

Abstract
We report here on the salient role of protein mobility in accessing conformational landscapes that enable efficient enzyme catalysis. We are focused on yeast enolase, a highly conserved lyase with a TIM barrel domain and catalytic loop, as part of a larger study of an expected correlation of protein motions with chemical reactivity within superfamilies. Enthalpically-hindered variants were developed by replacement of a conserved hydrophobic side chain (Leu 343) with smaller side chains. Leu343 is proximal to the active site base in enolase, and comparative pH rate profiles for the valine and alanine variants indicate a role for side chain hydrophobicity in tuning the pKa of the catalytic base. However, the magnitude of a substrate deuterium isotope effect is almost identical for wild type (WT) and Leu343Ala, supporting an unchanged rate-determining proton abstraction step. The introduced hydrophobic side chains at position 343 lead to a discontinuous break in both activity and activation energy as a function of side chain volume. Hydrogen-deuterium exchange mass spectrometry (HDX-MS) experiments were performed as a function of time and temperature for WT and Leu343Ala, and provide a spatially resolved map of changes in protein flexibility following mutation. Impacts on protein flexibility are localized to specific networks that arise at the protein-solvent interface and terminate in a loop that has been shown by X-ray crystallography to close over the active site. These interrelated effects are discussed in the context of long-range, solvent-accessible and thermally activated networks that play key roles in tuning the precise distances and interactions among reactants.
Graphical Abstract
Introduction.
Over millennia proteins have developed complex structures that sequester catalytic sites and achieve rate enhancements of up to 1026 over uncatalyzed reactions.1 Extensive studies focused on enzyme active sites have been unable to fully explain this rate acceleration, and de novo designed enzymes cannot match their native counterparts.2–4 There is a growing recognition of the role of the macromolecular structure of the protein as a flexible and dynamic entity in the evolution of optimal enzyme function. Experimental evidence has shown that protein motions occur across a hierarchy of timescales, and it is hypothesized that enzymes select for an amalgamation of conformational substates, known as the conformational landscape, that facilitates catalysis.5–9 Historically, research into structure-function relationships has primarily been focused on the static structures of dominant, stable conformers, providing less useful information regarding a protein’s intrinsic flexibility that could be the source of functionally related dynamical fluctuations. Contemporary scientific efforts are increasingly focused on the development of biophysical methods to examine and quantify protein motions during all stages of enzyme reactivity, with the ultimate goal of linking such behavior to the evolution of substrate specificity, chemical reactivity and catalytic rate enhancement.6,9–16 In our laboratory, we have been focused on the application of hydrogen-deuterium exchange mass spectrometry (HDX-MS) which, when paired with the interrogation of activity-impairing site-specific mutants, elucidates relationships between changes in protein flexibility and chemical catalysis.17–19 Herein, we apply this framework for uncovering protein motions that are catalytically relevant to the enzyme enolase from Saccharomyces cerevisiae, a model TIM barrel system that has been shown to undergo a functionally important loop closure upon substrate binding (Figure 1).20
Figure 1.
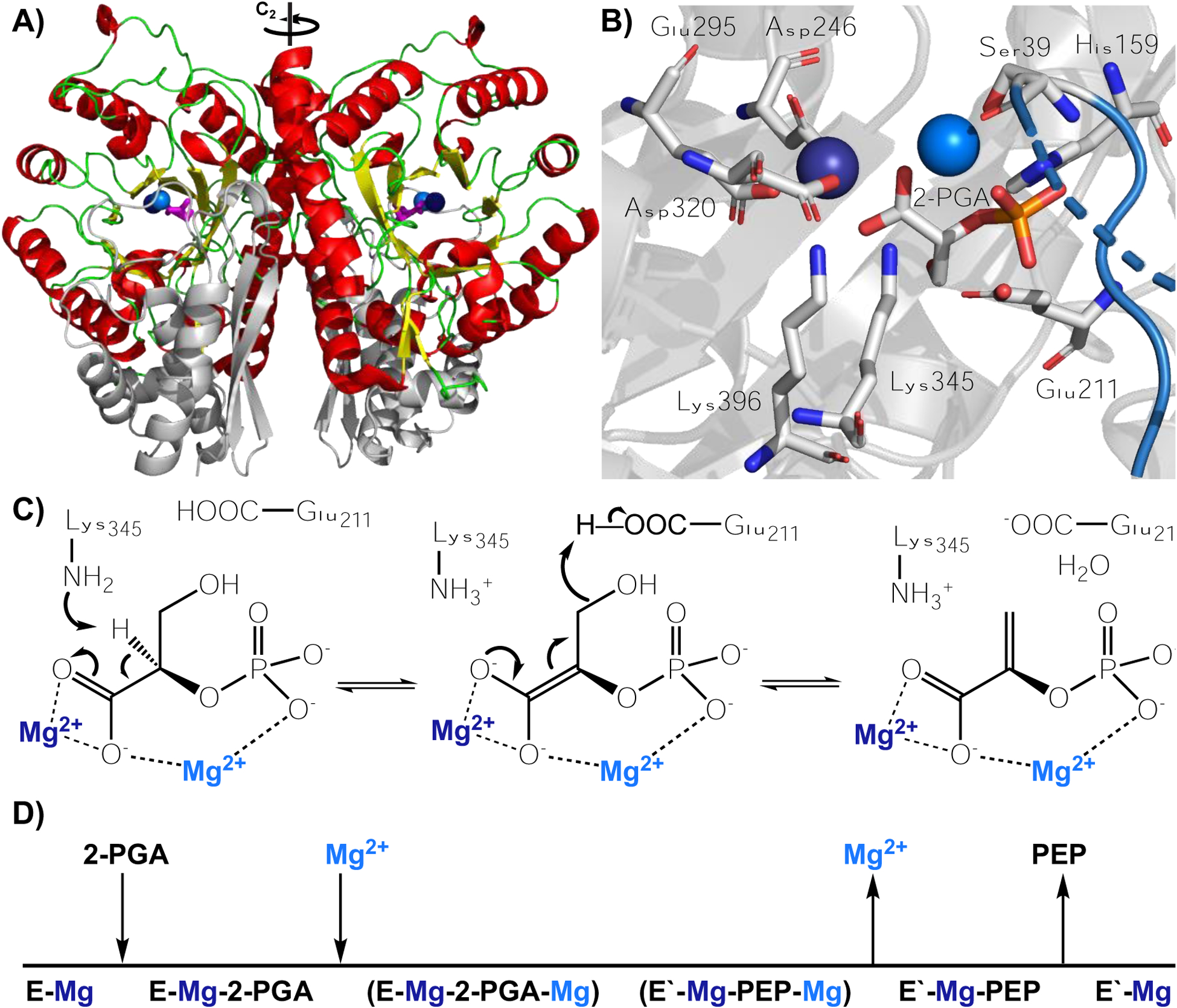
A) Homodimer of yeast enolase with helices, loops, sheets, and N-terminal domain indicated in red, green, yellow, and light grey, respectively. Substrate is magenta and the two Mg2+ ions, Mg(1) and Mg(2) are denoted as dark and light blue spheres; C2 rotation axis is indicated. B) Active site of Saccharomyces cerevisiae enolase with substrate (2-PGA), catalytic acid (Glu211), catalytic base (Lys345), metal binding residues (Asp320, Glu295, Asp246, Ser39), and 2-PGA binding residues (Lys396, His159) labeled.33 L1 is shown in blue; atoms uniquely colored (carbon is grey, oxygen is red, nitrogen is blue, and phosphorus is orange). C) Mechanism of reaction with magnesium ions colored dark and light blue to match Figure 1B. D) Order of binding. Mg(1) (dark blue) is initially bound and Mg(2) (light blue) binds in the process of substrate binding and loop closure.34 PDB 1ONE is a crystal grown with an equilibrium mixture of substrate and product.26
To date, there are more than 180,000 predicted members of the enolase superfamily, which is divided into seven subgroups based on the acid-base residues involved in catalysis.21–24 These enzymes are comprised of a C-terminal TIM barrel domain and an N-terminal α+ß capping domain. The barrel is responsible for acid-base chemistry and initial metal binding; the N-terminal capping domain is critical for substrate specificity and contains a second metal binding site. The primary reaction catalyzed is the abstraction of an α-proton from a carboxylate substrate, with the incipient anionic intermediate undergoing stabilization by a divalent metal cation (usually Mg2+).21 As the defining member of its superfamily, enolase is particularly well-studied, with research documented in the 1940s22 and continuing to the present time.23–25 The enzyme is part of the glycolytic pathway, responsible for converting 2-phospho-D-glycerate (2-PGA) to phosphoenol pyruvate (PEP), and is found across all domains of life. The reaction catalyzed by enolase is reversible and an X-ray structure has been solved for enolase in the presence of an equilibrium mixture of substrate and product.26 Figure 1 shows the structure of the S. cerevisiae enolase in complex with substrate, together with the proposed reaction mechanism.27–30 Enolase undergoes a conformational change upon substrate binding, where three loops contract on the front side of the barrel (Figure 2).26 Upon the closure of L1, a loop comprised of residues 37–42, Ser39 coordinates a second Mg2+ ion. Lys345 abstracts a proton from the C2 carbon of 2-PGA, forming an enolate intermediate which is stabilized by the two Mg2+ ions.26,31 Loss of the hydroxyl group at C3 of substrate is facilitated by a general acid, Glu211. At pH 7.5 the initial abstraction of the C2 proton is at least partially rate limiting under conditions of substrate saturation according to Figure 1C.27,32
Figure 2.
