

Abstract
Adenosquamous cancer of the pancreas (ASCP) is a subtype of pancreatic cancer that has a worse prognosis and greater metastatic potential than the more common pancreatic ductal adenocarcinoma (PDAC) subtype. In order to distinguish the genomic landscape of ASCP and identify actionable targets for this lethal cancer we applied DNA content flow cytometry to a series of 15 tumor samples including five patient derived xenografts (PDXs). We interrogated purified sorted tumor fractions from these samples with whole genome copy number variant (CNV), whole exome sequencing (WES) and ATAC-seq analyses. These identified a variety of somatic genomic lesions targeting chromatin regulators in ASCP genomes that were superimposed on well characterized genomic lesions including mutations in TP53 (87%) and KRAS (73%), amplification of MYC (47%), and homozygous deletion of CDKN2A (40%) that are common in PDACs. Furthermore, a comparison of ATAC-seq profiles of three ASCP and three PDAC genomes using flow-sorted PDX models identified genes with accessible chromatin unique to the ASCP genomes, including the lysine methyltransferase SMYD2 and the pancreatic cancer stem cell regulator RORC in all three ASCPs, and a FGFR1-ERLIN2 fusion associated with focal CNVs in both genes in a single ASCP. Finally, we demonstrate significant activity of a pan FGFR inhibitor against organoids derived from the FGFR1-ERLIN2 fusion-positive ASCP PDX model. Our results suggest that the genomic and epigenomic landscape of ASCP provide new strategies for targeting this aggressive subtype of pancreatic cancer.
Keywords: adenosquamous carcinoma of the pancreas, flow sorting, somatic genomics, ATAC-seq, therapeutics
Introduction
It is estimated that in 2020, 57,600 Americans will be diagnosed with pancreatic cancer and 47,050 will die from the disease, making pancreatic cancer the third most common cause of cancer death (1). Adenosquamous cancer of the pancreas (ASCP) is a rare subtype of pancreatic cancer representing 1–4% of all pancreatic cancers (2). Strikingly, ASCP displays a higher metastatic potential and a worse clinical outcome than the more common pancreatic ductal adenocarcinoma (PDAC) (4). Yet, a large population-based analysis did not detect any differences in tumor stage at the time of diagnosis between PDAC and ASCP (5). There are various theories as to why this histological subtype exists, as normal pancreas tissue has no benign squamous epithelium. These include the induction of a squamous epithelium by inflammation, the collision of histologically distinct tumor lineages in the same tissue, and the aberrant differentiation and enrichment of tumor stem cells that acquire features of one or both subtypes (2). However, the rarity of ASCP, the scarcity of tissue samples suitable for high resolution genomic analyses, and the lack of validated preclinical models has limited the study of this deadly subtype of pancreatic cancer. Thus, little is known about the genomic landscape of ASCP, its relationship with PDAC and potential driver aberrations responsible for the aggressive clinical phenotype that may be exploited for improved patient care.
We acquired both fresh frozen and formalin-fixed, paraffin-embedded (FFPE) ASCP samples, including primary and metastatic tissue from a rapid autopsy, from multiple institutions. We then applied DNA content flow cytometry, whole genome copy number and whole exome sequence analyses to the ASCP tumors to comprehensively profile the genomic landscape of this aggressive tumor. Notably, our integrated analyses of ASCP identified a variety of somatic genomic lesions targeting chromatin regulators in ASCP genomes that were superimposed on well characterized genomic lesions including mutations in KRAS and TP53, homozygous deletion of CDKN2A, and amplification of MYC, which are common to PDACs (6–8). We then compared the chromatin accessibility patterns of ASCP with PDACs using patient-derived xenograft (PDX) models with genomic lesions and mutations seen in patient samples. Of significant interest were genes with open chromatin regions unique to ASCP that included a FGFR1-ERLIN2 fusion, and the pancreas cancer stem cell regulator RORC. The latter is a nuclear hormone receptor known for its role in Th17 cell specification and regulation of inflammatory cytokine production and has been shown to be enriched during the emergence of chemorefractory tumors, a feature shared with ASCP, in the KPC mouse PDAC model (9). ASCP organoids were used to test the activity and functional significance of candidate therapeutic targets in ASCP. Specifically, organoids carrying the FGFR1-ERLIN2 fusion show a significant response to pharmacological FGFR inhibition. These results advance our understanding of this lethal subtype of pancreas cancer and provide new candidate targets for developing effective therapies for patients with ASCP and potentially refractory PDAC.
Materials and Methods
Tumor Samples:
All patients gave written informed consent for collection and use of the samples. All tissue was collected for this study under Mayo Clinic IRB 14–003050. All tumor samples were histopathologically evaluated by a board certified GI pathologist at each institution prior to genomic analysis. As per the 2019 World Health Organization (WHO) Classification of Tumors, Digestive System Tumours, ASCP samples in this study both primary and metastatic, including patient matched samples from a rapid autopsy case, and patient derived xenograft (PDX) tissues (Supplemental Table 1), were defined by the presence of at least 30% squamous differentiation (Supplemental Figure 1). PDXs from resected ASCP tumors were obtained from the Mayo Clinic Institutional PDX program in Hepatobiliary and Pancreatic Tumors. All established tumors undergo extensive histologic comparison and DNA fingerprinting to confirm that each PDX is derived from each specific patient. The ASCP histology was confirmed for each PDX using WHO criteria. All research conformed to the Helsinki Declaration (http://www.wma.net/en/30publications/10policies/b3/).
Flow Cytometry:
Excess paraffin was removed from each FFPE sample with a scalpel from either side of 40–60 μm scrolls then processed according to our published methods (10,11). We used one to three 50 µm scroll(s) from each FFPE tissue block to obtain sufficient numbers of intact nuclei for sorting and molecular assays. Frozen tissue samples were minced in the presence of NST buffer and DAPI according to published protocols (10,12,13). Nuclei from each sample were disaggregated then filtered through a 40 μm mesh prior to flow sorting with an Influx cytometer (Becton-Dickinson, San Jose, CA) with ultraviolet excitation and DAPI emission collected at >450 nm. DNA content and cell cycle were analyzed using the software program MultiCycle (Phoenix Flow Systems, San Diego, CA).
For sorting single nuclei into 96 well plates, organoid cultures were resuspended in DAPI (10 µg/ml) then disaggregated using a 1mL 18g syringe followed by filtering through a 40 µm mesh. Samples were examined under a microscope to confirm single nucleus suspensions. The forward scatter (FSC) and side scatter (SSC) signal and the fluorescence signals of the 488 nm Blue Laser and 355 nm UV laser were aligned and optimized using Ultra Rainbow fluorescent particles (3.0–3.4 µm) containing a mixture of fluorophores that enable the Ultra Rainbow particles to be excited at any wavelength from 356 to 650 nm. The coefficient of variation (CV) was adjusted to a range of 1.9–2.9. A four- way sort was set up using a 100 µl nozzle tip, 20 psi with a frequency 29.4 kHz. Nuclei were sorted in a one drop pure mode (high purity and high recovery of the sample) at 1500–2000 events per second to maintain purity and intactness of sorted material. In addition we deposited empty droplets in two wells and 50 nuclei and 100 nuclei in two additional wells as negative and positive controls for downstream amplification and sequencing.
Copy Number Analysis:
DNAs from frozen tissue and FFPE samples were treated with DNAse 1 prior to Klenow-based labeling. High molecular weight templates were digested for 30 minutes while DNAs from FFPE samples were digested for only 1 minute. In each case 1 µl of 10x DNase 1 reaction buffer and 2 μl of DNase 1 dilution buffer were added to 7 μl of DNA sample and incubated at room temperature then transferred to 70°C for 30 minutes to deactivate DNase 1. Sample and reference templates were then labeled with Cy-5 dUTP and Cy-3 dUTP respectively using a BioPrime labeling kit (Invitrogen, Carlsbad, CA) according to our published protocols (14). All labeling reactions were assessed using a Nanodrop assay (Nanodrop, Wilmington, DE) prior to mixing and hybridization to 400k CGH arrays (Agilent Technologies, Santa Clara, CA) for 40 hours in a rotating 65°C oven. All microarray slides were scanned using an Agilent 2565C DNA scanner and the images were analyzed with Agilent Feature Extraction version 11.0 using default settings. The aCGH data was assessed with a series of QC metrics then analyzed using an aberration detection algorithm (ADM2) (15). The latter identifies all aberrant intervals in a given sample with consistently high or low log ratios based on the statistical score derived from the average normalized log ratios of all probes in the genomic interval multiplied by the square root of the number of these probes. This score represents the deviation of the average of the normalized log ratios from its expected value of zero and is proportional to the height h (absolute average log ratio) of the genomic interval, and to the square root of the number of probes in the interval. All data have been deposited to GEO repository (accession GSE143256).
