

Abstract
Virtually all aspects of cell biology are regulated by a ubiquitin code where distinct ubiquitin chain architectures guide the binding events and itineraries of modified substrates. Various combinations of E2 and E3 enzymes accomplish chain formation by forging isopeptide bonds between the C-terminus of their transiently-linked donor ubiquitin and a specific nucleophilic amino acid on the acceptor ubiquitin, yet it is unknown whether the fundamental feature of most acceptors - the lysine side-chain - affects catalysis. Here, use of synthetic ubiquitins with non-natural acceptor site replacements reveals that the aliphatic side-chain specifying reactive amine geometry is a determinant of the ubiquitin code, through unanticipated and complex reliance of many distinct ubiquitin carrying enzymes on a canonical acceptor lysine.
Introduction
Ubiquitin (UB) chains are a major post-translational modification controlling protein function in eukaryotic cells. Eight distinct chain-types result from linkage of UB’s C-terminus to an amino group acceptor (seven lysines and the N-terminus) on another UB. Different UB chains formulate a “UB code” read by cognate binding domains that control the fates of modified proteins1–4. Studies of endogenous and recombinant proteins have shed light on this code, showing that K48-linked chains often direct proteasomal degradation while K63-linked chains mediate diverse regulation through modulating multi-subunit complex assembly1–4. Structural studies have shown how specificity is determined by distinct spacing between hydrophobic patches displayed from the UB molecules linked in various chain types1–4. In some cases, the actual isopeptide linkages between UBs, and surrounding residues, also dictate recognition of specific UB chains.
Recently, chemical biology approaches have also elucidated principles governing important aspects of UB biology5. Indeed, synthetic UB chains with defined linkages and chemically unique properties have illuminated mechanisms underlying protein degradation by the proteasome6,7 and revealed the potential of hundreds of UB-binding domains to partner with their cognate chain types8.
Despite this progress in deciphering how the code is “read” by downstream machineries recognizing UB chains, the mechanisms underlying generation of specific UB chain linkages remain incompletely understood. Chains are forged by combinations of UB-conjugating enzymes (E2s) and UB ligases (E3s). In humans, various pairings amongst ≈30 E2s and ≈600 E3s mediate UB ligation to selected target proteins, and determine the generation of UB chains with specific linkages. Different E2s and E3s employ distinct enzymatic mechanisms to achieve polyubiquitylation9,10. Some E2 enzymes can generate chains themselves, whereby after enzymatic linkage of UB’s C-terminus to an E2 catalytic cysteine, UB is transferred from the resultant E2~UB intermediate (~ refers to thioester bond) to a lysine on an “acceptor UB”. The preferred UB acceptor lysine may be intrinsic to an E2, and/or may be influenced by an E2 partner protein11. In some cases, UB transfer from the E2 is stimulated by the hallmark “RING” domain in many E3s. An E2 may also transfer UB to an active site cysteine of some E3s, as in a “HECT” catalytic domain, from which the donor UB is linked to an acceptor UB to generate a chain.
Prior studies identified E2 or E3 residues critical for catalysis and presentating an acceptor UB to the active site, and roles of acceptor UB residues surrounding the targeted lysine11–17. However, whether features of a UB’s target lysine beyond its nucleophilic primary amino group - such as the distance between the primary amine and the UB polypeptide backbone - influence UB chain formation remains unknown. Within classes of UB carrying enzymes (e.g. E2 or HECT E3), catalytic domains adopt similar structures that have the capacity to catalyze covalent bond formation between the donor UB and assorted free amino acid acceptors - lysine, cysteine, serine, and threonine 9,10,18,19. Because substrates of the UB system are often degraded even after mutation of preferred lysines, one has the impression that targeting by some E2 and E3 enzymes is relatively lax. This would contrast from protein interaction domains or histone modifying enzymes that strictly depend on lysines for specific salt-bridge geometries or substrate targeting20,21.
To investigate if acceptor lysine side-chain features beyond the primary amino group influence UB chain formation, we employed a suite of synthetic UBs harboring replacements for K11, K48 or K63 with shorter or longer aliphatic side-chains, and tested their reactivities with a broad set of ubiquitylating enzymes. Our results demonstrate that the geometry between the polypeptide backbone and primary amine strongly influences chain formation for diverse polyubiquitylating enzymes. Thus, the lysine side-chain itself helps establish the UB code.
Results
Acceptor UB lysine geometry required by K63-specific E2
The simplest activity of an E2 involves UB transfer to a nucleophilic amino acid free in solution. For some E2s, such discharge onto an isolated amino acid acceptor (e.g. lysine, cysteine, threonine, etc.) correlates with preferred residue-type modified in the context of a protein target18,19. We examined reactivity of the well-characterized K63-linked UB chain forming enzyme, the human heterodimeric E2 UBE2N/UBE2V1 complex, which uniquely partners a canonical E2 subunit (UBE2N) with the dedicated catalytically-inactive E2-like UBE2V116,22,23. UBE2V1 guides K63 on an acceptor UB towards the thioester linkage between UBE2N’s active site cysteine and the donor UB’s C-terminus13,22. The rate of this K63-linked UB chain formation is accelerated by the RING domain of RNF4 E3 (hereafter referred to as RNF4). RNF4 stabilizes the active conformation of the donor UB thioester-bonded to the UBE2N active site24. Moreover, coupling with UBE2V1 and RNF4 stimulates intrinsic reactivity of the UBE2N~UB intermediate as monitored by UB discharge to free lysine23, albeit less efficiently than to an acceptor UB’s K63.
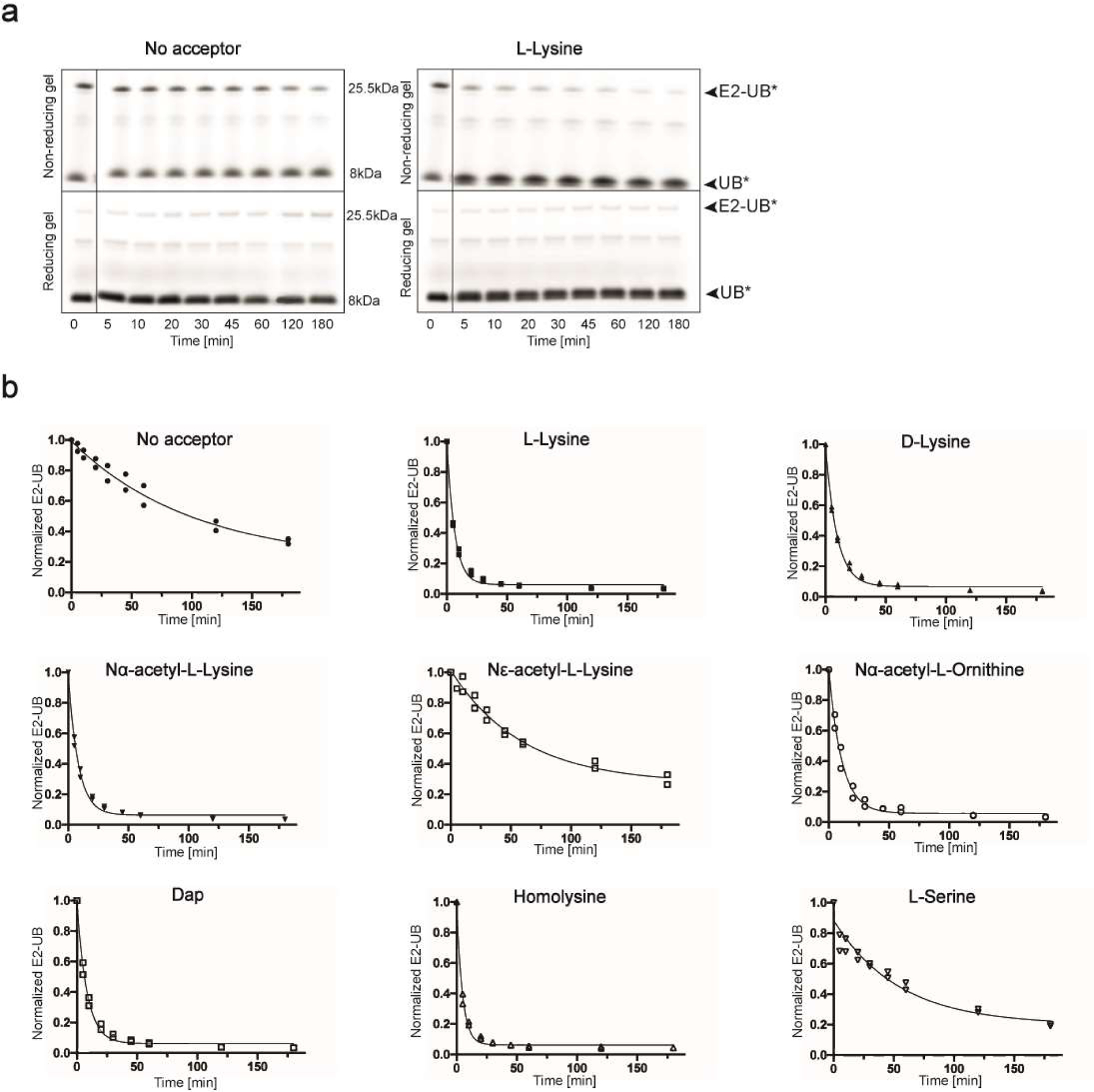
We examined transfer of the donor UB from RNF4-UBE2V1-activated UBE2N to various free amino acids using a pulse-chase assay (Fig. 1a). UBE2N was charged with fluorescent donor UB in the pulse reaction using E1 enzyme. After quenching this reaction, the resultant UBE2N~UB intermediate was incubated with RNF4, UBE2V1 and an amino acid. We initially tested L-lysine (four methylene units in the side-chain, referred to here as C4) and two controls: L-serine, not known to accept UB from RNF4-UBE2V1-UBE2N, and Nε-Acetyl-L-lysine with a blocked epsilon amino group. As expected, L-lysine had high reactivity compared to controls (Figure 1b). Reactivity of Nα-Acetyl-L-lysine, with a blocked alpha amino but available epsilon amino group, verified lysine’s Nε-amine as the preferred acceptor. With this established, we tested lysine analogs differing in side-chain length. “C1”, “C3” and “C5” analogs (L-2,3-diaminopropionic acid, Nα-Acetyl-L-ornithine, and L-homolysine, respectively) demonstrated robust reactivity (Fig. 1b, Extended Data Fig. 1a,b), indicating lack of absolute requirement for aliphatic chain length between the backbone and nucleophilic amino group of lysine analogs free in solution.
Figure 1 |. UBE2N~UB/UBE2V1/RNF4 RING E3 complex reacts preferentially with free amino acids harboring amine acceptors and various side-chain hydrocarbon linkers.
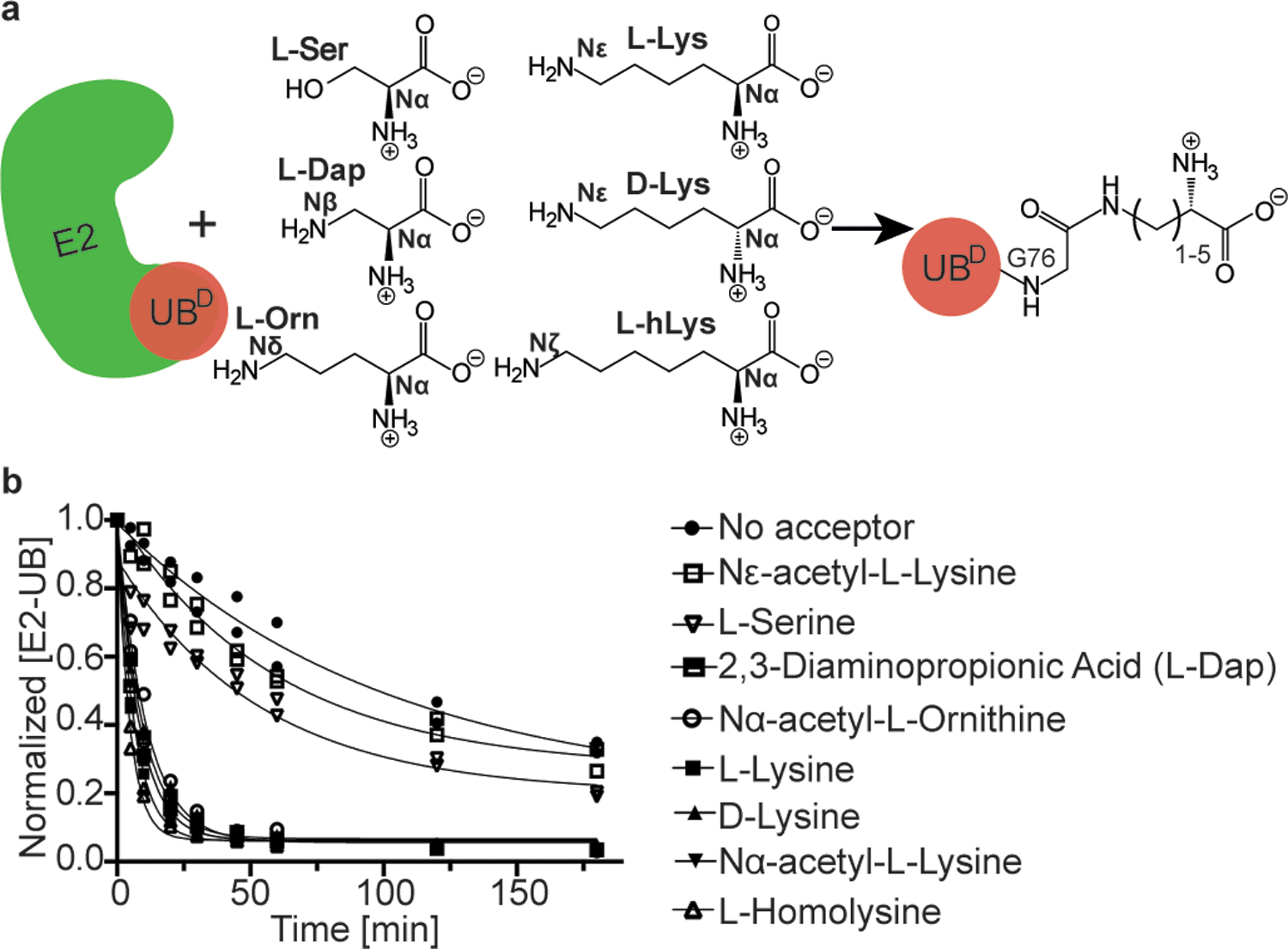
(a) Cartoon of experimental scheme, monitoring reactivity of E2~UB (D refers to the “donor” fluorescent UB to be transferred from E2) towards various free amino acids. (b) Time-course of fluorescent UB discharge from UBE2N~UB/UBE2V1/RNF4 RING E3 to the indicated amino acids, normalized to starting signal of fluorescent UB thioester-bonded to UBE2N. N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
We next wondered how L-lysine architecture within the context of an acceptor UB would affect UBE2N/UBE2V1 reactivity (Fig. 2a). Solid-phase peptide synthesis was employed to generate UBs with K63 analogs differing by number of methylene groups - one, two, three, four or five - between the alpha carbon and side-chain amino group: L-2,3-diaminopropionic acid (Dap, referred to here as K63UBC1 for one methylene group in the analog replacing native K63), L-2,4-diaminobutyric acid (Dab, referred to here as K63UBC2 for two methylene groups in the analog replacing K63), L-ornithine (Orn, referred to here as K63UBC3 for three methylene groups in the analog replacing K63), L-lysine (Lys, referred to here as K63UBC4 for the four methylene groups in the native acceptor), and L-homolysine (hLys, referred to here as K63UBC5 for five methylene groups in the analog replacing K63) (Fig. 2b).
