

This study represents a comprehensive community-wide genome-centered metagenome analysis of biological soil crust (BSC) communities in arid environments, providing insights into the distribution of genes encoding different energy generation mechanisms, as well as survival strategies, among populations in an arid soil ecosystem. It reveals the metabolic potential of several uncultured and previously unsequenced microbial genera, families, and orders, as well as differences in the metabolic potential between the most abundant BSC populations and their cultured relatives, highlighting once more the danger of inferring function on the basis of taxonomy.
KEYWORDS: biological soil crust, dormancy, metagenomics, mixotrophy, survival
ABSTRACT
Desert surface soils devoid of plant cover are populated by a variety of microorganisms, many with yet unresolved physiologies and lifestyles. Nevertheless, a common feature vital for these microorganisms inhabiting arid soils is their ability to survive long drought periods and reactivate rapidly in rare incidents of rain. Chemolithotrophic processes such as oxidation of atmospheric hydrogen and carbon monoxide are suggested to be a widespread energy source to support dormancy and resuscitation in desert soil microorganisms. Here, we assessed the distribution of chemolithotrophic, phototrophic, and desiccation-related metabolic potential among microbial populations in arid biological soil crusts (BSCs) from the Negev Desert, Israel, via population-resolved metagenomic analysis. While the potential to utilize light and atmospheric hydrogen as additional energy sources was widespread, carbon monoxide oxidation was less common than expected. The ability to utilize continuously available energy sources might decrease the dependency of mixotrophic populations on organic storage compounds and carbon provided by the BSC-founding cyanobacteria. Several populations from five different phyla besides the cyanobacteria encoded CO2 fixation potential, indicating further potential independence from photoautotrophs. However, we also found population genomes with a strictly heterotrophic genetic repertoire. The highly abundant Rubrobacteraceae (Actinobacteriota) genomes showed particular specialization for this extreme habitat, different from their closest cultured relatives. Besides the ability to use light and hydrogen as energy sources, they encoded extensive O2 stress protection and unique DNA repair potential. The uncovered differences in metabolic potential between individual, co-occurring microbial populations enable predictions of their ecological niches and generation of hypotheses on the dynamics and interactions among them.
IMPORTANCE This study represents a comprehensive community-wide genome-centered metagenome analysis of biological soil crust (BSC) communities in arid environments, providing insights into the distribution of genes encoding different energy generation mechanisms, as well as survival strategies, among populations in an arid soil ecosystem. It reveals the metabolic potential of several uncultured and previously unsequenced microbial genera, families, and orders, as well as differences in the metabolic potential between the most abundant BSC populations and their cultured relatives, highlighting once more the danger of inferring function on the basis of taxonomy. Assigning functional potential to individual populations allows for the generation of hypotheses on trophic interactions and activity patterns in arid soil microbial communities and represents the basis for future resuscitation and activity studies of the system, e.g., involving metatranscriptomics.
INTRODUCTION
Drylands constitute more than 35% of land surface and are currently expanding (1). They experience prolonged periods of drought interrupted by brief rain events, which limit vascular plant growth. Yet these environments are not devoid of life (2, 3). Dryland soils are inhabited by diverse microbial communities whose members are assumed to endure extended droughts in a largely inactive state often called “dormancy” but reactivate quickly once hydrated. Although the definitions of dormancy differ (reviewed in references 4 and 5), typical examples include sporulation performed by Gram-positive Firmicutes and some Actinobacteriota (6, 7), cysts with enforced cell envelopes formed by some Alphaproteobacteria (8), and specialized dormant cells termed akinetes formed by cyanobacteria of the orders Nostocales and Stigonematales (9). Yet, dryland soils are inhabited by a variety of other microbial clades (3, 10) whose mechanisms of surviving desiccation are unknown. It was recently suggested that many soil microorganisms might rely on inorganic energy sources such as light and oxidation of atmospheric trace gases (e.g., dihydrogen [H2] and carbon monoxide [CO]) to maintain the cell’s integrity in a dormant state (11). Furthermore, trace gas oxidation could potentially support primary production, as oxidation of atmospheric H2 has been shown to sustain CO2 fixation in an Antarctic desert soil (12). However, it is still unclear if these processes are present in other arid ecosystems and the extent of their distribution among the microbial populations within a given soil community, especially in taxonomic groups without cultured representatives.
The aim of this study was to assess the distribution of potential physiologies regarding carbon (C) metabolism, energy generation, and dormancy mechanisms across different populations of a dryland soil microbial community. We performed population-resolved metagenomics analysis of microbiota inhabiting biological soil crusts (BSCs) from the Negev Desert, Israel. BSCs cover the topmost layer of many arid soils and are formed by highly adapted microbial communities (1). Arid and hyperarid BSCs are characterized by the presence of filamentous cyanobacteria, which secrete ample extracellular polymeric substances (EPS) crucial for the structure and integrity of the crust (1, 13) and supply organic C to heterotrophic community members (14, 15). Their primary production and matrix secretion provide an optimized environment for soil microorganisms, resulting in significantly increased microbial abundance and diversity in BSC in comparison to that of the soil beneath (16, 17). In addition, BSC microbial communities are essential to this ecosystem, as they take over important ecological functions such as CO2 and N2 fixation and prevent the erosion of underlying soil (1, 18). Being populated by dryland soil microbiota yet containing more biomass than barren soil (10, 16) makes BSCs a good model system for in situ studies on microbial dormancy and resuscitation strategies.
Most of our functional knowledge on BSC native taxa stems from cultivation and gene amplification studies targeting specific metabolisms, such as nitrification (19), N2 fixation (20, 21), methanogenesis (22), or anoxygenic photosynthesis (23). However, gene amplification studies do not provide information on the abundances and metabolisms of other co-existing microbial community members. More recent metagenomic studies of soil crusts, or desert soils in general, identified broader categories of potential metabolic functions present in these communities (24–31). However, as these investigations were done mostly without assembly or genome binning, taxonomic assignments of the identified genomic potential were largely missing. Neither bulk metagenomics nor functional gene-centered studies can determine if, e.g., two metabolisms are performed by the same species or if two steps of a metabolism are divided between different species. Yet, gaining information on the potential metabolism of individual, co-occurring microbial populations is essential for predicting their ecological niches and for generating hypotheses on the dynamics and interactions among members of this BSC ecosystem. With the population genomes at hand, such hypotheses can now be tested by targeting specific microbial groups and their activities, for instance by employing transcriptomic investigations.
RESULTS
Microbial community composition.
Being collected from an arid site with less than 100 mm of annual rainfall (32), the samples represented light-colored Cyanobacteria-dominated BSCs (Fig. 1). Before choosing samples for metagenomic sequencing, we assessed the diversity and spatial heterogeneity of the BSC microbial communities at the Avdat long-term ecological research (LTER) site (Fig. 1) by sequencing 16S rRNA gene amplicons from 24 samples originating from eight different sampling positions labeled A to H within the LTER site (Fig. 1; see library statistics in Table S1 in the supplemental material). After quality filtering, denoising, and determination of amplicon sequence variants (ASVs), the average sequence count was 9,831 ± 3,690 reads per sample (Table S1). The 1,056 ASVs clustered into 921 operational taxonomic units (OTUs) via Swarm percentage identity-independent single-linkage clustering (33). The composition of BSC microbial communities appeared homogeneous across the site (Fig. 2). Between 37% and 68% of reads in any sample were attributed to the same ubiquitous 25 OTUs and thus represented taxa shared among all samples (Fig. 2C). An additional 17% to 35% of reads per sample belonged to 103 OTUs that were detected in more than half, but not all, samples. As expected, the community was composed of large proportions of Cyanobacteria and Actinobacteriota and smaller fractions of Alphaproteobacteria, Bacteroidota, Chloroflexota, Gemmatimonadota, and Acidobacteriota.
FIG 1.

Location of and sampling at the long-term ecological research (LTER) site in the Negev Desert, Israel. (A) Location of the Avdat LTER site. The map was generated with GeoMapApp and Global Multi-Resolution Topography data (http://www.geomapapp.org)/CC BY/CC BY (Ryan et al., 2009 [130]). (B) Landscape at the LTER site. Crust was sampled from shrub-free patches with a homogeneous surface appearance. (C) Close-up of soil crust pieces. The crusts were light in color and largely free of lichens or mosses. Coherent pieces of soil crust of ca. 5 mm thickness were collected and transported in petri dishes. (D) Surface of dry crust as seen through a stereoscope. (E) Surface of a rehydrated crust piece after 24 h as seen through a stereoscope. Large bundles of Cyanobacteria could be observed on the surface of soil crusts starting at around 2 h and reaching the largest extent by 12 h. (F) Areal distribution of sampling plots. From each plot of ca. 0.5 by 0.5 m, 20 petri dishes of crust pieces were sampled. For 16S rRNA amplicon sequencing, crust pieces were randomly chosen from three petri dishes per plot, as is exemplified for plot F.
FIG 2.

Microbial community composition of the biological soil crusts at Avdat LTER, Negev Desert, Israel. (A) Hierarchical clustering of samples based on the Bray-Curtis dissimilarity of the microbial community compositions at the Swarm-OTU level. (B) Taxonomic composition determined by amplicon sequencing of 16S rRNA gene fragments, unassembled metagenomic reads mapping to SILVA SSU database, and read mapping to GTDB-classified population genomes. Taxa represented by MAGs are highlighted in bold in the legend, and the number of corresponding MAGs is indicated next to the taxon name. (C) Relative sequence abundances of 25 OTUs present in every sample. The letters A to H in the sample names refer to the different sampling plots illustrated in Fig. 1F, while numbers indicate the randomly chosen subsamples (petri dishes) per plot. Outlined samples highlight different data originating from the very same DNA extracts (G19, B20, F08). Classification and relative abundances of OTUs and detailed composition of unassembled metagenomes based on read mapping to the SILVA database can be found in Data Set S1 in the supplemental material.
16S rRNA gene amplicon sequencing metrics. The table provides the numbers of quality-filtered reads obtained from each sample, the numbers of ASVs and Swarm-generated OTUs detected in each sample (richness), and the percentages of reads assigned to ubiquitous OTUs, OTUs appearing in more than 50% of the samples, and OTUs appearing only in fewer than four samples. Download Table S1, PDF file, 0.03 MB (35.1KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Large spreadsheet providing relative abundances and taxonomic classification of Swarm OTUs as well as relative abundances of microbial genera as determined by short read mapping to SILVA SSU132 database with PhyloFlash v.30 (13). Only genera with three or more reads mapping were considered as detected. Download Data Set S1, XLSX file, 0.2 MB (228.2KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of our metagenome libraries (see metrics in Table S2) with Nonpareil (34) indicated that by sequencing between 16 and 32 Gbp per sample, we covered between approximately 56% (sample C15) and 74% (sample B20) of the diversity. If taken together, the libraries covered 79% of the diversity. The Nonpareil diversity index for our samples was between 20.7 and 21.9 (Table S2, part A), which is similar to that of Atacama top soils (21.2) (3), higher than that of human-associated (17.5 to 18) or lake water (19) microbiomes, but lower than that of cornfield soil (23) (all reported in reference 34). Interestingly, based on unassembled read mapping to the SILVA SSU132 database (Data Set S1), the relative abundance of Cyanobacteria in the metagenome was much lower than in the amplicon data, although the libraries were prepared from the same DNA extract. This phenomenon was reported previously in BSCs (24), and possible explanations are discussed in Text S2 in the supplemental material.
