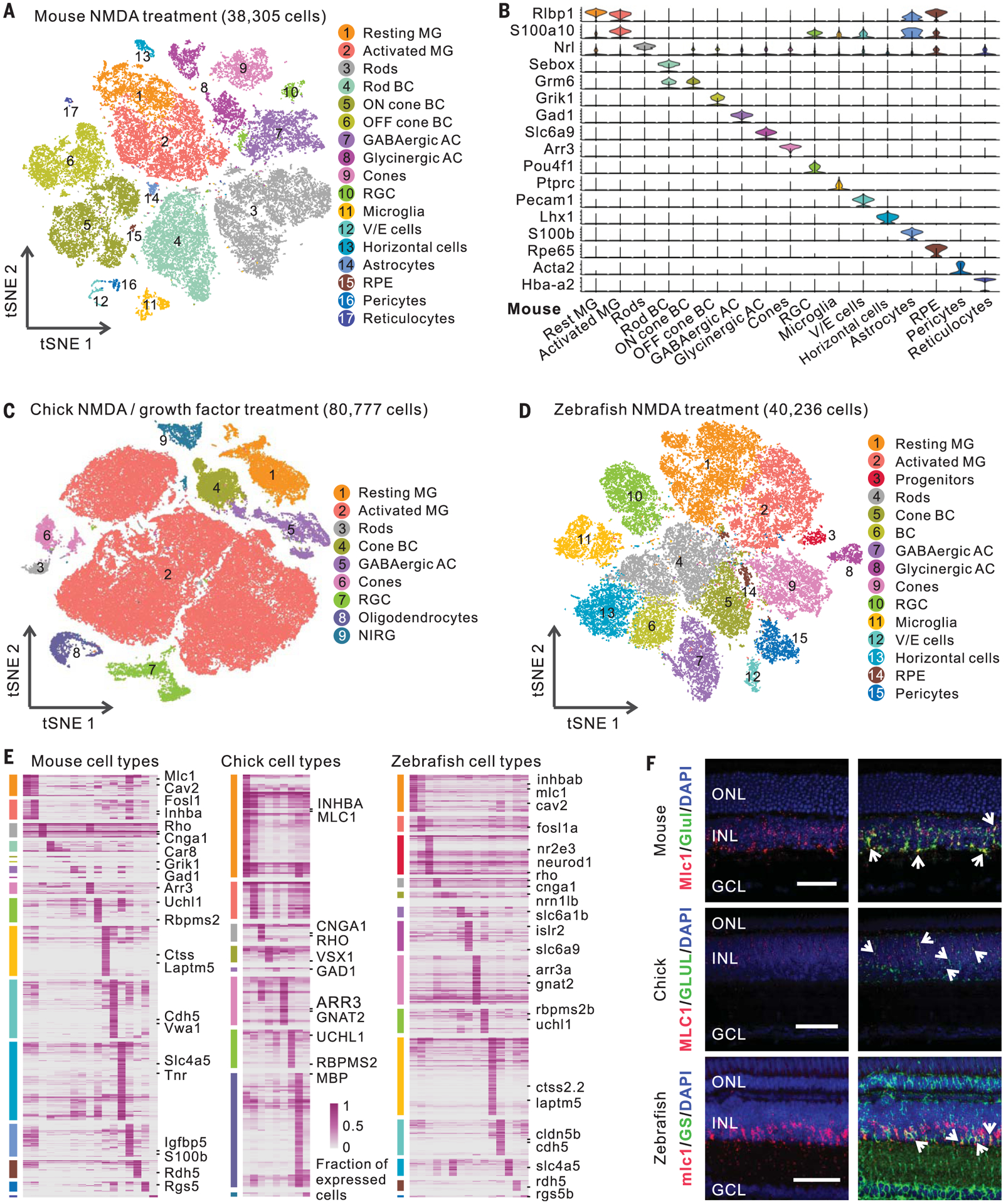
(A) Clustering of mouse retinal cells after NMDA treatment. (B) Expression of mouse retinal cell markers. (C and D) Clustering of chick and zebrafish retinal cells after NMDA or GF treatment. (E) Cell type–specific marker genes. Each column represents an individual cell type. (F) smFISH showing Mlc1/MLC1/mlc1 expression in zebrafish, chick, and mouse Müller glia (MG). Immunostaining of glutamine synthetase (GS, the coding gene glula/b) was used in zebrafish. Arrows indicate colocalized signals. Scale bars, 50 μm. BC, bipolar cells; GABAergic, γ-aminobutyric acid–releasing; AC, amacrine cells; RGC, retinal ganglion cells; V/E cells, vascular/endothelial cells; RPE, retinal pigment epithelium; NIRG, nonastrocytic inner retinal glial cells; DAPI, 4′,6-diamidino-2-phenylindole; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Finally, we compared transcriptomes and identified evolutionarily conserved and species-specific marker genes for each retinal cell type in all three species (Fig. 2E and table S5). We confirmed that Mlc1/MLC1/mlc1 and Inhba/INHBA/inhbab are selectively expressed in Müller glia of all three species using single molecule fluorescence in situ hybridization (smFISH) and that Cav2/cav2 are selectively expressed in mouse and zebrafish Müller glia (Fig. 2F and fig. S3E).
Trajectory analysis of Müller glia reprogramming
We conducted trajectory analysis of Müller glia after stimuli using scRNA-seq data. Consistent with bulk RNA-seq data (Fig. 1A), mouse Müller glia showed rapid changes in gene expression after NMDA treatment and light damage, then a gradual reversion toward a resting state (Fig. 3, A and B, and fig. S4). In contrast, zebrafish Müller glia showed progressive injury-induced transcriptional changes (Fig. 3C and fig. S5, A and B). T+R treatment produced changes in gene expression resembling both forms of injury (fig. S5, C and D). In zebrafish, we observed a side branch after both injury and T+R treatment (Fig. 3C and fig. S5, C and D), which was enriched for genes associated with the reactive state, such as manf, while the major branch showed increased expression of neurogenesis- and proliferation-related genes, such as ascl1a and pcna (fig. S5A). RNA velocity analysis (11) indicated that, after NMDA treatment, Müller glia transition from a resting state to a reactive state and then either proceed to a proliferative state or revert to a resting state (Fig. 3C). A similar response was observed in chick after NMDA or FGF2+insulin treatment (Fig. 3D and fig. S5, E to G). Furthermore, we calculated the dissimilarity of gene expression profiles in Müller glia after injury across species. We observed that both resting and activated Müller glia from mice and zebrafish were similar, as were proliferative Müller glia from zebrafish and chick (fig. S6, A and B). These results indicate that chick and zebrafish Müller glia undergo reprogramming by passing through a reactive state before reprogramming.
Fig. 3. Trajectory analysis of mouse, chick, and zebrafish Müller glia.
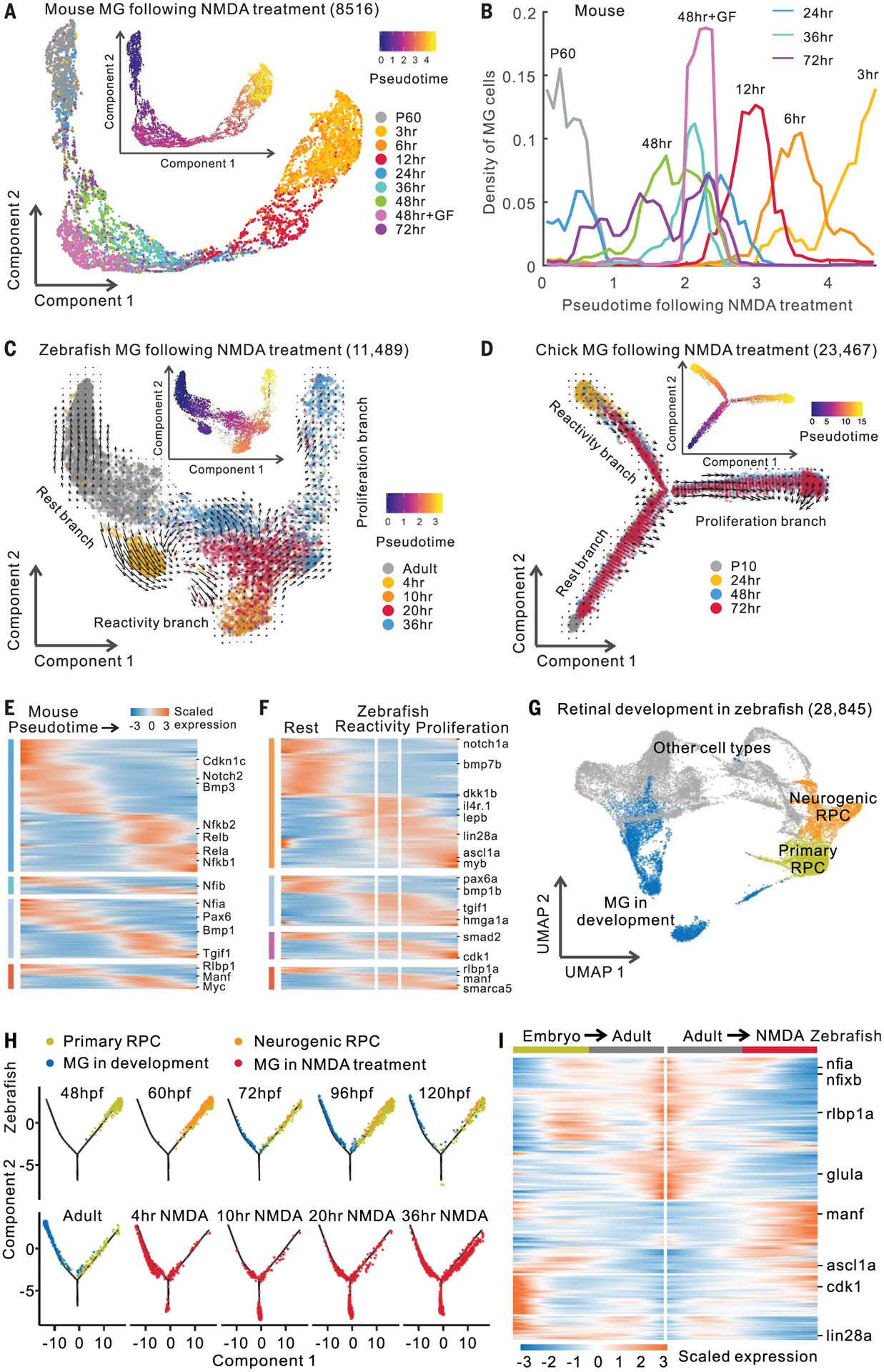
(A) Trajectory of mouse MG after NMDA treatment. Inserted plot shows pseudotime of MG. P60, postnatal day 60; GF, growth factor (FGF2+insulin) treatment. (B) Quantification of mouse MG across the pseudotime. (C and D) Trajectory of zebrafish and chick MG after NMDA treatment. Arrows indicate the direction of MG transitions identified through RNA velocity analysis. (E and F) Expression of DEGs in mouse and zebrafish. Color bars on the left indicate categories of conserved and species-specific DEGs in fig. S6D. (G) Clustering of retinal cells during zebrafish development. (H) Trajectory of RPCs and MG from zebrafish development and NMDA treatment. Hours post-fertilization (hpf) are shown. (I) Pseudotemporal expression of DEGs shared by developing and NMDA-treated MG. Columns represent cells in pseudotime. Rows show genes changed in expression during MG development (embryo to adult) and NMDA treatment (adult to NMDA treatment).
