

Abstract
Acute flaccid myelitis (AFM) is a disabling, polio-like illness mainly affecting children. Outbreaks of AFM have occurred across multiple global regions since 2012, and the disease appears to be caused by non-polio enterovirus infection, posing a major public health challenge. The clinical presentation of flaccid and often profound muscle weakness (which can invoke respiratory failure and other critical complications) can mimic several other acute neurological illnesses. There is no single sensitive and specific test for AFM, and the diagnosis relies on identification of several important clinical, neuroimaging, and cerebrospinal fluid characteristics. Following the acute phase of AFM, patients typically have substantial residual disability and unique long-term rehabilitation needs. In this Review we describe the epidemiology, clinical features, course, and outcomes of AFM to help to guide diagnosis, management, and rehabilitation. Future research directions include further studies evaluating host and pathogen factors, including investigations into genetic, viral, and immunological features of affected patients, host–virus interactions, and investigations of targeted therapeutic approaches to improve the long-term outcomes in this population.
Introduction
Unusual clusters of a disabling, polio-like illness, now termed acute flaccid myelitis (AFM), were recognised in California in 2012, and Colorado in 2014.1,2 AFM is now recognised as a global disease, with hundreds of cases reported across Europe,3,4 Asia,5–7 Australia,8 Africa,9 North America,10,11 and South America.12,13 Epidemic enteroviral infection is believed to be the main driver of AFM in recent years, particularly enterovirus D68 infection.14 Cases have usually occurred in geographical clusters, with a distinct seasonal biennial pattern in temperate regions.15 AFM most frequently affects young children, and is characterised by acute onset of flaccid weakness of one or more limbs, with MRI showing abnormalities of the spinal cord grey matter.5 Trunk, neck, respiratory, bulbar, facial, and extraocular muscles can also be affected. The clinical presentation of AFM may mimic other causes of acute weakness such as Guillain-Barré syndrome, demyelinating myelitis, and other infectious myelitis. The diagnosis of AFM can be informed by interpretation of the clinical features alongside findings of laboratory, neuroimaging, and electrophysiological tests.
Acute management of AFM is largely supportive because there is an absence of therapeutic agents proven to alter outcomes. A substantial proportion of patients with AFM will become critically ill during the acute illness, requiring intubation due to respiratory failure or severe bulbar weakness.16,17 Neurological recovery after AFM is usually incomplete, with many patients having substantial residual weakness and muscle atrophy. Over the long term, patients can be affected by a range of neurological, musculoskeletal, and psychological sequelae.18–20 Appropriate rehabilitation can improve functional status and quality of life after AFM.19 Additionally, surgical approaches including tendon or nerve transfer surgery have been used in individual cases to manage residual impairments.21,22 In this Review we describe the epidemiology, clinical features, course, and outcomes of AFM to help to guide diagnosis, management, and rehabilitation.
Epidemiology and cause
Several features support a viral link to AFM cases. Most individuals affected by AFM report a febrile prodrome accompanied by respiratory symptoms in the days before the onset of weakness.15,23 The primary sites of neurological involvement parallel poliomyelitis, with lesions targeting the anterior horn cells of the spinal cord and motor nuclei of the brainstem. To date, the virus suspected to be the predominant driver of the seasonal, biennial outbreaks of AFM observed in many global regions is enterovirus D68, although other enteroviruses (particularly enterovirus A71) and some coxsackie virus strains have also been implicated. Evidence specifically supporting the causal association of AFM with enterovirus D68 includes: (1) temporal and geographical correlations between AFM cases and enterovirus D68 circulation;1,24(2) enterovirus D68 predominating amongst pathogens identified in biological specimens (typically respiratory samples) from individuals with AFM, across many geographical regions;3,15,23 (3) recent emergence of strains of enterovirus D68 that could have acquired the ability to cause AFM;25–27 (4) a higher frequency of enterovirus-specific antibodies in the cerebrospinal fluid (CSF) of patients with AFM than in controls (albeit without definitive evidence of intrathecal synthesis);28,29 and (5) mouse models in which recent enterovirus D68 strains cause AFM-like limb paralysis with virus isolated from and visualised in the spinal cord.26,30,31
The specific mechanism by which infection with enterovirus D68 leads to AFM is not fully understood and represents a key question for future research. Enterovirus D68 most commonly causes respiratory disease,32,33 but there is ample precedent amongst other enteroviruses (particularly poliovirus) for occasional spread to the grey matter of the spinal cord, supported by evidence from autopsies.34–36 The pathogen, environmental, and host factors that can mediate progression to neurological disease are unknown. Mouse models and neuronal cell culture models suggest that recent strains of enterovirus D68 have evolved in terms of their capability of accessing the nervous system (neuroinvasion), their capacity to infect neurons (neurotropism), their ability to cause nervous system disease (neurovirulence), or any combination thereof.26,30,37,38 A lack of spinal cord or brain tissue specimens from affected patients has impeded direct confirmation of this possibility in humans. Also confounding characterisation of the recent AFM outbreaks is the infrequency of direct viral isolation or viral genome detection from the CSF at the time of clinical presentation, even with sensitive and unbiased pathogen discovery technologies.25,39 This difficulty nonetheless parallels similar experience with other viruses manifesting occasional neurotropic spread (eg, wild type poliovirus, vaccine-derived poliovirus, and West Nile virus).
The potential of other alternative viral causes as major contributors to recent AFM outbreaks would appear to be diminished by clinical features, reported investigations, and epidemiological characteristics. The first of these, enterovirus A71 is generally associated with outbreaks of hand-foot-mouth disease but also shows similar occasional tropism for the nervous system manifesting a poliomyelitis-like paralysis, thus meeting the AFM criteria. In regions reporting the recent increases of AFM cases, however, identified cases associated with enterovirus A71 have been less frequent than those associated with enterovirus D68, and could differ in clinical phenotype.3,5,16,40 Additionally, AFM cases associated with enterovirus A71 have been geographically restricted, with outbreaks mainly reported in the Asia-Pacific region (where the virus has been endemic since the 1990s),35,41 and more recently on a smaller scale in the USA and Europe.40,42,43 The other notable candidate viruses include the flaviviruses, whose members include the arboviruses, West Nile virus, and Japanese encephalitis virus, which can cause acute flaccid paralysis related to anterior horn cell involvement. Multiple epidemiological and clinical class characteristics of arboviral infections undermine the argument that they are a major cause of recent AFM outbreaks, including: (1) infections are vector-borne and occur seasonally in endemic regions, unlike the more ubiquitous recent worldwide distribution of AFM; (2) arboviral infections typically affect adults more commonly than they do children; (3) patients typically have characteristic systemic features such as rash or vomiting; and (4) when nervous system involvement is present it tends to include meningoencephalitis, with motor-neuron-limited presentations being less common.44 Thus, diagnosis of a specific arboviral infection in a patient manifesting a clinical syndrome of AFM will trigger diagnostic and management protocols already in existence.45
In reviewing the literature regarding AFM, some observational studies have been specifically restricted to patients with AFM associated with enterovirus D68,3 whereas other studies have not applied this criterion.23 For the purposes of clinical research, defining the disease by the associated organism provides a study population with more uniform pathophysiology, which is unlikely to include clinical mimics. Indeed, AFM cases occurring in years with epidemic peaks (ie, with clear-cut outbreaks driven by a single virus) show much greater clinical and paraclinical homogeneity than do AFM cases occurring in non-peak years.15 However, since enterovirus D68 may only be detectable in laboratory specimens in the early stage of the disease and other non-polio enteroviruses are likely to cause a small proportion of the AFM burden globally, defining the disease by the associated organism is not pragmatic for the purposes of clinical practice. Currently, the absence of sensitive confirmatory testing for specific non-polio enteroviruses (such as serological testing) represents a major barrier to aetiological confirmation, clinical assessment, and disease surveillance.