A detailed description of the metagenome analysis providing the reasoning behind the choice of methods and analysis steps performed. The scripts referenced in this section can be found in the following Github repository: https://github.com/meierdv/avdat_metagenome. Download Text S1, PDF file, 0.1 MB (130.9KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Discussion of possible causes for differences in relative abundances of microbial taxa between 16S rRNA gene amplicon data and metagenomic sequencing data, with a focus on difference in relative abundance of Cyanobacteria. Download Text S2, PDF file, 0.04 MB (42.5KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Metagenomic sequencing and assembly metrics. Part A provides the basic metrics of metagenomic sequencing efforts, such as sequencing depth, the estimation of genomic diversity covered (determined by Nonpareil), and percentages of reads represented by the assembly and analyzed MAGs. Part B provides the basic metrics of the initial metagenomic assembly. Download Table S2, PDF file, 0.04 MB (46.3KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
From sequence reads of five crust samples, we obtained 96 metagenome-assembled population genomes (MAGs) (main metrics in Table S3), representing most of the microbial taxa that we detected in the unassembled data (Fig. 2, short read mapping; Data Set S1) and 24% to 36% of the raw reads (mapped with identity of >95%). According to Genome Taxonomy Database classification (35, 36) (Table S3, GTDB classification data, including average nucleotide identities [ANIs]) and our phylogenetic analysis (Fig. S1), all 96 MAGs represented a previously unsequenced species, 59 MAGs belonged to 32 novel genera, 13 represented 10 novel families, and six belonged to four novel orders. The most diverse phylum by far was Actinobacteriota, represented by 54 MAGs from four different classes (Rubrobacteria, Thermoleophilia, Acidimicrobiia, and Actinobacteria). When we clustered the MAGs based on a presence-absence matrix of encoded functions according to functional annotation via EggNOG orthologs (Fig. 3), most of the MAGs clustered according to their taxonomic affiliation, with some notable exceptions. Instead of clustering with the rest of Actinobacteriota, the Rubrobacteria and Thermoleophilia MAGs grouped with MAGs from the Chloroflexota phylum and a Deinococcota MAG (Fig. 3).
FIG 3.
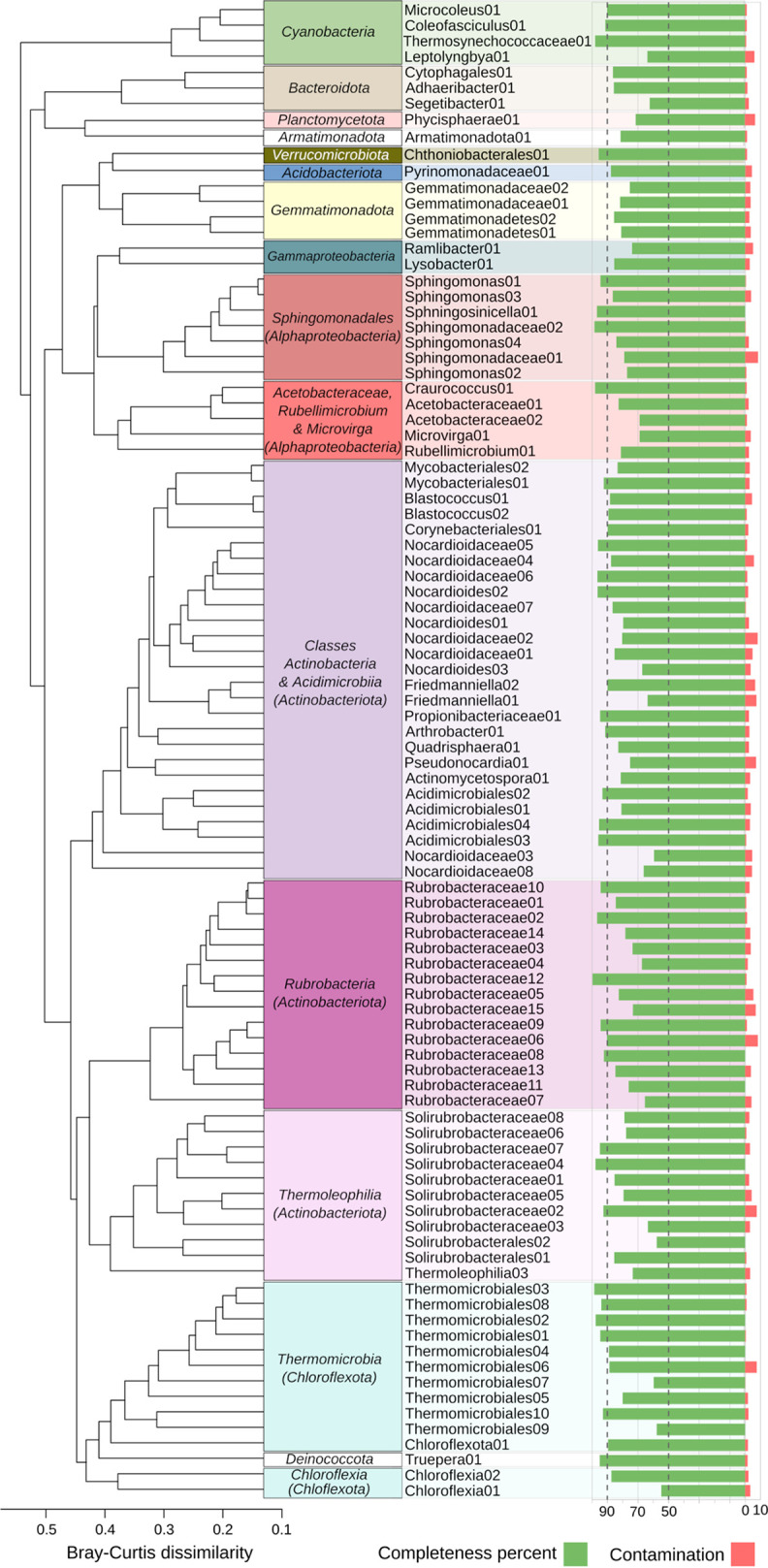
Clustering of the metagenome-assembled genomes (MAGs) based on presence-absence matrix of functions as assigned by EggNOGs. Different orthologous groups annotated with the same function were summarized into one functional category. In total, 8,279 functions were used for clustering. Functions appearing only in one MAG were removed from the matrix. Hierarchical clustering by average linkage was performed based on Bray-Curtis dissimilarity with the vegan package in R. Green bars indicate the percentage completeness of MAGs based on the presence of lineage-specific essential single-copy genes; red bars indicate the percentage of single-copy genes present in several different copies (according to CheckM).
Phylogenomic placement of MAGs representing previously unsequenced genera, families, and orders. Clade-specific trees were calculated based on concatenated amino acid alignment of 120 phylogenetic markers used in GTDB (8) using FastTree (starting tree, bio-neighbor-joining; model, Le-Gascuel). Alignment was filtered to contain sites conserved in 25% of sequences (4,911 positions for Sphingomonadales, 4,918 for Acetobacterales, 4,521 for Gemmatimonadota, 4,819 for Cytophagales, 4,118 positions for Phycisphaerae, 4,365 for Chtoniobacterales, 4,061 for Chloroflexota, 4,805 for Armatimonadota, 4,627 for Thermosynechococcales, 4,418 positions for Rubrobacteria and Thermoleophilia, 4,375 for Acidimicriia, 4,882 for Mycobacteriales, and 4,872 for Propionibacterales). Multifurcations have been introduced for branchings with less than 50% support. Branch support was calculated by 1,000 resamplings of site likelihood and the Shimodaira-Hasegawa test (FastTree default). Download FIG S1, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
MAG metrics and classification. This large table provides sequence-related metrics of the metagenome-assembled genomes (MAGs), estimated values of their completeness and duplication, relative abundance of the MAGs in the individual metagenome samples, estimated indices of replication (iRep values), and taxonomic classification of the MAGs by GTDB-Tk and the criteria on which the classification was based (e.g., ANI to closest-related genome, if applicable). Download Table S3, XLSX file, 0.03 MB (33.9KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Potential for use of C sources and energy generation mechanisms.
In order to predict ecological roles of microbial community members, we first investigated the distribution of autotrophic, mixotrophic, and heterotrophic metabolic potentials encoded in the population genomes (Fig. 4; Fig. S2).
FIG 4.

Selected metabolic traits encoded in the metagenome-assembled genomes. The genomes were grouped based on presence-absence of functions (see Fig. 2). The bar chart on top indicates the cumulative coverage of a given group of genomes in the whole data set. The number of genomes in a group is indicated in parentheses. On the left, the total number of enzyme subunits or pathway constituents is indicated in parentheses. No numbers are given for categories where a “completeness” criterion is not applicable, such as transporters of different specificity. The numbers in the table are the average number of different genes per MAG, like genes encoding (i) different subunits of an enzyme, (ii) different proteins of a pathway, or (iii) similar proteins of different specificity (e.g., different transporters, glycosyl hydrolases). Genes falling into shown categories can be found in Data Set S2.
Selected metabolic traits by MAG. The genomes were grouped based on a presence-absence matrix of EggNOG orthologs, which corresponds largely to taxonomic classification. For known pathways and enzyme subunits, numbers in parentheses indicate the number of different annotations in a given category, e.g., genes encoding different subunits of an enzyme or genes encoding different enzymes of a pathway. No numbers are given for categories where a “completeness” criterion is not applicable, such as transporters of different specificity. The numbers in the table indicate the number of genes per MAG. Several genes encoding the same function in a MAG, e.g., the same subunit of an enzyme, were counted only once. Genes falling into categories can be found in Data Set S2. Download FIG S2, TIF file, 1.3 MB (1.5MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Large spreadsheet listing the functional annotations, which were counted for the metabolisms depicted in Fig. 4 and Fig. S2. Download Data Set S2, XLSX file, 0.03 MB (33.2KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes encoding the large subunit of ribulose-1,5-bisphosphate carboxylase (RuBisCO) indicative of CO2 fixation via the Calvin-Benson-Bassham (CBB) pathway were found in the four cyanobacterial MAGs, three Alphaproteobacteria MAGs (two Acetobacteraceae MAGs and the Rubellimicrobium MAG), two Gemmatimonadota MAGs, two Thermoleophilia MAGs, one Rubrobacteria MAG, one Actinobacteria MAG (genus Pseudonocardia), one Acidimicrobiia MAG, and one Chloroflexia MAG (Fig. 4; Fig. S2). Also, Deinococcota and Armatimonadota MAGs contained RuBisCO genes. No evidence for other CO2 fixation pathways could be detected in our metagenomes (in MAGs or unassembled reads). Potential energy sources for CO2 fixation could be light or H2 oxidation.
In addition to the four Cyanobacteria MAGs, we found (bacterio)chlorophyll synthesis genes carried in three alphaproteobacterial MAGs belonging to the Acetobacteraceae family (two belonging to a novel genus and one belonging to Belnapia/Craurococcus) (Fig. 4; Fig. S2). Another two MAGs containing bacteriochlorophyll synthesis and RuBisCO genes were attributed to the Gemmatimonadaceae. Bacteriorhodopsins, which can be used to generate proton motive force or for light sensing, were found in 26 MAGs (Fig. S2). Genes for bacteriorhodopsin and ß-carotene di-oxygenase (a key enzyme in retinal synthesis) were present in most Rubrobacteria MAGs (in 11 and 12 MAGs, respectively), one Thermoleophilia MAG, four Actinobacteria MAGs (two Mycobacteriales MAGs, one Actinomycetospora MAG, and one Quadrisphaera MAG), and two Alphaproteobacteria MAGs (Sphingomonadales) (Fig. S2; Fig. 4). Seven various other MAGs encoded only the bacteriorhodopsin, and 12 MAGs encoded heliorhodopsins suggested to be involved in light sensing (37), among them five Thermomicrobia MAGs.
Apart from light, H2 oxidation seemed to be a common potential inorganic energy source. Of 27 detected hydrogenases, 23 were encoded by Actinobacteriota: Rubrobacteria (nine MAGs), Thermoleophilia (seven MAGs), Acidiimicrobia (two MAGs), and class Actinobacteria (five MAGs). Additionally, three hydrogenase-encoding genes were found in Chloroflexota MAGs. All hydrogenases belonged to the high-affinity group 1h [NiFe]-hydrogenase according to HydDB classification (38), indicative of the potential to oxidize atmospheric H2 (39). Genes encoding the form I CO dehydrogenase, experimentally shown to oxidize CO (40), were found in only four MAGs (Fig. S2). However, genes annotated as encoding CO dehydrogenase were widespread across Chloroflexota, Actinobacteriota, Proteobacteria, and Gemmatimonadota MAGs. Based on phylogenetic tree calculation (Fig. S3A), 5 of 59 annotated CO dehydrogenase genes were confirmed as encoding form II CO dehydrogenase. The function of the other CO dehydrogenase-like proteins could not be confidently determined, due to unresolved phylogeny of the functional domain. We have further identified genes annotated as encoding soluble methane monooxygenases (Fig. 4; Fig. S2). These genes (3 subunits) were found in seven Rubrobacteria MAGs, one Actinobacteria MAG, and one Thermoleophilia MAG. However, phylogenetic analysis suggests that they encode propane monooxygenases (Fig. S3B). Interestingly, two Acetobacteraceae, one Methylobacteriaceae (Microvirga, Alphaproteobacteria), and one Comamonadaceae (Ramlibacter, Gammaproteobacteria) MAG encoded the Sox multienzyme complex central to the oxidation of reduced sulfur species (Fig. 4; Fig. S2). While the bacteriochlorophyll-encoding Acetobacteraceae MAGs might potentially use sulfide as an electron donor in anoxygenic photosynthesis, the Microvirga and Ramlibacter MAGs did not carry genes indicative of phototrophy (Fig. S2).