ScRNA-seq analysis indicated that 67.7% of the zebrafish DEGs and 48.4% of the mouse DEGs overlapped between light damage and NMDA treatment (fig. S6C). T+R treatment resembled both injury models in zebrafish, as did NMDA and FGF2+insulin treatment in chick. We identified evolutionarily conserved DEGs, including Rlbp1/RLBP1/rlbp1a, and species-specific DEGs, such as Rela/b in mouse and lin28a in zebrafish (Fig. 3, E and F; fig. S6, D to F; and table S6). Gene Ontology analysis identified functions enriched in both conserved and species-specific DEGs (fig. S6G).
Comparing zebrafish Müller glia reprogramming and development
To determine whether Müller glia–derived progenitors transcriptionally resemble progenitors observed during normal retinal development, we compared scRNA-seq data from developing zebrafish retinas with reprogrammed Müller glia. As in our previous work in mouse (10), we found two transcriptionally distinct retinal progenitor cells (RPCs) in the developing zebrafish retina: primary and neurogenic RPCs (Fig. 3G and fig. S7, A to C). We combined all RPCs and Müller glia and performed trajectory analysis. NMDA treatment, light damage, and T+R treatment all induced Müller glia to a state resembling multipotent RPCs but also induced a separate branch consisting of reactive Müller glia (Fig. 3H and figs. S7, D to G, and S8). Significant overlap was observed between genes differentially expressed during development and after reprogramming (figs. S7D and S8, A and F). For most over-lapping genes (83.8%), there was a negative correlation between development and NMDA treatment, with decreasing developmental genes, such as ascl1a, increasing in expression in reprogrammed Müller glia and vice versa (Fig. 3I). These results imply that, after first passing through a reactive state, Müller glia reprogram by reversing the temporal pattern of gene expression associated with gliogenesis.
Gene regulatory networks controlling Müller glia reprogramming
To profile changes in chromatin accessibility in Müller glia, we performed ATAC-seq analysis after injury in zebrafish and mouse (fig. S9A). We identified differentially accessible chromatin regions (DARs) from NMDA-treated and light-damaged samples, of which 96% in mice and 90% in zebrafish are from promoter, intronic, and intergenic regions (fig. S9, B to D). Changes in chromatin accessibility correlated with changes in gene expression (Fig. 4, A and B, and fig. S9, E and F). We also observed species-specific changes in chromatin accessibility after injury (Fig. 4, A and B, and table S7). For instance, the ascl1a promoter became more accessible after injury in zebrafish but not in mice (fig. S10A). Likewise, we observed a mouse-specific increase in the promoter accessibility of genes that were induced after injury in mice but not zebrafish, such as Rel (fig. S10A).
Fig. 4. ATAC-seq analysis and gene regulatory networks in Müller glia.
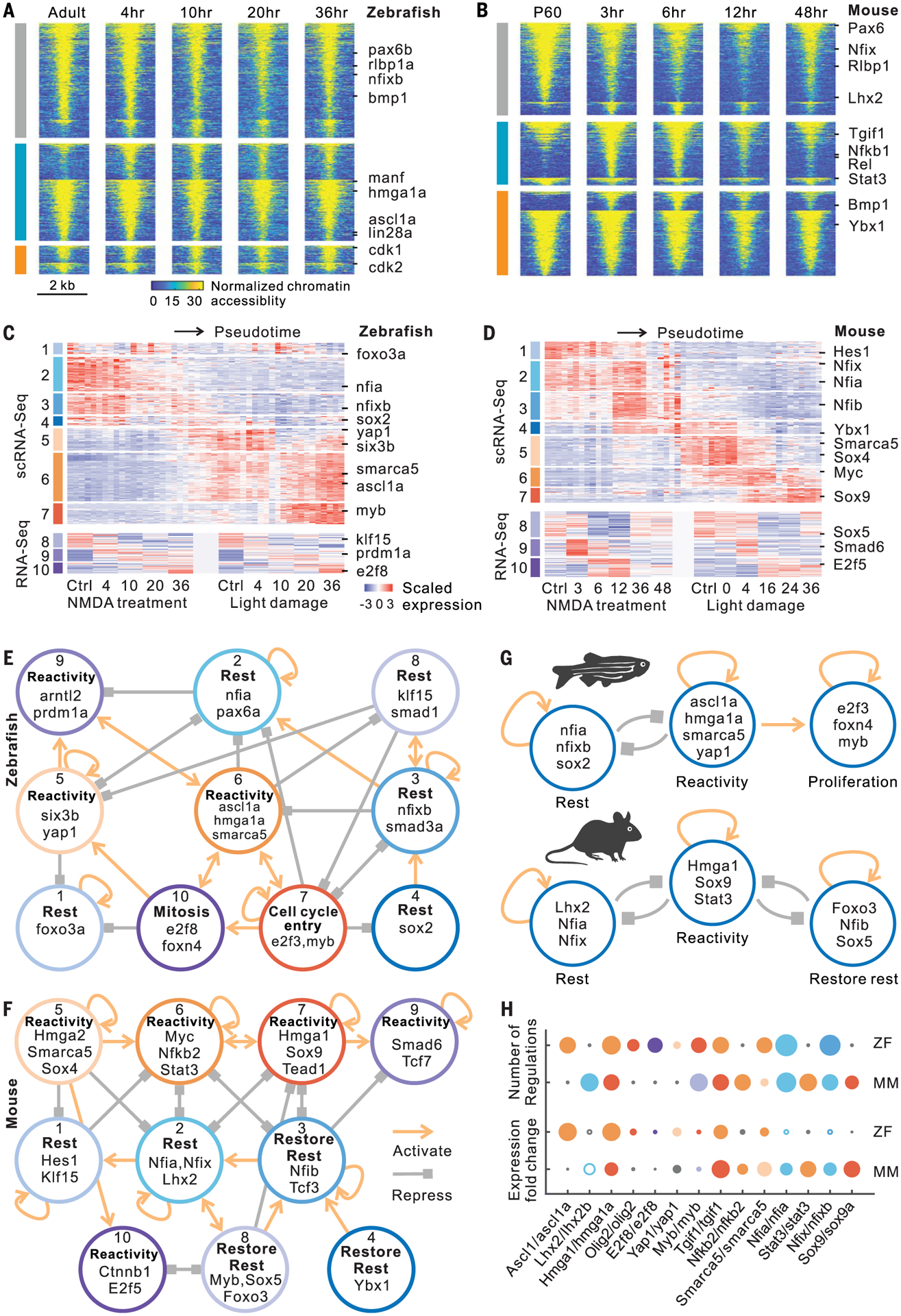
(A and B) Changes in chromatin accessibility of injury-repressed (gray), rapidly induced (cyan), and slowly induced (orange) genes after NMDA treatment in zebrafish and mouse. Lines indicate differentially accessible regions, which are separated by their positive and negative correlations with gene expression in each group. (C and D) Candidate genes including transcription factors were divided into 10 modules in zebrafish and mouse. ScRNA-seq expression profiles contain 50-bin divisions of MG from all stimuli studied. (E and F) Intramodular regulatory networks in zebrafish and mouse. Colored circles represent modules. Connections indicate statistically significant regulations among modules. (G) Regulatory models in zebrafish and mouse. (H) Comparison of gene features between zebrafish (ZF) and mouse (MM). Colors represent modules. Small black circle indicates that the gene is absent from the network in the indicated species. Circle size represents either fold change of gene expression or the number of gene regulations. Hollow circle indicates decreased expression after treatment.
To reconstruct gene regulatory networks controlling Müller glia reprogramming by integrating gene expression and chromatin accessibility data, we developed a method called integrated regulatory network analysis (IReNA; fig. S10B and see the Materials and methods section). We integrated bulk and single-cell RNA-seq data from zebrafish and mouse to obtain 10 modules of DEGs and Müller glia-expressed transcription factors (TFs) (Fig. 4, C and D; fig. S10, C and D; and table S8). In parallel, we analyzed ATAC-seq data to identify differentially accessible footprints that were used to infer regulatory relationships between TFs and DEGs. We constructed an integrated gene regulatory network for each module and generated an intramodular regulatory network for both zebrafish and mouse (Fig. 4, E and F; figs. S11 and S12; and table S9).
In zebrafish, we observed multiple active modules in resting Müller glia (rest-associated modules 1, 2, 3, 4, and 8), which are predicted to self-activate, promote expression of genes in other resting modules, and repress expression of genes in reactivity-associated modules (Fig. 4E, fig. S11, and table S10). TFs in these reactivity-associated modules (modules 5, 6, and 9) include ascl1a, tgif1, and six3b, which are necessary for zebrafish Müller glia reprogramming (2, 12). TFs in the reactivity network likewise activate their own expression and repress genes in the rest-associated network. Downstream of the reactivity network lies a third set of modules (modules 7 and 10), which regulate genes associated with cell cycle progression. These include TFs known to regulate cell cycle entry, such as myca and e2f3, and neurogenesis, such as olig2 and foxn4 (Fig. 4E and fig. S11) (13–15). In zebrafish, the transition of Müller glia to a reactive state triggers either return to quiescence or acquisition of neurogenic and proliferative competence and full conversion to neurogenic progenitors (Fig. 4G).