Figure 2 |. K48 and K63 chain-forming E2s show strong preferences for a native lysine acceptor on ubiquitin.
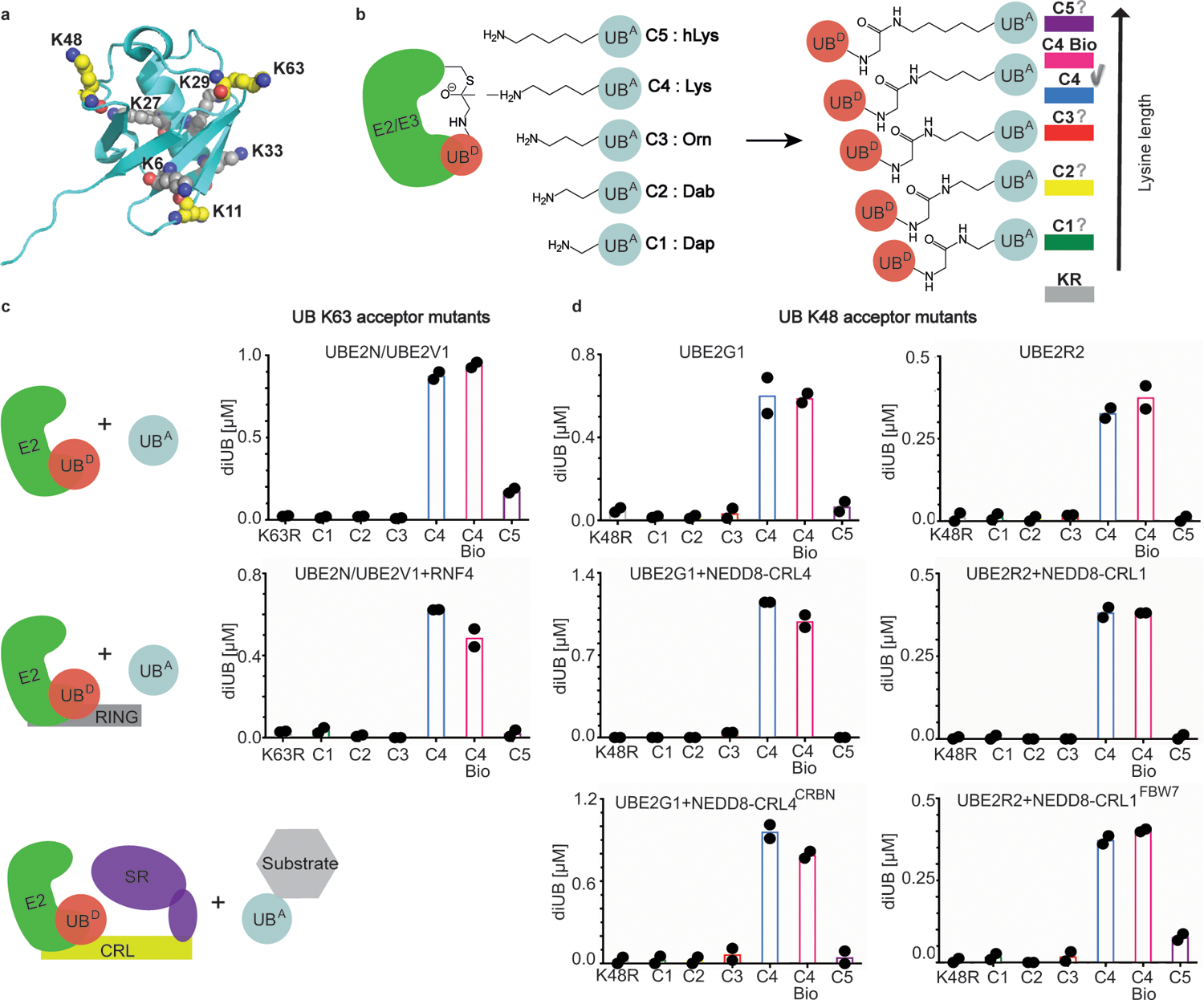
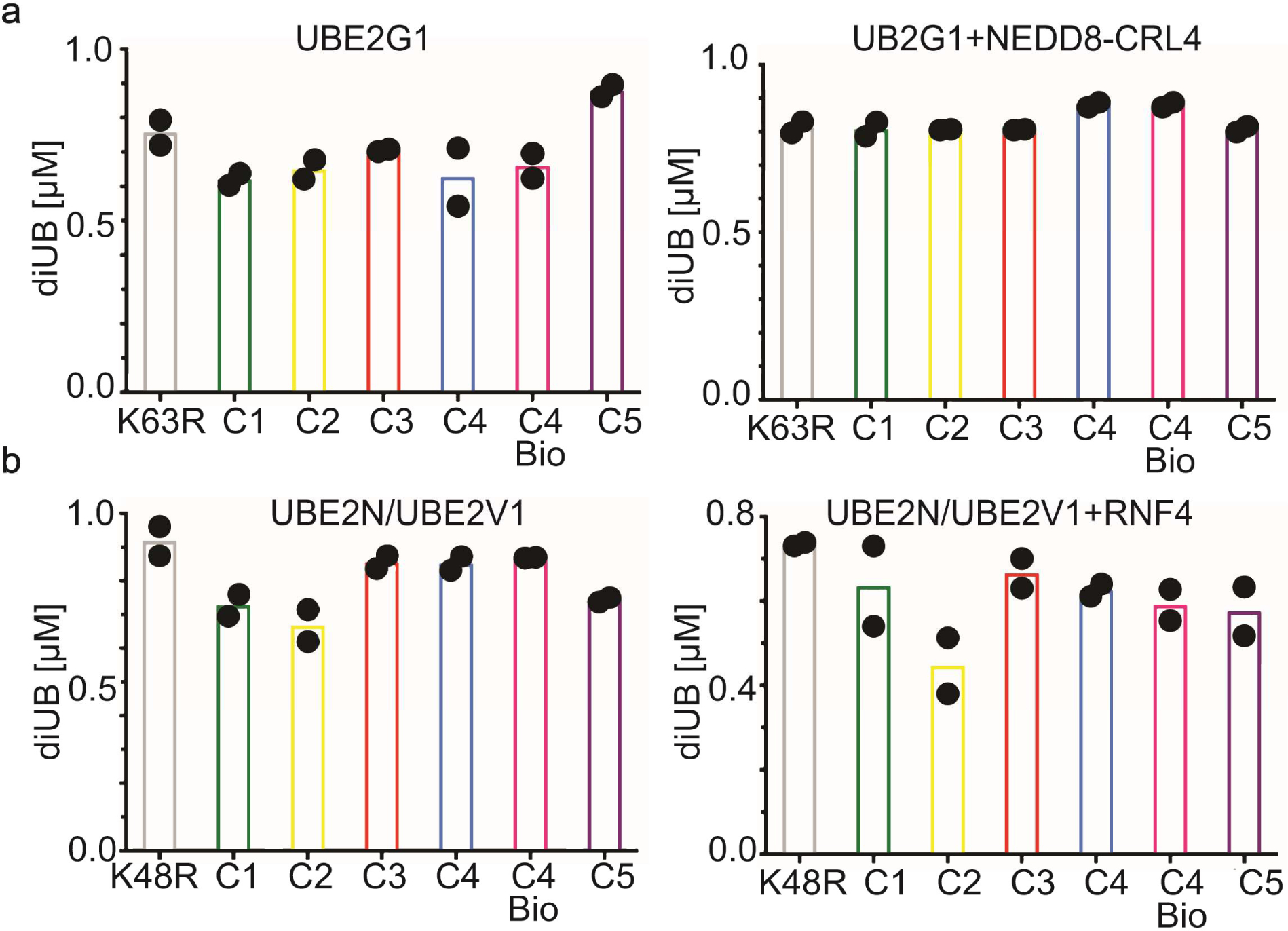
(a) Structure of UB (PDB:3CMM) showing lysines as spheres, highlighting carbon (yellow) and nitrogen atoms (blue) for K11, K48, and K63 where analogs were installed for this study. (b) Cartoon of experimental scheme, monitoring reactivity of E2~UB (D refers to the fluorescent “donor” UB to be transferred from E2) and formation of di-UB with various versions of acceptor UB (UBA). Color coding for acceptor lysine analogs denoted as C1-C5 based on 1–5 side-chain methylene groups, respectively. (c) Amount of di-UB chain produced by UBE2N/UBE2V1 with UBs harboring the indicated acceptor side-chain at position 63 in the absence (top) or presence (bottom) of the E3 RING domain from RNF4. (d) Amount of di-UB chain produced by E2s UBE2G1 and UBE2R2 in absence (top), or presence (middle) of cognate E3s, NEDD8-CRL4 or NEDD8-CRL1, respectively that activate di-UB synthesis. E2-dependent di-UB forming activity toward E3-bound substrates was tested with substrates (sortase-mediated K48UBC1-C5 linked to phospho-Cyclin E peptide or IKZF1 ZF2–3) of neddylated CRL1FBW7 or CRL4CRBN (bottom). For all plots, di-UB levels (μMol) represent the final time-points from the reactions (Source Data Fig. 2), N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
UBE2N/UBE2V1 activity was again measured using a pulse-chase assay, with the acceptor now being UB, and product a di-UB chain. Remarkably, unlike in the discharge to free amino acids, removal or addition of only a single methylene from or onto a canonical K63 side-chain greatly reduced di-UB chain formation. The striking preference for the native lysine persisted in reactions accelerated by the RNF4 E3 (Fig. 2c).
Lysine geometry impacts many di-UB forming E2s and E3s
Since acceptor UB placement for UBE2N is unique in depending on a partner (UBE2V1)23, UBE2V1’s grip may limit ability of the reactive amine to reposition in the active site upon addition or removal of a methylene. Thus, we wondered how changes to lysine architecture affects other E2s reliant on their own surfaces to orient an acceptor UB. Hence, activities of two K48 linkage-specific E2s, UBE2G1 and UBE2R2, were assayed toward a K48UBC1-C5 suite25,26,27. Significant di-UB product was only observed with K48UBC4 acceptor – for the E2s alone, and for UBE2R2 and UBE2G1 reactions stimulated by cullin-RING ligase E3s CRL1 or CRL4, respectively25–29, and for substrate-linked acceptors (Fig. 2d). These latter assays depended on CRL receptors recruiting specific substrate degron motifs. The CRL1 receptor FBW7, a tumor suppressor protein, recruits phosphopeptide motifs in targets including the cell cycle regulator Cyclin E30. For the CRL4 receptor CRBN, the chemotherapeutic agent Pomalidomide induces recognition of zinc finger motifs in neosubstrates including Ikaros family transcription factors31,32. CRL1FBW7 and CRL4CRBN substrates were generated by sortase-mediated transpeptidation of degron peptides (cyclin E phosphopeptide and IKZF1 zinc finger, respectively) with synthetic UBs. Only native lysine supported substantial UB-chain elongation onto CRL-bound substrates (Figure 2d)
To determine if the preference for native lysine is preserved for HECT E3 ligases - where UB is transferred from E2 to the HECT catalytic cysteine and then onto the substrate lysine - we assayed NEDD4 HECT domain33 and a version of its yeast ortholog Rsp5p harboring substrate-binding WW and catalytic domains34. Both forge K63-linked chains33–35. Again, robust di-UB formation was only observed with native lysine acceptor K63UBC4. Di-UB formation was greatly reduced with K63UBC1-C3, or K63UBC5 including for a substrate36 recruited to Rsp5p (Fig. 3a and 3c).
Figure 3 |. K63 chain forming HECT E3 ligases show strong preferences for a native lysine acceptor on ubiquitin.