Clinical presentation
AFM is predominantly a childhood disease (median age 6·3 years),16 with less than 15% of cases occurring in adults (more commonly in the immunocompromised), although AFM in adults could be under-recognised or under-reported.1,3–5,12,23,46 A slight predilection for males has been suggested.4,5,15,23 Most patients with AFM have a prodromal illness manifesting with fever and respiratory symptoms (cough, rhinorrhea, pharyngitis, or asthmalike illness). Gastrointestinal symptoms such as vomiting or diarrhoea are less frequent.15,23 Household contacts with similar prodromal illnesses are common; however, there have been no reported occurrences in the USA of multiple cases of AFM occurring in one household or family. The epidemiological context can provide useful clues, because known outbreaks of enterovirus D68 or A71 (or other confirmed AFM cases) in an area might prompt clinicians to consider AFM in patients presenting with acute weakness. Onset of neurological symptoms typically occurs 1–10 days after onset of the infectious prodrome, with many patients reporting improvement in prodromal symptoms before onset of neurological symptoms.5,23
The onset of neurological symptoms can be accompanied by headache, neck stiffness, or recurrence of fever (table 1). Meningism can be present in this early stage. In many patients, limb weakness is heralded by pain in the affected limb(s), neck, or lower back. Flaccid weakness is typically asymmetric and can affect one or more limbs, with predilection for the upper limbs and proximal muscle groups.23 Unlike many other causes of acute weakness, AFM can present with severe weakness in affected upper limb(s) and normal strength in the lower limbs, or marked asymmetry with a difference of more than 2 points on the medical research council (MRC) scale between right and left limbs.47 Affected limbs become hyporeflexic or areflexic. Weakness can also affect the neck, trunk, diaphragm, or other respiratory muscles. In addition to limb weakness, approximately 30% of patients also have motor deficits localising to the cranial nerve motor nuclei of the brainstem, primarily consisting of bulbar and facial weakness, and, less commonly, extraocular muscle weakness.2 Finally, although not meeting existing epidemiological criteria for AFM, in some patients weakness can be limited to the cranial nerve(s) or neck, in the absence of limb weakness.48 Given that presentations limited to the cranial nerves have been excluded from most published case series of AFM (which have required at least one weak limb for study inclusion), the frequency of these cases is unknown. In our experience, such presentations can occur within the syndrome of AFM, but represent a minority of cases.
Table 1:
Clinical presentation of acute flaccid myelitis
| Estimated frequency | |
|---|---|
| Age <21 years | 80–90% |
| Prodromal fever or viral illness | 85–95% |
| Neurological onset to nadir <10 days | 100% |
| Headache or neck stiffness at onset | 12–60% |
| Asymmetric onset of weakness | 65–95% |
| Limb weakness | 85–95% |
| Upper limb weakness | 60–85% |
| Flaccidity or hyporeflexia of affected limbs | 95–100% |
| Neck, face, extraocular, or bulbar weakness | 20–60% |
| Trunk weakness | 30–70% |
| Requirement for mechanical ventilation | 10–40% |
| Bladder or bowel dysfunction | 5–40% |
| Non-specific sensory symptoms (eg, paresthesia) | 10–20% |
| Cardiovascular autonomic dysfunction | <10% |
| CSF pleocytosis (with testing <5 days after | 85–95% |
| Grey-matter predominant spinal cord lesion(s) on MRI | 95–100% |
| Brainstem lesion(s) on MRI | 35–45% |
| Cerebral deep grey matter lesion(s) on MRI | <5% |
CSF=cerebrospinal fluid.
The severity of weakness in an individual patient can range from mild to moderate weakness of one limb to complete paralysis of all limbs, and axial and bulbar muscles. About a third of patients admitted to hospital require intubation and ventilation,39 either due to respiratory muscle weakness or bulbar muscle weakness (with inability to protect the airway). Respiratory failure can be precipitated by procedural sedation. Dysphagia might necessitate supplemental hydration and nutrition. Bladder and bowel dysfunction are common in the acute phase,23 and autonomic manifestations such as labile blood pressure or irregular heart rate and breathing patterns can occur.18 Sensory symptoms or deficits other than neuropathic pain or paraesthesia are atypical.49 Altered mental status is not common,5,23,49 and the contribution of factors such as metabolic or respiratory disturbances in cases of reported encephalopathy is uncertain. Especially notable in cases associated with enterovirus A71 infection, AFM can occur in conjunction with frank brainstem encephalitis, and common clinical features in this patient group are autonomic disturbance, myoclonus, ataxia, irritability, and drowsiness.42
Some clinical features of AFM overlap with other causes of acute flaccid paralysis, including Guillain-Barré syndrome, spinal cord stroke, demyelinating myelitis (eg, aquaporin-4-IgG seropositive or seronegative neuromyelitis optica spectrum disorder, myelin oligodendrocyte glycoprotein [MOG]-antibody associated myelitis, multiple sclerosis, and acute disseminated encephalomyelitis), poliomyelitis (wild type poliovirus or vaccine-derived poliovirus), other infectious myelitis (eg, Japanese encephalitis, West Nile virus, tick-borne encephalitis virus, and varicella zoster virus myelitis), acute plexopathy, periodic paralysis, botulism, toxic synovitis, and orthopaedic conditions including nursemaid’s elbow. Infections capable of causing acute weakness differ according to the endemic and epidemic organisms in each global region. Poliovirus remains an important consideration in areas where wild-type poliovirus has not been eradicated, or in areas where vaccine-derived poliovirus may circulate.50 Certain vector-borne infections that can affect the anterior horn cells have clear regional distributions, with West Nile virus occurring in North America, Europe, Africa, and West Asia; Japanese encephalitis occurring in Asia and west pacific regions; and tick-borne encephalitis occurring in Europe, Russia, and some countries in Asia.51–53 Important clinical clues for these infections include systemic features (eg, erythematous maculo-papular rash in West Nile virus) and neurological features accompanying acute weakness (eg, seizures and prominent neuroimaging involvement of the deep grey matter in Japanese encephalitis). Microbiological testing can be tailored according to these epidemiological and regional infectious considerations.
Guillain-Barré syndrome, particularly the acute motor axonal neuropathy subtype, can cause acute weakness in children (particularly in some regions);54–56 however, there are some clinical features that can help to distinguish AFM from Guillain-Barré syndrome (table 2). The weakness in AFM can be markedly asymmetrical, often completely sparing one or more limbs,23,47 and can appear in a descending pattern. Sensory symptoms are usually a prominent feature in Guillain-Barré syndrome (except in the acute motor axonal neuropathy subtype),57 unlike AFM. Additionally, the clinical and radiological features of MOG-antibody associated myelitis can be strikingly similar to AFM.58,59 A further overlapping characteristic of the two is the frequent triggering of MOG-antibody associated disease by a viral infection.59 Concurrent optic neuritis, an encephalopathy-predominant clinical presentation, clinical evolution over more than 10 days, or a history of previous CNS inflammatory events suggests an alternative diagnosis such as multiple sclerosis, neuromyelitis optica spectrum disorder, or MOG-antibody associated disease, rather than AFM. Spontaneous spinal cord infarction often presents with acute flaccid weakness with a grey-matter predominant MRI lesion, and is under-recognised in children;60 however, patients often report severe back or limb pain (eg, knife-like) at onset, progress to nadir within 4 h, and have symmetric weakness and a sensory level.61
Table 2:
Differentiating acute flaccid myelitis from clinical mimics
| Acute flaccid myelitis | Guillain-Barré syndrome | Acute transverse myelitis (demyelinating or idiopathic) | Spontaneous spinal cord infarction | |
|---|---|---|---|---|
| Prodromal illness | +++ | +++ | +/− | − |
| Temporal evolution | Hours to days | Days to weeks | Days to weeks | Minutes to hours |
| Pattern of weakness | Asymmetric, arms>legs | Symmetric, ascending | Variable | Symmetric, severe |
| Facial/bulbar weakness | ++ | ++ | +/− | +/− |
| Respiratory failure | ++ | ++ | +/− | +/− |
| Numbness/paraesthesia | +/− | +++ (except AMAN) | +++ | + |
| Sensory level | − | − | ++ | ++ |
| Encephalopathy | − | − | +/− (eg, ADEM) | − |
| Bowel/bladder dysfunction | +/− | +/− | ++ | +++ |
| Possible associated symptoms or syndromes | Headache, neck pain/stiffness, neuropathic pain | Neuropathic pain | Optic neuritis, encephalitis, seizures | Severe back/limb pain at onset |
| MRI spinal cord | Ill-defined grey-matter predominant lesion, +/− nerve root enhancement | Normal cord, +/− nerve root enhancement | Variable, but usually a well-defined enhancing white>grey matter lesion | Non-enhancing anterior cord or grey-matter lesion |
| CSF | Mild-moderate pleocytosis | Elevated protein | Mild-moderate pleocytosis | Sometimes elevated protein or mild pleocytosis |
| Microbiological tests | See panel 1 | Stool sample: bacterial culture, viral RT-PCR panel; respiratory sample: viral RT-PCR panel; serum: Campylobacter jejuni and Mycoplasma pneumoniae IgM/IgG; other organisms according to region and season | If indicated based on clinical presentation | Not usually indicated |
| Other useful tests | +/− EMG/NCS | EMG/NCS; serum: anti-ganglioside antibodies | Serum: MOG-IgG, aquaporin-4-IgG; CSF: oligoclonal bands | Angiography |
AMAN=acute motor axonal neuropathy subtype. CSF=cerebrospinal fluid. ADEM=acute disseminated encephalomyelitis. EMG/NCS=electromyography and nerve conduction studies. MOG=myelin oligodendrocyte glycoprotein.