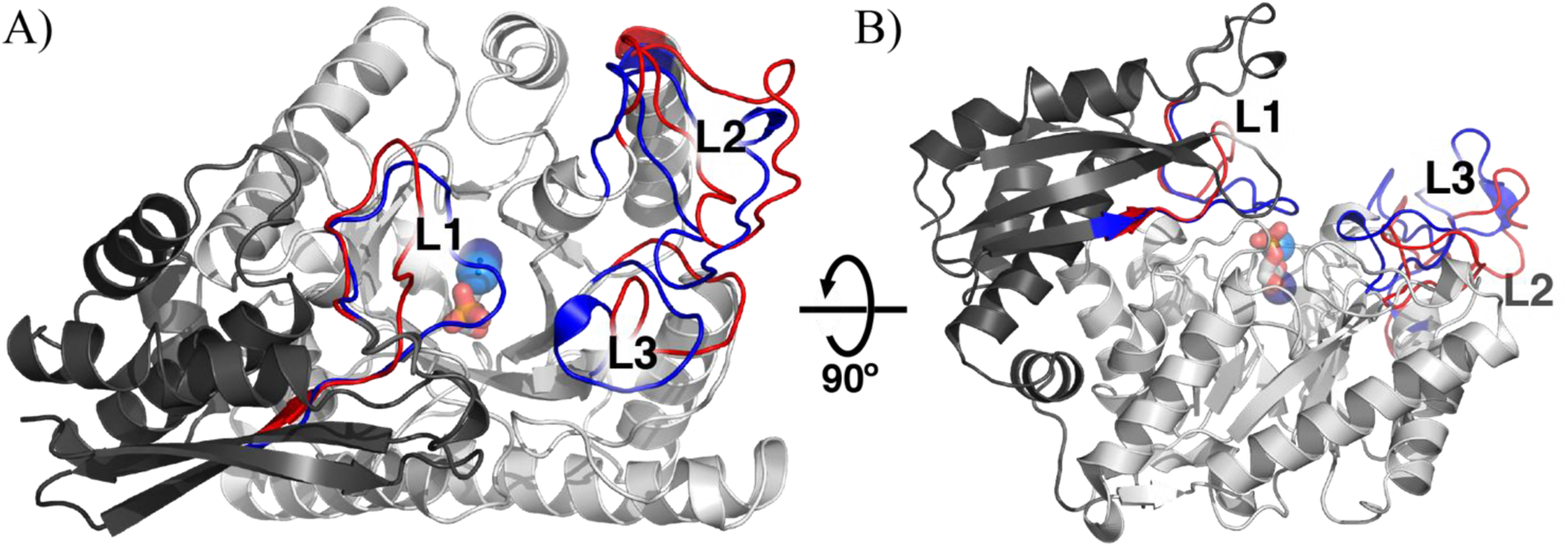
A single subunit of yeast enolase A) looking into the TIM barrel and B) rotated 90°, with open and closed conformational changes greater than 3 Å shown in red (open) and blue (closed), respectively. The N-terminal domain is colored dark grey. The C-terminal TIM barrel domain is colored light grey. Loops are labeled L1, L2, and L3 for residues 37–42, 156–165, and 252–276, respectively. From the closed conformation, the two Mg2+ ions are shown in dark and light blue and 2-PGA is shown as a stick colored by element (carbon is grey, oxygen is red, phosphorus is orange). Regions undergoing a change were identified using the Pymol script ColorByRMSD.36 PDB 1EBH (open) and 1ONE (closed).
The open structure has a single Mg2+ ion bound, while the closed structure has two Mg2+ ions and a substrate/product equilibrium mixture bound (PDB 1EBH and 1ONE, respectively). In addition to the closure of L1 over the active site concomitant with the binding of the second magnesium, closure of L2 enables His159 to coordinate the substrate. To understand the structural ramifications of these events, we compare the crystal structures of the open and closed structures (Figure 2). L1 (residues 37–42), that binds the second Mg2+ undergoes the largest shift, ca. 11 Å shift from the open to closed form (Table S4). The substrate binding loop (L2) undergoes a 6.5 Å shift and an additional third loop (L3), which is adjacent to L2, undergoes a 6.1 Å shift. Both structures were crystallized under nearly identical conditions.26,35
Hydrogen-deuterium exchange (HDX) is uniquely suited to provide a structure-based approach for our understanding of protein motions and flexibility.18,37 It measures the rate and extent of exchange of backbone amide protons with deuterons when a protein is solvated in D2O. This process has alternately been monitored either by NMR, in which it is necessary to first label a protein and resolve individual chemical shifts for each backbone amide, or by mass spectrometry, where the protein of interest is first fragmented into peptides that are subsequently analyzed by mass spectrometry (HDX-MS). The latter approach has been shown to be capable of resolving spatial patterns of protein flexibility in a relatively rapid manner under conditions that can mimic catalytic turnover.38 HDX is governed by a two-step process, where first the protein samples a distribution of open and closed states and protons undergo exchange based on their level of protection, a factor unique to the amide identity and environment:
| (1) |
Where kop, kcl, and kint are the rates of opening, closing, and intrinsic exchange, respectively. In the EX2 kinetic regime a transient, solvent-exposed open state of the protein returns to its closed, protected state much faster than the intrinsic chemical exchange step (kcl >> kint). This leads to a measured rate constant, kHDX, that is the product of kint and an equilibrium constant for the interconversion of the protein between locally open and closed states, kop:39
| (2) |
| (3) |
Within EX2 conditions the sensitivity of kHDX to differences arising from protein perturbation can be directly related to changes in local flexibility which are reflected in Kop. Many constant temperature studies have harnessed HDX-MS to understand protein-ligand or protein-protein interactions.40–43 Temperature-dependent HDX-MS is much less common due to the increased data collection and processing, but recent studies are employing temperature dependent HDX-MS to answer physiologically relevant questions and to understand the relationship of local and global dynamics in temperature dependent unfolding and melting.17,19,44–52 Through both constant and multi-temperature studies, it is increasingly clear that HDX-MS offers a powerful tool for interrogating the link between protein flexibility and catalytic function.19,38,43 As recently illustrated in studies of soybean lipoxygenase, human catechol O-methyltransferase and murine adenosine deaminase, the combination of temperature-dependent EX2 HDX-MS and site-specific mutagenesis makes it possible to uncover unique thermal networks that play an integral role in catalytic turnover.17–19 TIM barrel domains are estimated to be present in 10% of all enzymes and, despite being characterized by a highly conserved three-dimensional structure, catalyze a wide range of chemical reactions across five Enzyme Commission classes.53,54 This makes them ideal candidates for detecting regionally specific protein motions that have emerged in the course of catalytic adaptation.55 The present work is part of a series of studies17 aimed at understanding the manner in which TIM barrel enzymes may display unique dynamical properties that are specific to the reaction catalyzed.21,53,56
In this study we use temperature dependence as a sieve to sort the catalytically relevant motions from the background protein breathing. By focusing the study on variants with altered enthalpic barriers to catalysis, we can relate the enthalpically gated motions to catalysis.17–19 We began our investigation with Leu343, a large hydrophobic sidechain in the active site. We characterize changes in the rate and temperature dependence arising from a series of mutations at this site. This is followed by a mechanistic study and a comparative investigation of a representative variant to WT using time- and temperature-dependent HDX-MS experiments. Extensive visual and quantitative analyses reveal the nature of shifts in temperature-dependent dynamic motions associated with catalytic impairment. As shown herein, mutation at position 343 dramatically increases the flexibility of L1 and surrounding regions that connect L1 to the protein surface. We propose that the increased flexibility of L1 is detrimental to catalysis because it is directly coordinated to the active site via Ser39 and the second Mg2+, where the latter coordinates to substrate: increased flexibility is expected to affect the sampling of substates that are responsible for the correct positioning of metal ions, catalytic sidechains, and substrate as a prerequisite to efficient catalysis. The identified regions around L1 both support the hypothesis that the loop closure enables a thermal activation of catalysis and identify protein networks that reach from the protein/solvent interface to the active site. These networks are proposed to function as site-specific conduits for heat transfer from solvent to the reaction coordinate(s).
Results and Discussion.
Kinetic analyses of site selected mutants at position 343 in enolase.
Leu343 is part of a highly conserved motif in eukaryotic enolases, Leu-Leu-Leu-Lys, where the Lys345 is the catalytic base and Leu342 and Leu344 point away from the active site. In prokaryotic enolases Leu is the most prevalent residue at position 343, though Ile and Val are also found in some of these sequences. Leu343 was identified as one of the only hydrophobic residues in the active site of the yeast enolase whose side chain is directed toward the active site cavity. Leu343 is within van der Waals distance of key catalytic residues that include the catalytic base (Lys345), metal binding residues (Asp320, Glu295), and a substrate binding residue (Lys396) and as such, provides a likely location for mutagenesis to perturb active site geometries without introducing significant change to the catalytic mechanism (Figure 3). We generated and characterized four variants of yeast enolase: Leu343Ile, Leu343Val, Leu343Ala, and Leu343Gly.
Figure 3.
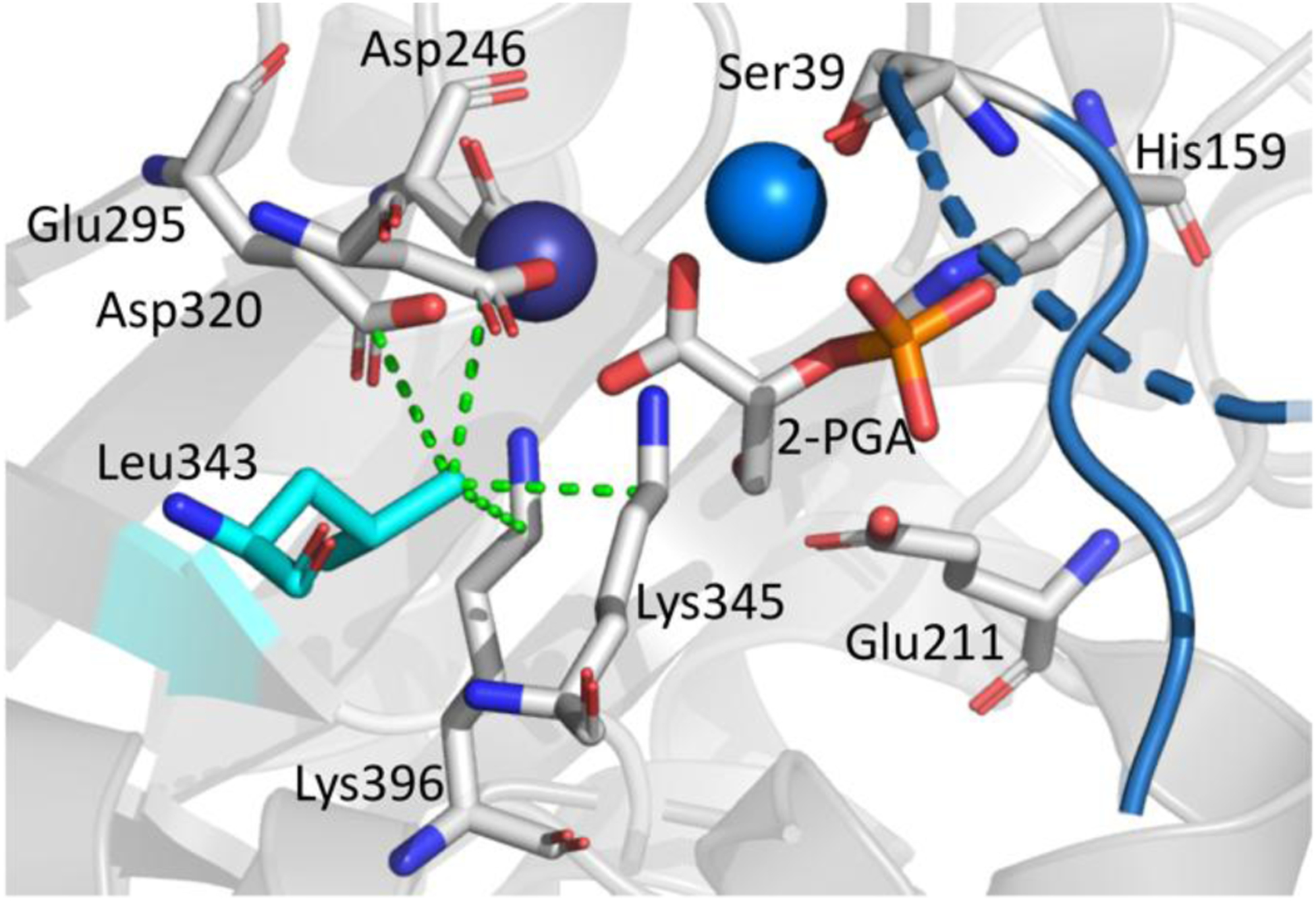
Active site geometry of enolase with bound substrate, 2-PGA (center). Residues are labeled. The dashed green lines indicate van der Waals contacts for Leu343 with the metal liganding residues, Asp320 and Glu295, the substrate binding residue, Lys396, and the catalytic base, Lys345. The catalytic acid, Glu211, the substrate binding residue, His159, and additional Mg2+ coordinating residues are also indicated. Mg2+ ions are shown as spheres. PDB 1ONE.