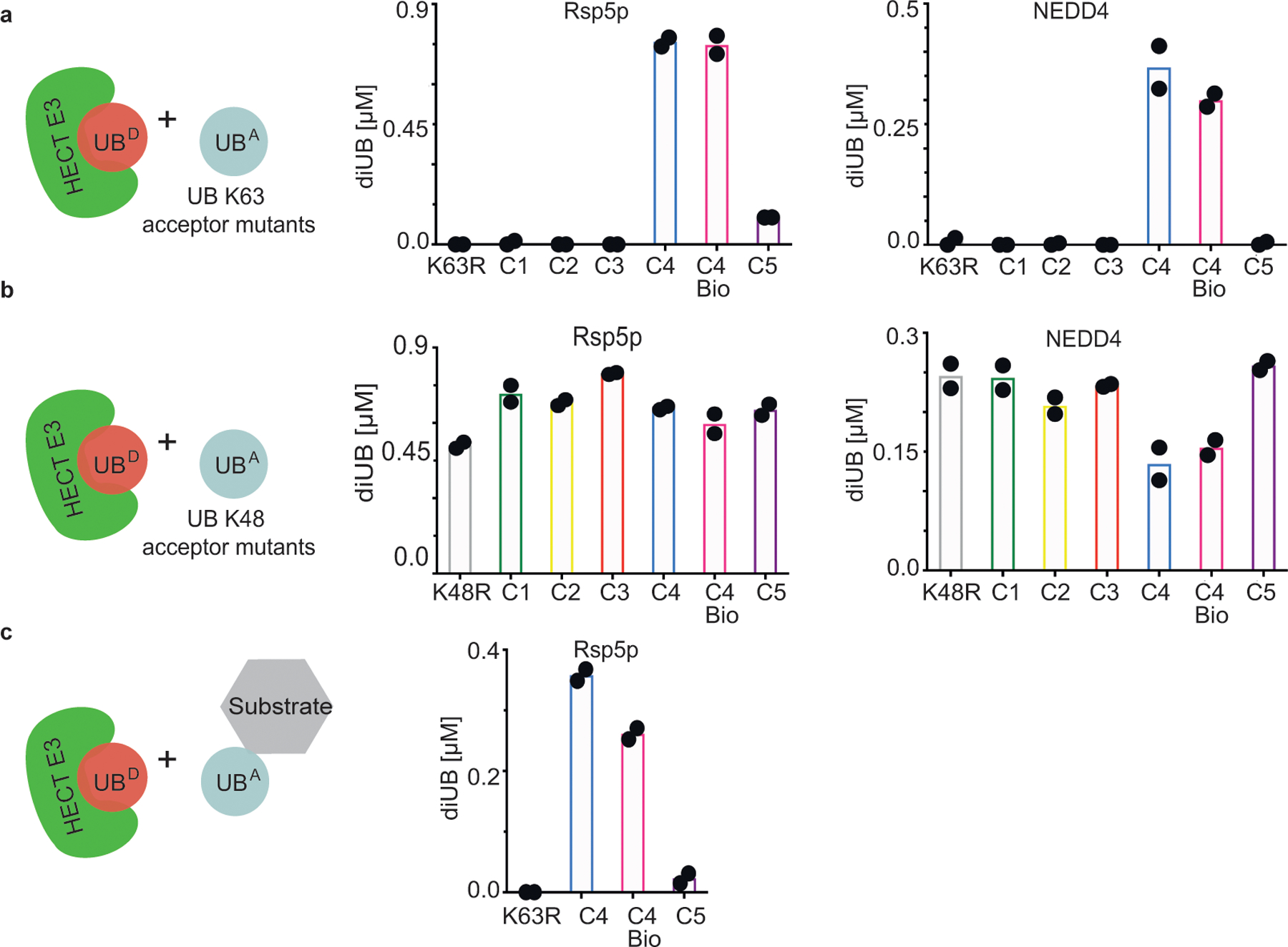
(a) Cartoon of experimental scheme (left), monitoring reactivity of the yeast HECT E3 Rsp5p (middle) or human HECT E3 NEDD4 (right) and formation of di-UB chains with K63UBC1-C5 acceptors (UBA). (b) same as (a), except with K48UBC1-C5 acceptors. (c) HECT E3 ligase-dependent di-UB forming activity in the context of an Rsp5p-bound substrate (sortase-mediated UB, UB K63R, or K63UBC5 linkage to the WW-domain-binding PPPY degron motif of the substrate Sna4p. For all plots, di-UB levels (μMol) represent the final time-points from the reactions (Source Data Fig.3), N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
As controls, the K63UBC1-C5 analogs served as acceptors with the K48-specific E2 UBE2G1, demonstrating proper folding for the synthetic UBs harboring K63 substitutions (Extended Data Fig. 2a). Similarly, UBE2N/UBE2V1, NEDD4 and Rsp5p also produce nearly wild-type amounts of di-UB chains with UBs harboring lysine analogs on the non-acceptor position 48 (K48UBC1–5, Fig. 3b and Extended Data Fig. 2b). Moreover, proton NMR spectra for recombinant UB (aka C4-bio), synthetic UB (aka C4), and K48UBC5 showed good dispersion and were superimposable except for a few resonances, presumably reflecting overall minor impact of sequence differences between them (Met1 in C4 bio substituted with NorLeu in synthetic UBs, and Lys48 versus C5 side-chain Extended Data Fig. 3).
Taken together, the data show that K63- and K48-specific E2 and E3 enzymes utilizing distinct modes of acceptor UB recruitment display exquisite specificity for the attacking lysine architecture in the context of an acceptor UB. Notably, E2~UB and HECT E3~UB active sites are structurally distinct. Thus, the demand for native lysine acceptor geometry for chain building seems to be a general property that could extend across many of the hundreds of E2/E3 ligation systems.
K48 side-chain impacts the multifunctional E2 UBE2D3
We pondered whether there may be exceptions to linkage specific ubiquitylation relying on native lysine (C4) acceptors. The E2 UBE2S, which generates K11 di-UB linkages, was an intriguing candidate, as UBE2S relies on acceptor UB-assisted catalysis14. UBE2S displays weak di-UB chain synthesis activity on its own, due to high Km for the acceptor14. This is overcome by fusing a UB-binding domain to UBE2S, or with the Anaphase-Promoting Complex/Cyclosome (APC/C) E3 whose RING domain recruits the acceptor UB37,38. Results from our qualitative assays suggest that UBE2S is less sensitive to lysine side-chain length, as di-UB formation occurred with K11UBC2-C5 acceptors, although K11UBC1 was relatively inactive (Extended Data Fig. 4). This is not due to an overt folding defect, as all K11UBC1-C5 analogs are acceptors for UBE2N/UBE2V1-dependent K63-linked di-UB formation.
We also examined the relatively promiscuous UB chain forming enzyme UBE2D3 (aka UBCH5C): UBE2D3 collaborates with numerous E3s, transfers UB to E3 catalytic cysteines and substrate lysines, generates several UB chain linkages, and forms branched UB chains in multiple turnover polyubiquitylation reactions39,40. Pulse-chase assays examining di-UB products of UBE2D3~UB revealed preferential targeting to K11 and K63, according to absolute quantitation by mass spectrometry (Fig. 4a). However, with a K48UBC5 acceptor added to UBE2D3~UB, the SDS-PAGE mobilities of di-UB products differed from those formed with a native UB acceptor. Because different UB chain linkages could impact electrophoretic migration, the result hinted at distinct products (Fig. 4b).
Figure 4 |. The location of lysine analogs on acceptor UB impacts the distribution of diUB chain linkage types generated by the E2 enzyme UBE2D3.
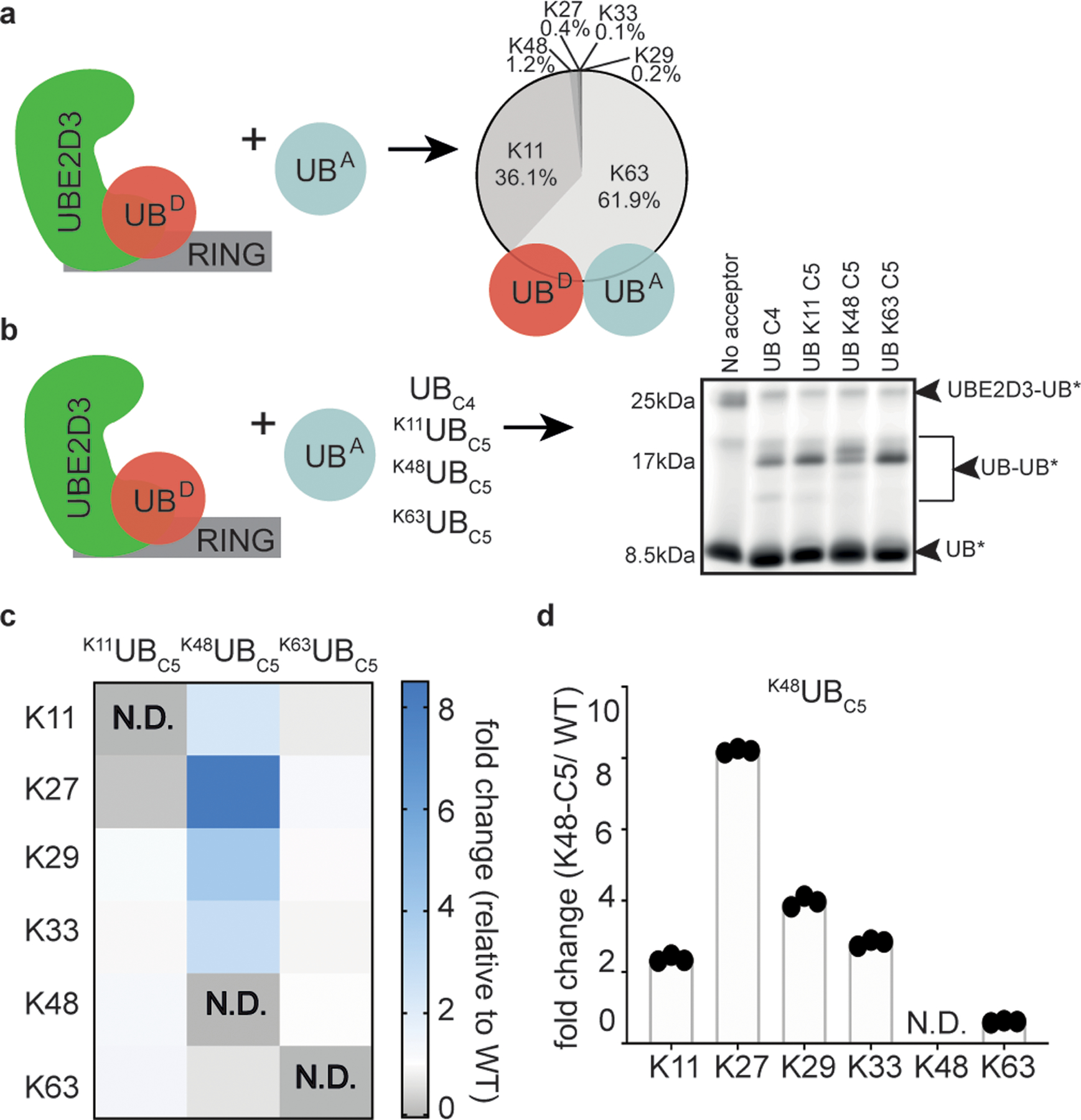
(a) Cartoon of experimental scheme, monitoring reactivity of UBE2D3~UB (D refers to the “donor” UB to be transferred from E2) in the presence of the E3 RING domain from RNF4 and formation of di-UB with UBC4 acceptor (UBA). The distribution of diUB linkage types generated is shown. (b) same as (a), except with fluorescent donor UB and various acceptor UBs (UBA). Notice that di-UB products with distinct electrophoretic mobilities were observed for each acceptor, indicating that the lysine analogs likely affect the di-UB chain linkage identity (N=2 independent experiments). (c) Relative fold changes of diUB linkage types for reactions with UBE2D3 and the E3 RING domain from RNF4 comparing K11UBC5, K48UBC5, or K63UBC5 acceptors with UBC4. N.D. is not-defined. (d) Plot showing the relative changes in UBE2D3/RNF4 generated di-UB chain linkages when comparing products containing K48UBC5 or UBC4 acceptors. For (c) and (d) N=3 technical replicates.
We developed a targeted mass spectrometry strategy to quantify distributions of UB chain linkages formed with native lysines. Although the method does not detect chains linked to the unnatural amino acid, it quantifies relative UB linkages to the remaining lysines in reactions with K11UBC5, K48UBC5, or K63UBC5 compared to reactions with C4 acceptor UB. With K11UBC5 or K63UBC5 acceptors, UBE2D3~UB generates di-UBs with linkage-type distributions similar to reactions with UBC4 (Fig. 4c,d and Extended Data Fig. 5). However, adding an extra methylene group to the side-chain at K48 alters the distribution of di-UB species formed. The change between preferred acceptors could be accounted for by two observations. First, there is a relative redistribution from K63- to K11-linkages. Second, although the di-UBs linked via K27, K29, and K33 remain a minor proportion of the total, utilization of these non-preferred acceptors increased compared to UBC4. Thus, the location on UB is a determinant of the requirement for a UBC4 by a multifunctional ubiquitylating enzyme.
Impact of side-chain architecture revealed by MD
Potential structural effects of adding a methylene group to the acceptor side-chain were revealed by Molecular Dynamics (MD) simulations on native UBC4 or UBC5 at position 11, 48, or 63. In two independent 50 ns simulations for UBs with native lysine or C5 at positions 11, 48, and 63, the overall UB globular fold (residues 1–70) was preserved, with 1.618, 1.271, 1.209, and 1.494 Å average Cα RMSD across the simulations, respectively. Nonetheless, relative differences for C5 - at all three sites - include: (1) increased potential range of distances between the alpha carbon and side-chain amine for C5, with limited overlap in the distribution of relative side-chain amine position, (Fig. 5a) - this would effectively impart a greater radius to the C5 side-chain when considering the backbone as the axis of rotation; (2) expanded number of potential rotamers from 81 to 273, with more accessed by the C5 side-chain in every simulation (C4:C5 rotamer ratios for residue 11 56:82; residue 48 65:117; residue 63 43:96); and (3) different dynamics for Chi angles, particularly Chi4, which oscillated more frequently between the three rotamer bins for C5 - this would cause more rapid fluctuation of relative side-chain amine positions (Fig. 5b and Extended Data Fig. 6a). With C5 at positions 48 and 63, there were also subtle but reproducible increases in fluctuations in phi and psi angles (Fig. 5c,d), and an increased number of allowable phi/psi combinations (C4:C5 ratios: residue 11 185:175; residue 48 138:169; residue 63 73:90). Collectively, between backbone and rotamer combinations, we typically observed more states accessible to the C5 residue, with a noticeable increase of over 1000 additional states at positions 48 and 63 (C4:C5 ratios: position 11 2942:3016; position 48 2942:4261; position 63 1188:2561).
