

Supplemental Digital Content is available in the text.
Keywords: aorta, carotid arteries, femoral artery, genetic therapy, lentivirus, spleen
Abstract
Objective:
Efficient gene transfer to the vascular wall via intravenous vector injection would be useful for experimental vascular biology and gene therapy. Initial studies of lentiviral vector tropism suggested that intravenously injected vectors do not transduce murine vascular tissue; however, there are also reports of highly efficient aortic transduction after jugular vein injection of high-titer lentiviral vectors. We sought to reproduce these results.
Approach and Results:
We injected high-titer preparations of GFP (green fluorescent protein)-expressing lentiviral vector into jugular veins of 8 mice; 6 mice received vehicle only. Four days later, samples of aorta (thoracic and abdominal), liver, spleen, and other tissues were harvested and processed for quantitative polymerase chain reaction detection of vector DNA and immunohistochemical detection of GFP. Our vector DNA assay did not detect transduction of any of the 16 aortic segments. This finding excludes an aortic transduction efficiency of >0.02 vector copies per cell. In contrast, vector DNA was detected in all 8 spleen and liver extracts (median, 0.8 and 0.1 vector copies per cell, respectively; P<0.001 versus vehicle controls). Quantitative polymerase chain reaction signals from DNA extracted from heart, lung, kidney, skeletal muscle, and femoral artery did not differ from background polymerase chain reaction signals from DNA extracted from tissues of vehicle-injected mice (P≥0.7 for all). Immunohistochemistry revealed GFP in scattered cells in spleen and liver, not in aorta.
Conclusions:
Injection of high-titer lentiviral vectors via the jugular vein transduces cells in the spleen and liver but does not efficiently transduce the aorta.
Highlights.
After injecting high-titer lentiviral vectors into the mouse jugular vein, we found no evidence of transduction of the aorta.
Injection of high-titer lentiviral vectors into the mouse jugular vein transduces cells in the spleen and liver.
Previous reports of efficient aortic gene transfer after jugular vein injection of lentiviral vectors were not reproduced.
See accompanying editorial on page 1156
A vector that achieved efficient gene transfer to artery-wall cells after intravenous injection would be extremely useful for experimental vascular biology and for gene therapy. Incorporation of a cell type–specific promoter in the vector would enable investigation of roles of gene products in specific types of artery-wall cells, eliminating time-consuming transgenic founder production and backcrossing. Varying the age of animals at the time of vector injection could initiate transgene expression at specific time points during postnatal life, eliminating the need to construct conditional alleles, generate bitransgenic mice, and activate expression of the alleles by injection of pharmacological activators. Although adeno-associated virus serotypes can efficiently deliver genes to the heart and liver after intravenous injection,1,2 no gene transfer vector has gained wide acceptance for its ability to transduce cells in the artery wall after intravenous injection.
Beginning in 2011, a series of articles reported highly efficient (essentially 100%) transgene expression throughout the mouse aortic wall after jugular vein injection of lentiviral vectors (LVs).3–6 Others reported transgene expression in the aorta—but not in non–smooth muscle–rich organs—after intravenous injection of an LV containing a smooth muscle–selective promoter.7 These reports suggest efficient intravenous gene transfer to aortic smooth muscle cells—and cell type–specific transgene expression within the aorta—is easily achieved with LV. However, these reports conflict with several rigorous investigations of in vivo LV tropism, published in 2000 to 2002.8–13 These investigations found abundant LV genomes and transgene expression in liver, spleen, and bone marrow after intravenous injection but virtually no vector genomes or transgene expression in smooth muscle–rich or highly vascularized organs such as duodenum, lung, bladder, kidney, or heart. These studies did not examine aortic transduction.
To address this discrepancy, we injected high-titer preparations of LV into jugular veins of mice, and we analyzed in vivo transduction using a highly sensitive and specific assay for vector genomes, complemented by immunohistochemical detection of recombinant protein in selected tissues. We found no evidence of intravenous gene transfer to the aorta or other large arteries.
Materials and Methods
The data that support this study are available from the corresponding author on reasonable request. Please see the Major Resources Table in the Data Supplement.
LV Preparation and Characterization
High-titer stock (1×109 transducing units [TU]/mL) of a second-generation LV in which expression of GFP (green fluorescent protein) is driven by the cytomegalovirus immediate-early promoter (LV-GFP) was a kind gift of Dr Juan Miguel Redondo (Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain). High-titer stock (2×109 TU/mL) of a third-generation LV-GFP vector—in which GFP expression is driven by the same cytomegalovirus promoter—was prepared by the viral vector core at the Fred Hutch Cancer Research Center (Seattle, WA). Both LVs are pseudotyped with vesicular stomatitis virus G protein. The features that differentiate second- and third-generation LV14 are not expected to alter either LV tropism or expression from the internal cytomegalovirus promoter. Moreover, the second- and third-generation LV yielded similar results, as described below.