A discontinuous trend in catalytic behavior with Leu343 variants was detected, in which change to the shape or size of residue 343 leads to a break in both kcat and the Ea(kcat), the activation energy required for catalysis (Table 1, Figure 4). A significant increase in Ea(kcat) with Leu343Ile, from 5.7 to 10.6 kcal mol−1 is observed, followed by a progressive diminishment as the size of the hydrophobic substitution becomes smaller; in the case of Leu343Gly, the Ea(kcat) is close to that of WT. This is not accompanied by a recovery of WT activity, indicating multiple effects that alter the rate of enzyme turnover versus the enthalpic barrier for catalysis (Figure 4). The increase of the activation energy for Leu343Ile by almost 5 kcal mol−1 is a surprising jump as the only distinction from WT is the relocation of a single methyl group to an adjacent carbon atom. Modeling of Ile at position 343 reveals that the primary carbon chain in Ile flips down and away from the substrate and metal binding residues, increasing the distances between residue 343 and the key metal binding residues Asp320 and Glu295, the substrate binding residue Lys396, and the catalytic base, Lys345 (Figure S1). The longer chain on the isoleucine disrupts hydrophobic packing, which further disturbs the location of two of the metal binding residues. This decrease in optimal packing likely leads distortion within the active site, requiring more energy imput to reach the correct geometry for reaction to occur.
Table 1.
Kinetics Parameters of Enolase and Mutantsa
| Enzyme | kcat (s−1) | KM (μM) | kcat/KM (μM−1 s−1) | Ea(kcat) (kcal mol−1)b | KIE |
|---|---|---|---|---|---|
| WT | 82 (5) | 17 (3) | 3.5 | 5.7 (0.3) | 2.04 (0.13) |
| Leu343Ile | 26 (1) | 23 (5) | 1.1 | 10.6 (0.5) | n.d. |
| Leu343Val | 14.4 (0.2) | 20 (5) | 0.72 | 8.6 (0.2) | n.d. |
| Leu343Ala | 0.127 (0.005) | 8 (1) | 0.016 | 9.3 (0.3) | 1.97 (0.07) |
| Leu343Gly | 0.188 (0.002) | 23 (5) | 0.0082 | 6.4 (0.3) | n.d. |
Initial rates for conversion of 2-PGA to PEP were measured using a continuous spectroscopic assay, in which the formation of PEP is monitored at 240 nm.57 All experiments were carried out in 50 mM HEPES, 2 mM MgCl2 at pH 7.5. The number in parentheses is the standard error. n.d. is not determined. Kinetic parameters kcat, KIE, and KM are calculated from experiments at 25°C and are an average of at least three experiments.
Figure 4.
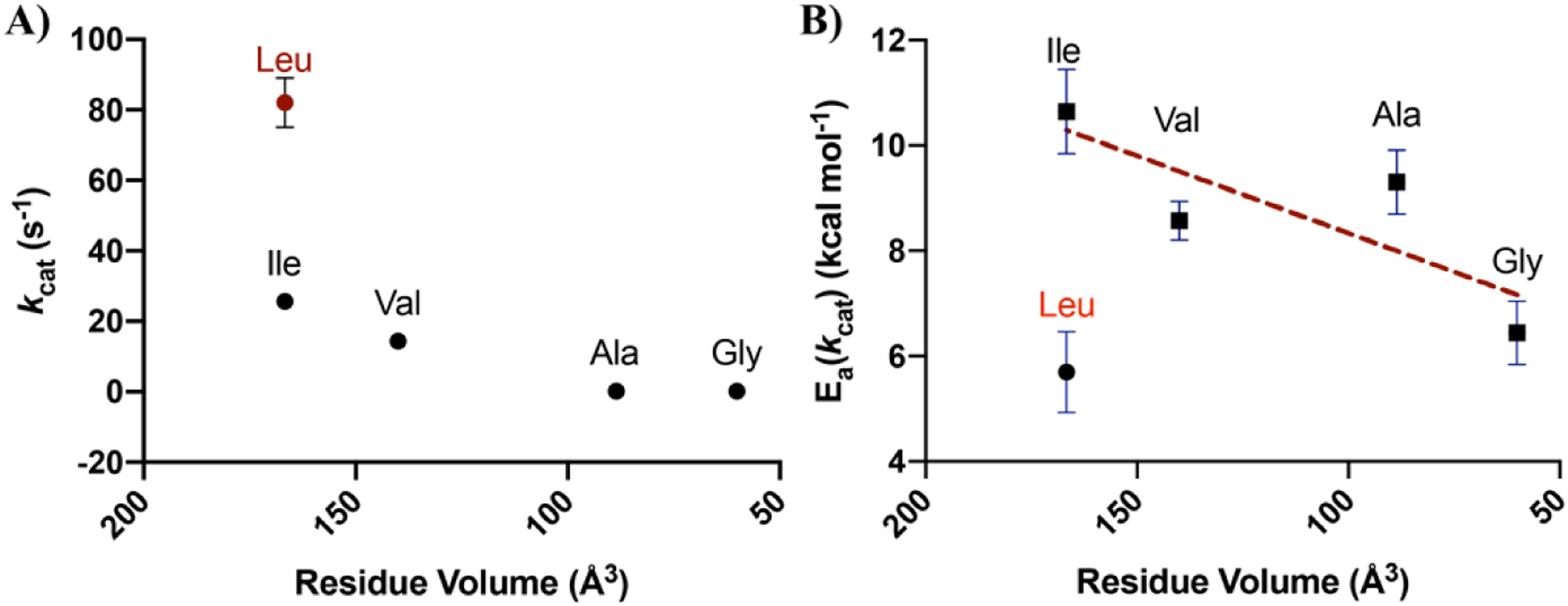
A) kcat, and B) Ea(kcat) plotted as a function of the volume of the mutated residue at Leu343. The native enzyme (Leu343) is noted in red. The x-axes go from large to small values. Error bars indicate the standard deviation. The dashed line indicate the trend in Ea(kcat) for variants at position 343. Plots of ΔH‡ vs. Residue Volume show the same trend (Figure S11).
We especially noted the lack of a one-to-one correspondence between changes in rate and Ea(kcat) in the case of the alanine and glycine substitutions, with unusually large impacts on kcat for these two substitutions while glycine shows an Ea approaching the WT. In order to better understand these trends, we undertook a study of both the pH profiles and substrate deuterium isotope effects in selected mutants (Figure 5). The two key mechanistic questions regarding mutants at Leu343 that we sought to determine were first, whether mutation at position 343 would influence the pKa of the adjacent base, Lys345, and second, whether a change in rate-determining step may have occurred for the alanine and glycine substitutions. The pH rate profiles for WT, Leu343Val and Leu343Ala are quite informative with regard to the first question. In the case of WT and Ile343Val, a bell-shaped profile is observed and the curves for WT and Leu343Val appear similar. Given the catalytic roles postulated for Glu211 and Lys345, the most straightforward interpretation of the ascending limb is deprotonation of GluH 211, with the descending limb reflecting LysH+ 345 ionization. At the plateau these species are expected to undergo a proton swap prior to catalysis, generating the neutral catalytic species, GluH and Lys, that can function as a general acid and base, respectively.58 Significantly, the Leu343Ala variant indicates a broader pH optimum, with a shift in the ascending and descending limbs to more acidic and basic behavior. This behavior of Leu343Ala can be ascribed to a decrease in hydrophobicity within this region upon mutation, possibly accompanied by an incursion of water. The impact of such behavior would be to reduce the concentration of the catalytically active species GluH and Lys, in equilibrium with the dominant active site species Glu- and LysH+, thereby accounting in part for the large fall off in rate relative to Leu343Val.
Figure 5.
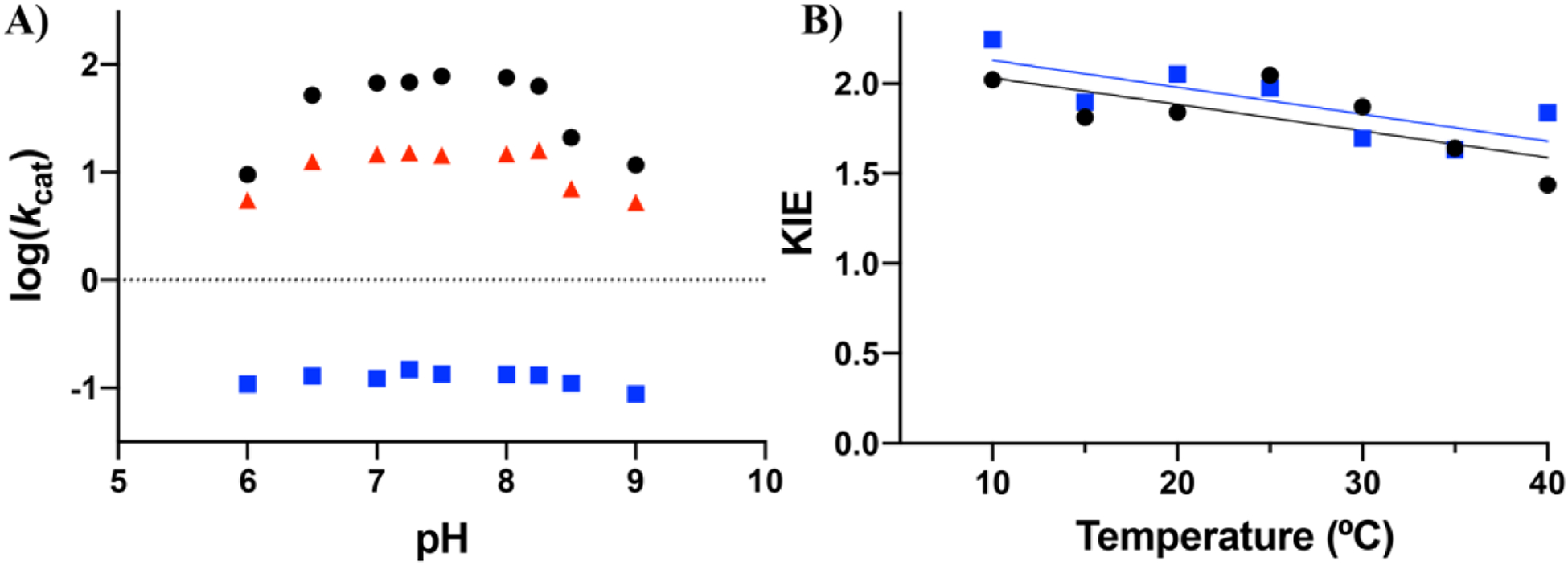
A) pH dependence of the catalytic rate for WT, Leu343Val, and Leu343Ala collected under conditions of saturating substrate (250 μM) where kcat approximates Vmax for WT and the variants at 25 °C. Black, blue, and red symbols represent WT, Leu343Ala, and Leu343Val respectively. B) Kinetic isotope effect of WT (black) and Leu343Ala (blue) at pH 7.5 across 10–40 °C. All experiments were carried out in 50mM HEPES, 2mM MgCl2. Error bars indicate the standard error for both plots; where error bars are not shown, they are smaller than the symbols.