Phylogenetic placements of proteins annotated as carbon monoxide dehydrogenase, large subunit (A), and methane monooxygenase (B). Trees were calculated with FastTree based on an HMM alignment to the respective Pfam motif (PF02738 for CO dehydrogenase catalytic domain and PF02332 for putative methane monooxygenase). Multifurcations have been introduced for branchings with less than 50% support. Branch support was calculated by 1,000 resamplings of site likelihood and the Shimodaira-Hasegawa test (FastTree default). Numbers in parentheses indicate the number of sequences contained in the cluster (note that one MAG can have multiple CoxL-encoding genes). For the putative CO dehydrogenases (A), a first tree was calculated for all proteins included in the seed alignment of the Pfam model (excluding eukaryotes), together with known form I and form II CO dehydrogenases (9) and MAG-derived sequences. Alignment was filtered to contain sites conserved in 25% of sequences (428 positions). As the Pfam motif includes several enzymes with different functions, a subtree was calculated just for the CO dehydrogenase-containing part, using glyceraldehyde dehydrogenase as an outgroup. Alignment was filtered to contain sites conserved in 25% of sequences (502 positions). Download FIG S3, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We have not found any indications for anaerobic respiration in any of the analyzed MAGs. Therefore, oxygen seems to be the only electron acceptor for respiratory processes. An overwhelming majority of the MAGs encoded heme-copper-based cytochrome c terminal oxidases, with a few MAGs additionally encoding cytochrome bd ubiquinol oxidases. Interestingly, Rubrobacteria MAGs did not encode any components of the cytochrome c oxidase, only the cytochrome bd ubiquinol oxidase. All genomes had genes encoding enzymes involved in at least one branch of mixed-acid fermentation. However, most enzymes involved in fermentation are bidirectional and also part of other metabolic pathways. Therefore, no prediction can be made about the ability of the populations to perform fermentation.
When looking for organic energy and C sources, we focused on transporters, as they determine the binding and uptake of organic substrates from the environment. MAGs of all groups encoded ATP-dependent sugar transporters of different specificities (Fig. 4; Fig. S2). Compared to other groups, Rubrobacteria MAGs had a high number of different phosphotransferase systems (on average, 2.7/MAG, up to a maximum of 8). The most common substrates targeted by the phosphotransferase systems were fructose, mannitol, sorbitol, and N-acetyl-d-glucosamine, according to RAST annotation. The mannitol-specific phosphotransferase system was found encoded in seven of the Rubrobacteria MAGs and only in five other MAGs (from the class Actinobacteria). At the same time, Rubrobacteria MAGs encoded strikingly few proteins with carbohydrate-binding motifs (on average, 0.5/MAG) (Fig. 4; Fig. S2) compared to other MAGs. High numbers of different carbohydrate-binding motifs and glycosyl hydrolases indicating an ability to utilize complex polymeric C substrates were detected in Bacteroidota, Armatimonadota, Planctomycetota, and Deinococcota MAGs (Fig. 4; Fig. S2). Amino acid and peptide transporters were relatively evenly distributed between different MAGs (Fig. 4; Fig. S2).
Genes involved in microbial dormancy and persistence.
We investigated the MAGs for the potential of actinobacterial sporulation by searching for genes essential for this sporulation type, such as those encoding sporulation-specific cell division activator SsgA and transcriptional regulators WhiA, WhiB, and WhiD (41, 42), as many other involved genes also can have roles beyond sporulation. As expected, the highest frequencies of these genes were detected in actinobacterial MAGs, more specifically in the classes Acidimicrobiia and Actinobacteria. In the MAGs belonging to these classes, we also found genes encoding histone-like proteins, possibly involved in DNA compaction and protection in a resting state (43, 44) (Fig. 4; Fig. S2). In contrast, MAGs belonging to the actinobacterial classes Thermoleophilia and Rubrobacteria were mostly lacking sporulation-related genes (Fig. 4; Fig. S2). Homologs of genes involved in the sporulation of Firmicutes were found in several MAGs (data not shown). However, their functions were not restricted to sporulation, their sets were far from complete, with essential key genes missing, and they seemed to be randomly distributed across MAGs. Therefore, we cannot conclude that the potential for Firmicutes-like sporulation was present in any of the MAGs. We further searched the MAGs for the potential to protect the cells with polysaccharide sheaths or capsules. Genes for exopolysaccharide production were present in several MAGs from various groups, most frequently in Deinococcota, Acetobacteraceae, and Cyanobacteria (Fig. 4; Fig. S2), but were lacking in all Rubrobacteria MAGs. Polymeric C storage compounds like glycogen or polyhydroxyalkanoates (PHA) are known to play an important role in successful formation of and resuscitation from resting stages (45–47). Rubrobacteria MAGs carried only genes for PHA synthesis, whereas most other MAGs carried genes for glycogen synthesis and modification. Among Alphaproteobacteria, Sphingomonadales MAGs were largely lacking glycogen synthesis-related genes.
Protein and DNA damage induced by reactive oxygen species (ROS) is considered the main mortality cause during desiccation (48, 49), and as such, we compared the genomic potentials for damage mitigation and repair. While many MAGs encoded heme-based catalases and peroxidases (KatE and KatG), Deinococcota, Rubrobacteria, and Planctomycetota encoded only manganese-containing catalases (Fig. 4; Fig. S2). Almost all Rubrobacteria and Thermoleophilia MAGs as well as one Thermomicrobia and the Verrucomicrobiota MAG additionally encoded a “DNA protection during starvation” (DPS) rubrerythrin-like protein, which contains a DNA-binding domain and has a peroxidase function (50). The exclusive use of cytochrome bd terminal oxidase, which has been shown to have significant catalase activity (51), might represent an additional ROS-reducing feature of the Rubrobacteria MAGs (Fig. 4; Fig. S2). The two key enzymes of the glyoxylate bypass of the tricarboxylic acid (TCA) cycle allowing the cells to utilize C2 compounds (here, malate synthase and isocitrate lyase) were encoded in most Rubrobacteria (11 MAGs encoded both enzymes, one MAG encoded only one) and Sphingomonadales MAGs (all encoded at least one enzyme, three MAGs encoded both enzymes) (Fig. 4; Fig. S2). Glyoxylate bypass was suggested to play a role in oxidative stress resistance, as its upregulation has been observed in oxygen stress conditions (52). Most of the MAGs had a similar potential for DNA repair, whereas Rubrobacteria and Acidobacteriota MAGs were missing genes for polymerase IV and exodeoxyribonuclease VII (Fig. 4; Fig. S2), both involved in “reckless degradation” of damaged DNA (53, 54). Furthermore, Rubrobacteria and Acidobacteriota encoded an additional DNA polymerase PolA protein that differs from the common PolA. PolA plays a crucial role in double-strand break repair (55).
Genomic potential for light sensing and motility.
We found genes encoding blue light sensor proteins in 19 MAGs, which were most frequent among the Proteobacteria (9 out of 14 MAGs). Twenty-three MAGs carried five or more genes for flagellum assembly (22 of them more than 10 genes), indicating the potential to move in liquid medium or attach to surfaces (Fig. S2; Fig. 4). Of these MAGs, six encoded both blue light sensor and flagellar proteins (one Actinobacteria, three Sphingomonadales, and two Acetobacteraceae MAGs). Three further MAGs (one Quadrisphaera and two Sphingomonadales) encoded flagellar proteins and a rhodopsin, which can be used for sensing light (56). The three putatively phototrophic Acetobacteraceae MAGs contained gliding motility genes (Fig. S2; Fig. 4), some of which were also carried by all cyanobacterial MAGs and two Bacteroidota MAGs. Notably, Rubrobacteria MAGs contained hardly any motility-related or blue light sensing genes (Fig. S2; Fig. 4).
Genome comparison of Rubrobacteria from BSC and aquatic habitats.
Due to the high abundance of Rubrobacteria MAGs (all closely related to genus Rubrobacter) in the investigated BSC metagenomes, we compared the functional potential encoded in these MAGs to that of previously sequenced genomes of Rubrobacter isolates (Fig. 5). We aimed to (i) investigate whether the special features we found in our BSC MAGs are common to all Rubrobacteraceae genomes and (ii) identify desert soil-specific adaptations, since all the previously sequenced Rubrobacter strains originated from aquatic environments (Fig. 5) (57–61).
FIG 5.

Comparison of selected metabolic traits between Rubrobacteraceae MAGs from this study and sequenced genomes of Rubrobacteraceae isolates. The analysis is based on comparison of EggNOG orthologous groups detected in the genomes. In cases where the EggNOG/COG functional annotation was too general, annotations were completed based on specific RAST, Pfam, and UniProt search results for the given genes. The asterisk indicates that the genes encoding light-independent protochlorophyllide reductase subunits B and N do not represent a potential for phototrophy, as they are the only bacteriochlorophyll synthesis genes found in Rubrobacter genomes. The isolate genomes stem from aquatic environments, such as a Mediterranean sponge (Rubrobacter aplysineae) (57), Indian Ocean sediment (Rubrobacter indicoceanii) (58), mud and thermo-mineral water of a radioactive spring (Rubrobacter radiotolerans) (59), hot spring water, and thermally heated mud and soil (Rubrobacter taiwanensis) (60), and biofilm of a thermally polluted runoff of a carpet factory (Rubrobacter xylanophilus) (61).
Among the common features we identified were the following: the presence of two chlorophyll synthesis-related proteins (light-independent protochlorophyllide reductase subunits B and N), the presence of a CO dehydrogenase-like protein, the absence of terminal cytochrome c oxidase and the use of the cytochrome bd ubiquinol oxidase instead, reliance on manganese catalase only, and the presence of the rubrerythrin-like DPS protein, the enzymes of the glyoxylate bypass, and the two copies of the DNA polymerase PolA essential for double-strand break repair (Fig. 5).
Differences between BSC MAGs and aquatic isolates were detected in the distribution of bacteriorhodopsins, which seemed to be more widespread in the BSC MAGs (11 in 15 MAGs compared to two in five isolate genomes). Likewise, a group 1h [NiFe]-hydrogenase was found encoded in only one isolate genome but in the majority of the BSC MAGs (Fig. 5). However, the most striking difference was the complete absence of respiratory nitrate reductase genes in the BSC MAGs, whereas these were present in every isolate genome. Further significant differences were the cell envelope-related proteins FstX-like permease, a Spo0M (a Firmicutes sporulation protein) homolog, and a fatty acid desaturase, which were found encoded only in the BSC MAGs. Unlike isolate genomes, the BSC MAGs were missing genes related to synthesis and modifications of glycogen and carried genes encoding phosphoenolpyruvate carboxylase. This anapleurotic enzyme replenishes oxaloacetate in the TCA cycle, thus accelerating utilization of organic C. Finally, the BSC MAGs encoded several proteins related to transcription regulation, DNA modification, and mobile elements, which were absent from almost all aquatic isolate genomes (Fig. 5).
Indices of replication.
We analyzed the contig coverage to identify potentially actively dividing populations using iRep (62). For any MAG that passed the criteria for iRep calculation in any sample, the indices suggested that at least 40% of the cells (iRep > 1.4) were dividing (Table S3). For many MAGs, replication with multiple replication forks was indicated (Table S3), and two Rubrobacteraceae and one Solirubrobacteraceae MAG reached values over 3. There was no significant difference between dry and hydrated samples.
