

Abstract
Single-stranded DNA-binding proteins (SSBs) play crucial roles in DNA replication, recombination and repair, and serve as key players in the maintenance of genomic stability. While a number of SSBs bind single-stranded DNA (ssDNA) non-specifically, the others recognize and bind specific ssDNA sequences. The mechanisms underlying this binding discrepancy, however, are largely unknown. Here, we present a comparative study of protein–ssDNA interactions by annotating specific and non-specific SSBs and comparing structural features such as DNA-binding propensities and secondary structure types of residues in SSB–ssDNA interactions, protein–ssDNA hydrogen bonding and π–π interactions between specific and non-specific SSBs. Our results suggest that protein side chain-DNA base hydrogen bonds are the major contributors to protein–ssDNA binding specificity, while π–π interactions may mainly contribute to binding affinity. We also found the enrichment of aspartate in the specific SSBs, a key feature in specific protein–double-stranded DNA (dsDNA) interactions as reported in our previous study. In addition, no significant differences between specific and non-specific groups with respect of conformational changes upon ssDNA binding were found, suggesting that the flexibility of SSBs plays a lesser role than that of dsDNA-binding proteins in conferring binding specificity.
INTRODUCTION
In many essential cellular processes such as DNA replication, recombination and repair, the double-stranded DNA (dsDNA) is unwound and exists transiently in a single-stranded form (1,2). Single-stranded DNA (ssDNA) is vulnerable to chemical and enzymatic attacks and is prone to forming secondary structures that can interfere with biological activity such as DNA replication. As a consequence, a specific group of proteins, single-stranded DNA-binding proteins (SSBs), has evolved to bind and stabilize ssDNA. SSBs are also essential in the maintenance of genomic stability, playing critical roles in telomere end protection, DNA damage repair, control of cell cycle checkpoints and the recruitment of partner proteins to regulate DNA metabolism (3–6). It has been demonstrated that aberrant ssDNA binding leads to genome instability and tumorigenesis (7,8). Therefore, knowledge of SSB–ssDNA interactions can help better understand the mechanisms underlying normal cellular processes and human malignancies. More importantly, it can help provide guidance for targeted drug design in therapeutics.
Despite the critical roles of ssDNA-binding proteins in essential biological processes, investigation of SSB–ssDNA interactions clearly lags far behind of other types of protein–nucleic acid interactions, such as protein–dsDNA and protein–RNA interactions. Our current understanding of SSB–ssDNA interactions mainly comes from several extensively studied individual SSBs, such as bacteriophage T4 gene 32 protein (gp32) (9–12), Escherichia coli SSB (13–19), replication factor A (RPA) (2,20–25) and human SSB1 and SSB2 (1,4). The first identified SSB is gp32, which exists as monomers in solution without DNA substrates (9). The central region of the gp32 monomer is the ssDNA-binding domain containing an oligonucleotide/oligosaccharide-binding fold (OB fold), while the terminal domains participate in the cooperative binding of gp32 monomers and protein–protein interactions (12,26–28). Escherichia coli SSB functions as a homotetramer with one OB fold in each subunit and the ssDNA-binding domain is in the N-terminal (29). gp32 and E. coli SSB are the two most widely studied SSBs and serve as the prototypes for many SSB studies in bacteria and higher organisms (5,10,30,31). Generally thought as a eukaryotic homolog of E. coli SSB, RPA is a heterotrimeric SSB. RPA is composed of three subunits with different molecular weights of 70, 32 and 14 kDa, named RPA70, RPA32 and RPA14 with four, one and one OB fold, respectively (2,32,33). One OB fold from each subunit interacts with each other to form a stable trimerization core (20,34). Besides studies of individual SSBs, researchers also investigated small groups of SSBs (4,35–37). Shi et al. compared the biological functions of a novel SSB derived from Thermococcus kodakarensis KOD1 with three known SSB proteins from Thermus thermophilus, E. coli and Sulfolobus Solfataricus P2 (37). They found all four SSBs bound to ssDNA and viral RNA, and affected viral RNA metabolism, but these SSBs showed different levels of resistance to heat treatment (37). Ashton et al. reviewed SSBs in the human genome, including RPA, hSSB1 and hSSB2, and their roles in cellular processes for maintaining genomic stability (35).
Of particular interest in SSB–ssDNA interactions is the specific SSB–ssDNA recognition, or the binding specificity between SSBs and ssDNA. Many SSBs bind ssDNA with high affinity but independent of sequences (36,38). They exist in different oligomeric states in solution, and present in stoichiometric quantities with respect to ssDNA substrates in different binding modes (30,39–41). These binding modes depend mainly on salt concentration and the length of ssDNA, and the stability of these modes is influenced by factors such as pH, protein-binding density and temperature (40,42,43). Some SSBs, on the other hand, bind ssDNA with high sequence specificity. Telomere-end protection (TEP) proteins, including Pot1, Cdc13 and TEBP (telomere end-binding protein), specifically bind short, repeated GT-rich ssDNA sequences, coordinate end protection and recruit telomerase at the telomere (44,45). The binding specificity of TEP proteins varies across different organisms. For instance, sequence specificity of the human POT1 is conferred by both OB folds in the dual-OB fold DNA-binding domain, while Schizosaccharomyces pombe Pot1 only relies on the first OB fold to achieve binding specificity (46,47). The mechanisms underlying this binding discrepancy, however, have not been clearly elucidated.
The binding specificity of SSBs is considered to be contributed by electrostatic, hydrogen-bonding and stacking interactions between SSBs and ssDNA, as well as the flexibility of SSB and/or ssDNA (36,38). However, the roles of each of these factors in ssDNA binding specificity seem to be different in various studies of individual SSBs. Shamoo suggested that hydrogen-bonding interactions and small pockets at the protein surface of TEBP contribute to sequence specificity, which is supplemented by the generalized stacking and electrostatic interactions (38). However, Dickey et al. found that despite the apparent base-specific hydrogen bonds, Pot1pC, one of the two OB folds in Schizosaccharomyces pombe Pot1, was able to bind various ssDNA sequences with little to no specificity (48). By comparing structures of Pot1pC in complex with different non-cognate ssDNA ligands, they suggested that the binding promiscuity of Pot1pC is achieved by new binding modes featured by different stacking interactions and new hydrogen-bonding networks (48). In addition to base-mediated hydrogen bonding, the binding specificity also relies on the flexibility of protein and/or ssDNA (36). The importance of protein and ssDNA flexibility is supported by the TEBP:(T4G4)2 complex structure, in which the protein and ssDNA bind in a cofolding mode to induce the formation of DNA-binding pockets (38,45). An analysis of crystal structures of 10 different non-cognate ssDNA ligands complexed with the Oxytricha nova telomere end-binding protein (OnTEBP) revealed that the overall protein conformation in all complexes remained nearly identical to that of the cognate complex, but the ssDNA exhibited subtle to dramatic conformational changes (49). Pal and Levy also found that the ssDNA molecules were more flexible than the proteins, but they suggested that the sequence specificity was mostly introduced by the stacking interactions between aromatic residues and DNA bases (50).
