

Abstract
The pathophysiology of neonatal stroke and adult stroke are distinct in many aspects, including the inflammatory response. We previously showed endogenously protective functions of microglial cells in acute neonatal stroke. We asked if galectin-3 (Gal3), a pleotropic molecule that mediates interactions between microglia/macrophages and the extracellular matrix (ECM), plays a role in early injury after transient middle cerebral occlusion (tMCAO) in postnatal day 9–10 mice. Compared to wild type (WT) pups, in Gal3 knockout pups injury was worse and cytokine/chemokine production altered, including further increase of MIP1α and MIP1β levels and reduced IL6 levels 72 h after tMCAO. Lack of Gal3 did not affect morphological transformation or proliferation of microglia but markedly attenuated accumulation of CD11b+/CD45med-high cells after injury, as determined by multi-color flow cytometry. tMCAO increased expression of αV and β3 integrin subunits in CD11b+/CD45low microglial cells and cells of non-monocyte lineage (CD11b−/CD45−), but not in CD11b+/CD45med-high cells within injured regions of WT mice or Gal3−/− mice. αV upregulated in areas occupied and not occupied by CD68+ cells, most prominently in the ECM, lining blood vessels, with expanded αV coverage in Gal3−/− mice. Cumulatively, these data show that lack of Gal3 worsens subchronic injury after neonatal focal stroke, likely by altering the neuroinflammatory milieu, including an imbalance between pro- and anti-inflammatory molecules, effects on microglial activation, and deregulation of the composition of the ECM.
Keywords: Microglia, Middle cerebral artery occlusion, Neonate, Cytokine, AlphaV beta3 integrin
1. Introduction
Perinatal arterial ischemic stroke is common—occurring in at least 1 in 2300 live term births—and produces significant morbidity and severe long-term neurological and cognitive deficits, including cerebral palsy and neurodevelopmental disabilities (Nelson, 2007; Nelson and Lynch, 2004; Raju et al., 2007). More than half of all children with cerebral palsy are born at term. There is now ample evidence that the mechanisms of ischemic injury differ greatly between the immature brain and adult brain (review in Fernandez-Lopez et al., 2014; Yager and Ashwal, 2009), including neuroimmune responses (Hagberg et al., 2015). Neuroinflammation plays a major modulatory role in the pathogenesis of stroke in the adult, both detrimental and beneficial (Iadecola and Anrather, 2011; Markowska et al., 2010; Shichita et al., 2014; Szalay et al., 2016). In perinatal stroke and hypoxic-ischemic encephalopathy (HIE), neuroinflammation also plays a key modulatory role (Hagberg et al., 2015) but the underlying signaling mechanisms are distinct, as demonstrated in mice with genetic manipulations of individual inflammatory mediators (Doverhag et al., 2008; Hedtjarn et al., 2005; Woo et al., 2012). The differing responses to stroke between neonates and adults are likely due to the CNS immaturity, including cells within the neurovascular unit, astrocyte and pericyte coverage of the brain vasculature, as well as microglial immaturity and the still present physiological neuronal programmed cell death during early postnatal brain development.
Historically, in adult stroke, microglia were viewed as purely injurious, in part due to production of inflammatory mediators and reactive oxygen species (reviewed in Vexler and Yenari, 2009). However, we discovered that after neonatal arterial stroke microglia protect neurovascular integrity, engulf and remove neuronal debris, and curb injury (Faustino et al., 2011; Fernandez-Lopez et al., 2016). Microglial cells can exert protection via many signaling mechanisms, including communication of microglia with the extracellular matrix (ECM) and direct interaction with endothelial cells. Our comparative analysis of the endothelial transcriptome following acute neonatal and adult stroke demonstrated marked differences in the expression of various ECM proteins produced in uninjured brain regions and distinct changes in injured regions (Fernandez-Lopez et al. 2012).
Galectin-3 (Gal3) has been demonstrated to modulate cell-cell interactions within the ECM (Danella Polli et al., 2013) and to play a modulatory role in models of adult stroke (Lalancette-Hebert et al., 2007; Lalancette-Hebert et al., 2012; Yan et al., 2009) and neonatal hypoxia-ischemia (H-I) (Doverhag et al., 2010). Considering that Gal3 is a pleotropic molecule that can exert context-dependent changes in the brain (Krzeslak and Lipinska, 2004) and exhibit brain maturation-dependent effects (Pasquini et al., 2011), in this study we focused on the role of genetic Gal3 deletion on sub-chronic injury after neonatal stroke. In a recently developed neonatal mouse arterial focal stroke model, a transient middle cerebral artery occlusion (tMCAO) model in postnatal day 9 pups (P9–P10), we show that genetic deletion of Gal3 affects microglial activation, alters the ECM-integrin communications and extends injury during the sub-chronic phase.
2. Materials and methods
All research conducted on animals was approved by the University of California San Francisco Institutional Animal Care and Use Committee and followed in accordance to the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services). Animals were given ad libitum access to food and water; housed with nesting material and shelters, and kept in rooms with temperature control and light/dark cycles.
2.1. Transient middle cerebral artery occlusion (tMCAO)
A transient 3 h MCAO was achieved by inserting a 6–0 coated filament into the internal carotid artery of male and female P7 rats, as previously described (Derugin et al., 2005; Derugin et al., 1998). Reperfusion of the MCA was achieved by the retraction of the filament. The pups were then returned to the dam until sacrifice.
Male and female P9–P10 C57Bl/6 (WT) and galectin-3 knockout (Gal3−/−) mice (on C57Bl/6 background; colony founders pur-chased from Jackson Laboratories, Bar Harbor, Maine) were anesthetized and subjected to 3 h tMCAO or sham surgery, followed by 24 or 72 h reperfusion as originally described for P7 rats (Derugin et al., 2005; Derugin et al., 1998) and modified for P9–P10 mice (Woo et al., 2012).
2.2. BrdU labeling, histological evaluation and immunofluorescence
To assess cell proliferation, mice were given Bromo-2-deoxyuridine (BrdU; Millipore) at 50 mg/kg of body weight by intraperitoneal injection (i.p.) twice daily with an interval of 10–12 h at 24 h, and 48 h, and one injection at 4 h before sacrifice at 72 h after MCAO.
Pups were deeply anesthetized with Euthasol (100 mg/kg; Virbac) and perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M PBS (pH 7.4). Brains were post-fixed in 4% PFA overnight at 4 °C, cryoprotected in 30% sucrose in 0.1 M PBS at 4 °C for 48 h, frozen and cut on cryostat (12 μm thick, 348 μm apart). The sizes of contralateral and ipsilateral hemispheres and the size of injured regions in 6 consecutive Nissl-stained coronal sections were traced by two investigators, one of them blinded to brain identity. Tissue loss was calculated as volumetric ratio of the remaining ipsilateral hemisphere compared to contralateral hemisphere. Injury volume is shown as ratio of injured region compared to the ipsilateral hemisphere.
For immunofluorescence, sections were blocked in 20% normal goat serum (NGS) in PBS containing 0.25% TritonX-100 followed by overnight incubation at 4 °C with rat anti-mouse Gal3 (gift from Dr. Jasna Kriz), rabbit polyclonal anti-ionized calcium binding adapter molecule 1 (Iba-1, 1:200; Wako), rat anti-mouse Cluster of Differentiation 68 (CD68, 1:100; Bio-Rad), rat anti-BrdU (1:200; Abcam), chicken anti-BrdU (1:500; Abcam), rabbit polyclonal anti-alphaV (αV, 1:300, Abcam), rabbit polyclonal anti-glucose transporter 1 (Glut-1, 1:500; Millipore), monoclonal anti-glial fibrillary acidic protein (GFAP; 1:300; Millipore), or monoclonal anti-Neuronal Nuclei (NeuN; 1:100; Millipore) following 1 h incubation with fluorochrome conjugated secondary antibodies raised in goat (1:500; Life Technologies). Some slides were co-stained with Alexa 647-conjugated Griffonia simplicifolia isolectin B4 (IB4; 1:150; Life Technologies) and with DAPI. Slides were coverslipped with Prolong Gold and mounted. Images were captured in four fields of view (FOV) in the peri-focal and ischemic core regions in the caudate and in the cortex, and in the corresponding contralateral regions using a Zeiss Axio Imager. Z2 microscope (Zeiss) equipped with Volocity Software (PerkinElmer). The number of Iba1+, CD68+ and BrdU+ cells per FOV, mean and total surface area and volume, were measured using custom-made threshold and size-exclusion protocols created in Volocity software, as we described (Faustino et al., 2011). The density, length, surface, volume of Glut-1+ vessels was determined using automated protocols for signal intensity threshold (Faustino et al., 2011).
2.3. Brain cell isolation and Multi-color Flow cytometry
Samples for multi-color flow cytometry were prepared as we described (Li et al., 2015), with modifications. Deeply anesthetized mice were transcardially perfused with 10 ml of Ca2+/Mg2+-free Hank’s Balance Salt Solution (HBSS) to eliminate peripheral cells, meninges were removed, and the cortices of injured and matching contralateral regions were dissected on ice and enzymatically digested to obtain single brain cells using Papain-containing Neural Tissue Dissociation Kit (Miltenyi Biotec, Germany). Myelin was removed with myelin-conjugated magnetic beads and was separated from the cell suspension by passing it through LS columns placed on a magnetic rack (Miltenyi Biotec) (Li et al., 2015).
