

SUMMARY
Macrophages are critical effector cells of the immune system, and understanding genes involved in their viability and function is essential for gaining insights into immune system dysregulation during disease. We use a high-throughput, pooled-based CRISPR-Cas screening approach to identify essential genes required for macrophage viability. In addition, we target 3′ UTRs to gain insights into previously unidentified cis-regulatory regions that control these essential genes. Next, using our recently generated nuclear factor κB (NF-κB) reporter line, we perform a fluorescence-activated cell sorting (FACS)-based high-throughput genetic screen and discover a number of previously unidentified positive and negative regulators of the NF-κB pathway. We unravel complexities of the TNF signaling cascade, showing that it can function in an autocrine manner in macrophages to negatively regulate the pathway. Utilizing a single complex library design, we are capable of interrogating various aspects of macrophage biology, thus generating a resource for future studies.
Graphical Abstract
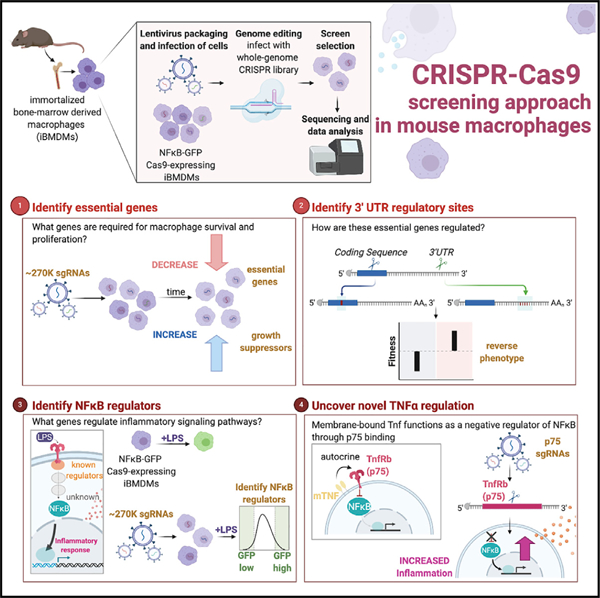
In Brief
Covarrubias et al. screen ~21,000 targets, generating a resource guide of genes required for macrophage viability as well as previously unidentified positive and negative regulators of NF-κB signaling. They identify regulatory elements within essential genes and show that membrane-bound TNF primarily functions in macrophages in an autocrine fashion to negatively regulate inflammation.
INTRODUCTION
Macrophages are critical cells of the innate immune system providing one of the first lines of defense against invading microbes. Macrophages arise from precursor monocyte cells that constitute ~10%–20% of the immune cells found in the blood (Kleiveland, 2015). Upon encountering a danger signal, monocytes differentiate into macrophages and rapidly move to the site of infection. Important aspects of macrophage function include their ability to proliferate and migrate, as well as their ability to induce the inflammatory program to aid in clearing infections and initiate tissue repair to maintain homeostasis (Wynn et al., 2013). While much work has been performed to understand the contribution of individual proteins to the processes that control macrophage biology, there has been no systematic approach adopted to study genes involved in macrophage viability and function simultaneously in a high-throughput manner.
Clustered regularly interspaced short palindromic repeat (CRISPR) technology has revolutionized the field of functional genomics, providing an easy-to-use method for disrupting specific genes (Knott and Doudna, 2018). The coupling of CRISPR technology with pooled single-guide RNA (sgRNA) screening allows simultaneous knockout of thousands of individual genes in a large population of cells (Shalem et al., 2014; Wang et al., 2014), enabling unbiased reconstruction of biological pathways. Numerous CRISPR screens have probed pathways ranging from cell viability (Tzelepis et al., 2016; Wang et al., 2015) to virus infection (Han et al., 2018; Park et al., 2017), supporting the use of this system for exploring a wide-range of biology. More recently, in macrophages, CRISPR screens have been performed to identify novel regulators of infection and inflammation. Schmid-Burgk et al. (2016) carried out a genome-wide CRISPR screen to uncover previously unidentified regulators of the NLRP3 inflammasome. They found that knockout of NEK7 rescued macrophages from lethality and was associated with activation of the NLRP3 inflammasome. Yeung et al. (2019) performed a screen in macrophages to identify regulators of Salmonella infection. They identified NHLRC2, showing that it can play a role both in Salmonella infection as well as macrophage differentiation. Interestingly, many of the hits identified in their screen are within pathways with known chemical inhibitors providing avenues for future therapeutic targeting. Finally, a recent screen was conducted to identify regulators of Shigella infection in macrophages (Lai et al., 2020). Lai et al. identified host factors modulated by Shigella flexniri infection. They could show that inhibiting acetyl-coenzyme A (CoA) production caused by the infection is beneficial to the function of macrophages and limits the infection. In addition, knockout of the TLR1/2 pathway reduced inflammation, enhanced macrophage survival, and limited infection (Lai et al., 2020).
Whereas these screens that have been performed, to date, in macrophages are focused on a single readout, we have utilized pooled screening to address three questions simultaneously: (1) What genes are required for macrophage survival and proliferation? (2) How are those genes regulated? (3) What genes contribute to the downstream inflammatory signaling processes?
Our screens provide a resource that identifies genes essential to macrophage viability and provide insights into potential cis-elements within the 3′ UTRs of these genes that may reveal important means of regulation. Lastly, we identify previously unidentified positive and negative regulators of nuclear factor κB (NF-κB) inflammatory signaling. Unexpectedly, our screen uncovers a role for tumor necrosis factor (TNF) as a negative regulator of NF-κB and shows that this is functioning in an autocrine manner in macrophages. In a single screen, we bring together decades of literature on the complex regulation of TNF. Here, we demonstrate the power of CRISPR pooled screening to identify a plethora of genes with varied and critical roles in macrophage biology.
RESULTS
Pooled CRISPR Screen Identifies Macrophage-Specific Genes Involved in Viability
To define all genes essential for macrophage survival, immortalized bone-marrow-derived macrophage (iBMDM)-Cas9 cells were transduced (MOI = 0.3) with pooled lentivirus generated from our custom whole-genome sgRNA library containing ~270K individual sgRNAs targeting all RefSeq annotated coding genes and ~500 microRNAs (miRNAs; 12 guides per gene), along with ~5K non-targeting controls (Figures 1A and S1A; Tables S1 and S2). Cells were maintained at >1,000 cells per sgRNA throughout the screen. Cells were cultured for 21 days collecting genomic DNA from cells at day 0 and day 21 (Figure 1A). The libraries were prepared as described previously (Boettcher et al., 2019). Using the Mann-Whitney (MW) U test, we compared the sgRNA repertoire from day 21 to that from day 0 and identified significant genes (Figure 1B; Tables S3 and S4). We identified expected viability-related genes with roles in spliceosome, proteasome, and cell-cycle functions (Figure 1C). The majority of the top significant hits were genes essential for viability, while only 1% of genes were growth suppressors (Figure 1D), consistent with previous findings (Gilbert et al., 2014). We also analyzed the data using the model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) analysis pipeline (Tables S5 and S6). We obtained a strong overlap (88% of genes) using both MW and MAGeCK analyses and no significant difference in the identified top hits (Figure S1B); however, the MW U test identifies a much larger set of hits, possibly due to its sensitivity and, therefore, identifying more true positives.
Figure 1. Screen Identifies Macrophage-Specific Genes Involved in Viability.