In mice, we likewise observed three inter-connected networks of modules (Fig. 4F, fig. S12, and table S10). As in zebrafish, the first network consists of TFs active in resting Müller glia (modules 1 and 2) (Fig. 4F). These include Lhx2, which represses reactive gliosis in mouse Müller glia (16), and Notch pathway genes such as Hes1. This rest-associated network is predicted to repress genes in a separate reactivity-associated network (modules 5, 6, 7, 9, and 10). TFs in the reactivity-associated network in turn are predicted to repress the rest-associated network, establishing a bistable regulatory relationship like that in zebrafish (Fig. 4, E and F). TFs in the reactivity-associated network include the NFκB pathway components (Rela/b and Nfkb1/2), which were also recently shown to repress Müller glia reprogramming in chick (17), as well as TFs that promote glial activation (Stat3 and Sox9) (18, 19) and the Hippo pathway target Tead1. Reversion to a resting state in mouse Müller glia is driven by a third network (modules 3, 4, and 8), which was not found in zebrafish (Fig. 4, E and F). This network is enriched for genes expressed in resting Müller glia, such as Nfib and Sox5, which are down-regulated immediately after injury but up-regulated at later stages (Fig. 4D). These modules are distinct from those predicted to maintain Müller glia in a resting state and may restore quiescence. The network analysis implies that the transition between rest, reactivity, and restoration of the resting state in mice is mediated by three separate bistable, autoactivating transcriptional regulatory networks (Fig. 4G).
Validation of genes regulating Müller glia reprogramming
To select candidates for functional analysis, we examined the degree of connectivity of each gene within regulatory networks and the fold change in gene expression during Müller glia reprogramming (Fig. 4H and table S11). We tested whether TFs predicted to regulate reactivity were required for Müller glia reprogramming in zebrafish. The high mobility group AT-hook 1 gene hmga1a/Hmga1 was induced in reactive Müller glia in zebrafish and mouse (Fig. 4, G and H, and fig. S13A). Zebrafish hmga1a morphants showed fewer proliferating Müller glia than did controls (Fig. 5, A and C, and fig. S13, B and C). We next tested yap1, which was induced in the reactive state (Fig. 4, G and H, and fig. S13A) and is required for Müller glia to exit quiescence in both Xenopus and mice (4, 5). Yap1 morphants showed fewer proliferating Müller glia (Fig. 5, A and C). Injection of verteporfin, which inhibits the interaction of Yap1 and TEAD (20), also reduced proliferating Müller glia (Fig. 5, B and C). Morphants of both smarca5, a SWI/SNF-related chromatin regulator expressed in reactive Müller glia, and myb, which is expressed in proliferating progenitors, showed less Müller glia proliferation (fig. S13, D and E). All treatments that reduced Müller glia proliferation also inhibited photoreceptor regeneration, with significant reductions seen in rods and both red and blue cones (Fig. 5, D to F, and fig. S13, F to I). Additionally, scRNA-seq analysis of both hmga1a morphant and verteporfin-treated retinas showed fewer proliferating Müller glia and a corresponding increase in reactive Müller glia (Fig. 5, G and H, and figs. S13, J and K, and S14, A and B).
Fig. 5. Validation of genes regulating Müller glia reprogramming in zebrafish and chick.
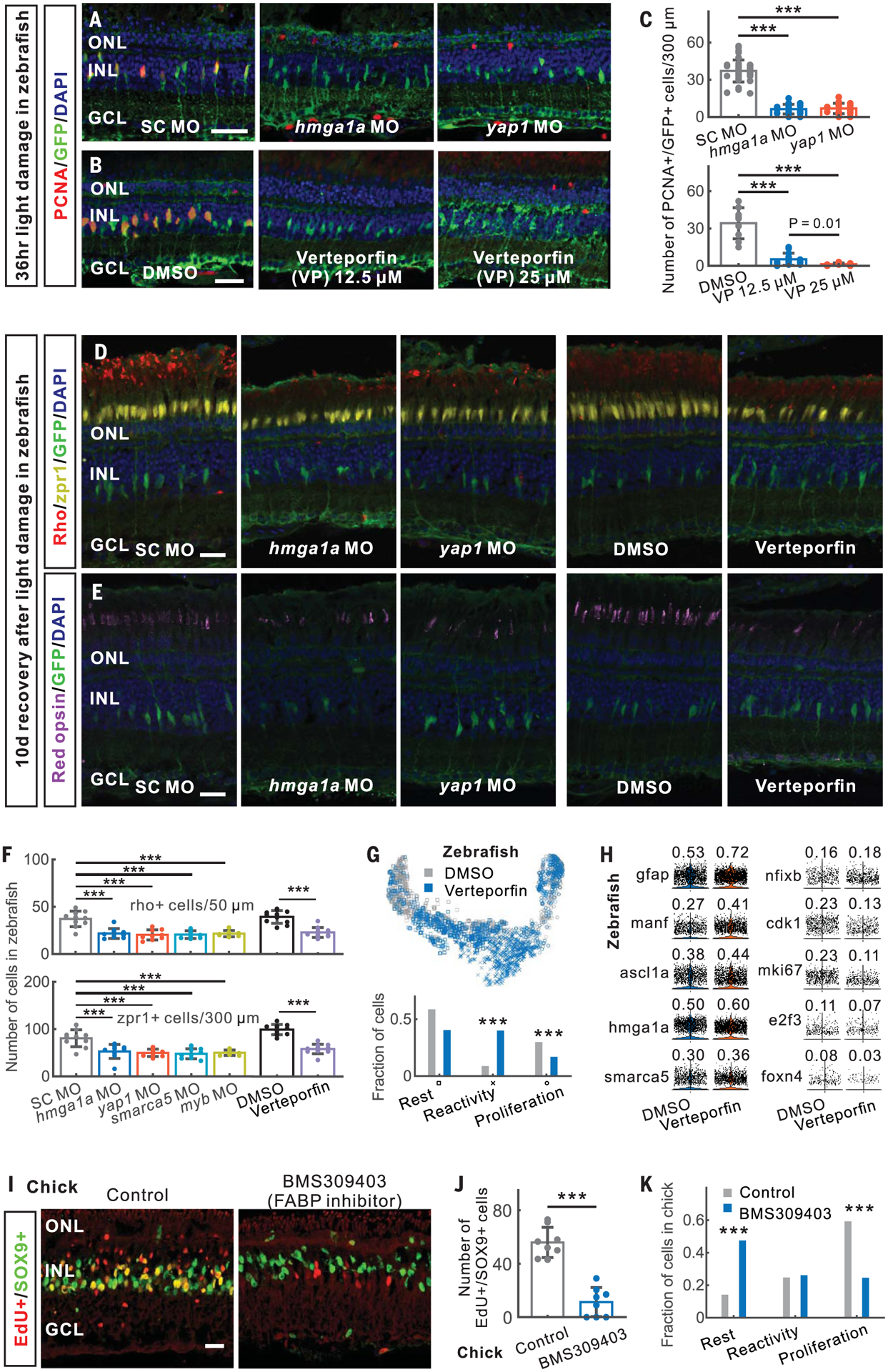
(A to F) In zebrafish, morpholino (MO)–mediated and verteporfin-treated knockdown of hmga1a and yap1 inhibited MG proliferation [(A) to (C)] and neurogenesis [(D) to (F)] after light damage. (G) Distribution of DMSO- and verteporfin-treated MG in the trajectory. Bottom panel shows the fraction of DMSO- and verteporfin-treated MG in three branches. (H) Expression of verteporfin-induced genes in MG. The fraction of expressed cells is shown. (I and J) Inhibition of FABP5/7/8 using BMS309403 reduced progenitor formation at 48 hours after NMDA injury in chick. (K) Distribution of three states of MG in scRNA-seq of control and BMS309403-treated chick retinas at 48 hours after NMDA injury. SC, standard control. Scale bars, 20 μm. Error bars indicate standard deviation. ***P < 0.001.
We identified both evolutionarily conserved genes and species-specific genes up-regulated in reactive Müller glia (table S6). For instance, fatty acid–binding proteins FABP5, FABP7, and FABP8/PMP2 (FABPs) are selectively expressed in reactive chick Müller glia (fig. S14, C to E). Inhibiting FABPs using BMS309403 (21) resulted in fewer proliferating chick Müller glia after NMDA treatment (Fig. 5, I and J). ScRNA-seq analysis also showed that BMS309403 treatment decreased the fraction of proliferative Müller glia and led to a concomitant increase in the fraction of resting Müller glia compared with NMDA treatment alone (Fig. 5K and fig. S14, F and G), indicating that FABPs promote injury-induced transition from rest to a reprogrammed state.
We next investigated the role of mouse TFs predicted to promote glial quiescence by targeting the nuclear factor I (NFI) factors Nfia, Nfib, and Nfix, which were expressed at rest, down-regulated immediately after injury, and elevated at later stages (fig. S15A). We examined tamoxifen-fed GlastCreER;CAG-lsl-Sun1-GFP;Nfia/b/xlox/lox mice, in which NFIs were selectively deleted from Müller glia (fig. S15, B and C). Loss of Nfia/b/x reduced expression of genes enriched in resting glia, such as Glul and Rlbp1, but up-regulated genes associated with cell cycle regulation (Ccnd1 and Ccnd3) and neurogenesis (Ascl1) (fig. S15, D to F). Nfia/b/x-deficient Müller glia proliferated more after injury than did controls, both with and without FGF2+insulin, with EdU+/GFP+ cells (EdU, 5-ethynyl-2′-deoxyuridine) observed in both inner and outer nuclear layers at 3 days after injury (Fig. 6A and fig. S15G). EdU+ Müller glia in the outer nuclear layer disappear over time, indicating possible interkinetic nuclear migration of mitotic Müller glia. Müller glia–derived GFP+ cells show the expression of the photoreceptor and bipolar cell marker CRX and amacrine, ganglion, and horizontal cell markers HuC/D and NeuN at 14 days after treatment (Fig. 6, B and C).
Fig. 6. Loss of Nfia/b/x enables mouse Müller glia proliferation and neurogenesis after injury.