We remeasured the titers of both virus stocks in our own laboratory, using flow cytometry performed on LV-GFP–exposed Jurkat cell clone E6-1 (TIB-152; American Type Culture Collection, Manassas, VA). This method was chosen because it is used by Dr Redondo’s group, and he kindly provided us with his laboratory protocol. Cells were seeded at a density of 50 000 cells/well in a 96-well plate in Iscove Modified Dulbecco Medium (Thermo Fisher Scientific, Waltham, MA) containing penicillin, streptomycin, and 10% fetal bovine serum. Ten microliters of serially diluted LV-GFP stock was immediately added to each well, and the cells were incubated at 37 °C, 5% CO2 for 48 hours. The cells were harvested, centrifuged, and suspended in 300 µL of cold PBS, and 10 µL of propidium iodide (8 ng/µL stock; Invitrogen, Carlsbad, CA) was added to each sample. The percentage of propidium iodide–negative (live) cells from each well that express GFP was measured by flow cytometry (LSR II flow cytometer; BD Bioscience, San Jose, CA), and the concentration of LV-GFP stock was calculated using the following formula: concentration (TU/mL)=(50 000×fold dilution of virus stock×percentage GFP+ cells). Gates for determining GFP positivity were set—conservatively based on flow cytometry results obtained at higher dilutions—by the director of the University of Washington flow cytometry core. The director was unaware of the nature of the experiment or the identity of the preparations. The LV-GFP stock from Dr Redondo was thawed only once and was used for titration and animal experiments on the same day.
Animal Procedures
All animal studies were approved by the University of Washington Office of Animal Welfare. We injected LV-GFP or vehicle into 14 mice. Specific pathogen-free, C57BL/6J male mice aged 3 to 7 weeks were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were injected preoperatively with subcutaneous buprenorphine (0.05 mg/kg body weight) and anesthetized with inhaled 1% to 5% isoflurane. A 0.6-cm incision was made parallel to the midline at the lower third of the right anterior neck, exposing the right external jugular vein. An insulin syringe with a 28-gauge needle was used to inject 100 µL of either vehicle (Iscove Modified Dulbecco Medium) or undiluted LV-GFP stock into the vein, over 2 seconds. Hemostasis was achieved with gentle pressure, and the skin wound was closed with 4-0 nonabsorbable silk suture. We injected six 8-week-old mice with vehicle. We injected two 10-week-old mice with LV-GFP provided by Dr Redondo. We injected four 10-week-old mice and two 17-week-old mice with LV-GFP prepared by the Fred Hutch. Aortas were removed from 6 unoperated mice aged 8 to 10 weeks, to generate positive and negative control tissue (see below).
Four days after LV-GFP or vehicle injection, mice were anesthetized with intraperitoneal ketamine/xylazine (100 and 8.8 mg/kg, respectively), after which they were euthanized by thoracotomy and exsanguination by intracardiac saline perfusion. The heart, lungs, carotid arteries, aorta, liver, spleen, kidneys, femoral arteries, quadriceps muscles, and gastrocnemius muscles were removed in that order. The aorta was divided into 5 segments (Figure I in the Data Supplement), with 3 segments processed for histology and 2 segments processed for DNA extraction. The apical half of the cardiac ventricles was used for histology, and the remainder of the heart was used for DNA extraction. Other tissues were divided for analysis as follows. The cranial half of the right lung, caudal half of the left lung, caudal halves of the common carotid and femoral arteries, right half of the liver, caudal half of the spleen, cranial half of the right kidney, caudal half of the left kidney, cranial halves of the right quadriceps and right gastrocnemius, and the caudal halves of the left quadriceps and left gastrocnemius were processed for histological analyses. The other halves of all of these organs and tissues were processed for DNA extraction. Tissues destined for DNA extraction were rinsed in saline, snap-frozen in liquid nitrogen, and stored at −80 °C. From 1 to 3 months later, frozen tissues were processed for DNA extraction as described below. Organs and tissues destined for histological analyses were placed in 10% formalin (in PBS) for 48 hours and then embedded in optimal cutting temperature medium.
