

SUMMARY
The phosphorylation of mitotic proteins is bistable, which contributes to the decisiveness of the transitions into and out of M phase. The bistability in substrate phosphorylation has been attributed to bistability in the activation of the cyclin-dependent kinase Cdk1. However, more recently it has been suggested that bistability also arises from positive feedback in the regulation of the Cdk1-counteracting phosphatase PP2A-B55. Here, we demonstrate biochemically using Xenopus laevis egg extracts that the Cdk1-counter-acting phosphatase PP2A-B55 functions as a bistable switch, even when the bistability of Cdk1 activation is suppressed. In addition, Cdk1 regulates PP2A-B55 in a biphasic manner; low concentrations of Cdk1 activate PP2A-B55 and high concentrations inactivate it. As a consequence of this incoherent feedforward regulation, PP2A-B55 activity rises concurrently with Cdk1 activity during interphase and suppresses substrate phosphorylation. PP2A-B55 activity is then sharply downregulated at the onset of mitosis. During mitotic exit, Cdk1 activity initially falls with no obvious change in substrate phosphorylation; dephosphorylation then commences once PP2A-B55 spikes in activity. These findings suggest that changes in Cdk1 activity are permissive for mitotic entry and exit but that the changes in PP2A-B55 activity are the ultimate trigger.
In Brief
Mitotic transitions are accompanied by drastic changes in the phosphorylation state of proteins. Kamenz et al. demonstrate biochemically that the major mitotic phosphatase PP2A-B55 is regulated by incoherent feedforward and double-negative feedback loops to promote rapid and switch-like mitotic entry and exit.
Graphical Abstract
INTRODUCTION
The dramatic events of mitosis, including chromosome condensation, nuclear envelope breakdown, and spindle assembly, are driven by the collective phosphorylation of hundreds of proteins1. Dephosphorylation of these proteins promotes the transition out of mitosis. The majority of mitotic phosphorylations are mediated by cyclin-dependent kinase 1 (Cdk1)2–4 and counteracted by a number of phosphatases, of which PP1 and PP2A-B55 have emerged as particularly important.5–8
Until recently, the changes in substrate phosphorylation that drive mitotic entry and exit were mainly viewed in terms of the changing activity of Cdk1. Cdk1 gradually rises in activity during interphase due to cyclin synthesis.9–12 Cdk1 activity then spikes at the onset of M phase as a result of the toggling of a bistable circuit13–15 involving Wee1/Myt1, negative regulators of Cdk1,16–20 and the Cdk1 activator Cdc25,21,22 from a high Wee1/low Cdc25 state to a low Wee1/high Cdc25 state. Once Cdk1 is fully activated, mitotic entry occurs. Shortly thereafter, the Cdc20-bound form of the anaphase-promoting complex/cyclosome (APC/CCdc20)23,24 becomes activated and tags the mitotic cyclins for rapid degradation by the proteasome.25–27 This inactivates Cdk1 and mitotic exit occurs.
However, it is now appreciated that along with these changes in kinase activity there are marked changes in the activities of PP1 and PP2A-B55; both are negatively regulated by Cdk1 activity and therefore decrease in activity during M phase.6,8,28–31 Moreover, the regulation of both phosphatases involves positive or double-negative feedback that could make them act as bistable switches. Indeed, based on computational modeling, Vinod and Novak proposed that at least PP2A-B55 functions as a bistable switch.32 This hypothesis has been supported by in vitro reconstitution experiments. A system comprising Cdk1, PP2A-B55, and proteins that are part of the double-negative feedback loop regulating PP2A-B55 activity produces bistability in the phosphorylation of a model substrate even if Cdk1 activity does not exhibit bistability.33 Furthermore, bistability in mitotic entry and exit was observed in human cells in response to the titration of Cdk1 activity. This bistability was only abolished when both the Cdk1 and PP2A-B55 feedback loops were impaired.34
In view of these findings, we have set out to determine the precise roles of kinase and phosphatase regulation in triggering the dramatic events of mitotic entry and exit. To this end, we have used Xenopus laevis egg extracts, which can be made to proceed through the cell cycle ex cellulo and which are unusually amenable to biochemical analysis and manipulation. Our main findings are that (1) PP2A-B55 gradually increases in activity during interphase, suppressing the phosphorylation of mitotic phosphoproteins; (2) PP2A-B55 does, as hypothesized, function as a bistable switch, with its activity changing about 20-fold between its on state and its off state; (3) in quantitative terms, mitotic entry is driven primarily by phosphatase inactivation; and (4) mitotic exit is driven primarily by phosphatase reactivation. These findings suggest that changes in Cdk1 activity are permissive for mitotic entry and exit but changes in phosphatase activity are instructive.
RESULTS
Cdk1 Substrate Phosphorylation Is Bistable Even When Cdk1 Activity Is Not
Previous work in Xenopus extracts has shown that the activity of Cdk1 and the phosphorylation states of its substrates exhibit bistability and hysteresis.35,36 At low concentrations of cyclin B the system settles into a state of low Cdk1 activity and low substrate phosphorylation independent of the initial condition—the system is monostable. Similarly, high concentrations of cyclin B always force the system to settle into a state of high Cdk1 activity and high substrate phosphorylation—again the system is monostable. But at intermediate cyclin B concentrations, either of two alternative stable steady states is possible—the system is bistable. At these intermediate concentrations, the initial condition (the history of the system) determines which state the system is going to settle into: low Cdk1 activity/low phosphorylation if the system comes from interphase, or high Cdk1 activity/high phosphorylation if the system comes from M phase.35,36 It seemed reasonable to assume that the observed bistability in Cdk1 activity is causal to the observed bistability in substrate phosphorylation.
Yet, there is evidence suggesting that bistability in mitotic substrate phosphorylation may persist even when bistability in Cdk1 activity is eliminated. Wee1 activity is only essential for the first mitotic division in the model organism X. laevis and the Wee1-mediated inhibitory phosphorylations on Cdk1 are sharply downregulated in the later cell cycles, implying that bistability in Cdk1 activity is dispensable for the later embryonic cell cycles.37–39 Mitotic trigger waves, which in theory require positive feedback, continue to propagate through X. laevis egg extracts when Wee1 is maximally inhibited.40 In fission yeast, Wee1—essential in the wild-type strain—becomes dispensable in strains that are driven by the expression of a cyclin B-Cdk1 fusion protein.41,42 In addition, several Cdk1 substrates exhibit sharper phosphorylation thresholds during mitotic entry in extracts, where presumably all of the physiological regulators of their phosphorylation are present, than they do in vitro with purified Cdk1 and a constitutive phosphatase.43,44 The sharp phosphorylation thresholds persist even when Wee1 and Myt1 are inhibited.33,43–45
These findings, together with the reported potential of PP2A-B55 to exhibit bistability in vitro33, suggested that an additional bistable circuit acting on one or more of the Cdk1-opposing phosphatases helps to keep the G2/M transition switch-like even when Cdk1 activity is forced to be graded and monostable.
To deconvolve these possibilities, we set out to study substrate phosphorylation in the absence of Cdk1 bistability. To eliminate bistability in Cdk1 activation, we abolished the double-negative feedback regulation of Cdk1 by blocking the Wee1/Myt1-mediated inhibitory phosphorylations of Cdk1 at Thr 14 and Tyr 1516–20 using the Wee1/Myt1 inhibitor PD0166825 (Figure 1A).46 Because Cdc25 acts only on Thr/Tyr-phosphorylated Cdk1, the positive feedback loop is inhibited as well (Figure 1A). We began with titration experiments to determine the concentration of PD0166285 required to make Cdk1 activation become graded and monostable. The half-maximal inhibitor concentration (IC50) value for Wee1/Myt1 inactivation in extracts was found to be 1.4–3 μM, similar to values reported previously (Figures S1A–S1F).40 Based on these results, we used inhibitor concentrations between 5 and 25 μM whenever we aimed to eliminate the bistability of Cdk1 regulation.
Figure 1. Cdk1 Substrate Phosphorylation Is Bistable in the Absence of Cdk1 Bistability.

(A) Schematic of the regulation of substrate phosphorylation by Cdk1 and opposing phosphatases. Note that the Wee1 inhibitor PD0166285 compromises the positive and double-negative feedback loops that regulate the activity of Cdk1.
(B) Schematic of the hysteresis experiment. Steady-state Cdk1 activity and substrate phosphorylation were measured as a function of non-degradable cyclin B1 (ΔN-Cyc B1) in the presence of the Wee1/Myt1 inhibitor PD0166285. The steady state was approached either starting from a state of low (interphase up; purple) or of high (M phase down; green) cyclin B concentration/Cdk1 activity.
(C and D) Cdk1 activity as a function of ΔN-Cyc B1 concentration (as monitored by histone H1 phosphorylation) is graded and monostable in the presence of the Wee1/Myt1 inhibitor (5 μM PD0166285). Autoradiograph of the histone phosphorylation is shown in (C). Quantification of two technical duplicates (circles) for the measurement and the mean of the duplicates (connecting lines) is shown in (D).
(E) The phosphorylation state of three Cdk1 substrates (APC3, Cdc25, and Nup53), monitored by mobility shift as a function of ΔN-Cyc B1 concentration. Note that despite the monostable response in Cdk1 activity shown in (C) and (D), these substrates still exhibit hysteretic responses.
(F and G) Quantification of the APC3 hyperphosphorylation for the experiment in (E), plotted as a function of ΔN-Cyc B1 concentration (F) and as a function of the corresponding Cdk1 activities (G) as measured in (D). Figure S1E shows a summary of multiple experiments for this measurement.
All activity and phosphorylation state analyses (C)–(G) were performed from the same experiment.
See Figure S1 for a detailed characterization of the Wee1 inhibitor (Figures S1A–S1F) and linearity tests for the antibodies (Figure S1H).
To look for hysteresis in substrate phosphorylation, we carried out experiments based on the general strategies that previously established the bistability of Cdk1 activation.35,36 We obtained M-phase-arrested egg extracts (cytostatic factor [CSF]-arrested extracts), added cycloheximide to abolish cyclin synthesis and PD0166285 to inhibit Wee1 and Myt1, and treated the extracts in one of two ways. We supplemented one portion of the extract with 0.8 mM CaCl2 to induce the degradation of the endogenous cyclin B and reach low Cdk1 activity, and then added back different concentrations of non-degradable Δ65-cyclin B1. This way, the system approaches steady state from a point of low Cdk1 activity (Figure 1B; referred to as “interphase up”). Alternatively, we first added the Δ65-cyclin B1, which binds to excess Cdk1 in the extract and adopts the active phosphorylation state, and only afterward added CaCl2 to induce the selective degradation of the endogenous cyclin B. These extracts therefore approach steady state from initially high Cdk1 activity (Figure 1B; referred to as “M phase down”). After allowing the system to reach steady state, we measured Cdk1 activity and assessed the phosphorylation state of a set of well-characterized Cdk1 substrates. If the interphase up and M phase down conditions yielded two different steady-state levels of substrate phosphorylation despite having a single steady-state level of Cdk1 activity, it would indicate that some phosphatase was bistable.