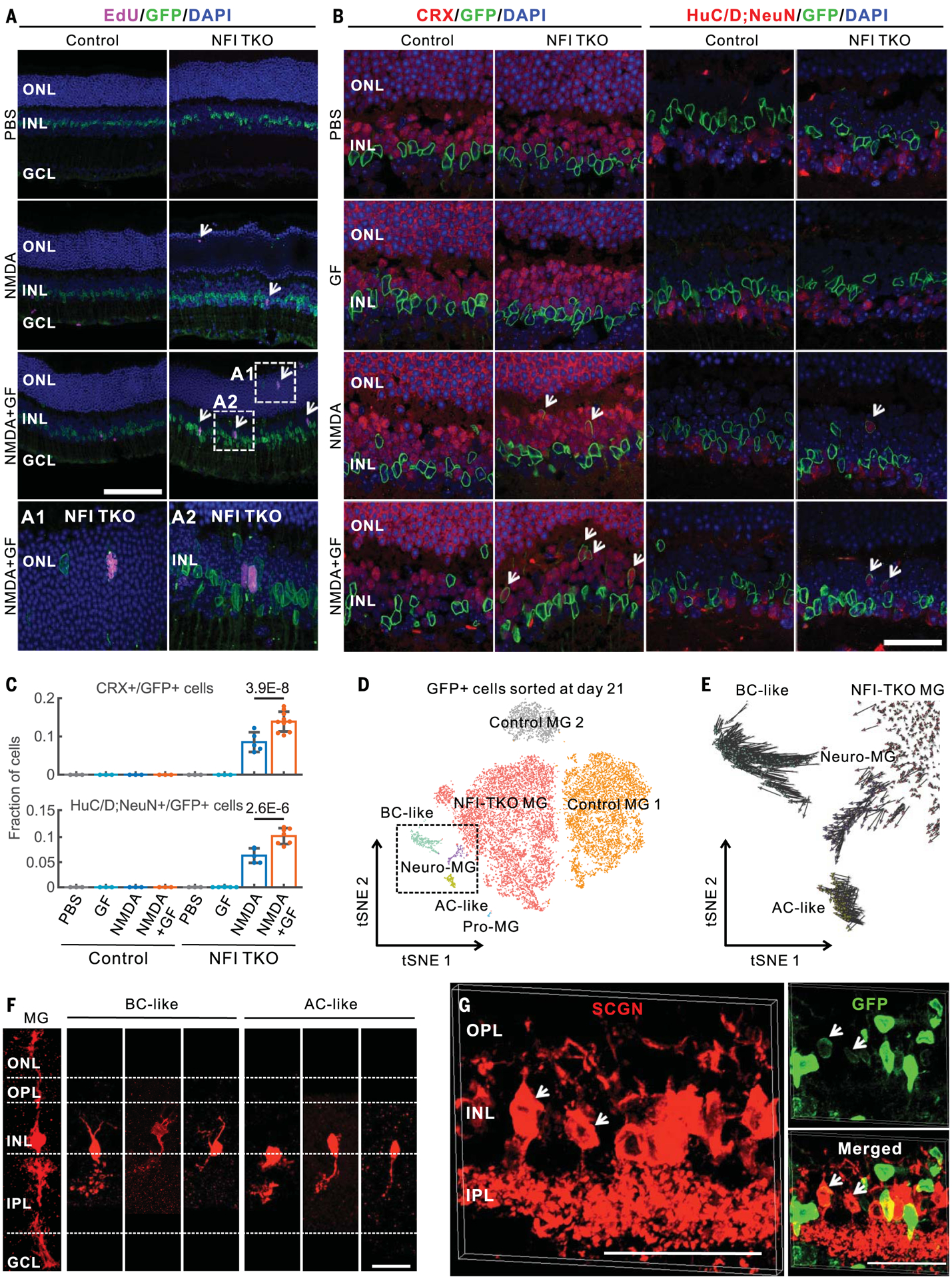
(A) EdU immunostainings of adult control and MG-specific Nfia/b/x knockout mice at 3 days after intravitreal injection with PBS, NMDA, or NMDA with growth factors (A1 and A2). (B) Nfia/b/x-deficient MG generated CRX+ and HuC/D;NeuN+ neurons (white arrows) at 14 days after injury. HuC/D and NeuN antibodies were mixed for these immunostainings. (C) Quantifications of CRX+ and HuC/D;NeuN+ neurons derived from Nfia/b/x-deficient MG glia at 14 days after injury. Error bars indicate standard deviation. (D) Clustering of GFP+ cells flow-sorted from control and MG-specific Nfia/b/x knockout retina at 21 days after NMDA+GF treatment. Neuro-MG, neurogenic Müller glia; Pro-MG, proliferative Müller glia. (E) RNA velocity analysis of MG-derived cells in the dotted box from (D). (F) Morphological characterization of MG-derived neurons in Nfia/b/x-deficient retinas using AAV9-pCAG-Flex-TdTomato after 14 days of NMDA+GF treatment. (G) MG-derived bipolar cells in Nfia/b/x-deficient retinas at 2 months after NMDA +GF treatment were immunolabeled for secretagogin (SCGN). Bipolar cells extend processes to the OPL and IPL retinal layers (white arrows). Scale bars, 20 μm.
We analyzed cells generated from Nfia/b/x-deficient Müller glia using scRNA-seq 21 days after NMDA+FGF2+insulin treatment (fig. S15H). Nfia/b/x-deficient Müller glia produced several distinct cell populations, including proliferative Müller glia, neurogenic Müller glia, and bipolar- and amacrine-like cells (Fig. 6D). Nfia/b/x-deficient Müller glia significantly up-regulated genes associated with the reactive state, such as Manf (fig. S15I). Neurogenic Müller glia did not express cell cycle regulators. Conversely, proliferative Nfia/b/x-deficient Müller glia did not express neurogenic markers. RNA velocity analysis indicated that neurogenic Müller glia gave rise to bipolar and amacrine cells (Fig. 6E). These Müller glia–derived neurons expressed markers typical of mature bipolar and amacrine cells but did not express markers for other retinal cell types (fig. S15I). Expression of the on-center bipolar cell marker PCP2 in the Müller glia–derived bipolar cells was confirmed by immunohistochemistry (fig. S15J). We observed 8.5% of CRX+ neurons and 6.1% of HuC/D;NeuN+ neurons derived from Müller glia colabeled with EdU (fig. S16, A and B). These data suggest that neurons arose from Nfia/b/x-deficient Müller glia through direct transdifferentiation. Native bipolar and amacrine cells showed similar expression profiles to neurons derived from Nfia/b/x-deficient Müller glia (fig. S16C). Verteporfin-mediated inhibition of YAP resulted in reduced proliferation and neurogenesis (fig. S16, D and E). Infection with AAV9-Flex-TdTomato showed that Müller glia–derived neurons had bipolar and amacrine-like morphologies (Fig. 6F and fig. S16F). Immunostaining for secretagogin (SCGN), a cone bipolar cell marker, indicates that Müller glia–derived bipolar cells extend processes to the inner plexiform layer at 2 months after injury (Fig. 6G and fig. S16G).
Discussion
This study reveals that Müller glia in all three species enter a reactive state after injury, in which mouse Müller glia arrest before returning to quiescence. However, most reactive Müller glia in zebrafish, and some in chick, pass through this state to become progenitors. Zebrafish genes essential for reprogramming, such as six3b, tgif1, and lepb (12, 22, 23), are up-regulated in reactive Müller glia. Inhibition of genes that are up-regulated in reactive Müller glia, such as hmga1a, smarca5, and yap1 in zebrafish and FABPs in chick, blocks proliferation and neurogenesis. Whereas loss of function of hmga1a and yap1 disrupts the transition from reactive to progenitor state, FABP inhibition blocks reprogramming by inhibiting transition from resting to reactive state. Some of these genes, including yap1, regulate neurogenic competence in the spinal cord (24, 25), suggesting that these same mechanisms may broadly regulate central nervous system (CNS) regeneration.
Although activated mouse Müller glia transiently express cell cycle regulators and neurogenic factors, they rapidly return to quiescence. This is mediated by a dedicated gene regulatory network that includes NFI factors, which are up-regulated at later stages after injury, and restore reactive Müller glia to a resting state in mice. Deletion of Nfia/b/x in mouse Müller glia resulted in proliferation and formation of Müller glia–derived inner retinal neurons, which was suppressed by YAP inhibition. NFI factors thus maintain Müller glia quiescence, prevent expression of genes associated with transition to reactivity and/or progenitor-like stages, and suppress neurogenesis, much like in developing retina (10). While it has been previously shown that down-regulation of Notch signaling is sufficient for zebrafish Müller glia reprogramming (26) and that MYT1L-dependent repression of non-neuronal genes is important for direct reprogramming to neuronal cell fate (27, 28), our study highlights the importance of repressing cell type–specific genes in the context of cellular reprogramming.
Recently, it has been reported that down-regulation of the RNA binding protein Ptbp1 can convert Müller glia into retinal ganglion cells in adult mouse retina (29). However, we found that Ptbp1 expression was maintained in reprogramed Müller glia after injury in both zebrafish and chick (fig. S17). Likewise, no significant change in Ptbp1 expression was observed between control and neurogenic Nfia/b/x-deficient mouse Müller glia. These results do not support the hypothesis that dynamic expression of Ptbp1 directly regulates Müller glia reprogramming.
Multiple independent mechanisms strongly repress neurogenic competence in mice, making it challenging to experimentally identify regulators of Müller glia reprogramming. In contrast, the limited neurogenic competence retained by the post-hatch chick facilitates functional analysis of both positive and negative candidate regulators of Müller glia reprogramming, making it an attractive system for analyzing regulators of retinal regeneration.
Injury-induced neurogenesis is limited or absent in the CNS of warm-blooded animals (30), in part as the result of active repression of regenerative competence. It has been hypothesized that this may reflect an evolutionary trade-off between regenerative competence and parasite resistance (31). Remaining in an activated state may allow CNS glia to efficiently restrict spread of infection, whereas regeneration would facilitate spread. This interpretation is supported by the fact that proinflammatory signals repress Müller glia reprogramming (17) and that evolutionary loss of injury-induced axonal regeneration typically tracks with loss of injury-induced neurogenic neurogenesis (30).
Materials and methods
Fish maintenance
Zebrafish (Danio rerio) lines AB, albino;Tg [gfap:GFP]mi2001 (32) and albino;Tg[gfap:EGFP] nt11 (33) were maintained in the Center for Zebrafish Research at the University of Notre Dame Freimann Life Science Center. Six- to twelve-month-old adult zebrafish were maintained under a 14 hours light/10 hours dark cycle at 28.5°C. All experimental protocols were approved by the University of Notre Dame Institutional Animal Care and Use Committee (IACUC) committee and are in compliance with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in vision research.
Chick maintenance
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and the Ohio State University. Newly hatched wild-type leghorn chickens (Gallus gallus domesticus) were obtained from Meyer Hatchery (Polk, OH). Postnatal chicks were kept on a cycle of 12 hours light/12 hours dark (lights on at 8:00 a.m.). Chicks were housed in a stainless steel brooder at about 25°C and received water and Purina chick starter ad libitum.
