

ABSTRACT
Traditionally, non-specific chemical conjugations, such as acylation of amines on lysine or alkylation of thiols on cysteines, are widely used; however, they have several shortcomings. First, the lack of site-specificity results in heterogeneous products and irreproducible processes. Second, potential modifications near the complementarity-determining region may reduce binding affinity and specificity. Conversely, site-specific methods produce well-defined and more homogenous antibody conjugates, ensuring developability and clinical applications. Moreover, several recent side-by-side comparisons of site-specific and stochastic methods have demonstrated that site-specific approaches are more likely to achieve their desired properties and functions, such as increased plasma stability, less variability in dose-dependent studies (particularly at low concentrations), enhanced binding efficiency, as well as increased tumor uptake. Herein, we review several standard and practical site-specific bioconjugation methods for native antibodies, i.e., those without recombinant engineering. First, chemo-enzymatic techniques, namely transglutaminase (TGase)-mediated transamidation of a conserved glutamine residue and glycan remodeling of a conserved asparagine N-glycan (GlyCLICK), both in the Fc region. Second, chemical approaches such as selective reduction of disulfides (ThioBridge) and N-terminal amine modifications. Furthermore, we list site-specific antibody–drug conjugates in clinical trials along with the future perspectives of these site-specific methods.
Keywords: hybrid modality, bioconjugation, transglutaminase, glycan remodeling, antibody–drug conjugate (ADC)
Statement of Significance: Compared with their non-specific counterpart, site-specific bioconjugation produces more homogenous, well-defined, and developable antibody conjugates that are likely to achieve their desired properties and functions. In this review, we present practical site-specific methods to conjugate native (non-engineered) antibodies, as well as highlight their clinical applications and discuss future directions.
INTRODUCTION
Hybrid Modality Engineering of Protein is a platform that introduces non-canonical chemical moieties or scaffolds, i.e., effectors—and thus functions—into peptides and proteins, e.g., native antibodies, to confer novel and improved functions otherwise unavailable via recombinant technology. Common protein engineering approaches include mutagenesis, insertion or deletions of peptides or incorporation of unnatural amino acids. These approaches have several shortcomings, such as providing only a linear architecture, and in some cases, it requires laborious optimization and low yield results. As reviewed herein, a powerful alternative is the site-specific conjugation of native antibodies, i.e., those without recombinant engineering.
Site-specific vs. non-specific (stochastic or random)
Until recently, most antibody conjugates were constructed through traditional chemical methods, such as acylation of amines on lysine residues and N-termini, as well as alkylation of thiols on cysteine residues. These processes have been used to construct antibody–drug conjugates (ADC) approved by the food and drug administration (FDA) [2, 3]. Unfortunately, these strategies are typically not site-specific, which result in heterogeneous mixtures, produce positional isomers, and suffer poor reproducibility. For example, Lou et al. [4] have shown that all amines in an IgG (76 lysine residues in both light and heavy chains [HCs]) were modified via N-hydroxysuccinimide (NHS) chemistry to observable degrees. These reactions yield heterogeneous constructs with a widely distributed drug-to-antibody ratio (DAR) of 0–8. The multiple sites of modifications may cause structural changes to the antibody and have been shown to perturb their underlying biological function [1, 5–7]; for instance, modifications at or near the complementarity-determining region (CDR) in antibodies may reduce antigen binding and specificity.
Further complications arise from the challenges in the characterization of conjugates. To start, antibodies themselves consist of various post-translational modifications (e.g., charge variants) such as deamidation, oxidation, glycosylation, or other reactive metabolites [8–17]. Therefore, the analysis of antibody conjugates constructed via the non-specific approaches is rather complicated, and the detection of all modifications can be difficult, thereby underestimating the sites of modifications [18]. For example, the modification sites are missed due to false negatives’ prevalence during analysis [18]. Furthermore, non-specific methodologies can produce side reactions that are often underappreciated [18]. Overall, these species—both unmodified and modified antibody conjugates—exponentially increase the complexity of the analysis after non-specific chemical modifications and further increase the resulting product’s heterogeneity.
To overcome non-specific conjugation hurdles, site-specific methods to modify antibodies produce homogenous and well-defined conjugate and do not compromise the conjugate’s biological activity (Fig. 1). For instance, these chemistries avoid modifications to the antigen-binding region (Fab) and have been shown to increase the binding efficiency compared with their non-specific counterpart [1]. With the advent of click and other bioorthogonal chemistries [19–21]—which are fast, high yielding, and with commercially available reagents—various antibody conjugates have been constructed via site-specific conjugation with a wide range of applications in analysis, imaging, diagnosis, and therapy.
Figure 1.
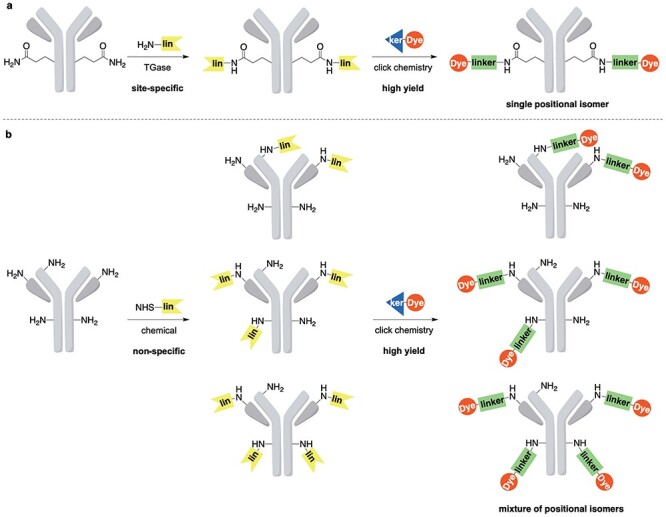
Antibody conjugates constructed via a convergent assembly [1]. (a) Site-specific, transglutaminase (TGase)-catalyzed conjugation leads to homogenous constructs that preserve binding affinity. (b) Non-specific chemical modification, such as acylation of amines, yields a heterogenous mixture that ultimately reduces binding affinity. Adapted with permission from Sadiki et al. [1]. Copyright © 2020, John Wiley, and Sons.
Some of the previously reported site-specific conjugation technologies include linchpin directed modification, which conjugates drugs and fluorescent tags to the Fab and monoclonal antibodies, and regioselective and chemo-selective lysine modification with sulfonyl acrylate reagents [22–24]. In 2019, a novel proprietary technology—affinity peptide-mediated regiodivergent functionalization (AJICAP™) was reported, which utilizes Fc affinity reagents to target selective lysine residues, onto which the payloads are attached [25].
This review will focus on both chemical and enzymatic site-specific methods for the conjugation of native antibodies without the need for genetic engineering. In this context, native antibodies refer to antibodies produced or naturally existing in various species (e.g., human, mouse or goat) and antibodies that are recombinantly produced, where the conserved sequences are preserved and not engineered.
Improved properties and functions
Site-specific methods offer advantages over their non-specific counterparts in the conjugate’s properties and functions, such as enhanced plasma stability, increased tumor uptake, enhanced binding efficiency, less variability in dose studies, and a higher level of binding (Fig. 2). The work conducted by Kristensen et al. [6], measuring the percentage of intact tracer present in plasma, demonstrated increased stability for site-specifically labeled conjugates compared with their stochastically labeled counterparts. After 168 h, the site specifically labeled conjugates, ß- [1–4] galactosidase and endoglycosidase S2, were stable in plasma with >99% and 91% intact tracer, respectively. On the other hand, random labeling showed a decrease to 80% intact tracer in plasma (Fig. 2a) [6]. Moreover, site-specific conjugates exhibited a higher tumor uptake than non-specific conjugates (Fig. 2a) [6]. The tumor uptake was “twice as high for the site-specific conjugates, 14.2 ± 1.7 %ID/g (β-Gal) and 16.7 ± 1.5 %ID/g (endoS2), compared with 6.5 ± 0.5 %ID/g for the randomly labeled trastuzumab 120-h post-injection” (Fig. 2a) [6]. Sadiki et al. [1] reported a two-fold higher binding efficiency for the site-specific compared with the non-specific constructs (Fig. 2b). Less variability of site-specific conjugates (center and right panels) at low concentrations (between 10 nM and 10 pM) compared with the non-specific conjugates (left panel) was observed by Pellegrino et al. [5] (Fig. 2c). Randomly labeled conjugates (left panel) showed an impaired level of binding compared with the site specifically labeled conjugates (center and right panels) (Fig. 2d).
Figure 2.