In the presence of the Wee1/Myt1 inhibitor, Cdk1 activity showed a graded, almost linear response to the titration of non-degradable cyclin B1 (Figures 1C and 1D). We consistently noted a small (~5–10 nM) threshold in the response (Figures 1D and S1F). This may at least in part result from the presence of the Cdk inhibitor Kix1/Xic1, which has a reported concentration of ~2 nM.47 More importantly, the system now settled into a state of similar Cdk1 activity irrespective of the starting point, arguing that abolishing the positive and double-negative feedback eliminated the bistability of Cdk1 activity (Figures 1C and 1D), as expected.
We then asked whether hysteresis still persisted in the phosphorylation of three Cdk1 substrates (Figures 1E): APC3/Cdc27, a subunit of the APC/C considered a late substrate of Cdk1 during mitotic entry; the phosphatase Cdc25, an early substrate; and a subunit of the nuclear pore complex, Nup53/MP44, an intermediate (or middle) substrate.48 All of these proteins exhibit phosphorylation-dependent changes in their electrophoretic mobility between interphase and M phase.
Although Cdk1 activity showed no hysteresis (Figures 1C and 1D), the three Cdk1 substrate proteins did (Figures 1E–1G and S1G). The cyclin B1 thresholds at which the substrates transitioned between hypo- and hyperphosphorylation differed from substrate to substrate, but in all cases the responses were hysteretic: higher cyclin B1 concentrations were needed to obtain hyperphosphorylation in the interphase up condition than were needed to maintain hyperphosphorylation in the M phase down condition. Therefore, substrate phosphorylation is hysteretic and bistable even when Cdk1 activity is not.
APC/C Activity Is Bistable in the Absence of Cdk1 Bistability
The Cdk1-mediated activation of APC/CCdc20 is a crucial feature of the negative feedback at the core of the embryonic cell-cycle oscillator (Figures 1A and 2A).45,49 An ultrasensitive or even bistable behavior in the activation of APC/CCdc20 in response to Cdk1 activity could contribute to the robustness of this oscillator.45 We had observed bistability in the hyperphosphorylation of the APC/C subunit APC3 (Figures 1E–1G and S1G), and this phosphorylation may be important for regulating the activity of the APC/C.49–51 However, because the hyperphosphorylation of APC3 does not always correlate with the activity of the APC/C,45 we set out to directly measure the dose response of APC/C activity as a function of the non-degradable cyclin B1 concentration.
Figure 2. APC/C Activity Is Bistable in the Absence of Cdk1 Bistability.

(A) Schematic of the system used to assess APC/C activity. ΔN-Cyc B1 titration experiments were performed as described in Figure 1B and steady-state APC/C activity was measured by following the degradation of a fluorescently labeled APC/C substrate (securin-CFP) using a plate reader.
(B) APC/C is activated during mitosis. Significant loss of fluorescent signal was only detected in an M phase extract (red) but not in an interphase extract (blue) or in an extract not supplemented with securin-CFP (black). Shown is the mean (circles with connecting line) and the standard deviation (error band) of a technical triplicate.
(C) Securin-CFP degradation dynamics after adding different concentrations of ΔN-Cyc B1, approaching steady state starting from either a state of high (M phase down; green) or low (interphase up; purple) cyclin B concentration/Cdk1 activity. Shown are data from two technical replicates (circles or squares; the dashed or solid lines, respectively, show the exponential fit of the data). Note that at intermediate concentrations of ΔN-Cyc B1 (10 or 15 nM), the steady-state level of APC/C activity depends upon whether the system has come from interphase or M phase.
(D) Quantitation of the apparent first-order rate constant for APC/C activity plotted as a function of ΔN-Cyc B1 for the experiment shown in (C), including additional ΔN-Cyc B1 concentrations. Shown is the average of a technical duplicate with standard deviation.
(E) Quantitation of the apparent first-order rate constant for APC/C activity (the same activities shown in D) plotted as a function of Cdk1 activity rather than non-degradable cyclin B concentration. Shown is the average of a technical duplicate with standard deviation.
(F) Quantification of the apparent first-order rate constant for APC/C activity as a function of ΔN-Cyc B1. Shown are mean and standard error of the mean from 4 independent experiments (for 20 nM ΔN-Cyc B1: n = 3; for 40 nM ΔN-Cyc B1: n = 2). Note that due to variability between experiments, the switch-like transition between low and high APC/C activity is less obvious in the averaged data than it is in the given single experiment (D and E).
All experiments were performed in the presence of 5 μM PD0166285.
To this end, we performed a titration experiment similar to that described in Figure 1 but, after allowing the extract to reach steady state, we added a fluorescently labeled substrate of the APC/C, securin-CFP, and followed substrate degradation using a fluorescence plate reader (Figures 2A and 2B).45 The apparent rate constant for degradation serves as a gauge of APC/C activity.
As previously reported, in the absence of non-degradable Δ65-cyclin B1 (interphase), APC/C activity was low, whereas at high concentrations of Δ65-cyclin B1 (M phase), APC/C activity was high (Figure 2B). At low (0 nM) and high (40 nM) concentrations of Δ65-cyclin B1, the degradation kinetics did not depend on the initial starting conditions (Figures 2C–2F). However, at intermediate concentrations of Δ65-cyclin B1 (e.g., 10 and 15 nM in Figures 2C and 2D), APC/C activity was high if the extract came from M phase and low if it came from interphase. This suggests that not only the phosphorylation state of APC/C but also the activity of APC/C is regulated in a hysteretic, bistable fashion even when Cdk1 activity is forced to be graded and monostable.
Note that protein synthesis is blocked in these experiments and therefore Emi2/XErp1, an additional regulator of APC/C activity that normally becomes synthesized during the first embryonic interphase52 and could be a source for bistability in APC/C activity,53 is absent.
PP1 Does Not Exhibit Bistability
Because substrate phosphorylation remained bistable even when Cdk1 activation was graded, we inferred it must result from bistability in the regulation of a counteracting phosphatase. Theoretically, Cdk1-mediated feedback regulation of either PP1 or PP2A-B55 could give rise to bistability (Figures 3A and 4A). In the case of PP1, two interlinked loops raise the possibility of bistability: Cdk1 inhibits PP1 by phosphorylating threonine 320 (T320)28,54,55 and PP1 may activate itself by dephosphorylating T320 in an autocatalytic circuit. PP1 is further inhibited through binding to phosphorylated inhibitor-1 (I1) and can release itself from I1-mediated inhibition by dephosphorylation8.
Figure 3. PP1 T320 Phosphorylation Does Not Exhibit Bistability.

(A) Schematic of the regulation of PP1 activity by Cdk1 and inhibitor-1 (I1).
(B and C) Two independent experiments examining whether the phosphorylation of the C-terminal regulatory tail of PP1 at T320, which can be taken as a measure of PP1 activity, is hysteretic. Although Nup53 phosphorylation showed bistable behavior in both experiments, PP1 T320 phosphorylation was graded and monostable. 10 μM PD0166285 was used to inhibit Wee1/Myt1 in (B) and 5 μM in (C).
Figure 4. PP2A-B55 Activity, but Not Gwl Activity, Exhibits Bistability When Cdk1 Activity Is Graded and Monostable.

(A) Schematic of the regulation of PP2A-B55 activity by Cdk1 via Greatwall kinase (Gwl) and ENSA/Arpp19. Two double-negative feedback loops, a shorter one involving only PP2A-B55 and Arpp19/ENSA and a longer one involving Gwl kinase and PP2A-B55, could give rise to bistability.
(B and C) The steady-state activity of PP2A-B55 is hysteretic in the presence of the Wee1/Myt1 inhibitor (10 μM PD0166285). Mitotic substrate phosphorylation (monitored by the mobility shift of Nup53) in (B) and PP2A-B55 activity in (C) are shown as a function of non-degradable cyclin B1 (ΔN-Cyc B1), approaching steady state from either a state of high (M phase down; green) or low (interphase up; purple) cyclin B concentration/Cdk1 activity (mean of technical duplicates with connecting line). Two additional experiments are shown in Figures S2B–S2E.
(D) Gwl and ENSA phosphorylation exhibit bistability in the presence of the Wee1/Myt1 inhibitor (5 μM PD0166285). Phosphorylation states of several Cdk1 substrates including Gwl and ENSA were analyzed by immunoblotting. Figure S3A shows an additional experiment. 10 μM Phos-tag was used to enhance the mobility shift upon ENSA phosphorylation. A shorter exposure for the ENSA immunoblot and a quantification of the phosphorylated form of ENSA are shown in Figures S3A and S3B. An additional experiment is shown in Figures S3C and S3D and an experiment detecting ENSA phosphorylation using a phospho-specific antibody against the phosphorylated S67 epitope of ENSA is shown in Figures S3I and S3J.
(E and F) Gwl kinase activity is ultrasensitive but not bistable. Cdk1 and Gwl kinase activities were measured for the experiment shown in (D) and are depicted as the dose response of Gwl kinase activity as a function of ΔN-Cyc B1 (E) or Cdk1 activity (F). Shown is the mean of a technical duplicate (circles). Although not bistable, the dose response of Gwl kinase activity exhibits significant ultrasensitivity as demonstrated by the large Hill exponent (nH) necessary to fit the data (solid lines in E and F). Additional experiments are shown in Figures S3G–S3L.
Thus, we set out to determine whether the response of PP1 to Cdk1 was bistable or monostable by using the phosphorylation state of T320 as a proxy for PP1 activity; the higher the T320 phosphorylation, the lower the activity. In contrast to the Cdk1 substrates shown in Figures 1 and 2, PP1 T320 phosphorylation showed a linear, monostable response to Δ65-cyclin B1 concentration and Cdk1 activity, with little if any hysteresis (Figures 3B and 3C). This finding suggests that PP1 is not the phosphatase responsible for the observed bistability in substrate phosphorylation and APC/C activity (Figures 1 and 2). The lack of hysteresis in PP1 phosphorylation underscores the fact that the presence of positive feedback and double-negative feedback does not guarantee that a system will exhibit bistability.
PP2A-B55 Is Regulated in a Bistable, Biphasic Fashion
We next investigated whether PP2A-B55 exhibits bistability (Figure 4A) as hypothesized.32–34 In mitosis, Cdk1 phosphorylates and activates Greatwall (Gwl).30,56–58 Active Gwl then phosphorylates two closely related proteins, Arpp19 and ENSA,59,60 which in their phosphorylated state bind and inhibit PP2A-B55 by an “unfair competition” mechanism.61 PP2A-B55 promotes its own release from this inhibition by dephosphorylating Arpp19/ENSA and by dephosphorylating Gwl,62–64 although PP1 also significantly contributes to Gwl dephosphorylation.63–65 Therefore, there are at least two double-negative feedback loops that regulate PP2A-B55 and could potentially generate bistability: one between PP2A-B55 and Gwl, and one between PP2A-B55 and the stoichiometric inhibitors ENSA and Arpp19 (Figure 4A).