The above interpretation suggests that the reaction mechanism might also be impacted in the case of the alanine and glycine replacements. Based on the substantial enolase mechanism literature, we chose kinetic isotope effect experiments to examine whether the proton abstraction step is partially rate-limiting to the sam extent for WT and Leu343Ala27,32,59 The enolase mechanism is dependent on pH, temperature, and metal concentration, so particular care was taken in choosing experimental conditions that were already in the literature (Ref31, see SI for details) as well as being optimal for the HDX-MS studies described below. Following the synthesis of deuterated substrate, 2H-PGA, according to literature protocols,28 the magnitude and temperature dependence of the deuterium kinetic isotope effect on kcat was compared for Leu343Ala to WT (Figure 5B). As shown, C-H bond cleavage remains at least partially rate determining to a similar extent in Leu343Ala at all temperatures interrogated. Overall these mechanistic interrogations support a conclusion that the large reductions in rate for Leu343Ala and Leu343Gly are likely due to a reduction in the concentration of the catalytically active, neutral GluH and Lys pair, while the reaction mechanism of WT is maintained.
The impact of mutations at position 343 on the rate and activation energy can be rationalized in part from X-ray structures of the closed enzyme form in which both a substrate/product mixture and the second metal are bound. As shown in Figure 3, the methyl group on leucine is tightly packed between the catalytic base Lys345, a substrate binding residue, Lys396, and two of the ligands to the tightly bound magnesium, Asp320 and Glu295. We note that Leu343 is not located along the principal reaction coordinate (e.g., behind the active site base) and, as such, may not have been expected to exert a large impact on catalytic rate. However, Leu343 is expected to be important in achieving precise alignments of both the catalytic base Lys345 and substrate, for the initial proton abstraction from the substrate α-carbon (Figure 3). Extensive studies of C-H activation in enzyme reactions have shown a dominance of hydrogen tunneling between the donor and acceptor atoms that is critically linked to the distance between these atoms as well as the environmental reorganization term that transiently creates energetic degeneracy between reactant and product wells as a prerequisite for wave function overlap.60–63 Such effects are expected to be operative in yeast enolase where mutational disruption of the donor acceptor distance and positioning is expected to alter the dynamical tuning and electrostatic properties of the active site.
Temperature-dependent hydrogen deuterium exchange-mass spectrometry (HDX-MS) to assess mutational alterations in protein flexibility.
HDX-MS enables peptide-level resolution of changes in a protein’s flexibility in response to the protein’s alteration via mutagenesis, allosteric regulation, and complexation to ligands and/or other proteins.37,64 As has been developed in this laboratory, temperature-dependent HDX-MS can be used to obtain the impact of similar mutations on the enthalpic barrier for regional HDX, which can then be compared to the impact of mutation on the Ea(kcat).19,65 This approach is a powerful tool for the detection of protein networks that may facilitate the transfer of thermal energy from solvent to the active site.17–19 Herein we harness this technique to investigate the impact of mutation at Leu343 on thermally activated protein motions within the native S. cerevisiae enolase. The variant Leu343Ala was chosen for in-depth HDX-MS studies, based on the evidence for a rate-limiting proton abstraction step similar to WT (Figure 5), and the observed increase in its energy of activation by 3.6 kcal mol−1 (Table 1).
HDX experiments were conducted in the presence of Mg2+ but in the absence of substrate, to enable the observation of conformational substates that are inherent to the enzyme structure, while avoiding complications that would arise from substrate binding and reactivity. The EX2 regime was maintained, as confirmed by the patterns in the mass spectrometric analyses,37,39,66 and analysis was carried out using HDX Workbench (Figure S3).67 Throughout the course of these studies, peptide retention times did not vary by more than ±0.1 min (Figure S4). An average of 180 unique peptides resulted from the digestion of both the native enzyme and Leu343Ala. A set of 31 non-overlapping peptides was chosen for the analysis herein, with an average peptide length of 12.6 measurable amides, which reports on 88% of the protein sequence (details on the choice of these sample peptides are given in the SI and Figure S5). HDX-MS results are corrected for back exchange based on peptide-specific back exchange controls (discussed extensively in the SI, Table S2). Fifteen time points (0s – 4 h) were collected at five separate temperatures (10 – 40 °C). The data represent the average of at least two replicates and each dataset was collected in a random order over three days to account for day-to-day variability. Additional details are in the SI and are consistent with those recommended by the international HDX-MS community.68
Peptides are classified based on the HDX uptake plots, which show distinct patterns of exchange as well as the extents of deuteration out to the longest time point. While all amides are assumed to undergo temperature dependent exchange, some peptides do not exhibit such exchange within our experimental conditions and time points collected. As defined in previous works,17,18 peptides that do not show temperature dependent exchange of any amplitude in our experimental protocol are classified as Type I. Peptides that display temperature-dependent exchange in the experimental time and temperature regimes are classified as Type II.
In yeast enolase approximately 6% of the protein, 4 of 31 peptides, displays Type I behavior, exhibiting little to no temperature dependence of HDX within the experimental timescale (e.g., Figure 6A). The remaining 26 peptides, approximately 81%, show a strong dependence of HDX on temperature, Type II, (e.g., Figure 6B, C). Mapping the type II regions (white) and the type I regions (violet), the vast majority of the enzyme is seen to display temperature-dependent exchange (Figure 6D). Comparison of the temperature-dependent HDX for the Leu343Ala variant to the native enzyme shows that the temperature-dependent and independent regions remain the same, but that the magnitude of the temperature dependence is diminished in Leu343Ala.
Figure 6.
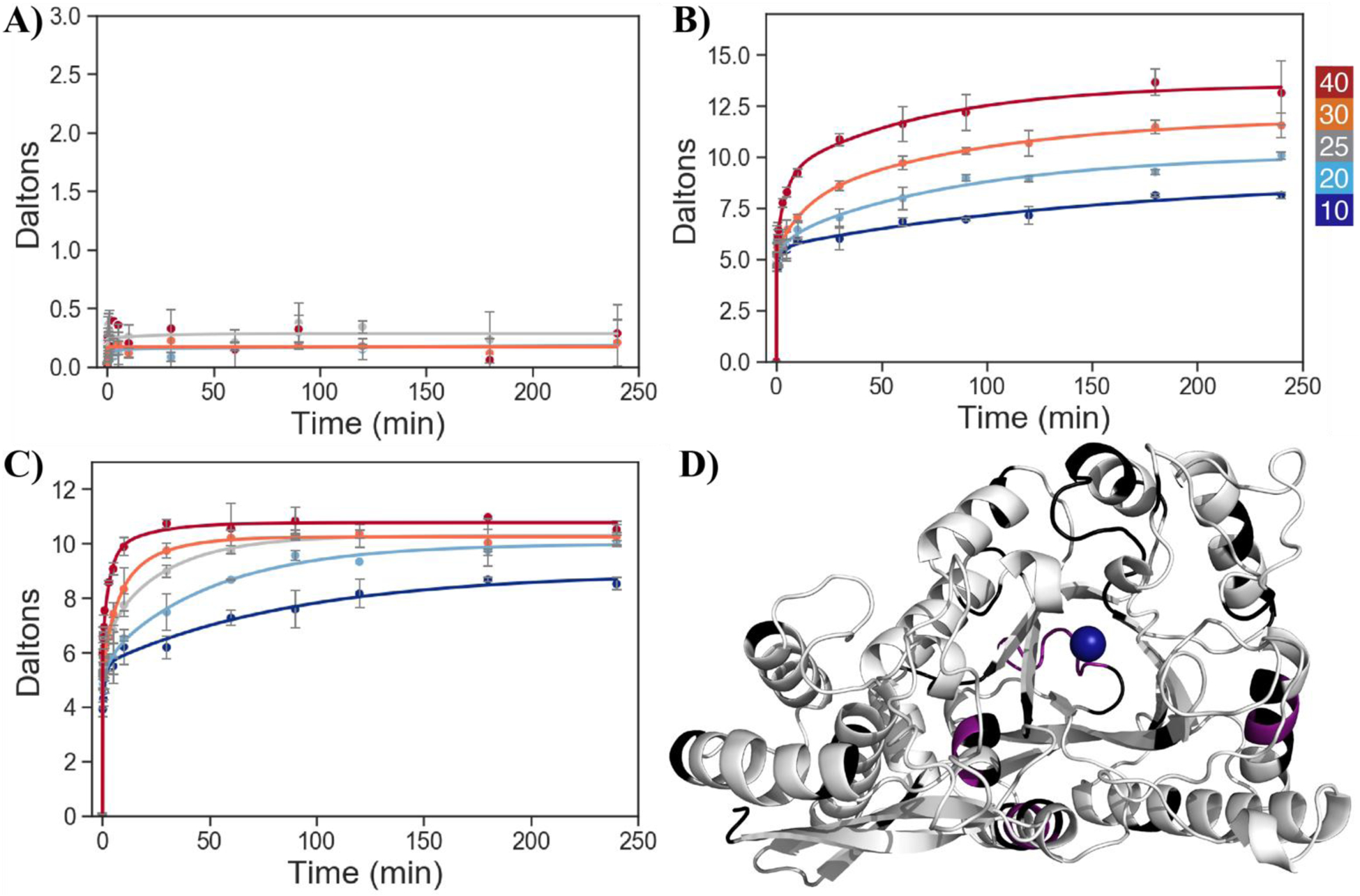
Representative HDX uptake plots showing the time dependence of peptide-specific deuteration at different temperatures for WT peptides in the open structure; color scale for temperature at top right. A) 224–228, Type I, which shows temperature-independent exchange in the HDX experimental timescale, B) 263–279, Type II, where the different temperatures approach a common plateau, and C) 94–112, Type II, where HDX traces do not converge even at the longest times and high temperature. D) Crystal structure of holo-enolase with Type I, Type II, and non-reporting regions shown in violet, white, and black, respectively. The tightly bound magnesium ion is shown as a blue sphere. Yeast enolase is a homodimer with a C2 axis of symmetry; for the orientation of the monomer above, the dimer interface is horizontally located along the very bottom of the subunit. PDB 1EBH.
In order to proceed to a quantitative assessment of the impact of Leu343Ala on the temperature dependence of HDX for enolase, a rate constant for HDX is written in terms of the traditionally defined time regimes of exchange,42 kfast, kmed, and kslow, according to Eq 3:
| (4) |
where A, B, and C are the amplitudes for each regime of exchange, and Ntot and NNE represent the total number of amides and the number of non-exchanging amides, respectively. The time restraints of these handheld HDX experiments do not permit accurate measurement of kfast; therefore, we limited our rate analysis to the measurable regime that includes kmed and kslow. As presented by Gao et al.,17 we employed a weighted average rate constant, kHDX, to report on the general trends of amide exchange according to:
| (5) |
Type II peptides were analyzed via Arrhenius plots to generate the respective values for Ea(HDX) = ΔH‡(HDX) + RT (where R is the gas constant and T is temperature) representing the temperature dependence of the rates of amide proton exchange for WT and Leu343Ala (Dataset SI). These reveal intricacies in the data that are not apparent from deuterium uptake plots at constant temperatures, reflecting the sum of the temperature dependences of kint (ΔH‡) and Kop (ΔH°) components. Since kint is expected to be unaffected by a single site mutation, with the possible exception of the peptide containing Leu343Ala, the impact of mutation will reside in Kop. The observed activation energies for kHDX are most often found to be smaller than the estimated ΔH‡ for kint, indicating that the ΔH° of Kop favors the open conformation. This indicates that the uphill and transient local protein unfolding that is a prerequisite for HDX must be largely determined by unfavorable entropic changes, analogous to the main thermodynamic factor controlling global protein unfolding.