Figure 5 |. Molecular Dynamics simulations reveal pleiotropic structural effects on UBs harboring lysine analogs.
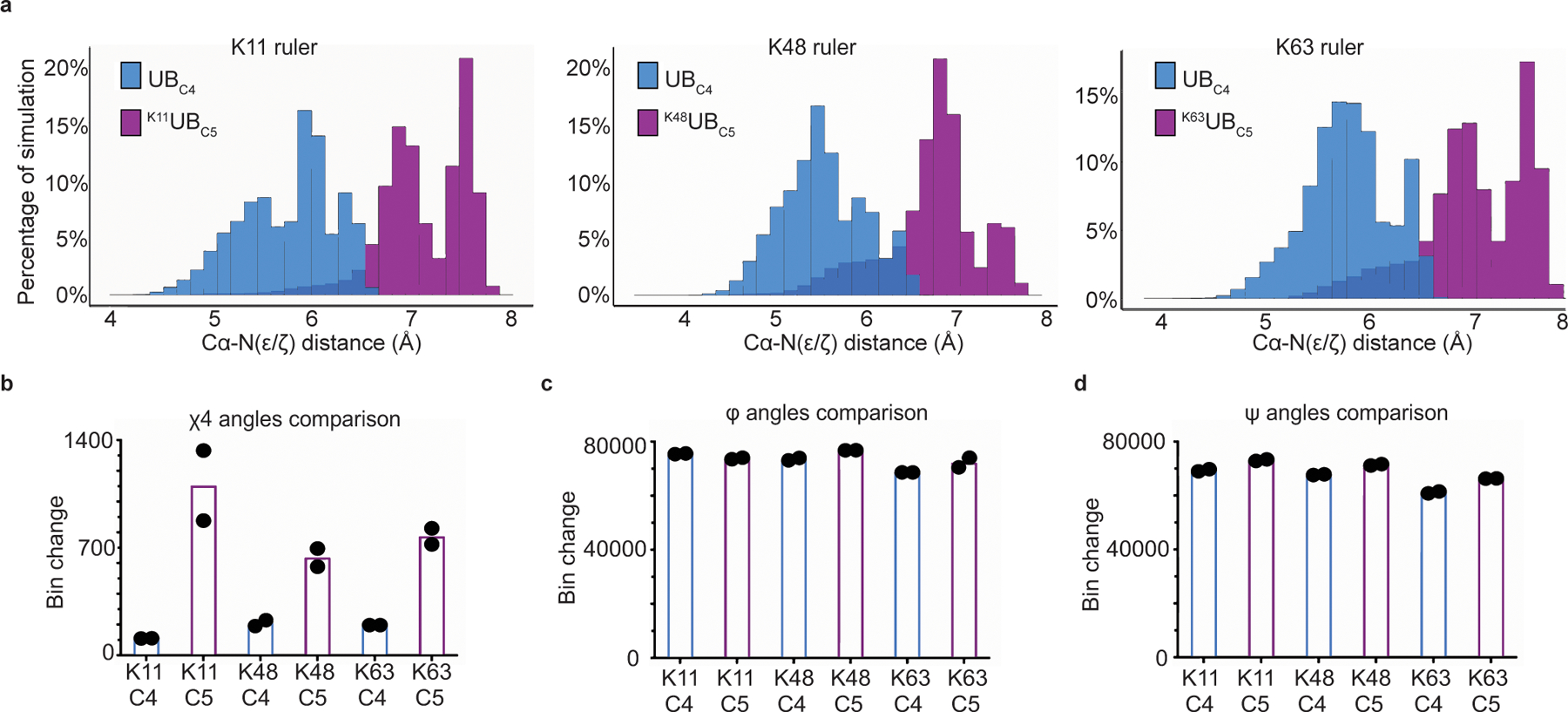
(a) Distribution of the distances between lysine acceptor amine and Cα atoms for UBC4 versus UBC5 in molecular dynamics (MD) simulations performed of UB. (b) Plot showing the dynamics of Chi4 side-chain rotomers for various UBC5 acceptors compared with UBC4. Bins are divided by 120° intervals. (c) same as (b), except for the psi main-chain torsion angle. Bins are divided by 10° intervals. (d) same as (b), except for the phi main-chain torsion angle. For all plots graphs, N=2 independent experiments.
We wished to further probe potential effects of the C5 side-chain as an acceptor in di-UB chain formation. The only structurally-characterized reaction is a donor UB~UBE2N/UBE2V1/acceptor UB complex, where the acceptor UB’s K63 points toward, but is 12.5 Å from, the donor UB’s carbonyl to which it becomes linked during di-UB synthesis23. Nonetheless, we adapted an intermediate of the the acceptor K63 based on modeling and on constraints from enzymology and crystal structures of wild-type UBE2N/UBE2V141,42. Three independent MD simulations (25 nanoseconds each) showed the C5 acceptor side-chain preferentially adopting extended conformers, and more frequently fluctuated between rotamers, as in the simulations of UB alone. While both C4 and C5 side-chain amines maintained a similar distance to the UBE2N~UB active site (Extended Data Fig. 6b–d). closer inspection revealed two appreciable differences between the simulations: (1) lysine occupied a favorable trajectory toward the active site for a greater proportion of the simulations, whereas C5 more frequently rotated between rotamer bins and approached the active site from different angles (Extended Data Fig. 6e,f); (2) greater deviation in the conformation of UBE2N’s so-called “active site gate loop” (residues 115–120). Interestingly, this gate loop is important for stabilizing noncovalent interactions between the donor UB tail and UBE2N, configuring catalytic residues, and positioning the acceptor lysine relative to the thioester bond for catalysis41,43. Distortion of the gate loop conformation, as observed with the C5 side-chain, could reduce the probability of adopting a structure favoring ligation (Extended Data Fig. 6g,h).
UB acceptor lysine geometry impact on kinetic parameters
To illuminate mechanistic roles for lysine, quantitative biochemical experiments were performed. Substantially increasing reaction time and protein levels under steady-state conditions enabled quantification for C5 as acceptor for di-UB formation by the E2s UBE2N/UBE2V1 (with or without RNF4 E3), UBE2R2 and by the HECT E3 Rsp5p.
The reactions with both E2s showed overall similar profiles: kcat values were lower with C5 replacements for acceptor lysines, 16-fold and 14-fold, respectively (Table 1), consistent with the striking results from the pulse-chase assays (Fig. 2). While defects in enzyme activity can manifest themselves through various perturbations, failure to activate the acceptor lysine amine or decreasing affinity of the acceptor UB for the E2 are quite common13,14,44. Pioneering investigation of the related modification SUMOylation suggested that E2s catalyze ubiquitylation at least in part through the active site complementing the acceptor lysine to achieve pKa suppression44. Despite being unable to estimate apparent pKa for reactions with E3s due to loss of enzyme activity at high pH, we were able to determine apparent pKa values in the reactions with E2s.
Table 1|. kcat, pKaapp and KM for ubiquitin carrying enzymes with native vs. homolysine acceptor UBs.
Kinetic parameters for several ubiquitin carrying enzymes, including the apparent pKa (pKaapp), the rate of di-UB formation, kobs, for UBE2N/UBE2V1 and UBE2R2 at pH 10.1 or 9.7, respectively (kobs (hr−1) top pH), and the KM and kcat of UBC4 or UBC5 acceptors for E2 or HECT E3. Each value represents the mean of duplicate data points (Source Data).
| E2/E3 | UB | Lys | pKaapp | kobs (hr −1) top pH | KM (10−6 M) | kcat (hr −1) |
|---|---|---|---|---|---|---|
| UBE2N/V1 | C4bio | K63 | 190 | 6.1 | ||
| UBE2N/V1 | C4 | K63 | 8.9 | 15.8 | 398 | 3.4 |
| UBE2N/V1 | C5 | K63 | 9.0 | 0.58 | 284 | 0.21 |
| UBE2N/V1 + RNF4 | C4 | K63 | 23 | 39.1 | ||
| UBE2N/V1 + RNF4 | C5 | K63 | 58 | 9.3 | ||
| UBE2R2 | C4 | K48 | 6.6 | 2.67 | 528 | 15.8 |
| UBE2R2 | C5 | K48 | 7.3 | 0.028 | 1940 | 1.1 |
| Rsp5p | C4 | K63 | 21 | 1.11 | ||
| Rsp5p | C5 | K63 | 335 | 0.44 |
UBE2N/UBE2V1 activity (with a K92R mutation to decrease auto-ubiquitylation at high pH23) was measured in the presence of K63UBC4 or K63UBC5 across varying pHs (Table 1, Extended Data Fig. 7 a,b). While caution should be taken when interpreting apparent pKa values, since both kcat and KM may display pH dependencies of their own, the data fit best to a model where a single ionizing species is responsible for pH dependency of kobs (Extended Data Fig. 7b). Surprisingly, pKa values were similar in reactions with K63UBC4 or K63UBC5 (8.9 and 9.0, respectively; Table 1). Parallel experiments with UBE2R2 showed apparent pKa values of 6.6 and 7.3 for K48UBC4 and K48UBC5, respectively (Table 1, Extended Data Fig. 7e). For both E2s, differences in pKa values are insufficient to account for those between rates of di-UB formation with acceptor lysine or C5 side-chains in reactions at elevated pH (nearly 100-fold for UBE2R2 at pH 9.7, Table 1). The estimated KM values of K63UBC4 or K63UBC5 for UBE2N/UBE2V1 were within 2-fold, and those of K48UBC4 or K48UBC5 for UBE2R2 within 4-fold, suggesting similar affinities for lysine- and C5-bearing acceptor UBs and their respective E2s (Table 1 and Extended Data Figs.7c,f). Thus, defective catalysis seemingly arises from other effects of the additional methylene in the acceptor UB side-chain.
An E3 may affect mechanisms underlying acceptor UB lysine specificity. Although the RNF4 RING domain greatly impacted UBE2N/UBE2V1-catalyzed di-UB formation (lowering the KM of acceptor UB for E2 and increasing kcat by approximately 17-fold and 11-fold respectively (Table 1 and Extended Data Fig. 7d)), only modest effects were observed for KM (~2.5-fold) as well as kcat (~4-fold) in the presence of K63UBC5. In combination, these effects are not greater than those observed without E3.
In contrast, kinetic experiments performed on the HECT E3 Rsp5p showed a remarkable 16-fold lower KM for the acceptor K63UBC4 compared with K63UBC5, with only a ~2.5-fold difference in kcat (Table 1 and Extended Data Fig. 7g). Overall, the kinetic results unveiled a diverse spectrum of effects of the lysine side-chain ruler on ubiquitin-carrying enzyme activities.
Discussion
Our data show that many different UB chain forming enzymes are strikingly sensitive to the lysine side-chain hydrocarbon linker at the Ångstrom length-scale as determined by a single methylene. Biochemical assays show that UBC5 can affect KM, kcat, and pKa (Table 1). Meanwhile, MD simulations unveiled pleiotropic structural effects of C5, including additional degrees of freedom, more side-chain flexibility, and more dynamics in the backbone in UB itself (Fig. 5). It might stand to reason that side-chains that are too short simply could not span the distance between the acceptor UB backbone and UB carrying enzyme active site. However, the fact that the UBC5 analogs impacted most enzymes tested indicates further roles of the acceptor side-chain.
For both UBE2N/UBE2V1 and UBE2R2, the mild effects on apparent pKa and/or KM are insufficient to explain the defects in kcat observed upon acceptor lysine substitution with C5 (Table 1). The MD simulations pointed toward several possible features of the lysine side-chain length that may be optimal for E2-catalyzed UB chain formation. For example, for enzymes where substrate binding and/or lysine positioning are rate-limiting, it seems that the increased entropy afforded by an extra methylene in the acceptor side-chain could decrease the frequency of catalytic encounter (Fig. 5). Interestingly, this mechanism would differ from that of another E2, UBE2W, for which a confluence of disorder between a flexible substrate N-terminus and a noncanonical E2 C-terminus guides ubiquitylation to a substrate’s N-terminal amine45. Rather than demanding disorder, the systems tested herein appear to favor a calibrated reach by the nucleophile that also must have restrained degrees of freedom.
In addition to entropic effects on the side-chain, the hydrocarbon linker length would also affect catalysis. For example, as shown for UBE2N (without UBE2V1-RING E3 partners), computational studies support a model where there is a precise “hole” fitting the lysine amine, and attack on the thioester carbonyl is rate-limiting46. Our data may suggest that the acceptor UB lysine itself is optimal not only for accessing the amine hole, but also for the chemistry of ubiquitylation. Indeed, the MD simulations of the UB~UBE2N/UBE2V1/UB complex pointed to multiple ways the acceptor lysine side-chain length could impact catalysis, including through optimal geometric approach to the active site, and through conformationally toggling the active site gate loop in the UB~E2 intermediate. Moreover, in agreement with prior studies suggesting this loop in UBE2N essentially closes around the acceptor K63 to promote formation of the transition state43, our MD simulations showed distortion of the active site gate loop with the suboptimal C5 side-chain. This would be consistent with UB discharge to free side-chain amine acceptors irrespective of hydrocarbon length, and a dramatic impact on kcat in the context of acceptor UB presented from UBE2V1. One would also predict little impact on KM in such a case, although lack of an effect on KM may also reflect that the additional methylene does not impact acceptor UB recruitment to this auxiliary UB-binding domain.
The impact of acceptor side-chain length on the HECT E3 Rsp5p represents the opposite extreme. The predominant effect on KM implies a role of the acceptor lysine itself in productive binding to the E3. It is possible that local interactions – awaiting elucidation by future structural studies - dominate acceptor UB recruitment47. It is also possible that placement of the acceptor lysine in the active site allosterically stabilizes the enzyme~UB conformation that binds the acceptor48.
While our study relied on installing side-chain chemical variants, it seems likely that in the cellular milieu, many natural factors – including linkage within a chain and binding to protein partners – could influence presentation of acceptor lysines resulting in specificity with E2 and E3 enzymes. Strong preferences for the lysine side-chain itself may contribute to robust ubiquitylation sufficient to elicit proteasomal degradation even when preferred targeting sites are unavailable. Such features may also influence successes or failures of targeted protein degradation strategies that rely on small molecules to direct proteins of therapeutic interest to ubiquitylating enzymes49,50.