While these studies provide some aspects of protein–ssDNA interactions, to our knowledge, there are no reports of larger scale structural studies of SSB–ssDNA interactions, especially comparative studies for understanding structural features in protein–ssDNA binding specificity as in protein–dsDNA interactions. DsDNA-binding proteins (DSBs) recognize their specific target sites with a combination of two readout mechanisms: base readout and shape readout (51–53). Comparative studies of DSB–dsDNA complexes demonstrated that hydrogen bonds between amino acid side chains and DNA bases, π-interactions between aromatic residues and DNA bases, and protein flexibility all play important roles in specific DSB-dsDNA binding (54–57). Compared with dsDNA, ssDNA is more flexible since there is no steric hindrance from a complementary DNA strand. It is interesting to see if there are differences between SSB–ssDNA and DSB–dsDNA interactions in terms of binding specificity.
The current number of available protein–ssDNA complexes in Protein Data Bank (PDB) is similar to the number that was used in the initial study of protein–dsDNA interactions about 20 years ago (58–61). To provide a general picture of SSB–ssDNA interactions, especially the mechanisms of SSB–ssDNA binding specificity, here we carried out a comparative analysis of SSB–ssDNA interactions between specific and non-specific SSB–ssDNA complexes. To do this, we first collected all protein–ssDNA complex structures from PDB (58,59) and the Nucleic Acid Database (NDB) (62,63) and assigned them into specific (SP) and non-specific (NS) groups. We then compared the key structural features in protein–ssDNA interaction. These features include the propensities and secondary structure types of ssDNA base-interacting residues, side chain-base hydrogen bonds and π–π interactions between SSB and ssDNA, interaction interface, and protein conformational changes upon ssDNA binding.
MATERIALS AND METHODS
Datasets
SSB–ssDNA complex structures, defined as any structures containing one or more protein chains and at least one single-stranded DNA, were collected from the November 2019 release of NDB (62,63) and PDB (58,59). Complex structures containing false ssDNA and the ones that lack primary evidence were filtered out first. The major source of false positive SSB–ssDNA complexes comes from complexes that contain only one strand of the double helix in the asymmetric unit, such as 4KMF, 3ER8 and 3HZI. These cases have been successfully annotated by NDB, where the coordinates for the complete structure have been reconstructed by applying the transformation matrices provided in the PDB files (62,63). A dataset of 214 protein–ssDNA complexes was generated (Supplementary Table S1).
For comparative analyses, only high-quality X-ray structures of SSB–ssDNA complexes with resolutions better than 3.0 Å and R-values <0.3, and NMR structures were selected. All ssDNA-contacting protein chains that have at least one heavy atom within 3.9 Å of any heavy atoms of a nucleotide of the ssDNA were first identified in these complexes as described in protein–dsDNA interaction studies (54,56,64,65). The atomic distances were calculated with pdb-tools (66). An ssDNA-contacting protein chain was filtered out if: (i) the ssDNA has nucleotides other than AGCT; (ii) the length of ssDNA is shorter than three nucleotides; (iii) ssDNA is engineered, such as aptamers; and (iv) there are mutated residues in the DNA-contacting chain.
Of these high-quality ssDNA-contacting protein chains, some only have ssDNA-binding domains, while others also contain signal-sensing domains or dimerization domains. To avoid any potential comparison biases, we chose ssDNA-binding domains and their target ssDNA as comparison units. CATH, one of the widely used structural classification databases, was used for protein structural domain annotation (67,68). An ssDNA-binding domain was selected for analysis if there are more than one protein–ssDNA contacts within 3.9 Å and the domain has at least 40 amino acids. A total of 458 ssDNA-binding domains were identified and used to generate two datasets, Dataset I and Dataset II, for comparative analyses (Figure 1).
Figure 1.

Flowchart for generation of the non-redundant specific (SP) and non-specific (NS) datasets. Dataset I: non-redundant ssDNA-binding domains in complex with their target ssDNA; Dataset II: non-redundant holo-apo pairs of ssDNA-binding domains.
Dataset I contains non-redundant complexes of SP and NS ssDNA-binding domains and their corresponding ssDNA, representing the specific and non-specific ssDNA-binding proteins, respectively. To generate this dataset, redundant ssDNA-binding domains were removed using PISCES with a 30% sequence identity cutoff (69). These non-redundant, domain-based SSB–ssDNA complexes were then assigned into two groups, SP and NS, based on their binding specificity. The ssDNA binding specificity was manually annotated by searching the primary references for these structures and their homologs in PDB (58,59), as well as relevant information in UniProt (70). The non-redundant, domain-based Dataset I has 22 SP and 42 NS SSB-ssDNA complexes (Supplementary Tables S2 and S3). Out of these 64 non-redundant ssDNA-binding domains, 8 of them have not been classified by CATH from the V4.3 release. The remaining 56 domains represent three major classes, 11 different Architectures, 29 Topologies and 35 Homologous Superfamilies (3, 7, 12, 13 and 3, 9, 21, 23 for the specific and non-specific groups, respectively), suggesting a good structural diversity and coverage (Supplementary Tables S2 and S3).
Dataset II includes non-redundant SP and NS ssDNA-binding domains paired with their corresponding apo structures. To get the apo structures, all 458 ssDNA-binding domains (holo forms) were searched against PDB using default settings in the NCBI BLAST Blastp program (71). All unbound structures (apo forms) that have 100% sequence identity and at least 80% coverage with the bound protein domain of the complex structures were selected. Low quality X-ray structures with resolutions worse than 3.0 Å and R-values >0.3 were filtered out. An NMR apo structure was used if there were no X-ray apo structures available. Redundant holo-apo structural pairs were then removed using PISCES with a 30% sequence identity cutoff. These non-redundant pairs were assigned into SP and NS holo-apo pairs based on their ssDNA binding specificity annotations. Dataset II has 14 SP and 29 NS holo-apo ssDNA-binding domain pairs (Supplementary Table S4).
Structural features of SSB–ssDNA interactions
Comparative analyses of structural features in SSB–ssDNA interactions include: (i) propensities of amino acids in contact with ssDNA; (ii) side chain-base hydrogen bonds and π–π interactions between proteins and ssDNA; (iii) protein–DNA contact area (PDCA) and the number of residue-base contacts (NRBC) (54,72); and (iv) secondary structure types of residues involved in protein–ssDNA interactions.
An amino acid is defined as a DNA base-contacting residue if it has at least one heavy atom of its side chain within 3.9 Å of any heavy atom of a DNA base (54,56). The propensity of an amino acid that interacts with ssDNA was calculated as the ratio of the percentage of this amino acid in contact with ssDNA over the percentage of this amino acid in the dataset (Equation 1),
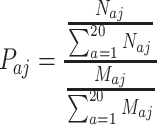 |
(1) |
where Paj is the propensity of amino acid a in dataset j; Naj represents the total number of amino acid a in contact with DNA in dataset j; Maj is the total number of amino acid a in dataset j. If Paj >1, amino acid a is said to be enriched in protein–ssDNA contacts in dataset j. If Paj <1, amino acid a is said to be depleted in protein–ssDNA contacts in dataset j. Since the datasets are relatively small, we applied both bootstrap and jackknife methods to estimate the sampling distributions, and the variances and potential bias of the data. The bootstrap samplings were repeated 1000 times with replacement. The mean propensities and ± 2SE (standard error) were plotted.