Mice maintenance
CD1 mice were purchased from Charles River Laboratories (Wilmington, MA). GLASTCreERT2 and Sun1-sGFP transgenic mice were provided by J. Nathans (16, 34). GLASTCreERT2;Sun1-sGFP were generated by breeding and subsequent backcrossing. To induce Cre recombination, GLASTCreERT2;Sun1-sGFP mice at ~2 weeks of age were intraperitoneally injected with tamoxifen in corn oil for two consecutive days at 1 mg per dose. To generate Müller glia-specific loss of function mutants of Nfia/b/x genes, 3-week-old GLASTCreERT2;Nfia/bxlox/lox; Sun1-sGFP mice were fed tamoxifen diet for 3 weeks, followed by 2 weeks on normal diet. All mice were housed in a climate-controlled pathogen-free facility on a 14 hours light/10 hours dark cycle. All experimental procedures were preapproved by the IACUC of the Johns Hopkins University School of Medicine.
Retinal damage paradigms in fish
To damage rods and cones, adult albino;Tg [gfap:EGFP]nt11 fish were dark-adapted for 14 days then placed in constant light (20,000 lux) at 32°C for up to 72 hours (35). Fish were euthanized by anesthetic overdose of 0.2% 2-phenoxyethanol in system water.
To damage amacrine and ganglion cells, adult Tg[gfap:GFP]mi2001 zebrafish were anesthetized in 0.1% 2-phenoxyethanol and intravitreally injected with 0.5 μl of 100 mM N-methyl-d-aspartic acid (NMDA) (Sigma-Aldrich; St. Louis, MO) (36). The fish were revived and placed in a 32°C dark incubator for up to 36 hours before euthanization.
TNFα and RO4929097 treatment in fish
Recombinant TNFα was expressed in Escherichia coli and purified as previously described (26). Undamaged adult Tg[gfap:GFP]mi2001 fish were anesthetized and intravitreally injected with 0.5 μl of TNFα (1.5 mg/ml) and intraperitoneally injected with 50 μl 0.75 mM RO4929097 (Selleckchem; Houston, TX) every 12 hours. The fish were revived and placed in a 32°C dark incubator for up to 72 hours before euthanization.
Intraocular injections in chicks
Chickens were anesthetized via inhalation of 2.5% isoflurane in oxygen. Intraocular injections performed as described previously (37). For all experiments, the right eyes of chicks were injected with the test compound and the contralateral left eyes were injected with a vehicle as a control. Compounds were injected in 20 μl sterile saline with 0.05 mg/ml bovine serum albumin added as a carrier. Compounds used in these studies included NMDA (38.5 or 154 g per dose; Sigma-Aldrich), FGF2 (200 ng per dose; R&D Systems), insulin (1 μg per dose; Sigma-Aldrich), and BMS309403 (500 ng per dose; Tocris). EdU (2 μg) was injected to label proliferating cells. Injection conditions are included in each figure.
NMDA treatment and light damage in mice
For NMDA treatment, adult mice, either CD1 or GLASTCreERT2;Sun1-sGFP mice at ~2 months of age, were anesthetized with isoflurane inhalation. Two microliters of 100 mM NMDA in phosphate-buffered saline (PBS) were intravitreally injected using a syringe with a 33G blunt-ended needle. Light damage was performed as previously described (18).
For NMDA and growth factor (GF) treatment of NFI triple-knockout (TKO) mice, adult GLASTCreERT2;Nfia/bxlox/lox;Sun1-sGFP and GLASTCreERT2;Sun1-sGFP control mice, both in CD1 background, were intravitreally injected with 2 μl of 100 mM NMDA or 2 μl of 100 mM NMDA with 100 ng/μl FGF2 and 1 μg/μl insulin (N+GF treatment), followed by 2 μl of 500 ng/μl EdU at 24 and 48 hours after the first injection. For the N+GF treatment group, 2 μl of 100 ng/μl FGF2 and 1 μg/μl Insulin was co-injected with each EdU injection. Contralateral eyes were injected with PBS and EdU as the controls. For long term EdU chasing, EdU was administered daily via intraperitoneal injection at 50 mg/kg in PBS for 10 days. Retina were collected at the indicated time points for immunohistochemistry and FACS.
For morphological examination of Müller glia and Müller glia–derived neurons, GLASTCreERT2;Nfia/b/xlox/lox;Sun1-sGFP and GLASTCreERT2;Sun1-sGFP control mice were intravitreally injected with 1 μl of AAV9-pCAG-Flex-Tdtomato (Addgene #28306) at 1 × 1013 vg/ml and treated with a tamoxifen diet for 3 weeks. Morphology of GFP/TdTomato+ Müller glia and Müller glia–derived neurons was examined on day 14 after N+GF treatment.
Zebrafish retinal dissociation, cell sorting, and methanol fixation
Zebrafish were euthanized in 0.2% 2-phenoxy-ethanol, and retinas were dissected and placed in Leibowitz medium (Thermo Fisher). Retinas were subsequently placed in 20 U/ml papain (10 retinas per 1 ml) (Worthington), and incubated at 28°C for 30 min with gentle agitation. Cells were pelleted and resuspended in PBS containing 0.1 mg/ml leupeptin (Sigma-Aldrich) and 10 U/ml DNaseI (Roche). Cells were filtered through a 70-μm filter (Miltenyi Biotec), kept on ice until sorting, and incubated with 5 μg/ml propidium iodide (PI; Life Technologies).
Cells were transferred into a 5-ml FACS tube and loaded into a FACS AriaII. Gating was set using dissociated retinas from albino zebrafish both unstained and PI stained and unstained Tg[gfap:EGFP]nt11 zebrafish. GFP+ and GFP− cells were sorted directly into Trizol LS (Life Technologies) and immediately flash frozen.
For scRNA-seq, cells were methanol-fixed after the 10x Genomics protocol. Dissociated cells were pelleted, resuspended in 100 μl Dulbecco’s PBS in cryovials and added with 900 μl −20°C methanol dropwise while slowly vortexing. Cells were left on ice for 15 min and then placed directly in −80°C.
Mouse retinal cell dissociation and FACS
Mice were euthanized by CO2, and eye globes were removed and kept in ice-cold 1x PBS. The neural retinas were dissected, and cells were dissociated using Papain Dissociation System (LK003150, Worthington). For FACS experiments, dissociated cell pellets were resuspended in ice-cold PBS containing 2% heat-inactivated fetal bovine serum (FBS) and 5 units/ml DNaseI). For scRNA-seq experiments, dissociated cells were then resuspended with ice-cold PBS containing 0.04% bovine serum albumin (BSA) and 0.5 U/μl RNase inhibitor. Cells were filtered through a 50-μm filter, and cell count and viability assessed by Trypan blue staining.
Cells were sorted using a Sony SH800S Cell Sorter. Retinal cells from nontransgenic mice were used to set the gating threshold for GFP+ cells. Cells were flow-sorted into GFP+ and GFP− fractions into ice-cold PBS containing a final concentration of 10% heat-activated FBS. For ATAC-seq, cells were kept on ice until use. For bulk RNA-seq, cells were centrifuged at 500g for 5 min, resuspended in 700 μl QIAzol lysis reagent (miRNAeasy Mini Kit), and stored at −80°C until RNA extraction.
Immunohistochemistry and imaging in mice
Mouse eye globes were collected and fixed in 4% paraformaldehyde in PBS for 2 hours at room temperature. Eyes were washed with PBS, and retina were placed in 30% sucrose overnight at 4°C. Retina were then embedded, cryosectioned at 20-μm thickness, and stored at −80°C. Sections were dried at 37°C for 20 min and washed 3 × 5 min with PBS before incubation with a blocking buffer (0.2% Triton, 5% horse serum in PBS) for 2 hours at room temperature. For CRX, HuC/D, and NeuN immuno-staining, retinal sections were treated with 100 mM sodium citrate pH 6.0 at −80°C for 30 min for antigen retrieval before the blocking step. Sections were then incubated with primary antibodies at indicated concentrations in the blocking buffer (table S12) overnight at 4°C. EdU staining was performed by following the manufacturer instructions. Images were acquired using the confocal Zeiss LSM 700. EdU+/GFP+ cells were counted and averaged from >6 random whole sections for each retina. For examining neurogenesis in NFI TKO mice, immunohistochemistry for CRX, a photoreceptor and bipolar cell marker (38) was performed. To maximize the possibility of detecting Müller glia–derived neurons, two pan-neuronal antibodies, HuC/D and NeuN, which are amacrine/ganglion/horizontal cell markers, were mixed for the immunostainings. CRX+/GFP+ and HuC/D; NeuN+/GFP+ cells were counted from six images taken across >4 random sections per retina. The fraction of CRX+/GFP+ and HuC/D;NeuN+/GFP+ cells was calculated from total GFP+ cells. Each data point in the quantification bar graphs was calculated from an individual retina. For morphological characterization of neurons, 10 to 20 z-stacked confocal images (0.15 to 0.35 μm per image) were taken for GFP/TdTomato+ cells and GFP/SCNG+ cells at day 14 and 2 months after N+GF treatment.
Injection and electroporation of morpholinos in zebrafish
Morpholino-mediated knockdown was performed in dark-adapted albino;Tg[gfap:EGFP]nt11 fish as previously described (39). After morpholino electroporation, fish were intravitreally injected with NMDA and placed in a 32°C dark incubator for 36 hours, or they were placed in constant intense light for either 36 or 72 hours. The following previously validated morpholinos were used in this study: Standard Control: 5′-CCTCTTACCTCAGTTACAATTTATA-3′ (Gene Tools); pcna: 5′-TGAACCAGACGTGCCTCAAA-CATTG-3′ (39); myb: 5′-GCCGCCTCGCCATCCC-GCTGTTCG-3′ (40); smarca5: 5′-CTTCTTCCC-GCTGCTGCTCCATGCT-3′ (41); hmga1a: 5′-CTGTGTCCTTGCCAGAATCACTCAT-3′ (42); and yap1: 5′-CTCTTCTTTCTATCCAACAGAAACC-3′ (43).
Verteporfin injection
Verteporfin (#SML0534, Millipore Sigma) was resuspended to 3.48 mM in 100% dimethyl sulfoxide (DMSO). For zebrafish, verteporfin was diluted with water to the desired working concentration (either 12.5 or 25 μM). Dark-adapted albino;Tg[gfap:EGFP]nt11 fish were anesthetized and 0.5 μl of either verteporfin or DMSO was intravitreally injected and the fish were exposed to constant light damage. Every 12 hours, the intravitreal injections were repeated until 36 hours, when fish were euthanized and retinal sections analyzed.
