

Abstract

We tested the susceptibility of 102 proanthocyanidin (PA)-rich plant extracts to oxidation under alkaline conditions and the possibility to produce chemically modified PAs via oxidation. Both the nonoxidized and the oxidized extracts were analyzed using group-specific ultrahigh-performance liquid chromatography–diode array detection–tandem mass spectrometry (UHPLC–DAD–MS/MS) methods capable of detecting procyanidin (PC) and prodelphinidin (PD) moieties along the two-dimensional (2D) chromatographic fingerprints of plant PAs. The results indicated different reactivities for PCs and PDs. When detected by UHPLC–DAD only, most of the PC-rich samples exhibited only a subtle change in their PA content, but the UHPLC–MS/MS quantitation showed that the decrease in the PC content varied by 0–100%. The main reaction route was concluded to be intramolecular. The PD-rich and galloylated PAs showed a different pattern with high reductions in the original PA content by both ultraviolet (UV) and MS/MS quantitation, accompanied by the shifted retention times of the chromatographic PA humps. In these samples, both intra- and intermolecular reactions were indicated.
Introduction
Proanthocyanidins (PAs, syn. Condensed tannins) are oligomers and polymers consisting of flavan-3-ol (Figure 1) units and are found almost in all plant families, both in woody and nonwoody plants.1−3 The most common PAs are procyanidins (PCs) consisting of catechin and/or epicatechin units, as well as prodelphinidins (PDs) consisting of gallocatechin and/or epigallocatechin units. PAs are often mixtures of PCs and PDs, but they can also contain rare monomers such as the propelargonidins that share afzelechin/epiafzelechin subunits.4 The subunits can be linked via C4 → C8 bonds or C4 → C6 bonds (B-type PAs) or additionally via C2 → O → C7 or C2 → O → C5 bonds (A-type PAs), thus increasing the potential number of oligomers and polymers easily to several hundreds and even thousands.2,4,5 The possible galloylation of the subunits increases this complexity even further.4
Figure 1.
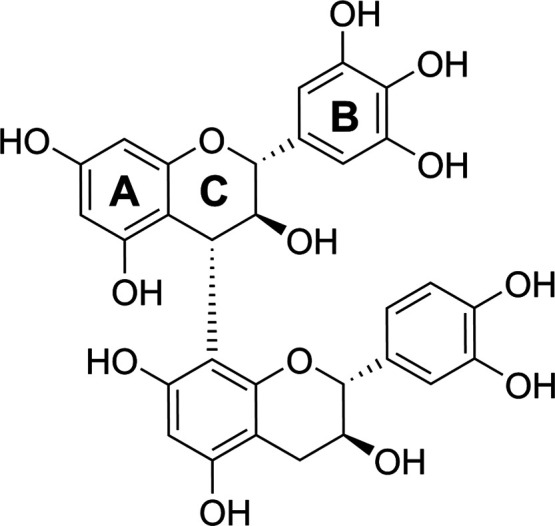
Dimeric PA, prodelphinidin B3, consisting of the gallocatechin extension unit and the catechin terminal unit.
Currently, the estimated annual worldwide production of PAs is several 100 kilotons.6−8 From this, majority is extracted from a few resources only (barks and/or woods of wattle, mimosa, quebracho, oak, chestnut, mangrove, sumach, myrabolans, tara, and several species of pines and firs), mainly for the demands of leather tanning industry, wine industry, animal nutrition, and for some other industrial uses such as mineral flotation and oil drilling.8−10 Meanwhile, a substantial part of the residues from the handling and processing of fruits, vegetables, and forest resources still comprises significant amounts of the original plant materials and accordingly, natural compounds, such as PAs, can be found in many of these leftovers.11−14 In addition to the possible use of these resources to extract tannins for the abovementioned industrial purposes, with some additional processing steps, these residues could be transformed from low-value leftovers into attractive high-value products, for example, as feedstock for the expanding markets in the sustainable food and feed, cosmetic, and pharmaceutical industries.
One possibility to increase the value of plant PAs is to modify the intact PAs via oxidation, thereby producing new types of molecules with modified, hopefully enhanced bioactivities. Our preliminary studies have indicated that for some PA sources, oxidation increases the protein precipitation capacities (Engström et al. unpublished). The oxidation reactions of flavan-3-ols and dimeric PAs are well known, and diverse oxidation products have been characterized and their mechanisms of formation proposed.15−21 However, although the oxidation reactions of oligomeric and polymeric PAs play an important role in, for example, wood adhesive13,22−25 and beverage industries,13,26−28 little is known about these reactions and the resulting structural changes. This is mostly due to the challenges in the analysis of the structurally diverse PAs in general, which becomes even more complex after oxidation. Thus far, most detailed studies concerning PA oxidation have utilized chemical depolymerization29,30 and/or focused on the characterization of oxidation markers20,26,31,32 The results have indicated two main reaction routes, intramolecular and intermolecular oxidation reactions; the main reaction route depends on the PA characteristics and their concentration in the sample oxidized.17,26,27,29,33 These pioneering studies have focused on specific PA sources, such as apple PAs or the PAs involved in wine-making.20,26,27,33 However, the identification of the reactivity of different types of PA sources, their possible reaction pathways, and the resulting structural changes due to oxidation is needed to establish solid knowledge to support the exploitation of various PA residues for possible applications.
In the present study, we tested the effects of nonspecific alkaline oxidation mimicking the often-used alkaline extraction process for bark waste23,34−37 on 102 PA-containing plant extracts. Both the original and oxidized extracts were analyzed using the Engström method.38−40 The method produces two-dimensional (2D) chromatographic PA fingerprints and provides both qualitative and quantitative information on the PA content, the PD/PC share and the mean degree of polymerization (mDP) of the samples, and how these are distributed along the chromatographic hump typically produced by large PA oligomers and polymers. To our knowledge, this is the first study to explore the oxidative modifications of the many PA sources with varying PA compositions. In this initial screening step, we aimed at gaining a better understanding on what types of PAs can tolerate these quite harsh conditions and, on the other hand, what new types of PAs could be produced by this process and how these are related to the original PA compositions of the plant samples.
Results and Discussion
Ultrahigh-Performance Liquid Chromatography–Diode Array Detection Fingerprints
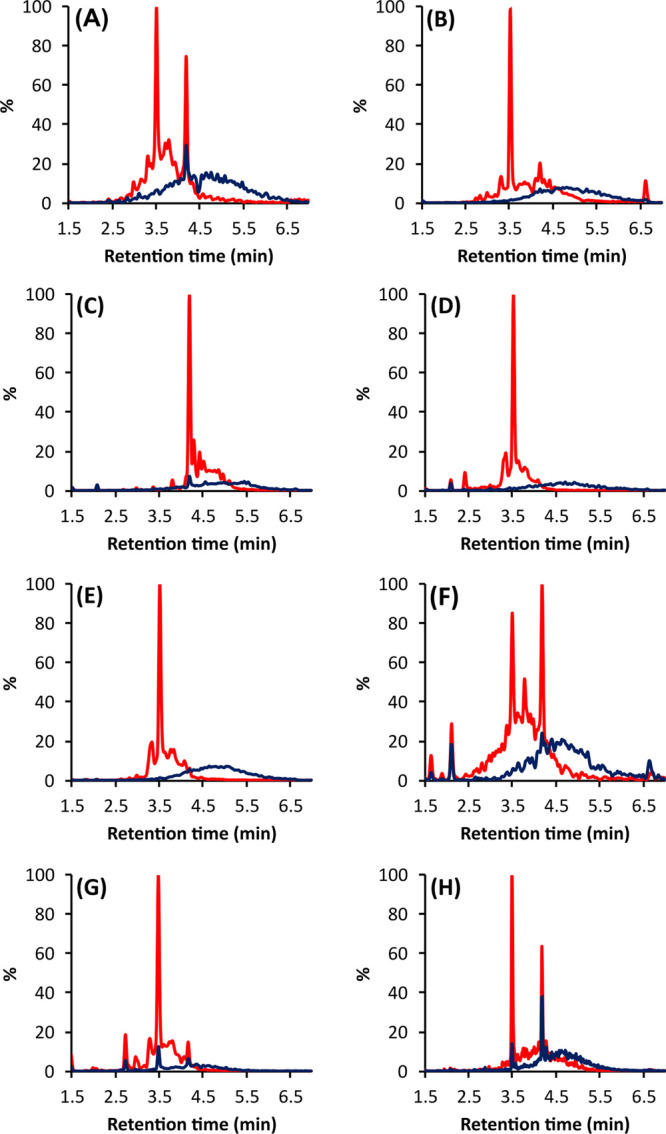
From the preliminary screening of 300 plant samples, we found 102 samples to contain PC- and PD-rich oligomers and polymers, resulting in a distinctive chromatographic hump at 280 nm and variable quantitative characteristics (PA content, PC/PD ratio, mDP). At this stage, the relevance of the plant species to agricultural usage or human consumption was not considered, but the focus was on the variability of quantitative measures. These 102 samples were taken forward to the oxidation tests at high pH. Based on the visual observations of the ultrahigh-performance liquid chromatography–diode array detection (UHPLC–DAD) chromatograms and the integration of the total chromatographic peak areas at 280 nm before and after oxidation (Figure S1, Supporting Information), we divided the 102 plant samples into four different categories (Table 1). These categories were as follows: (A) nonmodified PAs without a clear loss (< 20%) of PA concentration; (B) nonmodified PAs with a clear loss (≥ 20%) of PA concentration; (C) modified PAs without a clear loss (< 20%) of PA concentration; and(D) modified PAs with a clear loss (≥ 20%) of PA concentration. Herein, the word “nonmodified” is used for samples where no shift in the retention time window of the PA hump was observed. Examples of the PA chromatograms from each category are presented in Figure 2. In category A samples, as in Phoenix loureiroi leaflets (Figure 2A), the PA humps of the control and the oxidized samples were very similar, both peak area wise and retention time wise. This indicated very little or no changes in the PA structures and concentrations. A majority, 40 studied samples, belonged to this category. Twenty of the studied samples, such as Coffea arabica leaves (Figure 2B), belonged to category B. In these samples, the peak area of the PA hump significantly decreased because of the oxidation but did not change retention time wise. On the other hand, in categories C and D, with 13 and 30 samples in each, respectively, the chromatographic PA humps moved to later retention times. The difference between the two categories was that in the category C samples, as in Newtonia buchananii leaflets (Figure 2C), the peak area of the PA hump was the same (<20% decrease) before and after oxidation, but in the category D samples, as in Aeonium arboreum flowers (Figure 2D), the PA hump areas significantly decreased (≥20%). These findings indicated high reactivity and major structural changes, such as further polymerization reactions and other rearrangements, in the PAs. Regarding the decrease in the PA hump area in B and D categories, no precipitation was observed during the oxidation, and thus, the decrease in the ultraviolet (UV) peak areas was mainly caused by the structural modifications affecting the ability of the PAs to absorb light at 280 nm. Altogether, these modifications agreed with the previous studies showing changes in the chromatographic profiles of PAs during oxidation.19,36,41 To better understand how the reactivity/stability of the studied samples under alkaline conditions was reflected in the original PA compositions and concentrations, we analyzed both the nonoxidized and the oxidized samples by ultrahigh-performance liquid chromatography triple-quadrupole mass spectrometry (UHPLC–QqQ–MS/MS).