We measured PP2A-B55 activity based on the release of inorganic phosphate from a 32P-labeled peptide encompassing the serine 50 site of Cdc20 (Figure S2A). 66 The specificity of this assay has been established through immunodepletion experiments, which showed that PP2A-B55, and in particular PP2A-B55δ, accounts for most of the phosphatase activity toward this substrate in Xenopus extracts.6 As previously reported,6 PP2A-B55 activity was significantly higher in interphase than in M phase (Figure S2A).
We then carried out a hysteresis experiment and measured mitotic substrate phosphorylation (Figure 4B) as well as PP2A-B55 activity (Figure 4C). In the interphase up leg of the experiment, PP2A-B55 activity was found to increase about 4-fold as the Δ65-cyclin B1 was raised from 0 to 25 nM, and it then decreased about 20-fold once mitotic concentrations (40 nM) of cyclin were reached (Figure 4C). Thus, the response of PP2A-B55 to Δ65-cyclin B1 was biphasic, with low Cdk1 activities positively regulating the phosphatase and high Cdk1 activities negatively regulating it.
The response was also hysteretic, with a higher concentration of Δ65-cyclin B1 required to inhibit PP2A-B55 coming from interphase than was required to maintain its inactivation coming from M phase (Figure 4C, purple versus green curves). Note that the biphasic response was seen in both the interphase up and M phase down legs of the experiment (Figure 4C), and that both the biphasic response and the bistability were consistently observed (Figures S2B–S2E).
To determine whether the double-negative feedback between PP2A-B55 and Gwl and/or PP2A-B55 and ENSA/Arpp19 was important for the observed bistability in PP2A-B55 activity, we assessed the phosphorylation state of Gwl and ENSA/Arpp19. As shown in Figures 4D, S3C, and S3I, the hyperphosphorylation of Gwl was hysteretic under conditions where PD0166285 had eliminated hysteresis in Cdk1 activity, suggesting that the PP2A-B55-Gwl double-negative feedback might be critical for the bistability of PP2A-B55.
Similarly, we detected some hysteresis in the phosphorylation response of ENSA and Arpp19, either by resolving the different phosphorylation forms using Phos-tag gels (Figures 4D and S3A–S3D) or using a phospho-specific antibody (Figures S3I and S3J) raised against the Gwl target sites serine 67 and serine 62 in ENSA and Arpp19, respectively.
These findings suggested that the PP2A-B55-Gwl double-negative feedback might be critical for the bistability of PP2A-B55. However, although PP2A-B55 has been shown to dephosphorylate Gwl, it has more recently been shown that the activity of Gwl is mainly regulated by PP1 rather than PP2A-B55.63–65
Furthermore, the hyperphosphorylation of Gwl only weakly correlates with its activity.63 Therefore, we asked whether the observed bistability in Gwl phosphorylation actually translated into bistability in Gwl activity.
To measure Gwl activity, we used a recombinant Arpp19 protein that carried alanine mutations in two prominent non-Gwl phosphorylation sites (S28 and S109; Arpp19–2A) as an in vitro substrate and monitored incorporation of radioactive phosphate from [γ−32P]ATP, as previously described (Figures S3E and S3F).63 Although we had clearly detected hysteresis in the phosphorylation state of Gwl, we did not detect hysteresis in Gwl activity. The response curves were ultrasensitive but indistinguishable for extracts coming out of interphase versus M phase (Figures 4E, 4F, S3G, and S3H). Note that in our enzymatic assays to monitor Gwl activity we used okadaic acid (OA), a PP2A inhibitor. Although this is commonly done in order to preserve the phosphorylation and activity state of Gwl during the measurement, it could have altered the outcome of the measurement. However, even when we isolated Gwl from extracts and measured Gwl activity in the absence of OA, we did not detect bistability in Gwl activity (Figures S3I–S3L).
We cannot formally exclude the possibility that our assay is too noisy to detect some small degree of bistability in Gwl activity. However, given that we did not detect bistability in PP1 activity and PP1 is thought to be an important regulator of Gwl activity, we favor the possibility that bistability in ENSA/Arpp19 phosphorylation and PP2A-B55 activity does not arise from double-negative feedback between PP2A-B55 and Gwl but that instead the feedback interaction between PP2A-B55 and Arpp19/ENSA is responsible for the observed hysteresis in PP2A-B55 regulation.
Partially Inhibiting PP2A Activity Abolishes the Bistability in Cdk1 Substrate Phosphorylation
If indeed the bistability of PP2A-B55 activity is responsible for the observed bistability in substrate phosphorylation and APC/C activity, then partially inhibiting PP2A-B55 might abolish this bistable behavior. This could either be a result of disturbing the balance between the two legs of the PP2A-B55-Arpp19/ENSA double-negative feedback loop or because phosphatases other than PP2A become dominant in regulating the phosphorylation of these substrates when PP2A-B55 is inhibited. We first chose to decrease PP2A-B55 activity using OA (Figure 5A). OA is a high-affinity inhibitor of PP2A with an IC50 of about 0.14 nM in vitro67 but also inhibits PP4 and at higher concentrations PP1 and PP5.60 Therefore, we carried out titration experiments to identify a concentration of OA where the balance between Cdk1-mediated phosphorylation and PP2A-B55-mediated dephosphorylation was slightly shifted toward the phosphorylation reaction but PP1 activity was not impaired. Concentrations of 0.4–0.6 μM OA induced subtle changes in the phosphorylation states of several Cdk1 substrates (Cdc25, Nup53, Wee1, and Gwl but not APC3) in the presence of low amounts of non-degradable cyclin B1 but did not affect the phosphorylation state of T320 of PP1 (Figure S4A), suggesting that at these concentrations we had partially inhibited PP2A but not PP1. Accordingly, we set out to see whether these low concentrations of OA would have any effect on the hysteresis seen in Cdk1 substrate phosphorylation.
Figure 5. Dampened PP2A Activity Weakens the Bistability in Cdk1 Substrate Phosphorylation.

(A) Okadaic acid (OA) was used to partially inhibit the activity of PP2A-B55 and the feedback between PP2A-B55 and ENSA/Arpp19.
(B and C) The dose-response relationship of the phosphorylation state of the APC/C subunit APC3 as a function of ΔN-Cyc B1, approaching steady state starting from either a state of high (M phase down) or low (interphase up) cyclin B concentration/Cdk1 activity in the presence of the Wee1/Myt1 inhibitor (25 μM PD0166285) and in the absence (DMSO) or presence (OA) of different concentrations of OA. OA decreased the hysteresis seen in the DMSO control and made intermediate phosphorylation states more apparent. Note that the DMSO and OA comparisons were performed in parallel using the same extract, but (B) and (C) were performed using different extracts; biological variability may account for the differences in the overall response between the two DMSO control experiments.
(D) Thiophosphorylated Arpp19 (Arpp19-S) was used to partially inhibit the activity of PP2A-B55 and the feedback between PP2A-B55 and ENSA/Arpp19.
(E and F) The dose-response relationship of the phosphorylation state of Nup53 as a function of ΔN-Cyc B1, approaching steady state starting from either a state of high (M phase down) or low (interphase up) cyclin B concentration/Cdk1 activity in the presence of the Wee1/Myt1 inhibitor (10 μM PD0166285) and in the absence (DMSO) or presence (Arpp19-S) of different concentrations of Arpp19-S. Arpp19-S decreased the hysteresis seen in the DMSO control and made intermediate phosphorylation states more apparent. Note that the DMSO and Arpp19-S comparison was performed in parallel using the same extract, but for (E) and (F) was performed using different extracts; biological variability may account for the differences in the overall response between the two DMSO control experiments, and variability in the activity of the ΔN-Cyc B1 preparation might account for overall differences in the response curve when comparing (C) and (D) to (E) and (F).
See also Figure S4.
In the presence of the Wee1 inhibitor PD0166285, 0.5 μM OA changed the dose response of APC3 phosphorylation in three ways. First, the dose-response curve shifted toward lower non-degradable cyclin B1 concentrations; on the interphase up leg, half-maximal phosphorylation was obtained at a cyclin concentration of ~8 nM, compared with 30–40 nM in the absence of OA (Figure 5B, compare top versus bottom panels). Second, the phosphorylation of APC3 became more graded; whereas in the absence of OA, APC3 exhibited mainly two prominent phosphorylation states (a hypo- and a hyperphosphorylated state), addition of OA yielded a variety of APC3 forms of intermediate mobility in a dose-dependent manner (Figure 5B). Finally, the bistability in the dose-response relationship was almost completely abolished (Figure 5B); the phosphorylation responses starting in interphase and starting in M phase were very similar. Increasing the OA concentration to 0.6 μM potentiated all of these effects (Figure 5C).
The thiophosphorylated form of Arpp19 has been shown to be a specific, high-affinity inhibitor of PP2A-B55 with an IC50 of 1.47 nM in vitro.61,68 We therefore used thiophosphorylated Arpp19 (Arpp19-S) as an alternative way of inhibiting PP2A-B55 and perturbing the feedback regulation (Figure 5D). We used nominal concentrations of between 0.4 and 1.7 μM Arpp19-S to inhibit PP2A-B55 (although the actual inhibitor concentration was estimated to be about 50% lower due to incomplete thiophosphorylation of Arpp19). In the presence of 25 nM non-degradable cyclin B1, addition of Arpp19-S but not unphosphorylated Arpp19 induced significant changes in the phosphorylation states of Nup53 and Gwl (Figure S4B). These concentrations are consistent with stoichiometric inhibition of PP2A-B55 in the extract, where the catalytic subunit of PP2A is present at about 0.3 μM.69 At higher concentrations of non-degradable cyclin B1, both thiophosphorylated and unphosphorylated Arpp19 were able to induce changes in the phosphorylation states of Nup53 and Gwl with the thiophosphorylated Arpp19 being more potent. Similar results were reported previously.68
When comparing the dose response of substrate phosphorylation with or without Arpp19-S in the presence of the Wee1 inhibitor PD0166285, we observed effects similar to the ones we had observed with low doses of OA. In particular, we observed that in the presence of Arpp19-S, half-maximal substrate phosphorylation occurred at lower concentrations of cyclin B1, and that the response overall seemed more graded with more intermediate phosphorylation bands being detected (Figures 5E and 5F). The impact of Arpp19-S on bistability was less obvious in these experiments than it had been using OA. These findings leave open the possibility that some phosphatase in addition to PP2A-B55 contributes to the observed hysteresis.
Both the Cdk1 and the PP2A Switches Contribute to the Bistability in Substrate Phosphorylation
So far, we have shown that PP2A-B55 regulation is bistable even when Cdk1 activity is not bistable. However, in the absence of PD0166285 and in the first cell cycle of an embryo, both the Cdk1 switch and the PP2A-B55 switch should be operative. The relative contributions of the two switches to substrate phosphorylation are unclear; therefore, we asked whether any hysteresis would remain when the PP2A-B55 loop was compromised by OA treatment but the Cdk1/Wee1/Cdc25 system was not compromised by PD0166285 treatment.