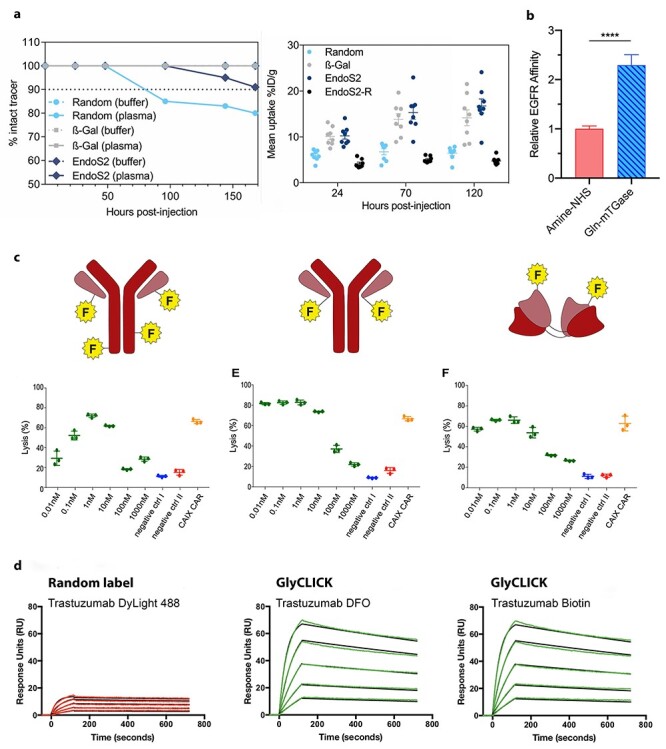
(a) Site-specific conjugates (modified via β-Gal and EndoS2) showed higher tracer stability in plasma compared with non-specific (random) labeled conjugates in plasma. Mean tumor uptake is more elevated for site-specific conjugates (β-Gal and EndoS2) compared to non-specific (random) conjugates post-injection in mice (right) [6]. (b) Site-specific conjugation (mediated by transglutaminase) demonstrated a two-fold increase in binding efficiency compared with its non-specific counterpart (i.e., acylation of amines) [1]. (c) Site-specific conjugates (center and right panels) show less variability at a low concentrations between 10 nM and 10 pM compared with non-specific conjugates (left panel) [5]. (d) SPR analysis of trastuzumab conjugates formed by random conjugation (left panel) and site-specific conjugation to introduce DFO (Deferoxamine) and Biotin (center and right panels) [26]. The randomly labeled conjugate showed impaired binding compared with the site-specifically conjugated antibody. Modified and adapted with permission from Kristensen et al. [6], Sadiki et al. [1], Pellegrino et al. [5], and Genovis. Copyright © 2020, John Wiley and Sons; Copyright © 2014, American Chemical Society.
Site-specific conjugation chemistries
In the following sections, chemo-enzymatic methods, namely transglutaminase (TGase) and glycan-mediated conjugation, and chemical methods such as selective reduction of disulfides (ThioBridge) and N-terminal modification are presented.
CHEMO-ENZYMATIC
Chemo-enzymatic methods are typically site-specific and generally proceed under mild reaction conditions. Furthermore, for a given family of enzymes, their activity and specificity can be tailored by tuning the reaction conditions, choosing multiple isoforms that confer different specificities, as well as protein engineering [27, 28]. In the next section, we introduce two of these approaches: transglutaminase-mediated modification of glutamine and glycan remodeling, which occur in the conserved Fc region of an antibody.
Transglutaminase
As early as 2000, antibodies were reported to be modified by transglutaminase (TGase, EC 2.3.2.13) [29]. Although over 60 glutamine (Gln) residues exist in an antibody, only a single conserved residue in the HC is typically modified, as reported by Schibli et al. [30, 31] [EEQ295YN*S; of note, the Asn (N*) residue is typically glycosylated]. This residue resides in the Fc region of deglycosylated antibodies and is efficiently modified by the corresponding amine substrate under the catalysis of TGase. The deglycosylation of the conserved asparagine (two residues C-terminal to the glutamine), ensure that the adjacent glutamine residue is accessible for enzymatic transformation. Interestingly, Marculescu et al. [32] recently reported that microbial transglutaminase (mTGase) also modifies another glutamine residue (Gln3) in the HC region, albeit to a lesser extent. We and others [1, 31, 33–35] have shown that desired, and commercially available amine reagents can be conjugated into antibodies via one-step (Fig. 3) or two-step (Fig. 1) convergent processes mediated by click and other bio-orthogonal chemistries. Various reports of one-pot dual conjugation [36], branched [37], heterobifunctional [34] and proteolytically cleavable [36, 38] linkers, as well as non-canonical substrates [35] such as hydrazines, hydrazides, and alkoxyamines, have been conjugated to antibodies and thus illustrates the versatility of this family of enzymes as a tool for bioconjugation. Moreover, recombinant peptide tags, as well as reactive glutamine and lysine residues, have been introduced into antibodies through genetic engineering; however, these systems are outside the scope of this review and have been summarized by others [39, 40]. Since only a conserved Gln295 is the primary modification site, in principle, IgG1, IgG2, IgG3, and IgG4 can also be modified without the need for genetic engineering. These methodologies have produced homogenous antibody conjugates for various applications, including imaging via radioimmunoconjugates [41], cytotoxicity using ADCs [38], PEGylation [40], and functional virus-based nanoparticles [42]. Lastly, Pfizer’s Trop-2 ADC constructed via transglutaminase-mediated conjugation showed promise in preclinical studies [43] and was further translated to a phase 1 (NCT02122146; Table 1) clinical trial in solid tumors [44]. The Phase 1 results show that the iso-peptide amide bond is stable in both in vitro and in vivo settings.
Figure 3.
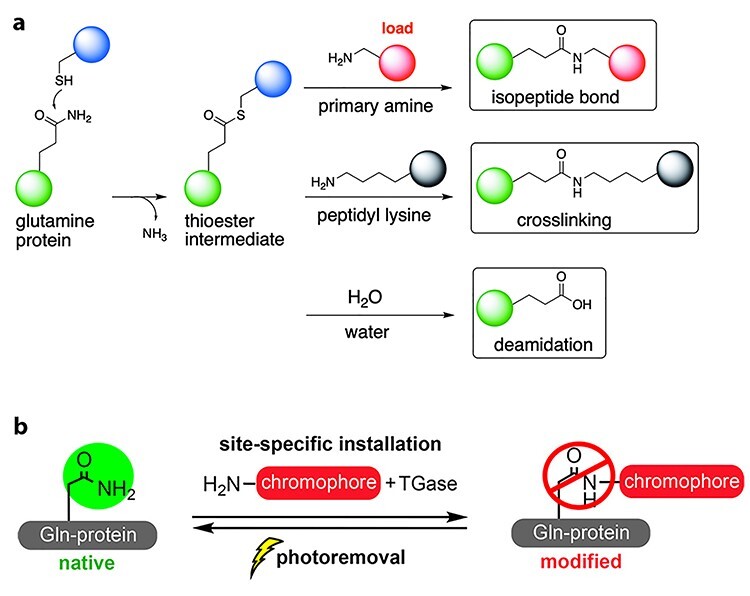
(a) Transglutaminase (TGase)-mediated transamidation of glutamine residues. First, a nucleophilic attack of the cysteine thiol in the active site of TGase to generate a thioester intermediate. Second, an amine-containing molecule nucleophilic attacks the thioester forging a new iso-peptide amide bond. Lastly, competing reactions include cross-linking of glutamines with lysine residues in peptides/proteins and deamidation of glutamine to generate glutamic acid via hydrolysis. These competing reactions can be entirely suppressed by a sub- or low-millimolar concentrations of exogenous amines. (b) TGase-mediated conjugation of protein to generate photoremovable conjugates. First, amine-containing chromophores are installed into glutamines by TGase. Second, light-mediated removal of the chromophores to regenerate the native peptide or proteins [51]. Modified and adapted with permission from Moulton et al. [51]. Copyright © 2019, American Chemical Society.
Table 1.