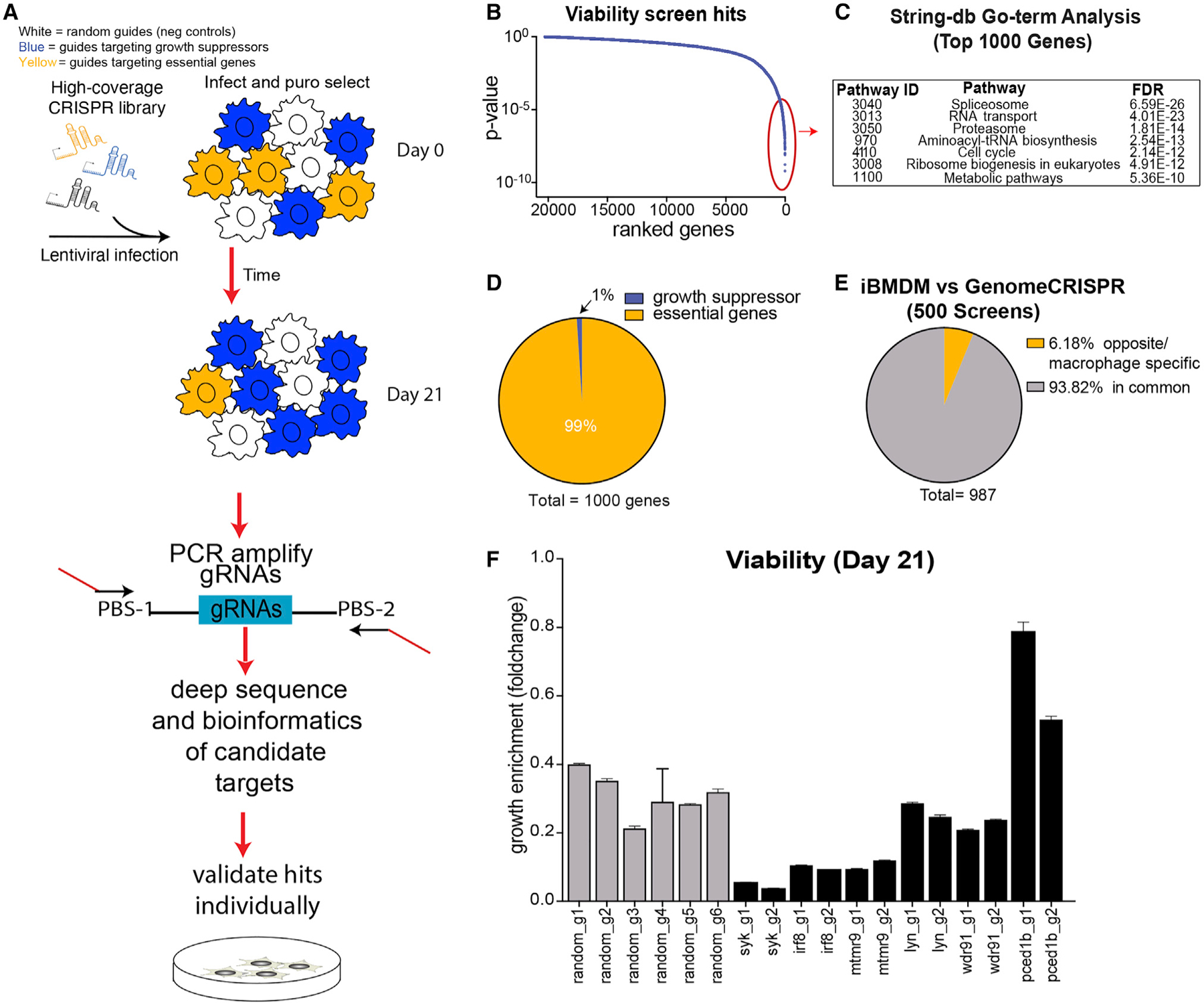
(A) Cas9-expressing iBMDM cells were infected with a whole-genome library targeting all RefSeq annotated coding genes (Table S1). Two days post-infection, cells were harvested for an initial day-0 time point and then again after 21 days in culture.
(B) The MW U test was performed comparing 12 sgRNAs targeting each gene to the nontargeting controls for samples collected at both day 21 and day 0 . Genes are displayed ranked by significance.
(C) GO-term analysis was performed on the top 1,000 significant hits using STRINGdb.
(D) We determined the number of essential genes (negative MW Z score) and total number of growth suppressor genes (positive MW Z score) and plotted them as a fraction of the total (total = 1,000 genes).
(E) Viability screen hits from our screen were compared to those from GenomeCRISPR, a collection of ~500 CRISPR screens (http://genomecrispr.dkfz.de/). Genes “in common” as well as genes showing “opposite/macrophage-specific” phenotypes are displayed.
(F) Cas9-expressing iBMDM cells were infected with sgRNAs targeting selected macrophage-specific viability genes. We combined cherry-positive cells (containing sgRNAs) with unedited cherry-negative cells at a 1:1 ratio and monitored growth of sgRNA-infected cells (cherry) relative to uninfected reference cells in a mix-cell growth assay for 21 days. Experiment was repeated 2 times, and a representative experiment is displayed.
Our library also contains within it random barcode sequences associated with each sgRNA (as described in Boettcher et al., 2019). Each sgRNA is associated with ~50 different barcodes. By analyzing the data with the barcodes, it enabled us to generate in-sample replicates providing greater statistical power to identify significant hits (Figure S1C; Tables S7 and S8). The barcodes were assigned to four bins, providing four in-sample replicates that were then used as the input into MAGeCK analysis. Figure S1C compares the single-sample analysis that identified 417 hits (false discovery rate [FDR] < 0.05) to the in-sample replicate analysis that identified 609 hits (FDR < 0.05), showing the power of utilizing the binning approach to increase statistical power.
We compared our viability screen hits to the GenomeCRISPR (http://genomecrispr.dkfz.de/) database, which includes a collection of ~500 CRISPR viability screens performed in ~421 cell types. Over 93% of genes from our screen overlapped those from the database (shown in gray, Figure 1E), suggesting that these are genes critical for a variety of biological processes common to all cell types. Interestingly, ~6% of genes identified showed the opposite MW Z-score phenotype in our screen compared to the database, suggesting that these genes have unique cell-type-specific functions in macrophages (shown in yellow, Figure 1E). Furthermore, these macrophage-specific essential hits included genes involved in NF-κB signaling (Figure S2), suggesting that these genes evolved functions involving not only survival but also inflammatory activation, a critical component of macrophage function. We individually cloned six candidate macrophage-specific essential guides and validated the phenotype using a mix-cell proliferation assay (Figure 1F). The mix-cell assay involved combining cherry-positive cells (containing sgRNAs) with cherry-negative cells at a 1:1 ratio and monitoring cell growth over time as assessed by changes in the ratio of cherry-positive to cherry-negative cells. As shown in Figure 1F, we confirmed four of the six selected candidate genes. We showed that tyrosine-protein kinase (Syk), Interferon regulatory factor 8 (Irf8), and myotubularin-related protein 9 (Mtmr9) are macrophage-specific essential genes, as targeting the coding sequence of these genes resulted in decreased fitness. While PC-esterase domain containing 1B (Pced1b) is a growth suppressor in macrophages, knocking it out resulted in increased fitness of the cells.
CRISPR TARGETING OF THE 3′ UTRs OF ESSENTIAL GENES IDENTIFIES CIS-REGULATORY ELEMENTS
Untranslated regions (UTRs) offer a critical source of regulation for messages through specific cis-elements, which can bind miRNAs and/or proteins to regulate pathways including RNA decay and translation (Mayr, 2019). To probe for novel cis-elements within 3′ UTRs, we specifically targeted the 3′ UTR within known essential genes. For these essential genes, we expected that guides targeting coding exons would cause a decrease in fitness. In contrast, guides targeting 3′ UTR cis-elements that result in an increase in fitness will represent regions containing regulatory cis-elements with negative roles on gene expression (miRNA binding sites, adenylate-uridylate-rich elements [AU-rich elements] affecting stability, etc.). We assessed the phenotypes of 3′ UTR-targeting sgRNAs and found an overall neutral average phenotype for these guides (Figure 2A), suggesting that the majority of the sites we targeted did not contain any cis-regulatory elements. However, a subset of these 3′ UTR-targeting guides demonstrated phenotypes of >3-fold change in both directions, suggestive of sites that contain regulatory elements that could improve or decrease fitness of the cells (Figures 2B–2D; Table S9). We focused on 3′ UTR guides that showed positive (>3-fold) enrichment for genes whose coding-targeting guides demonstrated significant negative enrichment (Figures 2B–2D, red stars). We reasoned that these 3′ UTR-targeting guides may disrupt significant cis-elements that help explain the opposing phenotype we observed (Figures 2B–2D, red stars). One possible concern with this analysis is that it is limited to a single sgRNA targeting the sites within the 3′ UTR (compared to 10–12 within the screen for each coding gene target), which could lead to false positives. To mitigate this, we made use of the internal barcodes associated with each individual sgRNA. Identifying a sgRNA hit associated with multiple individual barcodes reduces the possibility of the hit being a false positive. In one example, the viability phenotype could be caused by integration-specific effects of an sgRNA on neighboring genes. As a control, we analyzed all barcodes associated with all random control sgRNAs, comparing the day-0 to day-21 viability screens, and can show high correlation, with an R2 of 0.814. This shows that the distribution of reads for barcodes of random control genes do not change across the course of the experiment, making counting barcodes a valid approach (Figure 2E). Therefore, we counted all unique barcodes associated with sgRNAs in selected genes or random controls and compared the reads associated with these genes on day 21 to those on day 0. As expected, there is no difference in the read count from day 21 to day 0 for the random controls (shown in gray, Figure 2F). In contrast, we see positive enrichment for genes (shown in black, Figure 2F) whose coding-targeting guides demonstrated the opposite effect, which confirmed our previous observations in Figures 2B–2D.
Figure 2. Targeting of the 3′ UTRs of Essential Genes to Probe for Previously Unidentified cis-Regulatory Elements.