When computing the propensity for an amino acid interacting with a specific nucleotide, it is calculated as in Equation (2),
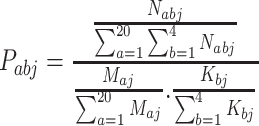 |
(2) |
where Pabj is the propensity of the interacting pair of amino acid a and nucleotide b in dataset j; Nabj represents the total number of amino acid a in contact with nucleotide b in dataset j; Maj is the total number of amino acid a in dataset j; and Kbj is the total number of nucleotide b in dataset j. If Pabj >1, contact between amino acid a and nucleotide b is said to be enriched in protein–ssDNA contacts in dataset j. If Pabj <1, contact between amino acid a and nucleotide b is said to be depleted in protein–ssDNA contacts in dataset j.
Hydrogen bonds between protein and ssDNA were identified using HBPLUS with default parameters (73). Percentages of side chain-base hydrogen bonds in all hydrogen bonds formed between protein and ssDNA and those of Watson–Crick atom-based side chain-base hydrogen bonds in all side chain-base hydrogen bonds were calculated for each complex to assess if there are any differences between SP and NS groups.
For π–π interactions, while previous studies used a vertical distance of 3.5 Å between DNA base-aromatic residue pairs, we applied a slightly relaxed distance cutoff of 3.9 Å for consistency with protein–ssDNA contact identification in this study and manually inspected to verify that the contact is π–π interaction using PyMOL (The PyMOL Molecular Graphics System, Version 2.3.2 Schrödinger, LLC) (74,75). In addition, the interplanar angle (ω) between the aromatic planes was measured with the angle_between_helices command in the PyMOL psico module. The geometry of π–π interactions were classified as stacked (0≤ ω ≤ 20°), inclined (20° < ω < 70°), and T-shaped (70° ≤ ω ≤90°) as described by Wilson et al. (76).
PDCA was calculated by subtracting the solvent accessible surface area (SASA) of a protein-ssDNA complex from the sum of solvent SASAs of its protein and ssDNA components and divided by two since the interface area is calculated twice from the protein and DNA components (Equation 3). The solvent accessible surface area was calculated with FreeSASA (77).
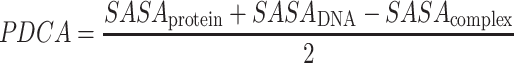 |
(3) |
Protein–ssDNA contacts were defined using a distance cutoff of 3.9 Å between side chain heavy atoms of an amino acid and all heavy atoms of a nucleotide. These protein–ssDNA contacts were further divided into two subsets depending on the atoms of DNA involved: (i) NRBC for the number of residue and DNA base contacts that represents specific interactions between protein and DNA as described in our previous studies (54,72), and (ii) NRBbC for the number of residue and DNA backbone contacts.
To investigate the roles of secondary structure types in protein–ssDNA binding specificity, DSSP program was applied to assign residues involved in protein–ssDNA interactions to three general secondary structure types: helix, strand and coil, where H (α-helix), G (310-helix) and I (π-helix) states from DSSP are assigned as helix type, E (extended strand) and B (residue in isolated β-bridge) states from DSSP are classified as strand type, and all the other states are considered as coil type (55,78–81).
Conformational changes of SSBs upon ssDNA binding were calculated by comparing both main chain root mean square deviation (RMSD) and interface RMSD (IRMSD) between bound (holo) ssDNA-binding domains and their unbound (apo) structures. Interface residues are residues that have at least one heavy atom within 10 Å of any heavy atoms of DNA. RMSD and IRMSD were calculated using the PyMOL align command for all heavy atoms in ssDNA-binding domains and in-house python scripts for heavy atoms of interface residues aligned with TM-align respectively (82).
Statistical analysis
For statistical analyses between two groups, Shapiro–Wilk test was performed first to test the normality of the data. If the data are normally distributed, a parametric Student’s t-test was carried out. Otherwise, a non-parametric Wilcoxon rank-sum test was applied. To test the association of interplanar angle distributions of protein–ssDNA π–π interactions between two groups, chi-square test or Fisher’s exact test was performed depending on the sample size and expected values.
RESULTS
Amino acid propensity for protein–ssDNA interaction
Overall propensity of residues involved in side chain-base contacts
The means and variances (2SE) of the amino acid propensities are shown in Figure 2 for bootstrap resampling and Supplementary Figure S1 for jackknife resampling, respectively. Both resampling methods are consistent and show very small variances (Figure 2 and Supplementary Figure S1). Aromatic and positively charged amino acids phenylalanine (F), histidine (H), tryptophan (W), tyrosine (Y), lysine (K) and arginine (R) are enriched in both SP and NS groups (Figure 2). Of these six residues, histidine, tyrosine and arginine are more enriched in the SP group than those in the NS group. The aromatic residues are likely involved in protein–ssDNA π–π interactions. The positively charged lysine and arginine can form hydrogen bonds with DNA bases and interact electronically with the negatively charged DNA backbone atoms. Aliphatic amino acids alanine (A), isoleucine (I), leucine (L) and valine (V), the negatively charged glutamate (E), and cysteine (C) show low propensity in both SP and NS groups (Figure 2). Three amino acids methionine (M), proline (P) and asparagine (N) are enriched in the NS group, but not in the SP group. The most interesting one is aspartate (D), a negatively charged small amino acid. It is enriched in the SP but depleted in the NS group. A similar pattern was found in the specific protein–dsDNA complexes (54), suggesting the importance of aspartate in both specific protein–ssDNA and protein–dsDNA interactions.
Figure 2.

Mean propensities and variances (±2SE) of amino acids that bind to ssDNA bases in the SP and NS groups from bootstrap resampling.
Propensity of aromatic residues involved in protein–ssDNA π–π interactions with different nucleotides
While aromatic residues in contact with ssDNA are enriched in both groups (Figure 2), those involved in protein–ssDNA π–π interactions show different preferences for nucleotides between the SP and NS groups. Figure 3 shows that while tryptophan–thymine is enriched in both NS and SP groups, it is even more enriched in the SP group. Tryptophan also shows preference to cytosine in the SP group. Another enriched residue histidine prefers guanine and adenine in the NS group but favors thymine in the SP group. To investigate if there are any differences in geometry types of π–π interactions between these two groups, we measured the interplanar angle (ω) between the aromatic planes of aromatic side chains and DNA bases. The π–π interactions were grouped into stacked, inclined, and T-shaped types as described in the ‘Materials and Methods’ section (76). The results are shown in Supplementary Table S5. Since the expected values of some cells are less than five, Fisher’s exact tests were carried out to compare the interplanar angle distributions between SP and NS groups for individual aromatic residues as well as group-wise comparisons. No significant differences were found between these two groups in terms of the geometry of protein–ssDNA π–π interactions (Supplementary Table S5).