Online methods
Constructs, protein expression and purification
All expression constructs were prepared using standard molecular biology methods. Modifications to protein amino acid sequences were accomplished using PCR and the Quikchange mutagenesis kit (Agilent). The human E2 constructs used in this study are GST-TEV-UBE2R2, GST-TEV-UBE2N, GST-TEV-UBE2N harboring a K92R mutation, GST-TEV-UBE2D2, GST-TEV-UBE2D3, His-GST-Ps3C-UBE2V1, UBE2G1-TEV-His, and GST-TEV-UBE2S (1–196) fused with the human USP5/IsoT (residues 173–289) domain - here called UBE2S_IsoT37. Human HECT E3 ligase NEDD4 was expressed as a GST-TEV-NEDD4 construct, and yeast HECT E3 Rsp5p containing residues 383-C (with WW-domain-binding PPPY degron motif of the substrate Sna4p) was expressed as a GST-TEV-Rsp5p construct34. All E2s, both HECT E3s and His-Sortase A were expressed in BL21-Gold(DE3) bacterial cells. Proteins were purified by either GST or Nickel affinity chromatography and cleaved on beads overnight with TEV or 3C Protease. Cleaved protein solutions were then subjected to ion exchange chromatography followed by size exclusion chromatography in 25mM HEPES pH 7.5, 150mM NaCl and 1mM DTT buffer. Human SKP1-FBW7 complex, NEDD8, APPBP1-UBA3 (the E1 to activate NEDD8), UBE2M (a NEDD8 E2-conjugating enzyme) and fluorescently labeled wild-type, K11R, K48R or K63R UB were generated as previously described48,51,52. APC/C and CDH1 were expressed and purified as previously described53. The RING-RING fusion version of RNF4 was expressed and purified as previously described54. Coding regions for CUL1, CUL4A (38-C), RBX1 (5-C) His-TEV-DDB1, CRBN, GST-TEV-IKZF1 (encompassing zinc fingers 2–3 containing amino acids 141–243Δ197–238; referred to as IKZF1 ZF 2–3)55 and UBA1 were all sub-cloned into pLIB vectors56. Baculoviruses for CUL1, GST-TEV-RBX1 5-C, CUL4A 38-C, HIS-TEV-DDB1 and CRBN were first prepared and isolated from Sf9 cells. followed by CUL1 and GST-TEV-RBX1 5-C, CUL4A 38-C and GST-TEV-RBX1 5-C, HIS-TEV-DDB1 and CRBN co-infection of Hi5 cells for co-expression51,52. Proteins were purified by His or GST affinity chromatography followed by overnight TEV cleavage. Cleaved protein solutions were then subjected to ion exchange chromatography followed by size exclusion chromatography in 25mM HEPES pH 7.5, 150mM NaCl and 1mM DTT buffer. The covalent modification of CUL1-RBX1 (CRL1) and CUL4A-RBX1 (CRL4) with the CRL activator protein NEDD8 (termed neddylation) was performed as previously described51,52,57. All variants of UB used in this study were generated as previously described53.
A plasmid for the bacterial expression of K63R human UB was prepared by using a previous construct for a GST fusion58 to wild-type human UB containing a consecutive N-terminal TEV cleavage site (ENLYFQG) and Protein Kinase A consensus sequence (RRASVG) for radio-labeling. Mutation of K63 to Arg was accomplished by the Quikchange method, using DNA oligo sequences 5’ GATTACAACATTCAGAGGGAGTCCACCTTACATC 3’ for the forward primer and 5’ GATGTAAGGTGGACTCCCTCTGAATGTTGTAATC 3’ for the reverse one. The construct DNA sequence was validated by Sanger sequencing. The plasmid was transformed into chemically competent BL21(DE3) E.coli bacteria for expression at 37°C. Protein purification was accomplished using standard approaches58, with the final step being gel filtration chromatography into a buffer containing 30 mM Tris, pH 8.0, 100 mM NaCl, 1 mM DTT, and 10% glycerol. Purified K63R UB was concentrated to approximately 250 µM based on an extinction coefficient of 1280 M−1cm−1 and flash frozen in liquid nitrogen prior to storage at −80°C. K63R UB (50 or 100 μM) was radiolabeled in the presence of 5 kU of cAMP-dependent protein kinase (New England Biolabs) and [γ32P]-ATP for 1 hour at 30°C.
All UB-conjugating enzymes (E2s) and their associated E3s employed in this study are listed in Supplementary Table 1.
Donor UB discharge assay (aka pulse-chase) to free amino acids (Fig. 1 and Extended Data Fig. 1)
20 μM UBE2N was loaded with 20 μM fluorescent UB K63R (UB*) in the presence of 0.3 μM UBA1 in a buffer containing 50 mM Hepes, pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 1.5 mM ATP and 0.05 mg/ml BSA. Loading reactions were incubated for 0.5 h and quenched by adding EDTA to a final concentration of 30 mM. The reaction was then initiated by adding UBE2N~UB* (0.5 μM final) to a substrate mix containing 0.5 μM UBE2V1, 0.5 μM RNF4 RING domain and 35mM amino acid acceptors (NƐ-acetyl-L-Lysine, L-Serine, L-Dap, Nɑ-acetyl-L-Ornithine, L-Lysine, D-Lysine, Nɑ-acetyl-L-Lysine, or L-Homolysine). Reactions were quenched with either non-reducing or reducing SDS-PAGE sample buffer after 0, 5, 10, 20, 30, 45, 60, 120 or 180 min, and substrates and products were separated by SDS-PAGE. Gels were scanned on an Amersham Typhoon (GE Healthcare) and the intensities of all fluorescent bands were quantified using ImageQuantTL (GE Healthcare). The E2~UB* band intensities were divided by the total fluorescent intensity in each lane and normalized to the 0 time point. Data were plotted in GraphPad Prism 8 (GraphPad Software) and fitted to an exponential decay function using non-linear regression. All reactions were performed in duplicate. Source Data Fig. 1 and Source Data Extended Data Fig. 1 contains all gels obtained from this experiment.
Transpeptidation reactions (Fig. 2d and Fig. 3c)
Sortase-mediated transpeptidation was utilized to link the C-terminus of various acceptor UBs to the N-terminus of a Cyclin E phosphopeptide (Nterm-GGGGLPSGLL(pT)PPQ(pS)GKKQSSDYKDDDDK-Cterm), IKZF1 ZF 2–3 or Sna4p peptide (Nterm-GGGGQSLVESPPPYVPENLYFQGDYKDDDDK-Cterm). UBs were synthesized or expressed recombinantly that contained a G76S mutation followed by the GSGSLPETGG sortase recognition sequence. Briefly, 50 μM UB was mixed with 100 μM substrate and 10 μM His-sortase on ice in a buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 10 mM CaCl2 for 1 h. Next, the reaction mixture was exposed to Nickel-agarose beads to remove His-sortase. Final products were purified by size-exclusion chromatography in 25 mM HEPES pH 7.5, 150 mM NaCl and 1 mM DTT buffer. For the UB-Sna4p fusions, an additional overnight incubation with TEV was included to remove a FLAG tag from Sna4p peptide, followed by size exclusion chromatography.
Donor UB discharge assay (aka pulse-chase) to UB analogs (Fig. 2, 3, 4b and Extended Data Fig. 2, 4)
20 μM E2s were loaded with 20 μM fluorescent donor UB (UB*) to form the E2-UB* complex in the presence of 0.3 μM UBA1 in a buffer containing 50 mM Hepes, pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 1.5 mM ATP and 0.05 mg/ml BSA. Loading reactions were quenched with EDTA (30 mM final) after a 0.5 h incubation period at room temperature. Reactions were initiated by the addition of various UB acceptors, and in some cases E3s (Supplementary Tables 2–4 report the final concentrations of these reagents for all pulse-chase reactions) in a buffer containing 25 mM Hepes, pH 7.5, 150 mM NaCl together with E2-UB* (approximately 0.5 μM final). All reactions were performed at room temperature for the indicated times and quenched with non-reducing SDS-PAGE sample buffer. Substrates and products were separated by SDS-PAGE and scanned on an Amersham Typhoon (GE Healthcare). The intensities of all fluorescent bands were quantified using ImageQuantTL (GE Healthcare). The amount of diUB chain was calculated by first dividing the diUB* band intensity by the total UB* intensity in each lane of the gel. The fraction of di-UB* product was then multiplied by the total amount of UB* (μMol) used in the reactions. All reactions were performed in duplicate.
For UBE2S, donor UB can be transfered to a lysine on the E2 surface (termed autoubiquitination). To minimize this, E1~UB* was prepared and added to UBE2S protein immediately prior to initiation of the reaction. Briefly, 10 μM UBA1 was first loaded with 20 μM UB* at room temperature for 0.5 h. E1-UB* was desalted twice, using Zeba desalting column, to quench loading into a buffer containing 25 mM HEPES pH 7.5 and 150 mM NaCl. Reactions were initiated as described above by the addition of UB acceptors and UBE2S with APC/C and its coactivator CDH1 to achieve final concentrations of approximately 5 μM E1-UB* and 0.2 μM E2. Reactions were processed as described above.
For reactions containing CRL1-bound substrate, the SKP1/FBW7 substrate receptor was utilized to reconstitute the full CRL (CRL1FBW7) which binds to phosphorylated cyclin E peptide. For reactions containing CRL4-bound substrate, the substrate adapter CRBN was utilized to reconstitute the full CRL (CRL4CRBN). Here the small molecule pomalidomide (2 μM final) facilitates complex formation between the sortased IKZF1 ZF 2–3-UB fusion and CRL4CRBN. All reactions for CRL-bound substrates had an approximate final concentration of 0.5 μM E2~UB that had been generated in the pulse step. All CRL-dependent reactions were processed as described above. Source Data Fig. 2,3 and Source Data Extended Data Fig. 2,4 contains all gels obtained from this experiments.
In-Gel digestion protocol for LC-MS/MS
UBE2D3 pulse-chase reactions were run as described above (see biochemical assay section). Briefly, UBE2D3 was loaded with either fluorescently labeled UB (UB*) or GST-UB. Note that GST-tagged donor UB was used to separate diUBD, which is formed during the pulse reaction as a side product, from the desired di-UB product between donor and acceptor UBs. Chase reactions contained 1 μM RNF4 and 100 μM UBC4, K11UBC5, K48UBC5 or K63UBC5. After a 15min incubation for UBC4 or 1h for UBC5, reactions were quenched with SDS-PAGE sample buffer. Reactants and products were separated by SDS-PAGE. Gels with samples containing UBE2D3-UB* were scanned on an Amersham Typhoon (GE Healthcare) and used to generate the image in Fig. 4b. Gels with samples containing UBE2D3-GST-UB were first stained with Coommassie Brilliant Blue to identify and excise the desired GST-UBD-UBA product band. After staining, the gel was subsequently destained by soaking for several hours in 10% acetic acid, 40% methanol, and 60% de-ionized water with at least two changes of the solvent to achieve a clear background. The gel band corresponding to GST-UBD-UBA was excised and chopped into smaller pieces (approximately 1 × 1 mm). Gel pieces were washed twice with 50% 50 mM Ammonium Bicarbonate, pH 8.0 (ABC) / 50% EtOH and then completely dehydrated by incubation in absolute EtOH. The gel pieces were then dried in a Speed-vac (Eppendorf, Concentrator plus), rehydrated in 200 µl of 1% (w/v) SDC buffer (10 mM TCEP, 40 mM CAA, 0.5 µg trypsin, 0.5 µg LysC in 100 mM Tris-HCL pH 8.5) and incubated at 37°C overnight. The next day, peptides were extracted from gel pieces by two consecutive rounds of adding isopropanol buffer (1% TFA in isopropanol) to the samples and subsequent collection of the liquid phase. At this step, stable isotope-labeled (SIL) analogs of chain specific native di-Gly peptide standards were added to the samples, which provided chromatographic orientation for the detection of endogenous (light) counterparts. For absolute quantification of different di-Gly peptides in UBC4 samples, SIL analog spike-in amounts were adjusted to yield peptide quantification ratios between 0.1 and 10 (20 and 2 fmol per injection for K11_GG, K48_GG, K63_GG and K27, K29, K33, respectively).
LC-MS/MS sample preparation
StageTips were prepared by inserting three layers of a SDB-RPS matrix (Empore) into a pipette tip using an in-house prepared syringe device as described previously59,60. The peptides mixed with isopropanol buffer were loaded onto the StageTips. The tips were washed with isopropanol buffer and subsequently with 2% ACN/ 0.2% TFA. Elution was performed using 80% ACN/ 1.25 % NH4OH. Eluates were collected in PCR tubes and dried using a Speed-vac centrifuge. Peptides were resuspended in buffer A* (2% ACN/ 0.2% TFA) and briefly sonicated (Branson Ultrasonics) before LC/MS-MS analysis.
LC-MS/MS measurements
Peptides were loaded on a 50 cm reversed phase column (75 µm inner diameter, packed in house with ReproSil-Pur C18-AQ 1.9 µm resin (Dr. Maisch GmbH)). Column temperature was maintained at 60°C using a homemade column oven. An EASY-nLC 1200 system (Thermo Fisher Scientific) was directly coupled online with the mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific) via a nano-electrospray source, and peptides were separated with a binary buffer system of buffer A (0.1% formic acid (FA)) and buffer B (80% acetonitrile, 0.1% FA), at a flow rate of 300 nl/min. Peptides were eluted with a gradient starting at 7% buffer B (0.1% (v/v) FA, 80% (v/v) ACN) and stepwise increased to 14% in 4 min and 26% in 22 min. After each gradient, buffer B concentration was increase to 95% in 2 min and maintained at this concentration for 6 min.
The mass spectrometer was programmed to acquire in targeted scan mode in which every full scan with resolution 60,000 at 200 m/z (3 × 106 ions accumulated with a maximum injection time of 20 ms) was followed by 20 multiplexed selected ion monitoring (SIM) scans employing multiplexing degree of four. Light (endogenous) and heavy counterpart peptides were always simultaneously recorded in the same scan. Each SIM scan covered a range of m/z 150–2000 with resolution 120,000 (5 × 104 ions accumulated with a maximum injection time of 65 ms, 1.4 m/z isolation window and 0.4 m/z isolation offset). The targeted peptides with m/z values are listed in Supplementary Table 5.
