

Abstract
A series of novel bis-imidazolium salts was synthesized, characterized, and evaluated in vitro against a panel of non-small cell lung cancer (NSCLC) cells. Two imidazolium cores were connected with alkyl chains of varying lengths to develop a structure activity relationship (SAR). Increasing the length of the connecting alkyl chain was shown to correlate to an increase in the anti-proliferative activity. The National Cancer Institute’s NCI-60 human tumor cell line screen confirmed this trend. The compound containing a decyl linker chain, 10, was chosen for further in vivo toxicity studies with C578BL/6 mice. The compound was well tolerated by the mice and all of the animals survived and gained weight over the course of the study.
Keywords: Imidazolium salt, Non-small cell lung cancer, Bis-imidazolium salt, SAR, in vivo toxicity
Graphical Abstract
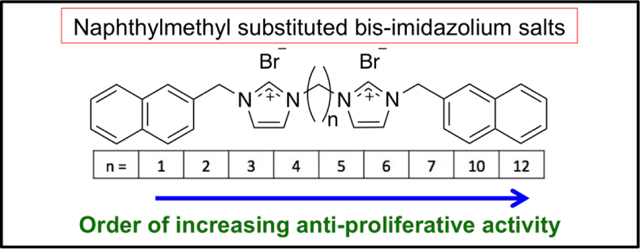
2. Introduction
Cancer is expected to kill over 600,000 Americans in the year 2020, accounting for one in every four deaths, with heart disease as the only more common cause of death.1 Lung cancer is the leading cause of all cancer-related deaths and treatment depends on the stage and type of cancer. The two major categories include small cell and non-small cell lung cancers (NSCLC), the latter of which makes up 84% of lung cancer cases.1 Treatment for NSCLC may include surgery, radiation, chemotherapy (first line treatments usually include a combination of platinum drugs i.e. cisplatin with a third-generation compound such as docetaxel or paclitaxel),2 and/or a targeted therapy such as Cetuximab.3 However, the five-year survival rate for NSCLC is only 16% for men and 23% for women,1 exhibiting that new forms of treatment are necessary for this devastating disease.
Imidazolium salts, such as IS-12 (Figure 1) are a class of compounds that have received significant attention in the literature for their in vitro anti-cancer properties against NSCLC.4–20 Many of these compounds exhibit IC50 values in the low micromolar to nanomolar range against a variety of NSCLC cell lines. The in vitro anti-cancer properties of these imidazolium salts are directly related to the number and type of substituents positioned around the central imidazolium core. For example IS-12 contains naphthylmethyl-substituents at both nitrogen atoms in the ring. A study done by Malhotra looked at imidazolium salts with a methyl group at the N1 position and aliphatic substituents ranging from one to fourteen carbons at the N3 position.13 Those with one to seven carbons at the N3 position showed no antiproliferative properties, whereas those with eight to fourteen carbons at the N3 position were active as determined by the National Cancer Institute’s (NCI) 60-human tumor cell line screen. However, highly lipophilic derivatives have minimal solubility in aqueous solution and therefore are limited in their clinical potential.5
Figure 1.

Structure of the naphthylmethyl-substituted imidazolium salt IS-12
Bis-imidazolium salts (Figure 2) have the advantage of two positively charged moieties to aid in increasing aqueous solubility and have been investigated as potential anti-cancer drugs due to their ability to inhibit human galactosyltransferase and glycosyltransferase enzymes.21 Many precursors to bis-metal-NHC complexes have also been investigated for a variety of applications including transmetallation agents and as catalysts.22,23 Bis-imidazolium salts with naphthylmethyl substituents at the N1 and N1’ positions are structurally similar to the depsipeptide echinomycin (Figure 3), with planar, aromatic groups separated by a linking moiety. Echinomycin is a bis-intercalator, meaning it can intercalate between DNA base pairs with both quinoxaline units.24 Though originally useful as an antibiotic, this depsipeptide has been investigated for its antitumor activity. It has been shown to bind a sequence of DNA that is recognized by the hypoxia inducible factor-1 (HIF-1) transcription factor and has the ability to inhibit the normal DNA-binding of HIF-1.24,25 Derivatives of echinomycin have also shown promise against various cancer cell lines while also retaining antimicrobial activity.26 Similar to echinomycin, IS-12 has two planar aromatic groups and significant anti-cancer activity against lung cancer cell lines.4 However, the lipophilicity of IS-12 causes challenges with administration.
Figure 2.

General depiction of a bis-imidazolium salt.
Figure 3.

1H NMR spectrum of 3 in DMSO-d6.
Presented herein is a series of bis-imidazolium salts with naphthylmethyl-substituents at both the N1 and N1’ positions. These complexes were designed with the purpose of increasing aqueous solubility by increasing the charge density of the molecule. In addition, the length of the alkyl chain bridging the two imidazolium units was varied from one to twelve carbons to determine whether increasing the distance between the two planar aromatic groups of IS-12, as seen in the structure of echinomycin, would alter anticancer activity. Ultimately this report seeks to establish a structure activity relationship (SAR) relating the length of the bridging alkyl chain to the anti-cancer properties of each compound.
3. Results and discussion
3.1. Synthesis and characterization
Compound 1 was synthesized by reacting 2.2 molar equivalents of 2-(bromomethyl)naphthalene with di(imidazole-1-yl)methane dissolved in acetonitrile (Equation 1). The solution was refluxed overnight and produced a white precipitate, 1. A mixture of water and ethanol (1:6) was used to recrystallize 1.
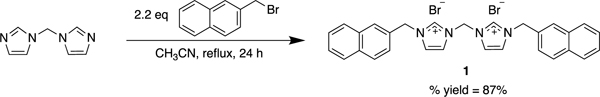 |
Equation 1. Synthesis of 1. |
1-(Naphthalen-2-ylmethyl)imidazole was synthesized by published procedures and used as starting material for the synthesis of 2-12.4 The appropriate di-bromoalkane was reacted with 2.2 molar equivalents of 1-(naphthalen-2-ylmethyl)imidazole to ensure no mono-cationic species would result. The di-bromoalkane was added to a solution of 1-(naphthalen-2-ylmethyl)imidazole in acetonitrile and refluxed overnight to synthesize 2–12 (Equation 2). The workup varied slightly for each compound, and specific details can be found with the synthetic procedures. Compounds 9 and 11 were difficult to isolate because they became viscous liquids under atmospheric conditions until they were isolated using a glove bag with a nitrogen atmosphere. The bis-cationic imidazolium salts were observed to be hygroscopic to varying degrees. Yields ranges from 16% to 87% with 1, 3, 4, 6, and 8 having high yields (87%, 84%, 76%, 77%, and 83% respectively); 5, 7, 9, 10, and 12 having moderate yields (52%, 64%, 53%, 49% and 48% respectively); and 2 and 11 having poor yields (16% and 18% respectively).