Table 1. Plant Species and Plant Parts Studied, their UV Categories, and Quantitative and Qualitative Measures before and after Oxidation.
| no. | plant family and species | plant parts | UV category | aPA content (mg/g) | PD % | mDP | GPAs |
|---|---|---|---|---|---|---|---|
| Adoxaceae | |||||||
| 1 | Viburnum tinus | leaves | A | 25 → 2 | 1 → 0 | 3 → 3 | |
| Apocynaceae | |||||||
| 2 | Mandevilla splendens | leaves | A | 28 → 26 | 0 → 1 | 10 → 11 | |
| Araceae | |||||||
| 3 | Aglaonema commutatum var.maculatum | leaves | A | 19 → 10 | 0 → 0 | 4 → 3 | |
| 4 | Aglaonema crispum | leaves | B | 13 → 1 | 0 → 0 | 4 → 2 | |
| 5 | Aglaonema treubii | leaves | B | 4 → 0 | 0 → ND | 3 → ND | |
| Araucariaceae | |||||||
| 6 | Araucaria bidwillii | leaves | D | 4 → 0 | 67 → ND | 23 → ND | |
| 7 | Wollemia nobilis | needles | A | 19 → 12 | 1 → 0 | 9 → 9 | |
| Asphodelaceae | |||||||
| 8 | Dianella ensifolia | leaves | D | 4 → 0 | 50 → ND | 12 → ND | |
| 9 | Dianella intermedia | leaves | C | 7 → 1 | 50 → 0 | 10 → 5 | |
| Balsaminaceae | |||||||
| 10 | Impatiens repens | flowers | D | 15 → 5 | 59 → 27 | 12 → 8 | |
| Begoniaceae | |||||||
| 11 | Begonia bowerae ’Nigra’ | leaves | A | 15 → 9 | 1 → 0 | 3 → 3 | |
| Casuarinaceae | |||||||
| 12 | Allocasuarina campestris | needles | B | 9 → 1 | 46 → 0 | 6 → 1 | |
| Cephalotaxaceae | |||||||
| 13 | Cephalotaxus harringtonia subsp. drupacea | leaflets | A | 55 → 22 | 8 → 8 | 3 → 3 | |
| Combretaceae | |||||||
| 14 | Callisia gentlei var. elegans | leaves | C | 20 → 1 | 92 → 65 | 10 → 8 | |
| 15 | Combretum bracteosum | leaves | B | 11 → 1 | 0 → 0 | 4 → 3 | |
| 16 | Combretum indicum | leaves | B | 4 → 1 | 0 → 0 | 3 → 4 | |
| Crassulaceae | |||||||
| 17 | Aeonium arboreum | flowers | D | 16 → 0 | 93 → ND | 17 → ND | x |
| 18 | Crassula ovata | leaves | D | 6 → 0 | 89 → ND | 37 → ND | x |
| 19 | Echeveria harmsii | leaves | D | 21 → 0 | 87 → ND | 21 → ND | x |
| 20 | Kalanchoë manginii | flowers | D | 29 → 1 | 87 → 0 | 17 → 3 | x |
| 21 | Pachyphytum hookeri | leaves | D | 36 → 0 | 89 → ND | 25 → ND | x |
| 22 | Sedum rubrotinctum | leaves | D | 14 → 0 | 92 → ND | 25 → ND | x |
| 23 | Villadia batesii | pieces | D | 26 → 0 | 87 → ND | 24 → ND | x |
| Cupressaceae | |||||||
| 24 | Cunninghamia lanceolata | leaves | A | 51 → 34 | 1 → 1 | 4 → 4 | |
| 25 | Cupressus bakeri | branches | C | 29 → 5 | 45 → 2 | 8 → 3 | |
| 26 | Sequoia sempervirens | branches | D | 25 → 1 | 82 → 0 | 11 → 2 | |
| 27 | Tetraclinis articulata | pieces | D | 14 → 4 | 46 → 8 | 7 → 3 | |
| Cyperaceae | |||||||
| 28 | Cyperus owanii | leaflets | A | 19 → 9 | 6 → 0 | 6 → 6 | |
| 29 | Cyperus owanii | flowers | A | 28 → 16 | 0 → 1 | 6 → 6 | |
| Davalliaceae | |||||||
| 30 | Davallia pyxidata | leaflets | C | 19 → 3 | 12 → 0 | 7 → 10 | |
| Dicksoniaceae | |||||||
| 31 | Dicksonia squarrosa | leaflets | A | 20 → 8 | 2 → 2 | 3 → 2 | |
| Dryopteridaceae | |||||||
| 32 | Cyrtomium falcatum | leaflets | D | 15 → 1 | 72 → 16 | 9 → 10 | |
| 33 | Polystichum proliferum | leaves | A | 44 → 25 | 1 → 2 | 13 → 13 | |
| Ebenaceae | |||||||
| 34 | Diospyros mespiliformis | leaves | D | 14 → 3 | 61 → 13 | 8 → 3 | x |
| Ericaceae | |||||||
| 35 | Rhododendron hemitrichotum | leaves | A | 63 → 25 | 3 → 7 | 3 → 4 | |
| 36 | Rhododendron hemitrichotum | flowers | A | 24 → 9 | 1 → 2 | 7 → 6 | |
| Euphorbiaceae | |||||||
| 37 | Euphorbia characias | leaves | B | 7 → 1 | 3 → 0 | 7 → 4 | |
| 38 | Euphorbia characias | flowers | B | 10 → 2 | 2 → 0 | 5 → 2 | |
| Fabaceae | |||||||
| 39 | Acacia karroo | leaves | D | 35 → 0 | 96 → ND | 10 → ND | x |
| 40 | Acacia melanoxylon | leaflets | C | 47 → 8 | 56 → 4 | 8 → 4 | x |
| 41 | Acacia victoriae | leaflets | D | 23 → 3 | 91 → 66 | 12 → 6 | x |
| 42 | Bauhinia variegata | leaves | A | 11 → 4 | 5 → 0 | 5 → 6 | |
| 43 | Calliandra haematocephala | leaflets | D | 24 → 4 | 83 → 72 | 9 → 6 | x |
| 44 | Ceratonia siliqua | leaves | D | 9 → 1 | 80 → 49 | 10 → 3 | x |
| 45 | Mimosa polycarpa | leaflets | D | 12 → 1 | 45 → 0 | 7 → 4 | x |
| 46 | Newtonia buchananii | leaflets | C | 68 → 1 | 88 → 0 | 4 → 2 | x |
| Fagaceae | |||||||
| 47 | Quercus ilex | leaves | C | 45 → 8 | 24 → 4 | 4 → 3 | |
| Geraniaceae | |||||||
| 48 | Pelargonium odoratissimum | leaves | B | 19 → 0 | 86 → ND | 10 → ND | |
| Lauraceae | |||||||
| 49 | Apollonias barbujana | leaves | A | 30 → 20 | 0 → 1 | 3 → 3 | |
| 50 | Laurus nobilis | leaves | A | 12 → 6 | 0 → 0 | 2 → 2 | |
| Malvaceae | |||||||
| 51 | Heritiera solomonensis | leaves | A | 76 → 50 | 0 → 1 | 7 → 7 | |
| 52 | Pavonia cauliflora | flowers | A | 25 → 12 | 1 → 1 | 6 → 6 | |
| Marcgraviaceae | |||||||
| 53 | Marcgravia umbellata | leaves | A | 29 → 13 | 0 → 2 | 3 → 3 | |
| Melastomataceae | |||||||
| 54 | Medinilla magnifica | leaves | D | 5 → 0 | 71 → ND | 9 → ND | |
| Moraceae | |||||||
| 55 | Artocarpus sp. | leaves | A | 15 → 3 | 0 → 0 | 2 → 4 | |
| Myrtaceae | |||||||