Representative native ADCs using site-specific conjugation methods that are currently in phase 1 clinical trials for cancer therapy [44, 95, 111, 112]
| Chemistry | Name | Developer | Drug to antibody ratio | Description | Clinical trial identifier |
|---|---|---|---|---|---|
| TGase-glutamine | PF-06664178 | Pfizer | 2 | • Anti-TROP2 IgG1 • Valine-citrulline cleavable linker • Auristatin-based cytotoxic payload |
NCT02122146 |
| Glycan remodeling | ADCT-601 | ADC Therapeutics | 2 | • Anti-Human AXL IgG1 • Valine-citrulline cleavable linker • PBD (pyrrolobenzodiazepine)—cytotoxic dimer payload |
NCT03700294 |
| XMT-1592 | Mersana Therapeutics | 6 | • Anti-NaPi2b IgG1 • Proprietary platform (Dolasynthen) • Auristatin-based cytotoxic payload |
NCT04396340 | |
| ThioBridge | OBI-999 | OBI-Pharmaceuticals | 4 | • Anti-Globo H IgG • Proteolytically cleavable linker • Auristatin-based cytotoxic payload |
NCT04084366 |
Transglutaminases (TGases, EC 2.3.2.13) are a family of enzymes that catalyze an iso-peptide amide bond formation between an unsubstituted side-chain amide group of glutamine residues (as acyl donor) and a nucleophilic amine substrate (as acyl acceptor), e.g., a primary amine (Fig. 3), under mild reaction conditions [45, 46]. The resulting amide bond on the glutamine side chain is as chemically stable as other amide bonds in proteins and appears to be stable in biological systems (i.e., no enzymatic hydrolysis has been reported). Numerous isoforms of TGases have been identified, produced, and engineered with tailored broad/narrow specificities, [47, 48]. The substrate specificity of TGases is governed primarily by solvent accessibility of the glutamine or lysine residues, dynamic and flexibility of the modified region, and flanking residues proximal to the modification site [49, 50]. Therefore, typically only a few glutamine or lysine residues are modified in any given protein. Competing reactions include cross-linking of glutamines with lysine residues in peptides/proteins and deamidation of glutamine to glutamic acid (Fig. 3); however, in practice, these reactions can be entirely suppressed by a sub- or low-millimolar concentrations of exogenous amines [1, 38, 46, 51].
Due to its site-specific nature, requiring no co-factor, robustness, commercial availability, and low cost, mTGase from Streptomyces mobaraense [52] is widely used to construct protein–DNA, protein–glycosylation, protein–polymer, protein–protein conjugates, surface immobilization, as well as cross-linking in the food industry, academic research, and pharmaceutical development [40]. mTGase displays broad specificity toward primary amine substrates [53, 54]; for example, various commercially available primary amines containing azides [1], strained alkenes and alkynes [1, 55], tetrazines [55], and bulkier substrates such as polyethylene glycol (PEG) [56], have been incorporated into proteins using mTGase mediated conjugation. Besides, experimental conditions can be tuned to enhance selectivity, e.g., addition of organic co-solvents [57]. Lastly, this enzyme has been immobilized on solid-matrices such as glass [58] and agarose [59] beads, facilitating easy removal from the reaction mixtures via simple filtration.
Native human antibodies are poor substrates for TGase and require deglycosylation for efficient modification. To ameliorate such restrictions, ongoing efforts on engineering TGases and other transferases [60] with altered specificity—e.g., modification of antibodies without the need to deglycosylate the conserved asparagine [61]—will undoubtedly facilitate clinical translation.
Our laboratory devised a novel photo-reversible conjugation chemistry based on TGase, which adds versatility to the field (as depicted in Fig. 3). The technology site-specifically installs photo-switches into peptides and proteins and generates photoremovable conjugates that allow the native proteins to be regenerated by light (i.e., photo-caging) [51]. This methodology is simple, robust, and easily adaptable and thus can be utilized to spatially and temporally control the biological functions of antibodies by light—e.g., binding to their respective antigen or effector functions such as antibody-dependent cellular cytotoxicity. Transglutaminase-mediated reactions are orthogonal to other site-specific approaches—i.e., genetic engineering such as unnatural amino acids, chemical methods, such as, thiol bridge or N-terminal modification, and enzymatic methods, such as, sortase. Therefore, combining these tools enables generations of novel conjugates that were previously unavailable, e.g., introducing two orthogonal proteolytically cleavable linkers like matrix metalloproteinase-2 and cathepsin-B using mTGase and recombinantly produced a tag for lipoate-acid ligase A [36].
Glycan-mediated, site-specific conjugation
Glycan-mediated conjugation offers a generally applicable and exclusively site-specific approach for antibodies’ bioconjugation due to several factors [62]. First, almost all IgG-type antibodies have one—and only one—glycan at a conserved site in the Fc region [63]. For this review, “glycan-mediated conjugation” refers to the chemo-enzymatic installation of payloads onto the conserved N-glycosylation sites of IgG-type antibodies. This installation is achieved via glycan remodeling, which entails the addition or removal of saccharides from these conserved sites, typically through galactose- or sialic acid-based chemistry.
The most common approach to glycan-mediated conjugation involves enzymatic transglycosylation and is composed of two steps: first, trimming of the antibody’s native glycan via β-galactosidase, which cleaves the terminal galactose moieties; or via endoglycosidase, which cleaves after the innermost N-acetylglucosamine or GlcNAc moiety; and second, rebuilding of the glycan by installing an unnatural sugar moiety containing a handle for further bioconjugation [64–74]. This glycan-mediated conjugation strategy is further discussed in the following sections. Other techniques also exist. For example, Zhou et al. [71] chemoenzymatically introduced terminal sialic acid moieties onto the antibody’s native glycan and functionalized them for conjugation.
GlyCLICK
GlyCLICK is a glycan-mediated conjugation technology marketed as a kit by Genovis AB (Sweden and Cambridge, MA, USA).
As illustrated in Fig. 4, the first step (typically performed at room temperature), involves a proprietary immobilized endoglycosidase that cleaves the native glycan after the innermost GlcNAc moiety; the trimmed antibody is readily separated from the immobilized enzyme. In the second step, β-1,4-galactosyltransferase-mediated installation of the azide-functionalized unnatural sugar occurs overnight at 30°C. Lastly, strained alkyne-functionalized payload reacts with the azide handle at room temperature without catalysts to afford conjugates containing fluorophores, affinity tags, chelators, or custom chemistries [64, 67].
Figure 4.
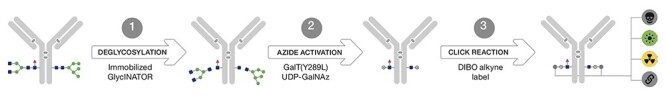
An overview of the steps for glycan-mediated conjugation using GlyCLICK: first, trimming the native antibody glycan using endoglycosidase; second, installation of the azide via β-1,4-galactosyltransferase; and third, click reaction using a payload with suitable chemistry [64]. Adapted with permission from Toftevall et al. [64]. Copyright © 2020, Springer Science Business Media, LLC, part of Springer Nature.
GlyCLICK produces homogeneous conjugates with well-controlled DARs. It is worth noting that the two attachment points per Fc domain can afford DARs greater than two if branched or dendrimeric linkers are used. For example, Thompson et al. [75] employed strategies similar to elements of GlyCLICK to produce glycan-remodeled conjugates with DARs of 2 and 4.
Because the Fc glycan is distant from the CDRs, glycan-mediated conjugation typically preserves the antibody’s antigen-binding capacity. Indeed, surface plasmon resonance (SPR) studies of trastuzumab conjugates produced via GlyCLICK show overlapping curves for native trastuzumab, whereas the product of stochastic conjugation exhibits impaired binding levels [64].
Scalability, scope, and limitations. Wide and robust applications have been demonstrated for glycan-mediated conjugation, including human IgG1–IgG4s and IgGs from goats, mice, rabbits, rats, monkeys, sheep, cows, and horses [64]. van Geel et al. [76] successfully produced conjugates of several human or humanized IgG1s, IgG2s, and IgG4s, including a rituximab-BCN-PEG8-doxorubicin conjugate up to the 5 g scale. All conjugates demonstrated site-specific incorporation of the linker and payload, negligible aggregation, hydrolytic stability, and homogeneous DARs. Also, trastuzumab-maytansine conjugate developed via glycan remodeling showed more favorable in vitro and in vivo efficacy data than Kadcyla, a commercial ADC composed of the same components but generated through stochastic conjugation methods [76].
Applications: preclinical stage. Preclinical use of glycan-mediated conjugates is growing [6, 77–79]. Recently, Kristensen et al. [6] demonstrated that conjugates of trastuzumab to 89Zr-SCN-Bn-DFO exhibited increased positron emission tomography (PET) tumor uptake and substantially higher in vitro stability and immunoreactivity compared with conjugates generated via traditional lysine acylation, as seen in Fig. 2. Similarly, Christensen et al. [77] showed an anti-PD-L1 antibody-89Zr-DFO-6E11 conjugate for PET imaging increased relative tumor uptake of the imaging agent and enabled quantification of PD-L1 expression differences in tumors and spleens of irradiated tumor-bearing mice.