Figure 3.

Propensities of aromatic amino acids that form protein–ssDNA π–π interactions with different DNA bases in the SP and NS groups.
Propensity of residues involved in side chain-base hydrogen bonds with different nucleotides
We found two enriched residue-nucleotide pairs, aspartate-guanine (propensity = 5.620) and lysine-guanine (propensity = 5.439) in the SP group but none such pairs in the NS group (Supplementary Figure S2). Of all 11 side chain-base hydrogen bonds formed between seven aspartate–guanine pairs, six are bidentate (where two hydrogen bonds formed with a DNA base via two pairs of hydrogen bond donors and acceptors) and two are bifurcated (where one hydrogen bond acceptor/donor is shared by two hydrogen bonds) hydrogen bonds formed with the same guanine bases (56). Interestingly, the DNA base atoms involved in these hydrogen bonding are those that typically form Watson-Crick base pairs in dsDNA (termed as WC atoms in this study Figure 4A). For instance, OD2 atom (hydrogen bond acceptor) of ASP223 on chain A of OnTEBP (PDBID: 1OTC) forms bifurcated hydrogen bonds with two WC atoms, H(N1) and H(N2) (hydrogen bond donors) of guanine 4 on the single strand telomeric DNA (Figure 4B). On the other hand, ASP42 on chain A of the unwinding protein (UP1) forms bidentate hydrogen bonds using OD1 and OD2 atoms as acceptors with H(N2) and H(N1) atoms of guanine 205, respectively, on a human telomeric repeat (PDBID: 1PGZ, Figure 4C). The second enriched pair lysine-guanine also forms four bidentate hydrogen bonds while the remaining six are simple hydrogen bonds. Unlike the aspartate–guanine pair, most of DNA base atoms involved in side chain-base hydrogen bonding (N7, 7 out of 10) in these lysine-guanine pairs are non-WC atoms.
Figure 4.

Watson-Crick (WC) atoms (A) and examples of aspartate forming bifurcate (B) and bidentate (C) hydrogen bonds with the same guanine in the SP group. Hydrogen bonds between atoms are represented by dashed lines. (A) Dashed red boxes indicate WC atoms involved in hydrogen bonds. Arrows represent atoms that can form hydrogen bonds with proteins: green arrow for hydrogen bond acceptor and blue arrow for hydrogen bond donor. (B) OD2 atom (acceptor) of ASP223 in OnTEBP (PDBID: 1OTC; protein chain: A; DNA chain: D) forms bifurcated hydrogen bonds with H(N1) and H(N2) atoms (donors, WC atoms) of guanine 4. (C) OD1 and OD2 atoms (acceptors) of ASP42 in the unwinding protein (UP1) form bidentate hydrogen bonds with H(N2) and H(N1) atoms (donors) of guanine 205 on a human telomeric repeat (PDBID: 1PGZ; protein chain: A; DNA chain: B).
It is not surprising that the number of side chain-base hydrogen bonds in the NS group is scarce. For example, there are only one histidine–guanine, one asparagine–guanine, one histidine–adenine and two arginine–guanine pairs observed in the NS group even though their propensity values are quite large (Supplementary Figure S2). Similarly, in the SP group, the tryptophan–thymine pair only has one case. Therefore, in these cases, both the raw count and propensity need to be considered for a fair and meaningful comparison.
Protein–ssDNA side chain-base hydrogen bonds
Unlike protein–dsDNA interactions, where the WC atoms of DNA bases are involved in A-T and C-G base pairing and are not available for interaction with proteins (Figure 4A), all base atoms on ssDNA have potential to interact with protein and form hydrogen bonds. To test if there are any differences between the SP and NS protein–ssDNA complexes, we compared the percentages of side chain-base hydrogen bonds among all protein–ssDNA hydrogen bonds and the percentages of WC atom-based side chain-base hydrogen bonds between these two groups (Figure 5). The percentages of side chain-base hydrogen bonds in each complex are shown in Figure 5A for the non-specific group and 5B for the specific group, with the percentages of side chain-base hydrogen bonds shown at the bottom in a descending order. Complexes in the SP group generally show larger contributions of side chain-base hydrogen bonds to the total number of protein–ssDNA hydrogen bonds. About 82% (18 of 22) of the SP complexes form side chain-base hydrogen bonds and ∼55% (12 of 22) of complexes have percentages of side chain-base hydrogen bonds equal to or above 50% (Figure 5B). The NS group, on the other hand, only has ∼47% (18 of the total 38 complexes that have at least one protein–DNA hydrogen bond) of the cases form side chain-base hydrogen bonds and ∼13% (5 of 38) of the complexes have percentages of side chain-base hydrogen bonds equal to or above 50% (Figure 5A). Even though the two NS complexes, domains 3kqlA03 and 4j1jA02 (the left two columns in Figure 5A), have exclusively side chain-base hydrogen bonds with their bound ssDNA, their raw counts are very small with only one for each complex. Wilcoxon rank-sum test shows the difference between these two groups is significant with a P-value of 0.00023 (Figure 5C).
Figure 5.

Comparisons of percentages of the side chain-base hydrogen bonds (A–C) and Watson-Crick atom-based side chain-base hydrogen bonds (D–F) between the SP and NS protein–ssDNA complexes. Percentages of side chain-base hydrogen bonds (SCBS, colored in blue) in all protein–DNA hydrogen bonds in NS complexes (A) and in SP complexes (B) are shown at the bottom in a descending order, while all other protein–DNA hydrogen bonds (other, colored in red) are on the top. Percentages of Watson-Crick atom-based SCBS hydrogen bonds (WC, colored in blue) in all side chain-base hydrogen bonds in NS complexes (D) and in SP complexes (E) are shown at the bottom in a descending order, and all other side chain-base hydrogen bonds (OSCBS, colored in red) are on the top. (C and F) Boxplots and statistical analyses for comparisons between the NS and SP groups. P-values are shown on top of the boxplots.
We also compared the percentages of WC atom-based hydrogen bonds in all side chain-base hydrogen bonds between these two groups. Percentages of WC atom-based hydrogen bonds in each complex are shown in Figure 5 (D for the NS group and E for the SP group), with percentages of WC atom-based hydrogen bonds shown at the bottom in a descending order. Overall, complexes in the SP group show larger percentages of WC atom-based hydrogen bonds. About 94% (17 of the total 18 complexes that have side chain-base hydrogen bonds) of the SP complexes form WC atom-based hydrogen bonds and all these 17 complexes (100%) have percentages of side chain-base hydrogen bonds larger than or equal to 50% (Figure 5E). The NS group only has ∼39% (7 of the total 18 complexes that have at least one side chain-base hydrogen bond) of the cases form WC atom-based hydrogen bonds and ∼28% (5 of 18) of the complexes have percentages of side chain-base hydrogen bonds no less than 50% (Figure 5D). Wilcoxon rank-sum test shows that the percentages of WC atom-based hydrogen bonds between the SP and NS groups are significantly different (P-value = 0.013) (Figure 5F).