To this end, we attenuated PP2A activity using 0.5 μM OA and compared the dose response of substrate phosphorylation in the presence or absence of Wee1 inhibition. In the presence of OA (but no PD0166285), substrate phosphorylation was hysteretic (Figures 6A and 6B), whereas OA plus PD0166285 abolished hysteresis (Figures 6C and 6D) as we had observed previously (Figure 5B). We conclude that both the kinase switch and the phosphatase switch contribute to the bistability of substrate phosphorylation.
Figure 6. The Cdk1 and PP2A Switches Both Contribute to Bistability.

The dose-response relationship of the phosphorylation state of several Cdk1 substrates shown in (A) and (C) as well as the Cdk1 activity shown in (B) and (D) were measured in the presence of 0.5 μM OA and in the absence (A and B) or presence (C and D) of the Wee1/Myt1 inhibitor (10 μM PD0166285). Hysteresis was still detectable if only PP2A activity was partially inhibited, but was almost abolished if Wee1 was inhibited in addition. The measurements were performed in parallel using the same extract. Cdk1 activity is shown as the mean of a technical duplicate with connecting lines.
Substrate Dephosphorylation Lags behind Cdk1 Inactivation during Mitotic Exit
In order to investigate how the steady-state properties of Cdk1 and PP2A contribute to the temporal dynamics of substrate phosphorylation, we turned to cycling Xenopus egg extracts. These extracts carry out cell-cycle transitions fairly homogeneously and can be sampled with good temporal resolution, and thus allow for a detailed dynamic description of when the phosphorylation and dephosphorylation of various Cdk1 substrates occur.
We first focused on Cdk1 activity and the phosphorylation dynamics of a number of substrates. In the experiment shown in Figures 7A and 7B, at the first time point (45 min into the first mitotic cycle) the cyclin B2 levels were already ~75% maximal and the Cdk1 activity was ~50% maximal but there was no detectable phosphorylation of the 7 mitotic substrates shown. The onset of mitosis occurred at 47–49 min, and was marked by the phosphorylation of all 7 substrate proteins. Phosphorylation was maximal by ~52 min, with the late substrate APC3 lagging slightly behind the other substrates (Figure 7A). Over the same time interval, cyclin B2 levels were maximal and Cdk1 activity increased 2-fold (Figure 7B).
Figure 7. PP2A-B55 Activity Peaks prior to Mitotic Entry and during Mitotic Exit.

(A–C) Phosphorylation and dephosphorylation of Cdk1 substrates show a distinct time lag compared to the increase and decrease in cyclin B concentration and Cdk1 activity. The changes in Cdk1 activity (B and C; mean of a technical duplicate), cyclin B2 concentration (A and C), and the phosphorylation of several substrates (B) were measured with high temporal resolution in a cycling extract progressing through mitosis. Note that cyclin B2 and Cdk1 activity are minimal at 61 min whereas most substrates are still hyperphosphorylated (B and C; for Cdk1 activity and the phosphospecies [as a fraction of total signal], exponential decays were fitted to the declining part of the time course).
(D and E) PP2A-B55 activity peaks prior to mitotic entry and during mitotic exit. The phosphorylation of two mitotic substrates (D; quantification for Nup53 and PP1 T320 is shown in E in orange and green, respectively) as well as the concentration of cyclin B2 (D) and the activity of Cdk1 and PP2A-B55 (E; black and blue, respectively) were measured in a cycling extract progressing through mitosis. For the PP2A-B55 measurements, the assays were carried out on undiluted extracts; thus, the PP2A-B55 activity could be changing during the 3 min the phosphatase assay is performed. Accordingly, we have plotted the time of each PP2A-B55 measurement as the middle of this incubation period and show the range of the assay time as a horizontal line. Note that in contrast to PP2A-B55 activity, PP1 T320 phosphorylation closely follows the activity of Cdk1 (D and E).
An additional independent experiment is shown in Figures S5A, S5B, S6, and S7.
This increase was likely caused by flipping the Cdk1 switch, as Cdc25 hyperphosphorylation and Wee1 T150 phosphorylation became detectable at that time (Figures 7A and 7B). Gwl and ENSA also became phosphorylated around this time, suggesting that during mitotic entry the Cdk1 and the PP2A-B55 switches are engaged roughly contemporaneously.
Cyclin B2 levels and Cdk1 activity began to fall at ~54 min, were half-maximal by ~57 min, and were back to basal levels by ~61–62 min (Figures 7B and 7C). In contrast, most of the mitotic substrates did not begin to be dephosphorylated until about 63 min, and then decayed with half-times of ~2 min (Figures 7A and 7C).
PP1 and ENSA appeared to begin being dephosphorylated slightly before the other mitotic substrates at around 57–58 min (Figure 7C). Thus, there was a lag of about 6 min between the time when Cdk1 activity had fallen to 50% maximal—an activity that was apparently insufficient to induce substantial substrate phosphorylation during M phase entry—and when the dephosphorylation of most mitotic substrates began. This suggests that Cdk1 inactivation alone is insufficient to induce detectable substrate dephosphorylation.
The Dynamics of PP2A-B55 Activity Is a Major Determinant of the Timing of Substrate Phosphorylation during Mitotic Entry and Exit
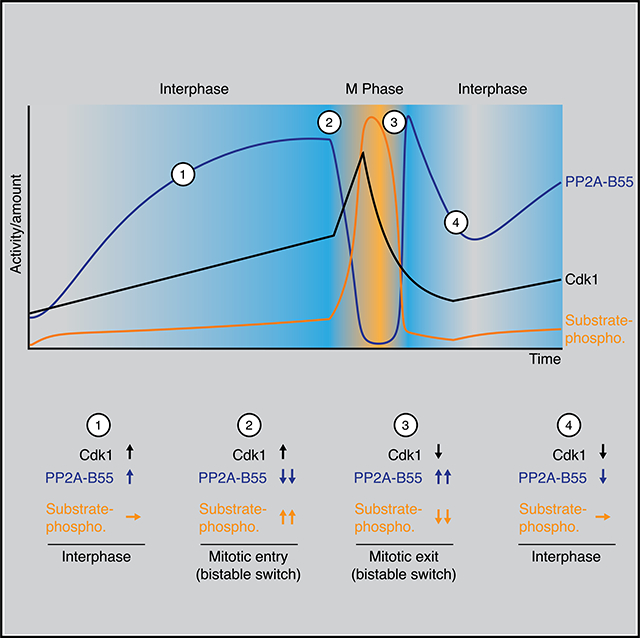
We also directly measured PP2A-B55 activity together with cyclin B2 levels, Cdk1 activity, and substrate phosphorylation during mitotic entry and exit. During interphase, as the cyclin B2 concentration and Cdk1 activity gradually rose about 2-fold, PP2A-B55 activity rose by ~4-fold (Figures 7E and S5B). This is consistent with the finding that sub-threshold levels of Cdk1 activity increase the steady-state activity of PP2A-B55 (Figure 4C). To our knowledge, this activation of PP2A-B55 during interphase has not been remarked upon previously.
However, it can be seen in time course data presented by Mochida and Hunt,66 but was not observed in a later publication by the same authors and colleagues.29 The increase in PP2A-B55 activity appeared to completely suppress the phosphorylation of APC3 and Nup53 (Figures 7D, 7E, S5A, and S5B).
At 56 min, PP2A-B55 activity began to fall, and by 60 min it had fallen 17-fold. The decrease in PP2A-B55 was closely followed by an additional ~2-fold increase in Cdk1 activity and the rapid accumulation of mitotic substrate phosphorylation.
PP2A-B55 activity remained low for the duration of mitosis but then showed a pronounced spike in activity during mitotic exit. The magnitude of the spike in PP2A-B55 activity was comparable to the PP2A-B55 activity right before mitotic entry, about 14-fold, and coincided with the onset of substrate dephosphorylation. PP2A-B55 activity eventually returned back to its basal interphase level before starting to increase again during the next cycle (Figures 7E and S5B). Thus, PP2A-B55 activity peaks at two different points in the cell cycle, just prior to mitotic entry and during mitotic exit. Moreover, the dynamics of PP2A-B55 activity appear to play a large role in determining substrate phosphorylation; during entry, a small increase in Cdk1 activity (~2-fold) is able to cause a large change in substrate phosphorylation because PP2A-B55 activity decreases ~17-fold and, during exit, the large change in Cdk1 activity due to cyclin proteolysis is unable to cause substrate dephosphorylation until PP2A-B55 activity increases sharply.
DISCUSSION
Here we have investigated the steady-state and temporal behavior of mitotic substrate phosphorylation and the roles of Cdk1, PP1, and PP2A-B55 in generating the dramatic events of mitotic entry and exit. We found that even when Cdk1 activity is forced to be a graded, monostable function of the cyclin B1 concentration, several mitotic phosphoproteins still exhibit bistable responses (Figure 1), including the critical mitotic regulator APC/C (Figure 2). Thus, even though the Wee1/Cdc25/Cdk1 system is essentially inoperative during embryonic cycles 2–12,37–39 the Cdk1 system as a whole still includes a bistable trigger13,15,70 and can still be viewed as a relaxation oscillator—an oscillator built from toggle switches. This class of oscillator is common in biology and, compared with oscillator circuits that are not built out of switches, tends to be especially robust and tunable.71 The bistability can be attributed to PP2A-B55: its regulators ENSA and Arpp19 exhibit bistable phosphorylation, and PP2A-B55 (but not Gwl) exhibits bistable activity changes (Figures 4, S2, and S3). In addition, perturbing PP2A activity with low concentrations of OA abolished this bistability (Figures 5 and 6). These findings agree well with in vitro reconstitution studies of the Xenopus PP2A-B55/Gwl system33 and provide a molecular mechanism to explain why it takes lower concentrations of a Cdk1 inhibitor to block mitotic entry than to induce mitotic exit.34 In contrast, there was no evidence for bistability in the response of PP1 (Figure 3). Instead, PP1 T320 phosphorylation closely tracked Cdk1 activity (Figure 7), suggesting that PP1 activity simply varies inversely with Cdk1 during mitotic entry and exit.
What Accounts for PP2A-B55/ENSA Bistability?