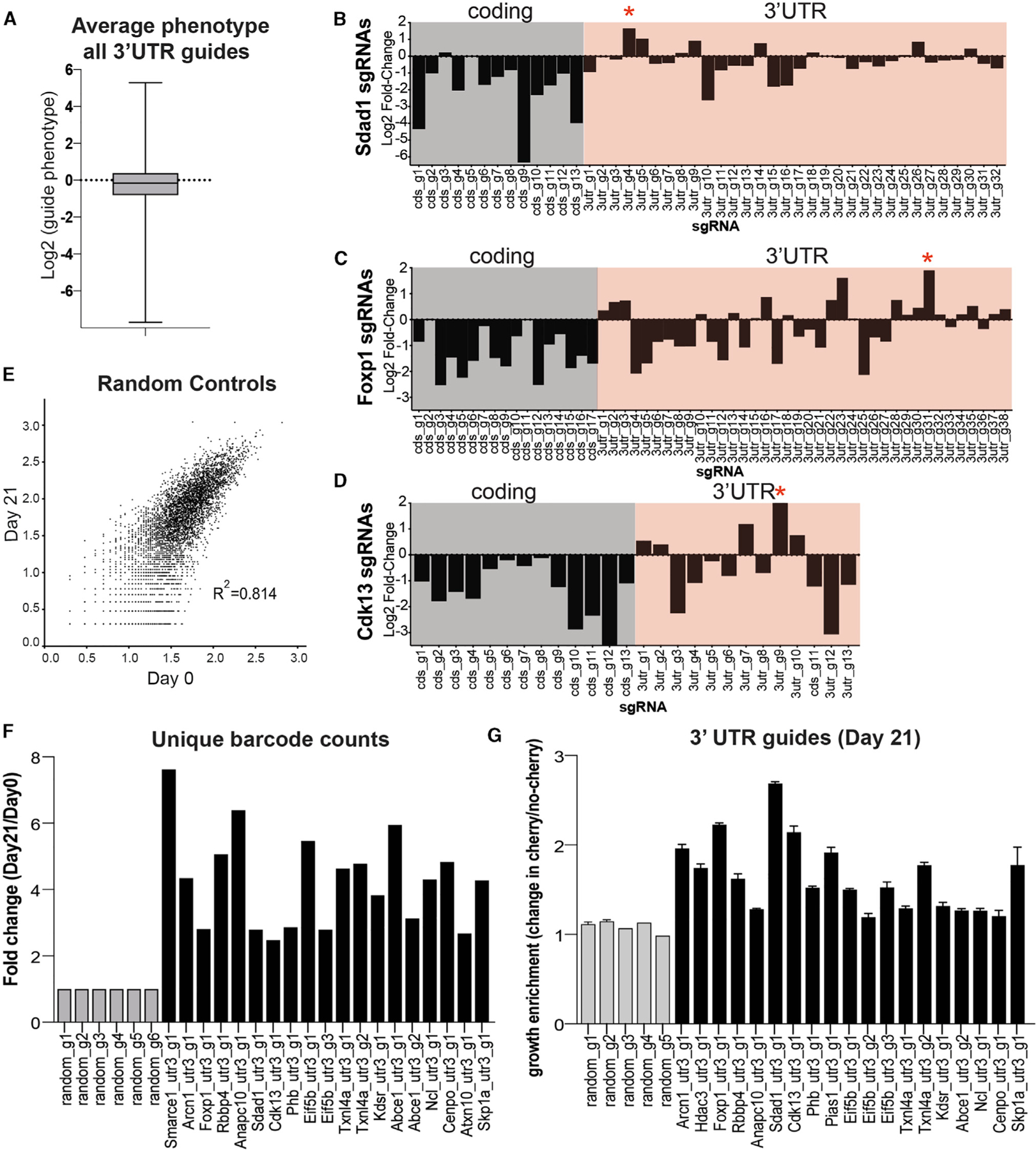
(A) Average phenotypes for all 3′ UTR-targeting guides was plotted. Error bars represent standard deviation of all sgRNAs.
(B–D) We summarize the phenotypes for coding-targeting guides (gray) and 3′ UTR-targeting guides (pink) for select genes: Sdad1, Foxp1, and Cdk13.
(E) Scatterplot. The x-coordinates represent the number of unique barcodes associated with each random sgRNA at day 0. The y-coordinates represent the number of unique barcodes associated with each random sgRNA at day 21 (R2 = 0.814).
(F) Unique barcode counts were obtained for the indicated genes, including random controls. The fold change of these counts from day 0 to day 21 was calculated.
(G) We selected 3′ UTR-targeting guides that demonstrated positive enrichment, opposite to their coding-targeting guides, which had negative enrichment. We validated select guides by monitoring growth of sgRNA-infected cells (cherry) relative to uninfected reference cells in a mix-cell growth assay. Error bars represent standard deviation of three technical replicates.
We individually cloned selected candidate 3′ UTR guides and validated the phenotype using a mix-cell proliferation assay as described previously (Figure 2G). In all the selected hits we could validate, we confirmed that elements targeted within 3′ UTRs resulted in an increase in fitness, while those targeting within the coding sequence resulted in a decrease in fitness. The phenotypes for these 3′ UTR-targeting guides provide a resource of potentially important cis-elements involved in mRNA stability, and further work could involve interrogating whether these sites are miRNA targets or targets of other proteins involved in RNA decay or translation processes.
FACS-Based Reporter Screen Identifies Positive and Negative Regulators of NF-κB
Macrophages are critical effectors of the inflammatory response, which involves transcription factors including NF-κB (Liu et al., 2017). We had previously developed NF-κB reporter iBMDMs, adding 5 × NF-κB-binding motifs (GGGAATTTCC) upstream of the minimal cytomegalovirus (CMV) promoter-driving green flourescent protein (GFP) and demonstrated lipopolysaccharide (LPS)-dependent activation of GFP fluorescence (Covarrubias et al., 2017). We lentivirally introduced Cas9 into these cells and confirmed its activity (iBMDM-NF-κB-Cas) (Covarrubias et al., 2017). Here, we performed a fluorescence-activated cell sorting (FACS)-based sorting screen using iBMDM-NF-κB-Cas cells and infected with the same library as outlined in Figure 1A. After the library was established in the cells for 7 days, we stimulated with LPS for 24 h (Figures 3A, S3A, and S3B; Table S10). We sorted the top/bottom 20% of GFP-expressing cells and collected approximately 100 cells per sgRNA (~27 million cells for each top/bottom sort), with the aim of identifying both positive (bottom 20%) and negative (top 20%) regulators of the pathway (Figures S3A and S3B; Table S11). Due to the inherently noisy nature of pooled screening, we chose to perform a MW U test to identify significant genes, comparing GFP-low to GFP-high sorted samples, and significant genes were ranked by p < 0.01 (Figure 3B) (Kampmann et al., 2013). As expected, we found several positive controls, including EGFP and known regulators myeloid differentiation primary response 88 (Myd88) and Rela (NF-κB/p65) in our top hits (Figure 3C). We performed Gene Ontology (GO)-term enrichment analysis for the top 150 significant positive regulators and found enrichment for pathways that included “NF-kappaB signaling” (Figure 3D). Within the top 150 genes, we identified numerous genes known to be involved in the Toll-like receptor (TLR)/NF-κB signaling pathway, which include Tlr4 (LPS receptor) and Rela (NF-κB/p65) (Figure 3E; Table S12), confirming that the screen was a success. We plotted the average phenotypes (top 3 guides) for our top 40 candidates, which showed that the average sgRNA enrichments were significant (Figure 3F). Top candidates were localized throughout the cell (Figure 3G; Table S13), including in the extracellular compartment. Numerous positive and negative regulators of NF-κB have been identified by their differential expression upon NF-κB activation (Bhatt and Ghosh, 2014). Using previously published data (Zhang et al., 2017), we examined the top 50 negative and positive regulators and found that the majority were not differentially expressed during LPS stimulation and, therefore, could have been missed by previous approaches as regulators of the pathway (Figure S3C). NF-κB has been demonstrated to activate genes that function in positive- or negative-feedback regulation of the pathway (Oeckinghaus and Ghosh, 2009). We assessed whether NF-κB (p65) bound to the promoters of our top candidates using published p65 chromatin immunoprecipitation sequencing (ChIP-seq) data (Lam et al., 2013) (Figures S3D and S3E). We found that p65 bound 42% and 54% of the top positive and negative regulators, respectively (Figures S3D and S3E), further supporting the idea that a significant number of regulators of NF-κB can, themselves, be regulated by NF-κB. We validated our top candidates by re-cloning the top two performing sgRNAs per candidate, generating individual cell lines for each sgRNA, followed by lentiviral infection, selection, and LPS stimulation for 6 h (Figure 3H; Table S14). The readout for our secondary validation experiments involved measuring Il6 by qPCR. Il6 is a well-known downstream target of NF-κB and a crucial gene in controlling inflammation (Figure 3H). As expected, ablation of our negative regulators resulted in increased Il6 expression, while targeting of positive regulators led to decreased levels of Il6 relative to non-targeting controls (Figure 3H). In summary, we performed a genome-wide screen and identified 50 previously unidentified positive and 65 negative regulators of NF-κB signaling. To date, there are 120 known regulators of NF-κB, and our screen has added 115 additional regulators (p < 0.01) to the pathway. These will provide a rich source of information going forward to better understand the complex pathways that control inflammation.