For mice, verteporfin was diluted to 30 μM in PBS and intravitreally co-injected with NMDA+FGF2+insulin into GLASTCreERT2; Nfia/b/xlox/lox;Sun1-sGFP mice at 2 μl per eye. This was followed by two doses of verteporfin with FGF2+insulin and EdU at 24 and 48 hours later, as described above. Mice also received daily intraperitoneal injection of verteporfin (6 mg/kg) for seven consecutive days starting from the first verteporfin injection. DMSO was used as the control. Retinal samples were colected at the indicated time points for immunohistochemistry.
Immunohistochemistry and imaging in zebrafish
Immunohistochemistry was performed as described previously (26). Cryosections (14 μm) were prepared, processed, and incubated with primary antibodies (table S12) at room temperature overnight. Confocal imaging was performed using a Nikon A1R laser scanning confocal microscope. Images were obtained from sections from the center of the dorsal retina containing the optic nerve. Quantification was performed using 6.5-μm z-stacks and cell counts normalized to either 50 or 300 μm. Data were statistically analyzed using one-way ANOVA with Tukey’s post-hoc test if more than two groups were compared, otherwise Student’s t test with unequal variances was used.
Immunohistochemistry and imaging in chick
Chicks were euthanized by CO2. Tissues were fixed, sectioned, and immunolabeled as described previously (37). Working dilutions and sources of antibodies are listed in table S12. Draq5 (10μM; Thermo Fisher) was added to the secondary antibody solution to label nuclei.
For EdU labeling, immunolabeled sections were fixed in 4% formaldehyde in PBS for 5 min at room temperature, washed for 5 min with PBS, permeabilized with 0.5% Triton X-100 in PBS for 1 min at room temperature, and washed twice for 5 min in PBS. Sections were incubated for 30 min at room temperature in 2M Tris, 50 mM CuSO4, Alexa Fluor 568 Azide (Thermo Fisher), and 0.5 M ascorbic acid in distilled H2O.
Confocal images were obtained using Leica SP8 imaging. Cell counts were performed on representative images. To avoid the possibility of region-specific differences within the retina, cell counts were consistently made from the same region of the retina for each dataset.
RNAscope in situ hybridization
RNA in situ hybridization was performed using RNA probes (table S13) and RNAscope Multiplex Fluorescent Reagent Kit V2 Assay (#323100-USM, ACDBio) by following the protocol for fixed-frozen tissue. Briefly, zebrafish, chick, and mouse retina were fixed and cryosectioned as described above for immunohistochemistry. Retinal sections were processed using standard RNAscope protocol with some modifications: Hydrogen peroxide treatment was reduced to 5 min, target retrieval step was omitted, and Protease III treatment was reduced to 15 min. Images were acquired using the confocal Zeiss LSM 700.
RNA extraction and RT-qPCR
RNA was extracted from both GFP+ and GFP− cell fractions using miRNAeasy Mini Kit (#217004, Qiagen). For quantitative reverse transcription polymerase chain reaction (RT-qPCR), RNA samples were first reverse-transcribed into cDNA using random primers and Superscript IV reverse transcriptase (#18091050, Thermo Fisher). The qPCR assays were performed on the cDNA using GoTaq Green Master Mix (#M7122, Promega) using a StepOnePlus Real-Time instrument (Thermo Fisher). Intron-spanning primers were designed to specifically quantify targeted mRNA transcripts. Glycer-aldehyde 3-phosphate dehydrogenase (Gapdh) expression was used as the endogenous control (table S13).
Bulk RNA-seq
Flow-sorted RNA samples were sent to the Deep Sequencing and Microarray Core (Johns Hopkins University) for library preparation and sequencing. Briefly, ribosomal RNA was depleted, and total RNA was captured from the RNA samples using Illumina TruSeq Stranded RNA LT kit Ribo-Zero Gold (#15032619, Illumina). Around 8 to 10 libraries were pooled and sequenced for paired-end 75 cycles using the NextSeq 500 system with ~400 million to 500 million reads per run, resulting in between ~45 million and 55 million reads per library.
Single-cell RNA-seq (scRNA-seq)
For fresh samples (table S1), cells were resuspended with ice-cold PBS containing 0.04% BSA and 0.5 U/μl RNase inhibitor. For methanol-fixed samples, fixed cells were processed according to 10x Genomics protocol for methanol-fixed cells. Briefly, methanol-fixed cells were centrifuged at 3000g for 10 min at 4°C and washed three times in an iced-cold resuspension buffer containing PBS, 1% BSA, and 0.5 U/μl RNAse inhibitors. Dissociated cells were resuspended in the resuspension buffer and filtered through a 50-μl cell filter. Cells (~10,000) were loaded into a 10x Genomics Chromium Single Cell system (10x Genomics, CA) using v2 chemistry following manufacturer’s instructions. Libraries were pooled and sequenced on Illumina NextSeq with ~200 million reads per library.
ATAC-seq
Sorted cells (~50,000 to 75,000) from GFP+ samples were used for ATAC library preparation. Nuclear isolation and all centrifugation steps were carried out on ice in a 4°C cold room. Briefly, cells were spun down at 500g for 5 min. Cells were rinsed with ice-cold PBS without disturbing cell pellets and centrifuged again at 500g for 5 min. Cells were lysed by adding 50 μl of ice-cold cell lysis buffer (10 mM Tris-Cl pH 7.4, 10 mM NaCl, 3 mM MgCl2) containing 0.03% octylphenoxy poly(ethyleneoxy) ethanol (IGEPAL) and protease inhibitors (one tablet per 7 ml of lysis buffer) and mixing three times by pipetting. Cells were then immediately spun down at 500g for 10 min and washed with 150 μl of ice-cold lysis buffer without IGEPAL and protease inhibitors. For tagmentation, cell nuclei were incubated with 2.5 μl enzyme in 50 μl total volume at 37°C in a thermo-cycler (Illumina Nextera DNA library prep kit, #FC1211030). DNA was cleaned up using MinElute PCR purification kit (#28006, Qiagen) and eluted in 10 μl of EB buffer.
ATAC-seq DNA was amplified, and the number of PCR cycles were calculated as described (44). PCR products (10 μl) were run on a 1.5% agarose gel for expected DNA pattern. PCR products were then cleaned up by double-sized selection using AMPure beads (Agencourt APure XP, Beckman Coulter, #A63880) to remove large and small DNA fragments. This was performed by using 1:0.5 and 1:1.6 ratio of sample to AMPure beads (v/v). Completed ATAC-seq libraries (8 to 10) were then analyzed by Fragment Bioanalyzer and sequenced for paired-end 75 cycles using the NextSeq 500 system with ~400 million to 500 million reads per run.
Bulk RNA-seq data analysis
Using bulk RNA-seq, we measured gene expression profiles of 50 GFP+ (GFP-positive, Müller glia, MG) and 50 GFP− (GFP-negative, non-Müller glia, non-MG) samples, with at least two biological replicates for each time point or condition (table S1). All raw sequencing data from mouse and zebrafish were separately mapped to the GRCm38/mm10 and GRCz10/danRer10 genome assembly using STAR (45). The mappability (the percentage of sequencing reads that were uniquely mapped to the reference genome) on average is 87.5% in mice and 60.7% in zebrafish. Lower mappability in zebrafish could be attributed to the relatively less well annotated zebrafish reference genome.
We calculated FPKM (fragments per kilobase of exon per million) using raw counts of genes. We performed t test of paired GFP+/GFP− samples from NMDA treatment and light damage to identify GFP+ or GFP− specific genes [fold change > 2 and false discovery rate (FDR) < 0.01, table S2]. To identify genes that are not differentially expressed after injury but that show significant differences in baseline expression between species, we ranked the genes on the basis of their expression levels in each species. We then performed Wilcoxon rank test between two species to identify species-specific expressed genes in Müller glia (fold change > 1.5 and FDR < 0.05).
Using EdgeR (46), we identified DEGs relative to control samples (fold change > 1.5 and FDR < 0.05). To identify model-independent DEGs (common DEGs) that are consistently changed between NMDA treatment and light damage, we calculated Pearson’s correlation coefficients between models for each species. Meanwhile, we shuffled the expression of each gene through randomly reordering the samples and obtained 0.5 correlation as the threshold of the correlation. We selected DEGs for further analysis if their expression correlation was >0.5.
To identify evolutionarily conserved and species-specific genes (table S3), we first identified orthologs between mouse and zebrafish (or chick) on the basis of vertebrate ortholog information from the Mouse Genome Informatics database (www.informatics.jax.org/). If multiple orthologs were mapped between species, we chose the DEG with the highest fold change. If no gene was differentially expressed, we selected genes that had the highest expression correlation between two models.
In addition, we performed K-means analysis of GFP+ samples to separate DEGs into three groups, including injury-repressed genes, rapidly induced genes, and slowly induced genes. Function enrichment analysis on genes was conducted in R package “clusterProfiler” (47). The principal components were obtained through principal component analysis (48).
Quality control and clustering of scRNA-seq data
We analyzed 19, 12, and 27 scRNA-seq libraries from mouse, chick, and zebrafish, respectively (table S1). Chick raw reads from scRNA-seq were mapped to Gallus_gallus–5.0/galGal5 genome. Gene-cell matrices were generated using Cell Ranger 2.1.1 from 10x Genomics. We calculated the number of genes and the number of unique molecular identifiers (UMIs). We removed low-quality cells (chick: <200 genes or <500 UMIs; mouse and zebrafish: <200 genes or <1000 UMIs). On average, we obtained ~1000 to 1500 genes and ~2500 to 3250 UMIs per cell.
For single cells from each treatment, we first identified variable genes that had 0.1 to 8 mean expression level and >1 dispersion. Using variable genes, we combined all samples from the same treatment and performed clustering analysis of single cells using Seurat (49). Clusters were identified through K-nearest neighbors and a shared nearest neighbor modularity optimization and then visualized using t-distributed stochastic neighbor embedding (tSNE) (50). No obvious batch effects were observed among samples (figs. S2 and S3). No clear evidence for molecular heterogeneity of Müller glia were observed in the unstimulated control retina from any species examined.
Identification of cell doublets in scRNA-seq
To identify cell doublets in scRNA-seq data, we developed a new algorithm. First, we performed single-cell clustering and identified marker genes for each cluster or cell type. For each marker, we next calculated the gene power (GP) using GP = 2 × abs(AUC – 0.5), where AUC is the area under the receiver operating characteristic curve of gene expression (49).
For each cell in cluster i, we then calculated the summed power (SPi)
where Ni is the number of markers in cluster i, and GPi,j is the gene power that indicates the capability of marker j to distinguish cluster i and the remaining clusters.