Diagnosis
MRI of the spinal cord is the most useful diagnostic test in AFM. T2 hyperintensity of the spinal cord grey matter is the hallmark of AFM (figure 1). Lesions in the early acute phase (hours to days) are typically confluent and ill defined, and affect the entire grey matter of the spinal cord when viewed axially,47,62,63 with a varying degree of surrounding white matter involvement and oedema.47 Spinal cord grey matter lesions are longitudinally extensive in most cases.62 The cervical cord is the most commonly, and often most prominently, affected, with marked oedema in some cases.47 T2 hyperintense lesions can also occur in the brainstem (most commonly in the dorsal pons).47,63 Spinal cord and brainstem lesions are usually non-enhancing or minimally enhancing. Swelling and hyperintensity of the brachial plexus in affected upper limbs has been identified in some patients in the acute stage using short tau inversion recovery MRI.64 Supratentorial lesions in the cortex and white matter generally do not occur,60 although T2 hyperintensity of deep grey matter structures has been recognised in a few patients (unpublished observations). Between 1 and 4 weeks after clinical onset, oedema improves and residual spinal cord lesions (present in many cases) become more focal, localising to the anterior horn region of the grey matter, and nerve root or cranial nerve enhancement frequently emerges,62 which can persist for weeks to months. MRI abnormalities can be subtle early in the acute course, which might even be interpreted as normal in the clinical setting. However, detailed retrospective MRI analysis by neuroradiologists with experience in AFM suggests that subtle lesions are invariably present on the initial MRI.47,60,62,63,65
Figure 1: Typical MRI findings in the acute phase of AFM.
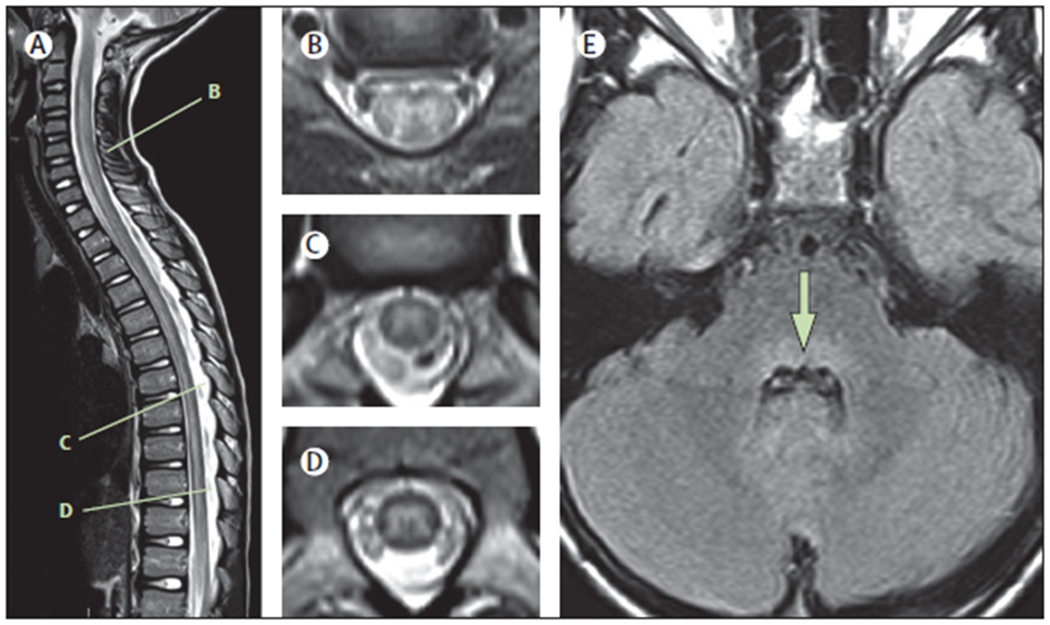
Spinal MRIs are shown of an 8-year-old child with AFM, acquired 24 h after onset of neurological symptoms.(A) Sagittal T2 image showing an ill-defined longitudinally extensive central/anterior spinal cord lesion. (B) Axial T2 image from C5–C6 shows hyperintensity of the entire grey matter of the spinal cord, with associated oedema and some surrounding white matter hyperintensity. (C) Axial T2 image from T7 shows asymmetric hyperintensity of the grey matter (right more than left). (D) Axial T2 image from T10 shows hyperintensity of the entire grey matter. (E) Axial FLAIR image at the level of the middle cerebellar peduncle demonstrates hyperintensity of the dorsal pons (arrow). AFM=acute flaccid myelitis.
CSF pleocytosis is identified in almost all patients with AFM undergoing lumbar puncture in the acute phase, with a mild to moderate elevation in white blood cell count (usually <100 per μL with lymphocytic predominance), which appears to resolve over subsequent weeks.1,5,23,47,60 How quickly pleocytosis evolves alongside the neurological syndrome is uncertain, and anecdotal reports suggest that if cell counts are normal very early in the course (within hours of neurological onset), pleocytosis can become apparent with repeat testing. A few patients with a clinical syndrome and imaging otherwise suggestive of AFM do not develop any CSF pleocytosis. The reason for this discrepancy is unclear but it can make differentiation of AFM from some clinical mimics more challenging. CSF protein can be mildly or moderately raised (usually <100 mg/dL), with occasional reports of values of almost 1000 mg/dL.5,23,39,47 CSF analysis can be helpful during the acute phase in differentiating AFM from other causes of flaccid paralysis less likely to produce pleocytosis (such as spinal cord infarction or Guillain-Barré syndrome).
Investigations outside the CNS or CSF are necessary to search for causes of AFM and its mimics (table 2). Respiratory (nasopharyngeal and oropharyngeal) and stool or rectal swab samples can show the presence of enterovirus D68, enterovirus A71, or other enterovirus RNA by reverse transcription polymerase chain reaction (RT-PCR), with detection most likely early in the clinical course. The highest yield for viral identification is in respiratory samples for enterovirus D68,39 and in rectal or stool samples for enterovirus A71.42 Additionally, stool viral culture for poliovirus (with RT-PCR of isolated virus to differentiate between wild-type and vaccine-derived virus) is indicated in some regions. Although not standard practice across all regions, the routine inclusion of enterovirus RT-PCR in viral respiratory and stool panels (as opposed to combined detection of enterovirus or rhinovirus species) would improve detection of these viruses in patients with AFM, and facilitate improved disease surveillance. Detection of enterovirus D68 or A71 in the CSF by RT-PCR is extremely rare.23 Serum testing for MOG-IgG (cell-based assay only) and aquaporin-4 (AQP4-IgG, cell-based assay preferable since low titre false-positive results can occur with ELISA testing) can identify these important treatable clinical mimics. Positive AQP4-IgG detected by cell-based assay is highly specific for a diagnosis of neuromyelitis optica spectrum disorder.66,67 The positive predictive value of MOG-IgG for a diagnosis of MOG-antibody associated disease is high (with high titres showing higher specificity and reproducibility than borderline or lowpositive titres).68,69 Identification of anti-ganglioside antibodies in the serum can support an alternative diagnosis of Guillain-Barré syndrome, although specificity is incomplete, and positive anti-ganglioside antibodies have been reported with other neuropathies (eg, diabetic neuropathy) and in some patients with AFM.5,70 Thus, detection of one or more anti-ganglioside antibodies (particularly at a low titer) does not exclude a diagnosis of AFM. Collection of serum samples for serological testing before intravenous immunoglobulin is administered will provide the most reliable results (given that intravenous immunoglobulin can alter sensitivity and specificity of auto-antibody tests).71
Electromyography or nerve conduction studies are often not required to make a diagnosis of AFM (in fact characteristic findings may only emerge 1 week after neurological onset); however, these studies can be a useful early investigation when differential diagnoses of Guillain-Barré syndrome or other acute neuromuscular disorders are being considered. Electromyography or nerve conduction studies can also have a role in the diagnosis of AFM in regions where MRI is not readily available, for patients for whom there is diagnostic uncertainty (eg, with equivocal MRI findings), or for patients with a delayed presentation (or initial misdiagnosis) in whom electrophysiologic changes are likely to be established by the time of assessment. Electro-physiological changes of AFM emerge over several weeks. Diminished or absent compound motor action potentials (CMAP) are an early finding, and can occur as soon as several days from symptom onset.1,72 By 2 weeks after onset, CMAP abnormalities tend to be evident, and lower CMAP amplitude appears to correlate with more severe injury (unpublished data). Decreased or absent F waves can also be detected.5,72 Sensory nerve conduction studies are normal.20,60,72 If electromyography or nerve conduction studies are completed early in AFM, reduced or absent recruitment of voluntary motor potentials might be the sole finding on needle electromyography. Fibrillations and positive sharp waves can develop as early as 1 week after symptom onset, followed by progressively increasing motor unit potential amplitude and duration consistent with denervation or reinnervation occurring over weeks to months or longer.1,3,60,72 Electromyography findings indicative of denervation can be seen even in limbs with apparently normal strength.20 Collectively, these findings are indicative of motor neuronopathy or axonal motor neuropathy, and may closely mimic the electrophysiologic changes seen in the acute motor axonal neuropathy subtype of Guillain-Barré syndrome.73 The symmetry and the relative length-dependence of the findings can be helpful clues in such cases; abnormalities that are asymmetrical and proximal>distal are characteristic of AFM. Alternatively, conduction block with early reversal of findings on serial studies is suggestive of acute motor axonal neuropathy.74,75
The poor availability of MRI or CSF analysis, or both, in resource-limited health-care settings is a particular challenge in the diagnosis of AFM. A typical prodromal illness and characteristic clinical presentation is suggestive of AFM even in the absence of advanced diagnostic testing, and electromyography or nerve conduction studies can be a useful adjunctive diagnostic tool when available. Epidemiological context can also be a useful clinical clue, because surveillance for enteroviral infections and AFM cases by regional public health systems may highlight seasonal periods and geographical locations associated with increased AFM risk. Although identification of a non-polio enterovirus species in respiratory or stool samples is not required to make a diagnosis of AFM, it can help to increase diagnostic certainty when MRI is not available. Serological testing for neuroimmune diseases with relapsing potential (specifically MOG IgG and AQP4 IgG) is not widely available in some resource-limited regions, and the onset of a second neurological event is an important flag for these treatable disorders. Clinical evolution that is atypical for AFM might also be a clue to these disorders—eg, a robust clinical response to any empirical steroid treatment if used, or development of upper motorneuron signs (spasticity and hyper-reflexia) during clinical recovery.