Isolated cells were plated at a density of 2 × 105 cells per well (96-well V-bottom plate, Falcon), blocked for 15 min with CD16/32 (1:70; Biolegend) in FACS buffer containing 2% fetal bovine serum to prevent unspecific binding by antibodies. Cells were washed once in FACS buffer and stained with a mixture of antibodies (30 min, 4 °C, protected from light), as we described (Li et al., 2015). The following antibodies conjugated to fluorochromes were used: anti-CD45-Pacific Blue (1:2000; Biolegend), anti-CD11b-APC-Cy7 (1:2000; Biolegend), anti-Ly-6C-APC (1:800; eBioscience), anti-CD51(αV)-PE (1:800; BD Pharmingen), and anti-CD61(β3)-AF647 (1:800; BD Pharmingen). Following antibody incubation cells were washed once with FACS buffer. The gating strategy was based on live single cells stained with Live/Dead Yellow reactive dye (Life Technologies). BD compensation beads (BD Bioscience) were used for compensation. All samples were run on BD LSRII flow cytometer (BD Bioscience) and data analysis was performed using FlowJo software (Tree Star).
2.4. Western blot
Tissue from injured and matching contralateral regions was collected from deeply anesthetized mice transcardially perfused with HBSS and flash frozen in 2-methyl-butane under dry-ice. Tissue was mechanically disrupted in lysis buffer (Cell Signaling) containing protease inhibitors (Roche), 1 mM PMSF (Roche), and 1ug/mL leupeptin (Roche) using glass homogenizer. 20 ng of protein per lane was loaded into a 4–12% Bis-Tris gradient gel (Life Technologies) and gel run under reduced conditions in MOPS buffer (Life Technologies). The blots were incubated in blocking buffer (Tris-HCl buffer containing 5% non-fat dry milk, 0.1% Tween-20, 1 h, RT) followed by overnight incubation with the following primary antibodies in blocking buffer at 4 °C: mouse anti-rat spectrin (1:1000, Millipore), rabbit polyclonal alphaV (1:1000, Abcam), monoclonal anti-beta-actin (1:8000; Sigma), and polyclonal anti-rat Gal3 (1:500; Cell Signaling). Blots were incubated with goat secondary antibodies conjugated to horse-radish peroxidase (1:2000; Santa Cruz) for 1 h at RT and signals were visualized by enhanced chemiluminescence (Pierce).
2.5. Multiplex ELISA assays for cytokines and endothelial antigens
Tissue from injured and matching contralateral regions was collected following HBSS-perfused deeply anesthetized mice, frozen, homogenized in cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM PMSF, 0.05% Tween-20, and a cocktail of protease inhibitors (Roche) (Li et al., 2015; Woo et al., 2012). Protein concentrations of cytokines: IL-1α, IL-1β, IL-6, G-CSF, MCP-1, MIP1α, MIP1β, TNFα, LIF and KC or of endothelial antigens: E-Selectin, P-Selectin, and ICAM-1, were determined using multiplex ELISA according to manufacturer’s instructions (Millipore). Measurements were performed using Bio-Plex System (Bio-Rad) and StatLIA® software (Brendan Scientific) with a 5-parameter logistic curve fitting, as described (Denker et al., 2007; Faustino et al., 2011). The data were normalized to protein concentration within the same sample.
2.6. Statistical analysis
Individual measurements were determined for each group and the Mean ± SD presented. Statistical analysis was performed on GraphPad Prism (GraphPad Software). One-way ANOVA with Bonferroni’s correction was performed to test for differences among groups and Student’s t test was used to test for differences between two groups.
3. Results
3.1. Galectin-3 is upregulated after neonatal stroke and affects injury
First, we asked if Gal3 is upregulated by transient MCAO in neonatal mice and affects injury. Consistent with published reports (Doverhag et al., 2010), Gal3 expression was undetectable in naïve neonatal brain and in the hemisphere contralateral to the lesion, but was gradually upregulated in ischemic-reperfused tissue between 24 and 72 h after reperfusion (Fig. 1A). The presence of injury was confirmed in the same brain lysates based on spectrin cleavage by calpain-mediated (150 kDa band) and caspase-3-mediated (120 kDa band) mechanisms (Fig. 1A). Double-immunofluorescence in the injured caudate of Gal3−/− (Suppl. Fig. 1A) or in the contralateral caudate of WT mice did not depict Gal3 expression (Suppl. Fig. 1B), but showed that Iba1+/IB4+ activated microglia/macrophages are the predominant source of Gal3 induction at 24–72 h after tMCAO (Fig. 1E and F). No Gal3+ cells were observed in injured regions of Gal3−/− mice (Suppl Fig. 1A). At the peri-infarct region of the cortex few reactive GFAP+ astrocytes expressed Gal3 (Suppl. Fig. 1C). Gal3 was not observed in NeuN+ neurons (data not shown).
Fig. 1.
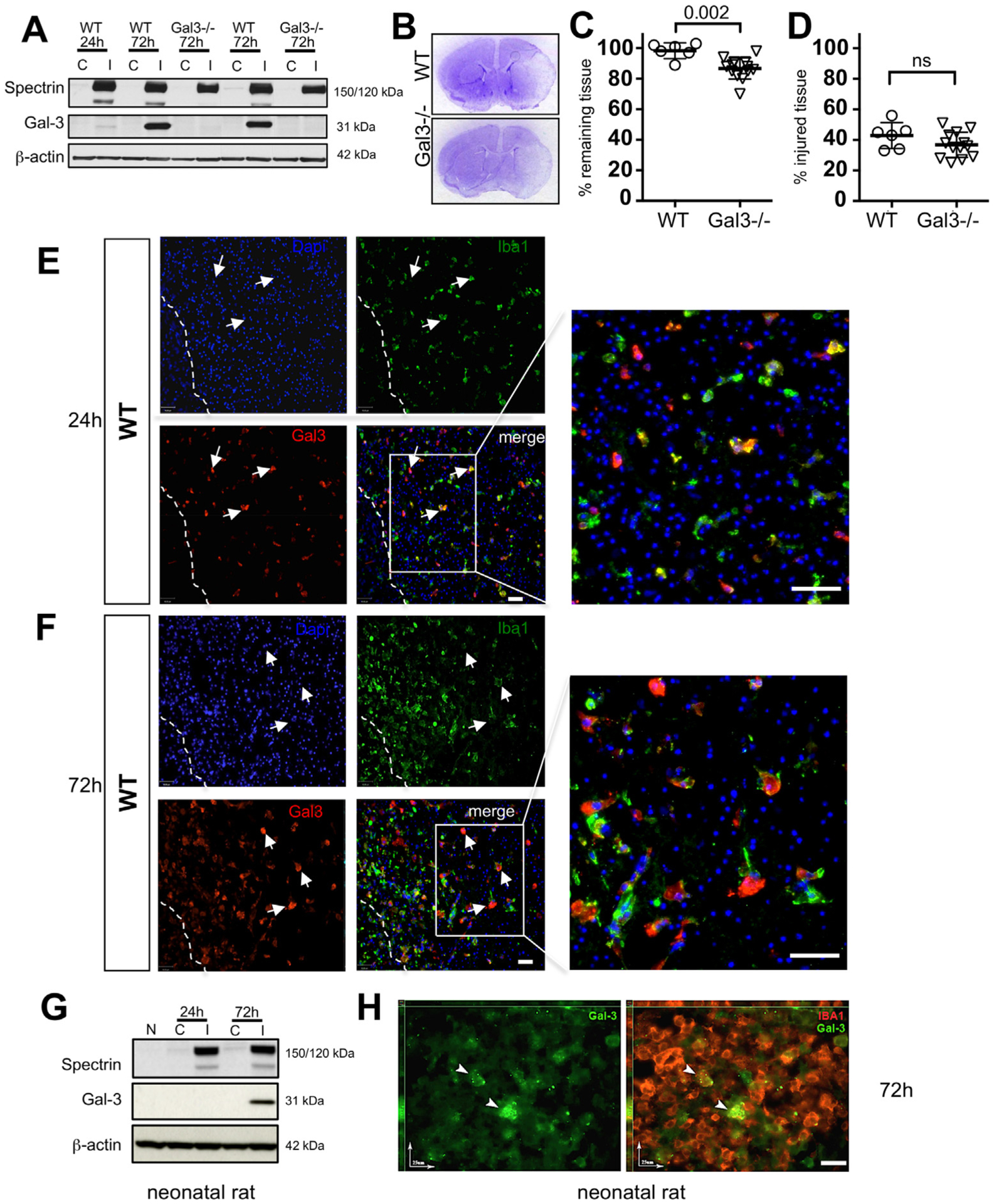
Gal3 is upregulated after tMCAO in neonatal mice and worsens injury. A. Top row: A representative Western Blot that demonstrates the presence of spectrin cleavage by calpain (150 kDa) and caspase-3 (120 kDa)-dependent mechanisms in injured (I) regions as compared to lack of cleavage in matching contralateral (C) region. Middle row: gradual upregulation of Gal3 expression in injured region of WT and not in Gal3−/− 72 h post-MCAO. Data are normalized to beta-actin protein expression (bottom row). B. Representative Nissl stained coronal sections of WT and Gal3−/− 72 h after MCAO C. Percent volume of remaining tissue in the ipsilateral hemisphere compared to that in contralateral hemisphere. D. Percent volume of injured tissue within the ipsilateral hemisphere. E–F. In WT mice, Gal3 is expressed in injured caudate 24 h and 72 h post-tMCAO. Injured and dying cells are identified by punctate nuclei stained with DAPI. Dashed line represents peri-infarct region at 72 h post-MCAO. Arrows indicate Gal3+ activated microglia and macrophages (scale bar: 40 μm). G. A representative Western Blot of spectrin cleavage (top), Gal3 expression (middle), and β-actin (bottom) expression in the rat brain. Shown is expression in naïve (N), contralateral (C) and injured (I) regions. H. Gal3 is upregulated in a subpopulation of Iba1+ microglial cells in injured rat brain, as evident from immunofluorescence at 72 h post-tMCAO (scale bar: 25 μm). Dots in C–D represent data from individual animals (n = 6 for WT, n = 14 for Gal3−/−). Mean ± SD. Significance levels as indicated on individual panels, ns: non-significant (Student’s t-test).