DISCUSSION
To the best of our knowledge, this is the first comprehensive, large-scale population-resolved metagenomics study linking metabolic potential to microbial populations in arid BSCs. Genomic bins were previously generated for support in a metabolomics-centered study (15) and in a study of the same data set addressing bacteriophages blooms (63), where the functional potential of the overall community based on generated MAGs was beyond the focus of the studies. Here, we analyzed the metabolic potential of MAGs representing the most abundant BSC microbial taxa (Fig. 2), including entirely uncultured and previously unsequenced orders, families, and genera (see Table S3 and Fig. S1 in the supplemental material). Thereby, we were able to not only identify potential physiologies regarding energy generation and dormancy mechanisms but to link these with the respective microbial populations. This study establishes a foundation that allows the generation of hypotheses about the activities and interactions of BSC community members and their future testing.
Widespread distribution of CO2 fixation genes in BSC populations—beyond the cyanobacterial realm.
We detected the potential for CO2 fixation via the Calvin-Benson-Bassham (CBB) cycle in several populations from six different phyla and a widespread potential for utilization of inorganic energy sources such as light and atmospheric gases among the MAGs (Fig. 4; Fig. S2). This widespread distribution suggests that trophic relations in the investigated BSC microbial community might extend beyond the dependency between cyanobacteria as primary producers and various heterotrophs consuming their products (14, 15). While an early metagenomic study of BSCs found cyanobacteria as the only CO2-fixing organisms (25), other culture- and marker gene-based studies of crusts and arid soils also detected anoxygenic phototrophic Alphaproteobacteria (23, 64). In addition to Alphaproteobacteria, we found potential for anoxygenic photosynthesis among Gemmatimonadota, a phylum that was only recently discovered to contain phototropic species (65). Further, we found populations with chemolithoautotrophic metabolic potential involving H2 oxidation among, e.g., Thermoleophilia, Acidimicrobiia, and other Actinobacteriota. The potential for chemolithoautotrophic CO2 fixation (e.g., by the aforementioned Thermoleophilia, Acidimicrobiia, and other Actinobacteriota) could make these organisms independent from cyanobacterial C input; however, it is unclear whether the energy from atmospheric gas oxidation is sufficient to produce surplus organic C that could support obligate heterotrophic community members.
Although previous studies reported indications of CO2 fixation pathways other than the CBB cycle in desert soil communities (28, 30, 66), we did not find any genomic potential for additional pathways, such as the reverse TCA cycle or the reductive acetyl coenzyme A (acetyl-CoA) pathway, encoded in our MAGs. Yet, there are likely low-abundance populations that use other pathways to fix CO2, e.g., sulfate-reducing Deltaproteobacteria and nitrifying or methanogenic Archaea. These groups were previously reported in BSCs based on process rate measurements and marker gene-based studies (19, 22, 67) and were detected as very low-abundance OTUs in the 16S rRNA amplicon data (Data Set S1).
Potential for atmospheric H2 oxidation is more widespread than for atmospheric CO oxidation.
Recent studies have shown that the oxidation of atmospheric trace gases can be an alternative energy source for soil microorganisms (39, 68, 69), and it has been hypothesized that it might be widely used to generate maintenance energy during dormancy (11). We detected MAGs with the potential for H2 oxidation in the phyla Actinobacteriota and Chloroflexota, consistent with previous investigations (70), and H2 oxidation was recently detected in BSCs from the same sampling site (133). As mentioned above, some actinobacterial MAGs also harbored the genes for CO2 fixation, suggesting that H2 oxidation may be a means to generate energy for CO2 fixation, as suggested by Ji et al. (12). However, we also detected the potential for H2 oxidation in populations without the potential to fix CO2, indicating its utilization solely as a supportive energy source. The genetic potential for CO oxidation (via form I CO dehydrogenase) was far less common than previously hypothesized (11, 68), as form I CO dehydrogenase genes were detected in only four MAGs within the phyla Actinobacteriota and Gemmatimonadota. Instead, genes homologous to the CO dehydrogenase genes were found in many populations within the Actinobacteriota and Gemmatimonadota, as well as genes encoding form II CO dehydrogenases in MAGs of Alphaproteobacteria and Gemmatimonadota. The form II CO dehydrogenase is homologous to form I and seems to differ in its affinity for CO: it has been suggested that CO oxidation might not be its primary function (40). As such, these putative CO dehydrogenase-oxidizing enzymes require follow-up investigations to ascertain if they are functioning as a true CO dehydrogenase. Taken together, it appears that the genomic potential for atmospheric H2 scavenging is more ubiquitous than for CO scavenging in this arid BSC.
Limited genomic potential for polymeric C degradation but widespread potential for use of small organic substrates in BSC genomes.
We detected the potential for degrading polymeric polysaccharides in MAGs from the phyla Bacteroidota, Armatimonadota, and Planctomycetota (Fig. 4; Fig. S2), which is consistent with previous investigations of representatives from these phyla in soils and other environments (71–73). Populations encoding this physiological potential were of relatively low abundance in the BSC (Fig. 4; Fig. S2), suggesting that this physiological potential is limited in the BSC. In contrast, the genes for uptake of small organic substrates, such as oligopeptides, amino acids, and sugars, appear to be widespread (98% of MAGs) in the community (Fig. 4; Fig. S2). It is noteworthy that the polysaccharide-specialized MAGs carried no genes for energy generation from inorganic energy sources (such as H2 or CO). Polysaccharides excreted by cyanobacteria and other microorganisms are one of the largest C pools in BSCs (13). The ability to utilize such complex substrates might provide the cell with ample organic C and energy during active phases, enabling them to build up storage compounds and alleviate the need for additional energy sources.
Most BSC populations use non-sporulation persistence strategies.
The majority of the genomes (based on MAG coverage) across many taxa in our data set (e.g., Thermoleophilia, all Chloroflexota, Gemmatimonadota) did not carry genes indicative of a classical resting-stage formation. Such genomic potential was encoded only in a few MAGs belonging to the classes Acidimicrobiia and Actinobacteria (phylum Actinobacteriota) that contain known spore-forming species (41, 42). Firmicutes, another prominent spore-forming phylum that was reported to bloom in BSCs upon hydration (63, 74), was present only at very low relative abundances (based on 16S rRNA gene amplicon and unassembled metagenome data) (Fig. 2), which likely precluded the generation of MAGs. The lack of a Firmicutes bloom might be explained by different rehydration conditions, e.g., only to ca. 75% water-holding capacity in this study (e.g., 350 μl added per 1 g crust), while crusts in which Firmicutes blooms were reported were inundated with water (1 ml added to 0.5 g crust) (15, 63). However, it is possible that genes involved in resting-stage formation in some of our MAGs were not homologous to sequences in public databases, and as such we were unable to detect them with confidence. Nevertheless, we found indications of desiccation-adapted cell envelopes in other MAGs, such as polysaccharide sheaths in Cyanobacteria (75), which was supported by detected exopolysaccharide synthesis genes (Fig. 4). In addition, exopolysaccharide synthesis genes found in Acetobacteraceae MAGs (Fig. 4) suggest the ability to form alginate-reinforced cysts, as known from some Alphaproteobacteria species (47). The missing sporulation genes in Thermoleophilia and Rubrobacteria, which are part of Actinobacteriota, suggest that they do not undergo morphological transformations in preparation for desiccation, as supported by Rubrobacter cultures (61, 76, 77). Accordingly, Rubrobacteria MAGs also did not contain the genes for glycogen storage buildup (Fig. 4). However, we found a high frequency of bacteriorhodopsin- and hydrogenase-encoding genes in the Rubrobacteria MAGs, indicating that they could use light and atmospheric H2 as alternative energy sources during starvation. Hydrogenase-encoding genes were more widespread among Rubrobacteria MAGs obtained in this study than in previously sequenced Rubrobacteria genomes stemming from aquatic environments (Fig. 5). This adaptation seems plausible, since the accessibility of atmospheric H2 is enhanced by increased gas exchange in dry, porous desert soil.
Rubrobacteria genomes in BSC are particularly equipped for desiccation survival.
Hereafter, we discuss the genomic potential of the most abundant group of MAGs in the data set, Rubrobacteraceae (Fig. 6), from the perspective of desiccation survival. While light and H2 oxidation may provide additional energy (Fig. 6) as water and organic C availability decreases, it is unclear if the low soil water content during the dry period allows any ongoing enzyme activity. While pockets of increased humidity have been proposed to exist in dry soil (5, 66), a study applying in situ X-ray tomography to BSCs from the Moab Desert suggested the absence of such pockets after complete desiccation (78). However, even the near complete absence of water does not have to be fatal for a microbial cell. The damage by low water content can be mitigated by small organic molecules, like trehalose or other sugars (Fig. 6), that replace water in the cell, maintaining hydrogen bonds and preserving the structure of proteins (79), a principle used for storage of freeze-dried microbial culture stocks. Rubrobacter xylanophilus has been shown to accumulate and maintain unusually high intracellular concentrations of the osmoprotectants trehalose and mannosylglycerate by default under any growth condition (80). Rubrobacteria cells could thus be permanently prepared for desiccation by maintaining these high concentrations of osmolytes (Fig. 6) and therefore do not need to transform into a dedicated resting stage. In our study, we found a large variety of sugar-importing phosphotransferase systems encoded in Rubrobacteria MAGs that could be beneficial for such a strategy. Extensive studies of extreme radiation and desiccation tolerance of Deinococcus radiodurans have shown that its cells reassemble the fragmented genome from multiple mutual-correcting copies upon rehydration (55, 81), while ROS-induced damage in the desiccated state is reduced by accumulating manganese ions and ROS-scavenging molecules (48). The Rubrobacteria MAGs showed several indications of a similar strategy. Most of their characteristic genomic features were related to minimizing ROS accumulation by using antioxidant proteins like manganese catalase, DPS rubrerythrin (Fig. 6), and a different terminal oxidase (Fig. 6) with an additional catalase function (51). BSC Rubrobacteria MAGs encoded an additional version of DNA double-strand break repair polymerase PolA (55). The lack of “reckless” DNA degradation enzymes (53) might be a way to exclude even the possibility of unnecessary degradation of damaged DNA that could still be repaired.
FIG 6.

Exemplary metabolic sketch of BSC Rubrobacteraceae species based on the most abundant MAG. The sketch illustrates basic energy generation and C acquisition mechanisms, as well as some stress resistance and survival mechanisms. Rubrobacteraceae encode genetic potential for a mixotrophic lifestyle with rhodopsin and a high-affinity hydrogenase to generate proton motive force. The exact functions of CO dehydrogenase and propane monooxygenase homologs are yet to be determined. The C metabolism is optimized for efficient C utilization by using phosphotransferase sugar import systems (PTS) and glyoxylate bypass of the TCA cycle. It is important to note the absence of terminal cytochrome c oxidase in the respiratory chain and exclusive reliance on the manganese-based catalase for combating reactive oxygen species (ROS). DNA-binding rubrerythrin might also contribute to preventing ROS damage to DNA. Also note the presence of an additional homolog of PolA polymerase (COG0749), key in double-stranded DNA break repair. For pathway evaluation, RAST annotations were analyzed with Pathway Tools v.24 (131), and missing enzymes, usually due to the absence of an EC number in the annotation or the use of an uncommon synonymous enzyme name, were searched manually in RAST and other annotations of the MAGs. The depicted pathways are simplified versions of pathway depictions in the MetaCyc database (132) used by Pathway Tools software.
Challenges in using the index of replication as a measure of activity in BSC microorganisms.
The index of replication (iRep) was suggested as a way to estimate the replication status of microbial genomes based on their coverage in metagenomes. It is based on the assumption that regions of the genome closer to the origin of replication should have a higher coverage when a population containing actively dividing cells is sequenced (62). Irrespective of whether BSCs were dry or rehydrated, all MAGs that passed the threshold for iRep calculation were indicated as actively dividing (Table S3). Similar values have been reported for the dry Atacama soil and interpreted as indicators of active cell division (3). However, in environments where the activity of cells is restricted by water availability, the principle of the iRep approach might be impaired. Possibly, cellular processes such as DNA replication pause when the cell dries out and resume upon a water pulse. In this scenario, high iRep values might reflect a state of paused DNA replication. Additionally, high iRep values may also be caused by high strain diversity in the samples, as genome regions conserved across all strains would have higher coverage than more divergent regions (62).