Acute management
Patients with AFM progress from neurological onset to nadir of weakness within hours to days.23 In 2018, 96% of identified AFM cases in the USA were admitted to hospital, and 58% to an intensive-care unit.16 Supportive treatment with careful monitoring focused upon potential emerging vital complications is the mainstay of early management. Although there is no specific evidence for optimal management of AFM, acute supportive management is similar to other causes of acute neuromuscular weakness. Supportive management includes optimising cardiorespiratory status including securing the airway and providing ventilatory support for respiratory failure when needed; treating bladder, bowel, or other autonomic dysfunction; managing pain; preventing complications of acute immobility (such as pressure ulcers and venous thromboembolism); and commencing early rehabilitation. AFM is a notifiable illness in many regions, requiring notification of the relevant public health authorities according to local protocols.
Since the pathophysiology of AFM is not fully understood, with the disease occurring in association with identified enterovirus infection in some patients or absent isolated virus in others, which biological process(es) should be targeted in acute disease to modify clinical outcomes is uncertain.76 There have been no prospective, controlled trials of specific medical therapies in AFM. Given that most experts believe neuroinvasive viral infection to be the primary cause of neurological disease in AFM, intravenous immunoglobulin (which has been shown to include neutralising antibodies against contemporary strains of enterovirus D68),77 is frequently used for its possible antiviral and immunomodulatory effects, along with a favourable adverse-effect profile. On the basis of the postulated mechanism of action, theoretical potential benefits of intravenous immunoglobulin treatment could be considered greatest when administered early in the course of the illness. Intravenous steroids and plasma exchange are sometimes used for their potential immunomodulatory effect, but the potential for therapeutic benefit versus harm remains controversial. Some physicians have used steroids in cases manifesting critical spinal cord oedema with secondary cord compression, although individual benefit in such cases is uncertain. In mouse models of enterovirus D68 nervous system infection, early administration of intravenous immunoglobulin reduced paralysis whereas steroid treatment resulted in increased viral titre in the spinal cord and worse outcomes.78 The applicability of these murine studies to a human disease of incompletely understood cause and pathogenesis such as AFM is uncertain and represents a key area of future research. In low-resource settings where insufficient advanced diagnostic testing precludes confirmation of AFM, and immune-mediated myelitis (ie, demyelinating or idiopathic myelitis) remains in the differential diagnosis, clinicians might need to consider a trial of treatment with high-dose steroids in individual cases. Antienteroviral and neuroprotective activity of fluoxetine has been shown in vitro,79 but not in a small retrospective uncontrolled cohort study in patients with AFM,80 or in the murine model.78 Small molecule antivirals and monoclonal antibodies against non-polio enteroviruses are being investigated as potential therapies.81–83
Recovery, rehabilitation, and long-term sequelae
AFM seems to be a monophasic disorder with high potential for residual impairment. Prognostication is challenging, but electromyography or nerve conduction studies and MRI could both have potential utility.20,62,65 Denervated muscles with severe neurogenic changes on electromyography or nerve conduction studies in the weeks to months after AFM onset are likely to experience residual weakness.5,20,72 Quantitative measures of grey matter MRI involvement during the acute phase of the illness show promise in predicting motor outcomes, based on findings from a small case series.65 Evolution of MRI abnormalities occurs in the weeks to months after onset of AFM, and the location of residual MRI lesions in the anterior horns, could correlate with the distribution of residual limb weakness.62 Localising these characteristic residual lesions could help to map areas of more severe injury in affected individuals. Future research to elucidate which combination of clinical or paraclinical factors (or both) best predicts long-term clinical outcomes in AFM is needed.
The extent of recovery in AFM is highly variable, although few patients (<10%) recover completely.3,5,18,20,48 After neurological nadir (which may last days to weeks), most patients show some improvement in motor strength, with recovery being most rapid in the first few months after onset. Cranial nerve dysfunction is more likely to improve and resolve than is limb weakness.20,48 In the limbs, early recovery appears to occur in a distal to proximal pattern.19 Profoundly affected muscle groups (particularly MRC grade 0 of 5) at neurological nadir are the least likely to recover, and thus recovery can be markedly asymmetrical.20,47 Respiratory muscle weakness can persist, although only a small proportion of patients remain ventilator dependent at approximately 1 year follow-up.19,47,48,84 Reports of death have been rare and are limited to immunocompromised adults, and children with early or late complications of respiratory failure.3,23
Nerve transfer surgery has been undertaken in some patients with poor clinical recovery of affected areas. Case series have shown generally positive outcomes from nerve transfers for restoration of elbow function in appropriately selected patients, with less positive outcomes for restoration of shoulder function.21,22,85 Muscle or tendon transfer, or both, has been reported in a few cases, with generally positive outcomes for restoration of elbow or hand function.21,85 Anecdotal reports indicate that lower limb nerve transfers, nerve transfer to the phrenic nerve, and diaphragmatic pacing have been undertaken in individual cases;86 however, there are few published data regarding outcomes in these cases.
Data for medium-term to long-term neurological and functional outcomes of patients with AFM since 2012 are limited to small cohorts followed up for 2 years or less. Some data suggest that severity and prognosis vary according to viral pathogen detected in relation to AFM, given that patients with AFM associated with enterovirus A71 can have milder muscle weakness and better recovery than patients with AFM associated with enterovirus D68.42 Patients engaged in multimodal rehabilitation can achieve functional improvements for years even after recovery of motor strength plateaus.19 However, many patients have substantial residual weakness, muscle atrophy, and functional impairment, with potential for a secondary broader range of developmental sequelae. Medium-term to long-term complications described to date in AFM include neurological sequelae (neuropathic pain, chronic constipation, chronic ventilator dependence, and dependence on artificial nutrition and hydration), musculoskeletal sequelae (joint subluxation and dislocation, particularly proximal joints with profound muscle weakness, limitation in range of joint motion, scoliosis, limb-length discrepancies, and chest wall abnormalities), and psychological sequelae (such as anxiety and depression).18–20,49 Given shared mechanisms, the known consequences of similar neurological disorders can provide some clues to potential complications even later in life, such as accelerated degenerative joint disease, reduced bone mineral density in affected limbs, cardiometabolic syndrome (including obesity, insulin resistance, hypertension, and dyslipi daemia), restrictive respiratory insufficiency, sleep disordered breathing, and nocturnal hypoventilation.87–92 Entrapment neuropathies can arise from the use of walking aids or wheelchairs,93,94 while scoliosis can predispose to later compressive myelopathy or radiculopathy.94 Finally, in other disorders causing substantial neurological disability in early life, such as poliomyelitis, some patients have reported deterioration in strength or function with ageing.95 Whether patients with AFM could have a similar decline later in life, or whether continued rehabilitation may mitigate this decline, is unclear.