Whole Exome Sequencing:
DNAs from each sorted tumor population and a patient matched control sample were sequenced within the Mayo Clinic Medical Genome Facility (MGF) using established protocols for whole exome analysis. Briefly, whole exon capture was carried out with Agilent’s SureSelect Human All Exon 71 MB v6 kit. 500 ng of the prepped library is incubated with whole exon biotinylated RNA capture baits supplied in the kit for 24 hours at 65 °C. The captured DNA:RNA hybrids are recovered using Dynabeads MyOne Streptavidin T1 (Invitrogen). The DNA was eluted from the beads and desalted using purified using Ampure XP beads (Beckman Coulter, Indianapolis IN).The purified capture products were then amplified using the SureSelect Post-Capture Indexing forward and Index PCR reverse primers (Agilent) for 12 cycles. Libraries were loaded onto paired end flow cells at concentrations of 4–5 pM to generate cluster densities of 600,000–800,000/mm2 using the Illumina cBot and HiSeq Paired end cluster kit version 3.The flow cells are sequenced as 101 X 2 paired end reads on an Illumina HiSeq 2500 or 4000 using TruSeq SBS sequencing kit version 3 and HiSeq data collection version 1.4.8 software. Base-calling was performed using Illumina’s RTA version 1.12.4.2.
Variant Calling and Annotation:
Aligned tumor and germline data (in bam format) were assembled for each patient. Tumor-specific variants were called using VarScan2 (version 2.3.9) (16) available on a high performance cluster computing environment. A minimum coverage of 10 reads in normal and tumor was used to call somatic variants, a minimum variant frequency of 0.08 to call a heterozygote, and a somatic p-value of 0.05 as a threshold to call a somatic site. The SNP calls were filtered to remove those near indel positions, and also removed likely false positives associated with common sequencing- and alignment-related artifacts (17). The variants were annotated functionally using Annovar (18) with hg19 reference genome.
Single cell sequencing:
We applied DNA content flow sorting to isolate single nuclei from PAX265-derived organoids and deposited them in individual wells of a 96 well plate. This approach includes fluorescence measures for each well that confirms the DNA content of each sorted nucleus and discriminates aggregates from single nuclei. DNA extraction and amplification of each sorted nucleus was performed using the REPLI-g Single Cell Kit (QIAGEN, Germantown, MD) according to protocols from the supplier. KRAS exon 2 was amplified using Platinum™ Taq DNA Polymerase (Thermo Fisher, Waltham, MA) according to the supplier’s protocol, then sequenced by M13 primed Sanger sequencing. Sequencing was done through the Arizona State University Genomics Core. Sequences were visually inspected using Sequence Scanner V2 software (ABI, Foster City, CA).
Omni-ATAC:
Approximately 50,000 flow sorted nuclei were utilized for OMNI ATAC-seq following the protocol by Corces et al. (19). Nuclei were subjected to transposition reaction following cell lysis and washing steps. The transposition reaction was performed at 37 °C for 30 min in a thermomixer with shaking at 1,000 rpm. DNA was purified and indexed ATAC-seq libraries amplified by PCR. The enrichment of accessible regions in each library was determined by real-time PCR targeting known highly accessible and closed chromatin sites and expressed as fold difference. Libraries passing this quality control step were sequenced to 51 base pairs from both ends on an Illumina HiSeq 4000 instrument at the Mayo Clinic Center for Individualized Medicine Medical Genomics Facility.
Omni-ATAC data were analyzed using the HiChIP pipeline developed by the Mayo Bioinformatics Core (20). Briefly, paired-end reads were mapped to the HG38 genome reference using Burrows-Wheeler Alignment tool (BWA) (21). Pairs of reads with one or both reads uniquely mapped were retained and duplicates removed using Picard MarkDuplicates command (http://broadinstitute.github.io/picard/). Peaks were identified using the Model-based Analysis of ChIP-Seq (MACS2) software package at FDR <=1% (22). For data visualization, BEDTools (23) in combination with in-house scripts were used to generate normalized tag density profiles at a window size of 200 bp and step size of 20 bp. Differential analysis of chromatin accessibility was performed using the DiffBind package (24). Raw read counts were normalized using the trimmed mean of M values (TMM) method, and sites with fold changes >=2 and FDR <=0.05 in the DiffBind test were extracted to represent differential chromatin accessibility sites. The ATAC-seq data are available in the GEO repository (accession GGSE143256).
Immunofluorescence:
Immunofluorescent staining was completed using FFPE sections. Slides were first deparaffinized using xylenes and a graded ethanol series, followed by permeabilization with Triton X-100. Antigen retrieval was completed using citrate buffer (pH 6) and steam heat. After blocking with 1 % BSA and 2% goat serum, slides were incubated with either anti-histone H3 acetyl K27 antibody (Abcam, Cambridge, UK) at 1:1000 dilution at 4 degrees overnight or anti-histone H3 mono methyl K4 antibody (Abcam), at 1:2000 dilution for 2 hours at room temperature. Slides were then incubated with goat anti-rabbit Alexa Fluor 594 secondary antibody (Life Technologies, Carlsbad CA) for one hour at room temperature and cover-slipped using ProlongGold antifade reagent with DAPI (Life Technologies) to preserve fluorescence and stain nuclei. Images were captured using a Zeiss Axio Oberserver Z1 microscope at 40x magnification.
Immunohistochemistry:
IHC staining was performed on a Leica BOND RX automated immunostainer using BOND primary antibody diluent and BOND Polymer Refine DAB Detection kit according to the manufacturer’s instructions (Leica Biosystems, Buffalo Grove, IL). Pre-treatment was performed using citrate buffer at 1000C for 30 min, and tissue was stained using rabbit anti-human RORC(t) (polyclonal, PA5–23148, Thermo Fisher Scientific, Waltham MA) at a dilution of 1:4000. Stained slides were scanned (20x magnification) using a Pannoramic P250 digital slide scanner (3DHistech, Thermo Fisher Scientific).
Organoid cultures and treatments:
Organoids were seeded in 96-well plate coated with Matrigel (5,000 cells /well) in organoid media. Fresh organoid media and DMSO or drug (10 wells/condition) was added every 2–3 days. MTT assay were performed on Day 7 to measure proliferation. The absorbance listed for each drug is the average of the 10 wells per condition. Each experiment was performed in triplicate and the average IC50 reported.
Results
Genomic landscape of ASCP identified alterations in chromatin regulators.
We screened 48 ASCP patient samples with DNA content flow cytometry. Tumor-specific fractions were detected in 15 samples. These included both fresh frozen and FFPE archival tissues. All tumor fractions were non-diploid by flow cytometry and genomically aberrant with both CNVs and somatic mutations. For example the sorted aneuploid fraction of sample A90–5 had multiple CNVs including gains of 18q11, 8q24, 7p12, 5p and 1p34.2 and multiple somatic mutations including in the lysine demethylase KDM3A (Figure 1). In contrast, the co-existing sorted diploid fraction was genomically normal at both the somatic CNV and mutation levels. The tumor fractions in the ASCP biopsies varied extensively (<5% to >50%) as a percentage of total cellular content (diploid, tetraploid and aneuploid fractions). However our sorting provided highly enriched (>90% tumor content) samples for analyses enabling the discrimination of homozygous deletions and mutations in these complex genomes. The samples that were diploid only by flow cytometry (n=20) were CNV neutral and lacked detectable somatic CNVs and mutations in their exomes (Supplemental Figure 2). In the remaining 13 cases there were insufficient intact nuclei for sorting due to extensive tissue degradation.