Preparation of Positive and Negative Control Aortic Tissue
Six unoperated mice were anesthetized and saline perfused as described above. Segments of aorta were removed and placed in wells of a 96-well plate containing DMEM with penicillin, streptomycin, and 10% fetal bovine serum. Two mice were used to generate positive control tissue (1 for histology and 1 for DNA analysis), and 4 mice were used to generate negative control tissue (1 for histology and 3 for DNA analysis). More mice were used to generate negative versus positive control tissue because vector-negative aortic DNA was needed for construction of all of the standard curves. To generate positive control tissue, we added LV-GFP (Fred Hutch preparation) to the wells, at a final concentration of 5×107 TU/mL (for LV DNA detection) and at both 1×107 and 5×107 TU/mL (for GFP immunohistochemistry). To generate negative control tissue, the virus was omitted. Two days later, the virus-containing medium was replaced with non–virus-containing medium. The segments were harvested 2 days later and were either snap-frozen in liquid nitrogen for DNA extraction or processed for histology as described for in vivo samples.
Real-Time Quantitative Polymerase Chain Reaction
Frozen tissue samples—except aortas, carotid arteries, and femoral arteries—were placed in liquid nitrogen and pulverized with a liquid nitrogen cooled mortar and pestle. The vascular segments were not pulverized because their limited volume and thickness impeded tissue recovery after pulverization and enabled DNA extraction without pulverization. DNA was extracted from all tissues using the DNeasy kit (Qiagen Sciences, Germantown, MD), and extracted DNA was quantified using a spectrophotometer (Nanodrop; Thermo Fisher Scientific). Yields of DNA per tissue sample ranged from 56 ng for one of the left carotid halves to ≈50 µg for one of the liver samples. Extracted DNA was frozen (8 months; −80 °C) until the quantitative polymerase chain reaction (PCR) assay was thoroughly tested.
We used real-time quantitative PCR to measure LV-GFP genomes in the tissue extracts. PCR primers targeting the GFP sequence (forward, 5′-GAACCGCATCGAGCTGAA-3′; reverse, 5′-TGCTTGTCGGCCATGATATAG-3′) were designed using IDT PrimerQuest tool and were each used at a final concentration of 250 nmol/L. This set of primers performed best among 5 sets suggested by PrimerQuest because standard curves were always linear, the lowest point on the curve was always detected above the no-template control value, and the quantification cycle (Cq) of the no-template control reaction was always undetermined (ie, >40). PCR reactions with these primers that were electrophoresed into 1% agarose gels revealed an amplicon of the expected size (≈111 base pairs). Moreover, when these primers are aligned with the mouse genome using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.gov/tools/primer-blast/), no target sequences are identified. Reactions were performed in a final volume of 20 µL using Powerup SYBR green PCR master mix (containing AmpliTaq Gold DNA polymerase, low DNA; Thermo Fisher Scientific). Amplification conditions were 2 minutes at 50 °C, 2 minutes at 95 °C, 40 cycles of 15 seconds at 95 °C, and 60 seconds at 60 °C using the CFX384 Touch Real-Time PCR Detection System with Bio-Rad CFX Manager 3.1 software (Bio-Rad Laboratories, Hercules, CA). Cq values are automatically determined by this software. All quantitative PCR reactions (other than the no-template control reactions) were performed with 10 ng of DNA.
We used the same quantitative PCR protocol to generate standard curves of GFP copies per cellular genome. The curves were constructed using DNA that was extracted from ex vivo mock-transduced mouse aorta and then spiked with a GFP-containing plasmid (LV-011-a; Applied Biological Materials, Inc, Richmond, BC, Canada) at 0.02 to 600 plasmid copies per cellular genome. The number of LV-GFP genomes per cell in experimental tissue extracts was calculated with reference to these standard curves, which were performed along with every assay of the experimental samples. The concentration of the stock GFP-containing plasmid sample that was diluted to construct the standard curves was measured using the Nanodrop spectrophotometer and was adjusted based on visualization of the plasmid after electrophoresis into an ethidium-stained agarose gel, using a known amount of HindIII-digested lambda phage DNA (Thermo Fisher Scientific) as a mass standard. DNA extracted from ex vivo mock-transduced aorta (without added plasmid) was a negative tissue control. DNA extracted from aortic segments transduced ex vivo with LV-GFP (see above) was a positive control for detection of LV-GFP genomes in tissue extracts and was most useful as a positive control for detection of LV-GFP genomes in aortic extracts. The concentration of LV-GFP DNA was measured twice in all tissue extracts, in separate assays that both included their own standard curves. Means of the 2 measurements were calculated and used to compare groups.