Population-resolved metagenomics as a stepping stone in BSC microbial ecology.
The high degree of genomic novelty revealed in this study of BSCs from the Negev Desert illustrates the importance of metagenomic sequencing of terrestrial environments. In contrast to well-catalogued human microbiomes, e.g., from the oral cavity (82) and gut (83), where DNA or RNA sequence reads can readily be mapped to existing genomes, reference genomes are still largely missing for the highly diverse soil communities (84). This missing information also hinders detailed studies of, e.g., microbial activity by transcriptomics. Additionally, assuming the function of an organism based on the closest cultured or sequenced relatives can be misleading, as illustrated by a couple of examples in our data set. First, based on taxonomy, many of the detected microorganisms could potentially be diazotrophs, such as Frankiales (Actinobacteria) (85), Microvirga (86), and Acetobacteraceae (Alphaproteobacteria) (87). But none of the MAGs assigned to these groups contained genes involved in N2 fixation, which does not exclude the possibility that other, lower-abundance MAGs in these groups might carry this genomic potential. However, the absence of such genes not only among the most abundant microbial populations represented by MAGs but also in the unassembled reads illustrates the low relative abundances of these important community members in the Microcoleus-dominated crusts. Similar observations on missing sequence coverage of diazotrophs have been made in metagenome libraries of ocean waters with known N2 fixation activity (88, 89). Another example is the process of denitrification that has been detected in BSCs, ranging from very low rates in North American BSCs (90) to high rates in BSCs from Oman (91). The ability to respire nitrate was previously found in all sequenced Rubrobacteria genomes stemming from aquatic environments (60, 61, 77, 92), and without genome information of BSC Rubrobacteria, one might have assumed this abundant group encoded this physiological potential. However, genes involved in denitrification were completely absent from Rubrobacteria MAGs and our complete data set, despite Rubrobacteria genomes being by far the most abundant ones.
The above-mentioned discrepancies show that population-resolved metagenomic analysis was essential to gain insights into the distribution of potential lifestyles and survival strategies within the microbial populations constituting the BSC communities.
Metagenomics as a tool for hypothesis generation.
Our extensive analysis of population-resolved metagenomic data from BSCs in the Negev Desert indicates that a spectrum of different desiccation survival potentials is simultaneously present among the BSC genomes. Components of this spectrum can exist in pure form or can be combined to various degrees in one organism. On the one hand, there are genomes with strictly heterotrophic genomic potential, e.g., those of Bacteroidota and Planctomycetota, and genomes encoding resting-stage formation, e.g., in some Actinobacteria and Alphaproteobacteria. On the other hand, there are genomes with the potential to use continuously available inorganic energy sources and/or to preserve the cell components with a matrix of small organic molecules that would accumulate upon evaporation of water. Such strategies could be used for survival instead of a morphological transformation into a dedicated resting stage. The majority of the microbial populations encode metabolic potential for a combination of both strategies. Potential energy sources of most members of the BSC microbial community include both inorganic and organic energy sources. The potential to use inorganic energy sources and the potential to fix CO2 found in several populations might at least partially decouple these microorganisms from the primary production of cyanobacteria. How these different metabolic potentials are implemented and what implications they have for resuscitation of individual populations and for the long-term maintenance of the microbial community structure can now be tested by activity-based studies targeting specific populations.
MATERIALS AND METHODS
Sample collection.
Biological soil crust samples were collected in June 2017 at the long-term ecological research site (LTER) Avdat (30°36′33″N, 34°44′48″E), Negev Desert, Israel. Descriptions of basic soil properties and microbial community composition can be found, e.g., in references 93 to 96. With approximately 100 mm of yearly precipitation, the crusts at the site are light in color and dominated by cyanobacteria and contain little to no lichens and mosses, which, having large genomes, might significantly reduce the coverage of bacterial and archaeal genomes.
Soil crust samples were collected from 0.5- by 0.5-m patches distributed across the LTER with a distance of over 20 m between patches (Fig. 1). The sampling positions were chosen in shrub-free areas with a homogeneous surface appearance of the crust. We defined the cohesive crust layer of ca. 5 mm thickness on top of the soil as the crust. The pieces were carefully collected by hand and placed into petri dishes, which were immediately sealed with Parafilm. The crust samples were shipped and stored at room temperature in the dark until DNA extraction (4 weeks in total). Before crust pieces were weighed for DNA extraction, loose soil, if present, was removed from below the crust with a spatula. The thickness of extracted pieces was ca. 3 to 5 mm.
DNA extraction and sequencing.
DNA was extracted from ca. 500 mg of soil crust with a harsh phenol-chloroform-based protocol, including three bead-beating and extraction steps and CTAB (cetyltrimethylammonium bromide)-based removal of organic polymeric substances (97).
Extracted DNA was first used for 16S rRNA gene fragment amplification and Illumina MiSeq sequencing as described by Herbold et al. (98).
16S rRNA gene amplicon analysis.
The sequences were error-corrected using the Bayes-Hammer module (99) of SPAdes assembler v.3.11 (100), paired reads were merged using BBmerge v.37.61 (101) with the “strict” setting and a minimum overlap of 50 bp after clipping 3′ ends with quality scores below 20, and amplicon sequence variants (ASVs) were determined using DADA2 (102) with standard settings. The ASVs were further grouped into percentage identity-independent “operational taxonomic units” with Swarm2 (33) in fastidious mode with the limit of a large swarm for grafting set at 20. Taxonomy was assigned to OTU centroids by the last common ancestor (LCA) algorithm using rRNA secondary structure-aware SINA aligner v.1.2.11 (103) and the SILVA SSU132 database (104). OTUs classified as mitochondria or chloroplasts (21 in total) were removed, since the primers are not designed and optimized to accurately capture eukaryotic diversity. Clustering and plotting of data were performed in R using the packages vegan (105) and ggplot2 (106).
Choosing samples for metagenomic sequencing.
Assignment of metagenomic contigs to genomes (binning) is to a large extent based on genome-specific patterns of varying relative abundance across samples. Thus, based on the community composition determined by 16S rRNA amplicon sequencing, we chose three DNA extracts for metagenome sequencing, with the aim to cover most of the diversity and to have different relative abundances of taxa in the samples to enable differential-coverage genome binning. Additionally, we wanted to evaluate the practicability of using read coverage of metagenome bins to estimate the proportion of dividing cells by calculating the indices of replication. Since we expected cell division to increase in the hydrated state (which should be detectable in a change in genome replication indices), we rehydrated two additional crust pieces from plots where we picked dry pieces for metagenomic sequencing. Water was added to crust pieces to reach up to 26% water content (v/mwet, 75% of water-holding capacity of these crusts), and crusts were then incubated for 24 h in a sealed petri dish under 12 h of light (27°C) and 10 h of darkness (19°C) with 1-h transition periods in between in a climate-controlled chamber (Aralab, Rio de Mouro, Portugal). DNA was extracted the same way as done from the dry crusts. DNA was sequenced on an Illumina HiSeq 2500 instrument at the Vienna Biocenter Core Facility in the 2 × 150 bp read mode.
Microbial community composition and diversity estimation based on metagenomic reads.
To estimate the diversity and coverage of our metagenomic libraries, we analyzed the reads with Nonpareil v.3.303 (34) with default settings using the k-mer-based overlap search and fastq input. To estimate the taxonomic diversity in the libraries, reads were mapped to the SILVA SSU132 database using PhyloFlash v.3.0 (107). Taxa were considered detected when three or more reads were collected. We consider this method more precise than to estimate taxa based on protein blast hits to the NCBI nr database, as the SILVA database contains many more taxa than the genome-based NCBI nr database does. The rRNA gene is a well-established phylogenetic marker, whereas not all proteins encoded on the contigs can serve as such and can create noise when used for classification of raw reads or contigs.
Metagenome assembly and binning.
A detailed description of the logic behind the assembly and binning process is provided in Text S1 in the supplemental material and on https://github.com/meierdv/avdat_metagenome. Bash and R scripts with the commands used can be found on github at https://github.com/meierdv/avdat_metagenome.
Sequence reads were trimmed using BBduk v.37.61 (101), error-corrected using the Bayes-Hammer module (99) of SPAdes assembler v.3.11 (100), normalized to a target k-mer depth of 33× with BBnorm, and coassembled using MEGAHIT v.1.1.2 (108) with k-mers ranging from 21 to 137 in steps of 10. Contigs below 1,000 bp were removed from the final assembly. Error-corrected, not normalized reads were mapped to the contigs with BBmap v.37.61 (101) using an identity cutoff of 95% to assess the true relative abundance of contigs in each data set. Contigs were taxonomically classified using diamond blastp v.0.9.10 (109) on the translated open reading frames (ORFs predicted by Prodigal v.2.6.2) (110) against the NCBI nr protein database. The contigs were binned with MetaBAT v.2.12.1 (111), MaxBin v.2.2.4 (112), CONCOCT v.0.4.1 (113), and Metawatt v.3.5.3 (114). The results were summarized using DAStool v.1.1.0 (115), and the bins were analyzed with CheckM v.1.0.7 (116). Bins were grouped based on their placement in the CheckM reference genome tree and used as references for read mapping in order to perform reassemblies using less-complex clade-specific read sets and the more computationally intensive metaSPAdes v.3.11 assembler (117), with k-mers ranging from 21 to 127 in steps of 10.
The idea of improving assembly and bins by targeted reassembly is based on the logic described by Albertsen et al. (118). Generated reassemblies of the different taxonomic groups were binned in the same way as the coassembly. The binning was inspected and refined manually in Anvi'o v.5.2 (114). Mainly, contigs clearly clustering apart from the bin, based on the combination of tetranucleotide frequencies and differential coverage patterns, were removed from the bins. New bins were compared to the corresponding initial bins with dRep v.1.4.3 (119), and the ones with the better metrics (bigger size, higher completeness, lower contamination) were kept. Bins were filtered based on completeness (>50%) and contamination (<10%) estimated with CheckM (116), resulting in a final set of 96 metagenome-assembled genomes (MAGs).
MAG annotation and taxonomic classification.
First, the MAGs were annotated using the RAST-Tk pipeline (120). The translated ORFs predicted by RAST-Tk were then searched against various databases as follows: diamond blastp versus UniProt (121), hmmscan (http://www.hmmer.org/) versus Pfam (122), EggNOG-mapper v.1.0.1 (123) versus EggNOG (124), hmmscan versus CAZY (125) and MEROPS (126). The results of all annotations combined in one table were loaded as a structured query language (SQL) database and searched with SQL queries. Annotations of large subunits of CO dehydrogenase and catalytic subunits of methane monooxygenase were verified by phylogeny. Briefly, sequences included in the seed alignment of the respective Pfam, together with further sequences with confirmed function or known classification, were obtained from UniProt. Reference and metagenome-derived sequences were aligned to the Pfam alignment with hmmalign (http://www.hmmer.org/), and phylogenetic trees were calculated using FastTree v.2.1.11 (127) starting from a bionj tree (128), using the Le-Gascuel substitution model (129) and gamma likelihood optimization.
The MAGs were taxonomically classified by GTDB-Tk (36) and by the 16S rRNA gene if it was present in a MAG. The GTDB-Tk classification is based on (i) placement in the phylogenomic tree, (ii) relative evolutionary divergence (RED) value as established by Parks et al. (35), and (iii) average nucleotide identity to the closest related genome within a genus. For novel genera, families, and orders, local phylogenetic trees were calculated de novo based on GTDB alignment (35) by using FastTree (same settings as described above).
Functional comparison.
MAGs were grouped using a presence-absence matrix of orthologous proteins as assigned by EggNOG-mapper v.1.0.1 (123, 124). When comparing all MAGs spanning several different phyla, we further summarized different EggNOGs with the same assigned function. MAGs were clustered based on present EggNOGs using the Bray-Curtis dissimilarity matrix and average linkage clustering (vegan package, R) (105).
When the genomic potentials of Rubrobacteraceae MAGs and isolate genomes were compared, the EggNOGs were not summarized by function.
The annotations resulting from other sources were checked to confirm the observation and to clarify the function as far as possible.
Index of replication.
Indices of replication were calculated with iRep (62) using standard settings. Only indices that passed all thresholds and corrections are reported.