Applications: clinical stage. At least two ADCs developed using GlycoConnect from SynAffix (a glycan-mediated conjugation approach similar to GlyCLICK) have advanced to early-stage clinical trials: ADCT-601 from ADC Therapeutics and XMT-1592 from Mersana Therapeutics. ADCT-601, an anti-human AXL-pyrrolobenzodiazepine-dimer toxin conjugate, is undergoing Phase I evaluation in patients with solid tumors (NCT03700294; Table 1), whereas XMT-1592, a NaPi2b-targeting antibody conjugated to an auristatin payload, recently initiated Phase I dose-escalation studies in patients with non-small cell lung cancer adenocarcinoma or ovarian cancer (NCT04396340; Table 1).
CHEMICAL CONJUGATION
Chemical conjugations, such as the acylation of amines and alkylation of thiols, are typically not site-specific (i.e., random or stochastic), and the solvent-accessible residues are typically modified. This method generates a mixture of ADC species with a variable DAR and mixed tethering sites. However, as detailed in the following section, two site-specific chemical methods are available: first, selective reduction of disulfide followed by alkylation (ThioBridge); and second, N-terminal transamination and imidazolidinone formation. Although there are several other approaches for selective reduction of disulfide bridging bonds such as bis-sulfone reagents, dibromo pyridazinediones (PDs), and next-generation maleimides acting as cysteine cross-linking reagents [80–83], this paper will focus specifically on ThioBridge.
Selective reduction of disulfide (ThioBridge)
Together with the acylation of amines (N-termini and lysine residues), alkylation of thiol (cysteine residues) is the most common and conventional bioconjugation chemistry. In human native antibodies, all cysteine residues exist as disulfides. There are four isoforms of IgG (IgG1, IgG2, IgG3, and IgG4), each of which contains 12 intra-chain disulfide bonds that are conserved. In addition, an IgG1 antibody has four interchain disulfide bonds typically used as potential sites for conjugation. To some degree, these interchain disulfides bonds can be selectively reduced by judiciously combining some reductants under certain conditions, such as tris (2-carboxyethyl) phosphine (TCEP) or dithiothreitol (DTT) [84–88]. Selective reduction of the interchain disulfide bonds is a vital step as it yields different DARs (e.g., 2, 4, 6, or 8) [85, 88].
Disulfide bonds, which are often referred as “disulfide bridges,” are crucial in holding different antibody domains together. Modification of disulfide bonds/cysteines can be achieved using salts such as vinyl sulfonium [89–91]. Reduction followed by a single alkylation of each resulting cysteinyl thiol like the one obtained via the venerable maleimide chemistry often leads to significant destabilization of antibody structures. Modification of the four inter-chain disulfide bonds in the antibody often lead to increased conjugation heterogeneity and reduced site-specificity. Furthermore, alkylation by maleimide is reversible (i.e., deconjugation), which has been observed in vivo [92]. These limitations are rectified by the rebridging approach detailed next.
In 2014, Badescu, Godwin, and co-authors reported the first disulfide re-bridging strategies with bis-alkylating reagents—namely ThioBridge. This technology was developed by PolyTherics (a subsidiary of Abzena) as a novel way to prepare more homogeneous and stable ADCs without protein engineering [85]. As depicted in Fig. 5, the two-step method uses bis-sulfone reagents that are selective for a pair of cysteine thiols in close proximity, which is the case for the thiols generated from the reduction of disulfides in antibodies.
Figure 5.
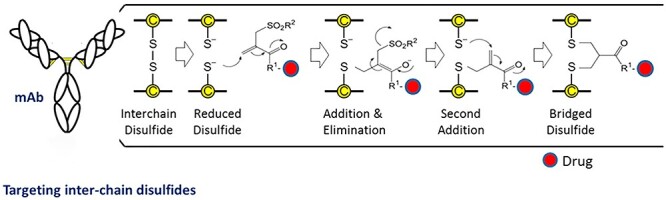
Conjugation of a drug to the site-selective reduced interchain disulfide bonds of an antibody, via bis-thiol reactive reagent that cross-links the reduced disulfide bonds, involving a sequence of Michael addition and elimination reactions [85]. Modified and adapted with permission from Badescu et al. [85]. Copyright © 2014, American Chemical Society.
Step 1: site-specific disulfide reduction to liberate free cysteine thiols for conjugation
Antibodies generally have minimum to no free thiols. The four accessible interchain disulfide bonds can be selectively reduced fully or partially, using DTT or TCEP, respectively. Moreover, different degrees and sites of reductions can be achieved under controlled conditions, e.g., tuning the equivalence of the reductants to the antibody, reaction time, and temperature. For example, DTT reduced all four interchain disulfide bonds generating eight free thiols that can be re-bridged to make up to four conjugation sites. Whereas, at 1–2 equivalent of TCEP per disulfide, varying loading ratios can be achieved (e.g., DAR of 1–4) [88].
Step 2: bridging of reduced antibody with drug-containing reagent
The pair of thiols (reduced disulfide) is then followed by bis-alkylation, proceeding through Michael addition and elimination, to conjugate both thiols derived from the original disulfide bond. This reaction results in covalent re-bridging of the disulfide bond via a three-carbon bridge leaving the antibody’s structure less perturbed compared with single alkylation [85]. Naturally, once the ThioBridge linkage is introduced, various conjugates (drugs or payloads) can be incorporated, such as PEG or clickable handles [85, 93].
Stability. In the ThioBridge technology, the disulfides are reannealed, resulting in greater stability and homogeneity. In contrast, ADCs produced using maleimide are unstable due to deconjugation and cross-conjunction to free thiols of albumin serum [94]. Shen et al. [85, 94] demonstrated that in rat serum, the conjugated fluorophore (i.e., Alexa Fluor 488) was lost, based on maleimide, yet, the ThioBridge counterpart was stable and showed no detectable cross conjunction to serum albumin. Due to its in vivo stability, ThioBridge technology is being developed by OBI Pharma to construct a glycolipid antigen targeting ADC, globohexaosylceramide (Globo H, OBI-999). This molecule is currently under clinical evaluation in Phase I/II study (NCT04084366; Table 1) [95].
N-terminal conjugation
The N-terminal transamination of antibodies and imidazolidinone formation are two site-specific methods that are not reactive toward the amine on the lysine side chain for two main reasons. First, only two N-termini exist in antibodies, one on the light and one of the HC. Second, the origin of chemical selectivity of the N-terminal amine stems from the adjacent carbonyl group of the amide [96, 97]; in comparison, the epsilon amine on the side-chain of lysine is adjacent to methylene (alkyl) groups. [98, 99].
N-terminal modification with pH control offers limited selectivity: Contrary to common assumption, even at low pHs, exclusive selectivity cannot be achieved for the N-terminal amines via acylation by NHS ester and reductive amination; in other words, under these conditions, amines on the lysine side chains can nonetheless be modified, albeit to a reduced degree. A contributing factor to this misconception is the incomplete characterization of bioconjugate products and side-products. [18, 100].
Transamination
N-Terminal modification via transamination reaction using carbonyl agents directly converts the N-terminal amine to a carbonyl group, which can be further conjugated via established chemistries, such as with hydrazines, hydrazides, or hydroxylamines. In 2006, Francis and co-authors reported a mild method based on biomimetic transamination reaction using various aldehydes, such as pyridoxal phosphate (PLP) (vitamin B6) [101, 103–105].
Conditions and by-products. Transamination reaction can occur in mild conditions, near physiological pH and temperature, and can thrive in various buffers, and does not require any particular catalysts. Of note, PLP-mediated transamination reaction often forms pyridoxal phosphate (PLP) aldol adducts as by-products, especially at high pyridoxal phosphate (PLP) concentrations and long reaction times (Fig. 6a); however, the aldol adducts can nevertheless be further conjugated as well [101].
Figure 6.
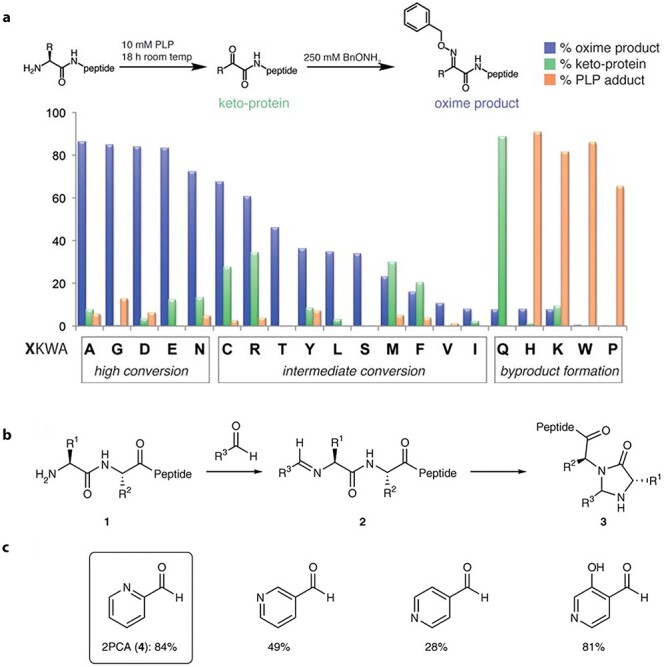
(a) Transamination of the N-terminal amine by pyridoxal phosphate (PLP) and reactivities of different amino acids [101]. (b, c) Modification of protein N-termini by pyridine aldehyde and the formation of imidazolidinone [102]. Adapted with permission from Witus et al. [101]; MacDonald et al. [102]. Copyright © 2014, John Wiley and Sons; Copyright © 2014, Springer Nature.