Figure 3. Screen Identifies Positive and Negative Regulators of NF-κB signaling.
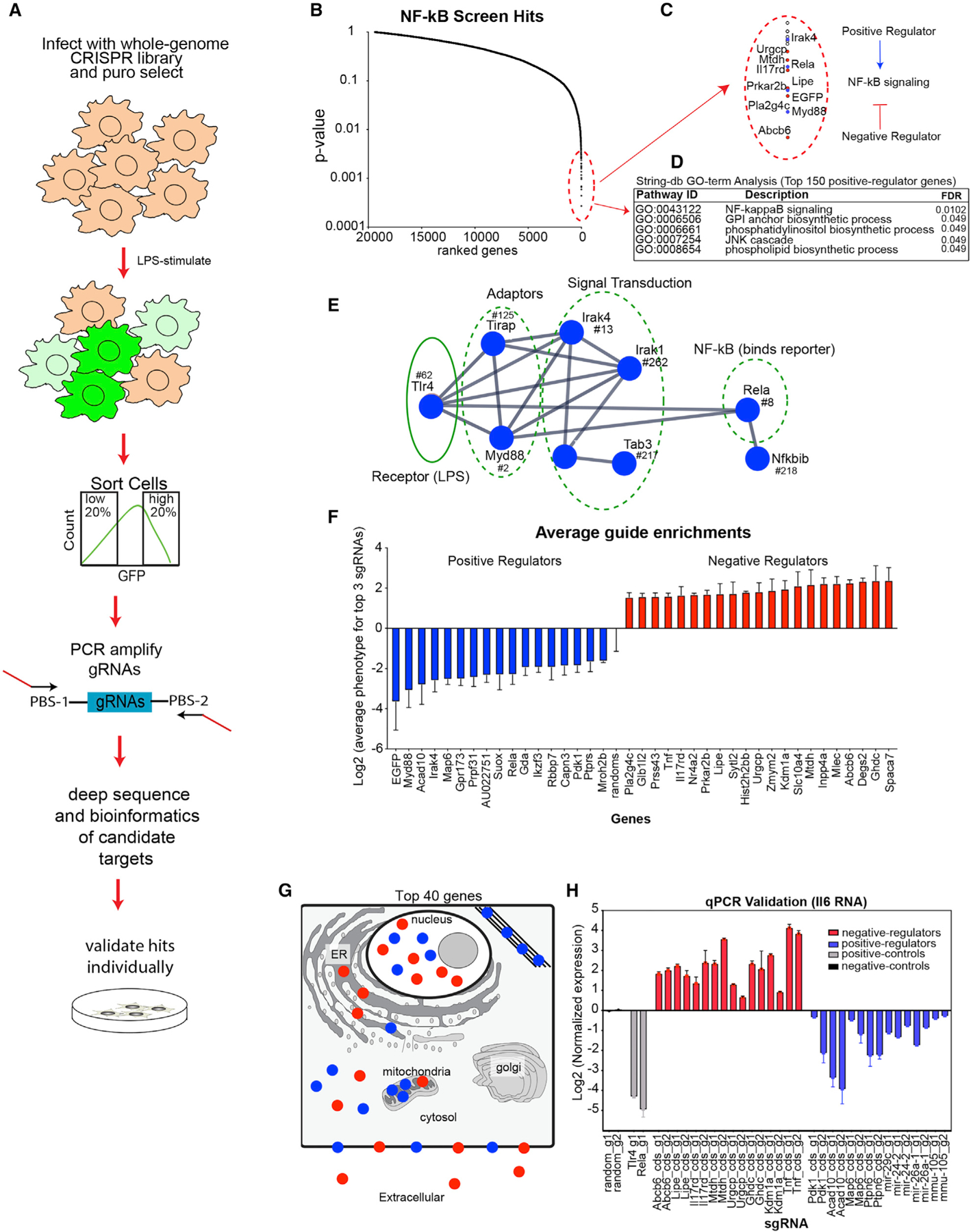
(A) Overview of the NF-κB screen: sgRNA-library-infected iBMDM-NF-κB-Cas9 cells were stimulated with LPS (200 ng/mL) for 24 h before sorting the top and bottom 20% of GFP-expressing cells. Cells were collected and processed as described in STAR Methods.
(B) MW U test was performed comparing 12 sgRNAs targeting each gene to the non-targeting controls for GFP-low versus GFP-high sorted samples. Significant genes are displayed, ranked by significance.
(C) Zoom-in of the top screen hits, displaying positive regulators (blue) and negative regulators (red). Diagram depicts positive- and negative-regulation NF-κB signaling.
(D) GO-term analysis was assessed for the top 150 positive regulators using STRINGdb.
(E) Connectivity was determined by STRINGdb for the top 150 positive-regulator candidates.
(F) Average sgRNA enrichment for the top 3 sgRNAs was calculated for the top 40 screen hits. Error bars represent standard deviation of three biological replicates.
(G) Predicted protein localization was determined for the top 40 most significant genes using UniProt’s COMPARTMENTS database (https://compartments.jensenlab.org/Downloads).
(H) Selected candidates were infected with either control (random) or candidate-specific sgRNAs and were stimulated for 6 h with LPS, before RNA harvest. qPCR-based validation was performed by conducting qRT-PCR for Il6 RNA relative to Gapdh. Experiment was repeated 3 times, and a representative experiment is displayed. Error bars represent standard deviation of three technical replicates.
MEMBRANE-BOUND TNF ALPHA (TNF-α) ACTS AS A STRONG NEGATIVE REGULATOR OF THE NF-κB PATHWAY
TNF-α is a well-known pro-inflammatory cytokine with established roles in driving NF-κB-related inflammation (Bradley, 2008). Furthermore, anti-TNF therapy is a proven method for the treatment of various inflammatory-related diseases (Ma and Xu, 2013). Given its established role as a soluble protein functioning as a positive regulator of NF-κB, it was surprising to discover that our pooled-based screen approach identified TNF as a strong negative regulator of inflammation (Figures 3F and 3H). We confirmed TNF editing by stimulating control or anti-TNF edited cells with LPS for 24 h before collecting supernatant and analyzing TNF protein via ELISA. We found that TNF was undetectable in the supernatant after LPS stimulation (Figure 4A). We also confirmed near-complete ablation of TNF via intracellular staining (Figure 4B). Our screen suggested a localized function for TNF, which would be incompatible with its soluble state. Interestingly, TNF can exist as both soluble and membrane bound (Ardestani et al., 2013). We confirmed the presence of membrane-bound TNF on the surface of our iBMDMs and found maximal surface TNF at 6 h post-LPS stimulation (Figure 4C). TNF mediates its inflammatory effect via binding to its receptor Tnfrsf1a (p55) (Bradley, 2008). Our screen confirmed Tnfrsf1a (p55) as a positive regulator of NF-κB in contrast to what was found for Tnf (Figures 4D and 4E). However, TNF can bind to two different receptors, Tnfrsf1a (p55) and Tnfrsf1b (p75), which may have opposing functions (Peschon et al., 1998). While the sgRNAs targeting p75 in our screen show a trend toward it being a negative regulator similar to TNF, it was not significant (Figure 4D). However, we were able to confirm by qPCR that targeting p75 can result in an increase in Il6 (Figure 4F) similar to that observed when TNF is knocked down (Figures 4D and 4E). Interestingly, 6 h post-LPS stimulation, levels of TNF and Tnfrsf1b (p75) increased 11-fold and 66-fold, respectively, while the levels of Tnfrsf1a (p55) increased a moderate 4-fold (Figure 4G). We evaluated whether the enhanced activation of NF-κB in TNF-edited cells could be rescued by mixing these cells with unedited (TNF-expressing) cells. We combined cherry-positive cells (containing TNF guide RNAs) with unedited cherry-negative cells at a 1:1 ratio. Mixed cells were LPS stimulated for 24 h before FACS analysis to measure GFP mean fluorescence intensity (MFI) (NF-κB activation) (Figure 4H). The enhanced NF-κB activation in TNF-edited cells could not be rescued by mixing these cells with unedited cells (even when we increased the cherry-negative cells to >75%). These data suggest that TNF has autocrine properties, acting within the cell from which it is produced, and neither soluble TNF production or membrane-bound TNF from neighboring cells can reverse the increase in inflammatory signaling (Figure 4I). The expression profiles of TNF, TNFRSF1A, and TNFRSF1B were mirrored in the human THP1 monocytic cell line stimulated with PAM3CSK4 (TLR1/2 agonist), suggesting that this specific regulation of TNF is conserved (Figure S4). Here, we confirm that, indeed, it is membrane bound TNF that is functioning as a negative regulator of NF-κB, presumably through interactions with p75. Remarkably, our single screening assay could provide insight into this complex signaling cascade and bring together decades of different approaches to reveal the complexity of TNF signaling in macrophages involving membrane-bound forms of TNF in addition to the roles of the respective receptors.