| 56 | Acca sellowiana | leaves | B | 18 → 4 | 9 → 0 | 4 → 2 | |
| 57 | Callistemon citrinus | leaves | B | 6 → 0 | 3 → ND | 2 → ND | |
| 58 | Melaleuca squarrosa | leaves | B | 8 → 3 | 32 → 0 | 6 → 2 | |
| 59 | Myrtus communis | leaves | B | 4 → 0 | 70 → ND | 6 → ND | |
| 60 | Psidium cattleianum | leaves | B | 17 → 2 | 27 → 0 | 4 → 2 | |
| 61 | Psidium cattleianum | leaflets | A | 33 → 20 | 17 → 6 | 5 → 4 | |
| Nepenthaceae | |||||||
| 62 | Nepenthes maxima | leaves | C | 22 → 6 | 2 → 5 | 4 → 3 | x |
| 63 | Nepenthes maxima | pitcher | C | 32 → 5 | 2 → 6 | 4 → 3 | x |
| Oxalidaceae | |||||||
| 64 | Biophytum sensitivum | leaves | B | 45 → 23 | 0 → 1 | 7 → 6 | |
| Phyllanthaceae | |||||||
| 65 | Phyllanthus juglandifolius/grandifolius | leaves | C | 25 → 3 | 82 → 21 | 4 → 2 | |
| Podocarpaceae | |||||||
| 66 | Podocarpus macrophyllus | leaves | C | 27 → 2 | 73 → 0 | 6 → 2 | |
| Polygonaceae | |||||||
| 67 | Coccoloba uvifera | leaves | D | 24 → 9 | 14 → 3 | 6 → 4 | x |
| 68 | Microgramma mauritiana | leaflets | A | 35 → 6 | 1 → 4 | 10 → 10 | |
| 69 | Microgramma vacciniifolia | leaves | B | 11 → 2 | 0 → 0 | 4 → 3 | |
| 70 | Ruprechtia salicifolia | leaves | D | 24 → 4 | 3 → 5 | 7 → 4 | x |
| Portulacaceae | |||||||
| 71 | Portulaca alata | leaves | A | 10 → 9 | 1 → 0 | 12 → 14 | |
| Primulaceae | |||||||
| 72 | Aegiceras corniculatum | leaves | D | 9 → 2 | 88 → 88 | 8 → 3 | x |
| 73 | Ardisia crenata | leaves | D | 13 → 0 | 82 → ND | 12 → ND | x |
| 74 | Cyclamen africanum | leaves | C | 10 → 3 | 6 → 0 | 8 → 7 | |
| Pteridaceae | |||||||
| 75 | Pellaea ovata | pieces | D | 47 → 2 | 85 → 6 | 9 → 8 | |
| 76 | Pellaea ovata | leaflets | D | 31 → 1 | 77 → 0 | 7 → 5 | |
| Rhizophoraceae | |||||||
| 77 | Rhizophora mangle | leaves | A | 35 → 9 | 4 → 3 | 6 → 6 | |
| Rosaceae | |||||||
| 78 | Eriobotrya japonica | leaves | A | 15 → 6 | 0 → 0 | 5 → 5 | |
| 79 | Osteomeles schwerinae | leaves | A | 37 → 17 | 1 → 1 | 8 → 8 | |
| 80 | Osteomeles schweriniae | flowers | A | 7 → 7 | 0 → 0 | 9 → 9 | |
| 81 | Rhaphiolepis indica var. umbellata | leaves | A | 47 → 15 | 1 → 2 | 5 → 5 | |
| Rubiaceae | |||||||
| 82 | Coffea arabica | leaves | B | 43 → 8 | 0 → 1 | 10 → 9 | |
| 83 | Hoffmannia refulgens | leaves | A | 41 → 15 | 0 → 1 | 5 → 5 | |
| 84 | Ixora coccinea | leaves | A | 31 → 19 | 1 → 1 | 6 → 5 | |
| 85 | Ixora coccinea | flowers | B | 16 → 10 | 0 → 0 | 7 → 6 | |
| Sapindaceae | |||||||
| 86 | Dimocarpus longan | leaves | A | 28 → 12 | 0 → 1 | 3 → 3 | |
| 87 | Nephelium connatum | leaves | C | 30 → 9 | 93 → 86 | 8 → 7 | |
| Sapotaceae | |||||||
| 88 | Sideroxylon inerme | leaves | D | 37 → 0 | 96 → ND | 8 → ND | x |
| Sarraceniaceae | |||||||
| 89 | Sarracenia purpurea | leaves | A | 29 → 18 | 3 → 2 | 8 → 7 | |
| Strelitziaceae | |||||||
| 90 | Strelitzia reginae | leaves | A | 9 → 6 | 0 → 0 | 5 → 5 | |
| 91 | Strelitzia reginae | flowers | A | 11 → 7 | 1 → 0 | 8 → 8 | |
| Tectariaceae | |||||||
| 92 | Tectaria macrodonta | leaflets | B | 9 → 4 | 0 → 0 | 5 → 4 | |
| Theaceae | |||||||
| 93 | Camellia japonica | leaves | A | 15 → 2 | 1 → 0 | 2 → 3 | |
| 94 | Camellia japonica | petals | A | 18 → 10 | 1 → 2 | 3 → 3 | |
| Vitaceae | |||||||
| 95 | Cissus javana | leaves | D | 22 → 2 | 80 → 60 | 17 → 10 | x |
| 96 | Leea guineense | leaves | D | 32 → 11 | 45 → 12 | 9 → 5 | x |
| 97 | Rhoicissus sp. | leaves | D | 9 → 0 | 71 → ND | 10 → ND | x |
| Zamiaceae | |||||||
| 98 | Encephalartos ferox | leaflets | B | 21 → 13 | 0 → 0 | 9 → 9 | |
| 99 | Macrozamia communis | leaflets | A | 41 → 30 | 1 → 1 | 9 → 9 | |
| 100 | Zamia furfuracea | leaves | A | 15 → 3 | 0 → 0 | 4 → 4 | |
| Zingiberaceae | |||||||
| 101 | Alpinia purpurata | leaves | A | 34 → 17 | 0 → 1 | 7 → 7 | |
| 102 | Elettaria cardamomun | leaflets | A | 21 → 8 | 0 → 2 | 5 → 5 |
PA = proanthocyanidin, PD = prodelphinidin, mDP = mean degree of polymerization, GPAs = galloylated PAs, ND = not detected. The quantitative measures are expressed as before oxidation → after oxidation. Based on the changes in UHPLC–DAD profiles, the UV categories were as follows: A = nonmodified PAs without a loss in the PA hump area; B = nonmodified PAs with a loss in the PA hump area; C = modified PAs without a loss in the PA hump area; and D = modified PAs with a loss in the PA hump area.
Figure 2.

Examples of the UHPLC–DAD profiles (λ = 280 nm) before and after the oxidation of PA containing plant samples from the four categories: (A) nonmodified PAs without a clear loss of PA concentration in the Heritiera solomonensis leaves (51), (B) nonmodified PAs with a clear loss of PA concentration in Coffea arabica leaves (82), (C) modified PAs without a clear loss of PA concentration in the Newtonia buchananii leaflets (46), and(D) modified PAs with a clear loss of PA concentration in Aeonium arboreum flowers (17). The upper panels show the nonoxidized samples, and the lower panels show the oxidized samples. Sample numbers refer to Table 1.