Spatial resolution of the impact of mutation on temperature-dependent protein flexibility.
For the temperature-dependent regions (Type II) of the native enzyme, the average Ea(HDX) is 12.1 kcal mol−1, but upon mutation of Leu343 to alanine, the average Ea(HDX) drops to 6.3 kcal mol−1, indicating an overall lower enthalpic barrier to access the partially unfolded, surface accessible states that enable HDX. In Figure 7, we illustrate the range of changes that have been observed in comparing Leu343Ala to WT, presenting examples for which the Ea(HDX) is very different (A and B) and virtually superimposable (D and E).
Figure 7.
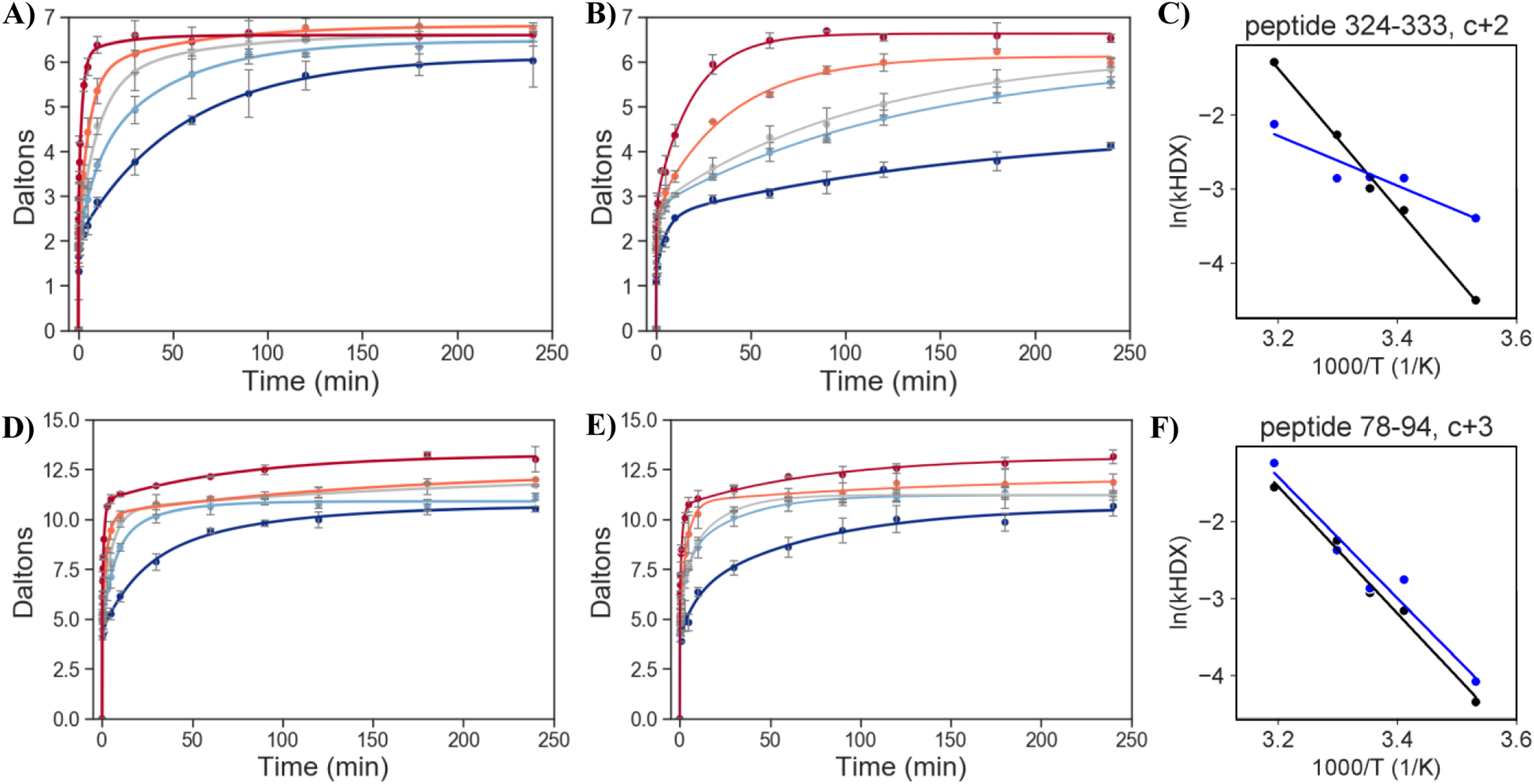
Examples of changes in Ea(HDX) for Leu343Ala in relation to WT. Peptide 324–333 (A-C) is an example of a large ΔEa(HDX) and peptide 78–94 (D-F) is an example of a negligible ΔEa(HDX). HDX uptake plots showing the increase in deuterated amide sites in peptide versus time at different temperatures for A,D) WT and B,E) Leu343Ala. Colors denote the different temperatures (40 °C, red; 30 °C, orange; 25 °C, grey; 20 °C, light blue; and 10 °C, dark blue), and data are fit to Eq 3. C, F). Comparative plots of ln[(Bkmed+Ckslow)/Ntot], as defined in Eq 4, versus inverse temperature, with WT and Leu343Ala indicated in black and blue, respectively. A compilation of the full set of HDX plots is presented in the Dataset SI.
To obtain a spatially resolved map of the impact of mutation on the flexibility of enolase, we focus on the individual peptide values, where we define ΔEa(HDX) = Ea(HDX)(Leu343Ala) – Ea(HDX)(WT). A total of nine peptides emerged as undergoing experimentally meaningful alterations in the temperature gradient of protein flexibility following mutation at position 343 (Table 2). From Table 2 it can be seen that in eight of the nine peptides, the magnitude of ΔEa(HDX) is significantly perturbed and in all cases outside of the corresponding experimental error. For the remaining Type II peptides, the majority displayed quite small ΔEa(HDX) values and large errors (see Table S6 and SI 1.p for a full discussion of the remainder of the peptides). Among the peptides included in Table 2, the sign of ΔEa(HDX) < 0 indicates an increase in the regional protein flexibility in the case of Leu343Ala, with the one exception being the N-terminal peptide, 22–31, where the ΔEa(HDX) > 0. Half of the identified peptides are in the N-terminus which also contains L1. The remainder of the peptides, all with large, negative values for ΔEa(HDX), reside within the TIM barrel and two are proximal to the active site base and metal binding residues (peptides 324–333 and 345–364).
Table 2.
Ea(HDX) values for WT and Leu343Ala peptides and the resulting ΔEa(HDX) in kcal mol−1a
| Peptide | WT Ea(HDX) | Leu343Ala Ea(HDX) | ΔEa(HDX) | Region | Properties |
|---|---|---|---|---|---|
| 324–333 | 18.9 (0.9) | 7 (1) | −12 (2) | A | TIMb, loop & helix, near Asp320 |
| 345–364 | 14 (2) | 2 (4) | −12 (5) | A | TIMb, loop & helix, near Lys 345 |
| 32–49 | 10.1 (0.4) | 1 (4) | −9 (4) | B | N-terminal L1 |
| 2–23 | 12 (1) | 4 (4) | −8 (4) | B | N-terminal sheet & loop |
| 64–77 | 11 (3) | 5 (2) | −6 (4) | B | N-terminal helix |
| 113–137 | 21 (3) | 6 (5) | −15 (6) | B | N-terminal helix & loop |
| 372–383 | 13 (1) | 11 (1) | −2 (1) | B | TIMb, loop |
| 263–279 | 15 (2) | 2 (4) | −12 (4) | C | TIMb, loop & helix |
| 22–31 | 2 (2) | 7 (2) | 5 (3) | D | N-terminal sheet & loop |
ΔEa(HDX) = Ea(HDX)(Leu343Ala) – Ea(HDX)(WT), where ΔEa(HDX) is larger than the error. Values for all Type II peptides are given in Table S6, with four outliers further discussed in SI p6. Activation energies were calculated from the temperature-dependent plots of ln(kHDX) versus inverse temperature (Section S6).
TIM refers to the canonical TIM barrel region of enolase. Regions A-D as shown in Figure 8.
Spatial mapping of the mutation-induced, temperature-dependent changes in protein flexibility within enolase.
We mapped the regions of enolase that undergo mutation-induced changes in the temperature gradient of HDX, using blue to represent regions that are increased in rigidity and raspberry to indicate regions with greater protein flexibility (Figure 8). Each of these regions is discussed in turn below, beginning with the seven peptides that display large negative ΔEa(HDX) values and are annotated as A, B, and C in the enolase structure.
Figure 8.
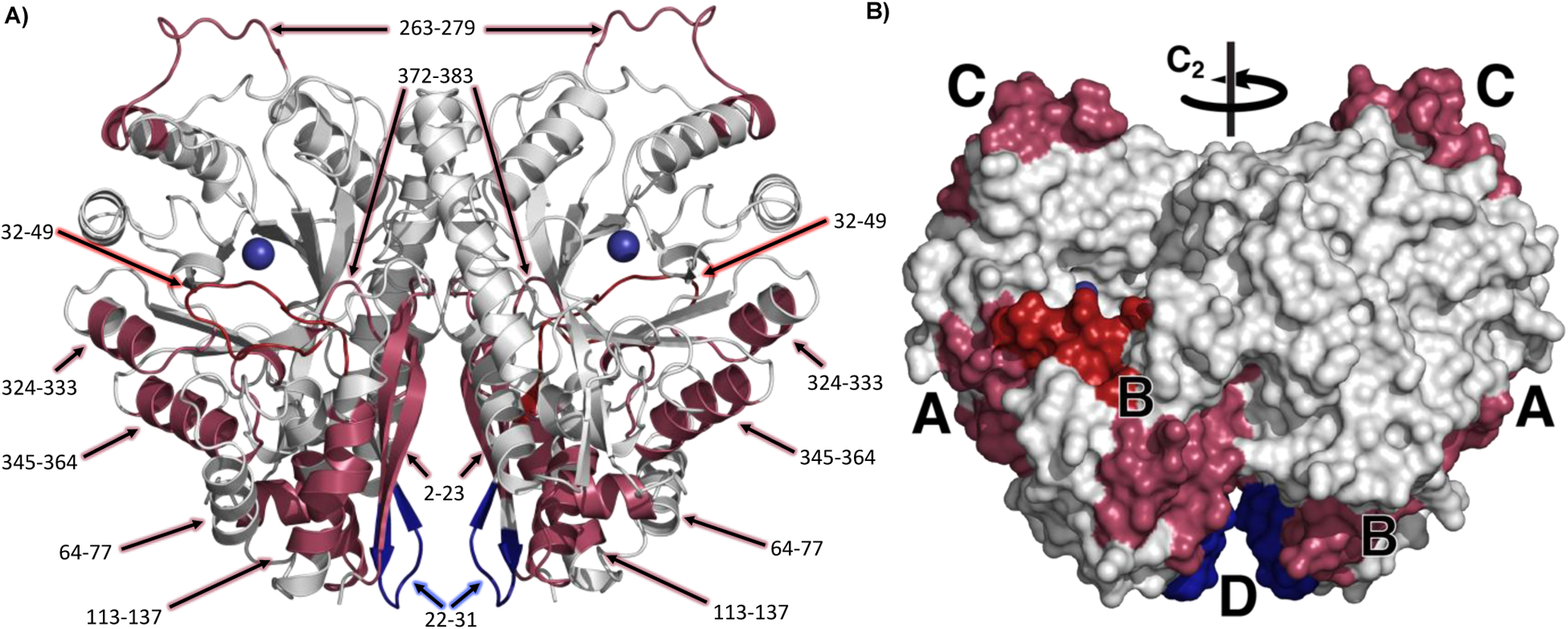
Homodimer of yeast enolase A) in cartoon form with peptides labeled and B) as a space filling model, with peptides with a ΔEa(HDX) > 0 colored in dark blue, ΔEa(HDX) < 0 colored in raspberry, and the peptide containing L1 distinguished in dark red (32,49). Mg(1) is blue. C2 symmetry axis for homodimer is indicated on the space filling model. Region labels are consistent with Table 2. PDB 1EBH.