Protein–ssDNA interaction interface
There is no significant PDCA difference between the SP and NS groups (Figure 6A, P-value = 0.22). PDCA represents the overall interface area with combined contacts among side chain, base and backbone atoms. When only the number of residue-base contacts is considered, the SP complexes show larger number of NRBC than the NS cases and the difference is statistically significant (Figure 6B, P-value = 0.012). After the NRBC is normalized with the overall interaction interface PDCA, the SP group shows more residue-base contacts per 1000 Å2 contact area, suggesting a larger percentage of specific interactions among all interactions (Figure 6C, P-value = 0.0032).
Figure 6.

Comparison of protein–ssDNA interface interactions. (A) Protein–DNA contact area (PDCA); (B) number of residue-base contacts (NRBC); (C) NRBC density, NRBC normalized to PDCA; and (D) percentage of NRBC in all protein–DNA contacts, the sum of NRBC and NRBbC (number of residue-DNA backbone contacts). P-values are displayed on top of the boxplots.
Compared with our previous study of protein–dsDNA binding specificity (54), the PDCAs between dsDNA-binding proteins and dsDNA and those between ssDNA-binding proteins and ssDNA are similar, but SSB–ssDNA interactions have more contacts between residues and bases, in terms of both raw and normalized NRBC counts (54). For instance, the majority of the normalized NRBC counts in specific ssDNA binding proteins are >10 (Figure 6C), while most of the normalized NRBC counts in specific dsDNA-binding proteins are <10 (54). One major difference between protein–dsDNA and protein–ssDNA interactions is that in protein–ssDNA complexes, residue–base contacts dominate the interaction between residues and ssDNA (Figure 6D), with median values >55 in non-specific ssDNA-binding proteins and >75 for the specific ssDNA-binding proteins, while protein–dsDNA interactions show the opposite trends that most of residue–DNA contacts are formed between residues and DNA backbone, with percentages of about 88% and 66% for non-specific and specific dsDNA-binding proteins, respectively (54). These differences are largely due to the increased accessibility of ssDNA base atoms compared to dsDNA.
Protein conformational changes upon ssDNA binding
To explore protein conformational changes upon ssDNA binding, we first calculated all heavy-atom RMSDs between the ssDNA-binding domains and their corresponding apo conformations. Most ssDNA-binding domains do not change conformation dramatically upon ssDNA binding, and majority of the RMSD values are <2 Å (Figure 7A). There are a few individual cases that show relatively large conformational changes. The largest change comes from a pair in the SP group, domain 3c2pA07 in Coliphage N4 virion-encapsidated RNA polymerase (vRNAP) and its apo structure 2po4 (RMSD: 6.532 Å). Figure 7B shows that the motif B of N4 vRNAP rearranges its structure from a loop (apo form, green) to a short antiparallel β-hairpin (holo form, magenta). This change and other conformational changes transit the polymerase from the inactive state to an active form to accommodate the binding of incoming DNA (83). Statistical analysis shows no significant RMSD difference between these two groups (Figure 7A, P-value = 0.37). Similarly, no significant IRMSD difference was found between these two groups (P-value = 0.89) (Figure 7C).
Figure 7.

Conformational changes upon ssDNA binding. (A) RMSD of heavy atoms of all residues between bound (holo) and unbound (apo) structures. (B) Structural alignment of domain 3c2pA07 (magenta) in Coliphage N4 virion-encapsidated RNA polymerase (vRNAP) and its apo structure 2po4 (green). (C) RMSD of heavy atoms of all interface residues (IRMSD) between bound (holo) and unbound (apo) structures.
Secondary structure types of ssDNA interacting residues
We recently compared the secondary structure types of residues involved in side chain-base hydrogen bonds in different types of dsDNA-binding proteins and found distinct patterns (55). To investigate the roles of secondary structure types of amino acids in specific ssDNA-binding proteins, the secondary structure type propensities of amino acids that are in contact with ssDNA bases were calculated against the relative frequencies of secondary structure types of all residues in the respective group of ssDNA-binding domains. SsDNA base-contacting residues in both groups are enriched in strand conformations with a higher enrichment in the SP group, while coil secondary structure types are also preferred in the NS group (Figure 8A).
Figure 8.

Propensities of secondary structure types in the SP and NS groups. (A) Propensities of secondary structure types of DNA base-contacting residues. The background distributions are based on all residues in the ssDNA-binding domains in the SP or NS group. (B) Propensities of secondary structure types of residues involved in side chain-base hydrogen bonds. The background distributions are based on all the base-contacting residues. (C) Propensities of secondary structure types of residues involved in side chain-base hydrogen bonds. The background distributions are based on all the DNA hydrogen-bonding residues (C).
For residues that form hydrogen bonds between their side chains and ssDNA bases, we used two different background distributions to calculate the propensities: one is the secondary structure type distribution of all base-contacting residues (Figure 8B) and the other is the secondary structure type distribution of all residues that form hydrogen bonds with DNA including bases and backbone atoms (Figure 8C). A similar trend is found in both cases. Residues involved in side chain-base hydrogen bonds in both groups are enriched in strand conformations. However, between these two groups, these residues in the NS group have higher propensity for strands than those in the SP group. The propensity for coil types in the SP group is larger than that in the NS group (Figure 8B and C). These results suggest that for residues involved in side chain-base hydrogen bonds, relatively more such residues in the SP group are adopting coil conformation, which may represent more flexible conformations. However, results in Figure 8B and C need to be interpreted with caution as the raw counts of secondary structure types in the NS group are relatively small (Helix: 6, strand: 13, and coil: 7).
DISCUSSION
We performed a comparative study of SSB–ssDNA interactions with a focus on the binding specificity. Our results suggest that side chain-base hydrogen bonds play a major role in protein–ssDNA binding specificity, while protein–ssDNA π–π interactions may mainly contribute to binding affinity. Without a complementary strand in ssDNA, atoms normally forming Watson-Crick base pairs in dsDNA are available to serve as additional hydrogen bond donors/acceptors to facilitate binding specificity and/or affinity. Significantly larger percentages of overall and WC atom-based side chain-base hydrogen bonds were found in the SP group than the NS group (Figure 5). Unlike specific dsDNA-binding domains, which are more flexible and undergo larger conformational changes after binding dsDNA than non-specific dsDNA-binding domains, there is no apparent difference between the specific and non-specific ssDNA-binding domains (54).