Figure 4. Screening Identifies Negative-Feedback Regulatory Loop Used by the Tumor Necrosis Factor (TNF) to Regulate Inflammation.
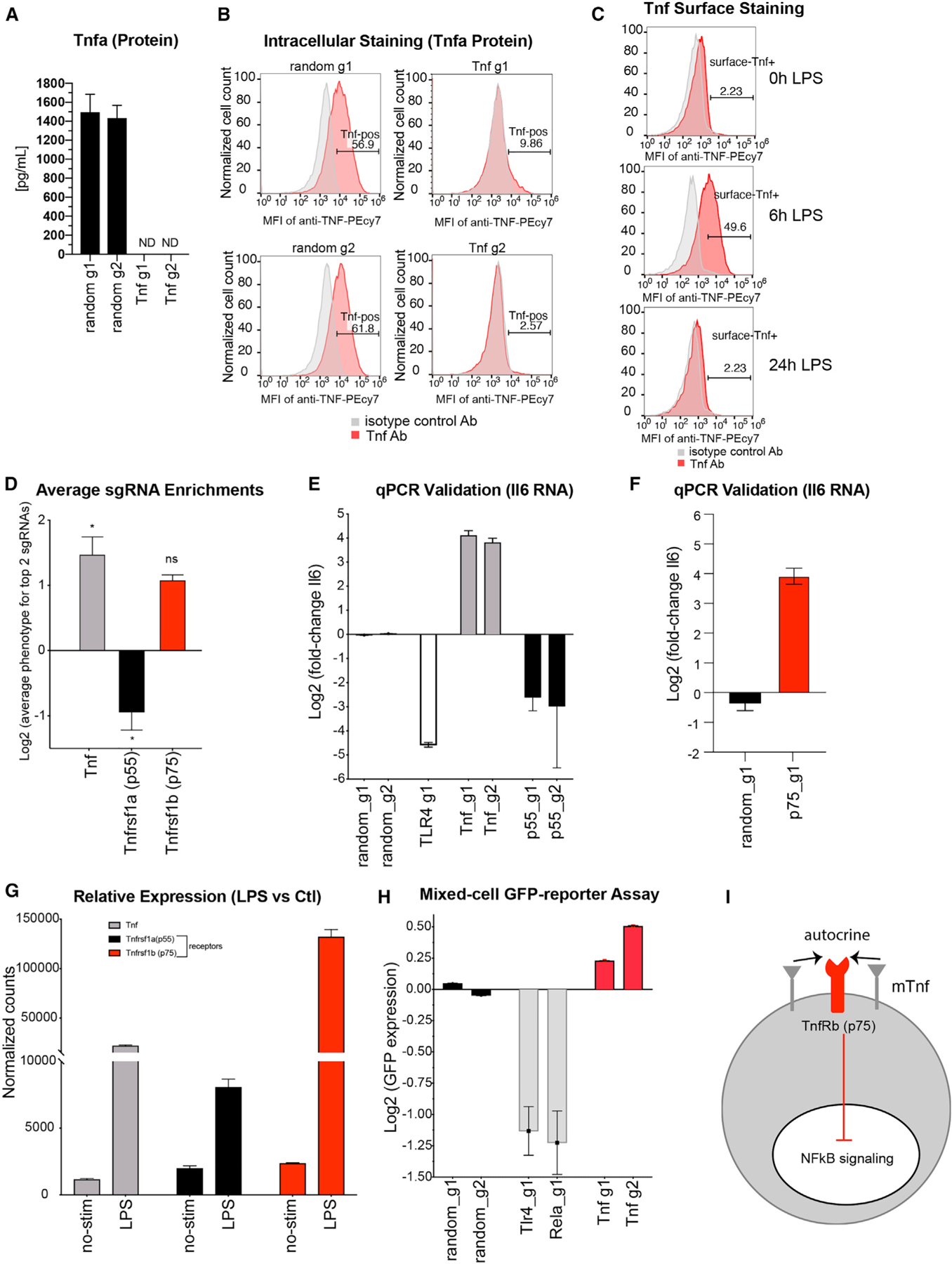
(A) ELISA was performed in control or TNF-edited iBMDMs stimulated with LPS for 24 h. Supernatant was harvested, and levels of TNF were quantified. Error bars represent standard deviation of three biological replicates.
(B) Control or TNF-edited iBMDMs were stimulated with LPS followed by brefeldin A treatment and stained to assess intracellular TNF levels by flow cytometry.
(C) iBMDMs were stimulated for the indicated time points (LPS for 0, 6, and 24 h), and cell surface expression of TNF was measured by flow cytometry.
(D) Average sgRNA enrichment for the top 2 sgRNAs was calculated for TNF and its receptors: p55 and p75. Error bars represent standard deviation of two biological replicates.
(E and F) qRT-PCR was performed for Il6 RNA in iBMDMs edited with either control, TNF, p55 (E), or p75 (F) sgRNAs and stimulated with LPS for 6 h. Error bars represent standard deviation of three biological replicates.
(G) Mean fragments per kilobase of transcript per million mapped reads (FPKM) values (4 biological replicates) are plotted for genes Tnf, Tnfrsf1a, and Tnfrsf1b for both un-stimulated (“no-stim”) or LPS-stimulated (24 h) cells. Error bars represent standard deviation of four biological replicates.
(H) We combined cherry-positive cells (containing Tnf sgRNAs) with unedited cherry-negative cells at a 1:1 ratio. Mixed cells were stimulated with LPS for 24 h before FACS analysis to measure GFP mean fluorescence intensity (MFI) as proxy for NF-κB activation. Error bars represent standard deviation of three biological replicates.
(I) Model of localized TNF negative regulation of NF-κB signaling.
DISCUSSION
Here, we utilize CRISPR-based pooled genetic screening to reveal genes important for both macrophage viability and inflammation. For the viability screen, we utilized both MW U test analysis as well as the MAGeCK analysis pipelines to identify significant hits. 88% of significant genes identified in the MAGeCK analysis were also identified in the MW U test. A larger proportion of significant hits were identified by MW test, which could represent true positives or that the more stringent MAGeCK pipeline identifies fewer false positives. We identified macrophage-specific viability genes enriched in NF-κB signaling (Figures 1F and S2). Additionally, we identified and validated viability phenotypes for 3′ UTR-targeting guides, which may reveal important cis-elements involved in mRNA stability (discussed further later). In a separate screen, we utilized our NF-κB -reporter cells to perform a FACS-based screen, resulting in the identification of positive and negative regulators of NF-κB signaling. We go on to describe an unexpected role for TNF as a locally acting negative regulator of NF-κB. Very few miRNAs were identified as hits in either screen (Tables S3, S4, S9, S10, and S11). Although we targeted ~500 miRNAs, it is possible that many of these are not expressed in macrophages or that the sgRNAs targeting the miRNAs did not work.
Macrophage-Specific Genes Involved in Viability
Numerous viability screens have been performed in a wide range of cell types and have demonstrated that the catalog of essential genes can vary significantly among distinct cell types (Wang et al., 2014, 2017). Cell-type-specific differences in viability can be due to genetic differences that could render some pathways inactive, forcing dependency on other pathways. Indeed, Wang et al. (2017) compared essential genes across 14 acute myeloid leukemia (AML) lines and found certain genes to be essential only for a specific subset of the AML lines with certain genotypes. Moreover, a typical cell expresses approximately two thirds of its genes (Hart et al., 2013). Therefore, whether a gene or set of genes is essential can be context specific, varying upon different growth conditions or treatments, etc. (Hart et al., 2015; Viswanatha et al., 2018). In our screen, we identified 61 genes with distinct essentiality in macrophages compared to a database collection of ~421 cell-line CRISPR screens (Figure 1E). We selected 6 of the top candidates that acted as either essential genes or growth suppressors and could validate 4. Not surprisingly, we identified and validated the myeloid-specific transcription factor Irf8 as a macrophage-specific essential gene (Figure 1F). Interestingly, Irf8 has roles in both myeloid cell viability and inflammatory response (Langlais et al., 2016). Additionally, the macrophage-specific essential genes included ones involved in NF-κB signaling, suggesting that this pathway, similar to Irf8, may be important for both viability and inflammatory activation (Figure S2). Indeed, a significant fraction of them (~10 proteins)—including Syk and Mtmr9, which we validated (Figure 1F)—are predicted to physically interact supporting a macrophage-specific regulation of protein complexes that control cell viability. In summary, understanding the pathways that control viability in macrophages is essential for development of novel gene targets to control macrophage proliferation in scenarios where the inflammatory response is not properly controlled.