Data availability.
rRNA gene amplicon reads, unassembled metagenomic reads, metagenome assemblies, and annotated MAGs have been deposited in the European Nucleotide Archive under project number PRJEB36534. Bash and R code of all sequence analysis steps can be found at https://github.com/meierdv/avdat_metagenome.
ACKNOWLEDGMENTS
This work was funded by an ERC Starting grant (grant agreement number 636928 to D.W.) from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program.
We thank the Division of Computational Systems Biology and University of Vienna, especially Thomas Rattei and Florian Goldenberg, for providing and maintaining excellent computational resources (Vienna Life Science Compute Cluster). We also thank Sean Bay, Capucine Baubin, and Nimrod Wieler for helping hands during the crust sampling campaign.
We declare no conflicts of interest.
REFERENCES
- 1.Pointing SB, Belnap J. 2012. Microbial colonization and controls in dryland systems. Nat Rev Microbiol 10:551–562. doi: 10.1038/nrmicro2831. [DOI] [PubMed] [Google Scholar]
- 2.Warren-Rhodes KA, Rhodes KL, Pointing SB, Ewing SA, Lacap DC, Gómez-Silva B, Amundson R, Friedmann EI, McKay CP. 2006. Hypolithic cyanobacteria, dry limit of photosynthesis, and microbial ecology in the hyperarid Atacama Desert. Microb Ecol 52:389–398. doi: 10.1007/s00248-006-9055-7. [DOI] [PubMed] [Google Scholar]
- 3.Schulze-Makuch D, Wagner D, Kounaves SP, Mangelsdorf K, Devine KG, de Vera J-P, Schmitt-Kopplin P, Grossart H-P, Parro V, Kaupenjohann M, Galy A, Schneider B, Airo A, Frösler J, Davila AF, Arens FL, Cáceres L, Cornejo FS, Carrizo D, Dartnell L, DiRuggiero J, Flury M, Ganzert L, Gessner MO, Grathwohl P, Guan L, Heinz J, Hess M, Keppler F, Maus D, McKay CP, Meckenstock RU, Montgomery W, Oberlin EA, Probst AJ, Sáenz JS, Sattler T, Schirmack J, Sephton MA, Schloter M, Uhl J, Valenzuela B, Vestergaard G, Wörmer L, Zamorano P. 2018. Transitory microbial habitat in the hyperarid Atacama Desert. Proc Natl Acad Sci U S A 115:2670–2675. doi: 10.1073/pnas.1714341115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebre PH, De Maayer P, Cowan DA. 2017. Xerotolerant bacteria: surviving through a dry spell. Nat Rev Microbiol 15:285–296. doi: 10.1038/nrmicro.2017.16. [DOI] [PubMed] [Google Scholar]
- 5.Schimel JP. 2018. Life in dry soils: effects of drought on soil microbial communities and processes. Annu Rev Ecol Evol Syst 49:409–432. doi: 10.1146/annurev-ecolsys-110617-062614. [DOI] [Google Scholar]
- 6.Onyenwoke R, Brill J, Farahi K, Wiegel J. 2004. Sporulation genes in members of the low G+C Gram-type-positive phylogenetic branch (Firmicutes). Arch Microbiol 182:182–192. doi: 10.1007/s00203-004-0696-y. [DOI] [PubMed] [Google Scholar]
- 7.Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Meier-Kolthoff JP, Klenk H-P, Clément C, Ouhdouch Y, van Wezel GP. 2016. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev 80:1–43. doi: 10.1128/MMBR.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imhoff JF. 2006. The phototrophic Alpha-Proteobacteria, p 41–64. In The prokaryotes. Springer, New York, NY. [Google Scholar]
- 9.Kaplan-Levy RN, Hadas O, Summers ML, Rücker J, Sukenik A. 2010. Akinetes: dormant cells of cyanobacteria, p 5–27. In Lubzens E, Cerda J, Clark M (ed), Dormancy and resistance in harsh environments. Springer, Berlin, Germany. [Google Scholar]
- 10.Steven B, Gallegos-Graves LV, Belnap J, Kuske CR. 2013. Dryland soil microbial communities display spatial biogeographic patterns associated with soil depth and soil parent material. FEMS Microbiol Ecol 86:101–113. doi: 10.1111/1574-6941.12143. [DOI] [PubMed] [Google Scholar]
- 11.Leung PM, Bay SK, Meier DV, Chiri E, Cowan DA, Gillor O, Woebken D, Greening C. 2020. Energetic basis of microbial growth and persistence in desert ecosystems. mSystems 5:e00495-19. doi: 10.1128/mSystems.00495-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji M, Greening C, Vanwonterghem I, Carere CR, Bay SK, Steen JA, Montgomery K, Lines T, Beardall J, Van Dorst J, Snape I, Stott MB, Hugenholtz P, Ferrari BC. 2017. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature 552:400–403. doi: 10.1038/nature25014. [DOI] [PubMed] [Google Scholar]
- 13.Mager DM, Thomas AD. 2011. Extracellular polysaccharides from cyanobacterial soil crusts: a review of their role in dryland soil processes. J Arid Environ 75:91–97. doi: 10.1016/j.jaridenv.2010.10.001. [DOI] [Google Scholar]
- 14.Baran R, Brodie EL, Mayberry-Lewis J, Hummel E, Nunes U, Rocha D, Chakraborty R, Bowen BP, Karaoz U, Cadillo-Quiroz H, Garcia-Pichel F, Northen TR. 2015. Exometabolite niche partitioning among sympatric soil bacteria. Nat Commun 6:8289. doi: 10.1038/ncomms9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swenson TL, Karaoz U, Swenson JM, Bowen BP, Northen TR. 2018. Linking soil biology and chemistry in biological soil crust using isolate exometabolomics. Nat Commun 9:19. doi: 10.1038/s41467-017-02356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Pichel F, Johnson SL, Youngkin D, Belnap J. 2003. Small-scale vertical distribution of bacterial biomass and diversity in biological soil crusts from arid lands in the Colorado Plateau. Microb Ecol 46:312–321. doi: 10.1007/s00248-003-1004-0. [DOI] [PubMed] [Google Scholar]
- 17.Maier S, Tamm A, Wu D, Caesar J, Grube M, Weber B. 2018. Photoautotrophic organisms control microbial abundance, diversity, and physiology in different types of biological soil crusts. ISME J 12:1032–1046. doi: 10.1038/s41396-018-0062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowker MA, Belnap J, Bala Chaudhary V, Johnson NC. 2008. Revisiting classic water erosion models in drylands: the strong impact of biological soil crusts. Soil Biol Biochem 40:2309–2316. doi: 10.1016/j.soilbio.2008.05.008. [DOI] [Google Scholar]
- 19.Marusenko Y, Bates ST, Anderson I, Johnson SL, Soule T, Garcia-Pichel F. 2013. Ammonia-oxidizing archaea and bacteria are structured by geography in biological soil crusts across North American arid lands. Ecol Process 2:9–10. doi: 10.1186/2192-1709-2-9. [DOI] [Google Scholar]
- 20.Yeager CM, Kornosky JL, Morgan RE, Cain EC, Garcia-Pichel F, Housman DC, Belnap J, Kuske CR. 2007. Three distinct clades of cultured heterocystous cyanobacteria constitute the dominant N2-fixing members of biological soil crusts of the Colorado Plateau, USA. FEMS Microbiol Ecol 60:85–97. doi: 10.1111/j.1574-6941.2006.00265.x. [DOI] [PubMed] [Google Scholar]
- 21.Yeager CM, Kornosky JL, Housman DC, Grote EE, Belnap J, Kuske CR. 2004. Diazotrophic community structure and function in two successional stages of biological soil crusts from the Colorado Plateau and Chihuahuan Desert. Appl Environ Microbiol 70:973–983. doi: 10.1128/aem.70.2.973-983.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angel R, Matthies D, Conrad R. 2011. Activation of methanogenesis in arid biological soil crusts despite the presence of oxygen. PLoS One 6:e20453. doi: 10.1371/journal.pone.0020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csotonyi JT, Swiderski J, Stackebrandt E, Yurkov V. 2010. A new environment for aerobic anoxygenic phototrophic bacteria: biological soil crusts. Environ Microbiol Rep 2:651–656. doi: 10.1111/j.1758-2229.2010.00151.x. [DOI] [PubMed] [Google Scholar]
- 24.Steven B, Gallegos-Graves LV, Starkenburg SR, Chain PS, Kuske CR. 2012. Targeted and shotgun metagenomic approaches provide different descriptions of dryland soil microbial communities in a manipulated field study. Environ Microbiol Rep 4:248–256. doi: 10.1111/j.1758-2229.2012.00328.x. [DOI] [PubMed] [Google Scholar]
- 25.Steven B, Kuske CR, Gallegos-Graves LV, Reed SC, Belnap J. 2015. Climate change and physical disturbance manipulations result in distinct biological soil crust communities. Appl Environ Microbiol 81:7448–7459. doi: 10.1128/AEM.01443-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steven B, Belnap J, Kuske CR. 2018. Chronic physical disturbance substantially alters the response of biological soil crusts to a wetting pulse, as characterized by metatranscriptomic sequencing. Front Microbiol 9:2382. doi: 10.3389/fmicb.2018.02382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. 2012. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci U S A 109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vikram S, Guerrero LD, Makhalanyane TP, Le PT, Seely M, Cowan DA. 2016. Metagenomic analysis provides insights into functional capacity in a hyperarid desert soil niche community. Environ Microbiol 18:1875–1888. doi: 10.1111/1462-2920.13088. [DOI] [PubMed] [Google Scholar]
- 29.Le PT, Makhalanyane TP, Guerrero LD, Vikram S, Van de Peer Y, Cowan DA. 2016. Comparative metagenomic analysis reveals mechanisms for stress response in hypoliths from extreme hyperarid deserts. Genome Biol Evol 8:2737–2747. doi: 10.1093/gbe/evw189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JY, Jin XY, Zhang XC, Chen L, Liu JL, Zhang HM, Zhang X, Zhang YF, Zhao JH, Ma ZS, Jin D. 2020. Comparative metagenomics of two distinct biological soil crusts in the Tengger Desert, China. Soil Biol Biochem 140:107637. doi: 10.1016/j.soilbio.2019.107637. [DOI] [Google Scholar]
- 31.Steven B, Gallegos-Graves LV, Yeager C, Belnap J, Kuske CR. 2014. Common and distinguishing features of the bacterial and fungal communities in biological soil crusts and shrub root zone soils. Soil Biol Biochem 69:302–312. doi: 10.1016/j.soilbio.2013.11.008. [DOI] [Google Scholar]
- 32.Angel R, Conrad R. 2013. Elucidating the microbial resuscitation cascade in biological soil crusts following a simulated rain event. Environ Microbiol 15:2799–2815. doi: 10.1111/1462-2920.12140. [DOI] [PubMed] [Google Scholar]
- 33.Mahe F, Rognes T, Quince C, de Vargas C, Dunthorn M. 2015. Swarm v2: highly-scalable and high-resolution amplicon clustering. PeerJ 3:e1420. doi: 10.7717/peerj.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-R LM, Gunturu S, Tiedje JM, Cole JR, Konstantinidis KT. 2018. Nonpareil 3: fast estimation of metagenomic coverage and sequence diversity. mSystems 3:e00039-18. doi: 10.1128/mSystems.00039-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, Hugenholtz P. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 36.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pushkarev A, Inoue K, Larom S, Flores-Uribe J, Singh M, Konno M, Tomida S, Ito S, Nakamura R, Tsunoda SP, Philosof A, Sharon I, Yutin N, Koonin EV, Kandori H, Béjà O. 2018. A distinct abundant group of microbial rhodopsins discovered using functional metagenomics. Nature 558:595–599. doi: 10.1038/s41586-018-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Søndergaard D, Pedersen CNS, Greening C. 2016. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep 6:34212. doi: 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greening C, Constant P, Hards K, Morales SE, Oakeshott JG, Russell RJ, Taylor MC, Berney M, Conrad R, Cook GM. 2015. Atmospheric hydrogen scavenging: from enzymes to ecosystems. Appl Environ Microbiol 81:1190–1199. doi: 10.1128/AEM.03364-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King GM, Weber CF. 2007. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol 5:107–118. doi: 10.1038/nrmicro1595. [DOI] [PubMed] [Google Scholar]
- 41.Chater KF. 1993. Genetics of differentiation in Streptomyces. Annu Rev Microbiol 47:685–711. doi: 10.1146/annurev.mi.47.100193.003345. [DOI] [PubMed] [Google Scholar]
- 42.Chater KF. 2001. Regulation of sporulation in Streptomyces coelicolor A32: a checkpoint multiplex? Curr Opin Microbiol 4:667–673. doi: 10.1016/s1369-5274(01)00267-3. [DOI] [PubMed] [Google Scholar]
- 43.Salerno P, Larsson J, Bucca G, Laing E, Smith CP, Flärdh K. 2009. One of the two genes encoding nucleoid-associated HU proteins in Streptomyces coelicolor is developmentally regulated and specifically involved in spore maturation. J Bacteriol 191:6489–6500. doi: 10.1128/JB.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grove A. 2011. Functional evolution of bacterial histone-like HU proteins. Curr Issues Mol Biol 13:1–12. [PubMed] [Google Scholar]