Data analysis
Raw MS data were processed using Skyline which is an open source software project57,58. Graphical displays of chromatographic traces were manually inspected for proper peak picking of MS1 filtered endogenous peptides based on co-eluting SIL peptides. All quantification was done on the precursor ion level, based on area. Only the most abundant peak of the isotope cluster was used for quantitation.
Bioinformatics analysis in this study were performed with Microsoft Excel and data visualized using GraphPad Prism (GraphPad Software). The background signal detected in the sample lacking acceptor UB was substracted from the corresponding signals of samples containing acceptor UBs. Next, each chain peptide was normalized to the first tryptic peptide of the N-terminaly modified UBA (M1Nle) sequence: ‘NleQIFVK’. Because this peptide was used for normalization, K6-linked diUB was not measured in our protocol. Finally, fold changes of each chain peptide (relative to WT) are calculated and shown in figures: Fig. 4c,d Extended Data Fig. 5a,b and Methods Equations (see below). All gels used in this experiment are shown in Source Data Fig. 4.
Methods Equations
Equation 1:
Correction for Background signal by substraction of signal detected in “No Acceptor” reactions
Equation 2:
Normalization of GlyGly peptides to NleQIFVK
Equation 3:
Foldchange calculation of UBC5 to UBC4.
Estimating the apparent pKa (pKaapp) values for ubiquitylation reactions containing UBE2N/UBE2V1 complex and K63UBC4 or K63UBC5 acceptor UBs or UBE2R2 and K48UBC4 or K48UBC5 acceptor UBs.
For UBE2N/UBE2V1, the pKaapp values for synthetic UBs (K63UBC4 or K63UBC5) were measured by a steady-state kinetics assay that detects isopeptide bond formation between radio-labeled donor UB and unlabeled acceptor. First, a titration series was created using Bis-Tris propane buffer with pH values of 5.7, 6.1, 6.5, 6.9, 7.3, 7.7, 8.1, 8.5, 8.9, 9.3, 9.7, and 10.1. Stocks of Bis-Tris propane buffer, 10x reaction buffer (20 mM ATP, 10 mM DTT, 50 mM MgCl2, and 500 mM NaCl), radio-labeled K63R donor UB, UBE2V1 and UBE2N (WT or K92R) proteins were added in the above order to autoclaved individual Eppendorf tubes. The final concentrations in the ubiquitylation reactions were 50 mM Bis-Tris Propane, 1X reaction buffer, 0.25 μM human UBA1, 5 μM radio-labeled K63R donor UB, and 2 μM UBE2V1/ K92R UBE2N complex. Following a 1-minute incubation period, either synthetic K63UBC4 or K63UBC5 were added to initiate the reaction (100 μM final concentration). Reactions with K63UBC4 were quenched after 2 minutes and 45 seconds and reactions with synthetic K63UBC5 were quenched after 15 minutes in either non-reducing or reducing 2x SDS-PAGE buffer (100 mM Tris-HCl, pH 6.8, 20% glycerol, 30 mM EDTA, 4% SDS, and 0.02% bromophenol blue). The reaction products and substrate were resolved by SDS-PAGE on 18% Tris-glycine poly-acrylamide gels, followed by autoradiography and detection on a Typhoon 9410 Imager. Quantification of substrate and products were performed using Image Quant (GE Healthcare). The fraction of di-UB product for each reaction was measured by normalizing the signal for product over the total signal in the lane. These fractions were then multiplied by the donor UB concentration and divided by both the UBE2N/UBE2V1 complex concentration and the time of incubation. The velocities were plotted as a function of the pH of the reaction and fit to a sigmoidal four-parameter logistic curve with the Hill slope constrained to 1 (GraphPad Prism software, version 8.3). Note that this model assumes that the reaction velocities are dependent on a single ionizing species that becomes activated at high pH. A similar procedure was followed for UBE2R2 and K48UBC4 or K48UBC5, except for the following modifications. The final concentrations in the ubiquitylation reactions were 0.5 μM human UBA1, 15 μM radio-labeled K63R donor UB, and 10 μM UBE2R2 protein. Reactions with K63UBC4 were quenched after 5 minutes and reactions with K48UBC5 were quenched after 60 minutes. The times of incubation were selected to ensure that all reaction velocities were within the linear range and that donor UB consumption was not sufficient to result in a lower concentration than E2. All reactions were performed in duplicate. All gels used in this experiment are shown in Source Data Table 1 and Source Data Extended Data Fig. 7.
Estimating the KM of UBC4 (E.coli produced and synthetic) or K63UBC5 acceptors for UBE2N/UBE2V1 complex.
The KM of acceptor UBs were measured by a steady-state kinetics assay that detects isopeptide bond formation between radio-labeled donor UB and unlabeled acceptor. A 10x reaction buffer was prepared with 500 mM Bis-Tris Propane, pH 7.3, 20 mM ATP, 10 mM DTT, 50 mM MgCl2, and 500 mM NaCl. First, a 2-fold dilution series was established for acceptor UB proteins that had first been dialyzed into a buffer containing 30 mM Bis-Tris Propane, pH 7.3. The starting concentrations of the dilution series were 1.4 mM for bacterial K63UBC4, 1.6 mM for synthetic K63UBC4, and 1.3 mM K63UBC5 (note that initiation of the reaction results in a further 2-fold dilution of each acceptor UB). Next, the follow reagents were added from stock solutions to an Eppendorf tube to achieve final concentrations in each reaction of 1X reaction buffer, 0.25 μM human UBA1, 5 μM K63R donor UB and 2 μM UBE2N/UBE2V1 for bacterial or synthetic UBC4, or 15 μM K63R donor UB and 10 μM UBE2N/UBE2V1 for K63UBC5. After a two-minute incubation period, aliquots of the master mix were evenly disbursed to clean Eppendorf tubes. Ubiquitylation reactions were then initiated by adding an equal volume of acceptor UB to the Eppendorf tubes containing the master mix. Reactions were incubated for either 15 or 30 minutes (K63UBC4or K63UBC5, respectively) prior to quenching in either non-reducing or reducing 2x SDS-PAGE buffer containing 100 mM Tris-HCl, pH 6.8, 20% glycerol, 30 mM EDTA, 4% SDS, and 0.02% bromophenol blue. The processing of reactions and estimation of velocities was performed as described in the previous section. The reaction velocities were fit to the Michaelis-Menten equation to estimate Km (GraphPad Prism software, version 8.3). Reactions containing the RING domain of RNF4 were performed similarly as above with the following changes. All reactions contained 0.5 μM human UBA1, 15 μM K63R donor UB, 10 μM UBE2N/UBE2V1 and 1 μM RNF4. The starting concentrations of the dilution series were 1.3 mM for UBC4 and 1.25 mM for K63UBC5. Reactions were incubated for either 0.5 or 2.5 minutes (UBC4 or K63UBC5, respectively) prior to quenching. The times of incubation were selected to ensure that all reaction velocities were within the linear range and that donor UB consumption was not sufficient to result in a lower concentration than E2. Reactions were performed in duplicate. All gels used in this experiment are shown in Source Data Table 1.
Estimating the KM of UBC4 or K48UBC5 acceptors for UBE2R2 and the KM of K63UBC4 or K63UBC5 acceptor UBs for Rsp5p.
The KM of acceptor UBs K48UBC4 and K48UBC5 for UBE2R2 and Rsp5p were measured similarly as described in the previous section with the following modifications. For UBE2R2, a 2-fold dilution series was established for acceptor UB proteins that had first been dialyzed into a buffer containing 30 mM Bis-Tris-Propane, pH 7.3 with starting concentrations of 8.4 mM for UBC4 and 12.5 mM for K48UBC5. The final concentrations in each reaction contained 0.5 μM human UBA1, 15 μM K48R donor UB and 10 μM UBE2R2 protein. Reactions were incubated for either 1 or 2.5 minutes for each replicate for the UBC4 titration series and for either 15 or 16 minutes for each replicate of the K48UBC5 titration series prior to quenching. For Rsp5p, the starting concentration of the acceptor UB dilution series was 1.6 mM for both UBC4 and K63UBC5. The final concentrations in each reaction contained 0.5 μM human UBA1, 7 μM K63R donor UB and 5 μM UBE2D2 and Rsp5p proteins. Reactions were incubated for either 5 or 30 minutes (UBC4 or K63UBC5, respectively) prior to quenching. The times of incubation were selected to ensure that all reaction velocities were within the linear range and that donor UB consumption was not sufficient to result in a lower concentration than E2 or E3. Reactions were performed in duplicate. All gels used in this experiment are shown in Source Data Table 1.
Extended Data
Extended Data Fig. 1. UBE2N~UB/UBE2V1/RNF4 RING E3 complex reacts preferentially with free amino acids harboring amine acceptors and various side-chain.
(a) Fluorescence scan of SDS-PAGE gels demonstrating the discharge of labeled UB (UB*) to L-lysine compared with the absence of amino acid acceptor using wild-type UBE2N. Electrophoresis was performed under both reducing and non-reducing conditions to differentiate thioester bonded complexes from isopeptide bonded E2-donor UB ones. (b) Time-courses of fluorescent UB discharge from UBE2N K92R~UB/UBE2V1/RNF4 RING E3 to the indicated amino acids, normalized to starting signal of fluorescent UB thioester-bonded to UBE2N. For all, N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
Extended Data Fig. 2. K48 and K63 chain-forming E2s equally discharge to K63UBC1-C5 and K48UBC1-C5 acceptors respectively.
(a) Di-UB formed by K48 UB chain-forming E2 UBE2G1 with K63UBC1-C5 acceptors in the absence (left) or presence (right) of neddylated CRL4 (N8CRL4). (b) Di-UB formed by the K63 UB chain-forming E2 UBE2N/UBE2V1 complex with the K48UBC1-C5 acceptors in the absence (left) or presence (right) of the E3 RNF4 RING domain. For all plots graphs, di-UB levels (μMol) represent the final time-points from the reactions (Source Data Extended Data Fig. 2), N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
Extended Data Fig. 3. 1D and 2D proton NMR spectra for synthetic UBC4, recombinant UB, and K48UBC5 are highly superimposabe.
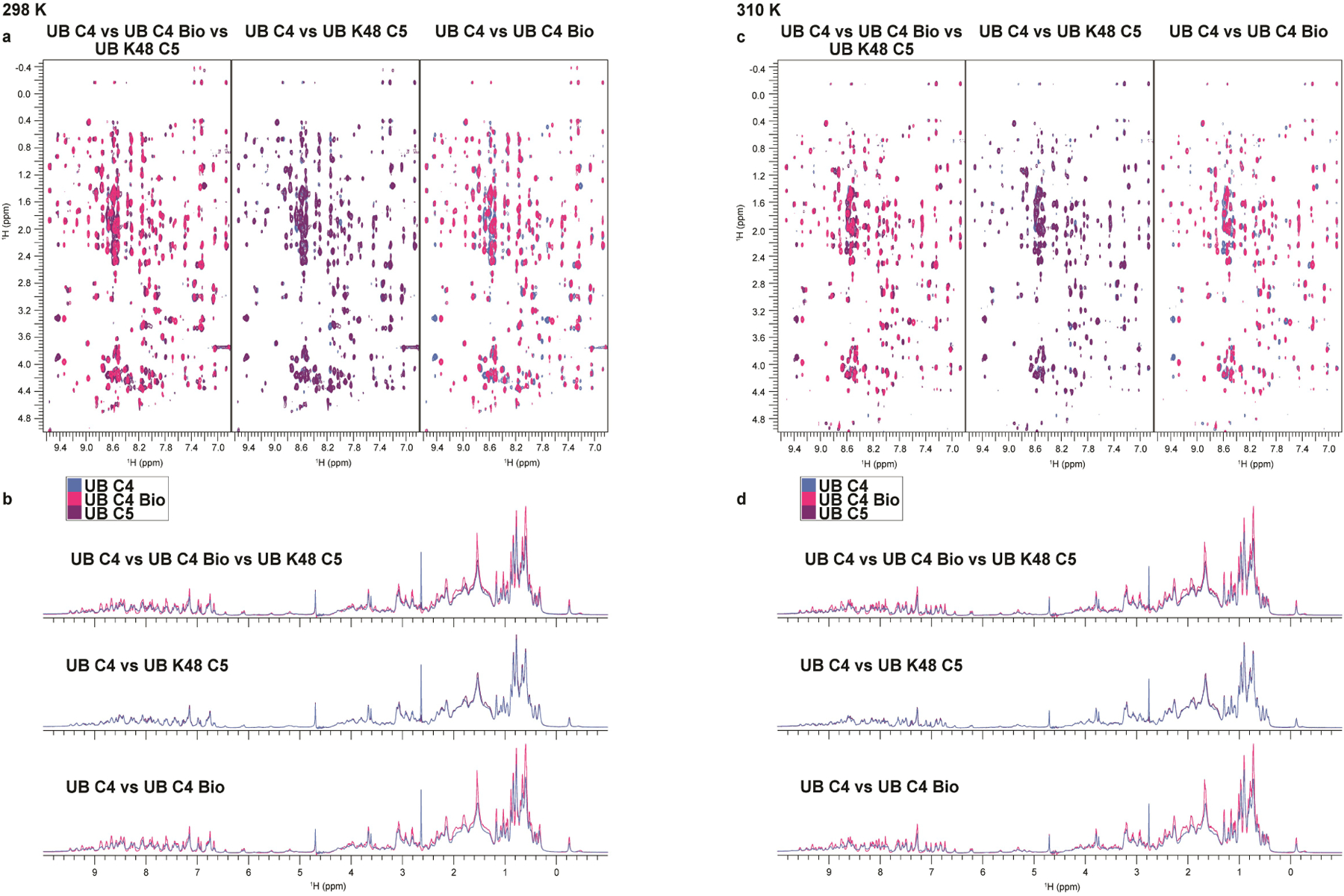
(a) 2D Nuclear Overhauser effect spectroscopy (NOESY) recorded at 298 K and 1D spectra (b) for UBC4 (blue), recombinant UB (C4 Bio; pink) and K48UBC5 (purple). The 2D NOESY spectra show NOE interactions between amide protons (x-axis) and amino acid side-chain protons (y-axis), whereas the 1D spectra show signals from methyl protons (−0.5 – 1.0 ppm), Cα-protons (3.5 – 6 ppm) and amide protons (6 – 10 ppm). The signal from water is at 4.7 ppm1. The observed dispersion of signals demonstrates that all three UBs are well folded, while the comparable overlays indicate that the UBs share a highly similar fold. (c) same as (a), except data were recorded at 310 K. (d) same as (b), except at 310 K.