To explore whether the effect is species-specific, we then examined Gal3 expression in neonatal rats subjected to a 3 h tMCAO. Similar to the effects observed in injured neonatal mice, in injured neonatal rats, Gal3 induction occured gradually in injured regions (Fig. 1G) and activated microglia/macrophages were the source of Gal3 (Fig. 1H). Thus, Gal3 upregulation occurs predominantly in activated microglia in two species after neonatal stroke.
Gal3 deficiency exacerbated severity of histologically defined injury 72 h after tMCAO (Fig. 1B and C). Compared to WT, a significant loss of tissue occurred in Gal3−/− mice (Fig. 1C). At the same time, the volume of injured tissue within a remaining tissue was similar in Gal3−/− and WT mice (Fig. 1D). Considering that sex may affect injury severity after stroke, we compared injury in male and female pups within the same cohort. In WT mice, the volumes of remaining tissue were 96.7 ± 7.1% in males and 100 ± 1.2% in females (ns). In Gal3−/− mice, volumes of remaining tissue were 89.9 ± 1.2% in males and 86.7 ± 5.5% in females (ns). Injured tissue occupied 40.7 ± 6.8% in WT males and 45.0 ± 1.1% in females, whereas in Gal3−/− the respective volumes were 36.4 ± 8.9% in males and 39.1 ± 9.1% (ns), indicating the absence of sex differences. Therefore, we combined data for males and females in the rest of the assays.
3.2. Gal3 deficiency produces unsynchronized changes in levels of individual cytokines and chemokines in injured neonatal brain
Several studies suggested that Gal3 is involved in inflammation (Burguillos et al., 2015; Rabinovich et al., 2002; Rotshenker, 2009). Therefore, we examined the effects of genetic deletion of Gal3 on protein levels of cytokines and chemokines after tMCAO. At 72 h, tMCAO induced IL-1α, IL-6, G-CSF, KC, LIF, MIP1α, MIP1β, and MCP-1 in injured regions of WT, whereas the levels of IL-1β and TNFα were not significantly different between injured and matching contralateral regions. Levels of IL-6 and G-CSF were significantly lower in injured regions of Gal3−/− mice as compared to WT (Fig. 2B and C), while levels of chemokines MIP-1α and MIP-1β were increased (Fig. 2D and E). The levels of MCP-1 (Fig. 2F), IL-1α, KC, and LIF were similar in both groups (data not shown).
Fig. 2.
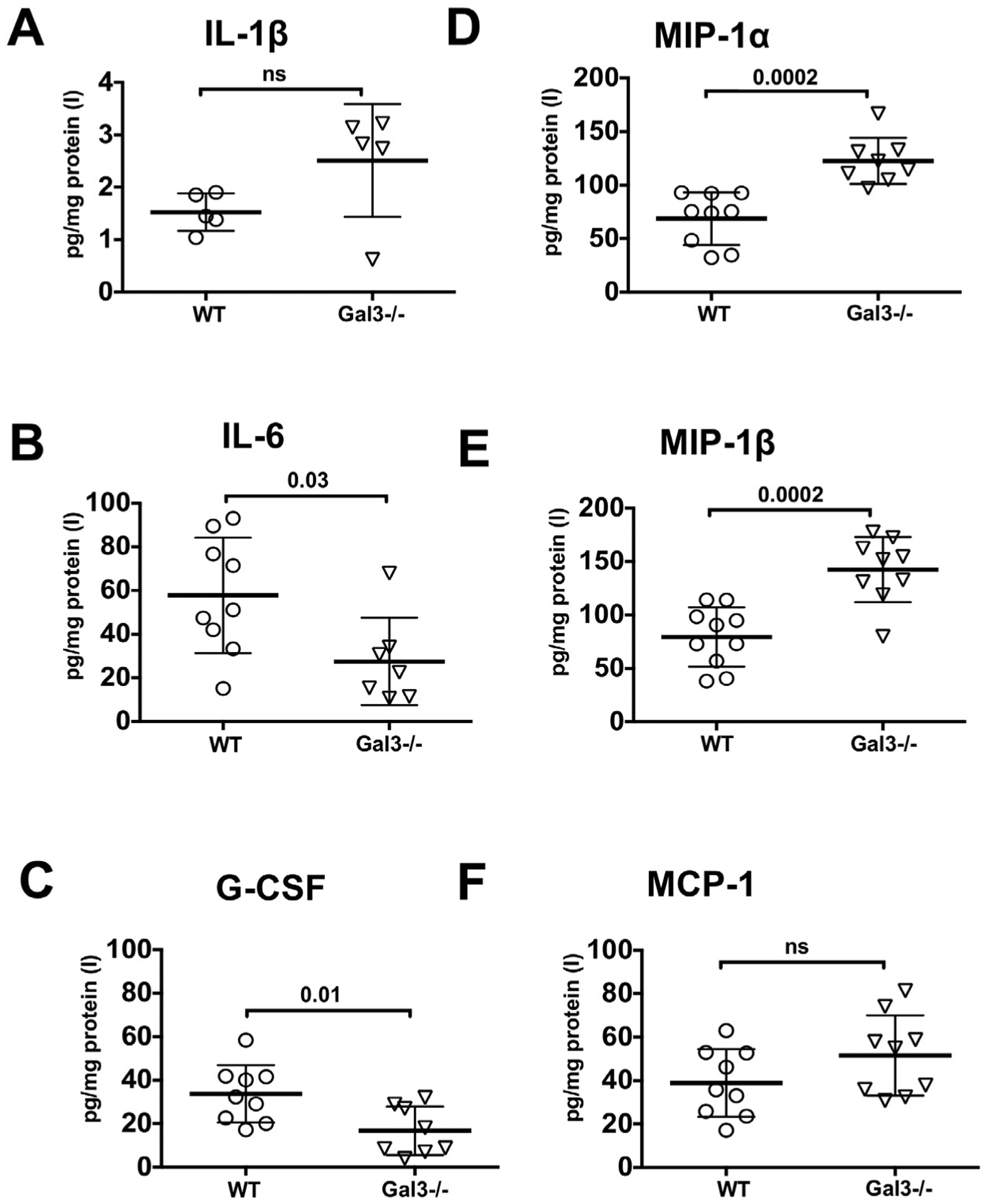
Effects of Gal3 genetic deletion on levels of individual cytokines and chemokines in injured regions of neonatal brain 72 h after tMCAO. A–F. Protein expression of IL-1β (A), IL-6 (B) G-CSF (C), MIP1α (D), MIP1β (E) and MCP-1 (F) measured by multiplex. Dots in A–F represent data from individual animals (n = 9 for WT, n = 10 for Gal3−/−). Mean ± SD. Significance levels as indicated on individual panels. ns: non-significant (Student’s t-test). (C) – contralateral, (I) – injured.
3.3. Gal3 deficiency does not affect morphological transformation but attenuates microglial activation after injury
To determine if lack of Gal3 changes the microglial response after tMCAO, we first examined morphological transformation and coverage of Iba1+ microglia in the cortex and in the caudate; the latter region is exclusively supplied by the MCA and, thus, most prone to injury after MCAO. Compared to contralateral regions, the number of Iba1+ microglia increased significantly in the cortex (Fig. 3A) and caudate (Fig. 3B) of both Gal3−/− and WT 72 h post-tMCAO, with similar changes in both groups.
Fig. 3.