Region A
Region A (raspberry) represents peptides 324–333 and 345–364, which lie directly outside of the mutation site (Leu343). This region also contains the catalytic base Lys345 and one of the ligands, Asp320, to the innermost Mg2+. The large impact of mutation within this part of the protein, with ΔEa(HDX) values of ca. −12 kcal mol−1, indicates the importance of a well packed, fairly rigid structure for optimal catalysis. The mutation of Leu343 was expected to have a direct impact on the positioning of the adjacent Lys345 and its proper positioning to act as the catalytic base; additionally, changes in mobility of a ligand to Mg(1), which together with Mg(2) bind the carboxylate of 2-PGA, may alter the ability of the enzyme to modulate the negative charge at the C1 of substrate and, hence, the pKa of the reactive carbon at C2. Two hydrophobic networks extend from the backbone of Leu343 out to the solvent interface, and we propose that in the native enzyme the precision of this structural motif must be maintained in order to facilitate a transfer of thermal energy from the protein solvent-interface toward the catalytic base as well as Mg(1), both of which will control the ability of substrate to undergo deprotonation.
Region B
Region B (raspberry and dark red) includes peptides 2–23, 32–49, 64–77, and 113–137 at the N-terminal domain, with ΔEa(HDX) values of −8, −9, −6, and −15 kcal mol−1, respectively, and peptide 373–383 which is a loop in the TIM barrel with an ΔEa(HDX) value of −2 kcal mol−1 (Table 2, Figure 8). This region includes the loop (L1, dark red) that closes over the active site pocket in the presence of substrate and Mg(2). The enthalpic barriers measured reflect the barrier to the positioning of key components within the enzyme. The meticulous flexibility of this region is likely salient to accessing the reactive conformation once substrate is bound, where the loop dynamics, through Ser39, will directly tune the donor-acceptor distances and electrostatic interactions among the catalytic residues and the substrate in response to thermal transfer arising at the protein/solvent interface from regions represented by peptides 64–77, and 113–137 and 2–23. As both the substrate and Ser39 coordinate the Mg2+ ions, dynamical motions at Ser39 will affect the Lewis acidity of the metal ions that influence the acidity of 2-PGA at C2 (see Figure 1b). This acidity is expected to be one of the key catalytic features controlling the (partially) rate-determining transfer of a proton from substrate to the active site base.
Peptides 2–23 and 64–77 in the N-terminal domain surround peptide 32–49, with multiple contact points. Peptide 372–383 in the TIM barrel domain is hydrogen bonded to the L1 loop via Gly376 to Ser36 in both the open and closed forms of the crystal structure (Figure S6). We hypothesize that the small increased flexibility observed in 372–383 for L343Ala is a reverberation of the enhanced flexibility of the surrounding region through the hydrogen bonding residues and additional close contacts. Based on the large overall increase in flexibility in region B upon mutation, we propose that these peptides need to be well structured in WT, as a prerequisite for optimal catalysis to ensue. The connectivity with L1 enables a thermally activated positioning and electrostatic optimization of the carboxylate of substrate toward rate-limiting proton loss from its α-carbon (see Figure 3 for the structure-based positioning of substrate).
Region C
Region C (raspberry) is located on the front of the TIM barrel, opposite the N terminal domain, and includes 15 amino acids in a flexible loop that undergoes a conformational shift upon substrate binding (Figure 2, L3). The peptide that comprises this loop, 263–279, has an ΔEa(HDX) value of −12 kcal mol−1, yet is ca. 30 Å away from Leu343Ala. We are currently uncertain how this region is triggered to increase its flexibility upon mutation, though it is part of L3 which undergoes a ca. 6 Å shift upon substrate binding. Its behavior is possibly related to the adjacent loop (L2) which contains His159 that binds the substrate, however L2 itself shows no detectable difference in flexibility.
Region D
Region D (dark blue) is located in the dimer interface, at the connection of two ß sheets by a short loop (peptide 22–31) and over 10 Å from the other monomer. D undergoes a large increase in rigidity upon the Leu343Ala mutation and is the only region to show a significant decrease in flexibility, ΔEa(HDX) = 5.0 kcal mol−1. This rigidification, over 30 Å from the Leu343Ala mutation site, may represent a structural compensation for the increased flexibility within the adjacent region B.
The remainder of the protein, shown in white in Figure 8, shows little to no change in Ea(HDX) in our experimental time regime upon the introduction of the catalytically impairing mutation Leu343Ala. This region includes the TIM barrel strands in the center of the protein adjacent to the active site, and the back side of the barrel. It is made up of 17 Type II peptides and four Type I peptides. This region demonstrates clearly that not all HDX-detected motions will be impacted by activity-altering mutations. Interestingly, the differences seen in enolase, between the front and back of the TIM barrel, are similar to those observed in murine adenosine deaminase, where the back of the TIM barrel showed minimal ΔEa(HDX) perturbation with a variant that increased the Ea(kcat) by 2.5 kcal mol−1.17 One emerging feature of TIM barrels is the presence of a “stable” surface on the rear of the barrel that “holds” some of the key active site residues.17,69 The front of the barrel contains the loops with catalytic residues and that control substrate specificity. From this study and the earlier findings with adenosine deaminase,17 the front of the barrel also contains the thermally activated protein networks that arise at the protein/solvent interface in a reaction specific manner.
Linking loop closures in proteins to solvent-exposed thermal networks.
In this work we used temperature dependent HDX-MS to analyze an enzyme for which X-ray crystallography had previously detected a substrate-initiated loop closure that consolidates key active site elements to initiate catalysis.26,35 Comparison of the regions of temperature dependent HDX perturbation in enolase with the regions of conformational change, as seen in the substrate-bound and holo X-ray structures (PDB 1ONE and 1EBH), shows similarities in the TIM barrel region. The primary conformational change seen from X-ray crystallography is the opening and closing of L1 with additional effects arising in L2 and L3 (Figure 2). Two of the nine peptides that show temperature-dependent perturbations upon the Leu343Ala mutation contain the L1 and L3 regions: peptide 32–49 (L1) and peptide 263–279 (L3). As discussed above, the changes in L3 that arise in Leu343Ala are far from the active site and do not appear to be engaged in a thermal network that connects the solvent-accessible surface to the active site (as seen for regions A and B, Figure 8). The role of this region of protein in catalysis is not well understood at the moment. The importance of the primary loop closure in enolase is much better understood and L1 has recently been examined through molecular dynamics. In a study that differentiates substrate- and product-liganded complexes, Li and Hammes-Schiffer found that the substrate-liganded enolase strongly prefers a closed conformation, whereas in the presence of product the loop is more like that of the holo-enzyme, with a high degree of flexibility and a preference for the open conformation. These differences are attributed primarily to changes in the hydrogen bonding network in the active site and to the interactions of Ser39 with Mg(2).70
In other TIM barrel enzymes with catalytic loop closures, Richard has proposed that the loops function as a switch, driving the protein from a flexible open conformation to a rigid and active enzyme form that can access the “transition-state binding energy” that enables catalysis.71 In the present work, which is focused on the unliganded enolase, the enzyme exhibits enthalpically rigid regions even in the absence of substrate binding. While the importance of loop flexibility in enolase is in general agreement with Richard and others, the observed temperature dependences of the L1 and surrounding regions indicate a different function. Based on the enthalpic sieve that we have introduced to isolate catalytically relevant protein motions, this loop is seen to be proximal to two primary thermal networks (A and B) that reach from the protein/solvent interface toward the region enveloping the active site. An ensuing transfer of thermal activation from solvent is expected to become distributed throughout L1, playing the role of dynamical active site tuning, that in enzymes lacking such loops must arise from more direct contacts between catalytically active side chains and their respective thermal networks (see also Ref. 17). Kurkcuoglu et al. have conducted an extensive computational study on catalytic loops that undergo a conformational shift to enable catalysis and similarly concluded that global enzyme dynamics are coupled to and define the catalytic loop motions.72 With the relaxation of regions A and B in Leu343Ala enolase, the thermal network becomes disrupted and the enzyme’s ability to convey thermal activation to the active site in an efficient and catalytically relevant manner through controlled motions is eroded. At the same time, the dynamic control that L1 is expected to exert on the active site is diminished.
Conclusion.
Regions distant from the active site are essential and play a role in efficient catalytic function through protein motions.9,15,42,71,73–79 Temperature-dependent HDX studies across diverse families of enzymes have found unique combinations of flexibility and rigidity that arise in spatially resolved ways and can be linked to active site chemistry.17–19 From the results documented herein with the enzyme enolase, we propose that the primary loop closure within L1 is tightly coupled to the dynamics of several thermal networks that originate at the protein/solvent interface (Figure 8). The closing of L1 fine tunes the distribution of thermal activation that initiates catalysis via transient adjustments to the distance and electrostatic interactions between substrate and the active site base (Region A in Figure 8). Catalysis is also significantly modulated by dynamical sampling within the Ser39-Mg(2)-2-PGA-Mg(1) (Region B in Figure 8) that transiently adjusts the pKa of the C2 proton of 2-PGA to enhance substrate reactivity. These mechanisms contrast with some hypotheses that point toward motions that originate at the active site, and agree with emerging studies showing the importance of evolved thermal conduits that reach from solvent-accessible surfaces, to enable rate accelerations that cannot yet be accessed in designed enzymes.5,71,75 We posit that understanding regional flexibility and the interconnectivity of protein motions is essential for the future engineering and design of enzymes capable of accessing the rate accelerations displayed by native enzymes. Ongoing research is focused on identifying thermal conduits in other TIM barrel systems, as well as building a library of conduits to inform design principles and to understand the origins of biological catalysis.