The observed bistability of the PP2A-B55/ENSA system presents a mechanistic conundrum. During M phase, Gwl-phosphorylated ENSA and Arpp19 act as stoichiometric inhibitors of PP2A-B55 by binding to the active site of the catalytic subunit of PP2A-B55.61 PP2A-B55 has an unusually high affinity for phospho-ENSA (~0.1 nM) and a low but non-negligible turnover rate (~0.03 s−1), so that once Gwl is inactivated during mitotic exit, PP2A-B55 can dephosphorylate all of the pENSA and pArpp19 (which are present at a concentration of ~1,000 nM, versus ~300 nM for the catalytic subunits of PP2A69) on a time-scale of ~100 s. This allows PP2A-B55 to gain access to lower-affinity mitotic phosphoproteins. This mechanism has been dubbed unfair competition, and other examples of unfair competition in biological regulation have now been found.43,72
Taken together, the PP2A-B55/ENSA system constitutes a double-negative feedback loop—pENSA/pArpp19 stoichiometrically inhibits PP2A-B55 and PP2A-B55 enzymatically inhibits pENSA/pArpp19—and double-negative feedback loops generally can give rise to bistability. But this particular type of double-negative feedback cannot.32,73–75 This led to the suggestion that double-negative feedback between PP2A-B55 and Gwl, rather than feedback between PP2A-B55 and pENSA/pArpp19, generates the bistability in the system.32
However, it appears that although PP2A-B55 dephosphorylates some residue(s) on Gwl, PP1 is responsible for inactivating Gw,l63 and we do not detect any bistability in Gwl activity (as opposed to Gwl hyperphosphorylation) (Figure 4). If feedback between PP2A-B55 and Gwl is not responsible for the bistability of the PP2A-B55/ENSA system, then what is?
Thron faced a similar issue in his theoretical studies of the interplay between the p21 and p27 Cdk inhibitors and the cyclin-Cdk complex.73,75 These inhibitors block the phosphorylation of other Cdk substrates through what could be called unfair competition and, conversely, Cdks can phosphorylate the inhibitors and tag them for proteolysis. The mechanism is thus similar to the situation with PP2A-B55 and ENSA/Arpp19: a double-negative feedback system that is not, at least in its simplest form, able to generate bistability.32,73,74 However, Thron was able to rescue bistability in his model by assuming that free cyclin-Cdk can phosphorylate the inhibitor both in cis and in trans.73,75 In STAR Methods and Figures S6 and S7, we implement an analogous model for the PP2A-B55/ENSA system.
The key to the model is an assumption that when phosphorylated ENSA/Arpp19 is bound to PP2A-B55, it can be dephosphorylated either in cis by the PP2A-B55 molecule it is bound to—in accordance with the unfair competition model61—or in trans, with PP2A-B55-bound ENSA/Arpp19 dephosphorylated by a second free PP2A-B55 molecule. How might trans dephosphorylation occur? Perhaps the pENSA phosphorylation site is not always buried in the PP2A-B55 active site. Indeed, the fact the pENSA inhibits PP2A-B55 but not other PP2A complexes suggests that pENSA might bind the specificity-determining B55 subunit (as, presumably, other PP2A-B55 substrates do) as well as the catalytic subunit.
Admittedly, though, the assumption that dephosphorylation can occur in trans is ad hoc; it is not required to account for the kinetics of pENSA dephosphorylation seen in vitro.61 However, trans dephosphorylation could have been obscured by the large excess of substrate used in these experiments.61 It may be interesting to re-examine this issue with both the enzyme and substrate present at physiological concentrations (hundreds nanomolar), if it is possible to achieve this.
There are at least three alternative possibilities to reconcile the bistability observed here for PP2A-B55 activity with the apparent lack of bistability in Gwl activity. (1) Some phosphatase other than PP2A-B55 could contribute to ENSA/Arpp19 inactivation, although Goldberg and co-workers have provided direct experimental evidence against this possibility.61 (2) There could actually be some small degree of bistability in the regulation of Gwl despite our inability to detect it. (3) The reactivation of PP2A-B55 after a decrease in Cdk1 activity could be too slow to allow PP2A-B55 to reach steady state in the M phase down legs of our hysteresis experiments although, if so, it is not clear how PP2A-B55 could be reactivated in a timely fashion during mitotic exit in vivo.
Cdk1 as an Activator of PP2A-B55
In addition, we found that Cdk1 activity at sub-threshold levels activates PP2A-B55. This is apparent in both the steady-state response of PP2A-B55 (Figures 4 and S2) and in the time course studies (Figures 7 and S5). This incoherent feedforward regulation has a number of significant consequences for the systems-level behavior:
Because PP2A-B55 activity gradually increases in parallel to Cdk1 activity when approaching mitosis, the kinase-to-phosphatase ratio remains low and substrates remain dephosphorylated during this time. Indeed, no mitotic substrate phosphorylation is detectable until PP2A-B55 is inactivated (Figures 7 and S5). It has been pointed out that the reciprocal regulation of Cdk1 and its counteracting phosphatases avoids energy-intensive futile cycles of phosphorylation and dephosphorylation.60 Although this energy-saving measure may be beneficial when keeping the system in a stable state, by itself it would render the system slower to respond.76 Upregulating the phosphatase in addition to the kinase activity prior to mitotic entry increases futile cycling and the flux in the system, without changing the net phosphorylation, which allows for a rapid and abrupt transition from hypo- to hyperphosphorylation once the phosphatase is inactivated. Consistent with this idea, mitotic entry has recently been associated with high energetic costs during the embryonic cell cycle of zebrafish.77
Whereas the differences between the basal and mitotic activities of PP2A-B55 are 4- to 5-fold, the additional activation of PP2A-B55 during the ramp up to mitosis results in an overall 15- to 20-fold change in PP2A-B55 activity at the time of mitotic entry. Note that by comparison, the activity of Cdk1 changes in a relatively subtle fashion during mitotic entry, with the activity increasing about 2-fold in X. laevis (here and in Solomon et al.,11 Pomerening et al.,78 Murray et al.,79 and Kumagi and Dunphy,80 and in other organisms as well81,82). Thus, the changes in PP2A-B55 appear to be the main determinants of the quantitative changes in substrate phosphorylation during mitotic entry.
The spike in PP2A-B55 activity during mitotic exit ensures that substrates are rapidly dephosphorylated. Indeed, although there is a lag between the time when Cdk1 falls to sub-mitotic levels and substrate dephosphorylation commences, once it begins, substrate dephosphorylation is essentially complete within 2–3 min (Figure 7). This allows substrate phosphorylation to be reversed rapidly and abruptly during mitotic exit. The mechanism of activation is unknown, but we suspect that it is indirect. This is based on the fact that such an activation has not been observed in in vitro reconstitution assays.33 Direct phosphorylation of threonine 304 of the catalytic subunit of the PP2A-B55 trimer by Cdk1 has recently been reported in human cells in cell culture.31 However, this phosphorylation has been implicated in a negative rather than positive regulation of PP2A-B55 activity by destabilizing the trimeric complex during mitosis.31,83 In X. laevis egg extracts, on the other hand, cell-cycle-dependent changes of PP2A-B55 complex formation have not been observed.6
Flipping the Bistable Switch
PP2A-B55 is thought to directly dephosphorylate Wee1 and Cdc25,6 thereby negatively regulating Cdk1 activity via tyrosine 15 phosphorylation. This raises the question of how such a system could switch into a state of high Cdk1 activity if Cdk1 steadily upregulates PP2A-B55. One explanation could be that an increase in the basal activity of Y15-phosphorylated cyclin B-Cdk1 complex due to increasing cyclin B levels suffices for flipping the switch. Such an increase is consistent with the slow but steady increase in Cdk1 activity during interphase that we observe in Figure 7E and with previous reports from human cells (e.g., Akopyan et al.9). Other activities might also contribute to the flipping of the switch, for example cyclin A-Cdk1, which is not inhibited by Wee1/Myt1.84
Note that during both mitotic entry and mitotic exit the changes in Cdk1 appear to be permissive, whereas the changes in PP2A-B55 activity appear to be instructive. Nevertheless, in the Xenopus system, Cdk1 does appear to be the master regulator of M phase; cyclin accumulation drives Cdk1 activation, and then Cdk1 activation drives PP1 and PP2A-B55 inactivation. However, in other systems, it is possible that the trigger for mitosis could initially act upon any of these components—the central players Cdk1, PP1, and PP2A-B55, or the intermediaries Wee1, Cdc25, Gwl, and ENSA/Arpp19—because they are all interconnected by feedback loops.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Enquiries on reagents and resources should be directed to, and will be fulfilled by the lead contact, Julia Kamenz (j.l.kamenz@rug.nl).
Materials Availability
Plasmids generated in this study will be made freely available upon request.
Data and Code Availability
No datasets or code accompany this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Egg extract preparation
Eggs were collected from female Xenopus laevis frogs 16–18 hours after egg laying was induced by injection of 500 U of human chorionic gonadotropin (Chorulon from Merck, Sigma #CG10–10VL). Cytostatic factor- (CSF-) arrested and cycling extracts were prepared as described previously,89 except that for cycling extracts eggs were activated with the calcium ionophore A23187 (200 ng/μL, Sigma Aldrich # C7522) rather than electric shock. CSF extracts were stored at −80°C until use; cycling extracts were used directly after preparation.
Animal work was conducted in accordance with relevant national and international guidelines and all animal protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care (APLAC protocol 13307).
METHOD DETAILS
Titration of non-degradable cyclin B1
CSF extracts were thawed at 20° C for 5 min. Extracts were supplemented with cycloheximide (100 μg/mL) and where applicable with the Wee1/Myt1 inhibitor PD0166285 (Abcam, ab219507) or okadaic acid (EMD Millipore, #495609). The extract was then treated in one of two ways: (1) Aliquots of the extract were supplemented with different concentrations of recombinant non-degradable cyclin B1 (ΔN-Cyc B1) purified from insect cells, incubated for 1 h at 20°C and then supplemented with CaCl2 to a final concentration of 0.8 mM and incubated for another hour (‘M phase down’) or (2) aliquots of the extracts were first treated with 0.8 mM CaCl2 for 1 h at 20°C and then supplemented with different concentrations of non-degradable cyclin B1 and incubated for another hour (‘Interphase up’). All samples were mixed regularly during the incubation.
Immunoblotting and antibodies
Protein samples were resolved on 10% Criterion Tris-HCl precast gels (Biorad, #3450011) or in the case of the phosphorylated species of Arpp19 and ENSA on a 10% Tris-HCl SDS-PAGE supplemented with 10 μM Phos-tag reagent (NARD Institute, AAL-107) and then transferred onto PVDF membranes. The following antibodies were used for detection of the respective proteins: mouse α-Cdc27 (BD Biosciences, #610455), mouse α-cyclin B2 (Santa Cruz Biotechnology, #sc-53239), rabbit α-Nup53 serum,85 rabbit α-PPP1A pT320 (Abcam, #ab62334), rabbit α-Cdc25C,44 rabbit α-Wee1 pT150,86 rabbit α-Greatwall serum,60 rabbit α-ENSA serum,60 rabbit α-Arpp19,63 rabbit α-ENSA pS67/Arpp19 pS62 (Cell Signaling Technology, #5240) and rabbit α-Cdk1 pY15 (Cell Signaling Technology, #9111L). For the detection of the primary antibody, peroxidase-linked sheep α-mouse IgG (GE Healthcare, #NA931V) and AMDEX goat α-rabbit IgG (GE Healthcare, RPN4301) was used. Chemiluminescence was detected on a BioRad ChemiDoc MP Imaging system using SuperSignal West Maximum Sensitivity Substrate (ThermoFisher Scientific, #34095) or Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, #WBLKS0500).