- 45.Preiss J. 1984. Bacterial glycogen synthesis and its Regulation. Annu Rev Microbiol 38:419–458. doi: 10.1146/annurev.mi.38.100184.002223. [DOI] [PubMed] [Google Scholar]
- 46.Nariya H, Inouye S. 2003. An effective sporulation of Myxococcus xanthus requires glycogen consumption via Pkn4-activated 6-phosphofructokinase. Mol Microbiol 49:517–528. doi: 10.1046/j.1365-2958.2003.03572.x. [DOI] [PubMed] [Google Scholar]
- 47.Espín G. 2016. Genes involved in the formation of desiccation-resistant cysts in Azotobacter vinelandii, p 709–715. In de Bruijn F (ed), Stress and environmental regulation of gene expression and adaptation in bacteria. John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- 48.Slade D, Radman M. 2011. Oxidative stress resistance in Deinococcus radiodurans. Microbiol Mol Biol Rev 75:133–191. doi: 10.1128/MMBR.00015-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daly MJ, Gaidamakova EK, Matrosova VY, Vasilenko A, Zhai M, Leapman RD, Lai B, Ravel B, Li SMW, Kemner KM, Fredrickson JK. 2007. Protein oxidation implicated as the primary determinant of bacterial radioresistance. PLoS Biol 5:e92. doi: 10.1371/journal.pbio.0050092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrews SC. 2010. The ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim Biophys Acta 1800:691–705. doi: 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 51.Borisov VB, Forte E, Davletshin A, Mastronicola D, Sarti P, Giuffrè A. 2013. Cytochrome bd oxidase from Escherichia coli displays high catalase activity: an additional defense against oxidative stress. FEBS Lett 587:2214–2218. doi: 10.1016/j.febslet.2013.05.047. [DOI] [PubMed] [Google Scholar]
- 52.Ahn S, Jung J, Jang I-A, Madsen EL, Park W. 2016. Role of glyoxylate shunt in oxidative stress response. J Biol Chem 291:11928–11938. doi: 10.1074/jbc.M115.708149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Repar J, Briški N, Buljubašić M, Zahradka K, Zahradka D. 2013. Exonuclease VII is involved in “reckless” DNA degradation in UV-irradiated Escherichia coli. Mutat Res 750:96–104. doi: 10.1016/j.mrgentox.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Henrikus SS, Wood EA, McDonald JP, Cox MM, Woodgate R, Goodman MF, van Oijen AM, Robinson A. 2018. DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli. PLoS Genet 14:e1007161. doi: 10.1371/journal.pgen.1007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, Lindner AB, Radman M. 2006. Reassembly of shattered chromosomes in Deinococcus radiodurans. Nature 443:569–573. doi: 10.1038/nature05160. [DOI] [PubMed] [Google Scholar]
- 56.Govorunova EG, Sineshchekov OA, Li H, Spudich JL. 2017. Microbial rhodopsins: diversity, mechanisms, and optogenetic applications. Annu Rev Biochem 86:845–872. doi: 10.1146/annurev-biochem-101910-144233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kämpfer P, Glaeser SP, Busse HJ, Abdelmohsen UR, Hentschel U. 2014. Rubrobacter aplysinae sp. nov., isolated from the marine sponge Aplysina aerophoba. Int J Syst Evol Microbiol 64:705–709. doi: 10.1099/ijs.0.055152-0. [DOI] [PubMed] [Google Scholar]
- 58.Chen RW, Wang KX, Wang FZ, He YQ, Long LJ, Tian XP. 2018. Rubrobacter indicoceani sp. nov., a new marine actinobacterium isolated from Indian Ocean sediment. Int J Syst Evol Microbiol 68:3487–3493. doi: 10.1099/ijsem.0.003018. [DOI] [PubMed] [Google Scholar]
- 59.Yoshinaka T, Yano K, Yamaguchi H. 1973. Isolation of highly radioresistant bacterium, Arthrobacter radiotolerans nov. sp. Agric Biol Chem 37:2269–2275. doi: 10.1080/00021369.1973.10861003. [DOI] [Google Scholar]
- 60.Chen MY, Wu SH, Lin GH, Lu CP, Lin YT, Chang WC, Tsay SS. 2004. Rubrobacter taiwanensis sp. nov., a novel thermophilic, radiation-resistant species isolated from hot springs. Int J Syst Evol Microbiol 54:1849–1855. doi: 10.1099/ijs.0.63109-0. [DOI] [PubMed] [Google Scholar]
- 61.Carreto L, Moore E, Nobre MF, Wait R, Riley PW, Sharp RJ, Da Costa MS. 1996. Rubrobacter xylanophilus sp. nov., a new thermophilic species isolated from a thermally polluted effluent. Int J Syst Bacteriol 46:460–465. doi: 10.1099/00207713-46-2-460. [DOI] [Google Scholar]
- 62.Brown CT, Olm MR, Thomas BC, Banfield JF. 2016. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol 34:1256–1263. doi: 10.1038/nbt.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Goethem MW, Swenson TL, Trubl G, Roux S, Northen TR. 2019. Characteristics of wetting-induced bacteriophage blooms in biological soil crust. mBio 10:e02287-19. doi: 10.1128/mBio.02287-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tahon G, Tytgat B, Stragier P, Willems A. 2016. Analysis of cbbL, nifH, and pufLM in soils from the Sør Rondane Mountains, Antarctica, reveals a large diversity of autotrophic and phototrophic bacteria. Microb Ecol 71:131–149. doi: 10.1007/s00248-015-0704-6. [DOI] [PubMed] [Google Scholar]
- 65.Zeng Y, Koblížek M. 2017. Phototrophic Gemmatimonadetes: a new “purple” branch on the bacterial tree of life, p 163–192. In Hallenbeck P (ed), Modern topics in the phototrophic prokaryotes. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 66.León-Sobrino C, Ramond JB, Maggs-Kölling G, Cowan DA. 2019. Nutrient acquisition, rather than stress response over diel cycles, drives microbial transcription in a hyper-arid Namib Desert soil. Front Microbiol 10:1054. doi: 10.3389/fmicb.2019.01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Angel R, Claus P, Conrad R. 2012. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J 6:847–862. doi: 10.1038/ismej.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cordero PRF, Bayly K, Man Leung P, Huang C, Islam ZF, Schittenhelm RB, King GM, Greening C. 2019. Atmospheric carbon monoxide oxidation is a widespread mechanism supporting microbial survival. ISME J 13:2868–2881. doi: 10.1038/s41396-019-0479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tveit AT, Hestnes AG, Robinson SL, Schintlmeister A, Dedysh SN, Jehmlich N, von Bergen M, Herbold C, Wagner M, Richter A, Svenning MM. 2019. Widespread soil bacterium that oxidizes atmospheric methane. Proc Natl Acad Sci U S A 116:8515–8524. doi: 10.1073/pnas.1817812116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, Cook GM, Morales SE. 2016. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J 10:761–777. doi: 10.1038/ismej.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas F, Hehemann J-H, Rebuffet E, Czjzek M, Michel G. 2011. Environmental and gut Bacteroidetes: the food connection. Front Microbiol 2:93. doi: 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang X, Sharp CE, Jones GM, Grasby SE, Brady AL, Dunfield PF. 2015. Stable-isotope probing identifies uncultured Planctomycetes as primary degraders of a complex heteropolysaccharide in soil. Appl Environ Microbiol 81:4607–4615. doi: 10.1128/AEM.00055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reintjes G, Arnosti C, Fuchs B, Amann R. 2019. Selfish, sharing and scavenging bacteria in the Atlantic Ocean: a biogeographical study of bacterial substrate utilisation. ISME J 13:1119–1132. doi: 10.1038/s41396-018-0326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karaoz U, Couradeau E, da Rocha UN, Lim H-C, Northen T, Garcia-Pichel F, Brodie EL. 2018. Large blooms of Bacillales (Firmicutes) underlie the response to wetting of cyanobacterial biocrusts at various stages of maturity. mBio 9:e01366-16. doi: 10.1128/mBio.01366-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mazor G, Kidron GJ, Vonshak A, Abeliovich A. 1996. The role of cyanobacterial exopolysaccharides in structuring desert microbial crusts. FEMS Microbiol Ecol 21:121–130. doi: 10.1111/j.1574-6941.1996.tb00339.x. [DOI] [Google Scholar]
- 76.Ferreira AC, Nobre MF, Moore E, Rainey FA, Battista JR, Da Costa MS. 1999. Characterization and radiation resistance of new isolates of Rubrobacter radiotolerans and Rubrobacter xylanophilus. Extremophiles 3:235–238. doi: 10.1007/s007920050121. [DOI] [PubMed] [Google Scholar]
- 77.Suzuki K, Collins MD, Iijima E, Komagata K. 1988. Chemotaxonomic characterization of a radiotolerant bacterium, Arthrobacter radiotolerans: description of Rubrobacter radiotolerans gen. nov., comb. nov. FEMS Microbiol Lett 52:33–39. doi: 10.1111/j.1574-6968.1988.tb02568.x. [DOI] [Google Scholar]
- 78.Couradeau E, Felde V, Parkinson D, Uteau D, Rochet A, Cuellar C, Winegar G, Peth S, Northen TR, Garcia-Pichel F. 2018. In situ X-ray tomography imaging of soil water and cyanobacteria from biological soil crusts undergoing desiccation. Front Environ Sci 6. doi: 10.3389/fenvs.2018.00065. [DOI] [Google Scholar]
- 79.Billi D, Potts M. 2002. Life and death of dried prokaryotes. Res Microbiol 153:7–12. doi: 10.1016/s0923-2508(01)01279-7. [DOI] [PubMed] [Google Scholar]
- 80.Empadinhas N, Mendes V, Simões C, Santos MS, Mingote A, Lamosa P, Santos H, Costa MSD. 2007. Organic solutes in Rubrobacter xylanophilus: the first example of di-myo-inositol-phosphate in a thermophile. Extremophiles 11:667–673. doi: 10.1007/s00792-007-0084-z. [DOI] [PubMed] [Google Scholar]
- 81.Mattimore V, Battista JR. 1996. Radioresistance of Deinococcus radiodurans: functions necessary to survive ionizing radiation are also necessary to survive prolonged desiccation. J Bacteriol 178:633–637. doi: 10.1128/jb.178.3.633-637.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. 2010. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010:baq013. doi: 10.1093/database/baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, Sun H, Xia Y, Liang S, Dai Y, Wan D, Jiang R, Su L, Feng Q, Jie Z, Guo T, Xia Z, Liu C, Yu J, Lin Y, Tang S, Huo G, Xu X, Hou Y, Liu X, Wang J, Yang H, Kristiansen K, Li J, Jia H, Xiao L. 2019. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol 37:179–185. doi: 10.1038/s41587-018-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mende DR, Letunic I, Huerta-Cepas J, Li SS, Forslund K, Sunagawa S, Bork P. 2017. proGenomes: a resource for consistent functional and taxonomic annotations of prokaryotic genomes. Nucleic Acids Res 45:D529–D534. doi: 10.1093/nar/gkw989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sellstedt A, Richau KH. 2013. Aspects of nitrogen-fixing actinobacteria, in particular free-living and symbiotic Frankia. FEMS Microbiol Lett 342:179–186. doi: 10.1111/1574-6968.12116. [DOI] [PubMed] [Google Scholar]
- 86.Radl V, Simões-Araújo JL, Leite J, Passos SR, Martins LMV, Xavier GR, Rumjanek NG, Baldani JI, Zilli JE. 2014. Microvirga vignae sp. nov., a root nodule symbiotic bacterium isolated from cowpea grown in semi-arid Brazil. Int J Syst Evol Microbiol 64:725–730. doi: 10.1099/ijs.0.053082-0. [DOI] [PubMed] [Google Scholar]
- 87.Saravanan VS, Madhaiyan M, Osborne J, Thangaraju M, Sa TM. 2008. Ecological occurrence of Gluconacetobacter diazotrophicus and nitrogen-fixing Acetobacteraceae members: their possible role in plant growth promotion. Microb Ecol 55:130–140. doi: 10.1007/s00248-007-9258-6. [DOI] [PubMed] [Google Scholar]
- 88.Johnston AWB, Li Y, Ogilvie L. 2005. Metagenomic marine nitrogen fixation—feast or famine? Trends Microbiol 13:416–420. doi: 10.1016/j.tim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 89.Delmont TO, Quince C, Shaiber A, Esen ÖC, Lee ST, Rappé MS, McLellan SL, Lücker S, Eren AM. 2018. Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nat Microbiol 3:804–813. doi: 10.1038/s41564-018-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johnson SL, Neuer S, Garcia-Pichel F. 2007. Export of nitrogenous compounds due to incomplete cycling within biological soil crusts of arid lands. Environ Microbiol 9:680–689. doi: 10.1111/j.1462-2920.2006.01187.x. [DOI] [PubMed] [Google Scholar]