Immunohistochemistry
Tissue blocks were sectioned (6-μm thick) at 3 steps, each separated by 200 μm. Five serial sections were cut at each of the 3 steps and placed on 5 separate slides that were stored at −20 °C. For staining, slides were brought to room temperature (30–60 minutes), fixed in acetone (−20 °C; 10 minutes), and washed in PBS. Endogenous peroxidase activity was blocked with 0.3% H2O2 for 20 minutes. After washing, sections were blocked with 5% goat serum in PBS for 60 minutes. Endogenous biotin was blocked in liver sections by incubation with 0.001% avidin (A9275; Sigma, St. Louis, MO) in PBS for 15 minutes, followed by incubation with 0.001% biotin (B4501; Sigma) in PBS for 15 minutes. After washing, rabbit anti-GFP antibody was applied overnight at 4 °C. Slides were washed again, and biotinylated anti-rabbit secondary antibody in PBS with 2% goat serum was applied for 45 minutes at room temperature. Slides were again washed and then incubated with Vectastain ABC reagent (PK-4001; Vector Laboratories, Burlingame, CA) for 45 minutes at room temperature. Staining was visualized with ImmPACT NovaRED (Vector Laboratories) applied for 10 minutes. Slides were briefly rinsed with water and then washed in water for 5 minutes and air-dried, followed by two 3-minute xylene washes and cover-slipping with Permount (No. SP15; Thermo Fisher Scientific). Both omission of the primary antibody and replacement of the primary antibody with rabbit IgG were used as negative controls. Positive controls included sections of mouse aorta transduced ex vivo with LV-GFP (as described above) and sections of a heart of a tamoxifen-treated αMHC-Mer-Cre-Mer×red fluorescent proteinflox/flox/GFP bitransgenic mouse,15 kindly provided by Dr Yiqiang Zhang (University of Washington). On all slides, the entirety of every section was examined for stained cells. Representative images were taken using a microscope (DM4000 B; Leica Microsystems, Inc, Buffalo Grove, IL) and camera (DFC295; Leica Microsystems, Inc).
Statistics
We used Excel (Microsoft, Redmond, WA) to calculate group medians, Prism 6 (GraphPad, San Diego, CA) to perform Bland-Altman tests on duplicate quantitative PCR measurements of LV-GFP genomes in extracts of spleen and liver, and SigmaPlot 13.0 (Systat Software, San José, CA) to perform Mann-Whitney rank-sum tests that compared the number of LV-GFP genomes in tissues of LV-GFP–injected and vehicle-injected mice.
Results
Vector Titration
We titrated both LV-GFP preparations on Jurkat cells, using GFP fluorescence and flow cytometry to calculate TU/mL. The titer of the LV-GFP stock provided by Dr Redondo was 0.3×109 TU/mL (calculated from results obtained at 1:50 and 1:100 dilutions; Figure IIA in the Data Supplement). The titer of the LV-GFP from Fred Hutch was 0.9×109 TU/mL (calculated from results obtained at 1:100 and 1:1000 dilutions; Figure IIB in the Data Supplement).
Detection of LV-GFP Genomes in Mouse Tissues After Jugular Vein Injection
We injected mice with one of the undiluted LV-GFP stocks or with an equal volume of vehicle. Tissues were harvested 4 days later and frozen. DNA was later extracted from the frozen tissue. Extracted DNA was of high quality and purity based on spectrophotometry that consistently showed ratios of absorbance at 260/280 nm and 260/230 nm of ≈1.9. We measured LV-GFP genomes in the extracts using quantitative PCR with reference to standard curves. These curves were constructed from samples of DNA extracted from a mock-transduced aorta that were spiked (after extraction) with 0.02 to 600 GFP copies per cellular genome (Figure 1A). Analysis of the spiked aortic DNA samples reliably detected the GFP sequence throughout the 0.02 to 600 copies/cell range. Cq values for the 0.02 copies per cellular genome samples ranged from 33.4 to 36.4. Cq values for the negative control samples (extracts of mock-transduced aorta) were undetermined (>40) except for one assay in which Cq was 34.2. However, this Cq value was greater than the Cq=33.4 value for the 0.02 copies/cell standard run in parallel. Cq values for the no-template control samples were undetermined (>40) in all assays except one (Cq=39.2). The standard-curve slopes were −1.3 to −1.7, y intercepts were 27.3 to 32.1, and r2 values were 0.96 to 1.0, yielding calculated PCR efficiencies of 80% to 117%.
Figure 1.