Supplementary Material
Acknowledgements.
We thank Prof. Adam R. Offenbacher, Dr. Shuaihua Gao, and Prof. Natalie G. Ahn for helpful and insightful discussions. We thank Prof. Birte Höcker for providing the enolase construct, eno1 from Saccharomyces cerevisiae, and we appreciate the work of Dr. Sooruban Shanmugaratnam in preparing and sending us the construct. We thank Prof. George Reed for helping us locate the plasmid for glycerate-2-kinase and thank Prof. Paul Sims for sending us the plasmid. EJT would like to thank the Berkeley Institute for Data Science for very helpful training modules in Python and Dr. Christopher T. Brown for additional Python discussions. This work was supported by the National Institutes of Health (NIH) grant (R35 GM118117). EJT was supported by an NIH National Institute of General Medicine F32 Fellowship (F32 GM130031). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the UC Berkeley’s College of Chemistry’s NMR facility for resources provided and the staff for their assistance in training. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We thank the Marqusee Lab at UC Berkeley for the use of the circular dichroism spectrometer and Dr. Charlotte Nixon for training and assistance. A mass spectrometer was purchased with NIH support (1S10OD020062-01).
Footnotes
Supporting Information
Experimental procedures, HDX calculation discussion, additional kinetic data, ΔEa table and discussion, HDX tables and graphs.
References.
- (1).Edwards DR; Lohman DC; Wolfenden R Catalytic Proficiency: the Extreme Case of S–O Cleaving Sulfatases. J. Am. Chem. Soc 2011, 134 (1), 525–531. [DOI] [PubMed] [Google Scholar]
- (2).Parker MFL; Osuna S; Bollot G; Vaddypally S; Zdilla MJ; Houk KN; Schafmeister CE Acceleration of an Aromatic Claisen Rearrangement via a Designed Spiroligozyme Catalyst That Mimics the Ketosteroid Isomerase Catalytic Dyad. J. Am. Chem. Soc 2014, 136 (10), 3817–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jiang L; Althoff EA; Clemente FR; Doyle L; Röthlisberger D; Zanghellini A; Gallaher JL; Betker JL; Tanaka F; Barbas CF; Hilvert D; Houk KN; Stoddard BL; Baker D De Novo Computational Design of Retro-Aldol Enzymes. Science 2008, 319 (5868), 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lassila JK; Baker D; Herschlag D Origins of Catalysis by Computationally Designed Retroaldolase Enzymes. Proc. Natl. Acad. Sci. U.S.A 2010, 107 (11), 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Agarwal PK A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2018, 58 (6), 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Henzler-Wildman KA; Lei M; Thai V; Kerns SJ; Karplus M; Kern D A Hierarchy of Timescales in Protein Dynamics Is Linked to Enzyme Catalysis. Nature 2007, 450 (7171), 913–916. [DOI] [PubMed] [Google Scholar]
- (7).Zoi I; Suarez J; Antoniou D; Cameron SA; Schramm VL; Schwartz SD Modulating Enzyme Catalysis Through Mutations Designed to Alter Rapid Protein Dynamics. J. Am. Chem. Soc 2016, 138 (10), 3403–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Klinman JP; Kohen A Evolutionary Aspects of Enzyme Dynamics. J. Biol. Chem 2014, 289 (44), 30205–30212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Eisenmesser EZ; Bosco DA; Akke M; Kern D Enzyme Dynamics During Catalysis. Science 2002, 295 (5559), 1520–1523. [DOI] [PubMed] [Google Scholar]
- (10).Saleh T; Kalodimos CG Enzymes at Work Are Enzymes in Motion. Science 2017, 355 (6322), 247–248. [DOI] [PubMed] [Google Scholar]
- (11).Doshi U; McGowan LC; Ladani ST; Hamelberg D Resolving the Complex Role of Enzyme Conformational Dynamics in Catalytic Function. Proc. Natl. Acad. Sci. U.S.A 2012, 109 (15), 5699–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nagel ZD; Cun S; Klinman JP Identification of a Long-Range Protein Network That Modulates Active Site Dynamics in Extremophilic Alcohol Dehydrogenases. J. Biol. Chem 2013, 288 (20), 14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bhabha G; Lee J; Ekiert DC; Gam J; Wilson IA; Dyson HJ; Benkovic SJ; Wright PE A Dynamic Knockout Reveals That Conformational Fluctuations Influence the Chemical Step of Enzyme Catalysis. Science 2011, 332 (6026), 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Frauenfelder H; Chen G; Berendzen J; Fenimore PW; Jansson H; McMahon BH; Stroe IR; Swenson J; Young RD A Unified Model of Protein Dynamics. Proc. Natl. Acad. Sci. U.S.A 2009, 106 (13), 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Benkovic SJ; Hammes-Schiffer S A Perspective on Enzyme Catalysis. Science 2003, 301 (5637), 1196–1202. [DOI] [PubMed] [Google Scholar]
- (16).Stojković V; Perissinotti LL; Willmer D; Benkovic SJ; Kohen A Effects of the Donor–Acceptor Distance and Dynamics on Hydride Tunneling in the Dihydrofolate Reductase Catalyzed Reaction. J. Am. Chem. Soc 2012, 134 (3), 1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gao S; Thompson EJ; Barrow SL; Zhang W; Iavarone AT; Klinman JP Hydrogen Deuterium Exchange within Adenosine Deaminase, a TIM Barrel Hydrolase, Identifies Networks for Thermal Activation of Catalysis. J. Am. Chem. Soc 2020, 142 (47), 19936–19949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang J; Balsbaugh JL; Gao S; Ahn NG; Klinman JP Hydrogen Deuterium Exchange Defines Catalytically Linked Regions of Protein Flexibility in the Catechol O-Methyltransferase Reaction. Proc. Natl. Acad. Sci. U.S.A 2020, 117 (20), 10797–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Offenbacher AR; Hu S; Poss EM; Carr CAM; Scouras AD; Prigozhin DM; Iavarone AT; Palla A; Alber T; Fraser JS; Klinman JP Hydrogen–Deuterium Exchange of Lipoxygenase Uncovers a Relationship Between Distal, Solvent Exposed Protein Motions and the Thermal Activation Barrier for Catalytic Proton-Coupled Electron Tunneling. ACS Cent. Sci 2017, 3 (6), 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Banner DW; Bloomer AC; Petsko GA; Phillips DC; Pogson CI; Wilson IA; Corran PH; Furth AJ; Milman JD; Offord RE; Priddle JD; Waley SG Structure of Chicken Muscle Triose Phosphate Isomerase Determined Crystallographically at 2.5 Å Resolution: Using Amino Acid Sequence Data. Nature 1975, 255, 609–614. [DOI] [PubMed] [Google Scholar]
- (21).Gerlt JA; Babbitt PC; Jacobson MP; Almo SC Divergent Evolution in Enolase Superfamily: Strategies for Assigning Functions. J. Biol. Chem 2012, 287 (1), 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Warburg O; Christian W Enolase. Biochem. Z 1942, No. 310, 385. [Google Scholar]
- (23).Schreier B; Höcker B Engineering the Enolase Magnesium II Binding Site: Implications for Its Evolution. Biochemistry 2010, 49 (35), 7582–7589. [DOI] [PubMed] [Google Scholar]
- (24).Zadvornyy OA; Boyd ES; Posewitz MC; Zorin NA; Peters JW Biochemical and Structural Characterization of Enolase From Chloroflexus Aurantiacus: Evidence for a Thermophilic Origin. Front. Bioeng. Biotechnol 2015, 3, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kornblatt MJ; Richard Albert J; Mattie S; Zakaib J; Dayanandan S; Hanic-Joyce PJ; Joyce PBM The Saccharomyces Cerevisiae Enolase-Related Regions Encode Proteins That Are Active Enolases. Yeast 2013, 30 (2), 55–69. [DOI] [PubMed] [Google Scholar]
- (26).Larsen TM; Wedekind JE; Rayment I; Reed GH A Carboxylate Oxygen of the Substrate Bridges the Magnesium Ions at the Active Site of Enolase: Structure of the Yeast Enzyme Complexed with the Equilibrium Mixture of 2-Phosphoglycerate and Phosphoenolpyruvate at 1.8 a Resolution. Biochemistry 1996, 35 (14), 4349–4358. [DOI] [PubMed] [Google Scholar]
- (27).Poyner RR; Cleland WW; Reed GH Role of Metal Ions in Catalysis by Enolase: an Ordered Kinetic Mechanism for a Single Substrate Enzyme. Biochemistry 2001, 40 (27), 8009–8017. [DOI] [PubMed] [Google Scholar]
- (28).Poyner RR; Laughlin LT; Sowa GA; Reed GH Toward Identification of Acid/Base Catalysts in the Active Site of Enolase: Comparison of the Properties of K345A, E168Q, and E211Q Variants. Biochemistry 1996, 35 (5), 1692–1699. [DOI] [PubMed] [Google Scholar]
- (29).Wedekind JE; Poyner RR; Reed GH; Rayment I Chelation of Serine 39 to Mg2+ Latches a Gate at the Active Site of Enolase: Structure of the Bis(Mg2+) Complex of Yeast Enolase and the Intermediate Analog Phosphonoacetohydroxamate at 2.1-a Resolution. Biochemistry 1994, 33 (31), 9333–9342. [DOI] [PubMed] [Google Scholar]
- (30).Reed GH; Poyner RR; Larsen TM; Wedekind JE; Rayment I Structural and Mechanistic Studies of Enolase. Curr. Opin. Struct. Biol 1996, 6 (6), 736–743. [DOI] [PubMed] [Google Scholar]
- (31).Zhang E; Hatada M; Brewer JM; Lebioda L Catalytic Metal Ion Binding in Enolase: the Crystal Structure of an Enolase-Mn2+-Phosphonoacetohydroxamate Complex at 2.4-. ANG. Resolution. Biochemistry 1994, 33 (20), 6295–6300. [DOI] [PubMed] [Google Scholar]
- (32).Anderson SR; Anderson VE; Knowles JR Primary and Secondary Kinetic Isotope Effects as Probes of the Mechanism of Yeast Enolase. Biochemistry 1994, 33 (34), 10545–10555. [DOI] [PubMed] [Google Scholar]
- (33).Sims PA; Larsen TM; Poyner RR; Cleland WW; Reed GH Reverse Protonation Is the Key to General Acid-Base Catalysis in Enolase. Biochemistry 2003, 42 (27), 8298–8306. [DOI] [PubMed] [Google Scholar]
- (34).Wedekind JE; Poyner RR; Reed GH; Rayment I Chelation of Serine 39 to Mg2+ Latches a Gate at the Active Site of Enolase: Structure of the Bis(Mg2+) Complex of Yeast Enolase and the Intermediate Analog Phosphonoacetohydroxamate at 2.1-.ANG. Resolution. Biochemistry 2002, 33 (31), 9333–9342. [DOI] [PubMed] [Google Scholar]
- (35).Wedekind JE; Reed GH; Rayment I Octahedral Coordination at the High-Affinity Metal Site in Enolase: Crystallographic Analysis of the MgII-Enzyme Complex From Yeast at 1.9 .ANG. Resolution. Biochemistry 2002, 34 (13), 4325–4330. [DOI] [PubMed] [Google Scholar]