Implications of current evidence: diagnostic criteria and clinical care
Literature to date focused on AFM is limited by no uniform diagnostic criteria, which is a barrier to advances in knowledge about treatment and outcomes in patients with AFM. Additionally, management approaches have been variable and centre based. On the basis of best evidence from published knowledge from multiple cohorts, we provide pathogen-agnostic diagnostic criteria (figure 2), and an approach to the clinical assessment (panel 1), management (panel 2), and rehabilitation (panel 3) of patients with suspected AFM. The pathogenagnostic diagnostic criteria for AFM include elements of clinical history, examination, neuroimaging, and CSF analysis. The core clinical syndrome of AFM is defined by acute onset of limb weakness, with lower motor neuron findings evident on neurological examination. Prodromal fever or illness is supportive but not essential to diagnose AFM, because not all patients report prodromal symptoms, including those for whom enterovirus D68 is identified.5 The diagnosis of AFM can be considered definite when characteristic MRI findings and CSF pleocytosis are present in addition to the previously mentioned core clinical features. The diagnosis of AFM can be considered probable when the core clinical features and characteristic MRI findings are present, but CSF pleocytosis is absent (or not checked). The diagnosis of AFM can be considered possible in cases with a limited or milder clinical syndrome, with characteristic MRI findings; and uncertain when the core clinical features are present, but without adequate MRI studies to evaluate. Additionally, factors that suggest an alternative diagnosis are: (1) encephalopathy that cannot be explained by fever, illness, respiratory distress, metabolic abnormalities, or medications; (2) presence of sensory deficits on examination; (3) presence of lesions in supratentorial white matter or cortex, which should prompt consideration of ADEM, MOG-antibody associated disease, neuromyelitis optica spectrum disorder, encephalomyelitis, and others; (4) absence of CSF pleocytosis, which should prompt consideration of Guillain-Barré syndrome, botulism, ischaemic cord lesions, and others; (5) positive serum aquaporin-4 (AQP-4) antibody, which would exclude AFM; and (6) positive serum MOG antibody, which would suggest MOG-antibody associated disease.
Figure 2: Diagnostic criteria for AFM.
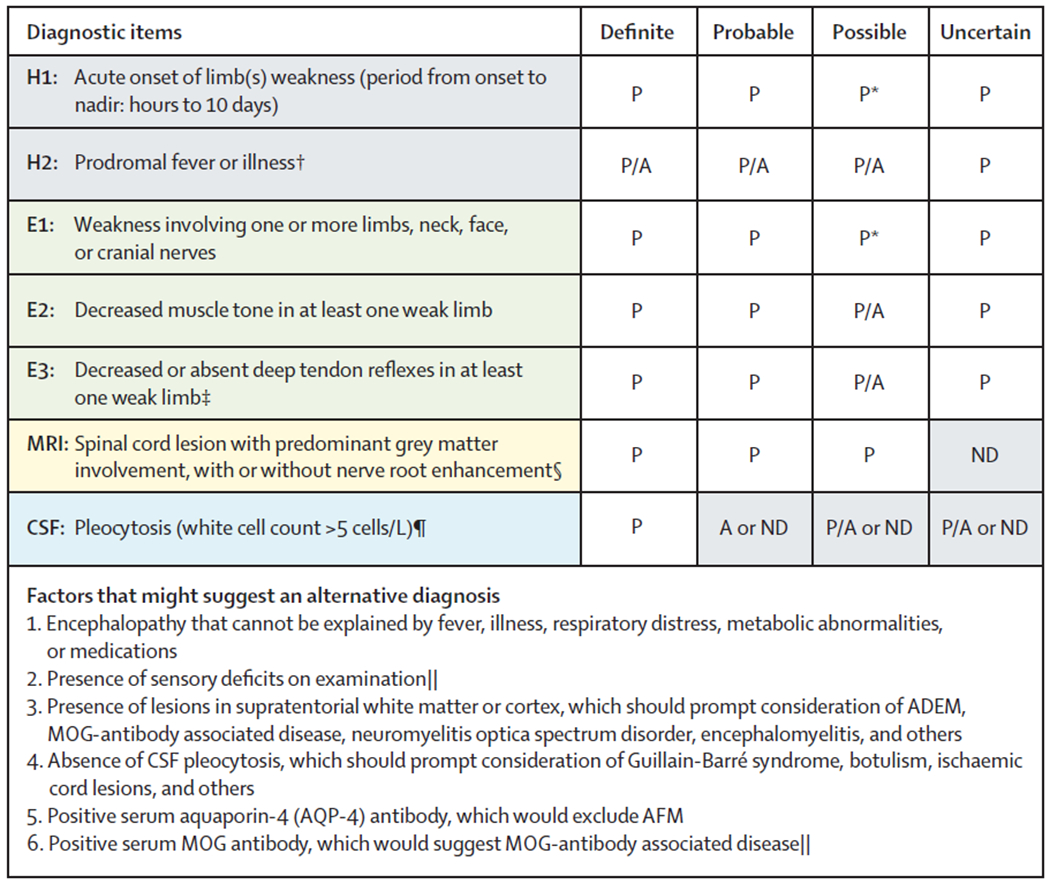
These criteria apply to the acute stage of the disease. AFM=acute flaccid myelitis. H=history. E=examination. CSF=cerebrospinal fluid. P=diagnostic element is present. A=diagnostic item is absent. P/A=presence of this diagnostic element is supportive but not required. ND=test was not done. ADEM=acute disseminated encephalomyelitis. MOG=myelin oligodendrocyte glycoprotein. *Subjective (H1) or objective (E1) weakness must be present in any of: limb(s), neck, or cranial nerves. †Prodromal illness can include respiratory, gastrointestinal, or other symptoms of viral illness. ‡Normal or increased reflexes can be found in other limbs. §If MRI obtained very early (within hours of neurological onset) appears normal, repeat MRI after clinical evolution might show diagnostic findings. MRI obtained at late stages (≥4 weeks) might be normal. ¶CSF may be normal at very early (hours) or late (≥4 weeks) stages of AFM. ||At present, there are no data describing the frequency of these features in patients with AFM.
Panel 1: Clinical and paraclinical evaluation of patients with suspected AFM.
Initial clinical assessment
Consider AFM in patients presenting with rapid-onset weakness, particularly when occurring during or shortly following a suspected viral illness.
Complete neurological examination should include specific tests for proximal muscle weakness (such as standing up from a seated position on the floor), axial weakness (neck and trunk flexion and extension), and cranial nerve abnormalities.
Clinical features atypical for AFM include encephalopathy unrelated to metabolic disturbance, seizures, extensive sensory abnormalities, or evolution to nadir over more than 10 days.
Neurology and infectious disease specialists should be consulted (where available) to help with diagnosis, evaluation, and treatment.
Admission to intensive care unit should be considered when indicated, and close monitoring for respiratory or autonomic deterioration, or both, is essential.
Radiological evaluation
MRI whole spine and brain should be prioritised, including T2 and T1 pre-contrast and post-contrast sequences in both axial and sagittal planes.
The characteristic MRI abnormality is grey-matter predominant T2 hyperintensity of the spinal cord with associated spinal cord oedema; lesion(s) are usually longitudinally extensive and non-enhancing. Nerve root enhancement might be present.
Repeat MRI can be considered after further clinical evolution in patients with a suggestive clinical presentation but in whom early MRI of the spinal cord is apparently normal.
Laboratory evaluation
Obtain specimens as soon as possible (ie, within hours of clinical presentation).
Respiratory samples (both nasopharyngeal and oropharyngeal): respiratory viral RT-PCR testing (to include enterovirus RT-PCR). When possible, a positive enterovirus RT-PCR result should be subtyped (to include enterovirus D68, enterovirus A71, and other common subtypes).
Stool samples or rectal swab: enterovirus RT-PCR, viral culture for poliovirus when epidemiologically relevant (with RT-PCR of isolated virus to differentiate between wild-type and vaccine-derived virus).
Blood sample: microbiological tests (enterovirus RT-PCR and other epidemiologically appropriate micro-organism tests—eg, West Nile virus serology), and testing for specific alternative myelopathy diagnoses to include MOG IgG and aquaporin-4 IgG.
CSF sample: cell counts, protein, glucose, oligoclonal bands, enterovirus RT-PCR (although yield is very low), and other epidemiologically appropriate micro-organism tests.
When RT-PCR is not readily available, samples can still be acquired and frozen for future analysis or transfer to public health authorities.
Respiratory, stool, serum, and CSF samples should also be sent to the relevant public health authorities, according to local protocols.
Low-resource settings
When MRI is not possible, rapid completion of available laboratory testing should be prioritised (CSF analysis, microbiological sampling), and EMG/NCS can be incorporated in the initial evaluation when available.
AFM=acute flaccid myelitis. MOG=myelin oligodendrocyte glycoprotein. CSF=cerebrospinal fluid. EMG/NCS=electromyography or nerve conduction studies.
Panel 2: Acute management of patients with suspected AFM.
Respiratory status
Patients with respiratory muscle weakness are at risk of hypoventilatory respiratory failure. Bulbar muscle weakness can lead to inability to protect the airway.
Poor head control, drooling, proximal upper limb weakness, neck weakness, or altered voice quality suggest a risk of respiratory failure. Settings without intensive care facilities should consider transfer of patients with risk of evolving respiratory failure to higher level of care institutions.
Respiratory function should be assessed every 4 h until clinical stabilisation. Monitoring may include testing of negative inspiratory force, vital capacity, oxygen saturation levels, and blood gas analysis (to detect evolving hypercarbia).
The possibility of concomitant respiratory tract infection should be considered and treated as appropriate.