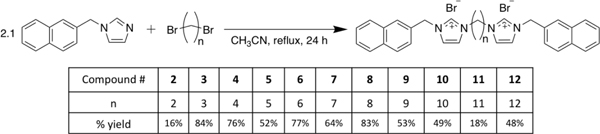 |
Equation 2. Synthesis of 2-12. |
Compounds were characterized by 1H NMR, 13C NMR, melting point analysis, and HRMS. In addition, several compounds were also characterized by X-ray crystallography. Difficulty in isolating and storing compounds 8, 9, and 11 led to incomplete characterization. It is important to note that compounds that are not easily stored lead to difficulties in completing in vitro studies. Therefore, only compounds that could be used for biological studies were further studied and fully characterized. Two singlet resonances in the 1H NMR spectra most notably indicated the formation of the bis-imidazolium salt (Figure 3). The first singlet resonance in each spectra ranged from 9.32 ppm to 9.70 ppm corresponding to the protons at the C2/C2’ positions (peak a, Figure 3) of the bisimidazolium salts. This is an indication of conversion from the starting material imidazole to the desired bis-cation system. The second most notable resonance suggesting the formation of the bis-imidazolium salt had chemical shifts that ranged from 5.57 ppm to 5.68 ppm corresponding with those of the methylene bridging the imidazole ring to the naphthalene moiety.4,5,27 Integrations of both distinctive resonances were consistent with the structure of the compound. The melting point analysis of 1-7, 10, and 12 revealed that these compounds went through a phase transition instead of melting. Also, the high-resolution mass spectrometry data suggested the parent ion was only singly charged, versus the doubly charged system we would expect to see. The C2 proton could be easily removed to form a carbene at one imidazole leaving the second imidazole positively charged. In addition, compounds 2, as the hexafluorophosphate salt (2-PF6), 3, 4, 7, as the hexafluorophosphate salt (7-PF6), and 8 were characterized by single crystal X-ray crystallography (Figures 4–8).
Figure 4.

Thermal ellipsoid plot of 2-PF6 with thermal ellipsoids drawn to the 50% probability level. Hydrogen atoms, carbon labels, and the PF6- anions were removed for clarity.
Figure 8.

Thermal ellipsoid plot of 8 with thermal ellipsoids drawn to the 50% probability level. Hydrogen atoms, carbon labels, disordered water solvent molecules, and bromide anions were removed for clarity.
3.2. MTT assay
The anti-cancer properties of 1-7, 10, and 12 were evaluated against several NSCLC cell lines (NCI-H460, NCI-H1975, HCC827, and A549) to determine their IC50 values (concentration that inhibits 50% growth of cells when compared to untreated control cells). This series of compounds was synthesized to establish a SAR of naphthylmethyl-substituted bis-imidazolium salts with increasing alkyl chain lengths bridging the two imidazolium cores. The original goal in synthesizing this series of compounds was to find the ideal chain length and determine the impact that the alkyl chain length has on anticancer properties. To obtain IC50 values, cells were plated at 5,000–7,000 cells per well, depending on the cell line, and allowed to adhere to 96-well plates overnight. Cells were exposed to compounds 1–7, 10, and 12 at concentrations of 1, 4, 16, and 32 μM for 72 hours at which time the MTT assay was utilized. For absorbance readings, DMSO was added at the end to each well. Results were compared to the well-known chemotherapeutic agent cisplatin and the previously published imidazolium salt IS-12.4
To prepare the aforementioned dilutions of each compound in growth medium, all compounds were dissolved in DMSO and diluted with water to prepare stock solutions at a concentration of 1 mM. Compounds 1 and 5 produced clear solutions with minor insolubles. Each compound was further diluted into growth medium to the final testing concentrations. The maximum amount of DMSO in the testing solution was 0.032% (v/v). The stock solution of cisplatin was prepared by stirring cisplatin in water at room temperature for several hours to completely solubilize the compound.
The resulting IC50 values from the MTT assay performed on 1–7, 10, 12, cisplatin, and previously published results for IS-12 are summarized in Table 1. The results follow the general trend of increased anti-proliferative effects being directly related to an increase in the alkyl chain length. Compound 1, with the shortest alkyl chain linker, has the lowest amount of anti-proliferative activity with IC50 values above the testing concentration for three of the four cell lines (16 μM for H460 as the only measurable value). Compound 12, the derivative with the longest alkyl chain (dodecyl) had the lowest IC50 values of < 1 μM for three of the four cell lines tested. For the bis-cationic systems presented here, lipophilicity would be increased as the alkyl chain is elongated. These results are consistent with SAR studies performed previously that show lipophilicity of imidazolium salts is essential for anti-cancer activity against NSCLC.4,5,28,29
Table 1.
Table of IC50 values for 1-7, 10, 12, cisplatin, and IS-12. ND = not determined.
| Compound | IC50 Value (μM) | |||
|---|---|---|---|---|
| NCI-H460 | NCI-H1975 | HCC827 | A549 | |
| 1 | 16 | > 30 | > 30 | > 30 |
| 2 | 14 | > 30 | > 30 | > 30 |
| 3 | 20 | > 30 | > 30 | 22 |
| 4 | 4 | 11 | 13 | 4 |
| 5 | 2 | 12 | > 30 | 4 |
| 6 | 2 | 6 | 12 | < 1 |
| 7 | 3 | 3 | 10 | 2 |
| 10 | < 1 | 2 | 7 | < 1 |
| 12 | < 1 | < 1 | 4 | < 1 |
| Cisplatin | 6 | 3 | 9 | 5 |
| IS-12 | 4 | 6 | 9 | ND |
3.3. NCI-60 human tumor cell line screen
The National Cancer Institute’s (NCI) Developmental Therapeutics Program (DTP) tested 1–7, 10, and 12 in their NCI-60 human tumor cell line screen one-dose assay. There are nine NSCLC lung cancer cell lines included in the 60-human tumor cell line screen and only results from these cell lines are discussed. Full experimental details can be found on the DTP’s website (https://dtp.cancer.gov/discovery_development/nci-https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm60/methodology.htm). Briefly, cells are seeded at a concentration depending on their doubling rate and incubated for 24 hours. The experimental drug is then exposed to the cells at one dose (10 μM) for 48 hours at which time growth percentage values are determined. Results are given as a single value, the growth percentage, relative to the initial cell population. A positive growth percentage value means there were more cells present at the end of the experiment than at the beginning. A negative growth percentage values means the compound was lethal at the concentration tested and less cell were present at the end of the experiment than at the beginning.
Results from the NCI-60 human tumor cell line screen one-dose assay against the nine NSCLC cell lines tested were consistent with IC50 values discussed above from the MTT assay performed in our lab (Table 2). The highest growth percentage values were from cells exposed to 1 and 2 with an average growth percentage of 79.55% and 80.88%, respectively. As the alkyl chain lengthens from 2 to 12, the average growth percentage values lower, meaning the compound was more effective at inhibiting the growth or was lethal against the NSCLC cell lines tested as in the case of 12. Compound 12 was lethal against all NSCLC lung cancer lines tested and was the most effective compound tested with an average growth rate of −60.87%. The anti-cancer activity of this series of bis-imidazolium salts is directly related to the alkyl chain length when considering the trend the average growth rate follows. The SAR established from the NCI-60 human tumor cell line screen further enhances the existing SARs described with imidazolium salts substituted at every position on the imidazole and benzimidazole ring with functional groups of varying hydrophilicities and lipophilicities.4,5,28,29
Table 2.