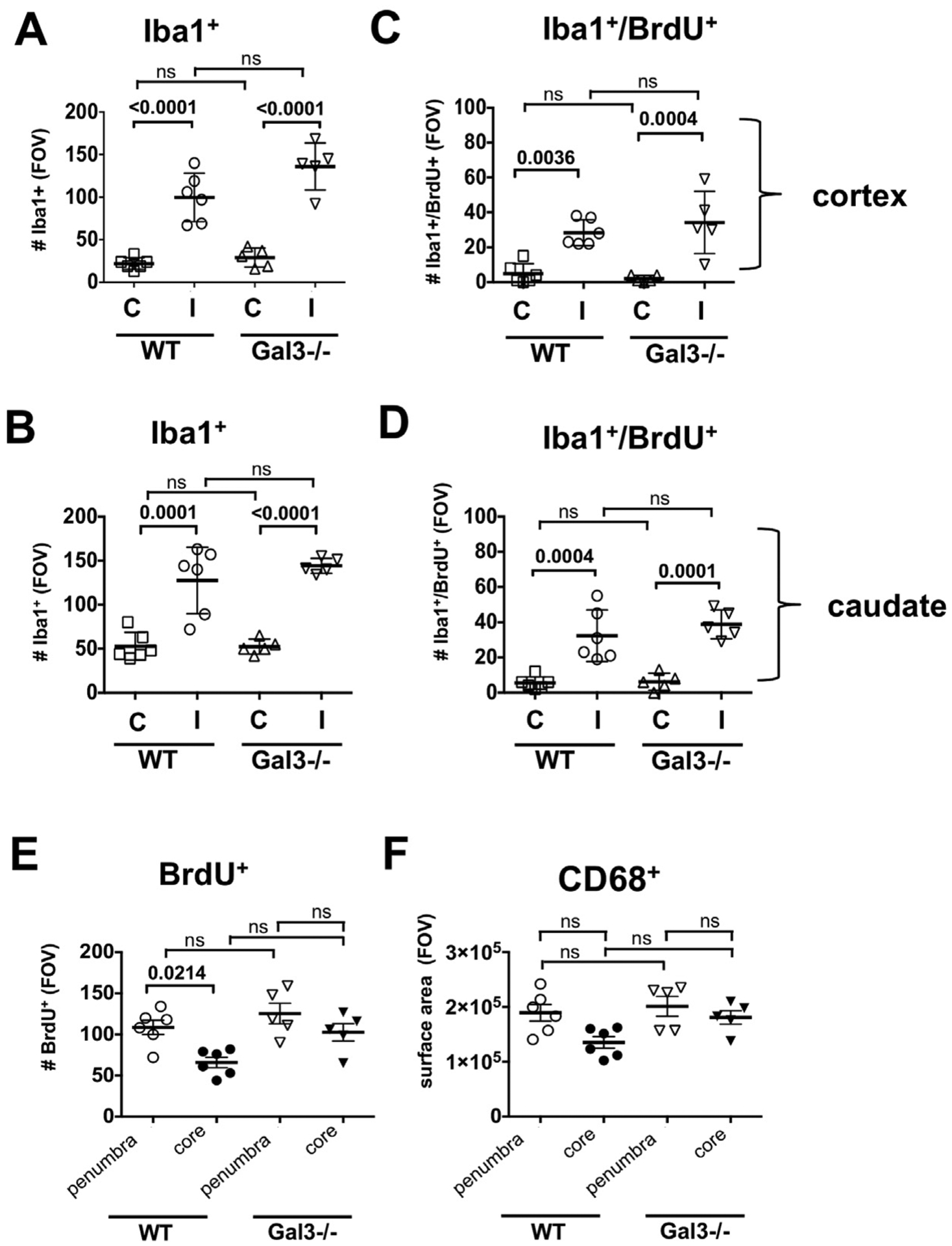
Microglial accumulation and proliferation are similar in injured cortex and caudate of Gal3−/− and WT mice 72h post-tMCAO. A–B. The number of Iba1+ in the cortex (A) and the caudate (B). C–D. Proliferation of Iba1+ cells in the cortex (C) and the caudate (D). E. The overall cell proliferation (BrdU+ cells) in the peri-focal injury and in the core. F. Acquisition of CD68+ cells in the peri-focal injury and in the core. FOV = 2.9 × 106 μm3. Dots in A–F represent data from individual animals (n = 6 for WT, n = 5 for Gal3−/−). Mean ± SD. Significance levels as indicated on individual panels, ns: non-significant (ANOVA with Bonferroni post hoc test). (C) – contralateral, (I) –injured.
Considering that proliferating and non-proliferating microglial cells contribute to the pathophysiology of adult stroke differently (Lalancette-Hebert et al., 2007), we then asked if Gal3 deficiency affects microglial proliferation. Microglial proliferation was low at 24 h (data not shown) but was significantly higher at 72 h in the injured cortex and caudate compared to respective matching contralateral regions (Fig. 3C and D). There was no significant difference in the overall number of BrdU+ cells (Fig. 3E) or the number of BrdU+/CD68+ cells between injured hemispheres of Gal3−/− and WT (Fig. 3F), indicating that lack of microglial Gal3 does not affect microglial proliferation.
We then used multi-color flow cytometry to characterize acquisition of microglial surface antigens after injury. Fig. 4 demonstrates the method we used to select viable single cell populations based on FSC/SSC plots (Fig. 4A) followed by gating on population of single cells (Fig. 4B). In some experiments, using Live/Dead staining, we confirmed that single cells used for analysis were viable.
Fig. 4.
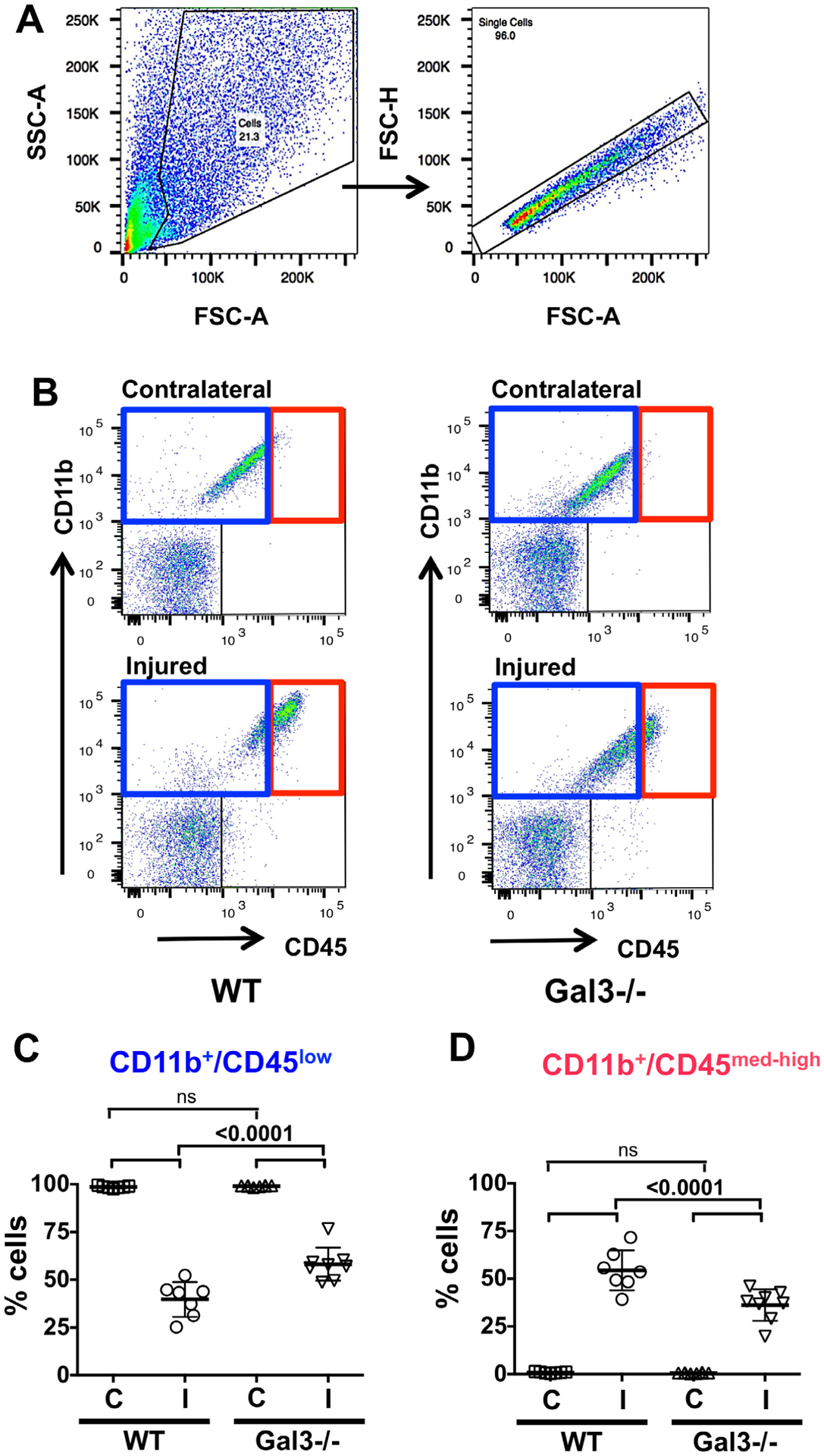
Lack of Gal3 attenuates CD11b+/CD45med-high acquisition 72 h after tMCAO. A. An example of gating strategy we use in flow cytometry data analysis to choose viable cells (left) and single cells (right). B. Representative plots of CD11b+/CD45+ cells in WT (left) and Gal3−/− mice (right) and selection of microglia (CD11b+/CD45low, blue rectangle; left upper quadrant) and activated microglia/macrophages (CD11b+/CD45med-high, red rectangle; right upper quadrant). Note corresponding blue/red color-coding in C–D. C–D. The number of CD11b+/CD45low cells (C) and CD11b+/CD45med-high (D). In injured regions, the number of CD11b+/CD45low is significantly higher in Gal3−/− whereas the number of CD11b+/CD45med-high is lower than in WT. Dots in C and D represent data from individual animals (n = 7 for WT, n = 8 for Gal3−/−). Mean ± SD. Significance levels as indicated on individual panels, ns: non-significant (ANOVA with Bonferroni post hoc test). (C) – contralateral, (I) – injured.