Reactivities of different amino acids. In 2010, Witus and Francis reported that high yields, up to 90% conversion, can be achieved for some amino acid residues such as alanine, glycine, asparagine aspartic acid, and glutamic acid, as shown in Fig. 6a, whereas others have much lower conversion due to the formation of various by-products (i.e., PLP adducts) [101]. Of note: aspartic acid undergoes decarboxylation before transamination [103, 106].
Cyclization: N-terminal modification of antibodies via one-step cyclization using pyridine carboxaldehyde (2PCA)
Another practical one-step site-specific N-terminal conjugation was first reported in 2015 by Francis, Macdonald, and co-workers, which included the reaction of 2-pyridinecarboxyaldehydes (2PCA) to form imidazolidinone by cyclization of the adjacent amide. It proceeds through an imine to afford stable imidazolidinone as the final product [102]. Thus, the reaction is selective against the amine on the lysine side chain, which does not have an amide, resulting in imidazolidinone formation with aldehydes, Fig. 6(b,c).
Potential issues and limitations: pyroglutamic acid formation. One potential limitation is the formation of pyroglutamic acid (PyroGlu or PyroE) at the N-termini of proteins, including antibodies. If the N-terminal residue is glutamine (Gln) or glutamic acid (Glu), pyroglutamic acid can be formed readily; the resulting product is an amide, which resists further reactions for amines. Moreover, both glutamine and glutamic acid are common at the N-termini of antibodies, hence Pyro-Glu formation. For instance, in 2006, Chelius et al. [107] reported pyroglutamic acid formation from N-terminal glutamic acid residues in both light and HCs of recombinant monoclonal antibodies. In 2011, Flynn et al. [108] showed the conversion of glutamate residues at the N-terminal of human IgG2 antibody into pyroglutamate in vivo. Since antibodies contain two N-termini, the formation of Pyro-Glu at one N-terminus may enhance the remaining N-terminus’ selectivity.
Clinical trials. To our best knowledge, at present, no ADCs are using N-terminal conjugation chemistry entering therapeutic studies. One reason may be the concern that N-termini’s close proximity to the antigen-binding sites could potentially perturb binding affinity or specificity. On the other hand, such circumstances can be readily addressed by standard tests.
ADCs IN CLINICAL TRIALS
Site-specific conjugation of native antibodies has several advantages that lend to their clinical translation (Table 1). First, these antibody conjugates are homogenous and well-defined constructs [1]. Second, these conjugates are more stable in plasma [6, 109], have enhanced binding efficiency [1], as well as increased cellular uptake [6] compared with non-specific methods. From 2011 to 2020, several site-specific ADCs have entered clinical trials, as illustrated in Fig. 7. As a general trend, the percentages of site-specific ADCs have been steadily increasing over the years, and is 100% in 2020.
Figure 7.
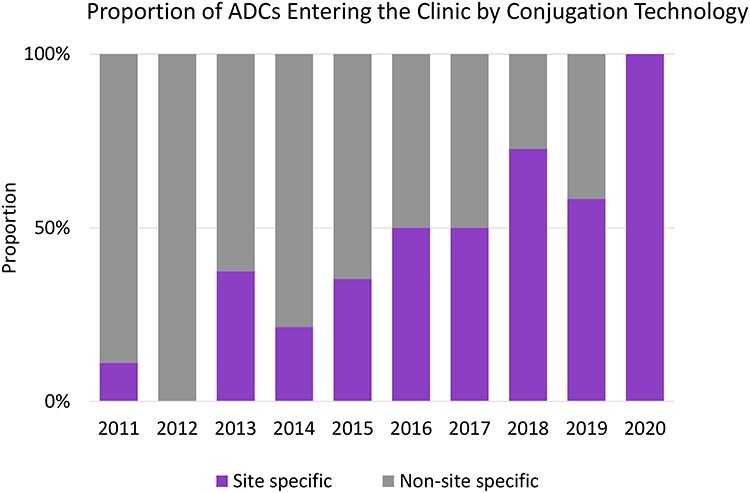
Site-specific ADCs entering clinical trials via conjugation technology over the years from 2011 to 2020. The percentage of site-specific antibody–drug conjugates (denoted in purple) entering clinical trials has steadily increased over the last decade. The upward trend of site-specific ADCs demonstrates a transition of prospective clinical ADCs to those constructed through site-specific conjugation. ADCs with undisclosed conjugation approaches are excluded. Cut-off date 3 September 2020. Insights provided by Beacon Targeted Therapies, Beacon-intelligence.com [110].
FUTURE PERSPECTIVE
Genetic engineering methods [1, 2] have been used to construct antibody conjugates. For instance, reactive cysteine (i.e., THIOMAB) was introduced into antibodies and evaluated in preclinical settings [94, 113, 114]. Other approaches include the incorporation of unnatural amino acids [115], which introduce a chemical handle for further conjugation; for example, an ADC based on para-acetyl phenylalanine (pAF) is being developed by Ambrx [116] and is currently in the clinical trial (NCT03255070). In addition, tags incorporated genetically to antibodies have been chemo-enzymatically conjugated using formyl glycine-generating enzymes (FGE; NCT03682796) and sortase [117–120]. These genetic approaches are orthogonal to the site-specific methods presented herein. Therefore, antibodies can be conjugated by both methods in a combinatorial fashion to produce multi-purpose conjugates.
Coupling site-specific conjugation and biorthogonal click chemistries, a diverse array of antibody conjugates can be envisioned. Various linkers can be coupled to native antibodies site-selectively, including exogenous cleavable linkers using chemical or light [51], endogenous cleavable linkers mediated by proteases, pH, or redox [121], as well as non-cleavable linkers. The design of these linkers can be branched (e.g., dendrimer) or polymers with higher payloads (e.g., branched polymers that are currently in the clinic) [122]. These advances will undoubtedly enable more efficacious and multi-functionalized antibody conjugates for many applications. Indeed, the future applications will combine different aspects and advantages of multiple approaches, as we outline in our Hybrid Modality Engineering of Protein concept.
ACKNOWLEDGMENTS
We would like to thank Tyler Dost and Olaf Tobias Bjornstal for providing their critical review of the manuscript and the reviewers for helpful discussion.
Contributor Information
Amissi Sadiki, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Shefali R Vaidya, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Mina Abdollahi, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Gunjan Bhardwaj, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Michael E Dolan, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA; Downstream Development, Biologics Process Development, Millennium Pharmaceuticals, Inc., (a wholly-owned subsidiary of Takeda Pharmaceuticals Company Limited), Cambridge, MA 02139, USA.
Harpreet Turna, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Varnika Arora, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Athul Sanjeev, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Timothy D Robinson, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Andrea Koid, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Aashka Amin, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
Zhaohui Sunny Zhou, Department of Chemistry and Chemical Biology, Northeastern University Boston, Boston, MA 02115-5000, USA; Barnett Institute of Chemical and Biological Analysis, Northeastern University Boston, Boston, MA 02115-5000, USA.
FUNDING
A.S thanks the support of Amgen Wolfgang Goetzinger Scholar Awards in Life Science Analysis and John Hatsopoulos Scholar Award. This work was supported by Alpha Fund (Center for Entrepreneurship Education) by Northeastern University, the National Science Foundation's Innovation Corps (I-Corps) program (NSF Award 1735260 to Marc H. Meyer at Northeastern University), and the National Institute of Health (National Institute of General Medical Sciences 1R01GM101396 to Z.S.Z.).
CONFLICTS OF INTEREST STATEMENT
M.E.D. is an employee of Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Z.S.Z. holds the position of Editorial Board Member for Antibody Therapeutics and is blinded from reviewing or making decisions for the manuscript.