Growth percentage values of compounds 1-7, 10, and 12 from NCI-60 human tumor cell line screen one-dose assay.
| Growth % | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | A549/ATCC | EKVX | HOP-62 | HOP92 | NCI-H226 | NCI-H23 | NCI-H322M | NCI-H460 | NCI-H522 | Average |
| 1 | 70.78 | 89.40 | 84.22 | 66.99 | 92.61 | 88.32 | 87.60 | 85.07 | 50.99 | 79.55 |
| 2 | 64.07 | 91.44 | 85.95 | 79.18 | 82.65 | 88.91 | 86.40 | 85.63 | 63.70 | 80.88 |
| 3 | 62.32 | 82.70 | 67.18 | 67.47 | 81.89 | 77.85 | 84.40 | 75.28 | 56.58 | 72.85 |
| 4 | 49.31 | 58.07 | 54.76 | 68.41 | 82.03 | 44.04 | 90.05 | 38.24 | 43.40 | 58.70 |
| 5 | 41.68 | 47.15 | 57.01 | 66.04 | 72.59 | 45.32 | 86.17 | 30.48 | 35.74 | 53.91 |
| 6 | 38.64 | 37.30 | 55.83 | 59.93 | 69.59 | 30.46 | 76.24 | 24.59 | 33.07 | 47.29 |
| 7 | 26.68 | 27.92 | 32.56 | n/a | 59.45 | 14.44 | 58.62 | 14.63 | 5.87 | 30.02 |
| 10 | 18.55 | 13.71 | −4.86 | −23.56 | 39.46 | 5.67 | 34.54 | 11.00 | −72.29 | 2.47 |
| 12 | −74.19 | −62.61 | −58.01 | −68.19 | −42.06 | −80.31 | −18.39 | −69.88 | −74.23 | −60.87 |
Compounds 10 and 12 were the only compounds that fulfilled the NCI’s DTP requirement and were graduated to the five-dose assay. The experimental procedures are the same for the five-dose assay as for the one-dose assay except the cells are exposed to the experimental drugs at five concentrations instead of one. Compound 10 was exposed to cells at 100 μM, 10 μM, 1 μM, 100 nM, and 10 nM; whereas, 12 was exposed to cells at 25 μM, 2.5 μM, 250 nM, 25 nM, and 2.5 nM. Results are given as three different values for the five-dose assay: (1) GI50 or growth inhibition of 50% of cells relative to control cells, (2) TGI or total growth inhibition, and (3) LC50 or lethal concentration of 50% of cells relative to control cells.
Results from the five-dose assay for 10 and 12 are summarized in Table 3 and growth percentage plots of the NSCLC cell lines exposed to 10 and 12 are shown in Figure 9 and Figure 10. Compound 10 had GI50 values in the range of 0.396 μM to 3.55 μM, TGI values ranging from 1.94 μM to 18.4 μM and LC50 values ranging from 15.2 μM to 58.4 μM. A value was not recorded with the NCI-H226 cell line because the LC50 concentration was higher than the testing conditions. A value was also not recorded for the A549/ATCC cell line. Compound 12 had GI50 values ranging from 0.470 μM to 0.984 μM (all in the nanomolar range), TGI values from 1.25 μM to 5.52 μM, and LC50 values ranging from 7.29 μM to 14.6 μM. An LC50 value was not recorded for the NCI-H226 cell line because the LC50 concentration was above the testing threshold. On average, 12 displayed higher anticancer activity than 10. All the GI50 values were in the nanomolar range for 12 again confirming the high anti-cancer activity of this derivative against NSCLC.
Table 3.
Results from the NCI-60 human tumor cell line screen five-dose assay for 10 and 12. Results are given as GI50, TGI, and LC50 values.
| Cell Line | Concentration (μM) | |||||
|---|---|---|---|---|---|---|
| 10 | 12 | |||||
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | |
| A549/ATCC | n/a | 13.3 | 50.3 | 0.744 | 3.57 | 14.2 |
| EKVX | 0.442 | 9.07 | 58.4 | 0.893 | 4.20 | 20.8 |
| HOP-62 | 1.09 | 7.24 | 44.5 | 0.815 | 2.65 | 12.2 |
| HOP-92 | 0.396 | 2.96 | 15.2 | 0.500 | 1.40 | 7.29 |
| NCI-H226 | 0.918 | 13.6 | > 100 | 0.984 | 5.52 | > 25 |
| NCI-H23 | 0.445 | 3.35 | 39.9 | 0.722 | 2.40 | 11.2 |
| NCI-H322M | 3.55 | 18.4 | 48.1 | 0.859 | 3.90 | 10.4 |
| NCI-H460 | 0.376 | 10.1 | 45.9 | 0.814 | 2.96 | 14.6 |
| NCI-H522 | 0.436 | 1.94 | 5.95 | 0.470 | 1.25 | 9.89 |
Figure 9.

Growth percentage plots from the NCI-60 human tumor cell line screen five-dose assay of compound 10 against all NSCLC cell lines tested by the DTP.
Figure 10.

Growth percentage plots from the NCI-60 human tumor cell line screen five-dose assay of compound 12 against all NSCLC cell lines tested by the DTP.
3.4. Preliminary in vivo toxicity study
A preliminary in vivo toxicity study using C57BL/6 mice was performed with compound 10. Compound 10 was chosen because of the high anti-cancer properties exhibited in the various in vitro studies described above and it could be solubilized by 2-hydroxypropyl-β-cyclodextrin (2-HPβCD), a chemical excipient that is FDA approved and used in several drug formulations.30 Eight week old C57BL/6 mice were allowed to acclimate in their cages for five-days prior to any injections. Animals were housed according to the experimental group (n = 3, with 3 animals per cage). The behavior and weight of each animal was closely monitored for the duration of the experiment. Experimental animals were injected on days zero, seven, fourteen, and twenty-nine (1 hour before being sacrificed) with 100 μL of a 20 mg/kg dose (assuming an average weight of 20 g per mouse) of 10 dissolved in a 20% (w/v) solution of 2-HPβCD by intraperitoneal (IP) injection. Vehicle control animals were injected with 100 μL of a 20% (w/v) solution of 2-HPβCD by IP injection on days zero, seven, fourteen, twenty-one, twenty-five, and twenty-nine. The weight and behavior of all animals were closely monitored each day for the duration of the experiment. The average weight gain for the mice is displayed in Figure 11. Error bars were removed for clarity. A weight gain chart displaying the error bars can be seen in SI 11.
Figure 11.

Average weight gain percent chart for mice treated with 10 and vehicle control. A chart displaying error bars can be found in the supplemental information.
All mice in the vehicle control group survived and gained weight over the course of the entire experiment. Mice treated with 10 had an average weight loss of 10% after the initial injection but significantly recovered by day six to an average weight loss of 4% and all animals survived for the entire duration of the experiment. There was not a drastic weight loss after the second and third injections and the mice continued to gain weight until the end of the experiment. The reason why drastic weight loss did not occur after the second and third injections is unclear. Overall, 10 was well tolerated at the treated dose regimen and provides a starting point for future toxicity studies and lung cancer xenograft models.