Because homeostatic microglial cells express low levels of CD45 and expression gradually increases during microglial activation (CD45med and CD45high), whereas monocytes constitutively express high CD45 levels (CD45high), we quantified microglial acquisition of CD11b+/CD45med-high. At 72 h, in the contralateral hemisphere of both groups, essentially all CD11b+ cells expressed low CD45 levels and the number of CD11b+/CD45low cells was similar. In injured regions of both groups, the number of CD11b+/CD45med-high significantly increased, in parallel to a marked reduction in the number of CD11b+/CD45low (Fig. 4C and D). Accumulation of CD11b+/CD45med-high cells was significantly more robust in WT mice than in Gal3−/− mice (Fig. 4D). To elucidate if lack of Gal3 alters upregulation of markers of alternatively activated microglia, we examined CD86+ or CD206+ expression in CD11b+/CD45low and CD11b+/CD45med-high cell populations. Importantly, upregulation of CD86+ or CD206+ was evident in injured regions, suggesting anti-inflammatory features of activated microglia/macrophages, but there was no difference in the number of CD86+ or CD206+ cells between WT and Gal3−/− (Suppl Fig. 2). To determine whether peripheral immune cells, such as monocytes, were affected by the lack of Gal3, Ly-6Clow and Ly-6Chigh cells of CD11b+/CD45med-high population were determined. In injured Gal3−/− pups, the number of Ly-6Clow was not affected but the number of Ly-6Chigh was significantly lower compared to WT (Suppl. Fig. 3).
3.4. Gal3 deficiency increases expression of integrin subunits αVβ3, but does not change vessel coverage or expression of several adhesion molecules 72h after tMCAO
Integrin αVβ3 is a major Gal3 binding protein. Gal3 mediates integrin-induced stabilization of focal adhesions and regulates cell motility (Goetz et al., 2008), contributing to cell-cell, cell-ECM interactions and angiogenesis (Boscher et al., 2011; Markowska et al., 2010; Wesley et al., 2013; Yan et al., 2009). Expression of αVβ3 integrin in the vasculature is affected by cerebral ischemia (Abumiya et al., 1999; Okada et al., 1996).
To test functional implications of genetic Gal3 deletion on the vasculature after neonatal stroke, we examined αV expression in injured regions. αV expression was below detectable levels in contralateral regions but upregulation of integrin αV was observed in both CD68+ cells and in areas not covered by CD68, most prominently in the ECM, lining the blood vessels in both Gal3−/− and WT (Fig. 5A). Brain volume covered by integrin αV was similar in perifocal regions in both groups, both in the caudate and cortex, but was larger in the ischemic core of Gal3−/− than in WT (Fig. 5B). Proliferation of BrdU+/CD68− cells was not affected in perifocal regions and in the ischemic core (Fig. 5C). Consistent with immunofluorescence data, significant increase of integrin αV expression was also evident in injured regions of Gal3−/− and WT mice by Western blot (Fig. 5D).
Fig. 5.
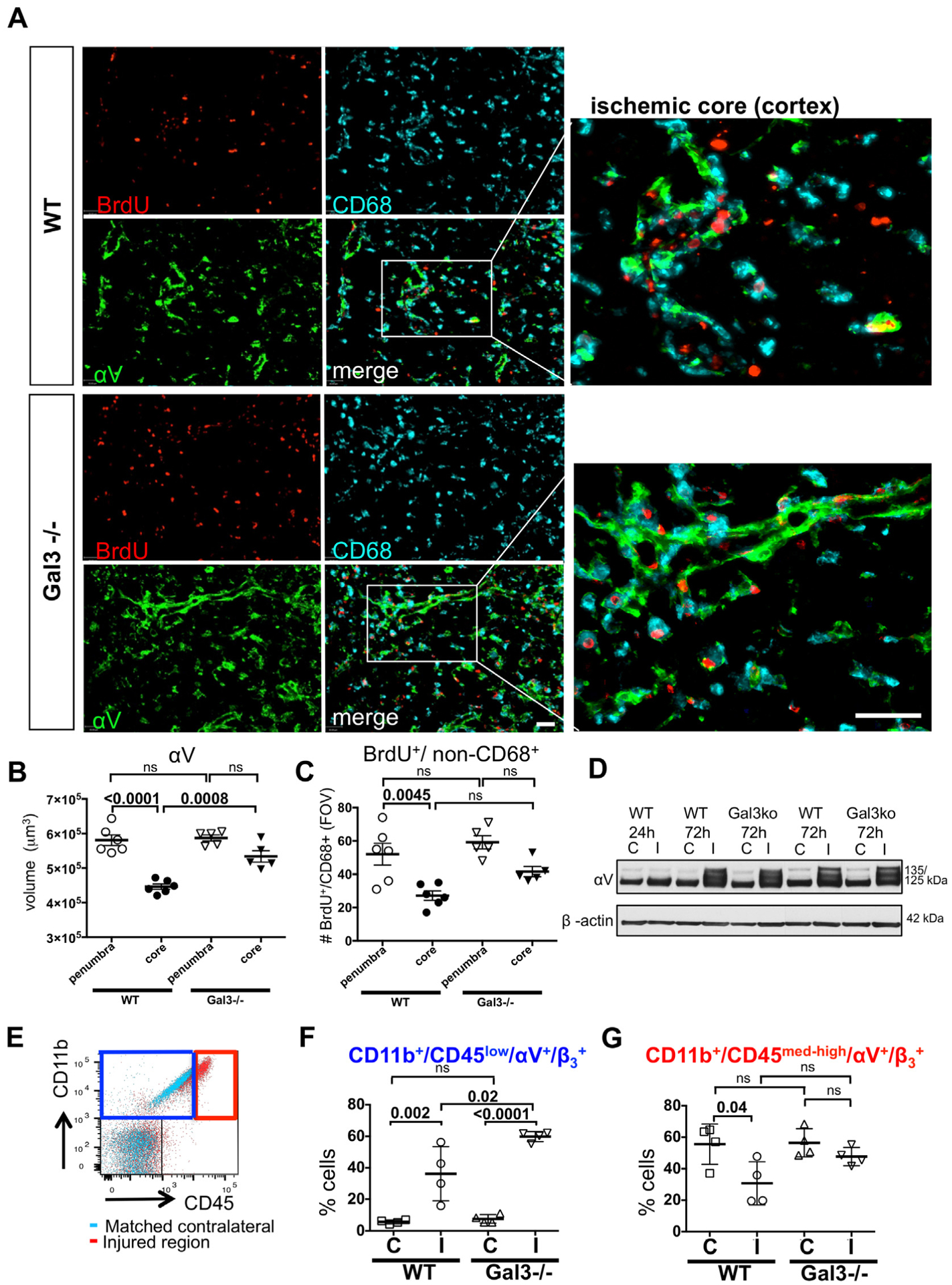
Microglia/macrophage coverage and expression of αV integrin in the penumbra and ischemic core. A. Examples of αV and CD68 expression in WT and Gal3−/− by immunofluorescence. B–C. Quantification of αV volume (B) and BrdU+/non-CD68+ cells (C) in the penumbra or ischemic core regions in the ischemic cortex or caudate (n = 6 for WT, n = 5 for Gal3−/−), Mean ± SD. Scale bar: 40 μm. FOV = 2.9 × 106 μm3. D. Representative example of Western blot of αV and β-actin expressed in WT and Gal3−/− at 24 h and 72 h post-MCAO. E. Overlay of dotplots of contralateral (aqua) and injured (red) regions. αV and β3 were gated from CD11b+/CD45low (blue square; left upper quadrant) or CD11b+/CD45med-high (red square; right upper quadrant). F. cell population in WT and Gal3−/− at 72 h post-MCAO. F–G. cells were gated from CD11b+/CD45low (in blue) or CD11b+/CD45med-high (in red) (n = 4 for WT, n = 4 for Gal3−/−). Dots in B, C, F and G represent data from individual animals. Results are shown as Mean ± SD. Significance levels are denoted by P values based on ANOVA with Bonferroni post hoc test. ns: non-significant. (C) – contralateral, (I) – injured.
FACS analysis showed increased expression of αV and β3 in CD11b+/CD45low cells of injured regions in WT and further increased expression in Gal3−/− mice (Fig. 5E and F). The increase of αV and β3 expression was more prominent in Gal3−/− than in WT mice (Fig. 5F). In contrast, in CD11b+/CD45med-high cells, αV and β3 expression was decreased in WT and unaffected in Gal3−/− mice (Fig. 5G). In injured regions of the same WT mice, αV and β3 were upregulated in cells of non-monocyte lineage (CD11b−/CD45− cells) and were further increased in injured regions of Gal3−/− mice (Fig. 6A).
Fig. 6.