Extended Data Fig. 4. Lack of preference for a native lysine on acceptor UBs for the K11 chain-forming E2 UBE2S.
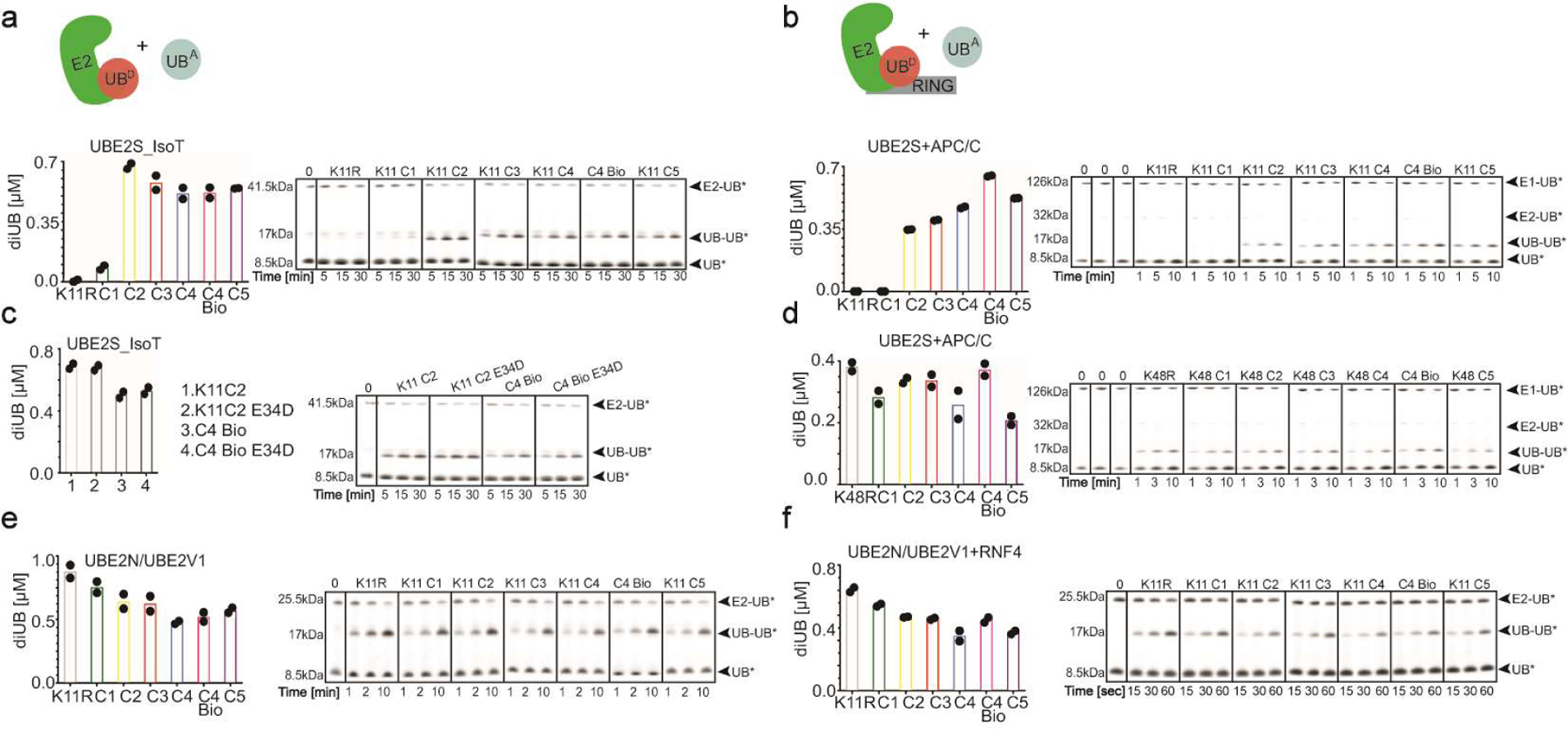
(a) Cartoon of experimental scheme monitoring the reactivity of E2s with K11UBC1-C5 acceptors (UBA). Plot of the discharge of labeled UB (UB*) from UBE2S_IsoT to K11RUB, UBC4 Bio or K11UBC1–5 acceptors (left) and representative fluorescence scans of SDS-PAGE gels representing the primary data (right). (b) same as (a), except in the presence of the E3 APC/C. (c) same as (a), except in the presence of K11UBC2 or UBC4 Bio acceptors or the same harboring an E34D mutation. (d) same as (b), except in the presence of K48RUB, UBC4 Bio or K48UBC1–5 acceptors. (e) same as (a), except with UBE2N/UBE2V1. (f) same as (b), except with UBE2N/UBE2V1 and the RING domain from the E3 RNF4. For all plots graphs, di-UB levels (μMol) represent the final time-points from the reactions (N=2 independent experiments). For samples derived from the same experiment, gels were processed in parallel.
Extended Data Fig. 5. The location of lysine analogues on acceptor UB impacts the distribution of diUB chain linkage types generated by the E2 enzyme UBE2D3.
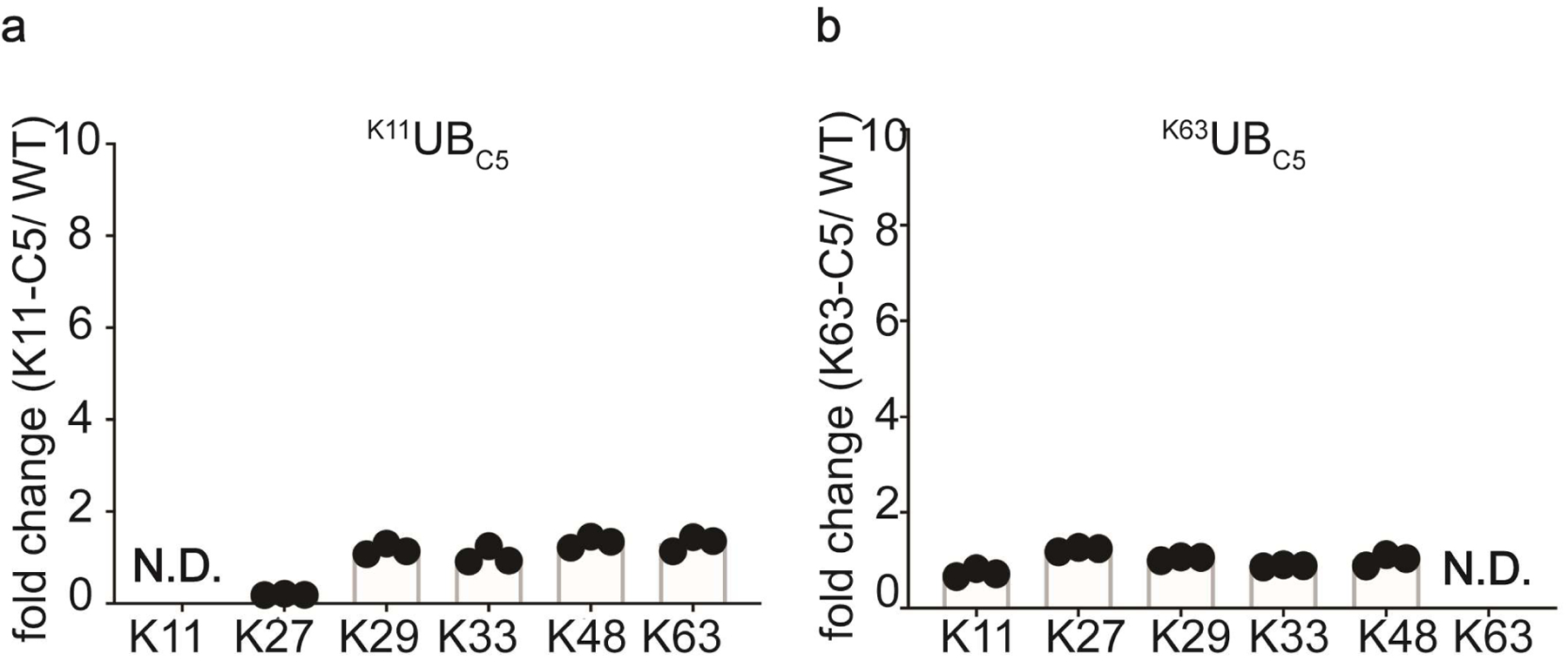
(a) Plots showing the relative changes in UBE2D3 generated di-UB chain linkages in the presence of the RING domain from the E3 RNF4, comparing products containing K11UBC5 or UBC4 acceptors (N=3 technical replicates). (b) Same as (a), except with K63UBC5 acceptor.
Extended Data Fig. 6. Molecular Dynamics simulations reveal pleiotropic structural effects on UBs harboring lysine analogs.
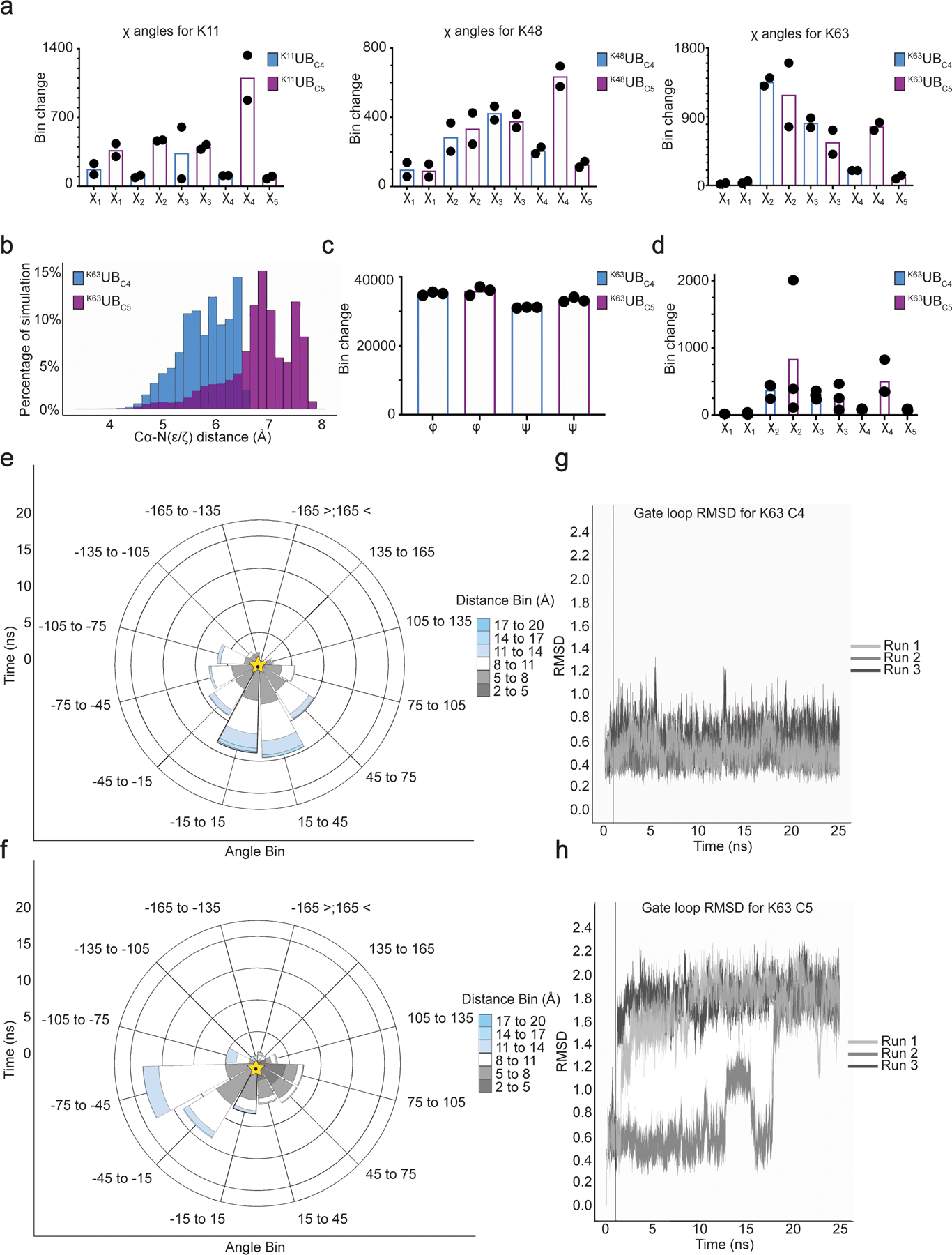
(a) Plot showing the degree of various side-chain rotamer interconversions for K11UBC5, K48UBC5, or K63UBC5 versus UBC4 acceptor UBs. Bins are divided by 120° intervals. (b) Distribution of the distances between lysine acceptor amine and Cα atoms for UBC4 versus UBC5 during 25 ns MD simulations (N=3 independent experiments) for the UBE2N~UB/UBE2V1/acceptor UB multi-subunit complex. Bins are divided by 10° intervals. (c) Plot showing the dynamics of phi and psi main-chain torsion angles for UBC4 or UBC5 acceptors in the UBE2N~UB/UBE2V1/acceptor UB multi-subunit complex. Bins are divided by 10° intervals. (d) Plot showing the dynamics of the side-chain rotamers for UBC4 or UBC5 acceptors in the UBE2N~UB/UBE2V1/acceptor UB multi-subunit complex. Bins are divided by 120° intervals. (e) Rose plot showing the distance and angle of the amine acceptor of UBC4 relative to the active-site during 25 ns MD simulations of the UBE2N~UB/UBE2V1/acceptor UB multi-subunit complex (N=3 independent experiments). Golden star indicated starting position. (f) same as (e), but with K63UBC5 (g) RMSD of gate loop during the trajectory for UBC4. (h) same as (g), except with K63UBC5.