4. Conclusions and future outlook
A series of bis-imidazolium salts was synthesized and assessed for anti-cancer properties against NSCLC by several methods to create a SAR. Each structure differed by the length of the alkyl chain that connected the two imidazolium cores. The anti-cancer properties were determined by the MTT assay in our lab and through the one-dose and five-dose NCI-60 human tumor cell line screen assays. Results from all in vitro experiments were consistent and suggested that the alkyl chain length is directly related to anti-cancer activity considering 1 had the lowest anti-cancer activity, whereas 12 had the highest. Based on previously published work about imidazolium salts, we conclude that longer alkyl chains correspond to an increase in lipophilicity and thus an increase in anti-cancer activity. This SAR will guide the synthesis of future derivatives to optimize the next generation of compounds. Compound 10, with a decyl linker, also displayed extremely high activity and was used for a preliminary in vivo toxicity study with C57BL/6 mice. All animals survived the study and gained weight over the course of the experiment. This study gave valuable information about compound 10 and will be used to guide future experiments including a more in-depth toxicity study and lung xenograft models, as well as the creation of further bis-imidazolium salts with longer alkyl chains.
7. Experimental Section
7.1. General Procedures
All reactions were conducted under aerobic conditions except where indicated. 2-(Bromomethyl)naphthalene was purchased from Waterstone Technologies. 1,2-Dibromoethane was purchased from Baker. 1,3-Dibromopropane, 1,4-dibromobutane, 1,6-dibromohexane, and 1,8-dibromooctane were purchased from Acros Organics. 1,5-Dibromopentane was purchased from Alfa Aesar. 1,7-Dibromoheptane and 1,10-dibromodecane were purchased from TCI. 1,11-Dibromoundecane was purchased from Aldrich. 1,12-Dibromododecane was purchased from Avocado. Imidazole was purchased from Acros Organics. Naphthylmethyl imidazole was synthesized according to literature procedures.11 Di(imidazol-1-yl)methane was synthesized according to the literature and recrystallized from chloroform.31 All solvents were purchased from Fisher Scientific. All reagents were used as received without further purification. 1H and 13C NMR spectra were obtained on a Varian 500 MHz instrument with all spectra referenced to residual deuterated solvent for compounds 1–8, 10, and 12 (DMSO-d6: 1H NMR: 2.50 ppm, 13C NMR: 39.5 ppm).
The human NSCLC cell lines NCI-H1975 and HCC827 were generously provided by Dr. Lindner from the Cleveland Clinic. The human NSCLC cell lines NCI-H460 and A549 were purchased from ATCC (Manassas, VA, USA). All cell lines were grown at 37 °C with 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum and passed every 2–3 days.
7.2. Single crystal X-ray crystallography procedures
Crystals of the compounds were coated in paratone oil, mounted on a CryoLoop and placed on a goniometer under a stream of nitrogen. Crystal structure data sets were collected on either a Bruker SMART APEX I CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) or a Bruker Kappa APEX II Duo CCD system equipped with a Mo ImuS source and a Cu ImuS micro-focus source equipped with QUAZAR optics (λ = 1.54178 Å). The unit cells were determined by using reflections from three different orientations. Data sets were collected using SMART or APEX II software packages. All data sets were processed using the APEX II software suite.32,33 The data sets were integrated using SAINT.34 An empirical absorption correction and other corrections were applied to the data sets using multi-scan SADABS.35 Structure solution, refinement, and modelling were accomplished by using the Bruker SHELXTL package.36 The structures were determined by full-matrix least-squares refinement of F2 and the selection of the appropriate atoms from the generated difference map. Hydrogen atom positions were calculated and Uiso(H) values were fixed according to a riding model.
CCDC #s 2015314–2015318 can be found on the Cambridge Crystallographic Data Center website. This material is available free of charge via the Internet at http://www.ccdc.cam.ac.uk/structures.
Compound 2-PF6 was obtained by adding ammonium hexafluorophosphate to a solution of 2 dissolved in water. Once converted to the hexafluorophosphate salt, 2-PF6 was insoluble in water and precipitated from solution to form a white powder, which was collected by filtration. A single crystal of 2-PF6 suitable for X-ray crystallography was obtained by slow evaporation of a solution of the white solid in a mixture of acetonitrile, chloroform, and 2-propanol (Figure 5). Single crystals of 3•H2O and 4•2(H2O) suitable for X-ray crystallography were grown from concentrated solutions of the compound dissolved in ethyl acetate and tetrahydrofuran (3), or methanol (4) (Figure 6 and Figure 7, respectively). Compound 3 co-crystallized with one water molecule and 4 co-crystallized with two water molecules. Compound 7 was converted to the hexafluorophosphate salt by the same route as 2. A single crystal of 7-PF6 suitable for X-ray analysis was obtained by slow evaporation of 7-PF6 in a concentrated solution of chloroform (Figure 8). A single crystal of 8 was obtained by slow evaporation of 8 dissolved in a mixture of acetonitrile, water, and diethyl ether (Figure 9).
Figure 5.

Thermal ellipsoid plot of 3•H2O with thermal ellipsoids drawn to the 50% probability level. Hydrogen atoms, carbon labels, water solvent molecule, and bromide anions were removed for clarity.
Figure 6.

Thermal ellipsoid plot of 4•2(H2O) with thermal ellipsoids drawn to the 50% probability level. Hydrogen atoms, carbon labels, water solvent molecules, and bromide anions were removed for clarity.
Figure 7.

Thermal ellipsoid plot of 7-PF6 with thermal ellipsoids drawn to the 50% probability level. Hydrogen atoms, carbon labels, and the PF6- anions were removed for clarity.
7.3. MTT assay
Cells were grown to confluence and plated in 96-well plates at 5,000–7,000 cells per well, depending on the cell line. Cells were incubated for 24 h prior to adding the compounds. All compounds were dissolved in a 1% DMSO solution and diluted in fresh medium to the desired concentrations of 1, 4, 16, and 32 μM. Compounds were added (6 replicates each) and cells were incubated for 72 h at which time the MTT assay protocol was followed. MTT reagent (10 μL) was added to each well and cells were incubated for 3–4 h, again depending on the cell line. Growth medium was removed by aspiration and DMSO (100 μL) was added to each well. Plates were incubated for 15 min. The optical density was read at 540 nm on a Biotek Epoch plate reader.
7.4. Preliminary in vivo toxicity study
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Akron. Eight-week-old male C57BL/6 mice were obtained from Charles River laboratories. Animals were housed in a 12 h light/dark cycle, and food and water were provided ad libitum (n = 3 animals per cage). Prior to the toxicity testing, animals were allowed to acclimate for five days. Vehicle control mice received IP injections on days 0, 7, 14, 21, 25, and 29 of 100 μL of a 20% 2-HPβCD sterile PBS solution. Experimental mice received 100 μL of a 4 mg/mL 20% 2-HPβCD sterile PBS solution (0.4 mg/100 μL, or ~ 20 mg/kg assuming an average mouse weight of 20 g) of 10 by IP injection on days 0, 7, 14, and 29 (1 hour before sacrificing). Animals were closely monitored and weighed on a daily basis. All animals were sacrificed on day 29.