Figure 1: DNA content histogram and genomic profile of flow sorted ASCP biopsy.
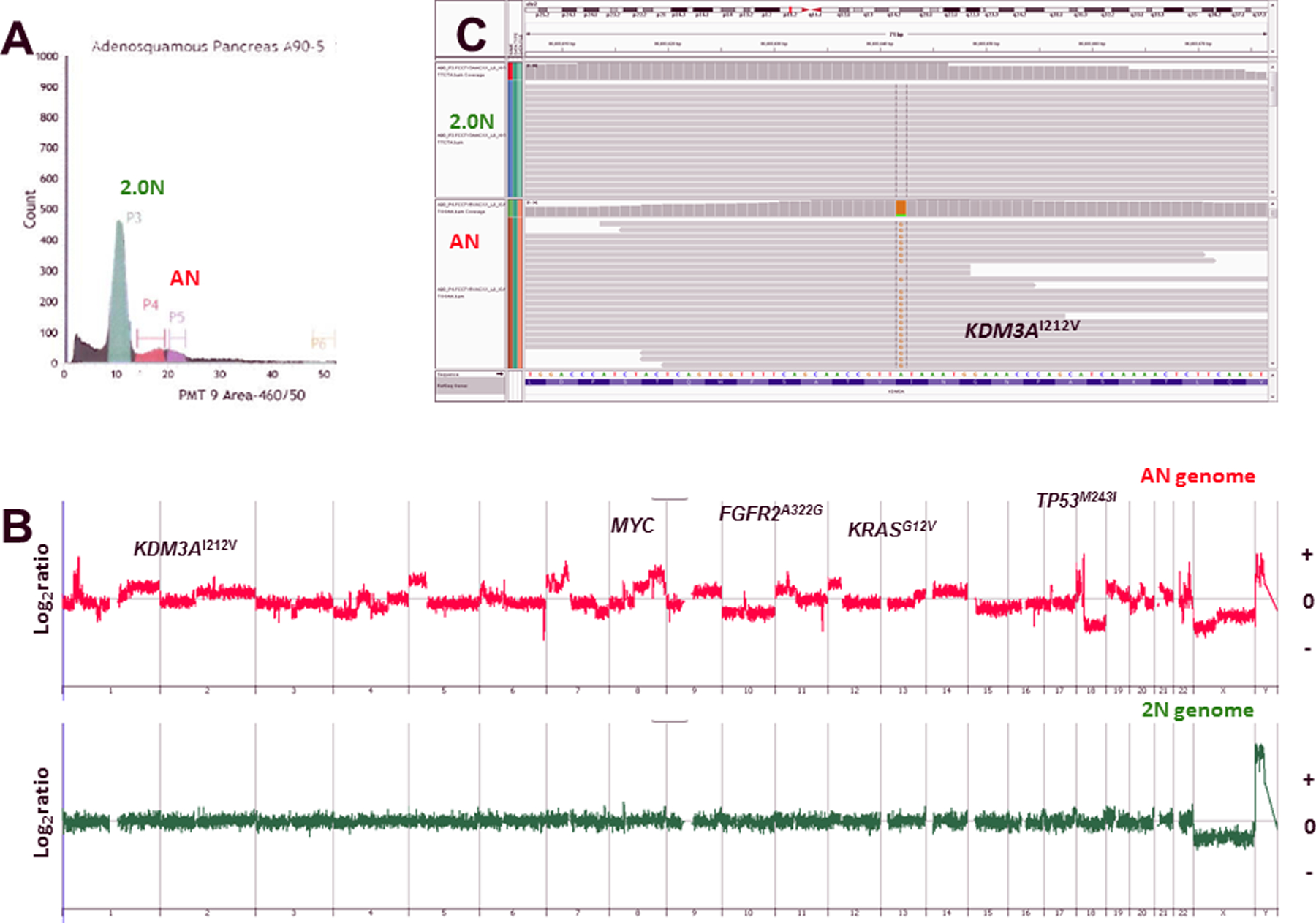
A) Diploid (2N) and aneuploid peaks were identified and sorted from FFPE tissue sample A90. B–C) CNV and mutation profiles of the normal 2N and tumor aneuploid populations. The X and Y axes in the CGH plots represent chromosome and log2 ratios for each genome.
Biopsies from PDX models contain variable amounts of host tissue. Studies have used bioinformatic approaches to subtract mouse sequences and to extract and enrich human tumor profiles from these samples (25). In contrast we used our DNA content sorting to prepare PDX samples for analyses. Near-diploid peaks representing mouse host nuclei were present in each sample (Supplemental Figure 3). We selected peaks that corresponded to human tumor nuclei based on their ploidy and lack of overlap with DNA content and cell cycle profiles of the host tissue. The DNAs from each sorted population of interest were interrogated with CGH arrays to confirm the human tumor content and to provide a CNV profile of each tumor genome. These sorted human tumor samples were subsequently used for whole exome NGS with a patient matched normal sample.
We detected common recurring PDAC driver events in each of the ASCP genomes. These included CDKN2A and SMAD4 homozygous deletions, KRAS and TP53 mutations, and MYC amplifications (Figure 2). Our results support a model whereby ASCPs evolve from the same tumor lineage as the more common PDACs. However, superimposed on ASCP genomes are “hits” targeting key regulators of chromatin organization. These hits were heterogeneous and included homozygous deletions in SMARCA2, ARID2, and ASXL2, and somatic mutations in the DNA demethylase TET1, the histone ubiquitin E3 ligase MSL2 , and the chromatin regulator KANSL1 (Figure 2; Supplemental Figures 4–6). We also identified a series of high level (log2 ratio >2) amplicons targeting genes associated with a variety of oncogenic signaling pathways. These included TLK2, ASH2L, MPL, and FRS2 (Supplemental Figures 7 and 8). The latter, (Fibroblast Growth Factor Receptor Substrate 2) is an adapter protein that plays an important role in the activation of MAP kinases and in the phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, in response to ligand-mediated activation of FGFR1 (26). In addition we detected a FGFR2A322G mutation that has been reported as a pathogenic variant in esophageal cancer (27). Notably this mutation targets exon 8 encoding the second half of one of the three Ig domains and part of the IIIb isoform associated with epithelial tumors (28).