Our comparative analyses show that the SP group proteins form more contacts with DNA bases than those in the NS group (Figure 6), and the propensities of amino acids that involved in protein–ssDNA contacts show that both groups prefer aromatic residues and positively charged residues, but residues H, Y and R are more enriched in the SP group (Figure 2). These findings are consistent with previous studies (50,84,85). The enrichment of all aromatic residues can be attributed to the increased accessibility of ssDNA. Without the steric hindrance from the complementary strand, ssDNA can change conformation relatively easily so that DNA bases become more accessible to aromatic residues to engage in π–π interactions with aromatic residues. The distributions of π–π geometry types between SP and NS groups do not show significant differences (Supplementary Table S5), suggesting that in general protein–ssDNA π–π interactions may mainly contribute to binding affinity although in individual cases π–π interactions can contribute to specific protein–ssDNA interactions. This is similar to the role of the minor groove interactions in specific protein–dsDNA recognition. Even though minor groove contacts are generally considered non-specific due to the lack of discriminative pattern for hydrogen bonds, several studies have demonstrated that some minor groove contacts can contribute to protein–dsDNA binding specificity through shape readout mechanism (51–54). While there are no π–π geometry studies in terms of protein-DNA binding specificity, based on a dataset with 428 protein–DNA complexes that include both protein–dsDNA and protein–ssDNA complexes, Wilson et al. found that the stacked orientation (58%) is more common than the inclined configuration (29%), with the T-shaped interaction as the least frequent one (13%) (76). For SSB–ssDNA complexes, by combining the frequencies of each geometric type of π–π interactions in both NS and SP groups (Supplementary Table S5), we found that the stacked orientation represents 39% of protein–ssDNA π–π interactions, similar to the inclined configuration (38.1%), with the T-shaped interaction at 22.9%.
The most interesting structural feature is the enrichment of amino acid aspartate in the SP group, but not in the NS group (Figure 2). This difference is more distinct at the side chain-base hydrogen bond level, where aspartate forms side chain-base hydrogen bonds with all nucleotides except for adenine in the SP group but none such hydrogen bonds were found in the NS group. Out of three types of contacting nucleotides, the preference of aspartate to guanine is the most dominant one. Structural inspections show this preference is achieved via the bidentate and/or bifurcate hydrogen bonds between aspartate and WC atoms of DNA bases of the same guanines (Figure 4). Aspartate was also enriched in specific protein–dsDNA binding but with different binding characteristics. In protein–dsDNA interactions, aspartate favors cytosine, and most (10 out of total 19) of aspartate-cytosine side chain-base hydrogen bonds are bidentate hydrogen bonds formed with two consecutive cytosines in the major groove (54).
In addition to guanine, aspartate also shows preferences to cytosine (propensity = 3.042) and thymine (propensity = 1.865). This may explain the critical role of aspartate in mutagenesis driven by the cytidine deaminase APOBEC in cancer (86). APOBEC-mediated mutations, especially those driven by APOBEC3A and APOBEC3B, are sensitive to cytosines in TpC sites in hairpin (stem-loop) DNA structures, potential hotspots for mutagenesis, formed while transiently single-stranded in a sequence specific manner (7,86,87). Particularly, Shi et al. found aspartate 131 (D131) strongly influenced the preference of the upstream nucleotide of the target cytosine at the TpC sites. A substitution by alanine (D131A) decreased selectivity, and a glutamate substitution (D131E) converted the preference to cytosine from thymine, while threonine substitution (D131T) retained selectivity (86) (Supplementary Figure S3). Additionally, they found that two neighbor tyrosine residues (Y130, Y132) were also important in conferring the selectivity (86) (Supplementary Figure S3). Despite all three APOBEC3(A/B)–ssDNA complex structures in PDB (PDBID: 5KEG, 5SWW, 5TD5) were excluded in this study due to different numbers of mutations in the protein, preferences of aspartate to cytosine and thymine only in the SP group suggest these patterns might be a general feature of the SP group rather than a unique feature of the APOBEC family.
Upon ssDNA binding, conformational changes of ssDNA-binding domains in terms of heavy atom RMSD between the SP and NS groups, either for all residues or interface residues only, do not show significant differences (Figure 7). This indicates that while specific protein–ssDNA recognition relies on the flexibility of protein and/or ssDNA in some cases (36), the conformational change of ssDNA may play a larger role than that of protein. This is consistent with what Theobald and Schultz found from structural and thermodynamics comparisons between the cognate OnTEBP–ssDNA complex and complexes with 10 different non-cognate ssDNA molecules (49). They found that while the protein conformation in all non-cognate complexes remained nearly identical to that in the cognate complex, the ssDNA exhibited dramatic differences in three non-cognate complexes and subtle conformational changes in the other seven non-cognate complexes (49). This study revealed the plasticity of the OnTEBP in accommodating non-cognate ssDNA sequences, especially via a phenomenon they named nucleotide shuffling—conformational rearrangements via shifts in the ssDNA register of various number of nucleotides (49).
DNA base-contacting residues in specific ssDNA-binding proteins are enriched in strands while non-specific ssDNA-binding proteins show preferences to strands and coils (Figure 8A). The secondary structure type preferences of specific ssDNA-binding proteins are different from those of specific dsDNA-binding proteins, including both highly specific and multi-specific dsDNA-binding proteins, where highly specific dsDNA-binding proteins prefer coils and multi-specific dsDNA-binding proteins favor helices (55). These results are largely in agreement with the conformational studies in both specific protein–dsDNA and protein–ssDNA interactions. In protein–ssDNA complexes, there are less conformational changes after binding DNA that is consistent with the larger strand propensity, while in specific protein–dsDNA interactions, proteins are more flexible and enriched relatively more in coil conformations. The SP group has a larger propensity of coils than the NS group (Figure 8B and C), indicating protein flexibility still plays a role in the binding specificity. Protein flexibility affects DNA recognition likely via speeding up locating DNA-binding proteins to their target sites (54,57,88–91). In addition, it is suggested that the loops/linkers connecting ssDNA-binding domains, especially their lengths, are responsible for binding specificity (36,85,92).
To our knowledge, this is the first comparative structural study of protein–ssDNA interactions, especially the mechanisms underlying the binding discrepancy between ssDNA binding proteins with different degrees of binding specificity. We believe findings from this study can help improve SSB–ssDNA binding prediction models (50). Moreover, this study can provide additional information, such as binding specificity to current databases about protein–DNA interactions (65).
Supplementary Material
Contributor Information
Maoxuan Lin, Department of Bioinformatics and Genomics, The University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
Fareeha K Malik, Department of Bioinformatics and Genomics, The University of North Carolina at Charlotte, Charlotte, NC 28223, USA; Research Center of Modeling and Simulation, National University of Science and Technology, Islamabad, 44000, Pakistan.
Jun-tao Guo, Department of Bioinformatics and Genomics, The University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institutes of Health [R15GM110618 to J.G]; National Science Foundation [DBI1356459 to J.G].
Conflict of interest statement. None declared.