7.5. General Synthesis
Synthesis of 1-(naphthalen-2-ylmethyl)-3-((3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)methyl)-imidazolium bromide (1) Di(1H-imidazol-1-yl)methane (0.12 g, 0.8 mmol) was dissolved in acetonitrile (7 mL) and 2-(bromomethyl)naphthalene (0.40 g, 1.8 mmol) was added. The reaction was heated and stirred at 70 °C overnight. The volatile components were removed under reduced pressure, and the white solid was washed with acetone (25 mL) and chloroform (15 mL), subsequently. The product was recrystallized in a solution of water and ethanol (1:6). After filtration, the crystals were ground to a fine powder by mortar and pestle and residual volatile components were removed under reduced pressure to purify the white solid, 1 (0.42 g, 87 % yield). Phase Transition, 283 – 285°C. HRMS (ESI2+) calcd for C29H26N42+ [M-2(Br)] of m/z = 215.1073, calcd for C26H24N4+ [M-2(Br)H] of m/z = 429.2074, found m/z = 429.2140. 1H NMR (500 MHz, DMSO-d6) δ = 9.70 (2H, s, Ar), 8.13–7.93 (12H, m, Ar), 7.59–7.57 (6H, m, Ar), 6.72 (2H, s, CH2), 5.68 (4H, s, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 137.8 (NCN), 132.8 (Ar), 132.6 (Ar), 131.5 (Ar), 128.7 (Ar), 128.0 (Ar), 127.8 (Ar), 127.6 (Ar), 126.8 (Ar), 126.7 (Ar), 125.9 (Ar), 123.3 (Ar), 122.5 (Ar), 58.3 (CH2), 52.5 (CH2).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(2-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)ethyl)-imidazolium bromide (2) 1,2-Dibromoethane (56 μL, 0.7 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.30 g, 1.4 mmol) were heated in acetonitrile (5 mL) overnight. The volatile components were removed under reduced pressure, and the white solid was triturated with acetone and filtered. Residual volatile components were removed under reduced pressure to yield a white solid, 2 (0.06 g, 16 % yield). Phase Transition, 285°C. HRMS (ESI2+) calcd for C30H28N42+ [M-2(Br)] of m/z = 222.1152, calcd for C30H27N4+ [M-2(Br)H] of m/z = 443.2230, found m/z = 443.2311.1H NMR (500 MHz, DMSO-d6) δ = 9.32 (2H, s, Ar), 7.98–7.91 (8H, m, Ar), 7.86 (2H, dd, Ar), 7.73 (2H, dd, Ar), 7.58–7.57 (4H, m, Ar), 7.48 (1H, d, Ar), 7.46 (1H, d, Ar), 5.57 (4H, s, CH2), 4.75 (4H, t, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.8 (NCN), 132.7 (Ar), 132.6 (Ar), 131.8 (Ar), 128.7 (Ar), 127.8 (Ar), 127.63 (Ar), 127.60 (Ar), 126.8 (Ar), 126.7 (Ar), 125.6 (Ar), 122.94 (Ar), 122.86 (Ar), 52.2 (CH2), 48.5 (CH2).
Crystal data for 2-PF6: C30H28F12N4P2, M = 734.50, monoclinic, a = 35.129(2) Å, b = 6.9070(4) Å, c = 12.5651(8) Å, β = 101.401(3)°, V = 2988.6(3) Å3, T = 100(2) K, space group C2/c, Z = 4, 10845 reflections measured, 3020 independent reflections (Rint = 0.0360). The final R1 values were 0.0448 (I > 2σ(I)). The final wR(F2) values were 0.0718 (I > 2σ(I)). The final R1 values were 0.0707 (all data). The final wR(F2) values were 0.0754 (all data). A single crystal of 2-PF6 was obtained by slow evaporation of a concentrated solution of 2-PF6 dissolved in acetonitrile, chloroform, and 2-propanol.
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(3-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)propyl)-imidazolium bromide (3) 1,3-Dibromopropane (66 μL, 0.7 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.30 g, 1.4 mmol) were heated in acetonitrile (5 mL) overnight. The clear solution was cooled (−20 °C) to induce precipitation. The white power was filtered and washed with cold (−20 °C) acetonitrile (25 mL). A fine, white powder, 3 was collected (0.34 g, 84 % yield). Phase Transition, 145 – 147°C. HRMS (ESI2+) calcd for C31H30N42+ [M-2(Br)] of m/z = 229.1230, calcd for C31H29N4+ [M-2(Br)H] of m/z = 457.2387, found m/z = 457.2438. 1H NMR (500 MHz, DMSO-d6) δ = 9.56 (2H, s, Ar), 8.01–7.89 (12H, m, Ar), 7.59–7.55 (6H, m, Ar), 5.64 (4H, s, CH2), 4.32 (4H, t, CH2), 2.46 (2H, p, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 137.0 (NCN), 133.2 (Ar), 133.17 (Ar), 132.5 (Ar), 129.2 (Ar), 128.3 (Ar), 128.2 (Ar), 128.1 (Ar), 127.2 (Ar), 127.17 (Ar), 126.3 (Ar), 123.2 (Ar), 123.15 (Ar), 52.6 (CH2), 46.5 (CH2), 29.8 (CH2).
Crystal data for 3•H2O: C31H32N4Br2O, M = 636.42, monoclinic, a = 21.4515(7) Å, b = 12.5776(4) Å, c = 10.6265(3) Å, β = 91.518(2)°, V = 2866.11(15) Å3, T = 100(2) K, space group P21/c, Z = 4, 51420 reflections measured, 5812 independent reflections (Rint = 0.0433). The final R1 values were 0.0382 (I > 2σ(I)). The final wR(F2) values were 0.0605 (I > 2σ(I)). The final R1 values were 0.0569 (all data). The final wR(F2) values were 0.0613 (all data). A single crystal of 3•H2O was obtained by slow evaporation of a concentrated solution of 3 dissolved in ethyl acetate and tetrahydrofuran.
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(4-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)butyl)-imidazolium bromide (4) 1,4-Dibromobutane (78 μL, 0.7 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.30 g, 1.4 mmol) were heated in acetonitrile (5 mL) overnight. A white precipitate formed, and the mixture was cooled (−20 °C) to induce further precipitation. The white power was filtered and washed with cold (−20 °C) acetonitrile (25 mL). A fine, white powder, 4 was collected (0.32 g, 76 % yield). Phase Transition, 223 – 225°C. HRMS (ESI2+) calcd for C32H32N42+ [M-2(Br)] of m/z = 236.1308, calcd for C32H31N4+ [M-2(Br)H] of m/z = 471.2543, found m/z = 471.2531. 1H NMR (500 MHz, DMSO-d6) δ = 9.37 (2H, s, Ar), 7.98–7.83 (12H, m, Ar), 7.58–7.52 (6H, m, Ar), 5.60 (4H, s, CH2), 4.23 (4H, t, CH2), 1.81 (4H, t, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.8 (NCN), 133.2 (Ar), 133.17 (Ar), 132.6 (Ar), 129.3 (Ar), 128.3 (Ar), 128.2 (Ar), 128.1 (Ar), 127.3 (Ar), 127.2 (Ar), 126.2 (Ar), 123.2 (Ar), 123.22 (Ar), 52.7 (CH2), 48.7 (CH2), 26.6 (CH2).