3′ UTR-Targeting Guides and Identification of Potential cis-Elements
UTRs of messages are important for regulating stability, translation, and localization (Mayr, 2019). Sequences within the UTRs (cis-elements) can function in miRNA binding, structure, and/or protein binding (Mayr, 2017). Furthermore, cell-type-specific expression differences in miRNAs and RNA binding proteins (RBPs) allow for specificity in regulating mRNAs (Erson-Bensan, 2016). Given the rich source of regulation that occurs within the 3′ UTR, we built into our library design sgRNAs targeting 3′ UTRs within known essential genes with the goal of systematically identifying cis-elements that contribute to the regulation of essential genes. For these genes, sgRNAs targeting coding sequences resulted in decreased fitness, as expected. Within these genes, we focused on 3′ UTR-sgRNAs that resulted in increased fitness, which could represent the disruption of destabilizing cis-elements, such as miRNA binding sites, sites of interaction with RBPs or AU-rich elements, etc. CRISPR targeting of these destabilizing cis-elements may result in stabilization of the mRNA. We determined whether there were any overlapping miRNA target sites using TargetScan, but we did not find any with significant overlap, suggesting that there are other mechanisms of regulation at play besides miRNA targeting. However, Cdk13-utr3-g1 and Pias1-utr3-g1 (Figure 2G) did target regions near (<50–80 bp) the miRNA binding sites for mir-124 and mir-10, respectively, which could potentially disrupt secondary structure, mir binding, or both (Table S9). Another 3′ UTR-targeting guide 1 for Arcn1, is predicted to target near a mushashi element, which is known to negatively regulate RNA stability (Bennett et al., 2016). Given that the majority of our 3′ UTR targeting guides resulted in no phenotype, an alternative method to try in future studies would be to use 2 sgRNAs to create larger deletions (Zhao et al., 2017). In summary, we have functionally validated potential 3′ UTR cis-elements and provided a rich resource for future work aimed at dissecting how these elements contribute to gene expression.
Dissection of the Complex Biology of TNF
We utilized the MW U analysis to identify hits from our FACS-based NF-κB reporter screen. Sorting screens are inherently noisier than viability screens, due to the fact that significant hits are going to be proportional to the sgRNA enrichment, and with our single (LPS) treatment of cells, there is no ability to enrich over time. Here, we activated the NF-κB iBMDM reporter cells for 24 h with LPS and then sorted for the GFP low and high hits (top and bottom 20%) that represent positive and negative regulators of the pathway. Newer tools are beginning to be developed to help with the analysis of these complex pooled sorting-based screens. Recently, de Boer et al. (2020) published a tool, “MAUDE,” that is specifically designed for sorting screens, which could be beneficial for future studies of these complex assays. Nevertheless, we could show, using MW analysis, that the GO terms for the top 150 hits included “NF-kappaB signaling,” and we confirmed 16 additional NF-κB regulators using individually cloned lines measuring Il6 production as a secondary readout.
One of the surprising findings from our NF-κB screen was the identification of TNF as a negative regulator of NF-κB (anti-inflammatory). We were surprised for two reasons: (1) TNF is an extensively studied pro-inflammatory cytokine with established roles in driving NF-κB-related inflammation (Bradley, 2008). (2) TNF is a secreted protein; therefore, a pooled CRISPR screen would not be expected to capture its biology. Numerous studies spanning decades of research have revealed that TNF biology is much more complex. TNF can exist as both soluble (17 kDa) and membrane-bound (26 kDa) forms, and multiple groups have shown that membrane-bound TNF functions as a negative regulator of inflammation in contrast to its soluble form (Alexopoulou et al., 2006; Ardestani et al., 2013). The dual functions of TNF can also be explained, in part, by its binding to two receptors: Tnfrsf1a (p55) and Tnfrsf1b (p75), which have opposing effects on inflammation (Peschon et al., 1998). Interestingly, the membrane-bound form of TNF has been shown to preferably bind to the inhibitory receptor, p75, allowing for localized regulation of inflammation (Grell et al., 1995). Here, we reveal the complex regulatory biology of TNF, providing evidence that membrane-bound TNF functions as a negative regulator of NF-κB likely through its binding to p75 in macrophages. Our results also support a model in which membrane-bound TNF can function in an autocrine manner, acting within the cell that produces it. In this study, we bring together decades of research on TNF biology and its role in inflammation using this single-screening approach. Anti-TNF therapy remains one of the most effective methods for the treatment of various autoimmune diseases, including rheumatoid arthritis (RA) and irritable bowel disease (IBD) (Hyrich et al., 2009; Peyrin-Biroulet, 2010), yet as many as 20%–40% of patients do not respond to treatment (Lopetuso et al., 2017). Our findings show that editing of TNF resulted in elevated Il6 levels, which is another important pro-inflammatory cytokine and might explain the lack of response to anti-TNF therapy. Yimin and Kohanawa (2006) demonstrated that a TNF‒/‒knockout mouse showed elevated levels of Il6, which is consistent with our findings. More importantly, they presented data showing that production of TNF and IL6 can be negatively regulated by each other (Yimin and Kohanawa, 2006). Therefore, the targeting of TNF could lead to elevated levels of Il6, which may result in elevated inflammation in patients. Our data showed that editing p55 inhibits NF-κB-driven Il6 production (Figures 4D and 4E), while editing p75 caused an increase in Il6 (Figure 4F). Blocking p55 (via antibody or small molecule) could be an alternative that could block the pro-inflammatory effects of p55 while allowing Tnf to, instead, bind to p75 to promote anti-inflammatory signaling (Yang et al., 2018). In summary, with one pooled CRISPR screen, we have revealed interesting complexities of TNF regulation and have presented evidence for alternative therapeutic strategies.
Future Directions
Here, we have demonstrated the power of CRISPR screening in revealing important macrophage biology probing both viability and inflammatory pathways. Macrophages are critical cells of the innate immune system providing one of the first lines of defense against invading microbes. The ability for macrophages to function optimally requires their ability to proliferate and migrate to reach the site of infection and appropriately engage their inflammatory program. Here, we found 60 macrophage-specific viability genes and uncovered 115 additional regulators of NF-κB. The future characterization of these genes will likely yield novel regulatory insights into the complex regulation of viability and inflammatory pathways. From a therapeutic point of view, it would be interesting to explore whether there are drugs that may target some or any of our screen candidates (Wishart et al., 2018). In conclusion, we have revealed important biology insights related to macrophage function and believe that this work represents a significant resource for the macrophage research community.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Susan Carpenter (sucarpen@ucsc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All sequencing data generated from CRISPR screens and RNA-seq reported in this paper have been deposited to GEO under accession codes GSE138786 and GSE138788.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Male immortalized bone-marrow-derived macrophages (iBMDMs) with the NF-κB reporter and Cas9 (iBMDM-NFKB-Cas9) cells (previously described in Covarrubias et al., 2017). Cas9 activity was validated via GFP kd (~75% kd) prior to beginning the screen. Cells were cultured in DMEM, supplemented with 10% low-endotoxin fetal bovine serum (ThermoFisher) and 1X penicillin/streptomycin and incubated at 37°C in 5% CO2.
THP-1 cells were cultured in RPMI 1640 supplemented with 10% low-endotoxin fetal bovine serum (ThermoFisher), 1X penicillin/ streptomycin and 2-mercaptoethanol (0.05 mM, Sigma-Aldrich, M6250), and incubated at 37°C in 5% CO2.
METHOD DETAILS
sgRNA library design and cloning
We created genome-scale sgRNA library consisting of over 270,000 total sgRNAs (12 sgRNAs per gene) targeting every RefSeq-an-notated (mm9) coding gene, as well as all microRNAs and select 3′UTRs. The library contains > 5,000 non-target control sequences (NTC). The earliest possible “constitutive” exon of each transcript variant was targeted. The criteria for sgRNA selection and the cloning strategy protocol have been previously described (Boettcher et al., 2018, 2019). All sgRNA sequences are shown in Table S1.
Lentiviral production
HEK293T cells were seeded at 6,000,000 cells per plate in 15 cm dishes in 20 mL media (DMEM, 10% FBS) and incubated overnight at 37°C, 5% CO2. The next morning, 8 µg sgRNA library plasmid, 4 µg psPAX2 (Addgene #12260), 4 µg pMD2.G (Addgene #12259) and 80 µL lipofectamine2000 (Invitrogen) were mixed into 1 mL serum-free OptiMEM (GIBCO), vortexed and incubated for 20 min at RT and added to the cells. At 72 h post-transfection, supernatant was harvested, passed through 0.45 um filters (Millipore, Stericup) and aliquots were stored at ‒80 C.
CRISPR Screen
iBMDM-NFKB-Cas9 cells were infected with the sgRNA genome-scale library at a at low multiplicity of infection (MOI = 0.3). Three days post infection, cells were puromycin-selected (10 µg/ml) for 5 days to obtain cherry-positive (sgRNA) cells and were maintained at > 1000X coverage at all times.