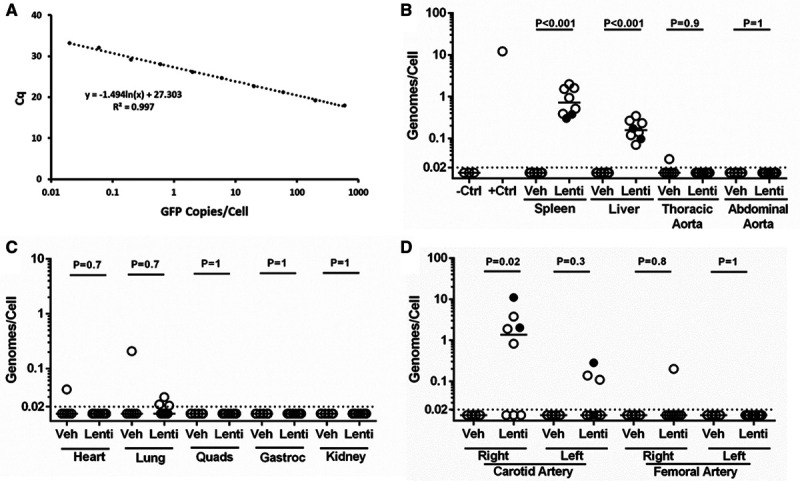
No aortic transduction after in vivo lentiviral vector (LV) injection. A, Standard curve made by mixing GFP (green fluorescent protein)-containing plasmid with genomic DNA extracted from ex vivo mock-transduced aorta and measuring quantification cycle (Cq) values of the GFP amplicon. B–D, LV genomes per diploid cell were measured by quantitative polymerase chain reaction (PCR) for the GFP sequence, performed on DNA extracted from indicated tissues, with reference to a standard curve (A). Filled circles indicate, among samples from mice injected with LV provided by the Redondo Laboratory (n=2), those with positive PCR reactions. B, The negative control (−Ctrl) samples are DNA extracted from mock-transduced aortic segments (n=3). The positive control (+Ctrl) sample is DNA extracted from an aortic segment transduced ex vivo (n=1). C, Data are from right-sided lung, muscles, and kidney; for left-sided results, see Figure IV in the Data Supplement. B–D, Dotted line: limit of detection; data points are from individual mice; bars are medians; P are from Mann-Whitney rank-sum test. Veh, n=6 for all tissues; Lenti, n=8 for all tissues. Gastroc indicates gastrocnemius; Lenti, lentivirus-GFP injected; Quads, quadriceps; and Veh, vehicle injected.
We also measured LV-GFP genomes per cell in an ex vivo LV-GFP–transduced aorta (a positive control for aortic transduction, vector genome extraction, and vector genome detection) and in tissues from experimental and control mice. The positive control aorta contained ≈12 LV-GFP genomes per cell (Figure 1B). In LV-GFP–injected mice, LV-GFP DNA was detected in 100% of spleen (8 of 8) and 100% of liver (8 of 8) extracts with a median 0.8 and 0.1 vector genomes per cell, respectively (P<0.001 for both organs compared with organs in vehicle-injected controls). In contrast, in these same mice, LV-GFP DNA was detected in 0 of 8 thoracic aorta extracts and in 0 of 8 abdominal aorta extracts (Figure 1B). To examine the reproducibility of LV-GFP genome quantification in spleen and liver (the only organs in which vector genomes were uniformly present), we compared the results of duplicate assays of each extract using the Bland-Altman test. All points were within the limits of agreement (Figure III in the Data Supplement).
Extracts of heart, lung, kidney, and muscles of the same LV-GFP–injected mice all had median LV-GFP genomes/cell <0.02, with signals barely above background in 3 of 16 lung samples but in none of the other samples. Low-level signals were also present in 1 of 6 hearts and 2 of 12 lung samples of vehicle-injected mice. For all of these tissues, there was no significant difference in LV-GFP genomes between LV-GFP–injected and vehicle-injected mice (P≥0.7 for all; Figure 1C; Figure IV in the Data Supplement).
We attempted to detect LV-GFP genomes in extracts of other large arteries. Median LV-GFP genomes/cell was <0.02 in both left and right femoral arteries of LV-GFP–injected mice, with a low-level signal in 1 of 8 right femoral arteries and 0 of 8 left femoral arteries. For both arteries, there was no significant difference in LV-GFP genomes between LV-GFP–injected and vehicle-injected mice (P≥0.8 for both; Figure 1D). Relatively high levels of LV-GFP genomes were detected in 5 of 8 right carotid arteries of LV-GFP–injected mice (median, 1 LV-GFP genome/cell for the group of 8; P=0.02 compared with right carotids of vehicle-injected mice), with lower levels of LV-GFP genomes in 3 of 8 left carotid arteries (median, <0.02 vector copies/cell; P=0.3 compared with left carotids of vehicle-injected mice; Figure 1D).