Crystal data for 4•2(H2O): C32H36N4O2Br2, M = 668.47, monoclinic, a = 11.8248(3) Å, b = 11.7188(3) Å, c = 10.6450(3) Å, β = 99.8570(10)°, V = 1453.33(7) Å3, T = 100(2) K, space group P21/c, Z = 2, 15227 reflections measured, 2951 independent reflections (Rint = 0.0324). The final R1 values were 0.0306 (I > 2σ(I)). The final wR(F2) values were 0.0819 (I > 2σ(I)). The final R1 values were 0.0375 (all data). The final wR(F2) values were 0.0859 (all data). A single crystal of 4•2(H2O) was obtained by slow evaporation of a concentrated solution of 4 dissolved in methanol.
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(5-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)pentyl)-imidazolium bromide (5) 1,5-Dibromopentane (178 μL, 1.3 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.60 g, 2.9 mmol) were heated in acetonitrile (4 mL) overnight. The volatile components were removed under reduced pressure, and the white solid was triturated with cold (−20 °C) acetone (25 mL). The white powder was recrystallized in a solution of water and ethanol (1:6). The crystals were filtered and washed with acetone (15 mL). The volatile components were removed under reduced pressure to yield a white powder, 5 (0.44 g, 52 % yield). Phase Transition, 226 – 228°C. HRMS (ESI2+) calcd for C33H34N42+ [M-2(Br)] of m/z = 243.1386, calcd for C33H33N4+ [M-2(Br)H] of m/z = 485.2700, found m/z = 485.2763.1H NMR (500 MHz, DMSO-d6) δ = 9.43 (2H, s, Ar), 8.00–7.87 (12H, m, Ar), 7.58–7.54 (6H, m, Ar), 5.61 (4H, s, CH2), 4.20 (4H, t, CH2), 1.85 (4H, tt, CH2), 1.24 (2H, p, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.2 (NCN), 132.7 (Ar), 132.65 (Ar), 132.1 (Ar), 128.8 (Ar), 127.8 (Ar), 127.6 (Ar), 127.58 (Ar), 126.7 (Ar), 126.71 (Ar), 125.7 (Ar), 122.8 (Ar), 122.6 (Ar), 52.1 (CH2), 48.6 (CH2), 28.5 (CH2), 22.1 (CH2).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(6-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)hexyl)-imidazolium bromide (6) 1,6-Dibromohexane (135 μL, 0.9 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.40 g, 1.9 mmol) were heated in acetonitrile (7 mL) overnight. A white precipitate formed, and the reaction mixture was cooled (−20 °C) to induce further precipitation. The white precipitate was filtered and washed with cold (−20 °C) acetonitrile (30 mL) to afford 6 (0.44 g, 77 % yield). Phase Transition, 233 – 234°C. HRMS (ESI2+) calcd for C34H36N42+ [M-2(Br)] of m/z = 250.1465, calcd for C34H35N4+ [M-2(Br)H] of m/z = 499.2856, found m/z = 499.2948. 1H NMR (500 MHz, DMSO-d6) δ = 9.57 (2H, s, Ar), 8.02–7.89 (12H, m, Ar), 7.59–7.54 (6H, m, Ar), 5.66 (4H, s, CH2), 4.20 (4H, t, CH2), 1.81 (4H, tt, CH2), 1.27 (4H, t, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.7 (NCN), 133.2 (Ar), 132.16 (Ar), 132.8 (Ar), 129.2 (Ar), 128.3 (Ar), 128.1 (Ar), 128.11 (Ar), 127.2 (Ar), 127.16 (Ar), 126.2 (Ar), 123.3 (Ar), 123.1 (Ar), 52.5 (CH2), 49.3 (CH2), 29.5 (CH2), 25.3 (CH2).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(7-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)heptyl)-imidazolium bromide (7) 1,7-Dibromoheptane (149 μL, 0.9 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.40 g, 1.9 mmol) were heated in acetonitrile (7 mL) overnight. The clear solution was cooled (−20 °C) and a white precipitate formed which was washed with cold (−20 °C) acetonitrile (25 mL). The white powder was dried via aspirator filtration to afford 7 (0.38 g, 64 % yield). Phase Transition, 198 – 200°C. HRMS (ESI2+) calcd for C35H38N42+ [M-2(Br)] of m/z = 257.1543, calcd for C35H37N4+ [M-2(Br)H] of m/z = 513.3013, found m/z = 513.3084. 1H NMR (500 MHz, DMSO-d6) δ = 9.55 (2H, s, Ar), 8.00–7.88 (12H, m, Ar), 7.58–7.54 (6H, m, Ar), 5.65 (4H, s, CH2), 4.19 (4H, t, CH2), 1.79 (4H, tt, CH2), 1.28 (2H, tt, CH2), 1.22 (2H, p, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.2 (NCN), 132.7 (Ar), 132.6 (Ar), 132.2 (Ar), 128.7 (Ar), 127.8 (Ar), 127.6 (Ar), 127.56 (Ar), 126.7 (Ar), 126.7 (Ar), 125.7 (Ar), 122.7 (Ar), 122.6 (Ar), 52.0 (CH2), 48.8 (CH2), 29.0 (CH2), 27.5 (CH2), 25.2 (CH2).
Crystal data for 7-PF6: C35H38N4F12P2, M = 804.63, monoclinic, a = 16.34(2) Å, b = 10.856(16) Å, c = 19.75(3) Å, β = 93.373(18)°, V = 3499(9) Å3, T = 100(2) K, space group P21/c, Z = 4, 27020 reflections measured, 7061 independent reflections (Rint = 0.0542). The final R1 values were 0.0676 (I > 2σ(I)). The final wR(F2) values were 0.1748 (I > 2σ(I)). The final R1 values were 0.0850 (all data). The final wR(F2) values were 0.1914 (all data). A single crystal of 7-PF6 was obtained by slow evaporation of a concentrated solution of 7 dissolved in acetonitrile.
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(8-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)octyl)-imidazolium bromide (8) 1,8-Dibromooctane (161 μL, 0.9 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.40 g, 1.9 mmol) were heated in acetonitrile (3 mL) overnight. The volatile components were removed via rotary evaporation under reduced pressure. The off-white powder was washed and filtered with cold (−20 °C) acetone (25 mL). The powder was recrystallized in an ethanol and water solution (5:1). The crystals were washed and filtered with cold (−20 °C) acetone (15 mL). The crystals were crushed to an off-white powder which was dried under reduced pressure to afford 8 (0.50 g, 83 % yield). 1H NMR (500 MHz, DMSO-d6) δ = 9.47 (2H, s, Ar), 7.99–7.86 (12H, m, Ar), 7.57–7.54 (6H, m, Ar), 5.63 (4H, s, CH2), 4.18 (4H, t, CH2), 1.78 (4H, tt, CH2), 1.24–1.20 (8H, m, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.7 (NCN), 133.2 (Ar), 132.7 (Ar), 129.3 (Ar), 128.3 (Ar), 128.1 (Ar), 128.0 (Ar), 127.2 (Ar), 127.2 (Ar), 126.2 (Ar), 123.3 (Ar), 122.7 (Ar), 123.1 (Ar), 52.6 (CH2), 49.4 (CH2), 29.7 (CH2), 28.6 (CH2), 25.9 (CH2).