Typical thresholds for non-invasive ventilation or intubation in patients with neuromuscular weakness or bulbar weakness should be applied.
Tracheostomy can be considered in patients requiring prolonged intubation.
Sedation
Sedation for procedures (eg, MRI) carries a risk of respiratory decompensation; patients should be closely monitored and short-acting agents used when possible.
Where intubation is required, medications with the least effect on respiration should be used—eg, dexmedetomidine.
Pain and autonomic dysfunction
Neuropathic pain is frequent and should be treated. In sedated patients or young children, pain might be recognised by irritability, tachycardia, and refusal to move.
Bladder function should be assessed with post-void residual volumes. Urinary catheterisation might be required.
Constipation is common and should be treated appropriately.
Autonomic involvement may manifest with hypertension, labile blood pressure, diaphoresis, and even cardiac arrhythmia, requiring close monitoring and treatment.
Immunomodulatory therapies or antiviral therapies
With little evidence regarding potential benefit or harm of therapies in humans, no standardised pharmacological treatment can be universally recommended.
A common, but unproven, approach is to provide intravenous immunoglobulin during the acute phase, which might provide anti-enteroviral neutralising antibodies, with minimal potential harm.
In low-resource settings where differentiating between AFM and immune-mediated myelitis can be challenging (eg, because of no MRI and a clinical presentation that is not wholly typical), a trial of steroids can be considered on an individualised basis.
Early rehabilitation
Physical, occupational, and speech therapy should be commenced early.
Consider early initiation of electrical stimulation therapy to minimise disuse muscle atrophy.
Psychological support should be provided to assist the child and family with coping and adjustment.
In settings with limited rehabilitation resources, early mobilisation and activity-based therapy should still be encouraged.
AFM=acute flaccid myelitis.
Panel 3: Rehabilitation and long-term clinical care of patients with AFM.
Inpatient rehabilitation
After the acute phase of AFM, medically stable children with significant residual neurological deficits should transfer to an inpatient rehabilitation programme with a multidisciplinary team.
Although specific evidence regarding rehabilitation in AFM is minimal, the approach can draw on methods used in other monophasic neurological injuries (eg, spinal cord injury) and in other motor neuronopathies (eg, poliomyelitis, Guillain-Barré syndrome).
Intensive rehabilitation should include short-term goals to facilitate developmentally appropriate functional independence and use of compensatory devices, while simultaneously working towards long-term goals for recovery of function and avoidance of musculoskeletal complications.
Intensive activity-based therapy can include weight loading of limbs, massed practice, and task-specific practice.
Locomotor gait training or functional electrical stimulation, or both, can be used when available, although data supporting the specific effect of these approaches on AFM outcomes are scarce.
Consider orthotic devices, mobility equipment, assistive technology, identification of home care needs, a plan for school and community re-entry, psychosocial support, and education for the child and family.
In low-resource settings with little access to skilled therapy, education of patients and care-givers regarding home-based activities is essential.
Nerve and tendon transfer surgery
Patients with poor recovery in an affected muscle group 3 months or longer after onset of AFM should be considered for potential nerve or tendon transfer surgery, or both, by a centre experienced in the relevant procedures (where available).
Experience with nerve and tendon transfers in the upper extremity has shown promising results in appropriately selected patients with AFM.
Phrenic nerve transfer, lower extremity nerve transfers, or pacing of the phrenic nerve or diaphragm can also be considered in selected patients, although data on outcomes are scarce.
The appropriate timing for nerve transfer surgery is uncertain, but a delay in consideration could result in a missed window of opportunity (because muscle viability wanes with extended periods of denervation). Tendon-transfer surgery is not time sensitive and can be completed months or years after the initial injury.
EMG/NCS can aid in the planning of nerve transfer surgery, and should include evaluation of the donor nerve and acceptor muscle.
Medium-term to long-term rehabilitation
After home discharge, continued rehabilitation with periodic skilled therapy should be provided to achieve acquisition of developmentally appropriate milestones and functional independence.
Educational and developmental transitions, age-appropriate self-advocacy skills, increasing independence in self-care, and responsibility for medical management will aid successful transition to adulthood.
Long-term medical management
Patients should continue vaccination protocols according to national guidelines (including delayed live vaccinations if intravenous immunoglobulin has been administered).
Long-term follow-up should be provided by neurology and physiatry or rehabilitation medicine services where available, alongside primary care.
Specialist input might be required to manage complications such as joint contracture, scoliosis, shoulder or hip subluxation, limb length difference, and loss of bone mineral density.
Children requiring long-term ventilatory support or artificial nutrition will require additional specialist input.
AFM=acute flaccid myelitis. EMG/NCS=electromyography or nerve conduction studies.
The frequency of each element of the diagnostic criteria as reported in the existing literature is outlined in the appendix (p 12), although these studies are notable for substantial heterogeneity of inclusion criteria (eg, inclusion or exclusion according to age or enterovirus detection). These diagnostic criteria are specific to the acute phase of the illness, and allow classification of the level of certainty of an AFM diagnosis, and to distinguish AFM from other causes of acute flaccid paralysis. The diagnostic criteria outlined do not replace epidemiological case definitions for acute flaccid paralysis or AFM that public health organisations (such as WHO or US Centers for Disease Control and Prevention) use for surveillance purposes. Furthermore, a clinical diagnosis of AFM to guide management of an individual patient remains nuanced and must take into account the particular characteristics of each case. These diagnostic criteria classify AFM cases using typical features, although clinicians might encounter patients with atypical features outside of the outlined criteria.
Conclusions and future directions
The increasing incidence, since 2012, of a likely enterovirus-driven severe paralytic disease with lifelong sequelae identifies AFM as a major global public health concern of high priority. Its relative rarity, widely disparate distribution, and resemblance to other causes of acute weakness argues for widespread education of clinicians and health-care providers on the characteristics necessary to appropriate diagnosis, acute management, and chronic rehabilitation. Whether the pattern of seasonal, biennial outbreaks of AFM will continue is uncertain, but preparedness for potential future increases in AFM cases is essential. Understanding of factors driving the seasonal, cyclic circulation of nonpolio enteroviruses could be key to predicting and preparing for future AFM outbreaks. The mechanism by which common exposures, such as enterovirus infections, lead to severe neurological disease in the few affected by AFM remains unknown. Potential host genetic and immunological factors, as well as virological or environmental determinants, need to be elucidated. To determine whether anti-infective therapies (ie, antivirals, monoclonal antibodies), immunomodulatory therapies (ie, intravenous immunoglobulin, steroids, plasma exchange), or a combined therapeutic approach may be most effective, there is a need to understand the pathophysiological role of direct viral infection, immune activation, and inflammation on neuronal damage. Ultimately, if cases of this disabling paralytic disease continue or increase, a preventative approach, including development of vaccine candidates against the leading suspected viral causes, might be necessary. All the above advances are dependent upon increased awareness of the presenting clinical features of AFM, allowing accurate case ascertainment to understand epidemiology and burden of disease, early recognition to allow prompt specimen collection and causal diagnosis, and early initiation of potential therapies.
Supplementary Material
Search strategy and selection criteria.
For this Review, we searched PubMed with the terms “acute flaccid myelitis”, “acute flaccid paralysis”, “polio-like”, “poliomyelitis”, and “enterovirus”, and sorted results into the following themes: epidemiology, clinical presentation, diagnosis, management, and outcomes. Search criteria were limited to publications in English from January, 2012, to July, 2020. In addition to identified primary research we included relevant materials such as published opinions and viewpoints, proposed case definitions, and other materials including conference abstracts, ongoing research work, and unpublished observations of AFM Working Group members based on their clinical experience.
Acknowledgments
We are grateful for the support provided by the Siegel Rare Neuroimmune Association (SRNA) for facilitating administrative support to the AFM Working Group, and the Bart McLean Fund for Neuroimmunology Research for AFM research. We thank Janell A Routh and Sarah E Kidd at the Centers for Disease Control and Prevention for sharing their expertise. OCM was funded through the SRNA James T Lubin Fellowship. CAP is supported by National Institutes of Health (R01 NS108358) and the Bart McLean Fund for Neuroimmunology Research. KM is supported by National Institutes of Health (K23 AI128069). KTT is supported by National Institutes of Health (1K23NS105935).