References
- 1. Sadiki, A, Kercher, EM, Lu, H et al. Site-specific bioconjugation and convergent click chemistry enhances antibody-chromophore conjugate binding efficiency. Photochem Photobiol 2020; 96: 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beck, A, Goetsch, L, Dumontet, C et al. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017; 16: 315–37. [DOI] [PubMed] [Google Scholar]
- 3. Chari, RV, Miller, ML, Widdison, WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 2014; 53: 3796–827. [DOI] [PubMed] [Google Scholar]
- 4. Luo, Q, Chung, HH, Borths, C et al. Structural characterization of a monoclonal antibody-Maytansinoid Immunoconjugate. Anal Chem 2016; 88: 695–702. [DOI] [PubMed] [Google Scholar]
- 5. Pellegrino, C, Favalli, N, Sandholzer, M et al. Impact of ligand size and conjugation chemistry on the performance of universal chimeric antigen receptor T-cells for tumor killing. Bioconjug Chem 2020; 31: 1775–83. [DOI] [PubMed] [Google Scholar]
- 6. Kristensen, LK, Christensen, C, Jensen, MM et al. Site-specifically labeled (89)Zr-DFO-trastuzumab improves immuno-reactivity and tumor uptake for immuno-PET in a subcutaneous HER2-positive xenograft mouse model. Theranostics 2019; 9: 4409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bai, C, Reid, EE, Wilhelm, A et al. Site-specific conjugation of the indolinobenzodiazepine DGN549 to antibodies affords antibody-drug conjugates with an improved therapeutic index as compared with lysine conjugation. Bioconjug Chem 2020; 31: 93–103. [DOI] [PubMed] [Google Scholar]
- 8. Manning, MC, Chou, DK, Murphy, BM et al. Stability of protein pharmaceuticals: an update. Pharm Res 2010; 27: 544–75. [DOI] [PubMed] [Google Scholar]
- 9. Liu, M, Cheetham, J, Cauchon, N et al. Protein isoaspartate methyltransferase-mediated 18O-labeling of isoaspartic acid for mass spectrometry analysis. Anal Chem 2012; 84: 1056–62. [DOI] [PubMed] [Google Scholar]
- 10. Chumsae, C, Gifford, K, Lian, W et al. Arginine modifications by methylglyoxal: discovery in a recombinant monoclonal antibody and contribution to acidic species. Anal Chem 2013; 85: 11401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu, M, Zhang, Z, Zang, T et al. Discovery of undefined protein cross-linking chemistry: a comprehensive methodology utilizing 18O-labeling and mass spectrometry. Anal Chem 2013; 85: 5900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chumsae, C, Zhou, LL, Shen, Y et al. Discovery of a chemical modification by citric acid in a recombinant monoclonal antibody. Anal Chem 2014; 86: 8932–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klaene, JJ, Ni, W, Alfaro, JF et al. Detection and quantitation of succinimide in intact protein via hydrazine trapping and chemical derivatization. J Pharm Sci 2014; 103: 3033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chumsae, C, Hossler, P, Raharimampionona, H et al. When good intentions go awry: modification of a recombinant monoclonal antibody in chemically defined cell culture by Xylosone, an oxidative product of ascorbic acid. Anal Chem 2015; 87: 7529–34. [DOI] [PubMed] [Google Scholar]
- 15. Liu, M, Zhang, Z, Cheetham, J et al. Discovery and characterization of a photo-oxidative histidine-histidine cross-link in IgG1 antibody utilizing (1)(8)O-labeling and mass spectrometry. Anal Chem 2014; 86: 4940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu, S, Moulton, KR, Auclair, JR et al. Mildly acidic conditions eliminate deamidation artifact during proteolysis: digestion with endoprotease Glu-C at pH 4.5. Amino Acids 2016; 48: 1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ni, W, Dai, S, Karger, BL et al. Analysis of isoaspartic acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal Chem 2010; 82: 7485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang, L, Chumsae, C, Kaplan, JB et al. Detection of alkynes via click chemistry with a brominated coumarin azide by simultaneous fluorescence and isotopic signatures in mass spectrometry. Bioconjug Chem 2017; 28: 2302–9. [DOI] [PubMed] [Google Scholar]
- 19. Kolb, HC, Finn, MG, Sharpless, KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl 2001; 40: 2004–21. [DOI] [PubMed] [Google Scholar]
- 20. Rostovtsev, VV, Green, LG, Fokin, VV et al. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl 2002; 41: 2596–9. [DOI] [PubMed] [Google Scholar]
- 21. Agard, NJ, Prescher, JA, Bertozzi, CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc 2004; 126: 15046–7. [DOI] [PubMed] [Google Scholar]
- 22. Adusumalli, SR, Rawale, DG, Singh, U et al. Single-site Labeling of native proteins enabled by a chemoselective and site-selective chemical technology. J Am Chem Soc 2018; 140: 15114–23. [DOI] [PubMed] [Google Scholar]
- 23. Matos, MJ, Jimenez-Oses, G, Bernardes, GJL. Lysine bioconjugation on native albumin with a sulfonyl acrylate reagent. Methods Mol Biol 2019; 2033: 25–37. [DOI] [PubMed] [Google Scholar]
- 24. Matos, MJ, Oliveira, BL, Martinez-Saez, N et al. Chemo- and regioselective lysine modification on native proteins. J Am Chem Soc 2018; 140: 4004–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamada, K, Shikida, N, Shimbo, K et al. AJICAP: affinity peptide mediated regiodivergent functionalization of native antibodies. Angew Chem Int Ed Engl 2019; 58: 5592–7. [DOI] [PubMed] [Google Scholar]
- 26. Genovis . Site-specific antibody conjugation technology. GlyClick. https://www.genovis.com/wp-content/uploads/GlyCLICK-product-brochure.pdf (29 August 2020, last accessed). [Google Scholar]
- 27. Cohen, JD, Thompson, S, Ting, AY. Structure-guided engineering of a Pacific blue fluorophore ligase for specific protein imaging in living cells. Biochemistry 2011; 50: 8221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cohen, JD, Zou, P, Ting, AY. Site-specific protein modification using lipoic acid ligase and Bis-Aryl hydrazone formation. Chembiochem 2012; 13: 888–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Josten, A, Haalck, L, Spener, F et al. Use of microbial transglutaminase for the enzymatic biotinylation of antibodies. J Immunol Methods 2000; 240: 47–54. [DOI] [PubMed] [Google Scholar]
- 30. Mindt, TL, Jungi, V, Wyss, S et al. Modification of different IgG1 antibodies via glutamine and lysine using bacterial and human tissue transglutaminase. Bioconjug Chem 2008; 19: 271–8. [DOI] [PubMed] [Google Scholar]
- 31. Jeger, S, Zimmermann, K, Blanc, A et al. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl 2010; 49: 9995–7. [DOI] [PubMed] [Google Scholar]
- 32. Marculescu, C, Lakshminarayanan, A, Gault, J et al. Probing the limits of Q-tag bioconjugation of antibodies. Chem Commun (Camb) 2019; 55: 11342–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benjamin, SR, Jackson, CP, Fang, S et al. Thiolation of Q295: site-specific conjugation of hydrophobic payloads without the need for genetic engineering. Mol Pharm 2019; 16: 2795–807. [DOI] [PubMed] [Google Scholar]
- 34. Walker, JA, Bohn, JJ, Ledesma, F et al. Substrate design enables heterobifunctional, dual "click" antibody modification via microbial transglutaminase. Bioconjug Chem 2019; 30: 2452–7. [DOI] [PubMed] [Google Scholar]
- 35. Chio, TI, Demestichas, BR, Brems, BM et al. Expanding the versatility of microbial transglutaminase using alpha-effect nucleophiles as noncanonical substrates. Angew Chem Int Ed Engl 2020; 59: 13814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thornlow, DN, Cox, EC, Walker, JA et al. Dual site-specific antibody conjugates for sequential and orthogonal cargo release. Bioconjug Chem 2019; 30: 1702–10. [DOI] [PubMed] [Google Scholar]
- 37. Anami, Y, Xiong, W, Gui, X et al. Enzymatic conjugation using branched linkers for constructing homogeneous antibody-drug conjugates with high potency. Org Biomol Chem 2017; 15: 5635–42. [DOI] [PubMed] [Google Scholar]
- 38. Dennler, P, Chiotellis, A, Fischer, E et al. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug Chem 2014; 25: 569–78. [DOI] [PubMed] [Google Scholar]
- 39. Zhang, Y, Park, KY, Suazo, KF et al. Recent progress in enzymatic protein labelling techniques and their applications. Chem Soc Rev 2018; 47: 9106–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider, H, Deweid, L, Avrutina, O et al. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal Biochem 2020; 595: 113615. [DOI] [PubMed] [Google Scholar]