Growth screen
Prior to puro-selection, we collected a day 0 time point, consisting of 1000X coverage (270 million cells). We then collected a day 21 time point (also 1000X coverage). Cells from both time points were cryo-preserved in 90% FBS, 10% DMSO for later processing.
FACS screen
Library infected and selected iBMDM-NFKB-Cas9 cells were expanded to 2000X coverage. Cells were stimulated with 200 ng/ml of LPS for 24 h to induce expression of GFP (NF-κB responsive). Prior to sorting, cells were collected in FACS buffer (1XPBS, 1%FBS, 5mM EDTA). Stimulated cells were analyzed by flow cytometry alongside unstimulated cells to ensure the mean fluorescence intensity (MFI) of stimulated cells was > 10-fold compared to unstimulated cells. All flow cytometry experiments and screening were conducted on a BD FACSAria II. GFP was excited using a 488-nm laser and detected using a 525/50-nm filter. Sorting was conducted using 4-way purity into 2 tubes and a 100-µm nozzle. Cells were gated by forward (FSC-A) and side scatter (SSC-A) for live cells, then for single cells using FSC-A/FSC-H. Lastly, we evaluated GFP expression (SSC versus GFP), FACS sorted and collected the top/bottom 20% into separate tubes. At least 100 cells/sgRNA (100X coverage) for each sorted population were collected and cryo-preserved in 90% FBS, 10% DMSO for later processing.
sgDNA processing, PCR and sequencing. Genomic DNA was collected from cell pellets (270 millions cells, 1000X coverage) or (27 millions cells, 100X coverage for sorted cells) and was extracted by methods described previously (Boettcher et al., 2018, 2019). A nested PCR strategy was used to 1) allow amplification sgRNA repertoire and 2) to add appropriate Illumina adapters for NGS (detailed protocol is described in Boettcher et al. (2019). For the 100X coverage sorted samples, we scaled the gDNA extraction volumes 1:5 (i.e., 2ml instead of 20ml). Quality and purity of the PCR product were assessed by bioanalyzer (Agilent), and sequencing was performed on an Illumina HiSeq 2500 platform using paired end 50 kits with the custom sequencing primer 5′-GAGACTATAAG-TATCCCTTGGAGAACCACCTTGTTGG-3′ for reading the sgRNA sequence. Data was submitted to GEO. All tables can be accessed through Mendeley Reserved DOI: 10.17632/vtvxykv2cr.1.
Macrophage specific viability genes and 3′-UTR guide validation (Mix-cell growth assay)
sgRNA-infected cells (cherry-pos) were mixed with uninfected cells (cherry-neg) at a 1:1 ratio in triplicate. We used Flow cytometry to monitor the ratio of cherry-pos to cherry-neg cells at 0- and 21-days post plating. All validation cytometry was performed on the Attune NxT Flow Cytometer.
NF-κB guide Validation (qRT-PCR)
iBMDM-NF-κB-Cas cells infected with indicated guide-expressing lentivirus and were stimulated with LPS (200 ng/ml) for 6 h prior to harvesting for RNA. Total cellular RNA from BMDM cell lines was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research) according to manufacturer’s instructions. RNA was quantified and controlled for purity with a nanodrop spectrometer. (Thermo Fisher). For RT-qPCR, 500–1000 ng were reversely transcribed (iScript Reverse Transcription Supermix, Biorad) followed by RT-PCR (iQ SYBRgreen Supermix, Biorad) using the cycling conditions as follows: 50C for 2 min, 95°C for 2 min followed by 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 45 s. qRT-PCR primer sequences are list below.
ELISA Analysis
For the ELISA, iBMDMs were stimulated with LPS for 24 h and supernatant was collected from triplicate wells. Supernatant was diluted 1:3 and Tnf-alpha levels were measured using the mouse TNF-alpha DuoSet ELISA (R&D Systems) kit following manufacturer’s protocol.
Antibody staining for FACS
For intracellular staining, iBMDMs were LPS-stimulated for 0 or 6 h and were treated with Brefeldin A for the last 5 hours of stimulation. Cells were then collected, fixed with 4% PFA and permeabilized with perm buffer (3% BSA, 0.2% Triton-X, 1XPBS), followed by antibody staining with PEcy7 anti-mouse Tnf-alpha (1:150, Thermofisher) or isotype control (1:150, Biolegend). For surface staining, iBMDMs were LPS-stimulated for 0, 6, 24 h. Cells were collected in sorting media (2% Fetal Calf Serum, 5mM EDTA, 1XPBS), treated with Fc receptor block (1:250, BD PharMingen) and were then stained with same Tnf and isotype antibodies used above, all done in sorting media.
RNA isolation and cDNA synthesis and RT-qPCR
Total cellular RNA from THP1 cell lines was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research) according to manufacturer’s instructions. RNA was quantified and controlled for purity with a nanodrop spectrometer. (Thermo Fisher). For RT-qPCR, 500–1000 ng were reversely transcribed (iScript Reverse Transcription Supermix, Biorad) followed by RT-PCR (iQ SYBRgreen Supermix, Biorad) using the cycling conditions as follows: 50°C for 2 min, 95°C for 2 min followed by 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 45 s. The melting curve was graphically analyzed to control for nonspecific amplification reactions.
RNA-Sequencing
For generation of RNA-Sequencing libraries the human THP1 cells, RNA was isolated from control or Pam3CSK40-stimulated cells as described above and the RNA integrity was tested with a BioAnalyzer (Agilent Technologies). For RNA-Sequencing target RIN score of input RNA (500–1000ng) usually had a minimum RIN score of 8. RNA-Sequencing libraries were prepared with TruSeq stranded RNA sample preparation kits (Illumina), depletion of ribosomal RNA was performed by positive selection of polyA+ RNA. Sequencing was performed on Illumina HighSeq or NextSeq machines.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Sequencing
RNA-seq 50 bp reads were aligned to the human genome (assembly GRCh37/hg19) using TopHat. The Gencode V32 gtf was used as the input annotation. Differential gene expression specific analyses were conducted with the DESeq R package. Specifically, DESeq was used to normalize gene counts, calculate fold change in gene expression, estimate p values and adjusted p values for change in gene expression values, and to perform a variance stabilized transformation on read counts to make them amenable to plotting.
Screen Analysis and generation of hit list
fastq.gz files were analyzed using the gRNA_tool: https://github.com/quasiben/gRNA_Tool. All guide RNA (sgRNA) + barcode reads were collapsed to obtain raw sgRNA counts. Counts were normalized to the median and fold-changes were calculated for each sgRNA. To identify significant genes for the growth screen, the Mann-Whitney U test was performed comparing fold-changes for sgRNAs targeting each gene to non-targeting controls (described in Gilbert et al., 2014) or by following the MAGeCK analysis pipeline (as described in Li et al., 2014). MAGeCK analysis was performed on the full dataset as well as on the data binned into four separate samples (insample replicates) based on the 1st basepair of the random barcode (bins A,T,G, and C). The data was then used as input into the MAGeCK analysis pipeline. For the growth screen, the Day 21 sample was compared to the Day 0 sample. To identify significant genes for the for the FACS screen, GFP low (bottom 20%) sorted cells were compared to GFP high (top 20%) sorted cells.