REFERENCES
- 1. Richard D.J., Bolderson E., Cubeddu L., Wadsworth R.I., Savage K., Sharma G.G., Nicolette M.L., Tsvetanov S., McIlwraith M.J., Pandita R.K. et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008; 453:677–681. [DOI] [PubMed] [Google Scholar]
- 2. Wold M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 3. Gu P., Deng W., Lei M., Chang S. Single strand DNA binding proteins 1 and 2 protect newly replicated telomeres. Cell Res. 2013; 23:705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richard D.J., Bolderson E., Khanna K.K. Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit. Rev. Biochem. Mol. Biol. 2009; 44:98–116. [DOI] [PubMed] [Google Scholar]
- 5. Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008; 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu Y., Lu J., Kang T. Human single-stranded DNA binding proteins: guardians of genome stability. Acta Biochim. Biophys. Sin. (Shanghai). 2016; 48:671–677. [DOI] [PubMed] [Google Scholar]
- 7. Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019; 364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z. et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alberts B.M., Frey L. T4 bacteriophage gene 32: a structural protein in the replication and recombination of DNA. Nature. 1970; 227:1313–1318. [DOI] [PubMed] [Google Scholar]
- 10. Jose D., Weitzel S.E., Baase W.A., von Hippel P.H. Mapping the interactions of the single-stranded DNA binding protein of bacteriophage T4 (gp32) with DNA lattices at single nucleotide resolution: gp32 monomer binding. Nucleic Acids Res. 2015; 43:9276–9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pant K., Karpel R.L., Rouzina I., Williams M.C. Salt dependent binding of T4 gene 32 protein to single and double-stranded DNA: single molecule force spectroscopy measurements. J. Mol. Biol. 2005; 349:317–330. [DOI] [PubMed] [Google Scholar]
- 12. Shamoo Y., Friedman A.M., Parsons M.R., Konigsberg W.H., Steitz T.A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature. 1995; 376:362–366. [DOI] [PubMed] [Google Scholar]
- 13. Antony E., Lohman T.M. Dynamics of E. coli single stranded DNA binding (SSB) protein-DNA complexes. Semin. Cell Dev. Biol. 2019; 86:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamon L., Pastre D., Dupaigne P., Le Breton C., Le Cam E., Pietrement O. High-resolution AFM imaging of single-stranded DNA-binding (SSB) protein–DNA complexes. Nucleic Acids Res. 2007; 35:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Overman L.B., Bujalowski W., Lohman T.M. Equilibrium binding of Escherichia coli single-strand binding protein to single-stranded nucleic acids in the (SSB)65 binding mode. Cation and anion effects and polynucleotide specificity. Biochemistry. 1988; 27:456–471. [DOI] [PubMed] [Google Scholar]
- 16. Raghunathan S., Ricard C.S., Lohman T.M., Waksman G. Crystal structure of the homo-tetrameric DNA binding domain of Escherichia coli single-stranded DNA-binding protein determined by multiwavelength x-ray diffraction on the selenomethionyl protein at 2.9-A resolution. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sigal N., Delius H., Kornberg T., Gefter M.L., Alberts B. A DNA-unwinding protein isolated from Escherichia coli: its interaction with DNA and with DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 1972; 69:3537–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spenkelink L.M., Lewis J.S., Jergic S., Xu Z.Q., Robinson A., Dixon N.E., van Oijen A.M. Recycling of single-stranded DNA-binding protein by the bacterial replisome. Nucleic Acids Res. 2019; 47:4111–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suksombat S., Khafizov R., Kozlov A.G., Lohman T.M., Chemla Y.R. Structural dynamics of E. coli single-stranded DNA-binding protein reveal DNA wrapping and unwrapping pathways. Elife. 2015; 4:e08193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bochkarev A., Bochkareva E. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 2004; 14:36–42. [DOI] [PubMed] [Google Scholar]
- 21. Bochkarev A., Pfuetzner R.A., Edwards A.M., Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997; 385:176–181. [DOI] [PubMed] [Google Scholar]
- 22. Chen R., Wold M.S. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 2014; 36:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fairman M.P., Stillman B. Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J. 1988; 7:1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wold M.S., Kelly T. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:2523–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yates L.A., Aramayo R.J., Pokhrel N., Caldwell C.C., Kaplan J.A., Perera R.L., Spies M., Antony E., Zhang X. A structural and dynamic model for the assembly of replication protein A on single-stranded DNA. Nat. Commun. 2018; 9:5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casas-Finet J.R., Fischer K.R., Karpel R.L. Structural basis for the nucleic acid binding cooperativity of bacteriophage T4 gene 32 protein: the (Lys/Arg)3(Ser/Thr)2 (LAST) motif. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lonberg N., Kowalczykowski S.C., Paul L.S., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. III. Binding properties of two specific proteolytic digestion products of the protein (G32P*I and G32P*III). J. Mol. Biol. 1981; 145:123–138. [DOI] [PubMed] [Google Scholar]
- 28. Murzin A.G. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993; 12:861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raghunathan S., Kozlov A.G., Lohman T.M., Waksman G. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Biol. 2000; 7:648–652. [DOI] [PubMed] [Google Scholar]
- 30. Lohman T.M., Ferrari M.E. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 1994; 63:527–570. [DOI] [PubMed] [Google Scholar]
- 31. Sun S., Shamoo Y. Biochemical characterization of interactions between DNA polymerase and single-stranded DNA-binding protein in bacteriophage RB69. J. Biol. Chem. 2003; 278:3876–3881. [DOI] [PubMed] [Google Scholar]
- 32. Fanning E., Klimovich V., Nager A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006; 34:4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iftode C., Borowiec J.A. Denaturation of the simian virus 40 origin of replication mediated by human replication protein A. Mol. Cell. Biol. 1997; 17:3876–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bochkareva E., Korolev S., Lees-Miller S.P., Bochkarev A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002; 21:1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ashton N.W., Bolderson E., Cubeddu L., O’Byrne K.J., Richard D.J. Human single-stranded DNA-binding proteins are essential for maintaining genomic stability. BMC Mol. Biol. 2013; 14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dickey T.H., Altschuler S.E., Wuttke D.S. Single-stranded DNA-binding proteins: multiple domains for multiple functions. Structure. 2013; 21:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi H., Zhang Y., Zhang G., Guo J., Zhang X., Song H., Lv J., Gao J., Wang Y., Chen L. et al. Systematic functional comparative analysis of four single-stranded DNA-binding proteins and their affection on viral RNA metabolism. PLoS One. 2013; 8:e55076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shamoo Y. Single-stranded DNA-binding proteins. Encyclopedia of Life sciences. 2002; London: Macmillan Publishers Ltd, Nature Publishing Group; 1–7. [Google Scholar]
- 39. Chase J.W., Williams K.R. Single-stranded DNA binding proteins required for DNA replication. Annu. Rev. Biochem. 1986; 55:103–136. [DOI] [PubMed] [Google Scholar]
- 40. Kur J., Olszewski M., Dlugolecka A., Filipkowski P. Single-stranded DNA-binding proteins (SSBs) – sources and applications in molecular biology. Acta Biochim. Pol. 2005; 52:569–574. [PubMed] [Google Scholar]
- 41. Perales C., Cava F., Meijer W.J., Berenguer J. Enhancement of DNA, cDNA synthesis and fidelity at high temperatures by a dimeric single-stranded DNA-binding protein. Nucleic Acids Res. 2003; 31:6473–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bujalowski W., Lohman T.M. Escherichia coli single-strand binding protein forms multiple, distinct complexes with single-stranded DNA. Biochemistry. 1986; 25:7799–7802. [DOI] [PubMed] [Google Scholar]