Crystal data for 8: C36H40N4Br2, M = 688.54, monoclinic, a = 10.9871(9) Å, b = 10.3592(8) Å, c = 15.1017(13) Å, β = 98.618(4)°, V = 1699.4(2) Å3, T = 100(2) K, space group P21/c, Z = 2, 14597 reflections measured, 3440 independent reflections (Rint = 0.0465). The final R1 values were 0.0698 (I > 2σ(I)). The final wR(F2) values were 0.2154 (I > 2σ(I)). The final R1 values were 0.0874 (all data). The final wR(F2) values were 0.2369 (all data). A single crystal of 8 was obtained by slow evaporation of a concentrated solution of 8 dissolved in acetonitrile, water, and diethylether.
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(9-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)nonyl)-imidazolium bromide (9) 1,9-Dibromoundecane (293 μL, 1.4 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.600 g, 2.9 mmol) were heated in acetonitrile (4 mL) overnight. The volatile components from the reaction were removed under reduced pressure. The resulting viscous, white oil was dissolved in deionized water (300 mL) and washed twice with diethyl ether (100 mL). The water layer was collected and the water was removed under reduced pressure. The viscous, transparent, and tan oil was triturated with methylene chloride (100 mL) which induced precipitation of a tan solid. The mixture was filtered in a glove bag with a nitrogen environment. The tan solid was collected and stored at ambient conditions in open atmosphere to afford what was presumed to be 9 (0.522 g, 53 % yield).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(10-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)decyl)-imidazolium bromide (10) 1,10-Dibromodecane (0.26 g, 0.9 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.40 g, 1.9 mmol) were heated in acetonitrile (3 mL) overnight. A white precipitate formed as the solution cooled to room temperature and was cooled (−20 °C) to induce further precipitation. The chilled mixture was filtered and the white solid was washed with acetone (25 mL). The white solid was subsequently washed with diethyl ether (25 mL) three times. The white solid was dried under reduced pressure to afford 10 (0.31 g, 49 % yield). Phase Transition, 66°C. HRMS (ESI2+) calcd for C38H44N42+ [M-2(Br)] of m/z = 278.1778, calcd for C38H43N4+ [M-2(Br)H] of m/z = 555.3482, found m/z = 555.3456.1H NMR (500 MHz, DMSO-d6) δ = 9.43 (2H, s, Ar), 7.99–7.85 (12H, m, Ar), 7.59–7.53 (6H, m, Ar), 5.62 (4H, s, CH2), 4.18 (4H, t, CH2), 1.78 (4H, tt, CH2), 1.22–1.20 (12H, m, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.7 (NCN), 133.2 (Ar), 133.17 (Ar), 132.2 (Ar), 132.7 (Ar), 129.3 (Ar), 128.3 (Ar), 128.1 (Ar), 127.2 (Ar), 127.20 (Ar), 126.1 (Ar), 123.3 (Ar), 123.2 (Ar), 52.6 (CH2), 49.5 (CH2), 29.7 (CH2), 29.2 (CH2), 28.8 (CH2), 26.0 (CH2).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(11-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)undecyl)-imidazolium bromide (11) 1,11-Dibromoundecane (340 μL, 1.4 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.601 g, 2.9 mmol) were heated in acetonitrile (4 mL) overnight. The volatile components from the reaction were removed under reduced pressure. The resulting viscous, white oil was dissolved in deionized water (300 mL) and extracted twice with diethyl ether (100 mL). The water layer was collected and the water was removed under reduced pressure. The viscous, clear oil was triturated with methylene chloride (100 mL) which induced precipitation of a white solid. The mixture was filtered in a glove bag with a nitrogen environment. The solid was taken out of the bag which caused it to phase transition to a clear oil again. The oil was dissolved in chloroform (40 mL) and evaporated at ambient conditions in open atmosphere. A white, crystalline solid, presumed to be 11 was collected (0.182 g, 18 % yield).
Synthesis of 1-(naphthalen-2-ylmethyl)-3-(12-(3-(naphthalen-2-ylmethyl)-imidazolium-1-yl)dodecyl)-imidazolium bromide (12) 1,12-Dibromododdecane (0.29 g, 0.9 mmol) and 1-(naphthalen-2-ylmethyl)-imidazole (0.40 g, 1.9 mmol) were heated in acetonitrile (3 mL) overnight. The volatile components were removed under reduced pressure and the resultant off-white solid was washed with acetone (25 mL). The solid became a highly-viscous gel on the filter paper. After a week of being undisturbed in a fume hood, the hardened, tan product was ground and collected to afford 12 (0.31 g, 48 % yield). Phase Transition, 137 – 138°C. HRMS (ESI2+) calcd for C40H48N42+ [M-2(Br)] of m/z = 292.1934, calcd for C40H47N4+ [M-2(Br)H] of m/z = 583.3795, found m/z = 583.3722. 1H NMR (500 MHz, DMSO-d6) δ = 9.47 (2H, s, Ar), 7.98–7.86 (12H, m, Ar), 7.58–7.54 (6H, m, Ar), 5.63 (4H, s, CH2), 4.19 (4H, t, CH2), 1.79 (4H, tt, CH2), 1.23–1.19 (12H, m, CH2). 13C NMR (125 MHz, DMSO-d6) δ = 136.2 (NCN), 132.6 (Ar), 132.2 (Ar), 128.7 (Ar), 127.8 (Ar), 127.6 (Ar), 127.5 (Ar), 126.69 (Ar), 126.66 (Ar), 125.6 (Ar), 122.7 (Ar), 122.6 (Ar), 52.0 (CH2), 48.9 (CH2), 29.2 (CH2), 28.8 (CH2), 28.76 (CH2), 28.3 (CH2), 25.5 (CH2).