Declaration of interests
CAP, LB, BG, KM, and CLS are unpaid advisors to the AFM Task Force of the US Centers for Disease Control and Prevention (CDC). ML serves (unpaid) on the UK AFP Task Force and is the lead for the Clinical Working Group. SEH, CO, and GYG receive salary support from the CDC for AFM surveillance. LB reports research support from Biogen outside the submitted work, and is serving as a consultant to the national vaccine injury compensation programme. RB reports grants from Akili interactive; and personal fees from EMD Serono, Roche Genentech, Novartis, Alexion, Sanofi Genzyme, and Biogen, outside the submitted work. JDes reports receiving funds from EFGLA, UCB, Novartis, Ovid, Aquestive, and Neurelis, and for serving on advisory boards as medico-legal expert. BG reports grants from National Institutes of Health, National MS Society, Siegel Rare Neuroimmune Association, Guthy Jackson Charitable Foundation, and CLENE Nanomedicine; personal fees from Novartis, Genentech, EMD Serono, and Alexion, outside the submitted work; and serves as board member to Siegel Rare Neuroimmune Association. RK reports grants from Ministry of Health, Labour and Welfare of Japan for AFM study, and personal fees from Eisai, Otsuka Pharmaceutical, and UCB Japan, outside the submitted work. ML receives research grants from Action Medical Research, the Dancing Eyes Syndrome society, Great Ormond Street Hospital Children’s (GOSH) charity, National Institute for Health Research, MS Society, and Sparks charity; receives research support grants from the London Clinical Research Network and Evelina Appeal; has received consultation fees from CSL Behring, Novartis, and Octapharma; has received travel grants from Merck Serono; and was awarded educational grants to organise meetings by Novartis, Biogen Idec, Merck Serono, and Bayer. KM reports grants from the National Institute of Allergy and Infectious Diseases, outside the submitted work. TLS reports to have received a stipend from the American Academy of Neurology for lecturing on AFM. KTT reports grants by the National Institute of Health. EAY reports grants from the Canadian Network of MS Clinics, during the conduct of the study; grants from Biogen, National MS society, Consortium of Multiple Sclerosis Centers, MS Society of Canada/MS Foundation, Ontario Institute for Regenerative Medicine, Stem Cell Network, Sickkids Foundation, Centre for Brain and Mental Health, TEVA Pharmaceuticals, and Guthy Jackson Foundation; and personal fees from ACI Clinical, US Food and Drug Administration, and Juno, outside the submitted work. CAP reports grants from the National Institute of Health and Bart McLean Fund for Neuroimmunology Research, serves as co-investigator in the Natural History Study of Acute Flaccid Myelitis, and serves as a member of the National Institute of Health Scientific Board of the Siegel Rare Neuroimmune Association. All other authors declare no competing interests.
Footnotes
See Online for appendix
References
- 1.Van Haren K, Ayscue P, Waubant E, et al. Acute flaccid myelitis of unknown etiology in California, 2012-2015. JAMA 2015; 314: 2663–71. [DOI] [PubMed] [Google Scholar]
- 2.Messacar K, Schreiner TL, Maloney JA, et al. A cluster of acute flaccid paralysis and cranial nerve dysfunction temporally associated with an outbreak of enterovirus D68 in children in Colorado, USA. Lancet 2015; 385: 1662–71. [DOI] [PubMed] [Google Scholar]
- 3.Knoester M, Helfferich J, Poelman R, Van Leer-Buter C, Brouwer OF, Niesters HGM. Twenty-nine cases of enterovirus-D68-associated acute flaccid myelitis in europe 2016: a case series and epidemiologic overview. Pediatr Infect Dis J 2019; 38: 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramsay M, Dunning J, Foulkes S, et al. An increase in reports of acute flaccid paralysis (AFP) in the United Kingdom, 1 January 2018–21 January 2019: early findings. Euro Surveill 2019; 24: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chong PF, Kira R, Mori H, et al. Clinical features of acute flaccid myelitis temporally associated with an enterovirus D68 outbreak: results of a nationwide survey of acute flaccid paralysis in Japan, August–December 2015. Clin Infect Dis 2018; 66: 653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarmast SN, Gowda VK, Ahmed M, Basvaraja GV, Saini J, Benakappa A. Acute flaccid myelitis-clustering of polio-like illness in the tertiary care centre in southern india. J Trop Pediatr 2018; 65: 309–14. [DOI] [PubMed] [Google Scholar]
- 7.Chen IJ, Hu SC, Hung KL, Lo CW. Acute flaccid myelitis associated with enterovirus D68 infection: a case report. Medicine (Baltimore) 2018; 97: e11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersen EW, Kornberg AJ, Freeman JL, Leventer RJ, Ryan MM. Acute flaccid myelitis in childhood: a retrospective cohort study. Eur J Neurol 2017; 24: 1077–83. [DOI] [PubMed] [Google Scholar]
- 9.Fall A, Ndiaye N, Jallow MM, et al. Enterovirus D68 subclade B3 circulation in senegal, 2016: detection from influenza-like illness and acute flaccid paralysis surveillance. Sci Rep 2019; 9: 13881–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crone M, Tellier R, Wei XC, et al. Polio-like illness associated with outbreak of upper respiratory tract infection in children. J Child Neurol 2016; 31: 409–14. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. AFM cases in the United States. October, 2020. https://www.cdc.gov/acute-flaccid-myelitis/afm-cases.html (accessed Aug 18, 2020).
- 12.Ruggieri V, Paz MI, Peretti MG, et al. Enterovirus D68 infection in a cluster of children with acute flaccid myelitis, Buenos Aires, Argentina, 2016. Eur J Paediatr Neurol 2017; 21: 884–90. [DOI] [PubMed] [Google Scholar]
- 13.Carballo CM, Erro MG, Sordelli N, et al. Acute flaccid myelitis associated with enterovirus D68 in children, Argentina, 2016. Emerg Infect Dis 2019; 25: 573–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messacar K, Asturias EJ, Hixon AM, et al. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect Dis 2018; 18: e239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaren N, Lopez A, Kidd S, et al. Characteristics of patients with acute flaccid myelitis, united states, 2015–2018. Emerg Infect Dis 2020; 26: 212–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Board of Scientific Counselors. National overview of acute flaccid myelitis —United States, 2014–2018. December, 2018. https://www.cdc.gov/ddid/bsc/afm-overview-2018.html (accessed Aug 18, 2020).
- 17.Messacar K, Schreiner TL, Van Haren K, et al. Acute flaccid myelitis: a clinical review of US cases 2012–2015. Ann Neurol 2016; 80: 326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bove R, Rowles W, Carleton M, et al. Unmet needs in the evaluation, treatment, and recovery for 167 children affected by acute flaccid myelitis reported by parents through social media. Pediatr Neurol 2020; 102: 20–27. [DOI] [PubMed] [Google Scholar]
- 19.Melicosta ME, Dean J, Hagen K, et al. Acute flaccid myelitis: rehabilitation challenges and outcomes in a pediatric cohort. J Pediatr Rehabil Med 2019; 12: 245–53. [DOI] [PubMed] [Google Scholar]
- 20.Martin JA, Messacar K, Yang ML, et al. Outcomes of Colorado children with acute flaccid myelitis at 1 year. Neurology 2017; 89: 129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saltzman EB, Rancy SK, Sneag DB, Feinberg Md JH, Lange DJ, Wolfe SW. Nerve transfers for enterovirus D68-associated acute flaccid myelitis: a case series. Pediatr Neurol 2018; 88: 25–30. [DOI] [PubMed] [Google Scholar]
- 22.Pino PA, Intravia J, Kozin SH, Zlotolow DA. Early results of nerve transfers for restoring function in severe cases of acute flaccid myelitis. Ann Neurol 2019; 86: 607–15. [DOI] [PubMed] [Google Scholar]
- 23.Messacar K, Schreiner TL, Van Haren K, et al. Acute flaccid myelitis: a clinical review of US cases 2012–2015. Ann Neurol 2016; 80: 326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messacar K, Robinson CC, Pretty K, Yuan J, Dominguez SR. Surveillance for enterovirus D68 in Colorado children reveals continued circulation. J Clin Virol 2017; 92: 39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greninger AL, Naccache SN, Messacar K, et al. A novel outbreak enterovirus D68 strain associated with acute flaccid myelitis cases in the USA (2012-14): a retrospective cohort study. Lancet Infect Dis 2015; 15: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown DM, Hixon AM, Oldfield LM, et al. Contemporary circulating enterovirus D68 strains have acquired the capacity for viral entry and replication in human neuronal cells. MBio 2018; 9: e01954–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uprety P, Curtis D, Elkan M, et al. Association of enterovirus D68 with acute flaccid myelitis, Philadelphia, Pennsylvania, USA, 2009–2018. Emerg Infect Dis 2019; 25: 1676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra N, Ng TFF, Marine RL, et al. Antibodies to enteroviruses in cerebrospinal fluid of patients with acute flaccid myelitis. MBio 2019; 10: e01903–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schubert RD, Hawes IA, Ramachandran PS, et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat Med 2019; 25: 1748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hixon AM, Yu G, Leser JS, et al. A mouse model of paralytic myelitis caused by enterovirus D68. PLoS Pathog 2017; 13: e1006199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun S, Bian L, Gao F, et al. A neonatal mouse model of Enterovirus D68 infection induces both interstitial pneumonia and acute flaccid myelitis. Antiviral Res 2019; 161: 108–15. [DOI] [PubMed] [Google Scholar]