Figure 2:
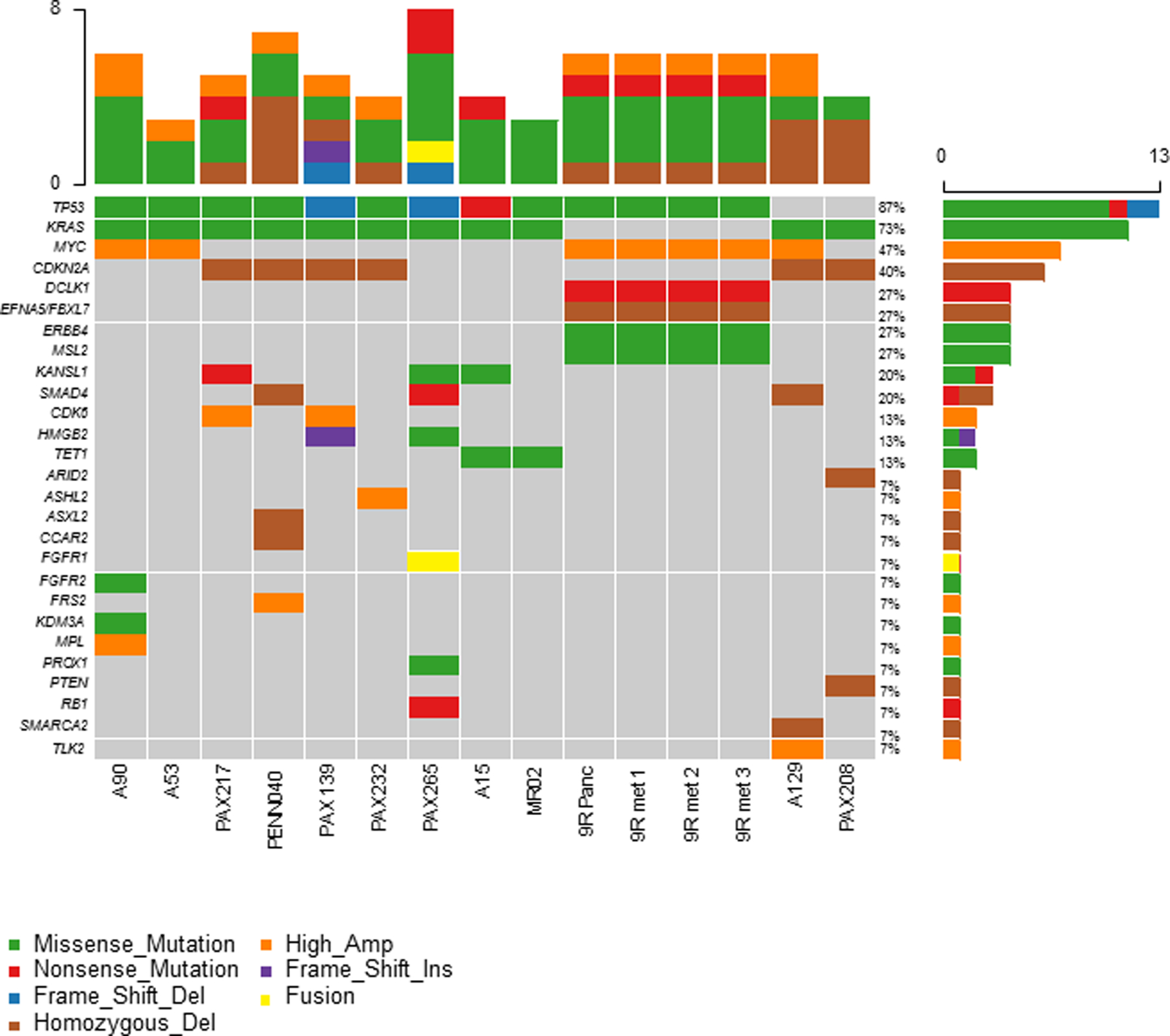
Oncomap summary of somatic lesions in ASCP genomes.
ASCP patient samples included a rapid autopsy case with a primary and multiple metastatic lesions. We detected small (<10% of total cell content) aneuploid tumor fractions in the primary tissue and each of three metastatic lesions (Figure 3). The CNV profiles of each flow-sorted aneuploid population overlapped and included gains of 8q21.3-qtel that contained the MYC locus, a focal amplicon at 15q23-q24.2 that included PKM and CD276, and a homozygous deletion at 5q21.3 targeting EFNA5 and FBXL17. The primary and metastatic lesions also shared pathogenic mutations including TP53Y205S and DCLK1R699X. Strikingly none of these tissues had a KRAS mutation. However, we detected a somatic ERBB4S183C mutation that was present in each sorted tumor population. Although not previously reported, this non-conserved variant is located adjacent to multiple pathogenic mutations in the Furin-like cysteine rich domain of ERBB4 (29).
Figure 3: Genomic profiles of primary and metastatic biopsies from ASCP rapid autopsy 9R.
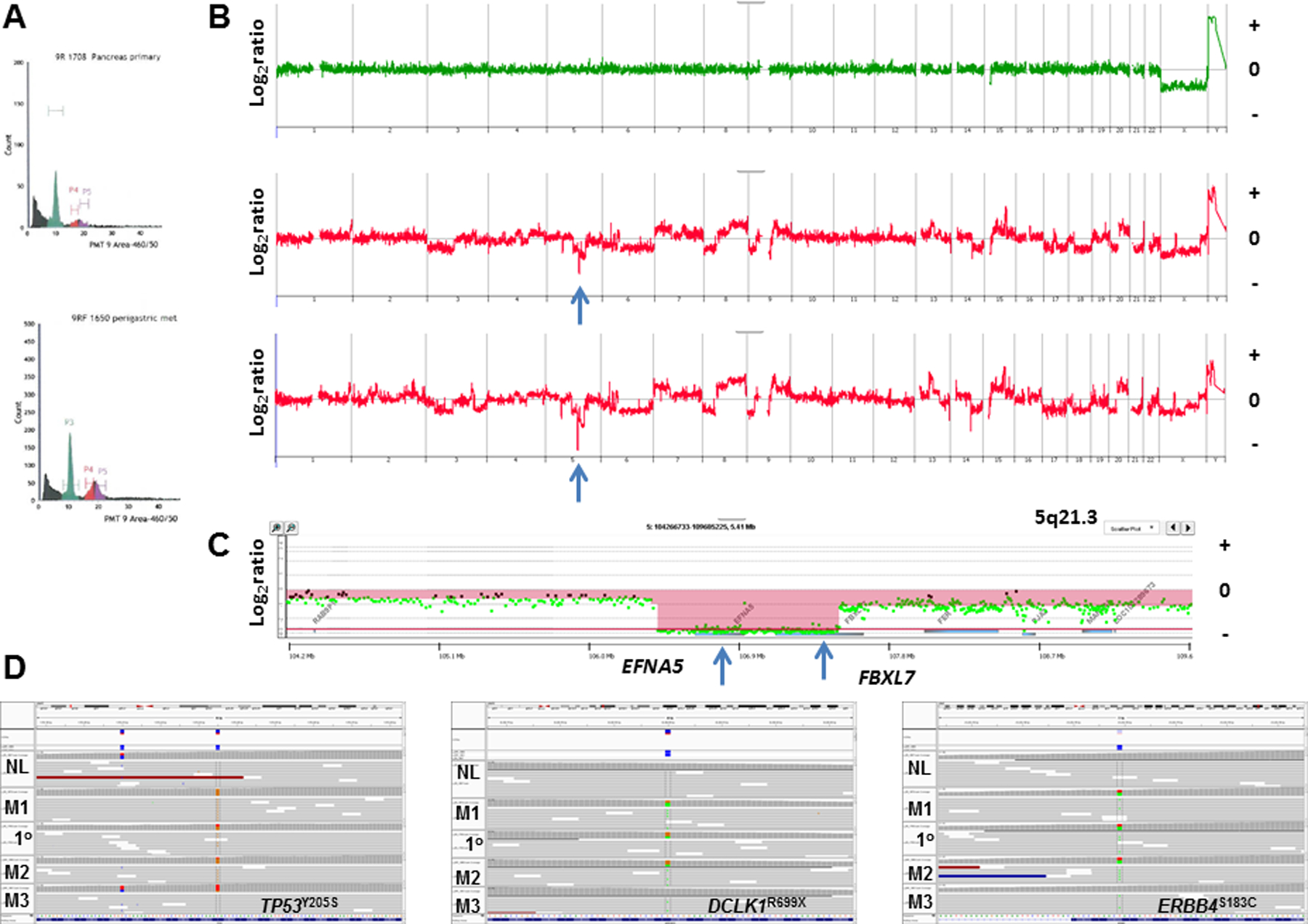
A) DNA content histograms of primary (top) and perigastric (bottom) biopsies. B) CNV profiles of flow sorted 2N and aneuploid populations. C) Homozygous deletion at 5q21.3 in sorted aneuploid population targeting EFNA5 and FBXL7. The X and Y axes in the CGH plots represent chromosome and log2 ratios for each genome. Red shaded areas denote homozygous deletions. D) IGV views of somatic mutations in TP53, DCLK1, and ERRB4 detected in the primary (1o), and metastases in the liver (M1), perigastric (M2) and lung (M3). The sorted diploid fraction from the primary was used as the patient matched control.