H1 kinase activity assay
H1 kinase assays followed the previously described protocol.89 In brief, 2 μL frozen extract sample were diluted in 98 μL EB buffer (80 mM β-glycerophosphate, 20 mM EGTA, 15 mM MgCl2, pH 7.4). Ten microliters of diluted extract were mixed with 10 μL reaction buffer (20 mM HEPES pH 7.5, 5 mM EGTA, 10 mM MgCl2, 200 mM ATP, 10 μg histone H1 (Millipore, #14–155), 20 μM PKA inhibitor IV (Santa Cruz Biotechnology, #sc-3010) and 2.5 μCi [γ−32P]-ATP) and immediately incubated for 3 min at 20°C. The reaction was stopped by adding 20 μL 3x SDS-PAGE loading dye. Five microliters of each sample were run on a 10% Criterion Tris-HCl precast gel (Biorad, #3450011), transferred onto a PVDF membrane and dried. The radiolabelled histone H1 was then detected using a BAS Storage Phosphor screen (GE Healthcare) and read out using a Phosphorimager Typhoon 8600 (Molecular Devices).
APC/C activity assay
APC/C activity assays were performed as described previously.45 Briefly, securin-CFP was in vitro translated using the TNT SP6 high-yield wheat germ protein expression system (Promega, #L3261). Three or 4 μL of in vitro translated securin-CFP was added to 50 or 80 μL extract. The extract was split into aliquots of 20 μL and added to a black 384-well plate (Greiner, #781076). Securin-CFP signal was followed using a plate reader (Molecular Devices Flexstation II). The degradation rate was calculated by normalizing the data to the starting value and the background control and fitting a single exponential decay. All measurements were performed in duplicate or triplicate.
PP2A-B55 activity assay
PP2A-B55 activity assays were performed as previously described66 with slight modifications. In brief, maltose-binding protein fused to amino acids 38 to 62 of Xenopus laevis Cdc20L (Fizzy) was recombinantly expressed in E. coli and affinity purified using amylose resin. Without elution, about 1 mg of protein was phosphorylated in 200 μL kinase reaction buffer (20 mM HEPES pH 7.7, 10 mM MgCl2, 15 mM KCl, 1 mM EGTA, 5 mM NaF, 20 mM β-glycerophosphate, 10 μM ATP, 2.5 μg cyclin A2/CDK2 (Sigma, #C0495), 60 μCi [γ−32P]-ATP) overnight at 37°C. The resin was extensively washed and the labeled substrate eluted with elution buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM maltose). The substrate was concentrated and stored at −20°C until use.
For measuring PP2A-B55 activity, 1 μL of substrate (> 15,000 cpm) was added to 5 μL of extract and incubated for 12 min at 20°C (3 min for the time course measurements). The reaction was stopped by adding 20 μL 10% ice-cold TCA and stored on ice until further processing. Samples were spun for 10 min at 14,000 g and 20 μL of the supernatant was transferred to a fresh tube. Thirty microliters of 5% ammonium molybdate in 0.5 M sulfuric acid was added and mixed. Fifty microliters of water-saturated heptane/butanol was added and the solution vortexed for 30 s. The solution was spun for 10 min at 14,000 g and 30 μL of the organic upper phase was used for detection of the inorganic phosphate using a scintillation counter.
Greatwall kinase activity assay
Greatwall kinase activity assays were performed following the previously described protocol.63 In brief, 1 μL egg extract was diluted in 16.5 μL reaction buffer (80 mM β-glycerophosphate, 20 mM EGTA, 15 mM MgCl2, 10 μM okadaic acid, 1x protease inhibitor (Roche, cOmplete #11873580001), 100 μM ATP, 10 μM PKA inhibitor IV (Santa Cruz Biotechnology, #sc-3010), 120 nCi/μL [γ−32P]ATP, 0.05 μg/μL Arpp19). The reaction was incubated at 20°C for 3 min and stopped by adding 17.5 μL of 3x SDS loading dye. Five microliters of each sample were run on a 4%–20% Criterion Tris-HCl precast gel (Biorad, #3450034), transferred onto a PVDF membrane and dried. The radio-labeled Arpp19 was then detected using a BAS Storage Phosphor screen (GE Healthcare) and read out using a Phosphorimager Typhoon 8600 (Molecular Devices). Alternatively, Gwl was first immunoprecipitated from 5 μL of extract using rabbit α-Greatwall serum60 coupled to magnetic Protein G beads (Invitrogen, #10004D) for 20 min at 20°C under shaking. The beads were washed three times with PBS and all supernatant was discarded. Gwl kinase activity was then measured as described above except that the beads were resuspended in 16.5 μL reaction buffer that did not contain okadaic acid.
Purification and thiophosphorylation of Arpp19
Purification and thiophosphorylation of Arpp19 were performed as described previously63 with slight modifications. In brief, N-terminally His-tagged Arpp19 (X. laevis) was overexpressed and purified from E. coli BL21(RIL) via affinity purification. mRNA encoding 3xFlag-Xl-Gwl (K71M, full length) was transcribed in vitro using an mMESSAGE mMACHINE kit (Invitrogen, #AM1344), poly(A)tailed using a Poly(A) tailing kit (Invitrogen, #AM1350), and purified using the MEGAclear Transcription Clean-Up kit (Invitrogen, #AM1908).
Freshly prepared CSF extract (200 μL) was supplemented with 2.5 μM okadaic acid, 1.4 μM non-degradable cyclin B (ΔN90-cyclin B from sea urchin) and 8 μg Gwl K71M mRNA in order to translate and phosphorylate 3xFlag-Gwl K71M. The translated protein was immunoprecipitated using 1.2 mg of anti-Flag coupled magnetic beads (Sigma-Aldrich, #F1804, covalently bound to Protein G beads (Invitrogen, #10004D)). Beads were washed 3 times briefly and 3 times for 5 min with CSF-XB buffer (10 mM HEPES pH 7.7, 100 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 50 mM sucrose, 0.5 mM EGTA,1 mM MgCl2). Full-length Arpp19 (100 μg) in phosphorylation buffer (10 mM Tris-HCl pH 7.4, 10 mM KCl, 30 mM NaCl, 0.5 mM EGTA, 20 mM beta-glycerophosphate, 5 mM MgCl2, 1 mM DTT, 1 mM MnCl2, 30 nM okadaic acid, 1 mM ATP-γ-S) was added to the beads and incubated overnight at 30° C. Subsequently, the beads were removed and the supernatant dialyzed against storage buffer (20 mM HEPES pH 7.7, 150 mM KCl, 10% glycerol, 1 mM DTT).
Mathematical modeling
Here we implement a model for the regulation of PP2A-B55 by phosphorylated ENSA/Arpp19, the regulation of ENSA/Arpp19 by PP2A-B55, and the regulation of the whole system by active cyclin B-Cdk1. The goal is to account for (1) the bistability of the PP2A-B55/ENSA system, (2) the biphasic dependence of PP2A-B55 activity on cyclin B-Cdk1, and (3) the temporal dynamics of PP2A-B55 regulation, with two peaks of activity per cell cycle.
Accounting for the observed bistability
We begin with a simpler model (Figure S6A), ignoring the activation of PP2A-B55 by Cdk1, because this makes it easier to understand how bistability can arise in the system. We focus on the total concentration of phosphorylated ENSA—free pENSA as well as pENSA complexed with PP2A-B55, which we denote pENSAtot. For pENSAtot to be at steady state, the rate of ENSA phosphorylation must equal the rate of pENSAtot dephosphorylation. The rate of phosphorylation is:
| (Equation 1) |
where ENSA is the concentration of free, unphosphorylated ENSA and Gwlact is the fraction of the Gwl that is activated at this concentration of Cdk1. At steady state, we assume that Gwlact is a Hill function of Cdk1 with a Hill coefficient of 5 (which is consistent with the range of Hill coefficients seen in Figure S3). And since ENSA is the total ENSA concentration minus pENSAtot, it follows that:
| (Equation 2) |
Thus the phosphorylation rate is maximal when pENSAtot is zero (and ENSA is maximal), and it falls to zero when pENSAtot = ENSAtot (and ENSA is zero). The phosphorylation rate curves are shown in Figure S6B in purple, for three assumed values of Cdk1 activity.
Following Thron,73,75 we assume that there are two ways for pENSAtot to be dephosphorylated: in cis and in trans. It follows that the rate of dephosphorylation of pENSAtot is given by:
| (Equation 3) |
where complex is the concentration of pENSA·PP2A-B55. The concentration of the complex at steady state can be written as a function of pENSAtot, PP2Atot, and the rate constants:
| (Equation 4) |
| (Equation 5) |
By substituting Equation 5 into Equation 3 we obtain an expression for the dephosphorylation rate as a function of pENSAtot and the parameters, which is plotted in green in Figure S6B. Note that the rate is independent of Cdk1. Note also that the shape of the curve is non-monotonic. As pENSAtot increases, the rate of pENSAtot dephosphorylation initially increases due mainly to the trans-dephosphorylation of the complexes, but as pENSAtot increases further, the amount of free PP2A-B55 available to decrease the complexes in trans falls to zero, and only the cis- contribution to the dephosphorylation remains. With a dephosphorylation rate curve shaped like this, one can easily find Cdk1 values that make the phosphorylation rate curve intersect the dephosphorylation curve at three points, yielding a bistable system with two stable steady states and an unstable steady state (Figure S6B). And if ENSA phosphorylation is bistable, then PP2A activity and substrate phosphorylation will be bistable as well, even if Cdk1 activity is monostable. See Table S1 for a description of the chosen parameters.
In summary, one way to account for the experimentally-observed bistability in PP2A-B55 under conditions where the bistability of Cdk1 has been eliminated (via the Wee1 inhibitor PD0166285) is to assume that phosphorylated ENSA can be dephosphorylated both in cis, as envisioned in the original unfair competition model,61 and in trans, as proposed by Thron in his analysis of the regulation of Cdk2 by p21 and p27.73
Accounting for Cdk1 activation of PP2A-B55 together with bistable inhibition of PP2A-B55
So far we have envisioned that there is only one type of PP2A-B55 and one type of PP2A-B55 complex. But experimentally it appears that Cdk1 can bring about activation of PP2A-B55. To account for this, we assume that Cdk1 can phosphorylate and activate PP2A-B55, as shown in Figure S7A, and that some constitutive phosphatase can return PP2A-B55 to its low activity state. We assume that there are then two types of PP2A-B55 complexes: one with PP2A-B55 in its low activity conformation (complex1) and one with it in its high activity conformation (complex2). We assume that complex2 dephosphorylates in cis 10x faster than complex1 (β = 0.1). We also assume that either complex can be dephosphorylated in trans by either type of free PP2A-B55, and that the activity of the free PP2A-B55 determines the rate of trans dephosphorylation.