- 91.Abed RMM, Lam P, De Beer D, Stief P. 2013. High rates of denitrification and nitrous oxide emission in arid biological soil crusts from the Sultanate of Oman. ISME J 7:1862–1875. doi: 10.1038/ismej.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Albuquerque L, Johnson MM, Schumann P, Rainey FA, Costa MS, Da Costa MS. 2014. Description of two new thermophilic species of the genus Rubrobacter, Rubrobacter calidifluminis sp. nov. and Rubrobacter naiadicus sp. nov., and emended description of the genus Rubrobacter and the species Rubrobacter bracarensis. Syst Appl Microbiol 37:235–243. doi: 10.1016/j.syapm.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 93.Angel R, Soares MIM, Ungar ED, Gillor O. 2010. Biogeography of soil archaea and bacteria along a steep precipitation gradient. ISME J 4:553–563. doi: 10.1038/ismej.2009.136. [DOI] [PubMed] [Google Scholar]
- 94.Bachar A, Al-Ashhab A, Soares MIM, Sklarz MY, Angel R, Ungar ED, Gillor O. 2010. Soil microbial abundance and diversity along a low precipitation gradient. Microb Ecol 60:453–461. doi: 10.1007/s00248-010-9727-1. [DOI] [PubMed] [Google Scholar]
- 95.Yu J, Glazer N, Steinberger Y. 2014. Carbon utilization, microbial biomass, and respiration in biological soil crusts in the Negev Desert. Biol Fertil Soils 50:285–293. doi: 10.1007/s00374-013-0856-9. [DOI] [Google Scholar]
- 96.Št'ovíček A, Kim M, Or D, Gillor O. 2017. Microbial community response to hydration-desiccation cycles in desert soil. Sci Rep 7:45735. doi: 10.1038/srep45735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Angel R. 23 October 2012. Total nucleic acid extraction from soil. Protoc Exch doi: 10.1038/protex.2012.046. [DOI] [Google Scholar]
- 98.Herbold CW, Pelikan C, Kuzyk O, Hausmann B, Angel R, Berry D, Loy A. 2015. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front Microbiol 6:731. doi: 10.3389/fmicb.2015.00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nikolenko SI, Korobeynikov AI, Alekseyev MA. 2013. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics 14:S7. doi: 10.1186/1471-2164-14-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bushnell B. 2017. BBtools. https://sourceforge.net/projects/bbmap/.
- 102.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pruesse E, Peplies J, Glockner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oksanen J, Blanchet FG, Kindt R, Legendre P, Michin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2017. vegan: community ecology package. R package version 2.0–10. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 106.Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer, New York, NY. [Google Scholar]
- 107.Gruber-Vodicka HR, Pruesse E, Seah Brandon K. 2017. phyloFlash. https://github.com/HRGV/phyloFlash/blob/master/docs/index.md. [DOI] [PMC free article] [PubMed]
- 108.Li D, Liu C, Luo R, Sadakane K, Lam T. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 109.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 110.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW. 2014. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2:26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. 2014. Binning metagenomic contigs by coverage and composition. Nat Methods 11:1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 114.Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. 2012. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front Microbiol 3:410. doi: 10.3389/fmicb.2012.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. 2018. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parks DH, Imelfort M, Skennerton CT, Hugenholz P, Tyson GW, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 119.Olm MR, Brown CT, Brooks B, Banfield JF. 2017. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11:2864–2868. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomason JA, Stevens R, Vonstein V, Wattam AR, Xia F, Xia F. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Magrane M, Consortium U . 2011. UniProt Knowledgebase: a hub of integrated protein data. Database. Database (Oxford) 2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, Rattei T, Mende DR, Sunagawa S, Kuhn M, Jensen LJ, von Mering C, Bork P. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44:D286–D293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lombard V, Golaconda RH, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzyme database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rawlings ND, Barrett AJ, Bateman A. 2010. MEROPS: the peptidase database. Nucleic Acids Res 38:D227–D233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gascuel O. 1997. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol 14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 129.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 130.Ryan WBF, Carbotte SM, Coplan J, O'Hara S, Melkonian A, Arko R, Weissel RA, Ferrini V, Goodwillie A, Nitsche F, Bonczkowski J, Zemsky R. 2009. Global multi-resolution topography (GMRT) synthesis data set. Geochem Geophys Geosyst 10:Q03014. doi: 10.1029/2008GC002332. [DOI] [Google Scholar]
- 131.Karp PD, Paley SM, Midford PE, Krummenacker M, Billington R, Kothari A, Ong WK, Subhraveti P, Keseler IM, Caspi R. 2020. Pathway Tools version 24.0: integrated software for pathway/genome informatics and systems biology. arXiv 1510.03964v4.
- 132.Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, Midford PE, Ong WK, Paley S, Subhraveti P, Karp PD. 2020. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res 48:D445–D453. doi: 10.1093/nar/gkz862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Giguere AT, Eichorst SA, Meier D, Herbold CW, Richter A, Greening C, Woebken D. 6 October 2020. Acidobacteria are active and abundant members of diverse atmospheric H2-oxidizing communities detected in temperate soils. ISME J. doi: 10.1038/s41396-020-00750-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
16S rRNA gene amplicon sequencing metrics. The table provides the numbers of quality-filtered reads obtained from each sample, the numbers of ASVs and Swarm-generated OTUs detected in each sample (richness), and the percentages of reads assigned to ubiquitous OTUs, OTUs appearing in more than 50% of the samples, and OTUs appearing only in fewer than four samples. Download Table S1, PDF file, 0.03 MB (35.1KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Large spreadsheet providing relative abundances and taxonomic classification of Swarm OTUs as well as relative abundances of microbial genera as determined by short read mapping to SILVA SSU132 database with PhyloFlash v.30 (13). Only genera with three or more reads mapping were considered as detected. Download Data Set S1, XLSX file, 0.2 MB (228.2KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A detailed description of the metagenome analysis providing the reasoning behind the choice of methods and analysis steps performed. The scripts referenced in this section can be found in the following Github repository: https://github.com/meierdv/avdat_metagenome. Download Text S1, PDF file, 0.1 MB (130.9KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Discussion of possible causes for differences in relative abundances of microbial taxa between 16S rRNA gene amplicon data and metagenomic sequencing data, with a focus on difference in relative abundance of Cyanobacteria. Download Text S2, PDF file, 0.04 MB (42.5KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Metagenomic sequencing and assembly metrics. Part A provides the basic metrics of metagenomic sequencing efforts, such as sequencing depth, the estimation of genomic diversity covered (determined by Nonpareil), and percentages of reads represented by the assembly and analyzed MAGs. Part B provides the basic metrics of the initial metagenomic assembly. Download Table S2, PDF file, 0.04 MB (46.3KB, pdf) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenomic placement of MAGs representing previously unsequenced genera, families, and orders. Clade-specific trees were calculated based on concatenated amino acid alignment of 120 phylogenetic markers used in GTDB (8) using FastTree (starting tree, bio-neighbor-joining; model, Le-Gascuel). Alignment was filtered to contain sites conserved in 25% of sequences (4,911 positions for Sphingomonadales, 4,918 for Acetobacterales, 4,521 for Gemmatimonadota, 4,819 for Cytophagales, 4,118 positions for Phycisphaerae, 4,365 for Chtoniobacterales, 4,061 for Chloroflexota, 4,805 for Armatimonadota, 4,627 for Thermosynechococcales, 4,418 positions for Rubrobacteria and Thermoleophilia, 4,375 for Acidimicriia, 4,882 for Mycobacteriales, and 4,872 for Propionibacterales). Multifurcations have been introduced for branchings with less than 50% support. Branch support was calculated by 1,000 resamplings of site likelihood and the Shimodaira-Hasegawa test (FastTree default). Download FIG S1, TIF file, 1.5 MB (1.5MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
MAG metrics and classification. This large table provides sequence-related metrics of the metagenome-assembled genomes (MAGs), estimated values of their completeness and duplication, relative abundance of the MAGs in the individual metagenome samples, estimated indices of replication (iRep values), and taxonomic classification of the MAGs by GTDB-Tk and the criteria on which the classification was based (e.g., ANI to closest-related genome, if applicable). Download Table S3, XLSX file, 0.03 MB (33.9KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Selected metabolic traits by MAG. The genomes were grouped based on a presence-absence matrix of EggNOG orthologs, which corresponds largely to taxonomic classification. For known pathways and enzyme subunits, numbers in parentheses indicate the number of different annotations in a given category, e.g., genes encoding different subunits of an enzyme or genes encoding different enzymes of a pathway. No numbers are given for categories where a “completeness” criterion is not applicable, such as transporters of different specificity. The numbers in the table indicate the number of genes per MAG. Several genes encoding the same function in a MAG, e.g., the same subunit of an enzyme, were counted only once. Genes falling into categories can be found in Data Set S2. Download FIG S2, TIF file, 1.3 MB (1.5MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Large spreadsheet listing the functional annotations, which were counted for the metabolisms depicted in Fig. 4 and Fig. S2. Download Data Set S2, XLSX file, 0.03 MB (33.2KB, xlsx) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic placements of proteins annotated as carbon monoxide dehydrogenase, large subunit (A), and methane monooxygenase (B). Trees were calculated with FastTree based on an HMM alignment to the respective Pfam motif (PF02738 for CO dehydrogenase catalytic domain and PF02332 for putative methane monooxygenase). Multifurcations have been introduced for branchings with less than 50% support. Branch support was calculated by 1,000 resamplings of site likelihood and the Shimodaira-Hasegawa test (FastTree default). Numbers in parentheses indicate the number of sequences contained in the cluster (note that one MAG can have multiple CoxL-encoding genes). For the putative CO dehydrogenases (A), a first tree was calculated for all proteins included in the seed alignment of the Pfam model (excluding eukaryotes), together with known form I and form II CO dehydrogenases (9) and MAG-derived sequences. Alignment was filtered to contain sites conserved in 25% of sequences (428 positions). As the Pfam motif includes several enzymes with different functions, a subtree was calculated just for the CO dehydrogenase-containing part, using glyceraldehyde dehydrogenase as an outgroup. Alignment was filtered to contain sites conserved in 25% of sequences (502 positions). Download FIG S3, TIF file, 1.2 MB (1.2MB, tif) .
Copyright © 2021 Meier et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
rRNA gene amplicon reads, unassembled metagenomic reads, metagenome assemblies, and annotated MAGs have been deposited in the European Nucleotide Archive under project number PRJEB36534. Bash and R code of all sequence analysis steps can be found at https://github.com/meierdv/avdat_metagenome.