The genomes of the five ASCP PDX models contain recurring pancreatic cancer lesions. These included pathogenic KRAS mutations in all 5 cases and homozygous CDKN2A deletions in 4/5 samples (Table 1). The one exception with an intact CDKN2A locus had a homozygous pathogenic RB1 nonsense mutation. Four of these models had TP53 mutations. The one TP53WT case (PAX208) had a homozygous deletion of PTEN a context associated with loss of TP53 protein and transcriptional activity (30). In addition, we detected a homozygous nonsense SMAD4 mutation in PAX208 and loss of 18q21.1 in the remaining four cases. Thus our preliminary data highlight the presence of prevalent genomic drivers of pancreatic cancer in our PDX models. Notably the KRAS mutations included the common G12D variant in three of the PDXs and the less frequent G12S and Q61H variants in each of the two additional samples. Although limited to 5 PDXs for this rare cancer these diverse KRAS genotypes provide favorable models to study the biology of ASCP and to evaluate therapeutic strategies against this aggressive subtype of pancreatic cancer. Similar to the patient samples, the PDXs had homozygous deletions and mutations targeting epigenomic regulators including ARID2, HMGB2 and KANSL1 (Figure 2, Supplemental Figure 6).
Table 1.
ASCP PAX Genotypes
| KRAS | TP53 | CDKN2A | SMAD4 | Epigenome “Hits” | |
|---|---|---|---|---|---|
| PAX139 | G12D | E204_L206delEYL | −/− | −/+ | HMGB249−50dupGC |
| PAX208 | Q61H | WT1 | −/− | W227X | ARID2−/− |
| PAX217 | G12D | G223V | −/− | −/+ | KANSL1Q396X |
| PAX232 | G12S | Q165K | −/− | −/+ | ASH2L+++ |
| PAX265 | G12D | P177_C182delPHHERC | +/+2 | −/+ | KANSL1T221I |
PTEN−/−
RB1R787X
ASCP shows a distinct chromatin accessibility pattern.
Given the presence of genomic lesions targeting key epigenomic regulators we hypothesized that ASCP and PDAC genomes would have shared and unique sites of active chromatin at key oncogenic drivers. We performed Omni-ATAC, an improved ATAC-seq protocol (19), on three of the ASCP PDXs and three PDAC PDXs that were also profiled with whole genome CNV analysis (Supplemental Figure 9). Active chromatin was present at the KRAS and the MYC loci in all six samples profiled (Supplemental Figure 10). Unique active chromatin sites included SMYD2, a lysine methyltransferase with specificity for the active chromatin mark H3K4me1, in all three ASCP samples (Figure 4A, B). Immunofluorescence analysis revealed higher H3K4me1 levels in ASCP PAX129 compared to PDAC PAX297 and normal pancreas tissue. In addition, all three ASCP PDX models had active chromatin at the CDK6 locus (Supplemental Figure 11). These included two, PAX139 and PAX217, with high level (log2 ratio >2) CDK6 amplicons. Given the presence of co-occurring CDKN2A deletions in these genomes we hypothesized that CDK4/6 inhibitors would have activity in these ASCP models. However we did not observe any response to either palbociclib or abemaciclib in organoid cultures derived from PAX139. The ASCP PDX genomes also had active chromatin at the RORC locus compared to PDACs (Figure 5A). Cytoplasmic and nuclear staining was observed in the ASCP and the PDAC PDX models, and in tissue from the rapid autopsy series (Figure 5B, Supplemental Figure 12). Notably there was an enrichment of nuclear staining in the metastatic lesions.
Figure 4: ATAC-seq profile of SMYD2 and active chromatin mark in ASCP PDX models.
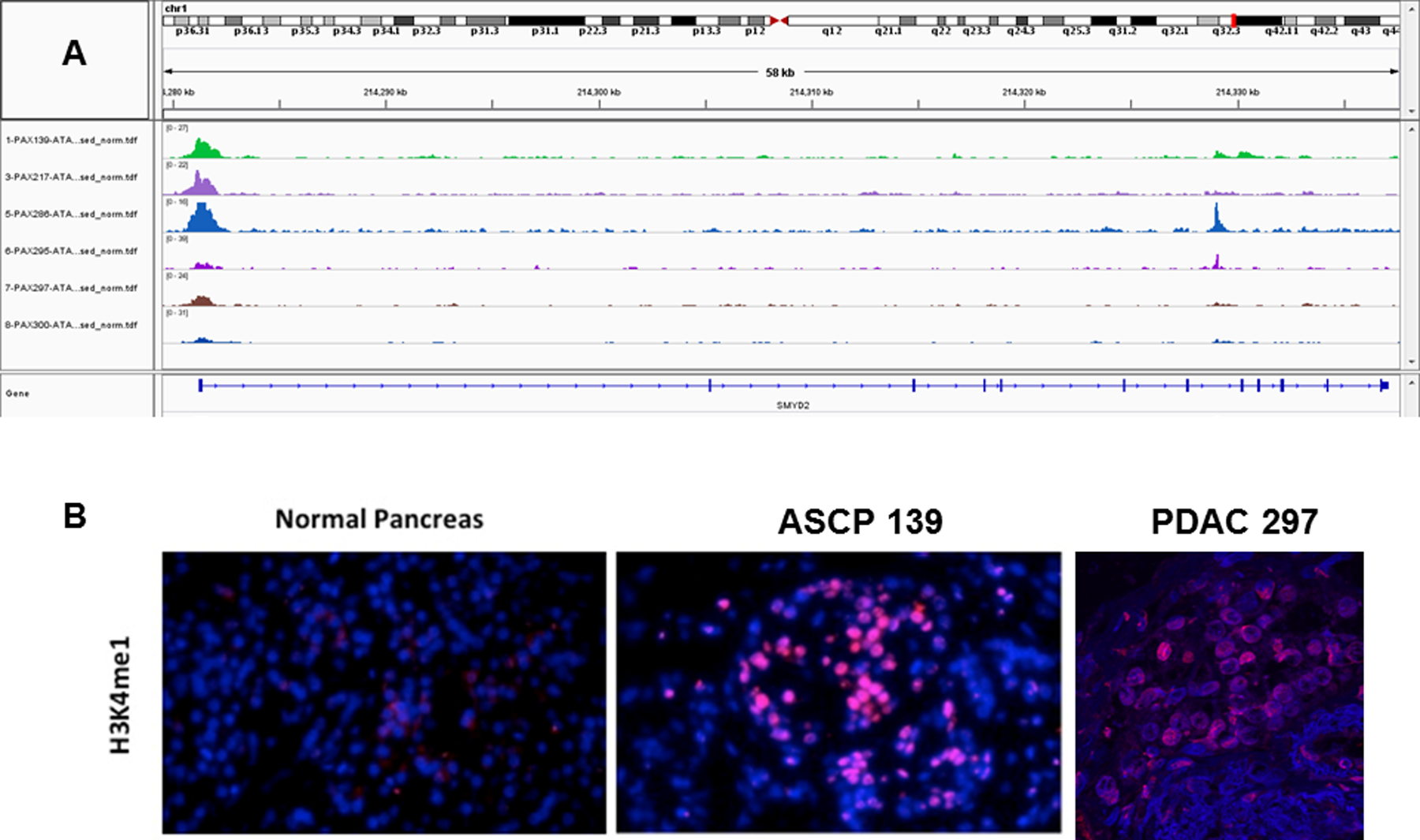
A) IGV views of chromatin accessibility in ASCP (top 3 rows) and PDAC (bottom 3 rows) PDX models. All samples were flow sorted prior to ATAC-seq analysis. B) Immunofluorescence analysis (40x magnification) of H3K4me expression in normal pancreas and tumor tissue (PAX 139).
Figure 5: ATAC-seq profile and expression of RORC in PDXs.