These assumptions yield a 7-ODE model:
| (Equation 6) |
| (Equation 7) |
| (Equation 8) |
| (Equation 9) |
| (Equation 10) |
| (Equation 11) |
| (Equation 12) |
To calculate steady-state responses (Figures S7B and S7C), we fixed the value of Cdk1, set the derivatives to zero, and numerically solved the system algebraic equations in Mathematica 12.0. To calculate time courses (Figure S7D), we assumed that Cdk1 activity increased linearly with time (at 0.007 activity units per min) over the first 53 min, then linearly but faster (at 0.1 activity units per min until 59 min), and then fell exponentially with a first-order rate constant of 0.2 units−1min−1 until 75 min, and then solved the ODEs numerically in Mathematica 12.0. The chosen parameters can be found in Table S2.
The resulting model accounts for the biphasic nature of PP2A regulation, the bistability of PP2A inactivation, and the qualitative features of the time course experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical analyses (e.g., mean, standard deviation, standard error) were performed using Prism 8.0.2 or 8.4 (Graphpad). Statistical parameters, e.g., number of experiments (n) are described in the figure legends of the respective figure. All experiments have been performed in at least two biological replicates (often many more) and for all main conclusions we show a replicate in the main or supplemental figures. Securin degradation rate was calculated by normalizing the data to the starting value and the background control and fitting a single exponential decay using Python 3. Hill exponents were fit using Prism 8.0.2 or 8.4 (Graphpad).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse α-Cdc27 | BD Biosciences | #610455, RRID:AB_397828 |
| mouse α-cyclin B2 | Santa Cruz Biotechnology | #sc-53239, RRID:AB_629356 |
| rabbit α-Nup53 serum | 85 | |
| rabbit α-PPP1A pT320 | Abcam | #ab62334, RRID:AB_956236 |
| rabbit α-Cdc25C | 44 | |
| rabbit α-Wee1 pT150 | 86 | |
| rabbit α-Greatwall serum | 60 | |
| rabbit α-ENSA serum | 60 | |
| rabbit α-Arpp19 | 63 | |
| rabbit α-Cdk1 pY15 | Cell Signaling Technology | #9111L, RRID:AB_331460 |
| rabbit α-ENSA pS67/Arpp19 pS62 | Cell Signaling Technology | #5240, RRID:AB_11220425 |
| mouse α-FLAG M2 | Sigma-Aldrich | #F1804, RRID:AB_262044 |
| peroxidase-linked sheep α-mouse IgG | GE Healthcare | #NA931V, RRID:AB_772210 |
| AMDEX goat α-rabbit IgG | GE Healthcare | RPN4301, RRID:AB_2650489 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PD0166285 | Abcam | ab219507 |
| Okadaic acid | EMD Millipore | #495609 |
| 10% Criterion Tris-HCl precast gel | Biorad | #3450011 |
| 4–20% Criterion Tris-HCl precast gel | Biorad | #3450034 |
| Phos-tag | NARD Institute | AAL-107 |
| Human chorionic gonadotropin | Merck | Chorulon |
| Human chorionic gonadotropin | Sigma-Aldrich | #CG10-10VL |
| A23187 | Sigma-Aldrich | # C7522 |
| Histone H1 | Millipore | #14-155 |
| PKA inhibitor IV | Santa Cruz Biotechnology | #sc-3010 |
| FLUOTRAC 200 384 well plates | Greiner | #781076 |
| Cyclin A2/CDK2 complex | Sigma | #C0495 |
| Protease inhibitor cOmplete | Roche | #11873580001 |
| Magnetic Protein G beads | Invitrogen | #10004D |
| Critical Commercial Assays | ||
| TNT SP6 high-yield wheat germ protein expression system | Promega | #L3261 |
| mMESSAGE mMACHINE kit | Invitrogen | #AM1344 |
| Poly(A) tailing kit | Invitrogen | #AM1350 |
| MEGAclear Transcription Clean-Up kit | Invitrogen | #AM1908 |
| Experimental Models: Organisms/Strains | ||
| Oocyte plus female Xenopus laevis | Nasco | LM00531 |
| Recombinant DNA | ||
| pQE80-6His-Arpp19 | 63 | |
| pQE80-6His-Arpp19 S28A S109A | 63 | |
| pCS2-T7-Flag3-Gwl K71M | 63 | |
| pMal-c5x-fizzy 38-62 aa | This study | |
| Software and Algorithms | ||
| Mathematica 10 and 12 | Wolfram Research | https://www.wolfram.com/mathematica/ |
| Graphpad Prism 8.0.2 and 8.4 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji (ImageJ, version 2.00-rc-49/1.51e) | 87 | https://imagej.net/Fiji |
| Excel | Microsoft | N/A |
| Python 3.7.3 | 88 | https://www.python.org/ |
| Other | ||
| Flexstation II | Molecular Devices | N/A |
| Phosphorimager Typhoon 8600 | Molecular Devices | N/A |
Highlights.
APC/C and PP2A-B55 activities are bistable, even when Cdk1 activity is not
Intermediate levels of Cdk1 activate PP2A-B55; high levels of Cdk1 inactivate it
PP2A-B55 activity peaks right before mitotic entry and spikes during mitotic exit
PP2A-B55 controls mitotic substrate phosphorylation more than Cdk1 does
ACKNOWLEDGMENTS
We would like to thank Renping Qiao Coudevylle and Jan Michael Peters for sharing purified non-degradable cyclin B1; Wolfram Antonin, Satoru Mochida, Andreas Heim, Thomas Mayer, Qiong Yang, Matthew Swaffer, and Connie Phong for reagents and helpful discussions; Satoru Mochida and Bela Novak for sharing their manuscript prior to publication; and Silke Hauf, Andreas Boland, Oshri Afanzar, Xianrui Cheng, William Huang, Connie Phong, and Ivan Zheludev for thoughtful comments on the manuscript. The work was supported by grants from the National Institutes of Health (R01 GM046383 and P50 GM107615 to J.E.F.), a postdoctoral fellowship from the German Research Foundation (KA 4476/1-1 to J.K.), Research Foundation Flanders (FWO; GOA5317N to L.G.), and the KU Leuven Research Fund (C14/18/084 to L.G.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cub.2020.11.058.
REFERENCES
- 1.Morgan DO (2007). The Cell Cycle: Principles of Control (New Science Press; ). [Google Scholar]
- 2.Swaffer MP, Jones AW, Flynn HR, Snijders AP, and Nurse P (2018). Quantitative phosphoproteomics reveals the signaling dynamics of cell-cycle kinases in the fission yeast Schizosaccharomyces pombe. Cell Rep. 24, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, and Gygi SP (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 105, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, and Morgan DO (2003). Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864. [DOI] [PubMed] [Google Scholar]
- 5.Healy AM, Zolnierowicz S, Stapleton AE, Goebl M, DePaoli-Roach AA, and Pringle JR (1991). CDC55, a Saccharomyces cerevisiae gene involved in cellular morphogenesis: identification, characterization, and homology to the B subunit of mammalian type 2A protein phosphatase. Mol. Cell. Biol 11, 5767–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mochida S, Ikeo S, Gannon J, and Hunt T (2009). Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 28, 2777–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz MH, Held M, Janssens V, Hutchins JR, Hudecz O, Ivanova E, Goris J, Trinkle-Mulcahy L, Lamond AI, Poser I, et al. (2010). Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat. Cell Biol 12, 886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu JQ, Guo JY, Tang W, Yang CS, Freel CD, Chen C, Nairn AC, and Kornbluth S (2009). PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat. Cell Biol 11, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akopyan K, Silva Cascales H, Hukasova E, Saurin AT, Müllers E, Jaiswal H, Hollman DA, Kops GJ, Medema RH, and Lindqvist A (2014). Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 53, 843–853. [DOI] [PubMed] [Google Scholar]
- 10.Murray AW, and Kirschner MW (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275–280. [DOI] [PubMed] [Google Scholar]
- 11.Solomon MJ, Glotzer M, Lee TH, Philippe M, and Kirschner MW (1990). Cyclin activation of p34cdc2. Cell 63, 1013–1024. [DOI] [PubMed] [Google Scholar]
- 12.Sunkara PS, Wright DA, and Rao PN (1979). Mitotic factors from mammalian cells induce germinal vesicle breakdown and chromosome condensation in amphibian oocytes. Proc. Natl. Acad. Sci. USA 76, 2799–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novak B, and Tyson JJ (1993). Modeling the cell division cycle: M-phase trigger, oscillations, and size control. J. Theor. Biol. 165, 101–134. [Google Scholar]
- 14.Goldbeter A (1993). Modeling the mitotic oscillator driving the cell division cycle. Comm. Theor. Biol 3, 75–107. [Google Scholar]
- 15.Novak B, and Tyson JJ (1993). Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J. Cell Sci 106, 1153–1168. [DOI] [PubMed] [Google Scholar]
- 16.Gould KL, and Nurse P (1989). Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature 342, 39–45. [DOI] [PubMed] [Google Scholar]
- 17.Krek W, and Nigg EA (1991). Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M transitions of the cell cycle: identification of major phosphorylation sites. EMBO J. 10, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGowan CH, and Russell P (1993). Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 12, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller PR, Coleman TR, Kumagai A, and Dunphy WG (1995). Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 270, 86–90. [DOI] [PubMed] [Google Scholar]
- 20.Parker LL, and Piwnica-Worms H (1992). Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 257, 1955–1957. [DOI] [PubMed] [Google Scholar]
- 21.Millar JB, McGowan CH, Lenaers G, Jones R, and Russell P (1991). p80cdc25 mitotic inducer is the tyrosine phosphatase that activates p34cdc2 kinase in fission yeast. EMBO J. 10, 4301–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strausfeld U, Labbé JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, Russell P, and Dorée M (1991). Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature 351, 242–245. [DOI] [PubMed] [Google Scholar]
- 23.Alfieri C, Zhang S, and Barford D (2017). Visualizing the complex functions and mechanisms of the anaphase promoting complex/cyclosome (APC/C). Open Biol 7, 170204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters JM (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol 7, 644–656. [DOI] [PubMed] [Google Scholar]
- 25.Glotzer M, Murray AW, and Kirschner MW (1991). Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138. [DOI] [PubMed] [Google Scholar]
- 26.Hershko A, Ganoth D, Pehrson J, Palazzo RE, and Cohen LH (1991). Methylated ubiquitin inhibits cyclin degradation in clam embryo extracts. J. Biol. Chem 266, 16376–16379. [PubMed] [Google Scholar]
- 27.King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, and Kirschner MW (1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279–288. [DOI] [PubMed] [Google Scholar]
- 28.Yamano H, Ishii K, and Yanagida M (1994). Phosphorylation of dis2 protein phosphatase at the C-terminal cdc2 consensus and its potential role in cell cycle regulation. EMBO J 13, 5310–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mochida S, Maslen SL, Skehel M, and Hunt T (2010). Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673. [DOI] [PubMed] [Google Scholar]
- 30.Vigneron S, Brioudes E, Burgess A, Labbé JC, Lorca T, and Castro A (2009). Greatwall maintains mitosis through regulation of PP2A. EMBO J 28, 2786–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasa I, Cressey LE, Kruse T, Hertz EPT, Gui J, Graves LM, Nilsson J, and Kettenbach AN (2020). Quantitative kinase and phosphatase profiling reveal that CDK1 phosphorylates PP2Ac to promote mitotic entry. Sci. Signal 13, eaba7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinod PK, and Novak B (2015). Model scenarios for switch-like mitotic transitions. FEBS Lett 589, 667–671. [DOI] [PubMed] [Google Scholar]