Detection of Transgenic GFP by Immunohistochemistry
We used an orthogonal approach—immunohistochemistry—to confirm the PCR results in spleen, liver, aorta, and carotid arteries. Staining for GFP was present in scattered cells in every section of spleen and liver from LV-GFP–infused mice (Figure 2A). There was no staining in sections of spleen and liver from vehicle-infused mice and no staining when the anti-GFP antibody was omitted or replaced with nonimmune IgG (Figure 2A; data not shown). No GFP staining was present in sections of aortas from either LV-GFP–infused or vehicle-infused mice. GFP staining was present in the adventitia of some of the carotid artery sections of LV-GFP–infused mice but not in carotid artery sections of vehicle-injected mice. No GFP staining was present in the intimal or medial layers of any of the carotid arteries. Carotid sections with the strongest adventitial staining were from mice that had high levels of carotid lentiviral transduction, as revealed by the quantitative PCR assay.
Figure 2.
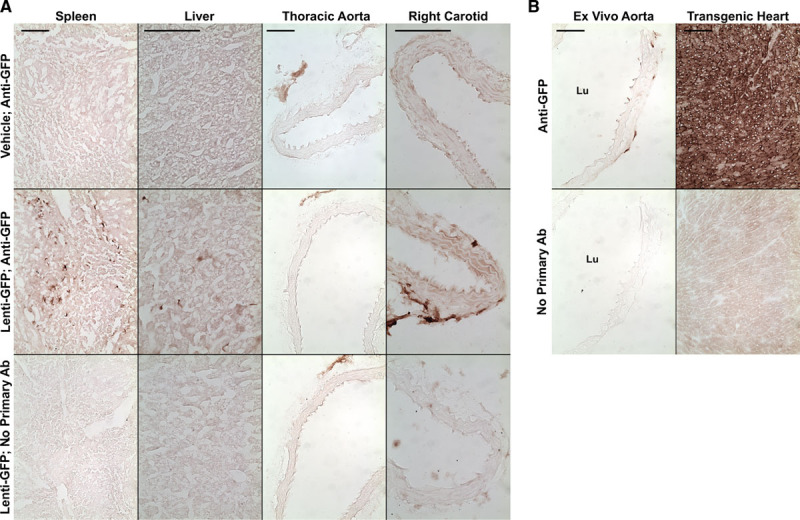
Immunohistochemical detection of GFP (green fluorescent protein). Mice were injected with either vehicle or with a lentiviral vector expressing GFP. A, Sections of the indicated tissues were immunostained with an antibody (Ab) to GFP (Anti-GFP). Sections from mice injected with GFP-expressing lentivirus (Lenti-GFP) were also stained without the primary Ab. B, Positive control sections from an ex vivo–transduced aorta and from the heart of a GFP-transgenic mouse, with controls. Size bars=100 μm and apply to all panels in each column. Lu indicates lumen.
Staining of positive control sections from ex vivo transduced aorta (transduced with 1×107 TU/mL LV-GFP) and from a transgenic mouse with cardiac-specific GFP expression—with and without the anti-GFP antibody—confirmed that our immunostaining protocol could detect GFP expression both in the aorta and in nonvascular tissue and that this staining was specific for GFP (Figure 2B).
Discussion
We tested whether intravenous injection of high-titer LV achieves efficient gene transfer to the aorta. We detected significant and reproducible transduction of the spleen and liver but not the aorta.
Our finding of highly reproducible transduction and transgene expression in spleen and liver—and not in heart, lung, kidney, or skeletal muscle—is congruent with results reported by several groups at the outset of LV development, ≈20 years ago.8–13 Some of these studies also found efficient transduction of bone marrow—a tissue that we did not examine. Several of these studies examined other tissues after intravenous LV injection including lung, heart, brain, duodenum, bladder, and pancreas; none reported more than low-to-undetectable levels of transduction in these tissues. Some of these tissues are highly vascularized (eg, lung and heart)16 or have large numbers of smooth muscle cells (eg, duodenum and bladder). Some of these studies used LV-expressed marker genes and either immunohistochemistry or immunofluorescence to detect transduction. These approaches can identify single transduced cells among numerous surrounding nontransduced cells and would be extremely unlikely to be falsely negative when applied by multiple investigators seeking to uncover the potential of a new gene transfer vector. Some of these studies measured LV DNA with highly quantitative approaches and report limits of detection as low as 1 LV genome in 105 cells.8,17 Taken together, although none of these studies specifically examined the aorta, none reported any evidence of efficient vascular cell transduction.