Supplementary Material
5. Acknowledgements
The authors would like to thank to NCI’s DTP for performing the 60 human tumor cell line screen with their one-dose and five-dose assays. The authors also would like to thank The University of Akron and Ashland University for support during the preparation of this manuscript. This project has been funded by The University of Akron, the Akron Research Commercialization Corporation, the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (R01-DK082546), and the OMNOVA Foundation. We thank the National Science Foundation (NSF) for providing funds for the purchase of the NMR instruments (Nos. CHE-0341701 and DMR-0414599), mass spectrometers (CHE-0821313 and CHE-1012636) and X-ray diffractometers (CHE-0116041 and CHE-0840446) used in this work
Footnotes
Supplementary Data
The 1H and 13C NMR spectra for compounds 1-12 (excluding 9 and 11) as well as the HRMS for compounds 1-12 (excluding 8, 9, and 11) can be found in the supplementary data. In addition, the average weight gain chart for the in vivo toxicity study with errors bars can also be found with the supplementary data. Crystallographic information files (CIF) for CCDC #s 2015314-2015318 can be found on the Cambridge Crystallographic Data Center website. This material is available free of charge via the Internet at http://www.ccdc.cam.ac.uk/structures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- (1).American Cancer Society. Cancer Facts and Figures 2020. Atlanta: American Cancer Society; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ramalingam S; Sandler AB Oncol. Lung Cancer 2006, 11, 655–665. [DOI] [PubMed] [Google Scholar]
- (3).Dempke WCM; Suto T; Reck M. Lung Cancer 2010, 67, 257–274. [DOI] [PubMed] [Google Scholar]
- (4).Wright BD; Deblock MC; Wagers PO; Duah E; Robishaw NK; Shelton KL; Southerland MR; DeBord MA; Kersten KM; McDonald LJ; Stiel J. a.; Panzner MJ; Tessier CA; Paruchuri S; Youngs WJ Med. Chem. Res 2015, 24 (7), 2838–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shelton KL; DeBord MA; Wagers PO; Southerland MR; Taraboletti A; Robishaw NK; Jackson DP; Tosanovic R; Kofron WG; Tessier CA; Paruchuri S; Shriver LP; Panzner MJ; Youngs WJ Tetrahedron 2016, 72 (38), 5729–5743. [Google Scholar]
- (6).Xu X; Yu C; Chen W; Li Y; Yang L-J; Li Y; Zhang H-B; Yang X-D Org. Biomol. Chem 2015, 13, 1550–1557. [DOI] [PubMed] [Google Scholar]
- (7).Zeng X; Yang X; Zhang Y; Qing C; Zhang H. Bioorganic Med. Chem. Lett 2010, 20 (6), 1844–1847. [DOI] [PubMed] [Google Scholar]
- (8).Sun C-J; Chen W; Li Y; Liu L-X; Wang X-Q; Li L-J; Zhang H-B; Yang X-D RSC Adv. 2014, 4 (31), 16312. [Google Scholar]
- (9).Chen W; Deng X-Y; Li Y; Yang L-J; Wan W-C; Wang X-Q; Zhang H-B; Yang X-D Bioorg. Med. Chem. Lett 2013, 23, 4297–4302. [DOI] [PubMed] [Google Scholar]
- (10).Chen W; Yang X-D; Li Y; Yang L-J; Wang X-Q; Zhang G-L; Zhang H-B Org. Biomol. Chem 2011, 9, 4250–4255. [DOI] [PubMed] [Google Scholar]
- (11).Cui B; Zheng BL; He K; Zheng QY J. Nat. Prod 2003, 66 (8), 1101–1103. [DOI] [PubMed] [Google Scholar]
- (12).Liu L; Wang X; Yan J; Li Y; Sun C; Chen W; Zhou B; Zhang H; Yang X. Eur. J. Med. Chem 2013, 66, 423–437. [DOI] [PubMed] [Google Scholar]
- (13).Malhotra SV; Kumar V. Bioorganic Med. Chem. Lett 2010, 20 (2), 581–585. [DOI] [PubMed] [Google Scholar]
- (14).Riduan SN; Zhang Y. Chem. Soc. Rev 2013, 42 (23), 9055–9070. [DOI] [PubMed] [Google Scholar]
- (15).Xu X; Wang J; Yu C; Chen W; Li Y; Li Y; Zhang H; Yang X. Bioorg. Med. Chem. Lett 2014, 24, 4926–4930. [DOI] [PubMed] [Google Scholar]
- (16).Yang J-L; Ma Y-H; Li Y-H; Zhang Y-P; Tian H-C; Huang Y-C; Li Y; Chen W; Yang L-J ACS Omega 2019, 4 (23), 20381–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Huang M; Duan S; Ma X; Cai B; Wu D; Li Y; Li L; Zhang H; Yang X. Medchemcomm 2019, 10 (6), 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Deng G; Zhou B; Wang J; Chen Z; Gong L; Gong Y; Wu D; Li Y; Zhang H; Yang X. Eur. J. Med. Chem 2019, 168, 232–252. [DOI] [PubMed] [Google Scholar]
- (19).Wang X-Q; Ye P-T; Bai M-J; Miu W-H; Yang Z-X; Duan S-Y; Li T-T; Li Y; Yang X-D Bioorg. Med. Chem. Lett 2020, 30 (13), 127210. [DOI] [PubMed] [Google Scholar]
- (20).Wang X-Q; Chen X-B; Ye P-T; Yang Z-X; Bai M-J; Duan S-Y; Li Y; Yang X-D Bioorg. Med. Chem. Lett 2020, 30 (4), 126896. [DOI] [PubMed] [Google Scholar]
- (21).Gao Y; Vlahakis JZ; Szarek WA; Brockhausen I. Bioorg. Med. Chem 2013, 21, 1305–1311. [DOI] [PubMed] [Google Scholar]
- (22).Jurgens E; Buys KN; Schmidt A-T; Furfari SK; Cole ML; Moser M; Rominger F; Kunz D. New J. Chem 2016. [Google Scholar]
- (23).Pinto MF; Cardoso B de P; Barroso S; Martins AM; Royo B. Dalton Trans 2016, 45, 13541–13546. [DOI] [PubMed] [Google Scholar]
- (24).Jarikote DV; Li W; Jiang T; Eriksson LA; Murphy PV Bioorg. Med. Chem 2011, 19, 826–835. [DOI] [PubMed] [Google Scholar]
- (25).Kong D; Park EJ; Stephen AG; Calvani M; Cardellina JH; Monks A; Fisher RJ; Shoemaker RH; Melillo G. Cancer Res. 2005, 65 (19), 9047–9055. [DOI] [PubMed] [Google Scholar]
- (26).Kim YB; Kim YH; Park JY; Kim SK Bioorg. Med. Chem. Lett 2004, 14 (2), 541–544. [DOI] [PubMed] [Google Scholar]
- (27).Wagers PO; Tiemann KM; Shelton KL; Kofron WG; Panzner MJ; Wooley KL; Youngs WJ; Hunstad D. a. Antimicrob. Agents Chemother 2015, 59 (9), AAC.00881–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shelton KL; Debord MA; Wagers PO; Southerland MR; Williams TM; Robishaw NK; Shriver LP; Tessier CA; Panzner MJ; Youngs WJ; Williams TM; Robishaw NK; Shriver LP; Tessier CA; Panzner MJ; Youngs WJ Bioorg. Med. Chem 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Debord MA; Wagers PO; Crabtree SR; Tessier CA; Panzner MJ; Youngs WJ Bioorg. Med. Chem. Lett 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brewster ME; Loftsson T. Adv. Drug Deliv. Rev 2007, 59 (7), 645–666. [DOI] [PubMed] [Google Scholar]
- (31).Haque RA; Hasanudin N; Iqbal MA; Ahmad A; Hashim S; Majid AA; Ahamed MBK J. Coord. Chem 2013, 66 (18), 3211–3228. [Google Scholar]
- (32).Bruker. SMART (Version 5.625). Madison, Wisconsin, USA: Bruker AXS Inc; 2012. [Google Scholar]
- (33).Bruker. APEX II. Madison, Wisconsin, USA: Bruker AXS Inc; 2012. [Google Scholar]
- (34).Bruker. SAINT. Madison, Wisconsin, USA: Bruker AXS Inc; 2012. [Google Scholar]
- (35).Bruker. SADABS. Madison, Wisconsin, USA: Bruker AXS Inc; 2012. [Google Scholar]
- (36).Sheldrick GM Acta Crystallogr. Sect. C Struct. Chem 2015, 71 (Md), 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.