For all cells in cluster i, we used the bottom 0.1% value of SPi as the threshold to determine whether the cell expresses markers from cluster i. The cell is regarded as a doublet if SPi of a cell is above the threshold for two or more clusters, suggesting that the cell expresses markers from multiple clusters. In each scRNA-seq sample, we identified and removed 5 to 10% of cells as cell doublets.
Identification of cell types and markers for each cluster
We used known marker genes to annotate cell type for each cluster of cells (table S4). Most known marker genes were reported in mice. For chick and zebrafish, we identified orthologs of mouse marker genes and used those orthologs to determine cell types of clusters. Meanwhile, we identified new markers of cell types in each injury model and species by setting >0.3 gene power. By comparing these marker genes across species, we identified species-specific and conserved markers for each cell type (table S5).
Trajectory and pseudotime analysis of single cells
We constructed the trajectory of Müller glia in response to retinal injury and performed pseudotime analysis using variable genes through Monocle (51). Pseudotime was calculated by setting unstimulated Müller glia as the root. Unstimulated Müller glia were from P60 mouse, P10 chick, and adult zebrafish. Furthermore, we identified significantly changed genes (or DEGs) along pseudotime. We used the following criteria to identify DEGs: fraction of expressed cells > 0.01, single-cell expression difference > 0.1, and q-value < 0.001. Single-cell expression difference was calculated as
where Q95-expression and Q5-expression separtely represent 0.95 and 0.05 quantile of log-transformed single-cell expression values across all bins of pseudotime. We divided cells into 50 bins across pseudotime with equal pseudotime intervals. Bin-derived expressions were obtained by averaging expressions of all cells within each bin.
To identify model-independent DEGs, we also used bin-derived expressions to calculate single-cell expression correlations between two models. Similar to bulk RNA-seq analysis, we identified evolutionarily conserved and species-specific DEGs through cross-species scRNA-seq analysis (table S6).
We aligned Müller glia trajectories from two species using cellAlign (52). In alignment, we separated and sorted different states of Müller glia on the basis of pseudotime. DEGs shared by two species were used for trajectory alignment.
RNA velocity analysis
To minimize batch effect, we chose branch-specific (zebrafish and chick) or cell type–specific (mouse) genes for performing RNA velocity analysis (11). First, in zebrafish, we identified branch-specific genes in the trajectory of Müller glia after NMDA treatment. We then selected the top 50 specifically expressed genes for each branch. Among 150 genes obtained from three branches, we selected 92 genes that satisfy the following criteria [the number of cells detected spliced UMIs >10, and the number of cells detected unspliced UMIs (U) is from 1 to 300]. Other parameters were set to the default. The same approach was applied to chick and mouse. The gene sets for RNA velocity analysis were listed in table S14.
Comparison of zebrafish Müller glia from development and injury models
We first used Seurat to identify cell subpopulations in zebrafish development. Single-cell clustering was visualized through uniform manifold approximation and projection (UMAP) (53). Using known marker genes of retinal cell types (table S4), we annotated cell types for clusters. We then selected 12,680 primary RPCs, neurogenic RPCs, and Müller glia to construct a trajectory and identify DEGs during Müller glia development. To compare each injury model with retinal development in zebrafish, we combined 12,680 developmental cells with Müller glia in NMDA treatment, light damage, and T+R treatment. Meanwhile, we identified overlapped DEGs between retinal development and injury model. Using overlapped DEGs, we reconstructed the Müller glia trajectory for each injury model combined with cells obtained from developing retina. We used the heatmap to visualize changes for all overlapped DEGs in retinal development and NMDA treatment. The heatmap includes left and right divisions, which represent the process from embryo to adult, and the transition from unstimulated adult Müller glia to NMDA treatment, respectively.
Mapping and normalization of ATAC-seq
We performed ATAC-seq for 20 zebrafish GFP+ and 20 mouse GFP+ samples (table S1). After removing adapters using cutadapt (54), raw reads from zebrafish and mouse were separately aligned to GRCz10/danRer10 and GRCm38/mm10 reference genome using Bowtie2 with default parameters (55). We performed a similar analysis as the previous report (56). Briefly, we filtered reads from chromosome M and Y and included reads with mapping quality (MAPQ) score > 10. After removing duplicate reads and blacklisted regions, we called ATAC-seq peaks using MACS2 with parameters -nomodel -shift −100 -extsize 200 (57). In total, we identified 248,761 peaks in zebrafish and 119,718 peaks in mice. We counted the raw fragment (CR) for each peak using high-throughput sequencing (HTSeq) (58). The normalized fragment (CN) was calculated as
where SL is the sequencing library size of the sample.
Differentially accessible regions and footprints from ATAC-seq
We identified DARs relative to controls using EdgeR (46). In total, we identified 56,939 DARs in zebrafish (fold change > 1.5 and FDR < 0.05) and 77,447 DARs in mice (fold change > 2 and FDR < 0.01).
To identify model-independent or common DARs, we calculated the correlation of chromatin accessibility between two injury models. We identified the cutoff of 0.5 on the basis of the correlation distribution of shuffled chromatin accessibility, which was obtained from randomly reordering samples. Finally, we separately identified 24,515 and 52,297 model-independent DARs in zebrafish and mouse (table S7). In DARs, we further identified footprints using DNase2TF (59).
Reconstructing integrated regulatory networks
To reconstruct regulatory networks by integrating bulk RNA-seq, scRNA-seq, and ATAC-seq data, we developed a method called integrated regulatory network analysis (IReNA) (fig. S10B). Common DEGs and common DARs across all treatments were used to identify core regulatory networks.
First, we identified Müller glia–expressed TFs using the following criteria: (i) highly expressed in Müller glia: expression level > 5 FPKM for bulk RNA-seq, or > 0.1 fraction of expressed cells for scRNA-seq; (ii) correlated expression patterns between NMDA and light damage: correlation > 0.5 in bulk RNA-seq, or > 0.2 in scRNA-seq; and (iii) differentially expressed in treatment versus control or species-specific expressed in Müller glia (identified from above RNA-seq and scRNA-seq analysis). We identified 192 and 212 candidate TFs in zebrafish and mouse, respectively.
Next, we used Müller glia–expressed TFs and significantly changed and species-specific expressed genes as candidate genes. Through K-means clustering, we separated candidate genes into 10 modules in each species, three modules of which consisted of genes only identified by bulk RNA-seq analysis. For each gene in the module, we used FIMO (find individual motif occurrences) to identify DNA motifs within its footprints (P value < 1.0 × 10−4) (60). Binding motifs of TFs were extracted from TRANSFAC 2018.3 version. Meanwhile, we calculated the footprint occupancy score (FOS) for each footprint in each ATAC-seq sample. FOS was calculated as previously described (56)
where NC indicates counts of ATAC-seq inserts in the central region of the motif. NL and NR are separately one-third counts of ATAC-seq inserts in left and right flanking regions of the motif, as a triple size of the central region was chosen as the size of the flanking region. To further refine regulatory relationships between TFs and target genes, we included binding motifs with FOS > 1 in any one of ATAC-seq samples.
Finally, we used expression correlations between TFs and target genes to determine active (positively correlated) or repressive (negatively correlated) regulations. Correlation coefficients >0.5 and <−0.5 were selected for active and repressive regulations, respectively. We constructed regulatory networks of candidate genes consisting of 26,083 and 30,177 regulatory relationships in zebrafish and mouse, respectively (table S9).
Identification of transcription factors regulating each gene module
For each gene module, we calculated the probability (Pr) of each TF regulating the module through hypergeometric test
where N is the total number of regulations, K is the number of all regulations targeting module A, n is the number of regulations from the TF, k is the number of regulations from the TF to module A, and the general form is a binomial coefficient. We separately calculated P values for active and repressive regulations. FDR was further calculated on the basis of P values.
Using FDR < 10 × 10−6 in zebrafish and FDR < 10 × 10−5 in mice, we separately identified 97 zebrafish TFs and mouse 156 TFs which significantly regulate at least one gene module. We further extracted regulatory networks consisting of these TFs from regulatory networks of candidate genes (figs. S11 and S12 and table S10).
Constructing intramodular regulatory networks
To determine regulatory relationships between two modules, we calculated the significance of regulations from module A to module B using hypergeometric test
where M is the total number of regulations, Q is the number of all regulations from TFs in module A, m is the number of all regulations targeting module B, q is the number of regulations from module A to module B, and the general form is a binomial coefficient. We assessed the significance of active and repressive regulations, respectively. We set FDR < 0.02 in zebrafish and FDR < 0.001 in mice as the cutoff to obtain significant regulations between modules (Fig. 4, E and F).
Features of transcription factors in regulatory networks
W calculated the degree of each enriched TF in the regulatory network. The degree includes out-degree (number of genes regulated by TFs of interest) and in-degree (number of TFs regulating TFs of interest). For the regulatory network of candidate genes, we calculated the normalized degree (DN) on the basis of the degree (DR)
where DT is the total number of regulations in the gene regulatory network. For the regulatory network of enriched TFs, the same analysis was performed to calculate the normalized degree.
Mapping of new single cells to the original trajectory
To map new single cells to the original trajectory, we first calculated Pearson’s correlation coefficients between new cells and original cells. To calculate the expression correlations, we chose the same set of variable genes that were used for constructing the trajectory. For each new cell, the most correlated cell from the original trajectory was then identified. Correspondingly, the location and state of the original cell were assigned to the new cell. We then obtained the distribution of new cells in the original trajectory.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Nathans, A. Kolodkin, and W. Yap for comments on the manuscript; B. Kulemeka and the Freimann Life Science Center staff for raising zebrafish; and X. Zhou and J. Wang for curation of sequencing data.
Funding: This work was supported by NIH grants U01EY027267 (D.R.H., S.B., J.Q., A.J.F., and J.A.), R01EY024519 (D.R.H.), R01EY029548 (J.Q.), K08EY027093 (F.R.), and R01EY020560 (S.B.). P.B. was partially supported by the Hiller Family Endowment for Stem Cell Research at the University of Notre Dame. M.L. was partially supported by the Center for Zebrafish Research at the University of Notre Dame.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: RNA-seq and ATAC-seq data are deposited in the Gene Expression Omnibus (GSE135406). All scRNA-seq data and source codes are available at GitHub https://github.com/jiewwwang/Single-cell-retinal-regeneration. RNA-seq, ATAC-seq, and scRNA-seq data can be queried interactively at https://proteinpaint.stjude.org/F/2019.retina.scRNA.html. All other data are available in the main manuscript or the supplementary materials. Materials requests should be directed to S.B..