Oxidation-Driven Changes across the Four Categories
To reveal possible qualitative and quantitative changes in the PA composition caused by oxidation, the samples were analyzed with the Engström method38−40 that allowed the quantitation of the PA content, the PC/PD ratio, and the mDP before and after oxidation (Table 1). In addition, the Engström method revealed a qualitative confirmation of whether the samples contained galloylated PAs or not (Table 1). First, when comparing the results from the UHPLC–DAD chromatograms and the quantitative MS/MS data for all the 102 samples, it was seen that due to oxidation, the PA content of all the samples but three decreased more than 25%, while the changes in the UV peak areas were versatile, and the correlation between the two measures was small (R2 = 0.29). Thus, in contrast to the UHPLC–DAD chromatograms, the MS/MS results indicated reactivity under alkaline conditions for most of the studied samples. Otherwise, comparisons between the different measures (UV peak area, PA content, PD% and mDP) showed strong positive correlations between the UV peak area before and after oxidation (R2 = 0.61), medium positive correlations between the original PD% and the percentage decrease in the PA content (R2 = 0.43), the original PA content and the PA content after oxidation (R2 = 0.42), and the original PD% and the original mDP (R2 = 0.35). Small positive correlations were obtained between the original PD% and percentage decrease in the UV peak area (R2 = 0.21) and between the original mDP and the percentage decrease in the UV peak area (R2 = 0.17). No significant correlations were seen between the original mDP and the original PA content, the original mDP and the percentage decrease in the PA content, the original PA content and the percentage decrease in the PA content, and the original PA content and the percentage decrease in the UV peak area (R2 < 0.1 for all comparisons). The correlation plots are presented in Figure S2 in the Supporting Information.
As suggested before,36,42 to some extent, these quantitative findings and correlations indicated higher reactivity for PD-rich than PC-rich PAs under alkaline conditions. The strong and medium positive correlations between the UV peak areas before and after oxidation and the PA contents by MS/MS before and after oxidation, respectively, suggested that the oxidation reaction was rather controlled in many of the samples. In addition, lower average decrease in the UV peak areas vs quantitative MS/MS levels and higher correlation between the UV peak areas before and after oxidation vs the PA content by MS/MS before and after oxidation were observed. The MS/MS method utilized in the work, that is, the Engström method, is based on the fragmentation of PC and PD units from the PA structures via quinone methide fragmentation.38,40 Thus, the results indicated that the structural changes caused by the oxidative reactions were such that the UV absorbance of the PAs was less and more homogenously affected than the quinone methide fragmentation of the PC and PD units from the oxidized PA structures in MS/MS. To further understand the reactivities/stabilities of the studied samples and the differences between the different categories, next, the results were compared between the categories and within each category.
Oxidation-Driven Changes in Category A Samples
In general, the average decrease in the quantitative MS/MS levels because of oxidation increased from category A to category D (A: -50%, B: -78%, C: −84%, and D: −91%) and so did the average PD percentage before oxidation (A: 2%, B: 15%, C: 48%, and D: 72%) and mDP before oxidation (A: 6, B: 6, C: 7, and D: 12). The most obvious finding was that all the samples in category A were PC-rich, almost PC-pure samples; the average PD % of the 40 samples being 2%. In addition, in most of the samples in this category, the mDP was the same before and after oxidation; in a few samples in which mDP changed, the increase or decrease was maximum two mDP units. The UV peak areas decreased less than 20% in all the samples in category A (on average 9%), indicating low reactivity or reactions that did not significantly affect the ability of the PAs to absorb light at 280 nm. In contrast, the decrease in the quantitative MS/MS results varied between 0 and 92% (on average 50%), indicating rather high reactivity and modifications that disabled the fragmentation of PC and PD units via quinone methide fragmentation in many of the samples. Accordingly, the correlation between the decrease in the UV peak areas and the decrease in the quantitative MS/MS levels was small (R2 = 0.15). Because of the low decrease rates of the PA humps in the UHPLC–DAD chromatograms, the UV peak areas before and after oxidation correlated well (R2 = 0.98). Interestingly, there was a strong positive correlation also between the PA content before and after oxidation (R2 = 0.69), but the percentage decrease did not correlate with the original PA content (R2 = 0.00). This suggests that oxidation was rather controlled in a way that the more the PAs were present in the original samples, the more PAs were detected after oxidation in most of the category A samples. In addition, there was a small negative correlation (R2 = 0.25) between the original mDP and a decrease in the quantitative MS/MS results, indicating that high mDP samples had slightly more unmodified units present in their structures after oxidation than low mDP samples. All the correlation plots are presented in Figure S3 in the Supporting Information.
Figure 3A–F shows examples of different types of UHPLC–MS/MS fingerprints before and after oxidation in category A. Some of the UHPLC–MS/MS fingerprints were almost identical before and after oxidation (e.g., Figure 3A,B), and the quantitative measures were unchanged; in these samples, the possibility of unsuccessful oxidation could be ruled out by the oxidation profiles of quinic acid derivatives.37 This indicated high stability for PCs in these samples under alkaline conditions. However, such samples were only a few, and in most of the category A samples, the UHPLC–MS/MS fingerprints were similar, but the intensity dropped or the intensity dropped and the shape changed, and/or a small shift was seen in the UHPLC–MS/MS fingerprints (e.g. Figure 3C,F). Interestingly, also in these samples, the mDP remained rather unmodified. This could be achieved in two ways: either the percentage decrease in both the extension and terminal units was the same, or alternatively, in comparison to the original PAs, both longer and shorter oligomers and polymers were formed with no percentual decrease in the detected units. To make a difference between the two possibilities, we compared the multiple reaction monitoring (MRM) detection of both the extension and terminal units separately. This confirmed that the percentage decrease in the MRM detection of the extension and terminal units of category A samples were very similar in most of the samples (R2 = 0.85, Figure S3, Supporting Information). The few outliers with greater percentage decrease in the extension units were samples in which the mDP decreased by one or two units (Table 1), and the few outliers with greater decrease in the terminal units were such samples in which the mDP increased by one or two units (Table 1). As the mDP was higher than two in most of the category A samples, it means that, on average, the extension units were more probable to react than the terminal units.
Figure 3.

Examples of UHPLC–DAD–MS/MS fingerprints of the studied plant samples in categories A and B before and after oxidation obtained using the Engström method. The solid red lines are PC units detected before oxidation, red dashed lines are PC units detected after oxidation, solid blue lines are PD units detected before oxidation, and blue dashed lines are PD units detected after oxidation. Shown plant species are as follows: (A) Mandevilla splendens leaves (2), (B) Osteomeles schweriniae flowers (80), (C) Pavonia cauliflora flowers (52), (D) Alpinia purpurata leaves (101), (E) Polystichum proliferum leaves (33), (F) Camellia japonica leaves (93), (G) Encephalartos ferox leaflets (98), (H) Aglaonema crispum leaves (4), (I) Coffea arabica leaves (82), (J) Acca sellowiana leaves (56), (K) Psidium cattleianum leaves (60), and(L) Pelargonium odoratissimum leaves (48). The sample numbers refer to Table 1.
The Engström method also enabled to compare if there was a difference between the average reactivity of the small oligomers, medium-size oligomers and polymers, and large polymers by comparing separately the change in the detection of the small oligomers, medium-size oligomers and polymers, and large polymers obtained by the three different cone voltages of the method, 75, 85, and 140 V for the PC units and 55, 80, and 130 V for the PD units, respectively. In category A, there was a small difference between the decrease in the detection with the lowest and highest cone voltage; the detection with the highest cone voltage decreased 4% more on average, while the correlation between the decrease in the quantitative MS/MS results was strong (R2 = 0.96, Figure S3, Supporting Information). This indicated slightly higher reactivity for the larger oligomers and polymers than that for the smaller oligomers. Altogether, in the category A samples, none of the obtained parameters (PA, content, PD share, and mDP) adequately explained the differences in the reactivities of the studied samples. However, based on the UV profiles before and after oxidation, the reaction routes were most likely similar in the category A samples.
Oxidation-Driven Changes in Category B Samples
In category B, all but two of the samples were PC-rich (Table 1). The decreases in the UV peak areas were versatile, varying between 21 and 96% (on average 45%), while the decreases in the PA content by MS/MS varied between 38 and 100% (on average 78%), and there was a medium positive correlation between the two measures (R2 = 0.32). The main differences in comparison to the category A samples were that the UV peak areas decreased more and so did the quantitative MS/MS results. In addition, the changes in the mDPs were more variable than in category A; in some samples, the mDP was the same before and after oxidation, while in other samples, the mDP decreased by one to five mDP units. In one sample, the mDP increased by one unit because of oxidation. Notably, all the samples with any PDs detected were such that the mDP decreased by at least two units, and the decrease in the quantitative MS/MS level was greater than 60%; this indicated a somewhat triggered reactivity for the PD-containing samples. In the PC-pure samples, the variation in reactivity was higher, and while the decrease in the UV peak area varied between 22 and 88%, the decrease in the quantitative MS/MS levels varied between 38 and 92%. The correlation between the PA contents before and after oxidation was strong (R2 = 0.62) and so was the correlation between the UV peak areas before and after oxidation (R = 0.67), a few outliers causing most of the variation. Again, this indicated a somewhat controlled oxidation reaction in most of the samples. There was a small negative correlation between the original PA content and the percentage decrease in the PA content (R2 = 0.14). The correlation between the original mDP and the decrease in the PA content was smaller (R2 = 0.11) than that in the category A samples. All the correlations presented above are presented in Figure S4 in the Supporting Information.