sgRNA selection for screen validation
For the macrophage specific viability genes, we selected 6 targets for validation out of possible 61 hits. We choose 5 essential genes and one growth suppressor. We chose the candidates based on their rankings according to P value. For the 3′utr-targeting guides, we selected guides with > 3-fold positive enrichment (Day 21 versus Day 0) that targeted the 3′utrs of essential genes with significant negative enrichment. The criteria for our NF-κB candidate selection was as follows: 1) Most significant: We selected candidates with the lowest Mann-Whitney U test p value (using a p value cut off of < 0.01). 2) Novelty: We focused on genes, which were not previously known to be involved in NF-κB signaling. We used several databases including KEGG, String-DB and Cell Signaling TLR-signaling gene list to determine novelty of gene. 3) Expression: We evaluated expression and confirmed > 10 FPKM for either un-stimulated or LPS-stimulation conditions. 4) Viability: We confirmed that our candidates did not have a significant viability phenotype. We targeted a total of 18 coding genes selecting guides with the strongest enrichment (2 guides/gene). We targeted microRNAs that showed > 3-fold positive enrichment for at least 2 gRNAs. For positive controls we used guides targeting Tlr4.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PEcy7 anti-mouse Tnf-alpha | ThermoFisher | Cat# 25–7321-82; RRID:AB_11042728 |
| Isotype control | Biolegend | Cat#400416; RRID:AB_326522 |
| Fc block | BD PharMingen | Cat#553142; RRID:AB_394657 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| E.coli LPS | InvivoGen | Cat#tlrl-3pelps |
| Lipofectamine 2000 | ThermoFisher | Cat#11668019 |
| iScript Reverse Transcription Supermix | Biorad | Cat#1708841 |
| iQ SYBR Green Supermix | Biorad | Cat#1725122 |
|
Critical Commercial Assays | ||
| Mouse TNF-alpha DuoSet ELISA | R&D Systems | Cat#DY410 |
| Direct-zol RNA MiniPrep Kit | Zymo | Cat#R2072 |
| TruSeq Stranded Total RNA Library Prep kit | Illumina | Cat#20020597 |
|
Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE138786 |
| Raw and analyzed data | This paper | GEO: GSE138788 |
|
Experimental Models: Cell Lines | ||
| immortalized bone-marrow-derived macrophages with an NFKB reporter and Cas9 (iBMDM-NFKB-Cas9) | Covarrubias et al., 2017 | PMID: 29051223 |
| THP-1 cells | ATCC | ATCC TIB202 |
|
Oligonucleotides | ||
| sgRNA sequences, see Table S1 | This paper | N/A |
| Custom sequencing primer: | This paper | N/A |
| 5′GAGACTATAAGTATCCCTTGGAGAACCACCTTGTTGG-3′ | ||
| Il6 qRT-PCR primer F: AACGATGATGCACTTGCAGA | IDT | N/A |
| Il6 qRT-PCR primer R: GAGCATTGGAAATTGGGGTA | IDT | N/A |
| Gapdh qRT-PCR primer F: CCAATGTGTCCGTCGTGGATCT | IDT | N/A |
| Gapdh qRT-PCR primer R: GTTGAAGTCGCAGGAGACAACC | IDT | N/A |
| Hprt qRT-PCR primer F: TGCTCGAGATGTCATGAAGG | IDT | N/A |
| Hprt qRT-PCR primer R: TATGTCCCCCGTTGACTGAT | IDT | N/A |
|
Recombinant DNA | ||
| Plasmid: psPAX2 | Addgene | Addgene Plasmid #12260; RRID:Addgene_12260 |
| Plasmid: pMD2.G | Addgene | Addgene Plasmid #12259; RRID:Addgene_12259 |
|
Software and Algorithms | ||
| FlowJo | BD | https://www.flowjo.com/ |
| MAGeCK | Li et al., 2014 | PMID: 25476604 |
| gRNA_Tool | https://github.com/quasiben/gRNA_Tool | |
| Mann-Whitney U test | Gilbert et al., 2014 | PMID: 25307932 |
| TopHat | Kim et al., 2013 | PMID: 23618408 |
| DESeq2 R package | Love et al., 2014 | PMID: 25516281 |
Highlights.
CRISPR screens to identify genes required for macrophage viability and function
Identification of cis-regulatory elements that control essential genes
TNF can function as a negative regulator of inflammation in a cell-intrinsic manner
ACKNOWLEDGMENTS
Thanks to all members of the Carpenter Lab and to Dr. Kate Fitzgerald and Dr. Maninjay Atianand for feedback on manuscript. This work was funded by NIH grants R03 AI131019–01 and R21 AI142165 to S. Carpenter and U01 CA217882 to M.T.M. A.C.V. was supported by an NIH Predoctoral Training grant (T32 GM008646), a Ford Predoctoral Fellowship, and a Howard Hughes Medical Institute Gilliam fellowship. E.K.R. was supported by an NIH predoctoral training grant (T32 GM008646).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108541.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alexopoulou L, Kranidioti K, Xanthoulea S, Denis M, Kotanidou A, Douni E, Blackshear PJ, Kontoyiannis DL, and Kollias G (2006). Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol 36, 2768–2780. [DOI] [PubMed] [Google Scholar]
- Ardestani S, Li B, Deskins DL, Wu H, Massion PP, and Young PP (2013). Membrane versus soluble isoforms of TNF-α exert opposing effects on tumor growth and survival of tumor-associated myeloid cells. Cancer Res 73, 3938–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CG, Riemondy K, Chapnick DA, Bunker E, Liu X, Kuersten S, and Yi R (2016). Genome-wide analysis of Musashi-2 targets reveals novel functions in governing epithelial cell migration. Nucleic Acids Res 44, 3788–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D, and Ghosh S (2014). Regulation of the NF-κB-Mediated Transcription of Inflammatory Genes. Front. Immunol 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher M, Tian R, Blau JA, Markegard E, Wagner RT, Wu D, Mo X, Biton A, Zaitlen N, Fu H, et al. (2018). Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat. Biotechnol 36, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher M, Covarrubias S, Biton A, Blau J, Wang H, Zaitlen N, and McManus MT (2019). Tracing cellular heterogeneity in pooled genetic screens via multi-level barcoding. BMC Genomics 20, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JR (2008). TNF-mediated inflammatory disease. J. Pathol 214, 149–160. [DOI] [PubMed] [Google Scholar]
- Covarrubias S, Robinson EK, Shapleigh B, Vollmers A, Katzman S, Hanley N, Fong N, McManus MT, and Carpenter S (2017). CRISPR/Cas9-based screening of long noncoding RNAs (lncRNAs) in macrophages with an NF-kappa B reporter. J. Biol. Chem 292, 20911–20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer CG, Ray JP, Hacohen N, and Regev A (2020). MAUDE: inferring expression changes in sorting-based CRISPR screens. Genome Biol 21, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erson-Bensan AE (2016). Alternative polyadenylation and RNA-binding proteins. J. Mol. Endocrinol 57, F29–F34. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgo-poulos S, Lesslauer W, Kollias G, Pfizenmaier K, and Scheurich P (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83, 793–802. [DOI] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- Hart T, Komori HK, LaMere S, Podshivalova K, and Salomon DR (2013). Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics 14, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Geno-type-Specific Cancer Liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Hyrich KL, Deighton C, Watson KD, Symmons DPM, and Lunt M BSRBR Control Centre Consortium; British Society for Rheumatology Biologics Register (2009). Benefit of anti-TNF therapy in rheumatoid arthritis patients with moderate disease activity. Rheumatology (Oxford) 48, 1323–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M, Bassik MC, and Weissman JS (2013). Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc. Natl. Acad. Sci. USA 110, E2317–E2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiveland CR (2015). Peripheral blood mononuclear cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models [Internet, Verhoeckx K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, and Wichers H, eds. (Springer; ). 10.1007/978-3-319-16104-4. https://www.ncbi.nlm.nih.gov/books/NBK500138/. [DOI] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Cui L, Doench JG, and Lu TK (2020). High-throughput CRISPR screens to dissect macrophage-Shigella interactions. BioRxiv 10.1101/2020.04.25.061671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MTY, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. (2013). Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Barreiro LB, and Gros P (2016). The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med 213, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zhang L, Joo D, and Sun S-C (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther 2, 17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopetuso LR, Gerardi V, Papa V, Scaldaferri F, Rapaccini GL, Gasbarrini A, and Papa A (2017). Can We Predict the Efficacy of Anti-TNF-α Agents? Int. J. Mol. Sci 18, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, and Xu S (2013). TNF inhibitor therapy for rheumatoid arthritis. Bio-med. Rep 1, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C (2017). Regulation by 3′-Untranslated Regions. Annu. Rev. Genet 51, 171–194. [DOI] [PubMed] [Google Scholar]
- Mayr C (2019). What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol 11, 034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, and Ghosh S (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-Beltran W, et al. (2017). A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet 49, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, and Mohler KM (1998). TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol 160, 943–952. [PubMed] [Google Scholar]
- Peyrin-Biroulet L (2010). Anti-TNF therapy in inflammatory bowel diseases: a huge review. Minerva Gastroenterol. Dietol 56, 233–243. [PubMed] [Google Scholar]
- Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, and Hornung V (2016). A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem 291, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, et al. (2016). A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep 17, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanatha R, Li Z, Hu Y, and Perrimon N (2018). Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. eLife 7, e36333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46 (D1), D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang J, Brand DD, and Zheng SG (2018). Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol 9, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung ATY, Choi YH, Lee AHY, Hale C, Ponstingl H, Pickard D, Goulding D, Thomas M, Gill E, Kim JK, et al. (2019). A Genome-Wide Knockout Screen in Human Macrophages Identified Host Factors Modulating Salmonella Infection. mBio 10, e02169–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimin, and Kohanawa M (2006). A Regulatory Effect of the Balance between TNF-α and IL-6 in the Granulomatous and Inflammatory Response to Rhodococcus aurantiacus Infection in Mice. J. Immunol 177, 642–650. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen X, Liu Q, Zhang S, and Hu W (2017). Translation repression via modulation of the cytoplasmic poly(A)-binding protein in the inflammatory response. eLife 6, e27786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Siegel D, Biton A, Tonqueze OL, Zaitlen N, Ahituv N, and Erle DJ (2017). CRISPR-Cas9-mediated functional dissection of 3′-UTRs. Nucleic Acids Res 45, 10800–10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated from CRISPR screens and RNA-seq reported in this paper have been deposited to GEO under accession codes GSE138786 and GSE138788.