Reports of highly efficient aortic transduction after intravenous LV injection are, therefore, surprising.3–6 Intravenously injected LV was reported to “efficiently transduce the three aortic layers,”6 and associated images of aortic tissue show evidence of transgene expression in essentially 100% of aortic wall cells.3,5,6 The authors attribute high-efficiency aortic transduction to their use of high viral titers (109 TU/mL) and to jugular versus tail vein injection.6 However, other groups have injected LV at this concentration and found transduction essentially limited to spleen and liver,10,11 and this group also reported success with a 10-fold lower LV concentration.4 It is unclear why results from this group differ so profoundly. It is also unclear why jugular versus tail vein injection would dramatically alter the efficiency of aortic transduction, and no mechanistic explanation is proposed.6
Because achievement of efficient percutaneous vascular gene transfer would have a major impact on vascular research and gene therapy, we attempted to reproduce the published results.3–6 We obtained LV-GFP and protocols from the authors and followed their protocols assiduously. However, in contrast to their work—and for several reasons—we relied primarily on measuring LV DNA rather than RNA or protein to detect transduction. First, LV DNA is measured with high sensitivity and specificity.8,17 Second, the single-stranded RNA genome in LV virions must be reverse transcribed into double-stranded DNA before it can serve as a template for transcription of virus-encoded genes. Third, conversion to double-stranded DNA is a prerequisite for function of any cell type–specific promoters placed in an LV.7 Fourth, RNA and protein can be generated remotely from sites of detection, whereas there is no known mechanism for efficient transfer of DNA—especially chromosomally integrated LV DNA—between tissues. Importantly, the method we used to extract tissue DNA efficiently recovers both integrated LV DNA and episomal, reverse-transcribed but nonintegrated LV genomes.18 If there were abundant episomal LV-GFP DNA in the aorta, our assay would have detected it.
Taken together, our DNA measurements and our immunohistochemistry studies provide strong evidence that jugular vein injection of LV does not achieve efficient gene transfer to the aorta or to other large arteries. Transduction of the right carotid adventitia (adjacent to the injection site) is almost certainly due to extravascular virus introduced during the vector injection procedure, as are the lower PCR signals in some of the left carotids. In evaluating reports of efficient aortic transduction,3–7 it should be considered that transgenic RNA and protein detected in the aorta after LV injection might have a splenic, hepatic, or bone marrow source, arriving via blood flow and diffusion and potentially transported by exosomes. Falsely positive assays must also be considered.
Our study has some limitations. We used a small number of mice. Our LV titration did not exactly match that of Dr Redondo’s laboratory. Our results do not exclude a low level of in vivo aortic transduction (<1 in 50 cells). Finally, it is impossible to exclude an impact of potentially uncontrolled variables when a procedure is performed by different individuals in different locations. These variables might include tissue processing and immunostaining protocols, genetic drift in colonies of C57BL/6 mice used in other studies,4,5 or differences in microbiomes. Nevertheless, we suggest that a rigorous approach to experiments that rely on efficient in vivo LV-mediated gene transfer to the aorta must include quantitative confirmation that the abundance of LV DNA correlates with any phenotypic effects.
Acknowledgments
We thank the Vector Core of the Cooperative Center for Excellence in Hematology at the Fred Hutch Cancer Research Center for help with vector preparation and Dr Yiqiang Zhang for providing transgenic mouse tissue.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01HL114541), the National Institute of Diabetes and Digestive and Kidney Diseases (U54DK106829), and by the John L. Locke Jr Charitable Trust.
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- Cq
- quantification cycle
- GFP
- green fluorescent protein
- LV
- lentiviral vector
- TU
- transducing unit
This article is part of the Null Hypothesis Collection, a collaborative effort between CBMRT, AHA Journals, and Wolters Kluwer, and has been made freely available through funds provided by the CBMRT. For more information, visit https://www.ahajournals.org/null-hypothesis.
These authors contributed equally to this article.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315125.
For Sources of Funding and Disclosures, see page 1155.
Contributor Information
Lianxiang Bi, Email: LBi@cardiology.washington.edu.
Bradley K. Wacker, Email: bwacker@cardiology.washington.edu.
Alexis Stamatikos, Email: adstama@clemson.edu.
Meena Sethuraman, Email: meenasethu@gmail.com.
Kaushik Komandur, Email: kaush@UW.edu.