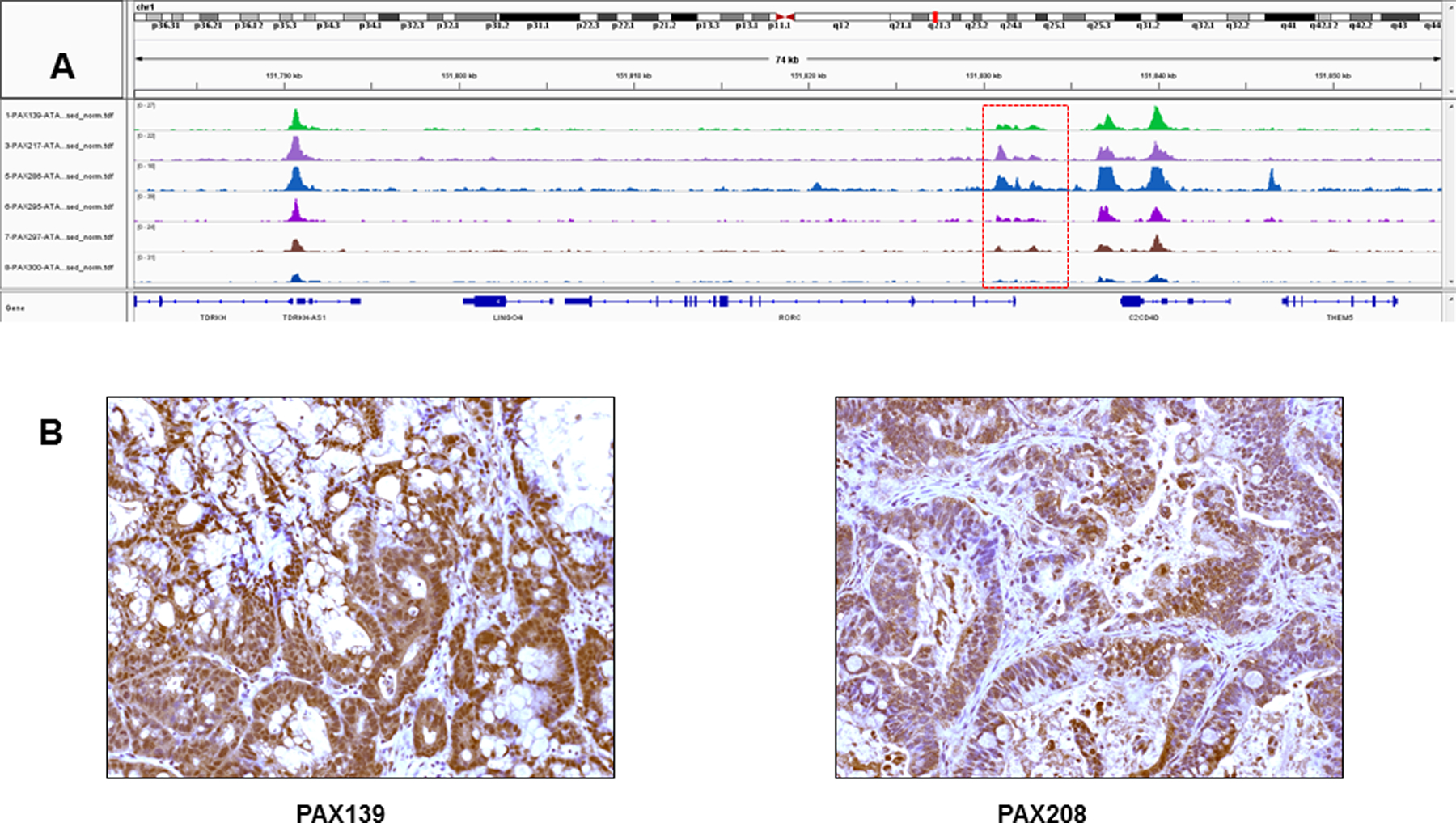
A) IGV views of chromatin accessibility in ASCP (top 3 rows) and PDAC (bottom 3 rows) PDX models. All samples were flow sorted prior to ATAC-seq analysis. B) IHC analysis (20x magnification) of RORC expression in ASCP (PAX139. PAX217, PAX265) and PDAC (PAX297, PAX295, PAX300) PDXs.
ASCP organoids carrying a FGFR1 fusion show sensitivity to pan FGFR inhibitor
We detected the CNV footprint of a FGFR1-ERLIN2 fusion on 8p12 and its ATAC-seq profile in PAX265 (Figure 6A–C). These were consistent with the presence of an active fusion that included the extracellular domains and the kinase domains of FGFR1 fused to ERLIN2. This fusion has been reported in breast cancer and its’ dimerization is believed to be sensitive to the FGF (31). Organoids developed from PAX265 with the FGFR1-ERLIN2 fusion were sensitive to the pan FGFR inhibitor infigratinib with an IC50 near 250nM in triplicate experiments (Figure 6D). To our knowledge this represents the first example of ASCP response to a targeted therapy. Notably this fusion co-occurred with a pathogenic KRASG12V mutation (Figure 6E). The KRASG12V variant was detected in 34/50 (68%) sorted single nuclei from the organoid model suggesting it did not affect the robust response to the pan FGFR inhibitor (Supplemental Figure 13). Given the presence of a pathogenic FGFR2A322G mutation and high level amplification of FRS2 in patient samples our data suggests that FGFR inhibition may provide a therapeutic window for a subset of ASCP similar to that observed in cholangiocarcinoma (32,33).
Figure 6: FGFR-ERLIN2 fusion in PAX265.
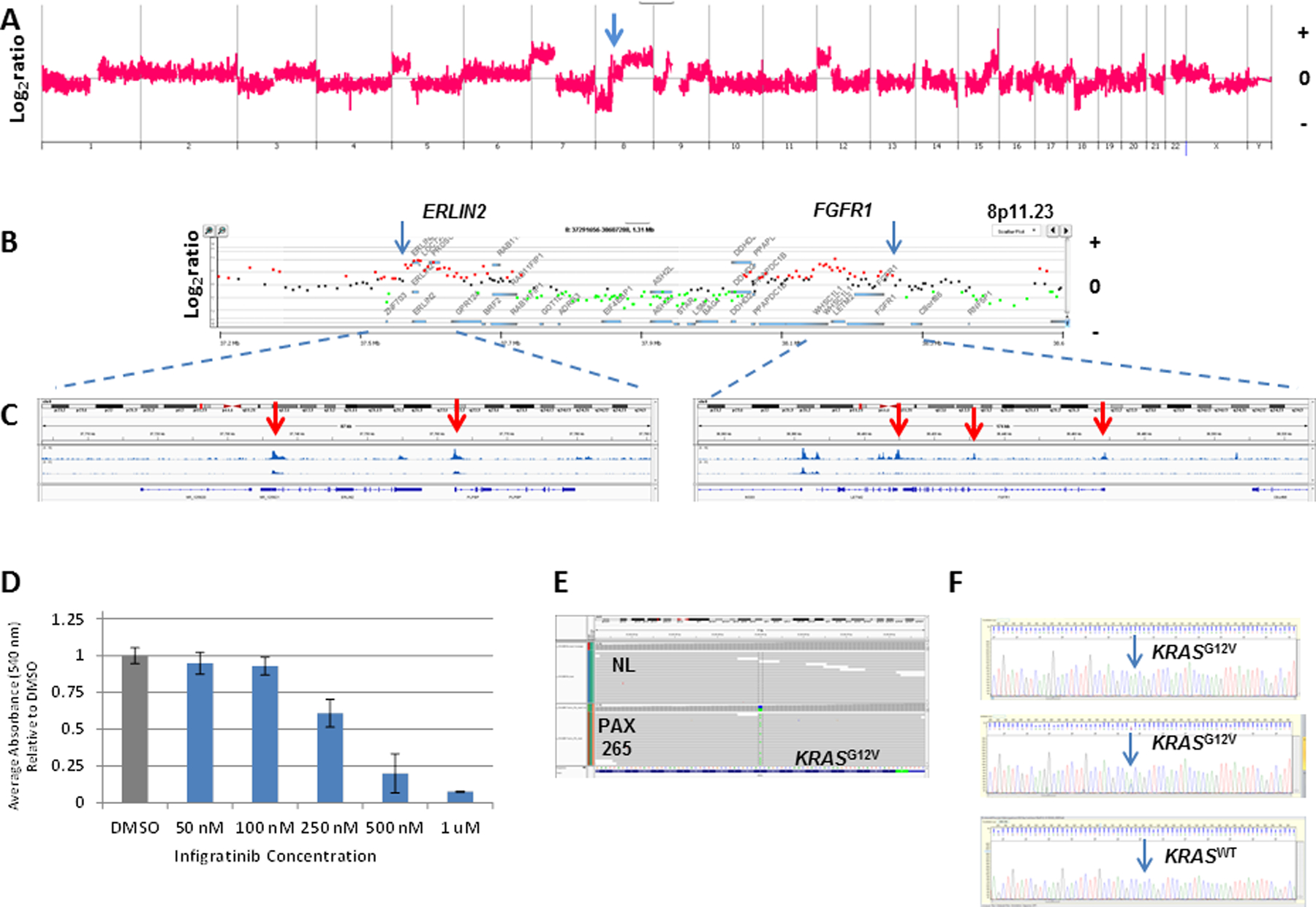
A) Whole genome and; B) locus specific analysis of CNV gains targeting ERLIN2 and FGFR1. C) IGV views of ATAC-seq profiles of ERLIN2 (left panel) and FGFR1 (right panel). Red arrows denote open chromatin peaks in each gene. D) Dose response profile of PAX265 organoids to Infigratinib. Organoids were seeded in 96-well plate coated with Matrigel (5,000 cells /well) in organoid media. After 2–3 days of growth, fresh organoid media and DMSO or drug (10 wells/condition) was added. Fresh media and drug was added every 2–3 days. MTT assay were done on day 7 to measure proliferation. The absorbance listed for each drug is the average of the 10 wells per condition. Experiment was performed in triplicate with an average IC50 near 250 nM. E) IGV view of KRASG12V mutation in flow sorted PAX265. F) Single cell sequencing of KRAS exon 2 in flow sorted PAX265 derived organoids. Sanger sequencing of single nuclei sorted from 50 individual organoids identified 19 homozygous (top) and 15 heterozygous (middle) KRASG12V variants. The other 16 were KRASG12G wild type (bottom).