- (36).Shandilya S; Vertrees J; Holder T ColorByRMSD. pymolwiki.org. March 6, 2012. [Google Scholar]
- (37).Englander SW Hydrogen Exchange and Mass Spectrometry: a Historical Perspective. J. Am. Soc. Mass Spectrom 2006, 17 (11), 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Habibi Y; Uggowitzer KA; Issak H; Thibodeaux CJ Insights Into the Dynamic Structural Properties of a Lanthipeptide Synthetase Using Hydrogen–Deuterium Exchange Mass Spectrometry. J. Am. Chem. Soc 2019, 141 (37), 14661–14672. [DOI] [PubMed] [Google Scholar]
- (39).Hoofnagle AN; Resing KA; Ahn NG Protein Analysis by Hydrogen Exchange Mass Spectrometry. Annu. Rev. Biophys. Biomol. Struct 2003, 32 (1), 1–25. [DOI] [PubMed] [Google Scholar]
- (40).Burkitt W; O’Connor G Assessment of the Repeatability and Reproducibility of Hydrogen/Deuterium Exchange Mass Spectrometry Measurements. Rapid Commun. Mass. SP 2008, 22 (23), 3893–3901. [DOI] [PubMed] [Google Scholar]
- (41).Bonnington L; Lindner I; Gilles U; Kailich T; Reusch D; Bulau P Application of Hydrogen/Deuterium Exchange-Mass Spectrometry to Biopharmaceutical Development Requirements: Improved Sensitivity to Detection of Conformational Changes. Anal. Chem 2017, 89 (16), 8233–8237. [DOI] [PubMed] [Google Scholar]
- (42).Hoofnagle AN; Resing KA; Goldsmith EJ; Ahn NG Changes in Protein Conformational Mobility Upon Activation of Extracellular Regulated Protein Kinase-2 as Detected by Hydrogen Exchange. Proc. Natl. Acad. Sci. U.S.A 2001, 98 (3), 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Balasubramaniam D; Komives EA Hydrogen-Exchange Mass Spectrometry for the Study of Intrinsic Disorder in Proteins. Biochim. Biophys. Acta 2013, 1834 (6), 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tajoddin NN; Konermann L Analysis of Temperature-Dependent H/D Exchange Mass Spectrometry Experiments. Anal. Chem 2020, 92 (14), 10058–10067. [DOI] [PubMed] [Google Scholar]
- (45).Deredge D; Wintrode PL; Tulapurkar ME; Nagarsekar A; Zhang Y; Weber DJ; Shapiro P; Hasday JD A Temperature-Dependent Conformational Shift in P38α MAPK Substrate-Binding Region Associated with Changes in Substrate Phosphorylation Profile. J. Biol. Chem 2019, 294 (34), 12624–12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Man P; Fábry M; Sieglová I; Kavan D; Novák P; Hnízda A Thiopurine Intolerance-Causing Mutations in NUDT15 Induce Temperature-Dependent Destabilization of the Catalytic Site. BBA Proteins Proteom 2019, 1867 (4), 376–381. [DOI] [PubMed] [Google Scholar]
- (47).van de Waterbeemd M; Llauró A; Snijder J; Valbuena A; Rodríguez-Huete A; Fuertes MA; de Pablo PJ; Mateu MG; Heck AJR Structural Analysis of a Temperature-Induced Transition in a Viral Capsid Probed by HDX-MS. Biophys J 2017, 112 (6), 1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rist W; Jørgensen TJD; Roepstorff P; Bukau B; Mayer MP Mapping Temperature-Induced Conformational Changes in the Escherichia Coli Heat Shock Transcription Factor Sigma 32 by Amide Hydrogen Exchange. J. Bio. Chem 2003, 278 (51), 51415–51421. [DOI] [PubMed] [Google Scholar]
- (49).Banks DD; Zhang J; Siska CC Relationship Between Native-State Solubility and Non-Native Aggregation of Recombinant Human Granulocyte Colony Stimulating Factor: Practical Implications for Protein Therapeutic Development. Mol. Pharmaceutics 2014, 11 (10), 3431–3442. [DOI] [PubMed] [Google Scholar]
- (50).Xiao P; Bolton D; Munro RA; Brown LS; Ladizhansky V Solid-State NMR Spectroscopy Based Atomistic View of a Membrane Protein Unfolding Pathway. Nat. Commun 2019, 10 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Liang Z-X; Lee T; Resing KA; Ahn NG; Klinman JP Thermal-Activated Protein Mobility and Its Correlation with Catalysis in Thermophilic Alcohol Dehydrogenase. Proc. Natl. Acad. Sci. U.S.A 2004, 101 (26), 9556–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lim XX; Chandramohan A; Lim X-YE; Crowe JE Jr.; Lok S-M; Anand GS Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 2017, 25 (9), 1391–1402.e1393. [DOI] [PubMed] [Google Scholar]
- (53).Reardon D; Farber GK The Structure and Evolution of Alpha/Beta Barrel Proteins. FASEB J 1995, 9 (7), 497–503. [DOI] [PubMed] [Google Scholar]
- (54).Farber GK An A/B-Barrel Full of Evolutionary Trouble. Curr Opin Struct Biol 1993, 3, 409–412. [Google Scholar]
- (55).Wierenga RK The TIM-Barrel Fold: a Versatile Framework for Efficient Enzymes. FEBS Lett 2001, 492 (3), 193–198. [DOI] [PubMed] [Google Scholar]
- (56).Wise EL; Rayment I Understanding the Importance of Protein Structure to Nature’s Routes for Divergent Evolution in TIM Barrel Enzymes. Acc. Chem. Res 2004, 37 (3), 149–158. [DOI] [PubMed] [Google Scholar]
- (57).Wold F; Ballou CE Studies on the Enzyme Enolase. II. Kinetic Studies. J. Bio. Chem 1957, 227 (1), 313–328. [PubMed] [Google Scholar]
- (58).Brocklehurst K; O’Driscoll M; Kowlessur D; Phillips IR; Templeton W; Thomas EW; Topham CM; Wharton CW The Interplay of Electrostatic and Binding Interactions Determining Active Centre Chemistry and Catalytic Activity in Actinidin and Papain. Biochem. J 1989, 257 (1), 309–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shen TY; Westhead EW Divalent Cation and pH Dependent Primary Isotope Effects in the Enolase Reaction. Biochemistry 1973, 12 (17), 3333–3337. [DOI] [PubMed] [Google Scholar]
- (60).Grant KL; Klinman JP Evidence That Both Protium and Deuterium Undergo Significant Tunneling in the Reaction Catalyzed by Bovine Serum Amine Oxidase. Biochemistry 1989, 28 (16), 6597–6605. [DOI] [PubMed] [Google Scholar]
- (61).Klinman JP; Offenbacher AR; Hu S Origins of Enzyme Catalysis: Experimental Findings for C–H Activation, New Models, and Their Relevance to Prevailing Theoretical Constructs. J. Am. Chem. Soc 2017, 139 (51), 18409–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Knapp MJ; Rickert K; Klinman JP Temperature-Dependent Isotope Effects in Soybean Lipoxygenase-1: Correlating Hydrogen Tunneling with Protein Dynamics. J. Am. Chem. Soc 2002, 124 (15), 3865–3874. [DOI] [PubMed] [Google Scholar]
- (63).Quantum Tunnelling in Enzyme-Catalysed Reactions; Allemann RK, Scrutton NS, Eds.; Royal Society of Chemistry, 2009. [Google Scholar]
- (64).Zhang Z; Smith DL Determination of Amide Hydrogen Exchange by Mass Spectrometry: a New Tool for Protein Structure Elucidation. Protein Sci 1993, 2 (4), 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Horitani M; Offenbacher AR; Carr CAM; Yu T; Hoeke V; Cutsail GE III; Hammes-Schiffer S; Klinman JP; Hoffman BM 13C ENDOR Spectroscopy of Lipoxygenase–Substrate Complexes Reveals the Structural Basis for C–H Activation by Tunneling. J. Am. Chem. Soc 2017, 139 (5), 1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wales TE; Engen JR Hydrogen Exchange Mass Spectrometry for the Analysis of Protein Dynamics. Mass Spec. Rev 2006, 25 (1), 158–170. [DOI] [PubMed] [Google Scholar]
- (67).Pascal BD; Willis S; Lauer JL; Landgraf RR; West GM; Marciano D; Novick S; Goswami D; Chalmers MJ; Griffin PR HDX Workbench: Software for the Analysis of H/D Exchange MS Data. J. Am. Soc. Mass Spectrom 2012, 23 (9), 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Masson GR; Burke JE; Ahn NG; Anand GS; Borchers C; Brier S; Bou-Assaf GM; Engen JR; Englander SW; Faber J; Garlish R; Griffin PR; Gross ML; Guttman M; Hamuro Y; Heck AJR; Houde D; Iacob RE; Jørgensen TJD; Kaltashov IA; Klinman JP; Konermann L; Man P; Mayne L; Pascal BD; Reichmann D; Skehel M; Snijder J; Strutzenberg TS; Underbakke ES; Wagner C; Wales TE; Walters BT; Weis DD; Wilson DJ; Wintrode PL; Zhang Z; Zheng J; Schriemer DC; Rand KD Recommendations for Performing, Interpreting and Reporting Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) Experiments. Nat. Methods 2019, 16 (7), 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tiwari SP; Reuter N Similarity in Shape Dictates Signature Intrinsic Dynamics Despite No Functional Conservation in TIM Barrel Enzymes. PLoS Comput. Biol 2016, 12 (3), e1004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Li P; Hammes-Schiffer S Substrate-to-Product Conversion Facilitates Active Site Loop Opening in Yeast Enolase: a Molecular Dynamics Study. ACS Catal. 2019, 9 (10), 8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Richard JP Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc 2019, 141 (8), 3320–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kurkcuoglu Z; Bakan A; Kocaman D; Bahar I; Doruker P Coupling Between Catalytic Loop Motions and Enzyme Global Dynamics. PLoS Comput. Biol 2012, 8 (9), e1002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Boehr DB; Dyson HJ; Wright PE An NMR Perspective on Enzyme Dynamics. Chem. Rev 2006, 106 (8), 3055–3079. [DOI] [PubMed] [Google Scholar]
- (74).Agarwal PK A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2018, 58 (6), 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Klinman JP Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc. Chem. Res 2015, 48 (2), 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tousignant A; Pelletier JN Protein Motions Promote Catalysis. Chem. Biol 2004, 11 (8), 1037–1042. [DOI] [PubMed] [Google Scholar]
- (77).Henzler-Wildman K; Kern D Dynamic Personalities of Proteins. Nature 2007, 450 (7172), 964–972. [DOI] [PubMed] [Google Scholar]
- (78).Xiao Y; Liddle JC; Pardi A; Ahn NG Dynamics of Protein Kinases: Insights From Nuclear Magnetic Resonance. Acc. Chem. Res 2015, 48 (4), 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Zimmermann J; Thielges MC; Yu W; Romesberg FE Protein Dynamics and the Evolution of Novel Protein Function; John Wiley & Sons, Ltd: Weinheim, Germany, 2011; Vol. 254, pp 147–185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.