Examples of the qualitative MRM fingerprints from the category B samples are presented in Figure 3G,L. As expected, because of the rather artificial category threshold value, some fingerprints were both visually and quantitatively very similar to category A fingerprints (e.g., Figure 3G). In other samples, the peak areas of the MRM fingerprints decreased markedly, but the shapes and the retention time windows were the same (e.g., Figure 3H,I). In samples with PDs present, the MS/MS levels decreased markedly: no PDs were detected after oxidation, and the shape of the PC fingerprints changed (e.g., Figure 3J,K). The change in the shape was caused by the greater decrease in the parts of the PC fingerprints where PDs were overlapping before oxidation; this further supported the higher reactivity of the PD units and also indicated that the PCs were more susceptible to modifications if PDs were present in the PA structure. In most extreme samples, no PAs were detected after oxidation (e.g., Figure 3L). When comparing the decrease in the extension and terminal units separately, it was noted that the correlation between them was weaker (R2 = 0.53, Figure S4, Supporting Information) than that in the category A samples, reflecting larger decreases in the mDPs. Regarding detection with different cone voltages, detection with a high cone voltage decreased in average 8% more than detection with a low cone voltage. The correlation between the two measures was smaller than that in category A but was still strong (R2 = 0.89, Figure S4, Supporting Information). Interestingly, the largest differences in the decrease in detection with the low and high cone voltages were for the PC units (a 13% average difference) in the PD-containing samples. The difference was smaller for the PC-pure samples (a 6% average difference) and for the detection of the PD units (a 6% average difference). Furthermore, in samples where both PC and PD units were detected, the decrease of the PD units was larger than the decrease of the PC units. Altogether, these findings further supported the higher oxidative activity PDs but, in addition, indicated an induced reactivity by the PD units in samples with both PCs and PDs present.
Oxidation-Driven Changes in Category C and D Samples
Based on the UHPLC–DAD chromatograms, categories C and D were very different in comparison to categories A and B: in categories C and D, the chromatographic PA humps of the samples eluted later, and the shapes of the humps were modified because of oxidation. The magnitude of both the change in the elution time and the change in the shape of the hump was significant: in some samples, the middle of the hump shifted only with a retention time of 0.2 min to the right, while in most extreme samples, the middle of the hump shifted with a retention time of 2.3 min, and part of the hump eluted after the actual gradient during the column wash. In general, category D contained more samples with large shifts in retention times than category C; however, this property could not be linked solely to either category.
As noted already between categories A and B, because of the 20% threshold value used, overlapping was detected in categories C and D regarding the quantitative MS/MS data. All the samples in categories C and D contained PDs, although the PD share varied in category C from 2 to 93% (average 48%) and in category D between 3 and 96% (average 72%). In both C and D categories, the decrease in the quantitative MS/MS levels was high because of oxidation, on average, 84 and 91%, respectively, and the correlations with the decrease in the UV peak areas were small (R2 = 0.05 and 0.22, respectively). In addition, while there was a strong correlation between the PA content before and after oxidation in categories A and B, in C and D categories, no correlation was observed (R2 = 0.07 and R2 = 0.05, respectively). Together, these findings indicated such oxidation reactions for categories C and D that most of the oxidation products were no longer detected with the Engström method.38 Regarding the detected UV peak areas at 280 nm before and after oxidation, in category C, the correlation was naturally strong (R2 = 0.82) as the changes in the peak areas were subtle. In category D, the correlation was smaller but was still strong (R2 = 0.64). Thus, in contrast to the MS/MS results, this supported somewhat controlled oxidation reactions to occur. In most of the samples, the detected mDP decreased, by two mDP units in category C and by seven mDP units in category D on average, indicating that many of the structural units participated in the reactions and that the extension units reacted more than the terminal units. The changes in the detection of the extension and terminal units separately and/or differences in detection by the low versus high cone voltage explained the decreases in the mDPs and confirmed the higher reactivity of the extension units. Also, the observation made with a few samples in category B containing both the PC and PD units was confirmed; the PD units decreased always more than the PC units, and when the PD units were in majority, the decrease in the PC units was higher than that if PCs were in majority. All the correlations discussed above are presented in Figure S5 in the Supporting Information.
Some UHPLC–MS/MS fingerprints from the category C and D samples are presented in Figure 4. These show the high decrease of both PDs and PCs in samples with a high PD share (Figure 4A,C,F,G,H,K). On the other hand, if the PD and PC shares were close to equal or PCs were in majority, the PCs decreased less (Figure 4B,D,E,I,J,L). As can be seen from the UHPLC–MS/MS fingerprints shown in Figure 4, the shifts were subtle in many of the samples and indeed, when plotted against the UV hump before and after oxidation, it was evident that in most of the samples, the UHPLC–MS/MS fingerprint only overlapped with the earlier parts of the UV hump after oxidation, indicating that other PA units than PCs and PDs were present in the oxidized samples. Interestingly, in the same samples, the intensities of the full-scan total ion chromatograms were low, and similarly to the UHPLC–MS/MS fingerprints, they only partially overlapped with the earlier parts of the UV hump, indicating low ionization. We have previously observed the same partial detection of the UV hump with the UHPLC–MS/MS fingerprint for unoxidized samples with a high degree of polymerization; when the mDP is high enough, the fragmentation and ionization of the largest polymers are attenuated, and, in general, this has been linked with the late retention times (data not shown). Thus, in addition to the structural changes in the PAs disabling their detection with the MS/MS method, the results indicated that the size of the PA polymers was increased; therefore, the fragmentation, ionization, and detection of the oxidized PAs were hampered. Interestingly, the same was not observed in the category A and B samples; even in samples with 90–100% decrease in the quantitative MS/MS levels, the total ion chromatograms showed clear signal humps accordingly to the UV hump. Again, this indicated different reaction routes to be dominant in categories A and B than in categories C and D.
Figure 4.

Some examples of the modified UHPLC–DAD–MS/MS fingerprints of polymeric PA extracts before and after oxidation and their combined patterns obtained using the Engström method. The solid red lines are the nonoxidized PC units, the red dashed lines are the oxidized PC units, the solid blue lines are the nonoxidized prodelphinidin (PD) units, and the blue dashed lines are the oxidized PD units. Shown plant species are as follows: (A) Callisia gentlei leaves (14), (B) Acacia melanoxylon leaflets (40), (C) Newtonia buchananii leaflets (46), (D) Nepenthes maxima pitcher (63), (E) Cyclamen africanum leaves (74), (F) Nephelium connatum leaves (87), (G) Aeonium arboretum flowers (17), (H) Kalanchoë manginii flowers (20), (I) Tetraclinis articulate pieces (27), (J) Diospyros mespiliformis leaves (34), (K) Cissus javana leaves (95), and(L) Leea guineense leaves (96) . The sample numbers refer to Table 1.
Effect of Galloylation
Based on the detection with the galloyl-specific MS/MS method, among the 102 samples studied, 25 samples contained galloylated PAs among which 21 were PD-rich and four were PC-rich. Interestingly, all these samples belonged to categories C and D; four samples belonged to category C, and 21 samples belonged to category D. Figure 5 shows the galloyl fingerprints before and after oxidation for the samples with galloylated PAs. In all the 25 galloylated samples, a general observation was that the galloyl fingerprint shifted to the right retention time wise, and the shape of the galloyl fingerprint changed markedly because of oxidation. In samples where a PA fingerprint was observed after oxidation, the galloyl fingerprint overlapped with the PA fingerprint and the UV hump. Most interestingly, even if no PAs were detected by the PC- and PD-specific MS/MS method after oxidation, a clear galloyl hump was detected with the galloyl-specific MS/MS method, and it had shifted accordingly to the observed UV hump. Thus, the original galloylated PAs were modified in ways that the galloyl groups could still be fragmented and detected by MS/MS, although the PC and PD units were not detected, that is, the quinone methide fragmentation route was disabled because of structural changes. The high decrease in both the UV peak areas and the MS/MS levels agreed with previous studies showing higher oxidation rates for galloylated PAs than for nongalloylated PAs.43−45 Interestingly, while the additional galloyl group increases the reactivity, studies with the monomeric PA units have shown that the principal site of oxidation is not the 3-galloyl group but the trihydroxyphenyl B-ring.44 This could explain why, in the present study, the galloyl fingerprints were detected although oxidized PAs were not detected with the Engström method.
Figure 5.
UHPLC–MS/MS galloyl fingerprints obtained before (red line) and after (blue line) oxidation using the Engström method for (A) Acacia melanoxylon leaflets (40), (B) Newtonia buchananii leaflets (46), (C) Nepenthes maxima pitcher (63), (D) Aeonium arboretum flowers (17), (E) Kalanchoë manginii flowers (20), (F) Diospyros mespiliformis leaves (34), (G) Cissus javana leaves (95), and (H) Leea guineense leaves (96). The sample numbers refer to Table 1.