Discussion
To our knowledge this is the first study to apply DNA content sorting to the genomic analysis of ASCP. Our methods yielded purified (>95%) tumor samples for whole genome analyses from a variety of clinical samples. These include fresh frozen and FFPE tissues with low tumor content (<10–20%) and high amounts (>90%) of necrosis and debris. By profiling flow-sorted tumor fractions from each sample of interest we identified multiple heterogeneous deletions, mutations and amplicons targeting epigenomic and stem cell regulators superimposed on common PDAC driver lesions. Notably, many of these can be classified as either Tier 1 or Tier 2 variants based on ASCO/CAP guidelines (Supplemental Table 2) (34). Our flow assays exploit a cancer cell hallmark, aneuploidy, present in >90% of solid tumors (35). The use of nuclei and DAPI provides discrimination of diploid, aneuploid, and tetraploid cells with C.V.s of ≥10% for DNA content (36,37). Thus, our assays can identify and sort tumor nuclei with DNA contents ranging from near diploid to hypertetraploid from clinical and preclinical samples of interest (10,14,38). In those relatively rare cases where DNA content overlaps with normal diploid content, the gating and collecting of proliferating fractions (e.g. 4N-G2/M) can enrich for tumor nuclei. Comparisons of ASCP with PDAC have been used to search for genomic lesions that account for the differences in appearance and clinical behaviors of these pancreas cancer subtypes. These include reports of enrichment for TP53 mutations and copy number losses at chromosome 3p in ASCPs relative to PDACs (39). However, we did not observe these differences in our small cohort of flow sorted samples. Additional studies have reported recurring mutations targeting UPF1, which encodes an RNA helicase essential for the highly conserved RNA degradation pathway, nonsense-mediated RNA decay (NMD), as a distinguishing feature of ASCP (40). The mutations were detected by targeted sequencing of archived tissues in 18/23 ASCPs and clustered in two regions spanning exon 10 through exon 11. Notably, we did not detect UPF1 variants in any of our flow sorted patient and PDX samples.
Currently there are chemotherapy regimens with significant clinical activity in patients with PDAC (41). However, a major clinical challenge is the emergence of chemorefractory tumors even after significant clinical responses. In contrast, ASCPs typically fail to respond and progress rapidly similar to relapsed PDACs. Our combined CNV and exome results demonstrate that ASCP genomes contain the common lesions seen in PDACs including KRAS and TP53 mutations, homozygous deletions of CDKN2A and SMAD4 and amplification of MYC. This supports previous reports and a model whereby ASCP shares a common origin with PDAC. However, our results highlight that ASCP acquire additional heterogeneous lesions including homozygous deletions, mutations and focal amplicons targeting key regulators of the epigenome. In addition, our ATAC-seq analyses of the epigenome in preclinical models identified active chromatin at the SMYD2 locus, a lysine methyltransferase with specificity for H3K4me1 as a distinguishing feature of ASCP compared to PDAC. This was associated with high expression of this active chromatin histone mark (Figure 4).
A genome-wide functional study of PDAC in the KPC mouse model identified a key role for immuno-regulatory genes in the self-renewal and maintenance of therapy resistant tumor stem cells in the pancreas (9). Notably, expression of RORC, a nuclear hormone receptor known for its role in Th17 cell specification and regulation of inflammatory cytokine production, increased with progression and its blockade via genetic or pharmacologic approaches depleted the pool of resistant cancer cells and profoundly inhibited tumor propagation in the KPC model. Our ATAC-seq data identified RORC as another distinguishing ASCP feature. Notably, IHC staining identified both cytoplasmic and nuclear RORC expression in all five PDX models and an enrichment of nuclear staining in metastatic lesions from the ASCP rapid autopsy (Figure 5, Supplemental Figure 12). This staining pattern compared to exclusive cytoplasmic expression, has been correlated with higher pathological tumor stages for PDACs at diagnosis (9).
The aggressive clinical behavior including lack of response to chemotherapy and an active RORC signature suggests that ASCPs are enriched for cell populations with cancer stem cell features. The presence of a clonal pathogenic DLCK1 mutation in the primary and metastatic lesions of a rapid autopsy, and a homozygous deletion of ASXL2 and a pathogenic PROX1 mutation in separate primary tumors provide additional support for the putative role of active pancreas cancer stem cells in ASCP (Figures 2 and 3, Supplemental Figure 5) (42).Thus, our analyses suggest that the differences between these two subtypes of pancreatic cancer may be driven through heterogeneous epigenomic lesions and cancer stem cell enrichment rather than selected ASCP-specific recurring genomic lesions.
Significantly, we also identified FGFR lesions including a focal FRS2 amplification, a FGFR2A322G activating mutation and a FGFR1-ERLIN2 fusion in ASCP samples (Figures 2 and 6, Supplemental Figure 7). Each of these lesions has been shown to activate FGFR/FRS2 signaling pathway in cancer (31,43–45). To our knowledge these have never been reported in this aggressive subtype of pancreatic cancer. Strikingly these lesions co-occur in tumors with pathogenic KRAS mutations. The co-occurrence of KRAS and FGFR activation suggests a unique ASCP-specific molecular context. The presence of KRAS activating mutations typically result in resistance to therapies targeting upstream signaling such as the EGFR (46). Nevertheless, organoids from RORC positive ASCP PDX models with a FGFR1-ERLIN2 fusion and a KRASG12V mutation were highly sensitive to single agent FGFR inhibition (Figure 6). KRAS mutations are early driver events in pancreatic cancer that can activate parallel signaling to bypass targeted inhibition of receptor tyrosine kinases. The KRAS mutant allele was present at ~60% frequency in the sorted PAX265 tumor nuclei and organoid. This is typical for flow-sorted primary and metastatic pancreatic tumors we have profiled where low level 12p copy number gain of a pathogenic driver KRAS variant co-exists with wild type alleles in the same genome (38). Single-cell sequencing confirmed the presence of the pathogenic G12V variant in 64% of sorted nuclei from the organoid. Although selective amplification of one allele resulting in allelic drop out of the other may occur in single cell analyses our results support a model whereby the fusion and the KRASG12V founder mutation co-existed in single cells. Future studies with these models, including DNA and RNA sequencing of sorted single cells and nuclei, will explore the basis of this response and investigate possible mechanisms of acquired resistance.