- 41. Grunberg, J, Jeger, S, Sarko, D et al. DOTA-functionalized polylysine: a high number of DOTA chelates positively influences the biodistribution of enzymatic conjugated anti-tumor antibody chCE7agl. PLoS One 2013; 8: e60350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park, J, Chariou, PL, Steinmetz, NF. Site-specific antibody conjugation strategy to functionalize virus-based nanoparticles. Bioconjug Chem 2020; 31: 1408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strop, P, Tran, TT, Dorywalska, M et al. RN927C, a site-specific Trop-2 antibody-drug conjugate (ADC) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol Cancer Ther 2016; 15: 2698–708. [DOI] [PubMed] [Google Scholar]
- 44. King, GT, Eaton, KD, Beagle, BR et al. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest New Drugs 2018; 36: 836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Folk, JE, Cole, PW. Mechanism of action of Guinea pig liver transglutaminase. I. Purification and properties of the enzyme: identification of a functional cysteine essential for activity. J Biol Chem 1966; 241: 5518–25. [PubMed] [Google Scholar]
- 46. Strop, P. Versatility of microbial transglutaminase. Bioconjug Chem 2014; 25: 855–62. [DOI] [PubMed] [Google Scholar]
- 47. Rachel, NM, Quaglia, D, Levesque, E et al. Engineered, highly reactive substrates of microbial transglutaminase enable protein labeling within various secondary structure elements. Protein Sci 2017; 26: 2268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steffen, W, Ko, FC, Patel, J et al. Discovery of a microbial transglutaminase enabling highly site-specific labeling of proteins. J Biol Chem 2017; 292: 15622–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spolaore, B, Raboni, S, Ramos Molina, A et al. Local unfolding is required for the site-specific protein modification by transglutaminase. Biochemistry 2012; 51: 8679–89. [DOI] [PubMed] [Google Scholar]
- 50. Rachel, NM, Pelletier, JN. Biotechnological applications of transglutaminases. Biomolecules 2013; 3: 870–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moulton, KR, Sadiki, A, Koleva, BN et al. Site-specific reversible protein and peptide modification: transglutaminase-catalyzed glutamine conjugation and bioorthogonal light-mediated removal. Bioconjug Chem 2019; 30: 1617–21. [DOI] [PubMed] [Google Scholar]
- 52. Ando, H, Adachi, M, Umeda, K et al. Purification and characteristics of a novel transglutaminase derived from microorganisms. Agric Biol Chem 1989; 53: 2613–7. [Google Scholar]
- 53. Gundersen, MT, Keillor, JW, Pelletier, JN. Microbial transglutaminase displays broad acyl-acceptor substrate specificity. Appl Microbiol Biotechnol 2014; 98: 219–30. [DOI] [PubMed] [Google Scholar]
- 54. Ohtsuka, T, Sawa, A, Kawabata, R et al. Substrate specificities of microbial transglutaminase for primary amines. J Agric Food Chem 2000; 48: 6230–3. [DOI] [PubMed] [Google Scholar]
- 55. Rachel, NM, Toulouse, JL, Pelletier, JN. Transglutaminase-catalyzed bioconjugation using one-pot metal-free bioorthogonal chemistry. Bioconjug Chem 2017; 28: 2518–23. [DOI] [PubMed] [Google Scholar]
- 56. Mero, A, Spolaore, B, Veronese, FM et al. Transglutaminase-mediated PEGylation of proteins: direct identification of the sites of protein modification by mass spectrometry using a novel monodisperse PEG. Bioconjug Chem 2009; 20: 384–9. [DOI] [PubMed] [Google Scholar]
- 57. Mero, A, Schiavon, M, Veronese, FM et al. A new method to increase selectivity of transglutaminase mediated PEGylation of salmon calcitonin and human growth hormone. J Control Release 2011; 154: 27–34. [DOI] [PubMed] [Google Scholar]
- 58. Spycher, PR, Amann, CA, Wehrmuller, JE et al. Dual, site-specific modification of antibodies by using solid-phase immobilized microbial transglutaminase. Chembiochem 2017; 18: 1923–7. [DOI] [PubMed] [Google Scholar]
- 59. Grigoletto, A, Mero, A, Yoshioka, H et al. Covalent immobilisation of transglutaminase: stability and applications in protein PEGylation. J Drug Target 2017; 25: 856–64. [DOI] [PubMed] [Google Scholar]
- 60. Lee, BW, Sun, HG, Zang, T et al. Enzyme-catalyzed transfer of a ketone group from an S-adenosylmethionine analogue: a tool for the functional analysis of methyltransferases. J Am Chem Soc 2010; 132: 3642–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dickgiesser, S, Rieker, M, Mueller-Pompalla, D et al. Site-specific conjugation of native antibodies using engineered microbial transglutaminases. Bioconjug Chem 2020; 31: 1070–6. [DOI] [PubMed] [Google Scholar]
- 62. Qasba, PK. Glycans of antibodies as a specific site for drug conjugation using glycosyltransferases. Bioconjug Chem 2016; 27: 264. [DOI] [PubMed] [Google Scholar]
- 63. Zhou, Q, Qiu, H. The mechanistic impact of N-glycosylation on stability, pharmacokinetics, and immunogenicity of therapeutic proteins. J Pharm Sci 2019; 108: 1366–77. [DOI] [PubMed] [Google Scholar]
- 64. Toftevall, H, Nyhlen, H, Olsson, F et al. Antibody conjugations via Glycosyl remodeling. Methods Mol Biol 2020; 2078: 131–45. [DOI] [PubMed] [Google Scholar]
- 65. Boeggeman, E, Ramakrishnan, B, Pasek, M et al. Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: application for cell surface antigen detection. Bioconjug Chem 2009; 20: 1228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ramakrishnan, B, Boeggeman, E, Pasek, M et al. Bioconjugation using mutant glycosyltransferases for the site-specific labeling of biomolecules with sugars carrying chemical handles. Methods Mol Biol 2011; 751: 281–96. [DOI] [PubMed] [Google Scholar]
- 67. Duivelshof, BL, Desligniere, E, Hernandez-Alba, O et al. Glycan-mediated technology for obtaining homogeneous site-specific conjugated antibody-drug conjugates: synthesis and analytical characterization by using complementary middle-up LC/HRMS analysis. Anal Chem 2020; 92: 8170–7. [DOI] [PubMed] [Google Scholar]
- 68. Boeggeman, E, Ramakrishnan, B, Kilgore, C et al. Direct identification of nonreducing GlcNAc residues on N-glycans of glycoproteins using a novel chemoenzymatic method. Bioconjug Chem 2007; 18: 806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ramakrishnan, B, Boeggeman, E, Qasba, PK. Applications of glycosyltransferases in the site-specific conjugation of biomolecules and the development of a targeted drug delivery system and contrast agents for MRI. Expert Opin Drug Deliv 2008; 5: 149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li, X, Fang, T, Boons, GJ. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed Engl 2014; 53: 7179–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhou, Q, Stefano, JE, Manning, C et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug Chem 2014; 25: 510–20. [DOI] [PubMed] [Google Scholar]
- 72. Zhu, Z, Ramakrishnan, B, Li, J et al. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs 2014; 6: 1190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hang, HC, Yu, C, Pratt, MR et al. Probing glycosyltransferase activities with the Staudinger ligation. J Am Chem Soc 2004; 126: 6–7. [DOI] [PubMed] [Google Scholar]
- 74. Wei, Y, Li, C, Huang, W et al. Glycoengineering of human IgG1-Fc through combined yeast expression and in vitro chemoenzymatic glycosylation. Biochemistry 2008; 47: 10294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thompson, P, Ezeadi, E, Hutchinson, I et al. Straightforward glycoengineering approach to site-specific antibody-pyrrolobenzodiazepine conjugates. ACS Med Chem Lett 2016; 7: 1005–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. van Geel, R, Wijdeven, MA, Heesbeen, R et al. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug Chem 2015; 26: 2233–42. [DOI] [PubMed] [Google Scholar]
- 77. Christensen, C, Kristensen, LK, Alfsen, MZ et al. Quantitative PET imaging of PD-L1 expression in xenograft and syngeneic tumour models using a site-specifically labelled PD-L1 antibody. Eur J Nucl Med Mol Imaging 2020; 47: 1302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zeglis, BM, Davis, CB, Aggeler, R et al. Enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconjug Chem 2013; 24: 1057–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang, LX, Tong, X, Li, C et al. Glycoengineering of antibodies for modulating functions. Annu Rev Biochem 2019; 88: 433–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chudasama, V, Smith, ME, Schumacher, FF et al. Bromopyridazinedione-mediated protein and peptide bioconjugation. Chem Commun (Camb) 2011; 47: 8781–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Forte, N, Chudasama, V, Baker, JR. Homogeneous antibody-drug conjugates via site-selective disulfide bridging. Drug Discov Today Technol 2018; 30: 11–20. [DOI] [PubMed] [Google Scholar]