- 43. Wei T.F., Bujalowski W., Lohman T.M. Cooperative binding of polyamines induces the Escherichia coli single-strand binding protein-DNA binding mode transitions. Biochemistry. 1992; 31:6166–6174. [DOI] [PubMed] [Google Scholar]
- 44. Horvath M.P. Structural anatomy of telomere OB proteins. Crit. Rev. Biochem. Mol. Biol. 2011; 46:409–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horvath M.P., Schweiker V.L., Bevilacqua J.M., Ruggles J.A., Schultz S.C. Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell. 1998; 95:963–974. [DOI] [PubMed] [Google Scholar]
- 46. Altschuler S.E., Dickey T.H., Wuttke D.S. Schizosaccharomyces pombe protection of telomeres 1 utilizes alternate binding modes to accommodate different telomeric sequences. Biochemistry. 2011; 50:7503–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lei M., Podell E.R., Cech T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004; 11:1223–1229. [DOI] [PubMed] [Google Scholar]
- 48. Dickey T.H., McKercher M.A., Wuttke D.S. Nonspecific recognition is achieved in Pot1pC through the use of multiple binding modes. Structure. 2013; 21:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Theobald D.L., Schultz S.C. Nucleotide shuffling and ssDNA recognition in Oxytricha nova telomere end-binding protein complexes. EMBO J. 2003; 22:4314–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pal A., Levy Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 2019; 15:e1006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joshi R., Passner J.M., Rohs R., Jain R., Sosinsky A., Crickmore M.A., Jacob V., Aggarwal A.K., Honig B., Mann R.S. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007; 131:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S. Origins of specificity in protein–DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B. The role of DNA shape in protein–DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Corona R.I., Guo J.T. Statistical analysis of structural determinants for protein–DNA-binding specificity. Proteins. 2016; 84:1147–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin M., Guo J.T. New insights into protein-DNA binding specificity from hydrogen bond based comparative study. Nucleic Acids Res. 2019; 47:11103–11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luscombe N.M., Laskowski R.A., Thornton J.M. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001; 29:2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Song W., Guo J.T. Investigation of arc repressor DNA-binding specificity by comparative molecular dynamics simulations. J. Biomol. Struct. Dyn. 2015; 33:2083–2093. [DOI] [PubMed] [Google Scholar]
- 58. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burley S.K., Berman H.M., Christie C., Duarte J.M., Feng Z., Westbrook J., Young J., Zardecki C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018; 27:316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999; 292:195–202. [DOI] [PubMed] [Google Scholar]
- 61. Luscombe N.M., Austin S.E., Berman H.M., Thornton J.M. An overview of the structures of protein–DNA complexes. Genome Biol. 2000; 1:REVIEWS001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berman H.M., Olson W.K., Beveridge D.L., Westbrook J., Gelbin A., Demeny T., Hsieh S.H., Srinivasan A.R., Schneider B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992; 63:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Coimbatore Narayanan B., Westbrook J., Ghosh S., Petrov A.I., Sweeney B., Zirbel C.L., Leontis N.B., Berman H.M. The nucleic acid database: new features and capabilities. Nucleic Acids Res. 2014; 42:D114–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sagendorf J.M., Berman H.M., Rohs R. DNAproDB: an interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2017; 45:W89–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sagendorf J.M., Markarian N., Berman H.M., Rohs R. DNAproDB: an expanded database and web-based tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2020; 48:D277–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rodrigues J., Teixeira J.M.C., Trellet M., Bonvin A pdb-tools: a swiss army knife for molecular structures. F1000Res. 2018; 7:1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dawson N.L., Lewis T.E., Das S., Lees J.G., Lee D., Ashford P., Orengo C.A., Sillitoe I. CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017; 45:D289–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lewis T.E., Sillitoe I., Dawson N., Lam S.D., Clarke T., Lee D., Orengo C., Lees J. Gene3D: extensive prediction of globular domains in proteins. Nucleic Acids Res. 2018; 46:D1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang G., Dunbrack R.L. Jr. PISCES: a protein sequence culling server. Bioinformatics. 2003; 19:1589–1591. [DOI] [PubMed] [Google Scholar]
- 70. UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008; 36:W5–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim R., Corona R.I., Hong B., Guo J.T. Benchmarks for flexible and rigid transcription factor-DNA docking. BMC Struct. Biol. 2011; 11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McDonald I.K., Thornton J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994; 238:777–793. [DOI] [PubMed] [Google Scholar]
- 74. Rutledge L.R., Campbell-Verduyn L.S., Wetmore S.D. Characterization of the stacking interactions between DNA or RNA nucleobases and the aromatic amino acids. Chem. Phys. Lett. 2007; 444:167–175. [Google Scholar]
- 75. Rutledge L.R., Durst H.F., Wetmore S.D. Evidence for stabilization of DNA/RNA-protein complexes arising from nucleobase-amino acid stacking and T-shaped interactions. J. Chem. Theory Comput. 2009; 5:1400–1410. [DOI] [PubMed] [Google Scholar]
- 76. Wilson K.A., Kellie J.L., Wetmore S.D. DNA-protein pi-interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res. 2014; 42:6726–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mitternacht S. FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Res. 2016; 5:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983; 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 79. Kim R., Guo J.T. Systematic analysis of short internal indels and their impact on protein folding. BMC Struct. Biol. 2010; 10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin M., Whitmire S., Chen J., Farrel A., Shi X., Guo J.T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017; 7:9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Touw W.G., Baakman C., Black J., te Beek T.A., Krieger E., Joosten R.P., Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015; 43:D364–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gleghorn M.L., Davydova E.K., Rothman-Denes L.B., Murakami K.S. Structural basis for DNA-hairpin promoter recognition by the bacteriophage N4 virion RNA polymerase. Mol. Cell. 2008; 32:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Maffeo C., Aksimentiev A. Molecular mechanism of DNA association with single-stranded DNA binding protein. Nucleic Acids Res. 2017; 45:12125–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mishra G., Levy Y. Molecular determinants of the interactions between proteins and ssDNA. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shi K., Carpenter M.A., Banerjee S., Shaban N.M., Kurahashi K., Salamango D.J., McCann J.L., Starrett G.J., Duffy J.V., Demir O. et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017; 24:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Faden D.L., Thomas S., Cantalupo P.G., Agrawal N., Myers J., DeRisi J. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral Oncol. 2017; 74:8–14. [DOI] [PubMed] [Google Scholar]
- 88. Fuxreiter M., Simon I., Bondos S. Dynamic protein-DNA recognition: beyond what can be seen. Trends Biochem. Sci. 2011; 36:415–423. [DOI] [PubMed] [Google Scholar]
- 89. Levy Y., Onuchic J.N., Wolynes P.G. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007; 129:738–739. [DOI] [PubMed] [Google Scholar]
- 90. Shoemaker B.A., Portman J.J., Wolynes P.G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhou H.X. Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 2012; 37:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Flynn J.M., Levchenko I., Sauer R.T., Baker T.A. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004; 18:2292–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.