Extended Data Fig. 7. Di-UB chain formation reactions with UBC4 or UBC5 acceptors produce distinct results depending on the identity of the ubiquitin carrying enzyme.
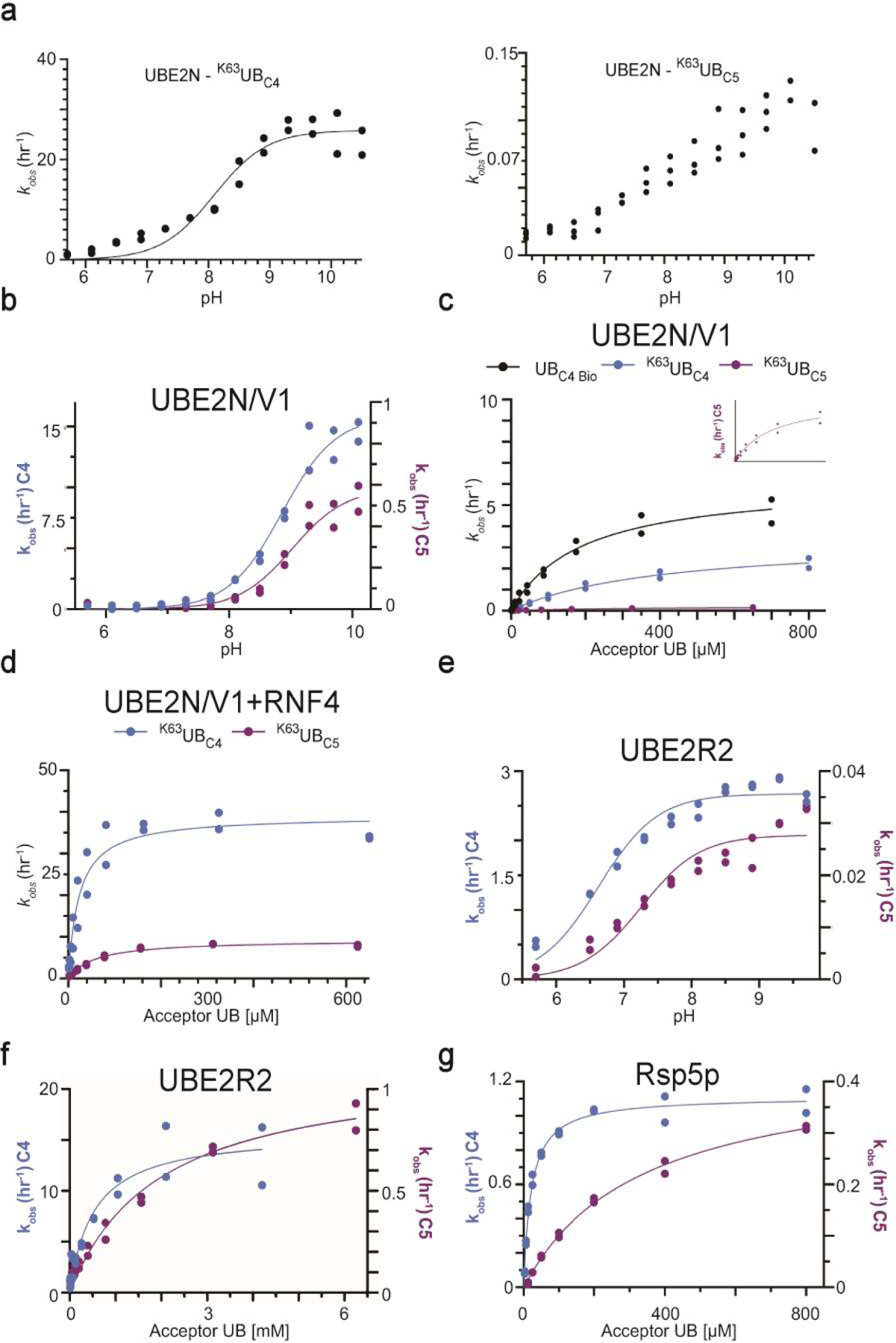
(a) Graph of the reactions velocities as a function of pH. performed in the presence of wild-type UBE2N/UBE2V1, radio-labeled K63R donor UB and either K63UBC4 or K63UBC5 acceptor UBs (b) same as (a), except with K92R UBE2N/UBE2V1. (c) Graph of the reaction velocities as a function of the acceptor UB concentration for UBE2N/UBE2V1. The inset shows the fit of the data to the model for reactions containing K63UBC5 acceptor UB since the magnitude of the velocities is far less than reactions containing UBC4. (d) same as (c) except in the presence of the RING domain of RNF4. (e) Graph of the reaction velocities performed as a function of pH, in the presence of UBE2R2 and radio-labeled K48R donor UB and either UBC4 or K48UBC5 acceptors. (f) Graph of the reaction velocities as a function of the acceptor UB concentration and their fit to the Michaelis-Menten model for UBE2R2. (g) same as (f), except in the presence of the yeast HECT E3 Rsp5p. N=2 independent experiments. For samples derived from the same experiment, gels were processed in parallel.
Supplementary Material
Acknowledgements
This study is dedicated the memory of our inspiring mentor, colleague, and beloved friend, Huib Ovaa, whom we miss dearly. We thank J.R. Prabu, J. Kellermann, S. von Gronau, D. Scott, S. Uebel, S. Pettera, V. Sanchez, K. Baek, D. Horn-Ghetko and S. Kostrhon for assistance, reagents, and helpful discussions. We also thank C. Talavera-Ormeño and P. Hekking for assistance in peptide synthesis. B.A.S has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 789016-NEDD8Activate), and from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation - SCHU 3196/1-1). B.A.S and M.M. are supported by the Max Planck Society. Also, N.P., D.H., N.B., and G.K. were supported by a grant from the National Institutes of Health (R15GM117555-02). G.J.v.d.H.v.N was supported by grants from NWO (VIDI and Off-Road). H.O. was supported by a VICI grant from the Netherlands Foundation for Scientific Research (NWO). Work from M.S. and M.B. was performed within the framework of SFB 1035 (German Research Foundation DFG, Sonderforschungsbereich 1035, Projektnummer 201302640, project Z01).
Footnotes
Competing Interests statement
H.O. was a shareholder of UbiqBio. All other authors declare no competing interests.
Code availability
ROSETTA software can be downloaded from www.rosettacommons.org and is available free to academic users.
Data availability
All raw gels are included in Source Data files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021286.
References
- 1.Dikic I, Wakatsuki S & Walters KJ Ubiquitin-binding domains — from structures to functions. Nature Reviews Molecular Cell Biology 10, 659–671 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komander D & Rape M The Ubiquitin Code. Annual Review of Biochemistry 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Yau R & Rape M The increasing complexity of the ubiquitin code. Nat Cell Biol 18, 579–586 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Kwon YT & Ciechanover A The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem Sci 42, 873–886 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Mulder MPC, Witting KF & Ovaa H Cracking the Ubiquitin Code: The Ubiquitin Toolbox. Curr Issues Mol Biol 37, 1–20 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Singh SK et al. Synthetic Uncleavable Ubiquitinated Proteins Dissect Proteasome Deubiquitination and Degradation, and Highlight Distinctive Fate of Tetraubiquitin. J Am Chem Soc 138, 16004–16015 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Sun H et al. Diverse fate of ubiquitin chain moieties: The proximal is degraded with the target, and the distal protects the proximal from removal and recycles. Proceedings of the National Academy of Sciences 116, 7805–7812 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X et al. An Interaction Landscape of Ubiquitin Signaling. Mol Cell 65, 941–955.e948 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Buetow L & Huang DT Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol 17, 626–642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng N & Shabek N Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 86, 129–157 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Mattiroli F & Sixma TK Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol 21, 308–316 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Victor Bernier-Villamor DAS, Michael J Matunis, Christopher D.Lima. Structural Basis for E2-Mediated SUMO Conjugation Revealed by a Complex between Ubiquitin-Conjugating Enzyme Ubc9 and RanGAP1. Cell 108, 345–356 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Eddins MJ, Carlile CM, Gomez KM, Pickart CM & Wolberger C Mms2–Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nature Structural & Molecular Biology 13, 915–920 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J & Rape M The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell 144, 769–781 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smit JJ et al. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. The EMBO journal 31, 3833–3844 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berndsen CE, Wiener R, Yu IW, Ringel AE & Wolberger C A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat Chem Biol 9, 154–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stieglitz B et al. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 503, 422–426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wenzel DM, Lissounov A, Brzovic PS & Klevit RE UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pao KC et al. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 556, 381–385 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Virdee S, Macmillan D & Waksman G Semisynthetic Src SH2 domains demonstrate altered phosphopeptide specificity induced by incorporation of unnatural lysine derivatives. Chem Biol 17, 274–284 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Temimi AHKA et al. Lysine Possesses the Optimal Chain Length for Histone Lysine Methyltransferase Catalysis. Scientific Reports 7, 16148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna S et al. Noncovalent interaction between ubiquitin and the human DNA repair protein Mms2 is required for Ubc13-mediated polyubiquitination. J Biol Chem 276, 40120–40126 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Branigan E, Plechanovová A, Jaffray EG, Naismith JH & Hay RT Structural basis for the RING-catalyzed synthesis of K63-linked ubiquitin chains. Nature Structural & Molecular Biology 22, 597–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branigan E, Carlos Penedo J & Hay RT Ubiquitin transfer by a RING E3 ligase occurs from a closed E2~ubiquitin conformation. Nature Communications 11, 2846 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petroski MD & Deshaies RJ Mechanism of Lysine 48-Linked Ubiquitin-Chain Synthesis by the Cullin-RING Ubiquitin-Ligase Complex SCF-Cdc34. Cell 123, 1107–1120 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Choi Y-S et al. Differential ubiquitin binding by the acidic loops of Ube2g1 and Ube2r1 enzymes distinguishes their Lys-48-ubiquitylation activities. J Biol Chem 290, 2251–2263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hill S, Harrison JS, Lewis SM, Kuhlman B & Kleiger G Mechanism of Lysine 48 Selectivity during Polyubiquitin Chain Formation by the Ube2R1/2 Ubiquitin-Conjugating Enzyme. Mol Cell Biol 36, 1720–1732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu G et al. UBE2G1 governs the destruction of cereblon neomorphic substrates. eLife 7, e40958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill S et al. Robust cullin-RING ligase function is established by a multiplicity of poly-ubiquitylation pathways. eLife 8, e51163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skaar JR, Pagan JK & Pagano M Mechanisms and function of substrate recruitment by F-box proteins. Nature Reviews Molecular Cell Biology 14, 369–381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kronke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maspero E et al. Structure of a ubiquitin-loaded HECT ligase reveals the molecular basis for catalytic priming. Nature Structural & Molecular Biology 20, 696–701 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Kamadurai HB et al. Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. eLife 2, e00828–e00828 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HC & Huibregtse JM Polyubiquitination by HECT E3s and the Determinants of Chain Type Specificity. Molecular and Cellular Biology 29, 3307 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta R et al. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol Syst Biol 3, 116–116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bremm A, Freund SMV & Komander D Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nature structural & molecular biology 17, 939–947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown NG et al. Dual RING E3 Architectures Regulate Multiubiquitination and Ubiquitin Chain Elongation by APC/C. Cell 165, 1440–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brzovic PS & Klevit RE Ubiquitin transfer from the E2 perspective: why is UbcH5 so promiscuous? Cell Cycle 5, 2867–2873 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Swatek KN et al. Insights into ubiquitin chain architecture using Ub-clipping. Nature 572, 533–537 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee BL, Singh A, Mark Glover JN, Hendzel MJ & Spyracopoulos L Molecular Basis for K63-Linked Ubiquitination Processes in Double-Strand DNA Break Repair: A Focus on Kinetics and Dynamics. J Mol Biol 429, 3409–3429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garg P et al. Structural and Functional Analysis of Ubiquitin-based Inhibitors That Target the Backsides of E2 Enzymes. J Mol Biol 432, 952–966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rout MK et al. Stochastic gate dynamics regulate the catalytic activity of ubiquitination enzymes. J Am Chem Soc 136, 17446–17458 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Yunus AA & Lima CD Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nature Structural & Molecular Biology 13, 491–499 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Vittal V et al. Intrinsic disorder drives N-terminal ubiquitination by Ube2w. Nat Chem Biol 11, 83–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones WM, Davis AG, Wilson RH, Elliott KL & Sumner I A conserved asparagine in a ubiquitin-conjugating enzyme positions the substrate for nucleophilic attack. J Comput Chem 40, 1969–1977 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Wang M, Cheng D, Peng J & Pickart CM Molecular determinants of polyubiquitin linkage selection by an HECT ubiquitin ligase. Embo j 25, 1710–1719 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott DC et al. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell 157, 1671–1684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell (2020). [DOI] [PMC free article] [PubMed]
- 50.Verma R, Mohl D & Deshaies RJ Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol Cell 77, 446–460 (2020). [DOI] [PubMed] [Google Scholar]
Additional references for online methods
- 51.Scott DC et al. Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell 166, 1198–1214.e1124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baek K et al. NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly. Nature (2020). [DOI] [PMC free article] [PubMed]
- 53.Brown NG et al. Mechanism of polyubiquitination by human anaphase-promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell 56, 246–260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plechanovova A et al. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat Struct Mol Biol 18, 1052–1059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sievers QL et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weissmann F et al. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc Natl Acad Sci U S A 113, E2564–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duda DM et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ziemba A et al. Multimodal mechanism of action for the Cdc34 acidic loop: a case study for why ubiquitin-conjugating enzymes have loops and tails. J Biol Chem 288, 34882–34896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rappsilber J, Ishihama Y & Mann M Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75, 663–670 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Kulak NA, Pichler G, Paron I, Nagaraj N & Mann M Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods 11, 319–324 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw gels are included in Source Data files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021286.