Intra- Versus Intermolecular Reactions
Previous studies have shown that the major oxidation reaction routes of PAs can be divided into intra- and intermolecular reactions.17,20,26,27 The intramolecular reactions include the epimerization of the monomeric units and other rearrangement reactions within each PA as well as additional links formed between the units.17,20,26 Regarding the UHPLC–DAD detection, the epimerization and formation of A-type linkages would have little effect on the absorbance,46 but the effect on retention times could be significant, depending on the different PAs present in the original versus oxidized samples. Studies with monomers and dimers have indicated large differences in retention times between PA epimers as well as their monomeric units, for example, epicatechin and its oligomers are more strongly retained in reverse-phase chromatography than catechin and its oligomers.46 In addition, PCs with the C4 → C6 linkage as well as doubly linked A-type PCs elute later than the ones with the C4 → C8 linkage.46,47
As the Engström method relies on the MS/MS detection of the ions formed via the quinone methide cleavage in the ion source,38,40 all modifications that disable this fragmentation route would concurrently affect their detection. Epimerization would affect little the detection of the PAs as the detection with the different cone voltages is summarized, and the sum would remain rather unmodified despite small changes in the detection efficiency of single PAs. However, other rearrangement reactions and formation of additional links, such as the formation of A-type PAs from B-type PAs, would alter the detection of the PAs and thus their quantitation and also the determination of the mDP using the Engström method. If mostly extension units are affected, the mDP would decrease, and if mostly terminal units are affected, the mDP would increase. In case the relative decrease of both units are close to equal, the mDP would remain unchanged.
Regarding intermolecular reactions, there are two main possibilities: either the PAs are linked end-to-end and they maintain a linear structure or they are linked via the middle extension units whereupon they become branched polymers.17,20,26,27 The effect on the detection of the PA units and thus the obtained mDP depends on the nature of oxidative coupling and if the PA units can still be fragmented and detected from the resulting oxidation products by the PC- and PD-specific MS/MS method. In case of linear structures with only C4 → C6 and/or C4 → C8 links formed, the mDP would increase. However, most likely, other oxidative couplings also occur, for example, some units get doubly linked with an additional C2 → O → C7 or C2 → O → C5 ether bond. These are yet rather simplified scenarios, and it should be noted that the interflavanyl bonds in PCs and PDs are extremely susceptible to cleavage at pH 10 with the release of the specific unit as an A-ring quinone methide. This may lead to an extensive scrambling of the interflavanyl bonds as well as releasing a powerful Michael acceptor capable of freely interacting with nucleophilic carbon and oxygen centers. This further complicates and multiplies the possible intermolecular reactions. Thus, the resulting mDP might increase, decrease, or be the same, depending on the original structures and the overall intermolecular reactions occurring.
Naturally, all the abovementioned reactions may occur simultaneously in a reaction mixture if the reaction conditions are favorable, and indeed, many studies have supported this.20,26,27 However, in most of these studies, the reaction times have been rather long, from days to weeks, enabling the occurrence of multiple reaction routes. In the present study, oxidation was rapid, and the total incubation time was only 1 h; this is prone to emphasize the most likely reaction routes and lessen the competition between intra- and intermolecular reactions.21,33 Previous studies have reported the formation of A-type PAs from B-type PAs under alkaline conditions, and interestingly, there is some evidence that this reaction route would be favored by PC-rich PAs, especially in dilute solutions.21,30,33 The unchanged UV chromatogram profiles, stable mDPs, and rather controlled oxidation reactions suggested that, on average, the favored reaction route in categories A and B would be intramolecular.
By combining data from the UHPLC–DAD chromatograms, the UHPLC–MS/MS chromatograms, the total ion chromatograms, and mass spectra, we were able to tentatively detect and identify some new peaks in the early part of the chromatographic PA humps of some of the category A and B samples. Although the intensities of individual peaks and corresponding ions were low, the most often appearing oxidation product could be identified as m/z 575 with fragments m/z 449 and m/z 423, as previously identified for the A-type PC dimer.32,48 In addition, the concurrent decrease of some peaks and increase of peaks with the same m/z value and identical UV spectrum indicated epimerization reactions to occur (data not shown). These further pointed toward the reaction route via intramolecular oxidation. However, as QqQ is an integer resolution mass spectrometer, these findings must be considered with caution and will be confirmed in future study, focusing on the identification of the nonoxidized and oxidized PAs and their possible reaction routes using a high-resolution mass spectrometer. Furthermore, as complex as the mass spectra of the PA hump area, the formation of new polymers via intermolecular reactions cannot be completely ruled out, but if these reactions occurred, they were rather subtle.
In categories C and D, the main findings were the shift in the retention times of the PA humps in the UHPLC–DAD chromatograms, high decrease rates in the MS/MS detection of PAs, and variable changes in the UV peak areas of the detected PA humps. In addition, the lower ionization efficiency of the oxidation products indicated the formation of numerous new oligomers and polymers, most likely with higher mDPs than the original PAs. Thus, we suggest that in C and D categories, both intra- and intermolecular oxidation reactions occurred with varying proportions, depending on the sample. However, these findings could not be even tentatively confirmed from the total ion chromatograms because of the low ionization efficiency and/or the low concentration of individual PAs.
Conclusions
Altogether, the results in the current study indicated two different main reaction routes for PAs under alkaline conditions; in most of the PC-rich samples, the main reaction route was such that the retention time of the detected PA hump by UHPLC–DAD remained unchanged, and in most of the samples, the decrease in the UV peak area was moderate, while the decrease in the MS/MS detection varied a lot. On the other hand, most of the PD-rich samples and the samples with galloylated PAs showed a different reaction route by UHPLC–DAD as the detected PA hump shifted retention time wise. Also, in these samples, the decrease in the quantitative MS/MS levels was high, while the decrease in the UV peak area varied a lot between the samples. Based on the results, we suggest the main reaction route in the category A and B samples to be intramolecular, while in the C and D categories, both intra- and intermolecular reactions were present. Unfortunately, the methods of the present study did not allow to make a difference between different PA epimers and other isomers, which have been shown to affect PA reactivity as well and could further explain some of the variations in the reactivities within the A/B and C/D categories in samples with otherwise similar quantitative parameters. Based on this preliminary screening, the next obvious step will be to use high-resolution MS to confirm the suggested reaction routes and findings from the present study. Additionally, as the studied samples expressed various stabilities, it will be of interest to define whether stability or significant modifications result in better bioactivities for an increased value of the original biomaterial.
Materials and Methods
Chemicals and Reagents
Analytical-grade acetone (VWR International S.A.S., France) was used during extraction. For oxidation, a pH 10 carbonate buffer (50 mM; sodium carbonate/sodium hydrogen carbonate, J.T. Baker, Deventer, Netherlands) and, for stopping the oxidation, formic acid (J.T. Baker, Deventer, Netherlands) were used. For the UHPLC analysis, LC–MS-grade acetonitrile was purchased from VWR International S.A.S. (USA), and LC–MS-grade formic acid was purchased from Sigma Aldrich (Seelze, Germany). The Milli-Q water used was purified with the Millipore Synergy UV (Merck KGaA, Darmstadt, Germany) system.
Plant Samples and Extraction
In total, 300 plant samples were collected from the botanical garden (60°26′0”N, 22°10′19″E) of the University of Turku, Finland. All the samples were identified with the herbarium of the botanical garden. After collection, the plant samples were freeze-dried, ground into fine powder, and stored in a freezer (−20 °C). For extraction, 20 mg of finely ground dried plant tissue was mixed with 1.4 mL of acetone/water (80/20, v/v), vortexed, and macerated overnight at 4 °C. The samples were then extracted twice for 3 h with 1400 μL of acetone–water (7:3, v/v), the extracts were combined and concentrated to remove acetone, and the aqueous phases were freeze-dried.
Sample Preparation
For the PA screening phase with UHPLC–MS/MS, the freeze-dried extracts were dissolved in 1 mL of Milli-Q water and vortexed for 15 min. The extracts were filtered with 0.2 μm polytetrafluoroethylene (PTFE) filters (VWR International, Radnor, PA, USA) to remove the lipophilic components. After fivefold dilution with water, the samples were analyzed by the UHPLC–DAD–QqQ-MS/MS protocol described below. For the aerial oxidation of the PA-containing extracts, 20 μL of each extract was oxidized with 180 μL of pH 10 buffer for 1 h at room temperature. Oxidation was stopped by adding 100 μL of 0.6% aq. HCOOH. The control extracts were prepared by taking 20 μL of each extract and adding 280 μL of the 180/100 (v/v) mixture of the pH 10 buffer and 0.6% aq. HCOOH. After further vortexing for 5 min, 100 μL of each sample (nonoxidized and oxidized) was inserted into separate UHPLC vials for the UHPLC–DAD–QqQ-MS/MS analysis.