- 33.Mochida S, Rata S, Hino H, Nagai T, and Novák B (2016). Two bistable switches govern M phase entry. Curr. Biol 26, 3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rata S, Suarez Peredo Rodriguez MF, Joseph S, Peter N, Echegaray Iturra F, Yang F, Madzvamuse A, Ruppert JG, Samejima K, Platani M, et al. (2018). Two interlinked bistable switches govern mitotic control in mammalian cells. Curr. Biol 28, 3824–3832.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pomerening JR, Sontag ED, and Ferrell JE Jr. (2003). Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol 5, 346–351. [DOI] [PubMed] [Google Scholar]
- 36.Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, and Sible JC (2003). Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl. Acad. Sci. USA 100, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrell JE Jr., Wu M, Gerhart JC, and Martin GS (1991). Cell cycle tyrosine phosphorylation of p34cdc2 and a microtubule-associated protein kinase homolog in Xenopus oocytes and eggs. Mol. Cell. Biol 11, 1965–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartley RS, Rempel RE, and Maller JL (1996). In vivo regulation of the early embryonic cell cycle in Xenopus. Dev. Biol 173, 408–419. [DOI] [PubMed] [Google Scholar]
- 39.Tsai TY, Theriot JA, and Ferrell JE Jr. (2014). Changes in oscillatory dynamics in the cell cycle of early Xenopus laevis embryos. PLoS Biol. 12, e1001788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang JB, and Ferrell JE Jr. (2013). Mitotic trigger waves and the spatial coordination of the Xenopus cell cycle. Nature 500, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coudreuse D, and Nurse P (2010). Driving the cell cycle with a minimal CDK control network. Nature 468, 1074–1079. [DOI] [PubMed] [Google Scholar]
- 42.Navarro FJ, and Nurse P (2012). A systematic screen reveals new elements acting at the G2/M cell cycle control. Genome Biol 13, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SY, and Ferrell JE Jr. (2007). Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell 128, 1133–1145. [DOI] [PubMed] [Google Scholar]
- 44.Trunnell NB, Poon AC, Kim SY, and Ferrell JE Jr. (2011). Ultrasensitivity in the regulation of Cdc25C by Cdk1. Mol. Cell 41, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Q, and Ferrell JE Jr. (2013). The Cdk1-APC/C cell cycle oscillator circuit functions as a time-delayed, ultrasensitive switch. Nat. Cell Biol 15, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, and Sun Y (2001). Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res 61, 8211–8217. [PubMed] [Google Scholar]
- 47.Shou W, and Dunphy WG (1996). Cell cycle control by Xenopus p28Kix1, a developmentally regulated inhibitor of cyclin-dependent kinases. Mol. Biol. Cell 7, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgi AB, Stukenberg PT, and Kirschner MW (2002). Timing of events in mitosis. Curr. Biol 12, 105–114. [DOI] [PubMed] [Google Scholar]
- 49.Fujimitsu K, Grimaldi M, and Yamano H (2016). Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 352, 1121–1124. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi M, Yu S, Qiao R, Weissmann F, Miller DJ, VanderLinden R, Brown NG, Frye JJ, Peters JM, and Schulman BA (2015). Structure of an APC3-APC16 complex: insights into assembly of the anaphase-promoting complex/cyclosome. J. Mol. Biol 427, 1748–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang S, Chang L, Alfieri C, Zhang Z, Yang J, Maslen S, Skehel M, and Barford D (2016). Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tischer T, Hörmanseder E, and Mayer TU (2012). The APC/C inhibitor XErp1/Emi2 is essential for Xenopus early embryonic divisions. Science 338, 520–524. [DOI] [PubMed] [Google Scholar]
- 53.Vinod PK, Zhou X, Zhang T, Mayer TU, and Novak B (2013). The role of APC/C inhibitor Emi2/XErp1 in oscillatory dynamics of early embryonic cell cycles. Biophys. Chem 177–178, 1–6. [DOI] [PubMed] [Google Scholar]
- 54.Dohadwala M, da Cruz e Silva EF, Hall FL, Williams RT, Carbonaro-Hall DA, Nairn AC, Greengard P, and Berndt N (1994). Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 91, 6408–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwon YG, Lee SY, Choi Y, Greengard P, and Nairn AC (1997). Cell cycle-dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc. Natl. Acad. Sci. USA 94, 2168–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, Zhao Y, Li Z, Galas S, and Goldberg ML (2006). Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell 22, 83–91. [DOI] [PubMed] [Google Scholar]
- 57.Zhao Y, Haccard O, Wang R, Yu J, Kuang J, Jessus C, and Goldberg ML (2008). Roles of Greatwall kinase in the regulation of cdc25 phosphatase. Mol. Biol. Cell 19, 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blake-Hodek KA, Williams BC, Zhao Y, Castilho PV, Chen W, Mao Y, Yamamoto TM, and Goldberg ML (2012). Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol. Cell. Biol. 32, 1337–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gharbi-Ayachi A, Labbé JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, and Lorca T (2010). The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330, 1673–1677. [DOI] [PubMed] [Google Scholar]
- 60.Mochida S, and Hunt T (2012). Protein phosphatases and their regulation in the control of mitosis. EMBO Rep. 13, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams BC, Filter JJ, Blake-Hodek KA, Wadzinski BE, Fuda NJ, Shalloway D, and Goldberg ML (2014). Greatwall-phosphorylated endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. eLife 3, e01695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hégarat N, Vesely C, Vinod PK, Ocasio C, Peter N, Gannon J, Oliver AW, Novák B, and Hochegger H (2014). PP2A/B55 and Fcp1 regulate Greatwall and Ensa dephosphorylation during mitotic exit. PLoS Genet 10, e1004004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heim A, Konietzny A, and Mayer TU (2015). Protein phosphatase 1 is essential for Greatwall inactivation at mitotic exit. EMBO Rep 16, 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma S, Vigneron S, Robert P, Strub JM, Cianferani S, Castro A, and Lorca T (2016). Greatwall dephosphorylation and inactivation upon mitotic exit is triggered by PP1. J. Cell Sci 129, 1329–1339. [DOI] [PubMed] [Google Scholar]
- 65.Rogers S, Fey D, McCloy RA, Parker BL, Mitchell NJ, Payne RJ, Daly RJ, James DE, Caldon CE, Watkins DN, et al. (2016). PP1 initiates the dephosphorylation of MASTL, triggering mitotic exit and bistability in human cells. J. Cell Sci 129, 1340–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mochida S, and Hunt T (2007). Calcineurin is required to release Xenopus egg extracts from meiotic M phase. Nature 449, 336–340. [DOI] [PubMed] [Google Scholar]
- 67.Bialojan C, and Takai A (1988). Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J 256, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mochida S (2014). Regulation of α-endosulfine, an inhibitor of protein phosphatase 2A, by multisite phosphorylation. FEBS J 281, 1159–1169. [DOI] [PubMed] [Google Scholar]
- 69.Wühr M, Freeman RM Jr., Presler M, Horb ME, Peshkin L, Gygi S, and Kirschner MW (2014). Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr. Biol 24, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas R (1981). On the relation between the logical structure of systems and their ability to generate multiple steady states or sustained oscillations In Numerical Methods in the Study of Critical Phenomena, Della Dora J, Demongeot J, and Lacolle B, eds. (Springer; ), pp. 180–193. [Google Scholar]
- 71.Tsai TY, Choi YS, Ma W, Pomerening JR, Tang C, and Ferrell JE Jr. (2008). Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science 321, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Filter JJ, Williams BC, Eto M, Shalloway D, and Goldberg ML (2017). Unfair competition governs the interaction of pCPI-17 with myosin phosphatase (PP1-MYPT1). eLife 6, e24665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thron CD (1999). Mathematical analysis of binary activation of a cell cycle kinase which down-regulates its own inhibitor. Biophys. Chem 79, 95–106. [DOI] [PubMed] [Google Scholar]
- 74.Hopkins M, Tyson JJ, and Novák B (2017). Cell-cycle transitions: a common role for stoichiometric inhibitors. Mol. Biol. Cell 28, 3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thron CD (1997). Bistable biochemical switching and the control of the events of the cell cycle. Oncogene 15, 317–325. [DOI] [PubMed] [Google Scholar]
- 76.Gelens L, and Saurin AT (2018). Exploring the function of dynamic phosphorylation-dephosphorylation cycles. Dev. Cell 44, 659–663. [DOI] [PubMed] [Google Scholar]
- 77.Rodenfels J, Neugebauer KM, and Howard J (2019). Heat oscillations driven by the embryonic cell cycle reveal the energetic costs of signaling. Dev. Cell 48, 646–658.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pomerening JR, Kim SY, and Ferrell JE Jr. (2005). Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell 122, 565–578. [DOI] [PubMed] [Google Scholar]
- 79.Murray AW, Solomon MJ, and Kirschner MW (1989). The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339, 280–286. [DOI] [PubMed] [Google Scholar]
- 80.Kumagai A, and Dunphy WG (1992). Regulation of the cdc25 protein during the cell cycle in Xenopus extracts. Cell 70, 139–151. [DOI] [PubMed] [Google Scholar]
- 81.Draetta G, and Beach D (1988). Activation of cdc2 protein kinase during mitosis in human cells: cell cycle-dependent phosphorylation and subunit rearrangement. Cell 54, 17–26. [DOI] [PubMed] [Google Scholar]
- 82.Swaffer MP, Jones AW, Flynn HR, Snijders AP, and Nurse P (2016). CDK substrate phosphorylation and ordering the cell cycle. Cell 167, 1750–1761.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, and Janssens V (2007). Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic subunit. J. Biol. Chem. 282, 26971–26980. [DOI] [PubMed] [Google Scholar]
- 84.Vigneron S, Sundermann L, Labbe JC, Pintard L, Radulescu O, Castro A, and Lorca T (2018). Cyclin A-cdk1-dependent phosphorylation of Bora is the triggering factor promoting mitotic entry. Dev. Cell 45, 637–650.e7. [DOI] [PubMed] [Google Scholar]
- 85.Vollmer B, Schooley A, Sachdev R, Eisenhardt N, Schneider AM, Sieverding C, Madlung J, Gerken U, Macek B, and Antonin W (2012). Dimerization and direct membrane interaction of Nup53 contribute to nuclear pore complex assembly. EMBO J 31, 4072–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim SY, Song EJ, Lee KJ, and Ferrell JE Jr. (2005). Multisite M-phase phosphorylation of Xenopus Wee1A. Mol. Cell. Biol 25, 10580–10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van Rossum G, and Drake FL (2009). Python 3 Reference Manual (Valley, CA: CreateSpace: Scotts; ). https://docs.python.org/3/reference/. [Google Scholar]
- 89.Murray AW (1991). Cell cycle extracts. Methods Cell Biol 36, 581–605. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets or code accompany this paper.