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/370/6519/eabb8598/suppl/DC1
Figs. S1 to S17
Tables S1 to S14
MDAR Reproducibility Checklist
REFERENCES AND NOTES
- 1.Lahne M, Nagashima M, Hyde DR, Hitchcock PF, Reprogramming Müller glia to regenerate retinal neurons. Annu. Rev. Vis. Sci 6, 171–193 (2020). doi: 10.1146/annurev-vision-121219-081808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fausett BV, Gumerson JD, Goldman D, The proneural basic helix-loop-helix gene Ascl1a is required for retina regeneration. J. Neurosci 28, 1109–1117 (2008). doi: 10.1523/JNEUROSCI.4853-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorstad NL et al. , Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548, 103–107 (2017). doi: 10.1038/nature23283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamon A et al. , Linking YAP to Müller glia quiescence exit in the degenerative retina. Cell Rep. 27, 1712–1725.e6 (2019). doi: 10.1016/j.celrep.2019.04.045 [DOI] [PubMed] [Google Scholar]
- 5.Rueda EM et al. , The Hippo pathway blocks mammalian retinal Müller glial cell reprogramming. Cell Rep. 27, 1637–1649.e6 (2019). doi: 10.1016/j.celrep.2019.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan J, Goldman D, Retina regeneration in zebrafish. Curr. Opin. Genet. Dev 40, 41–47 (2016). doi: 10.1016/j.gde.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson CM et al. , Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci 33, 6524–6539 (2013). doi: 10.1523/JNEUROSCI.3838-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer AJ, McGuire CR, Dierks BD, Reh TA, Insulin and fibroblast growth factor 2 activate a neurogenic program in Müller glia of the chicken retina. J. Neurosci 22, 9387–9398 (2002). doi: 10.1523/JNEUROSCI.22-21-09387.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schäfer P, Karl MO, Prospective purification and characterization of Müller glia in the mouse retina regeneration assay. Glia 65, 828–847 (2017). doi: 10.1002/glia.23130 [DOI] [PubMed] [Google Scholar]
- 10.Clark BS et al. , Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron 102, 1111–1126.e5 (2019). doi: 10.1016/j.neuron.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.La Manno G et al. , RNA velocity of single cells. Nature 560, 494–498 (2018). doi: 10.1038/s41586-018-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenkowski JR et al. , Retinal regeneration in adult zebrafish requires regulation of TGFβ signaling. Glia 61, 1687–1697 (2013). doi: 10.1002/glia.22549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esain V, Postlethwait JH, Charnay P, Ghislain J, FGF-receptor signalling controls neural cell diversity in the zebrafish hindbrain by regulating olig2 and sox9. Development 137, 33–42 (2010). doi: 10.1242/dev.038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor SM, Giuffre E, Moseley P, Hitchcock PF, The MicroRNA, miR-18a, regulates NeuroD and photoreceptor differentiation in the retina of zebrafish. Dev. Neurobiol 79, 202–219 (2019). doi: 10.1002/dneu.22666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vitorino M et al. , Vsx2 in the zebrafish retina: Restricted lineages through derepression. Neural Dev. 4, 14 (2009). doi: 10.1186/1749-8104-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Melo J et al. , Injury-independent induction of reactive gliosis in retina by loss of function of the LIM homeodomain transcription factor Lhx2. Proc. Natl. Acad. Sci. U.S.A 109, 4657–4662 (2012). doi: 10.1073/pnas.1107488109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palazzo I, Deistler K, Hoang TV, Blackshaw S, Fischer AJ, NF-κB signaling regulates the formation of proliferating Müller glia-derived progenitor cells in the avian retina. Development 147, dev183418 (2020). doi: 10.1242/dev.183418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueki Y, Wang J, Chollangi S, Ash JD, STAT3 activation in photoreceptors by leukemia inhibitory factor is associated with protection from light damage. J. Neurochem 105, 784–796 (2008). doi: 10.1111/j.1471-4159.2007.05180.x [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Shu Q, Ni Y, Xu G, CRISPR-mediated SOX9 knockout inhibits GFAP expression in retinal glial (Müller) cells. Neuroreport 29, 1504–1508 (2018). doi: 10.1097/WNR.0000000000001143 [DOI] [PubMed] [Google Scholar]
- 20.Liu-Chittenden Y et al. , Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305 (2012). doi: 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil GS, Bernlohr DA, Metabolic functions of FABPs—Mechanisms and therapeutic implications. Nat. Rev. Endocrinol 11, 592–605 (2015). doi: 10.1038/nrendo.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorsuch RA et al. , Sox2 regulates Müller glia reprogramming and proliferation in the regenerating zebrafish retina via Lin28 and Ascl1a. Exp. Eye Res 161, 174–192 (2017). doi: 10.1016/j.exer.2017.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X-F et al. , Leptin and IL-6 family cytokines synergize to stimulate Müller glia reprogramming and retina regeneration. Cell Rep. 9, 272–284 (2014). doi: 10.1016/j.celrep.2014.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mokalled MH et al. , Injury-induced ctgfa directs glial bridging and spinal cord regeneration in zebrafish. Science 354, 630–634 (2016). doi: 10.1126/science.aaf2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mokalled MH, Zhou L, Mcadow R, Burris B, Zebrafish-inspired strategies for spinal cord repair. FASEB J. 34, 1–1 (2020). doi: 10.1096/fasebj.2020.34.s1.04539 [DOI] [Google Scholar]
- 26.Conner C, Ackerman KM, Lahne M, Hobgood JS, Hyde DR, Repressing notch signaling and expressing TNFα are sufficient to mimic retinal regeneration by inducing Müller glial proliferation to generate committed progenitor cells. J. Neurosci 34, 14403–14419 (2014). doi: 10.1523/JNEUROSCI.0498-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mall M et al. , Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249 (2017). doi: 10.1038/nature21722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanda S et al. , Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Reports 3, 282–296 (2014). doi: 10.1016/j.stemcr.2014.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H et al. , Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 181, 590–603.e16 (2020). doi: 10.1016/j.cell.2020.03.024 [DOI] [PubMed] [Google Scholar]
- 30.Tanaka EM, Ferretti P, Considering the evolution of regeneration in the central nervous system. Nat. Rev. Neurosci 10, 713–723 (2009). doi: 10.1038/nrn2707 [DOI] [PubMed] [Google Scholar]
- 31.Rattner A, Nathans J, An evolutionary perspective on the photoreceptor damage response. Am. J. Ophthalmol 141, 558–562.e2 (2006). doi: 10.1016/j.ajo.2005.10.045 [DOI] [PubMed] [Google Scholar]
- 32.Bernardos RL, Raymond PA, GFAP transgenic zebrafish. Gene Expr. Patterns 6, 1007–1013 (2006). doi: 10.1016/j.modgep.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 33.Kassen SC et al. , Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev. Neurobiol 67, 1009–1031 (2007). doi: 10.1002/dneu.20362 [DOI] [PubMed] [Google Scholar]
- 34.Mo A et al. , Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015). doi: 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vihtelic TS, Soverly JE, Kassen SC, Hyde DR, Retinal regional differences in photoreceptor cell death and regeneration in light-lesioned albino zebrafish. Exp. Eye Res 82, 558–575 (2006). doi: 10.1016/j.exer.2005.08.015 [DOI] [PubMed] [Google Scholar]
- 36.Powell C, Cornblath E, Elsaeidi F, Wan J, Goldman D, Zebrafish Müller glia-derived progenitors are multipotent, exhibit proliferative biases and regenerate excess neurons. Sci. Rep 6, 24851 (2016). doi: 10.1038/srep24851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer AJ, Scott MA, Tuten W, Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia 57, 166–181 (2009). doi: 10.1002/glia.20743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glubrecht DD, Kim J-H, Russell L, Bamforth JS, Godbout R, Differential CRX and OTX2 expression in human retina and retinoblastoma. J. Neurochem 111, 250–263 (2009). doi: 10.1111/j.1471-4159.2009.06322.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thummel R et al. , Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res 87, 433–444 (2008). doi: 10.1016/j.exer.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Y-C et al. , c-Myb is an evolutionary conserved miR-150 target and miR-150/c-Myb interaction is important for embryonic development. Mol. Biol. Evol 25, 2189–2198 (2008). doi: 10.1093/molbev/msn165 [DOI] [PubMed] [Google Scholar]
- 41.Aguirre A et al. , In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15, 589–604 (2014). doi: 10.1016/j.stem.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moussavi Nik SH et al. , Alzheimer’s disease-related peptide PS2V plays ancient, conserved roles in suppression of the unfolded protein response under hypoxia and stimulation of g-secretase activity. Hum. Mol. Genet 24, 3662–3678 (2015). doi: 10.1093/hmg/ddv110 [DOI] [PubMed] [Google Scholar]
- 43.Jiang Q et al. , yap is required for the development of brain, eyes, and neural crest in zebrafish. Biochem. Biophys. Res. Commun 384, 114–119 (2009). doi: 10.1016/j.bbrc.2009.04.070 [DOI] [PubMed] [Google Scholar]
- 44.Zibetti C, Liu S, Wan J, Qian J, Blackshaw S, Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun. Biol 2, 142 (2019). doi: 10.1038/s42003-019-0375-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobin A, Gingeras TR, Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics 51, 11.14.1–11.14.19 (2015). doi: 10.1002/0471250953.bi1114s51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson MD, McCarthy DJ, Smyth GK, edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu G, Wang L-G, Han Y, He Q-Y, clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdi H, Williams LJ, Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat 2, 433–459 (2010). doi: 10.1002/wics.101 [DOI] [Google Scholar]
- 49.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linderman GC, Rachh M, Hoskins JG, Steinerberger S, Kluger Y, Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 16, 243–245 (2019). doi: 10.1038/s41592-018-0308-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu X et al. , Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). doi: 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alpert A, Moore LS, Dubovik T, Shen-Orr SS, Alignment of single-cell trajectories to compare cellular expression dynamics. Nat. Methods 15, 267–270 (2018). doi: 10.1038/nmeth.4628 [DOI] [PubMed] [Google Scholar]
- 53.Becht E et al. , Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2018). doi: 10.1038/nbt.4314 [DOI] [PubMed] [Google Scholar]
- 54.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 55.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J et al. , ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun 9, 1364 (2018). doi: 10.1038/s41467-018-03856-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng J, Liu T, Qin B, Zhang Y, Liu XS, Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740 (2012). doi: 10.1038/nprot.2012.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S, Pyl PT, Huber W, HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sung M-H, Guertin MJ, Baek S, Hager GL, DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol. Cell 56, 275–285 (2014). doi: 10.1016/j.molcel.2014.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grant CE, Bailey TL, Noble WS, FIMO: Scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). doi: 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.