UHPLC–DAD–QqQ-MS/MS Analysis
The UHPLC–DAD–QqQ-MS/MS analyses were performed with a Xevo TQ QqQ mass spectrometer (Waters Corp., Milford, MA, USA) coupled with an Aquity UPLC system (Waters Corp., Milford, MA, USA). The UPLC system consisted of a sample manager, a binary solvent manager, a column, and a diode array detector. The column used was a Waters Acquity UPLC BEH Phenyl (1.7 μm, 2.1 × 100 mm Waters Corp. Wexferd, Ireland). Similar elution protocol, ion source parameters, and detection at UV and MS/MS were utilized, as reported in Engström et al.38 The data collection of both UV and MS occurred continuously from 0 to 7 min. The stabilities of the UHPLC retention times and the m/z values of the MS detector were monitored with a flavonoid mix stock solution containing 4 μg mL–1 each of kaempferol-7-O-glucoside, kaempferol-7-O-neohesperoside, kaempferol-3-O-glucoside, quercetin-3-O-galactoside, and quercetin-3-O-glucoside in acetonitrile/0.1% aqueous formic acid (1:4 v/v). The stability of the MS/MS response was monitored by injecting 1 μg mL–1 catechin solution (in 1/4 acetonitrile/0.1% formic acid (v/v)) five times before and after every batch of 10 samples. Quantitative results were corrected for possible fluctuations in the system’s quantitative performance within each analysis set, as well as among different sets.
The detection and quantification of the PC and PD subunits and the mDP were obtained with the Engström method,38,40 with the exceptions that the PC and PD subunits were detected using three different cone voltages, which were 75, 85, and 140 V for the PCs and 55, 80, and 130 V for the PDs. The recorded PC and PD traces were smoothed (window size 30 scans × 5 smoothing iterations) and integrated with the TargetLynx software (V4.1 SCN876 SCN 917, 2012 Waters Inc.). Quantitative results were obtained from the calibration curves obtained separately for PC, PD, and mDP. The PC and PD calibration curves were obtained with two Sephadex LH-20 fractions: the PC standard from Tilia flowers and the PD standard from Ribes nigrum leaves. The concertation range of the calibration curves was 1.50–0.1875 mg mL–1 for the PC standard and 2.00–0.25 mg mL–1 for the PD standard. The mDP calibration curve (to fine-tune the equation for the calculation of mDP38) was obtained with six Sephadex LH-20 fractions from Vaccinium vitis-idaea leaves, Calluna vulgaris flowers, and Tilia flowers with known mDPs. All the calibration curve samples were prepared in 2/8 acetonitrile/0.1% formic acid (v/v).
The PA tannin fingerprints were produced from the MRM raw data by calculating the PA concentration at each time point with the PC and PD calibration curves, as explained by Salminen in 2018.40
Statistical Analysis
All the coefficients of determination, that is, the R2 values mentioned in the text, were obtained from simple linear regression models in Microsoft Excel software (Microsoft Office 365 ProPlus).
Funding
The project was funded by the ModiFeed project (part of Biofuture strategy), Department of Chemistry, University of Turku, Finland and by the Academy of Finland (grant number 298177 to J.-P.S.).
Acknowledgments
We thank Anne Koivuniemi and Jorma Kim for technical help. All members of NCRG are acknowledged for general help and discussion.
Glossary
Abbreviations
- ESI
electrospray ionization
- mDP
mean degree of polymerization
- MRM
multiple reaction monitoring
- PA
proanthocyanidin
- PC
procyanidin
- PD
prodelphinidin
- UHPLC
ultrahigh-performance liquid chromatography
- QqQ-MS
triple-quadrupole mass spectrometry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05515.
UHPLC–DAD profiles (λ = 280 nm) before and after oxidation. Correlations of different measured parameters before and after oxidation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Porter L.J.Condensed tannins. In Natural products of woody plants I; Rowe J.W., Ed. Springer-Verlag, Berlin, Germany, 1989, 651–690. [Google Scholar]
- Dixon R. A.; Xie D. Y.; Sharma S. B. Proanthocyanidins--a final frontier in flavonoid research?. New Phytol. 2005, 165, 9–28. 10.1111/j.1469-8137.2004.01217.x. [DOI] [PubMed] [Google Scholar]
- Ferreira D.; Slade D.; Marais P.J.. Flavans and proanthocyanidins. In Flavonoids: Chemistry, biochemistry and applications. Andersen O.M., Markham K.R., Eds.; Taylor and Francis, New York, US, 2005, 553–616. [Google Scholar]
- Ferreira D.; Slade D. Oligomeric proanthocyanidins: naturally occurring O-heterocycles. Nat. Prod. Rep. 2002, 19, 517–541. 10.1039/b008741f. [DOI] [PubMed] [Google Scholar]
- Haslam E. Symmetry and promiscuity in procyanidin biochemistry. Phytochemistry 1977, 16, 1625–1640. 10.1016/0031-9422(71)85060-4. [DOI] [Google Scholar]
- Yazaki Y. Utilization of flavonoid compounds from bark and wood: a review. Nat. Prod. Commun. 2015, 10, 513–520. 10.1177/1934578X1501000333. [DOI] [PubMed] [Google Scholar]
- Retrieved from https://www.grandviewresearch.com/industry-analysis/tannin-market
- Prigione V.; Spina F.; Tigini V.; Giovando S.; Varese G. C. Biotransformation of industrial tannins by filamentous fungi. Appl. Microbiol. Biotechnol. 2018, 102, 10361–10375. 10.1007/s00253-018-9408-4. [DOI] [PubMed] [Google Scholar]
- Pizzi A. Tannins: Prospectives and actual industrial applications. Biomolecules 2019, 9, 344. 10.3390/biom9080344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.P.; Kumar S. Applications of tannins in industry. In Tannins - structural properties, biological properties and current knowledge .Aires A., Ed.; IntechOpen, London, UK, 2020. 10.5772/intechopen.85984 [DOI] [Google Scholar]
- Kumar R.S.; Manimegalai G.. Fruit and vegetable processing industries and environment. In Industrial Pollution & Management. Kumar A., Ed.; APH Publishing Corporation, New Delhi, India, 2004, 97–117. [Google Scholar]
- Federici F.; Fava F.; Kalogerakis N.; Mantzavinos D. Valorisation of agro-industrial byproducts, effluents and waste: concept, opportunities and the case of olive mill wastewaters. J. Chem. Technol. Biotechnol. 2009, 84, 895–900. 10.1002/jctb.2165. [DOI] [Google Scholar]
- Aires A.; Carvalho R.; Saavedra M. J. Valorization of solid wastes from chestnut industry processing: Extraction and optimization of polyphenols, tannins and ellagitannins and its potential for adhesives, cosmetic and pharmaceutical industry. Waste Manage. 2016, 48, 457–464. 10.1016/j.wasman.2015.11.019. [DOI] [PubMed] [Google Scholar]
- de Hoyos-Martínez P. L.; Merle J.; Labidi J.; Charrier-El Bouhtoury F. Tannins extraction: a key point for their valorization and cleaner production. J. Cleaner Prod. 2019, 206, 1138–1155. 10.1016/j.jclepro.2018.09.243. [DOI] [Google Scholar]
- Laks P. E.; Hemingway R. W. Condensed tannins. Base-catalysed reactions of polymeric procyanidins with phloroglucinol: intramolecular rearrangements. J. Chem. Soc., Perkin Trans. 1 1987, 1875–1881. 10.1039/P19870001875. [DOI] [Google Scholar]
- Burger J. F. W.; Kolodziej H.; Hemingway R. W.; Steynberg J. P.; Young D. A.; Ferreira D. Oligomeric flavanoids. Part 15a. Base-catalyzed pyran rearrangements of procyanidin B-2, and evidence for the oxidative transformation of B- to A-type procyanidins. Tetrahedron 1990, 46, 5733–5740. 10.1016/S0040-4020(01)87771-1. [DOI] [Google Scholar]
- Ferreira D.; Steynberg J.P.; Burger J.F.W.; Bezuidenhoudt B.C.B. Oxidation and rearrangement reactions of condensed tannins. In: Plant polyphenols: synthesis, properties, significance. Hemingway R.W., Laks P.E., Branham S.J., Eds.; Plenum Press, New York, USA, 1992, 349–384. 10.1007/978-1-4615-3476-1_20 [DOI] [Google Scholar]
- Guyot S.; Vercauteren J.; Cheynier V. Structural determination of colourless and yellow dimers resulting from (+)-catechin coupling catalysed by grape polyphenoloxidase. Phytochemistry 1996, 42, 1279–1288. 10.1016/0031-9422(96)00127-6. [DOI] [Google Scholar]