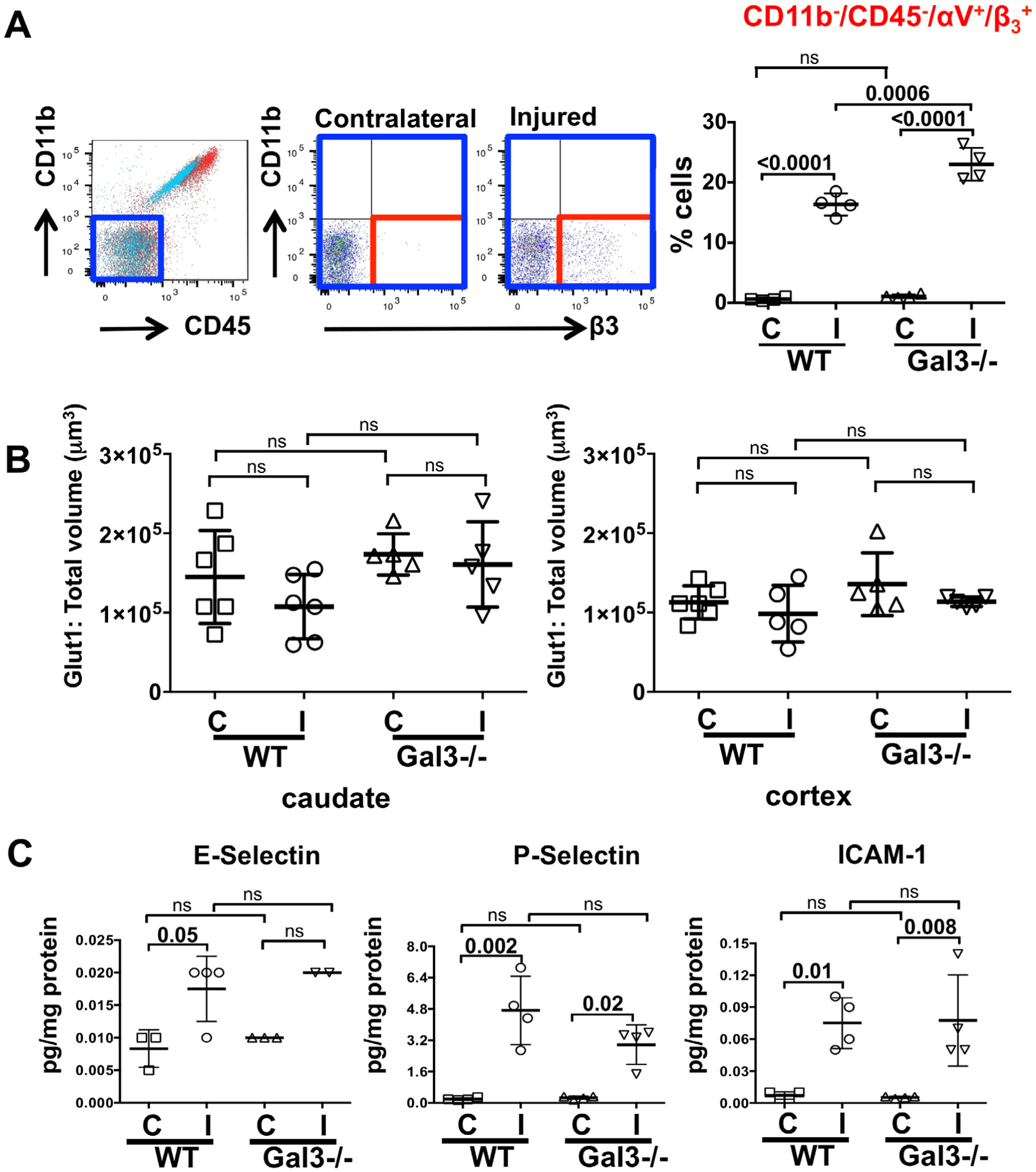
Lack of Gal3 does not change vessel coverage or acquisition of inducible endothelial antigens 72 h post-tMCAO. (A) A representative flow cytometry CD11b/CD45 plot (left) demonstrates the selection of CD11b−/CD45− cell population (blue rectangle; lower left quadrant), region chosen for nested analysis of cells in injured and contralateral regions (middle; lower right quadrant). Quantification of cells in the CD11b−/CD45− cell population is shown on the right. B. Total volume of Glut1+ blood vessels in ischemic and contralateral regions of WT and Gal3−/− mice (n = 6 for WT, n = 6 for Gal3−/−). C. Induction of E-selectin, P-selectin, ICAM-1 in the injured regions of both groups (n = 3–4 for WT, n = 2–4 for Gal3−/−). Dots in A–C represent data from individual animals. Mean ± SD. Significance levels as indicated on individual panels, ns: non-significant; ANOVA with Bonferroni post hoc test. (C) – contralateral, (I) – injured.
Considering that Gal3 affected integrin expression in the ECM, we evaluated vascular coverage and endothelial activation. The total volume of Glut1+ blood vessels within injured regions was similar in WT and Gal3−/− mice (Fig. 6B). As expected, tMCAO induced several endothelial antigens in injured regions, including E-selectin, P-selectin, and ICAM-1, but no significant differences in upregulation between groups were observed (Fig. 6C). Cumulatively, these data indicate that lack of Gal3 induces a compensatory increase in αVβ3 expression in injured regions, changes that occur in CD11b+/CD45low microglia and in cells of non-monocyte lineage, and that lack of Gal3 deregulates processes in the ECM.
4. Discussion
In the present study, we examined the role of Gal3 in injury after neonatal focal arterial stroke. We show that Gal3 deficiency affects levels of inflammatory mediators and worsens brain injury during the sub-chronic phase. Lack of Gal3 does not affect microglial morphological transformation or proliferation but lessens acquisition of microglial antigens and deregulates integrin expression by cells of both monocyte and non-monocyte lineage.
Galectins, a 15-member family of N-glycan-binding proteins, have functions in development, cancer, immunity, inflammation, as well as neurodegenerative diseases and stroke (Lalancette-Hebert et al., 2012; Rabinovich et al., 2002; Rotshenker, 2009; Schnaar, 2016; Yan et al., 2009). As do other lectins, Gal3 has a conserved sequence within the carbohydrate recognition domain (CRD) that has affinity for β-galactoside structures, a collagen-like internal R-domain, but Gal3 is unique within the family in that it has an amino-terminal non-lectin domain essential for its self-assembly and oligomerization (Boscher et al.,2011). Both the C- and N-termini play key roles for intracellular and extracellular signaling of Gal3, including glycosphingolipid-dependent membrane clustering and endocytosis of GPI-anchored proteins (Lakshminarayan et al., 2014) and formation of an extracellular galectin-glycoprotein lattice that regulates receptor tyrosine kinase signaling, cell migration and adhesion (Boscher et al., 2011). Gal3 can directly bind to key ECM glycoproteins and angiogenic mediators like integrin αVβ3, NG2 chondroitin-sulfate proteoglycan, collagen IV and laminin, contributing to tissue remodeling and angiogenesis (Yan et al., 2009; Yang et al., 2007). Localization of Gal3, whether intracellular, extracellular, cytoplasmic, or nuclear, can also influence inflammation and alternative macrophage activation (MacKinnon et al., 2008). Thus, Gal3 can serve as a pro-inflammatory and anti-inflammatory molecule in a context and time-dependent manner after injury.
In an adult stroke model, Gal3 expression increases gradually, with activated microglia/macrophages as the predominant cell types that upregulate Gal3 (Lalancette-Hebert et al., 2007; Lalancette-Hebert et al., 2012; Yan et al., 2009). Astrocytes were also shown to upregulate Gal3 but at later time points, during the angiogenic phase when repair occurs (Yan et al., 2009). Genetic deletion of Gal3 enhances apoptosis, affects microglial phenotypes, reduces microglial proliferation and exacerbates injury after adult stroke (Lalancette-Hebert et al., 2012). Decreased stroke-induced angiogenesis and proliferation of neural progenitors following infusion of a neutralizing anti-Gal3 antibody into the ischemic striatum is also consistent with Gal3 contribution to brain repair (Yan et al., 2009). In neonatal mice subjected to H-I, Gal3 exacerbates injury in the hippocampus and striatum (Doverhag et al., 2010). Like in injured adults after stroke, in Gal3−/− pups after H-I, the main source of upregulated Gal3 are activated microglia/- macrophages but, unlike in adult stroke, in neonates, the number of microglial cells is increased, while levels of toxic species (nitrotyrosine) are decreased and levels of trophic factors and apoptotic markers are unaffected (Doverhag et al., 2010). Reconciling these differing results is not straightforward because of brain immaturity, models used (focal stroke vs. systemic hypoxia combined with permanent CCA ligation), differences in the timing of histological outcomes, and distinct microglial markers used for analysis (Iba1 vs. CD11b).
In our study, lack of Gal3 extends sub-chronic injury induced by tMCAO, during the time frame when microglial activation and inflammation are evident (Denker et al., 2007; Li et al., 2015), neuronal apoptosis is profound (Manabat et al., 2003; Woo et al., 2012) and angiogenesis is low (Fernandez-Lopez et al., 2013; Shimotake et al., 2010). Consistent with previous reports in adult stroke (Lalancette-Hebert et al., 2012) and in neonates after H-I (Doverhag et al., 2010), Gal3 upregulation occurs gradually and almost exclusively in activated microglia/macrophages in both mouse and rat pups subjected to tMCAO. As in adult mice after tMCAO, lack of Gal3 leads to more severe injury, but in contrast to adult, microglial proliferation and morphological transformation caused by stroke does not depend on Gal3 in neonates.