References
- 1.Gray SJ, Samulski RJ. Optimizing gene delivery vectors for the treatment of heart disease. Expert Opin Biol Ther. 2008;8:911–922. doi: 10.1517/14712598.8.7.911 [DOI] [PubMed] [Google Scholar]
- 2.Kattenhorn LM, Tipper CH, Stoica L, Geraghty DS, Wright TL, Clark KR, Wadsworth SC. Adeno-associated virus gene therapy for liver disease. Hum Gene Ther. 2016;27:947–961. doi: 10.1089/hum.2016.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteban V, Méndez-Barbero N, Jiménez-Borreguero LJ, Roqué M, Novensá L, García-Redondo AB, Salaices M, Vila L, Arbonés ML, Campanero MR, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med. 2011;208:2125–2139. doi: 10.1084/jem.20110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martín-Alonso M, García-Redondo AB, Guo D, Camafeita E, Martínez F, Alfranca A, Méndez-Barbero N, Pollán Á, Sánchez-Camacho C, Denhardt DT, et al. Deficiency of MMP17/MT4-MMP proteolytic activity predisposes to aortic aneurysm in mice. Circ Res. 2015;117:e13–e26. doi: 10.1161/CIRCRESAHA.117.305108 [DOI] [PubMed] [Google Scholar]
- 5.Oller J, Méndez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, Briones AM, Alberca R, Lozano-Vidal N, Hurlé MA, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23:200–212. doi: 10.1038/nm.4266 [DOI] [PubMed] [Google Scholar]
- 6.Alfranca A, Campanero MR, Redondo JM. New methods for disease modeling using lentiviral vectors. Trends Mol Med. 2018;24:825–837. doi: 10.1016/j.molmed.2018.08.001 [DOI] [PubMed] [Google Scholar]
- 7.DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, et al. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight. 2016;1:e88942 doi: 10.1172/jci.insight.88942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park F, Ohashi K, Chiu W, Naldini L, Kay MA. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat Genet. 2000;24:49–52. doi: 10.1038/71673 [DOI] [PubMed] [Google Scholar]
- 9.Pan D, Gunther R, Duan W, Wendell S, Kaemmerer W, Kafri T, Verma IM, Whitley CB. Biodistribution and toxicity studies of VSVG-pseudotyped lentiviral vector after intravenous administration in mice with the observation of in vivo transduction of bone marrow. Mol Ther. 2002;6:19–29. doi: 10.1006/mthe.2002.0630 [DOI] [PubMed] [Google Scholar]
- 10.VandenDriessche T, Thorrez L, Naldini L, Follenzi A, Moons L, Berneman Z, Collen D, Chuah MK. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood. 2002;100:813–822. doi: 10.1182/blood.v100.3.813 [DOI] [PubMed] [Google Scholar]
- 11.Follenzi A, Sabatino G, Lombardo A, Boccaccio C, Naldini L. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243–260. doi: 10.1089/10430340252769770 [DOI] [PubMed] [Google Scholar]
- 12.Ohashi K, Park F, Kay MA. Role of hepatocyte direct hyperplasia in lentivirus-mediated liver transduction in vivo. Hum Gene Ther. 2002;13:653–663. doi: 10.1089/10430340252837242 [DOI] [PubMed] [Google Scholar]
- 13.Tsui LV, Kelly M, Zayek N, Rojas V, Ho K, Ge Y, Moskalenko M, Mondesire J, Davis J, Roey MV, et al. Production of human clotting Factor IX without toxicity in mice after vascular delivery of a lentiviral vector. Nat Biotechnol. 2002;20:53–57. doi: 10.1038/nbt0102-53 [DOI] [PubMed] [Google Scholar]
- 14.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/JVI.72.11.8463-8471.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Gago-Lopez N, Li N, Zhang Z, Alver N, Liu Y, Martinson AM, Mehri A, MacLellan WR. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019;5:30 doi: 10.1038/s41421-019-0095-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cleuren ACA, van der Ent MA, Jiang H, Hunker KL, Yee A, Siemieniak DR, Molema G, Aird WC, Ganesh SK, Ginsburg D. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc Natl Acad Sci U S A. 2019;116:23618–23624. doi: 10.1073/pnas.1912409116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi H, Carbonaro D, Pepper K, Petersen D, Ge S, Jackson H, Shimada H, Moats R, Kohn DB. Neonatal gene therapy of MPS I mice by intravenous injection of a lentiviral vector. Mol Ther. 2005;11:776–789. doi: 10.1016/j.ymthe.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 18.Yáñez-Muñoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ, Buch P, MacLaren RE, Anderson PN, Barker SE, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12:348–353. doi: 10.1038/nm1365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.