- Bernillon S.; Guyot S.; Renard C. M. G. C. Detection of phenolic oxidation products in cider apple juice by high-performance liquid chromatography electrospray ionisation ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 939–943. 10.1002/rcm.1430. [DOI] [PubMed] [Google Scholar]
- Mouls L.; Fulcrand H. UPLC-ESI-MS study of the oxidation markers released from tannin depolymerization: toward a better characterization of the tannin evolution over food and beverage processing. J. Mass Spectrom. 2012, 47, 1450–1457. 10.1002/jms.3098. [DOI] [PubMed] [Google Scholar]
- Hibi Y.; Yanase E. Oxidation of procyanidins with various degrees of condensation: Influence on the color-deepening phenomenon. J. Agric. Food Chem. 2019, 67, 4940–4946. 10.1021/acs.jafc.9b02085. [DOI] [PubMed] [Google Scholar]
- Pizzi A. Condensed tannins for adhesives. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 359–369. 10.1021/i300007a005. [DOI] [Google Scholar]
- Fradinho D. M.; Neto C. P.; Evtuguin D.; Jorge F. C.; Irle M. A.; Gil M. H.; de Jesus P. J. Chemical characterisation of bark and of alkaline bark extracts from maritime pine grown in Portugal. Ind. Crops Prod. 2002, 16, 23–32. 10.1016/S0926-6690(02)00004-3. [DOI] [Google Scholar]
- Pizzi A. Recent developments in eco-efficient bio-based adhesives for wood bonding: opportunities and issues. J. Adhes. Sci. Technol. 2006, 20, 829–846. 10.1163/156856106777638635. [DOI] [Google Scholar]
- Zhou X.; Du G.. Applications of Tannin Resin Adhesives in the Wood Industry. In Tannins - structural properties, biological properties and current knowledge .Aires A. Ed. IntechOpen, London, UK, 2020. 10.5772/intechopen.86424 [DOI] [Google Scholar]
- Poncet-Legrand C.; Cabane B.; Bautista-Ortin A.-B.; Carrillo S.; Fulcrand H.; Perez J.; Vernhet A. Tannin Oxidation: Intra- versus intermolecular reactions. Biomacromolecules 2010, 11, 2376––2386. 10.1021/bm100515e. [DOI] [PubMed] [Google Scholar]
- Vernhet A.; Carrillo S.; Poncet-Legrand C. Condensed tannin changes induced by autoxidation: effect of the initial degree of polymerization and concentration. J. Agric. Food Chem. 2014, 62, 7833–7842. 10.1021/jf501441j. [DOI] [PubMed] [Google Scholar]
- Vernhet A.; Carrillo S.; Rattier A.; Verbaere A.; Cheynier V.; Nguela J. M. Fate of anthocyanins and proanthocyanidins during the alcoholic fermentation of thermovinified red musts by different saccharomyces cerevisiae strains. J. Agric. Food Chem. 2020, 68, 3615–3625. 10.1021/acs.jafc.0c00413. [DOI] [PubMed] [Google Scholar]
- Jorgensen E. M.; Marin A. B.; Kennedy J. A. Analysis of the oxidative degradation of proanthocyanidins under basic conditions. J. Agric. Food Chem. 2004, 52, 2292–2296. 10.1021/jf035311i. [DOI] [PubMed] [Google Scholar]
- Vernhet A.; Dubascoux S.; Cabane B.; Fulcrand H.; Dubreucq E.; Poncet-Legrand C. Characterization of oxidized tannins: comparison of depolymerization methods, asymmetric flow field-flow fractionation and small-angle X-ray scattering. Anal. Bioanal. Chem. 2011, 401, 1559–1569. 10.1007/s00216-011-5076-2. [DOI] [PubMed] [Google Scholar]
- Mouls L.; Fulcrand H. Identification of new oxidation markers of grape-condensed tannins by UPLC-MS analysis after chemical depolymerization. Tetrahedron 2015, 71, 3012–3019. 10.1016/j.tet.2015.01.038. [DOI] [Google Scholar]
- Miranda-Hernández A. M.; Muñiz-Márquez D. B.; Wong-Paz J. E.; Aguilar-Zárate P.; de la Rosa-Hernández M.; Larios-Cruz R.; Aguilar C. N. Characterization by HPLC-ESI-MS2 of native and oxidized procyanidins from litchi (Litchi chinensis) pericarp. Food Chem. 2019, 291, 126–131. 10.1016/j.foodchem.2019.04.020. [DOI] [PubMed] [Google Scholar]
- Zanchi D.; Konarev P. V.; Tribet C.; Baron A.; Svergun D. I.; Guyot S. Rigidity, conformation, and solvation of native and oxidized tannin macromolecules in water-ethanol solution. J. Chem. Phys. 2009, 130, 245103. 10.1063/1.3156020. [DOI] [PubMed] [Google Scholar]
- Vázquez G.; Antorrena G.; Parajó J. C. Studies on the utilization of Pinus pinaster bark. Wood Sci. Technol. 1987, 21, 155–166. 10.1007/BF00376195. [DOI] [Google Scholar]
- Salminen J.-P.; Karonen M. Chemical ecology of tannins and other phenolics: we need a change in approach. Funct. Ecol. 2011, 25, 325–338. 10.1111/j.1365-2435.2010.01826.x. [DOI] [Google Scholar]
- Vihakas M.; Pälijärvi M.; Karonen M.; Roininen H.; Salminen J.-P. Rapid estimation of the oxidative activities of individual phenolics in crude plant extracts. Phytochemistry 2014, 103, 76–84. 10.1016/j.phytochem.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Kim J.; Pälijärvi M.; Karonen M.; Salminen J.-P. Oxidatively active plant phenolics detected by UHPLC-DAD-MS after enzymatic and alkaline oxidation. J. Chem. Ecol. 2018, 44, 483–496. 10.1007/s10886-018-0949-x. [DOI] [PubMed] [Google Scholar]
- Engström M. T.; Pälijärvi M.; Fryganas C.; Grabber J. H.; Mueller-Harvey I.; Salminen J.-P. Rapid qualitative and quantitative analyses of proanthocyanidin oligomers and polymers by UPLC-MS/MS.. J. Agric. Food Chem. 2014, 62, 3390–3399. 10.1021/jf500745y. [DOI] [PubMed] [Google Scholar]
- Engström M. T.; Pälijärvi M.; Salminen J.-P. Rapid fingerprint analysis of plant extracts for ellagitannins, gallic acid, and quinic acid derivatives and quercetin-, kaempferol- and myricetin-based flavonol glycosides by UPLC-QqQ-MS/MS.. J. Agric. Food Chem. 2015, 63, 4068–4079. 10.1021/acs.jafc.5b00595. [DOI] [PubMed] [Google Scholar]
- Salminen J.-P. Two-dimensional tannin fingerprints by liquid chromatography tandem mass spectrometry offer a new dimension to plant tannin analyses and help to visualize the tannin diversity in plants. J. Agric. Food Chem. 2018, 66, 9162–9171. 10.1021/acs.jafc.8b02115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami H.; Ohkoshi E.; Amano S.; Satoh K.; Kanamoto T.; Terakubo S.; Nakashima H.; Sunaga K.; Otsuki T.; Ikeda H.; Fukuda T. Efficient utilization of plant resources by alkaline extraction. Altern. Integ. Med. 2013, 02, 7. 10.4172/2327-5162.1000133. [DOI] [Google Scholar]
- Zhu Q. Y.; Holt R. R.; Lazarus S. A.; Ensunsa J. L.; Hammerstone J. F.; Schmitz H. H.; Keen C. L. Stability of the flavan-3-ols epicatechin and catechin and related dimeric procyanidins derived from cocoa. J. Agric. Food Chem. 2002, 50, 1700–1705. 10.1021/jf011228o. [DOI] [PubMed] [Google Scholar]
- Bors W.; Michel C.; Stettmaier K. Electron paramagnetic resonance studies of radical species of proanthocyanidins and gallate esters. Arch. Biochem. Biophys. 2000, 374, 347–355. 10.1006/abbi.1999.1606. [DOI] [PubMed] [Google Scholar]
- Valcic S.; Burr J. A.; Timmermann B. N.; Liebler D. C. Antioxidant chemistry of green tea catechins. New oxidation products of (−)-epigallocatechin gallate and (−)-epigallocatechin from their reactions with peroxyl radicals. Chem. Res. Toxicol. 2000, 13, 801–810. 10.1021/tx000080k. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Wei L.; Ge Z.; Zhu W.; Li C. Comparison of the degradation kinetics of A-type and B-type proanthocyanidins dimers as a function of pH and temperature. Eur. Food Res. Technol. 2015, 240, 707–717. 10.1007/s00217-014-2375-9. [DOI] [Google Scholar]
- Hümmer W.; Schreier P. Analysis of proanthocyanidins. Mol. Nutr. Food Res. 2008, 52, 1381–1398. 10.1002/mnfr.200700463. [DOI] [PubMed] [Google Scholar]
- Guyot S.Flavan-3-ols and proanthocyanidins. In: Handbook of analysis of active compounds in functional foods .Nollet L.M.L., Toldrá F., Eds.; CRC Press/Taylor & Francis, Boca Raton, FL, USA, 2012, 317–348. [Google Scholar]
- Rue E. A.; Rush M. D.; van Breemen R. B. Procyanidins: a comprehensive review encompassing structure elucidation via mass spectrometry. Phytochem. Rev. 2018, 17, 1–16. 10.1007/s11101-017-9507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.