Perinatal stroke and cerebral palsy are more common in males than in females (Johnston and Hagberg, 2007; Nunez, 2012), and sex and sex hormones may have independent effects in stroke in children (Normann et al., 2009; Vannucci and Hurn, 2009) and in immature mice (Herson et al., 2013). Consistent with the latter notion, Gal3 affects longer-term injury (7-days post-H-I) in male but not in female pups (Doverhag et al., 2010). Comparisons of injury severity in male and female mouse pups in our study show no evidence of sex differences in each group during the sub-chronic injury phase.
Cytokine and chemokine production contributes to neuroinflammation after neonatal H-I (Bona et al., 1999; Hedtjarn et al., 2004) and tMCAO (Denker et al. 2007; Woo et al. 2012), but pharmacologic depletion of microglial cells or disruption of phagocytosis of neuronal debris by activated microglia further enhance neuroinflammation and worsen histological outcome (Faustino et al., 2011; Woo et al., 2012), indicating that microglial cells serve as endogenous protectants after neonatal stroke. Lack of Gal3 further deregulates production of several cytokines and chemokines triggered by tMCAO, including an increase of two major microglial/monocyte chemoattractant proteins, MIP1α and MIP1β. However, the number of Iba1+ cells is not increased in spite of enhanced chemoattractant gradient in Gal3 deficient mice. Furthermore, within 72 h, tMCAO induces a similar extent of microglial morphological transformation, proliferation and acquisition of CD68 in both groups. At the same time, the number of CD11b+/CD45med-high microglia/macrophages is significantly lower in Gal3−/− pups in the absence of a CD45 ligand, Gal3. Consistent with previous data on the lack of TNFα induction, upregulation of arginase-1 and unaffected Toll-like-receptor expression in injured regions of WT mice (Li et al., 2015), nested analysis of CD11b+/CD45med-high cells shows abundantly present CD86+ and CD206+ cells in injured regions of WT, suggesting a non-toxic nature of microglial cells. In addition, Gal3 deficiency does not induce IL-1β or TNFα. Together, these data demonstrate a different pattern of microglial response to Gal3 deficiency than in adult stroke.
For decades microglia have been perceived as purely toxic in adult stroke because of production of cytokines and reactive oxygen species. However, more recent stroke studies have revealed the complexity of the effects of subpopulations of cells of the monocyte lineage. The source of inflammatory cytokines, microglial vs. monocytic, was shown to produce opposite effects in stroke (Lambertsen et al., 2009) and microglial cells were in fact recently shown to play a protective role in adult stroke (Szalay et al., 2016). Although by now endogenous protective role of microglia was demonstrated in adult stroke (Szalay et al., 2016) and in neonatal stroke (Faustino et al., 2011), the underlying mechanisms are likely dependent on brain maturation at the time of stroke. During the postnatal period, microglia play a key role in shaping-up brain connectivity by phagocytosis of overproduced neurons and weak synapses (Hoshiko et al., 2012; Paolicelli et al., 2011; Schafer et al., 2012). Microglial cells themselves undergo marked maturational changes in postnatal brain (Butovsky et al., 2014), in parallel with a decline of programmed neuronal death and a lesser contribution of neuronal apoptosis to cerebral ischemia injury with brain maturation (Hu et al., 2000).
We used several markers to distinguish microglia versus monocytes and show that the acquisition of CD45 by CD11b+ cells within injured regions depends on Gal3. The abundance of cells expressing CD86+ and CD206+, markers considered as indicators of non-toxic CD11b+/CD45+ cells, together with the presence of well-defined CD11b+/CD45+/Ly6Clow cell subpopulations in injured regions of both WT and Gal3−/−, are in agreement with our previous observation of endogenously neuroprotective nature of cells of the monocyte lineage early after neonatal stroke (Faustino et al., 2011) and with the data that migration and infiltration of toxic CCR2RFP/+ monocytes is low 24 h in our stroke model (Fernandez-Lopez et al., 2016). At the same time, considering the continuously evolving concepts on expression and associated functions of various antigens by individual leukocyte subsets after injury, additional data would be needed to confirm identities of the cells, microglia vs. monocyte-derived brain macrophages. Such information in general can be obtained by either using antibodies that are exclusive for microglia or monocytes (Butovsky et al., 2014), but such antibodies are not commercially available, or by the use of bone marrow chimeras, as was recently studied in adult stroke (Courties et al., 2015), but such studies aren’t executable in neonates.
Gal3 is part of a dynamic extracellular environment that modulates cellular behavior, regulates tissue function, processes that depend on the glycan profiles of surface-resident glycoproteins and glycolipids. Multiple integrin subunits bind to galectins, including Gal3. Heterodimers formed by alpha and beta integrin subunits guide cell-cell and cell-ECM interactions and activate signaling pathways important for cell survival, proliferation, differentiation, and migration (Giancotti and Ruoslahti, 1999; Miranti and Brugge, 2002). A major Gal3 binding protein, integrin αVβ3, is strongly expressed on activated endothelial cells but is weakly expressed on resting endothelial cells. Gal3 mediates integrin-induced stabilization of focal adhesions and regulates cell motility (Goetz et al., 2008). Together with intercellular and adhesion molecules expressed on endothelial and immune cells, integrins and chemokines are involved in regulating attachment, adhesion, and infiltration of peripheral cells (del Zoppo and Milner, 2006; Okada et al., 1994; Zhang et al., 1996). We focused on Gal3 interactions with αV and β3 integrins, as the latter integrin subunits have been demonstrated to be important in adult stroke, but other integrins, such as β1 integrin, also bind and mediate Gal3 signaling in endocytosis and focal adhesion turnover (Goetz et al., 2008). Composition of the ECM proteins produced by endothelial cells differs greatly between uninjured neonatal and adult brain and, additionally, after stroke (Fernandez-Lopez et al., 2012). Furthermore, individual ECM components and individual cell types, pericytes and astrocytes, mature at a different pace (Peppiatt et al., 2006; Zhang and Barres, 2010), with continuously increasing vessel coverage by pericytes and astrocytes during postnatal brain maturation (Daneman et al., 2010). The endfoot-basal lamina junctional complex is consolidated after the first postnatal week (Lunde et al., 2015), likely affecting protein interactions at the ECM-vascular-astrocyte interface. Consistent with data in adult stroke, tMCAO in WT pups upregulates αV and β3 subunits on microglia and cells of non-monocyte lineage, but, interestingly, Gal3 deficiency further upregulates αV and β3 subunits in injured pups, likely as a compensatory response via Gal3-independent mechanisms.
In this study, we focused on Gal3 effects in early injury after neonatal focal stroke, but as a pleotropic molecule, Gal3 may also exert a different signaling repertoire during brain repair. The Gal3 effects may also depend on the animal models used, species and genetic background, as is evident in adults (Mostacada et al., 2015; Nishihara et al., 2016) and neonates (Ek et al., 2015; Fernandez-Lopez et al., 2012).
To summarize, genetic deletion of Gal3 worsens injury, imbalances production of pro- and anti-inflammatory molecules and slows acquisition of microglial surface antigens. Gal3 deficiency also deregulates the composition of the ECM. However, opposite to reports in adult stroke, microglial proliferation is unaffected by Gal3 deficiency as is accumulation of CD68. Additional studies are needed to understand whether a hierarchy in Gal3-mediated signaling in immature microglial cells accounts for its role in neonatal stroke.
Supplementary Material
Acknowledgments
The authors acknowledge Dr. Jasna Kriz for useful discussions and for providing an anti-Gal3 antibody, and Shivani Mahuvakar for technical help. The work was supported by RO1 NS44025 (Z. S.V), RO1 NS76726 (Z.S.V), Fundación Ramón Areces, Madrid, Spain (D.F.L) and NSFC 31260242 (F.L).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bbi.2016.11.005.