In summary, our data provide the basis for the development of novel approaches to treat ASCP. We propose that ASCP evolve from the same lineage as PDACs yet consist of enriched levels of RORC positive cancer stem cells, a feature that may drive other tumors with mixed adenosquamous and adenocarcinoma features. Furthermore, in addition to an epigenome that may promote stem cell features we report that a subset of ASCP has activated FGFR signaling that can be targeted with current inhibitors. Although currently limited in numbers, the availability of PDXs and organoids that recapitulate the genomic and epigenomic lesions found in patient samples provides initial preclinical models to interrogate therapeutic targets in this lethal chemoresistant cancer. Of significant interest will be clinical trials with FGFR and RORC inhibitors that include correlative studies of genomic and epigenetic lesions in both ASCP and PDAC.
Supplementary Material
Significance:
These data provide a unique description of the ASCP genomic and epigenomic landscape and identify candidate therapeutic targets for this dismal cancer.
Acknowledgments
Funding for this work was provided by the Lee Hanley Foundation and a Stand up to Cancer (SU2C) Lustgarten-Cancer Research UK (CRUK) Pancreatic Cancer Dream Team Research grant (SU2C-AACR-DT-20-16), NIH R35 CA197699, the Pancreatic Cancer Collective New Therapies Challenge, an initiative of the Lustgarten Foundation and Stand Up To Cancer, grant number SU2C-AACR-PCC-05-18, and a fellowship (to N. Rajbhandari) through the Tobacco Related Disease Research Program (TRDRP): T29FT0280. Mayo Clinic Cancer Center is supported in part by an NCI Cancer Center Support grant 5P30 CA15083-36. The UNMC Tissue Bank Rapid Autopsy Program for Pancreas is supported by the following: SPORE in 19 Pancreatic Cancer, P50CA127297; Pancreatic Cancer Detection Consortium, U01CA210240; NCI Cancer Center Support Grant, P30CA36727; NCI Research Specialist, 5R50CA211462. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The abovementioned SU2C-supported research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. We thank Dr. Xianfeng Chen Department of Health Sciences Research & Center for Individualized Medicine, Mayo Clinic Arizona for assistance in figure preparation. R.M. Evans is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute and is supported by the NIH (DK057978, HL105278, ES010337), the Cancer Center (CA014195), a NOMIS Foundation Distinguished Scientist and Scholar Award. This work was funded by grants from the Lustgarten Foundation, the Don and Lorraine Freeberg Foundation, Ipsen Bioscience, and a Stand Up To Cancer-Cancer Research UK-Lustgarten Foundation Pancreatic Cancer Dream Team Research Grant (Grant Number: SU2C-AACR-DT-20-16). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. C.E. Antal was supported by the Damon Runyon Cancer Research Foundation (DRG-2244-16) as a Robert Black Fellow.
Footnotes
The authors declare no conflicts of interest
References
- 1.Society AC. Cancer Facts & Figures 2020. American Cancer Society 2020
- 2.Borazanci E, Millis SZ, Korn R, Han H, Whatcott CJ, Gatalica Z, et al. Adenosquamous carcinoma of the pancreas: Molecular characterization of 23 patients along with a literature review. World J Gastrointest Oncol 2015;7:132–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020;76:182–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyd CA, Benarroch-Gampel J, Sheffield KM, Cooksley CD, Riall TS. 415 patients with adenosquamous carcinoma of the pancreas: a population-based analysis of prognosis and survival. J Surg Res 2012;174:12–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hester CA, Augustine MM, Choti MA, Mansour JC, Minter RM, Polanco PM, et al. Comparative outcomes of adenosquamous carcinoma of the pancreas: An analysis of the National Cancer Database. J Surg Oncol 2018;118:21–30 [DOI] [PubMed] [Google Scholar]
- 6.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52 [DOI] [PubMed] [Google Scholar]
- 7.Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019;35:267–82 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lytle NK, Ferguson LP, Rajbhandari N, Gilroy K, Fox RG, Deshpande A, et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019;177:572–86 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holley T, Lenkiewicz E, Evers L, Tembe W, Ruiz C, Gsponer JR, et al. Deep clonal profiling of formalin fixed paraffin embedded clinical samples. PLoS One 2012;7:e50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett MT, Anderson KS, Lenkiewicz E, Andreozzi M, Cunliffe HE, Klassen CL, et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015;6:26483–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett’s esophagus III: baseline flow cytometric variables. Am J Gastroenterol 2001;96:3071–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett MT, Deiotte R, Lenkiewicz E, Malasi S, Holley T, Evers L, et al. Clinical study of genomic drivers in pancreatic ductal adenocarcinoma. Br J Cancer 2017;117:572–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz C, Lenkiewicz E, Evers L, Holley T, Robeson A, Kiefer J, et al. Advancing a clinically relevant perspective of the clonal nature of cancer. Proc Natl Acad Sci U S A 2011;108:12054–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipson D, Aumann Y, Ben-Dor A, Linial N, Yakhini Z. Efficient calculation of interval scores for DNA copy number data analysis. J Comput Biol 2006;13:215–28 [DOI] [PubMed] [Google Scholar]
- 16.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al] 2013;44:15 4 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature methods 2017;14:959–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan H, Evans J, Kalmbach M, Moore R, Middha S, Luban S, et al. HiChIP: a high-throughput pipeline for integrative analysis of ChIP-Seq data. BMC Bioinformatics 2014;15:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481:389–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo XY, Srivastava A, Graber JH, Yadav V, Sarsani VK, Simons A, et al. Genomic data analysis workflows for tumors from patient-derived xenografts (PDXs): challenges and guidelines. BMC medical genomics 2019;12:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A 2001;98:6074–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013;45:478–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine & growth factor reviews 2015;26:425–49 [DOI] [PubMed] [Google Scholar]
- 29.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47:D941–D7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 2003;3:117–30 [DOI] [PubMed] [Google Scholar]
- 31.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014;10:e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazzaferro V, El-Rayes BF, Droz Dit Busset M, Cotsoglou C, Harris WP, Damjanov N, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer 2019;120:165–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weaver BA, Cleveland DW. The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 2008;14:431–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merlo LM, Wang LS, Pepper JW, Rabinovitch PS, Maley CC. Polyploidy, aneuploidy and the evolution of cancer. Adv Exp Med Biol 2010;676:1–13 [DOI] [PubMed] [Google Scholar]
- 37.Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet 1999;22:106–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrett MT, Deiotte R, Lenkiewicz E, Malasi S, Holley T, Evers L, et al. Clinical study of genomic drivers in pancreatic ductal adenocarcinoma. Br J Cancer 2017 [DOI] [PMC free article] [PubMed]
- 39.Fang Y, Su Z, Xie J, Xue R, Ma Q, Li Y, et al. Genomic signatures of pancreatic adenosquamous carcinoma (PASC). J Pathol 2017;243:155–9 [DOI] [PubMed] [Google Scholar]
- 40.Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 2014;20:596–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jameson GS, Borazanci E, Babiker HM, Poplin E, Niewiarowska AA, Gordon MS, et al. Response Rate Following Albumin-Bound Paclitaxel Plus Gemcitabine Plus Cisplatin Treatment Among Patients With Advanced Pancreatic Cancer: A Phase 1b/2 Pilot Clinical Trial. JAMA Oncol 2019 [DOI] [PMC free article] [PubMed]
- 42.Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014;146:245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang K, Chu K, Wu X, Gao H, Wang J, Yuan YC, et al. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res 2013;73:1298–307 [DOI] [PubMed] [Google Scholar]
- 44.Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A 2001;98:8578–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo LY, Kim E, Cheung HW, Weir BA, Dunn GP, Shen RR, et al. The Tyrosine Kinase Adaptor Protein FRS2 Is Oncogenic and Amplified in High-Grade Serous Ovarian Cancer. Mol Cancer Res 2015;13:502–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.