- 82. Smith, ME, Schumacher, FF, Ryan, CP et al. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J Am Chem Soc 2010; 132: 1960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yamada, K, Ito, Y. Recent chemical approaches for site-specific conjugation of native antibodies: technologies toward next-generation antibody-drug conjugates. Chembiochem 2019; 20: 2729–37. [DOI] [PubMed] [Google Scholar]
- 84. Lee, MTW, Maruani, A, Baker, JR et al. Next-generation disulfide stapling: reduction and functional re-bridging all in one. Chem Sci 2016; 7: 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Badescu, G, Bryant, P, Bird, M et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem 2014; 25: 1124–36. [DOI] [PubMed] [Google Scholar]
- 86. Sun, MM, Beam, KS, Cerveny, CG et al. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 2005; 16: 1282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang, Z, Rejtar, T, Zhou, ZS et al. Desulfurization of cysteine-containing peptides resulting from sample preparation for protein characterization by mass spectrometry. Rapid Commun Mass Spectrom 2010; 24: 267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Godwin, A. Bridging the conjugation gap: maintaining Disulfide bridges leads to improved homogeneity and stability for ADCs. Genetic Engineering & Biotechnology News 2013; 33: 20–1. [Google Scholar]
- 89. Catcott, KC, Yan, J, Qu, W et al. Identifying unknown enzyme-substrate pairs from the cellular milieu with native mass spectrometry. Chembiochem 2017; 18: 613–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Qu, W, Catcott, KC, Zhang, K et al. Capturing unknown substrates via in situ formation of tightly bound bisubstrate adducts: S-Adenosyl-vinthionine as a functional probe for AdoMet-dependent methyltransferases. J Am Chem Soc 2016; 138: 2877–80. [DOI] [PubMed] [Google Scholar]
- 91. Zhao, G, Zhou, ZS. Vinyl sulfonium as novel proteolytic enzyme inhibitor. Bioorg Med Chem Lett 2001; 11: 2331–5. [DOI] [PubMed] [Google Scholar]
- 92. Wei, C, Zhang, G, Clark, T et al. Where did the linker-payload go? A quantitative investigation on the destination of the released linker-payload from an antibody-drug conjugate with a maleimide linker in plasma. Anal Chem 2016; 88: 4979–86. [DOI] [PubMed] [Google Scholar]
- 93. Balan, S, Choi, JW, Godwin, A et al. Site-specific PEGylation of protein disulfide bonds using a three-carbon bridge. Bioconjug Chem 2007; 18: 61–76. [DOI] [PubMed] [Google Scholar]
- 94. Shen, BQ, Xu, K, Liu, L et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012; 30: 184–9. [DOI] [PubMed] [Google Scholar]
- 95. OBI Pharma, I . Phase 1/2 Study of OBI-999 in Patients With Advanced Solid Tumors. ClinicalTrials.gov, 2019, Identifier: NCT04084366 [Google Scholar]
- 96. Scheck, RA, Francis, MB. Regioselective labeling of antibodies through N-terminal transamination. ACS Chem Biol 2007; 2: 247–51. [DOI] [PubMed] [Google Scholar]
- 97. Rosen, CB, Francis, MB. Targeting the N terminus for site-selective protein modification. Nat Chem Biol 2017; 13: 697–705. [DOI] [PubMed] [Google Scholar]
- 98. Schramm, VL. Chemical mechanisms in biochemical reactions. J Am Chem Soc 2011; 133: 13207–12. [DOI] [PubMed] [Google Scholar]
- 99. Qiu, H, Boudanova, E, Park, A et al. Site-specific PEGylation of human thyroid stimulating hormone to prolong duration of action. Bioconjug Chem 2013; 24: 408–18. [DOI] [PubMed] [Google Scholar]
- 100. Alfaro, JF, Gillies, LA, Sun, HG et al. Chemo-enzymatic detection of protein isoaspartate using protein isoaspartate methyltransferase and hydrazine trapping. Anal Chem 2008; 80: 3882–9. [DOI] [PubMed] [Google Scholar]
- 101. Witus, LS, Francis, M. Site-specific protein bioconjugation via a pyridoxal 5'-phosphate-mediated N-terminal transamination reaction. Curr Protoc Chem Biol 2010; 2: 125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. MacDonald, JI, Munch, HK, Moore, T et al. One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat Chem Biol 2015; 11: 326–31. [DOI] [PubMed] [Google Scholar]
- 103. Gilmore, JM, Scheck, RA, Esser-Kahn, AP et al. N-terminal protein modification through a biomimetic transamination reaction. Angew Chem Int Ed Engl 2006; 45: 5307–11. [DOI] [PubMed] [Google Scholar]
- 104. Zang, T, Dai, S, Chen, D et al. Chemical methods for the detection of protein N-homocysteinylation via selective reactions with aldehydes. Anal Chem 2009; 81: 9065–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Witus, LS, Netirojjanakul, C, Palla, KS et al. Site-specific protein transamination using N-methylpyridinium-4-carboxaldehyde. J Am Chem Soc 2013; 135: 17223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li, DZ, Han, BN, Wei, R et al. N-terminal alpha-amino group modification of antibodies using a site-selective click chemistry method. MAbs 2018; 10: 712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chelius, D, Jing, K, Lueras, A et al. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Anal Chem 2006; 78: 2370–6. [DOI] [PubMed] [Google Scholar]
- 108. Liu, YD, Goetze, AM, Bass, RB et al. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem 2011; 286: 11211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Martin, C, Brachet, G, Colas, C et al. In vitro characterization and stability profiles of antibody-fluorophore conjugates derived from interchain cysteine cross-linking or lysine bioconjugation. Pharmaceuticals (Basel) 2019; 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Beacon Targeted Therapies . (2020), www.beacon-intelligence.com (11 October 2020, last accessed).
- 111. van Berkel, SS, van Delft, FL. Enzymatic strategies for (near) clinical development of antibody-drug conjugates. Drug Discov Today Technol 2018; 30: 3–10. [DOI] [PubMed] [Google Scholar]
- 112. Li, J, Guo, Y, Xue, J et al. First-in-human phase I study of anti-HER2 ADC MRG002 in patients with relapsed/refractory solid tumors. J Clin Oncol 2020; 38: TPS1101–1. [Google Scholar]
- 113. Junutula, JR, Flagella, KM, Graham, RA et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res 2010; 16: 4769–78. [DOI] [PubMed] [Google Scholar]
- 114. Junutula, JR, Raab, H, Clark, S et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008; 26: 925–32. [DOI] [PubMed] [Google Scholar]
- 115. Axup, JY, Bajjuri, KM, Ritland, M et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A 2012; 109: 16101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Skidmore, L, Sakamuri, S, Knudsen, NA et al. ARX788, a site-specific anti-HER2 antibody-drug conjugate, demonstrates potent and selective activity in HER2-low and T-DM1-resistant breast and gastric cancers. Mol Cancer Ther 2020; 19: 1833–43. [DOI] [PubMed] [Google Scholar]
- 117. Carrico, IS, Carlson, BL, Bertozzi, CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol 2007; 3: 321–2. [DOI] [PubMed] [Google Scholar]
- 118. Albers, AE, Garofalo, AW, Drake, PM et al. Exploring the effects of linker composition on site-specifically modified antibody-drug conjugates. Eur J Med Chem 2014; 88: 3–9. [DOI] [PubMed] [Google Scholar]
- 119. Drake, PM, Albers, AE, Baker, J et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug Chem 2014; 25: 1331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Thompson, P, Bezabeh, B, Fleming, R et al. Hydrolytically stable site-specific conjugation at the N-terminus of an engineered antibody. Bioconjug Chem 2015; 26: 2085–96. [DOI] [PubMed] [Google Scholar]
- 121. Costoplus, JA, Veale, KH, Qiu, Q et al. Peptide-cleavable self-immolative Maytansinoid antibody-drug conjugates designed to provide improved bystander killing. ACS Med Chem Lett 2019; 10: 1393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yurkovetskiy, AV, Yin, M, Bodyak, N et al. A polymer-based antibody-Vinca drug conjugate platform: characterization and preclinical efficacy. Cancer Res 2015; 75: 3365–72. [DOI] [PubMed] [Google Scholar]