References
- Abumiya T, Lucero J, Heo JH, Tagaya M, Koziol JA, Copeland BR, del Zoppo GJ, 1999. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)beta3 during focal cerebral ischemia. J .Cereb. Blood Flow Metab 19, 1038–1050. [DOI] [PubMed] [Google Scholar]
- Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H, 1999. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr. Res 45, 500–509. [DOI] [PubMed] [Google Scholar]
- Boscher C, Dennis JW, Nabi IR, 2011. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol 23, 383–392. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E, Santiago M, Viceconte N, Oliva-Martin MJ, Osman AM, et al. , 2015. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, Sun Y, Severe N, Dutta P, Scharff J, 2015. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res 116, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danella Polli C, Alves Toledo K, Franco LH, Sammartino Mariano V, de Oliveira LL, Soares Bernardes E, Roque-Barreira MC, Pereira-da-Silva G, 2013. Monocyte migration driven by galectin-3 occurs through distinct mechanisms involving selective interactions with the extracellular matrix. ISRN Inflamm. 2013, 259256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA, 2010. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R, 2006. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler. Thromb. Vasc. Biol 26, 1966–1975. [DOI] [PubMed] [Google Scholar]
- Denker S, Ji S, Lee SY, Dingman A, Derugin N, Wendland M, Vexler ZS, 2007. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem 100, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derugin N, Ferriero DM, Vexler ZS, 1998. Neonatal reversible focal cerebral ischemia: a new model. Neurosci. Res 32, 349–353. [DOI] [PubMed] [Google Scholar]
- Derugin N, Dingman A, Wendland M, Fox C, Vexler ZS, 2005. Magnetic resonance imaging as a surrogate measure for histological sub-chronic endpoint in a neonatal rat stroke model. Brain Res. 1066, 49–56. [DOI] [PubMed] [Google Scholar]
- Doverhag C, Keller M, Karlsson A, Hedtjarn M, Nilsson U, Kapeller E, Sarkozy G, Klimaschewski L, Humpel C, Hagberg H, 2008. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol. Dis 31, 133–144. [DOI] [PubMed] [Google Scholar]
- Doverhag C, Hedtjarn M, Poirier F, Mallard C, Hagberg H, Karlsson A, Savman K, 2010. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol. Dis 38, 36–46. [DOI] [PubMed] [Google Scholar]
- Ek CJ, D’Angelo B, Baburamani AA, Lehner C, Leverin AL, Smith PL, Nilsson H, Svedin P, Hagberg H, Mallard C, 2015. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J. Cereb. Blood Flow Metab 35, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino J, Wang X, Jonhson C, Klibanov A, Derugin N, Wendland M, Vexler ZS, 2011. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci 31, 12992–13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Daneman R, Zhou L, Lee SY, Derugin N, Wendland MF, Vexler ZS, 2012. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J. Neurosci 32, 9588–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Derugin N, Vexler ZS, 2013. Acute and chronic vascular responses to experimental focal arterial stroke in the neonate rat. Transl. Stroke Res 4, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Natarajan N, Ashwal S, Vexler ZS, 2014. Mechanisms of perinatal arterial ischemic stroke. J. Cereb. Blood Flow Metab 34, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Klibanov AL, Derugin N, Blanchard E, Simon F, Leib SL, Vexler ZS, 2016. Microglial cells prevent hemorrhage in neonatal focal arterial stroke. J. Neurosci 36, 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E, 1999. Integrin signaling. Science 285, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Goetz JG, Joshi B, Lajoie P, Strugnell SS, Scudamore T, Kojic LD, Nabi IR, 2008. Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J. Cell Biol 180, 1261–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Ferriero D, Vannucci S, Levison S, Vexler Z, Gressens P, 2015. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol 11, 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Hagberg H, 2004. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab 24, 1333–1351. [DOI] [PubMed] [Google Scholar]
- Hedtjarn M, Mallard C, Iwakura Y, Hagberg H, 2005. Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia-ischemia in the immature brain. Dev. Neurosci 27, 143–148. [DOI] [PubMed] [Google Scholar]
- Herson PS, Bombardier CG, Parker SM, Shimizu T, Klawitter J, Quillinan N, Exo JL, Goldenberg NA, Traystman RJ, 2013. Experimental pediatric arterial ischemic stroke model reveals sex-specific estrogen signaling. Stroke 44, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko M, Arnoux I, Avignone E, Yamamoto N, Audinat E, 2012. Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci 32, 15106–15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjo BK, 2000. Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J. Cereb. Blood Flow Metab 20, 1294–1300. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J, 2011. The immunology of stroke: from mechanisms to translation. Nat. Med 17, 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Hagberg H, 2007. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol 49, 74–78. [DOI] [PubMed] [Google Scholar]
- Krzeslak A, Lipinska A, 2004. Galectin-3 as a multifunctional protein. Cell Mol. Biol. Lett 9, 305–328. [PubMed] [Google Scholar]
- Lakshminarayan R, Wunder C, Becken U, Howes MT, Benzing C, Arumugam S, Sales S, Ariotti N, Chambon V, Lamaze C, 2014. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat. Cell Biol 16, 595–606. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J, 2007. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci 27, 2596–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J, 2012. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci 32, 10383–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, 2009. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci 29, 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Faustino J, Woo M, Derugin N, Vexler ZS, 2015. Lack of the scavenger receptor CD36 alters microglial phenotypes after neonatal stroke. J. Neurochem 13, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde LK, Camassa LM, Hoddevik EH, Khan FH, Ottersen OP, Boldt HB, Amiry-Moghaddam M, 2015. Postnatal development of the molecular complex underlying astrocyte polarization. Brain Struct. Funct 220, 2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T, 2008. Regulation of alternative macrophage activation by galectin-3. J. Immunol 180, 2650–2658. [DOI] [PubMed] [Google Scholar]
- Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, Vexler ZS, 2003. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska AI, Liu FT, Panjwani N, 2010. Galectin-3 is an important mediator of VEGF-and bFGF-mediated angiogenic response. J. Exp. Med 207, 1981–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS, 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol 4, E83–E90. [DOI] [PubMed] [Google Scholar]
- Mostacada K, Oliveira FL, Villa-Verde DM, Martinez AM, 2015. Lack of galectin-3 improves the functional outcome and tissue sparing by modulating inflammatory response after a compressive spinal cord injury. Exp. Neurol 271, 390–400. [DOI] [PubMed] [Google Scholar]
- Nelson KB, 2007. Perinatal ischemic stroke. Stroke 38, 742–745. [DOI] [PubMed] [Google Scholar]
- Nelson KB, Lynch JK, 2004. Stroke in newborn infants. Lancet Neurol. 3, 150–158. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Shimizu F, Kitagawa T, Yamanaka N, Akada J, Kuramitsu Y, Sano Y, Takeshita Y, Maeda T, Abe M, et al. , 2016. Identification of galectin-3 as a possible antibody target for secondary progressive multiple sclerosis. Mult. Scler [DOI] [PubMed] [Google Scholar]
- Normann S, de Veber G, Fobker M, Langer C, Kenet G, Bernard TJ, Fiedler B, Strater R, Goldenberg NA, Nowak-Gottl U, 2009. Role of endogenous testosterone concentration in pediatric stroke. Ann. Neurol 66, 754–758. [DOI] [PubMed] [Google Scholar]
- Nunez J, 2012. Sex and steroid hormones in early brain injury. Rev. Endocrinol. Metab. Disord 13, 173–186. [DOI] [PubMed] [Google Scholar]
- Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, del Zoppo GJ, 1994. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke 25, 202–211. [DOI] [PubMed] [Google Scholar]
- Okada Y, Copeland BR, Hamann GF, Koziol JA, Cheresh DA, del Zoppo GJ, 1996. Integrin alphavbeta3 is expressed in selected microvessels after focal cerebral ischemia. Am. J. Pathol 149, 37–44. [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Pasquini LA, Millet V, Hoyos HC, Giannoni JP, Croci DO, Marder M, Liu FT, Rabinovich GA, Pasquini JM, 2011. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 18, 1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D, 2006. Bidirectional control of CNS capillary diameter by pericytes. Nature 443, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Baum LG, Tinari N, Paganelli R, Natoli C, Liu FT, Iacobelli S, 2002. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 23, 313–320. [DOI] [PubMed] [Google Scholar]
- Raju TN, Nelson KB, Ferriero D, Lynch JK, 2007. Ischemic perinatal stroke: summary of a workshop sponsored by the National Institute of Child Health and Human Development and the National Institute of Neurological Disorders and Stroke. Pediatrics 120, 609–616. [DOI] [PubMed] [Google Scholar]
- Rotshenker S, 2009. The role of Galectin-3/MAC-2 in the activation of the innate-immune function of phagocytosis in microglia in injury and disease. J. Mol. Neurosci 39, 99–103. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, 2016. Glycobiology simplified: diverse roles of glycan recognition in inflammation. J. Leukocyte Biol 99, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Ito M, Yoshimura A, 2014. Post-ischemic inflammation regulates neural damage and protection. Front. Cell Neurosci 8, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM, 2010. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke 41, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, Csaszar E, Fekete R, West BL, Katona G, et al. , 2016. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun 7, 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci SJ, Hurn PD, 2009. Gender differences in pediatric stroke: is elevated testosterone a risk factor for boys? Ann. Neurol 66, 713–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vexler ZS, Yenari MA, 2009. Does inflammation after stroke affect the developing brain differently than adult brain? Dev. Neurosci 31, 378–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley UV, Vemuganti R, Ayvaci ER, Dempsey RJ, 2013. Galectin-3 enhances angiogenic and migratory potential of microglial cells via modulation of integrin linked kinase signaling. Brain Res. 1496, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo MS, Wang X, Faustino J, Derugin N, Wendland MF, Zhou P, Iadecola C, Vexler ZS, 2012. Genetic deletion of CD36 enhances injury after acute neonatal stroke. Ann. Neurol 72, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager JY, Ashwal S, 2009. Animal models of perinatal hypoxic-ischemic brain damage. Pediatr. Neurol 40, 156–167. [DOI] [PubMed] [Google Scholar]
- Yan YP, Lang BT, Vemuganti R, Dempsey RJ, 2009. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 1288, 116–124. [DOI] [PubMed] [Google Scholar]
- Yang E, Shim JS, Woo HJ, Kim KW, Kwon HJ, 2007. Aminopeptidase N/CD13 induces angiogenesis through interaction with a pro-angiogenic protein, galectin-3. Biochem. Biophys. Res. Commun 363, 336–341. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Barres BA, 2010. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol 20, 588–594. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, Manning A, 1996. E-selectin in focal cerebral ischemia and reperfusion in the rat. J. Cereb. Blood Flow Metab 16, 1126–1136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.