- 32.Midgley CM, Watson JT, Nix WA, et al. Severe respiratory illness associated with a nationwide outbreak of enterovirus D68 in the USA (2014): a descriptive epidemiological investigation. Lancet Respir Med 2015; 3: 879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kramer R, Sabatier M, Wirth T, et al. Molecular diversity and biennial circulation of enterovirus D68: a systematic screening study in Lyon, France, 2010 to 2016. Euro Surveill 2018; 23: 1700711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menant JC, Gandevia SC. Poliomyelitis. In: Day BL, Lord SR, eds. Handbook of clinical neurology. Netherlands: Elsevier Health Sciences, 2018: 337–44. [DOI] [PubMed] [Google Scholar]
- 35.Chang LY, Lin HY, Gau SS, et al. Enterovirus A71 neurologic complications and long-term sequelae. J Biomed Sci 2019; 26: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodian D Histopathologic basis of clinical findings in poliomyelitis. Am J Med 1949; 6: 563–78. [DOI] [PubMed] [Google Scholar]
- 37.Hixon AM, Clarke P, Tyler KL. Contemporary circulating enterovirus D68 strains infect and undergo retrograde axonal transport in spinal motor neurons independent of sialic acid. J Virol 2019; 93: e00578–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenfeld AB, Warren AL, Racaniello VR. Neurotropism of enterovirus D68 isolates is independent of sialic acid and is not a recently acquired phenotype. MBio 2019; 10: e02370–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayers T, Lopez A, Lee A, et al. Acute flaccid myelitis in the United States: 2015–2017 Pediatrics 2019; 144: e20191619. [DOI] [PubMed] [Google Scholar]
- 40.Messacar K, Burakoff A, Nix WA, et al. Notes from the field: enterovirus A71 neurologic disease in children - Colorado, 2018. MMWR Morb Mortal Wkly Rep 2018; 67: 1017–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puenpa J, Wanlapakorn N, Vongpunsawad S, Poovorawan Y. The history of enterovirus A71 outbreaks and molecular epidemiology in the asia-pacific region. J Biomed Sci 2019; 26: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Messacar K, Spence-Davizon E, Osborne C, et al. Clinical characteristics of enterovirus A71 neurological disease during an outbreak in children in Colorado, USA, in 2018: an observational cohort study. Lancet Infect Dis 2020; 20: 230–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casas-Alba D, de Sevilla MF, Valero-Rello A, et al. Outbreak of brainstem encephalitis associated with enterovirus-A71 in Catalonia, Spain (2016): a clinical observational study in a children’s reference centre in Catalonia. Clin Microbiol Infect 2017; 23: 874–81. [DOI] [PubMed] [Google Scholar]
- 44.Sejvar JJ, Bode AV, Marfin AA, et al. West Nile virus-associated flaccid paralysis. Emerg Infect Dis 2005; 11: 1021–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Centers for Disease Control and Prevention. West Nile virus. December, 2018. https://www.cdc.gov/westnile/healthcareproviders/healthCareProviders-Diagnostic.html (accessed Aug 18, 2020).
- 46.Giombini E, Rueca M, Barberi W, et al. Enterovirus D68-associated acute flaccid myelitis in immunocompromised woman, Italy. Emerg Infect Dis 2017; 23: 1690–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordon-Lipkin E, Muñoz LS, Klein JL, Dean J, Izbudak I, Pardo CA. Comparative quantitative clinical, neuroimaging, and functional profiles in children with acute flaccid myelitis at acute and convalescent stages of disease. Dev Med Child Neurol 2019; 61: 366–75. [DOI] [PubMed] [Google Scholar]
- 48.Nelson GR, Bonkowsky JL, Doll E, et al. Recognition and management of acute flaccid myelitis in children. Pediatr Neurol 2016; 55: 17–21. [DOI] [PubMed] [Google Scholar]
- 49.Yea C, Bitnun A, Robinson J, et al. Longitudinal outcomes in the 2014 acute flaccid paralysis cluster in Canada. J Child Neurol 2017; 32: 301–07. [DOI] [PubMed] [Google Scholar]
- 50.World Health Organization. Global polio eradication initiative. October, 2020. http://polioeradication.org/polio-today/polio-now/this-week/ (accessed Aug 18, 2020).
- 51.Turtle L, Solomon T. Japanese encephalitis - the prospects for new treatments. Nat Rev Neurol 2018; 14: 298–313. [DOI] [PubMed] [Google Scholar]
- 52.Sejvar JJ. Clinical manifestations and outcomes of West Nile virus infection. Viruses 2014; 6: 606–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet 2008; 371: 1861–71. [DOI] [PubMed] [Google Scholar]
- 54.Islam Z, Jacobs BC, Islam MB, Mohammad QD, Diorditsa S, Endtz HP. High incidence of Guillain-Barre syndrome in children, Bangladesh. Emerg Infect Dis 2011; 17: 1317–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho TW, Mishu B, Li CY, et al. Guillain-Barré syndrome in northern china relationship to campylobacter jejuni infection and anti-glycolipid antibodies. Brain 1995; 118: 597–605. [DOI] [PubMed] [Google Scholar]
- 56.Doets AY, Verboon C, van den Berg B, et al. Regional variation of Guillain-Barré syndrome. Brain 2018; 141: 2866–77. [DOI] [PubMed] [Google Scholar]
- 57.Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. Lancet 2016; 388: 717–27. [DOI] [PubMed] [Google Scholar]
- 58.Wang C, Narayan R, Greenberg B. Anti-myelin oligodendrocyte glycoprotein antibody associated with gray matter predominant transverse myelitis mimicking acute flaccid myelitis: a presentation of two cases. Pediatr Neurol 2018; 86: 42–45. [DOI] [PubMed] [Google Scholar]
- 59.Dubey D, Pittock SJ, Krecke KN, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol 2019; 76: 301–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elrick MJ, Gordon-Lipkin E, Crawford TO, et al. Clinical subpopulations in a sample of north American children diagnosed with acute flaccid myelitis, 2012–2016. JAMA Pediatr 2019; 173: 134–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zalewski NL, Rabinstein AA, Krecke KN, et al. Characteristics of spontaneous spinal cord infarction and proposed diagnostic criteria. JAMA Neurol 2019; 76: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okumura A, Mori H, Fee Chong P, et al. Serial MRI findings of acute flaccid myelitis during an outbreak of enterovirus D68 infection in Japan. Brain Dev 2019; 41: 443–51. [DOI] [PubMed] [Google Scholar]
- 63.Maloney JA, Mirsky DM, Messacar K, Dominguez SR, Schreiner T, Stence NV. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. AJNR Am J Neuroradiol 2015; 36: 245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chong PF, Yoshida T, Yuasa S, Mori H, Tanaka-Taya K, Kira R. Acute flaccid myelitis with neuroradiological finding of brachial plexus swelling. Pediatr Neurol 2020; 109: 85–88. [DOI] [PubMed] [Google Scholar]
- 65.McCoy DB, Talbott JF, Wilson M, et al. MRI atlas-based measurement of spinal cord injury predicts outcome in acute flaccid myelitis. AJNR Am J Neuroradiol 2017; 38: 410–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012; 78: 665–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKeon A, Fryer JP, Apiwattanakul M, et al. Diagnosis of neuromyelitis spectrum disorders: comparative sensitivities and specificities of immunohistochemical and immunoprecipitation assays. Arch Neurol 2009; 66: 1134–38. [DOI] [PubMed] [Google Scholar]
- 68.Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol 2019; 15: 89–102. [DOI] [PubMed] [Google Scholar]
- 69.Waters PJ, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology 2019; 92: e1250–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matà S, Betti E, Masotti G, Pinto F, Lolli F. Motor nerve damage is associated with anti-ganglioside antibodies in diabetes. J Peripher Nerv Syst 2004; 9: 138–43. [DOI] [PubMed] [Google Scholar]
- 71.Grüter T, Ott A, Meyer W, et al. Effects of IVIg treatment on autoantibody testing in neurological patients: marked reduction in sensitivity but reliable specificity. J Neurol 2020; 267: 715–20. [DOI] [PubMed] [Google Scholar]
- 72.Hovden IAH, Pfeiffer HCV. Electrodiagnostic findings in acute flaccid myelitis related to enterovirus D68. Muscle Nerve 2015; 52: 909–10. [DOI] [PubMed] [Google Scholar]
- 73.Kuwabara S, Yuki N. Axonal Guillain-Barré syndrome: concepts and controversies. Lancet Neurol 2013; 12: 1180–88. [DOI] [PubMed] [Google Scholar]
- 74.Kokubun N, Nishibayashi M, Uncini A, Odaka M, Hirata K, Yuki N. Conduction block in acute motor axonal neuropathy. Brain 2010; 133: 2897–908. [DOI] [PubMed] [Google Scholar]
- 75.Peediackal S, Jose J, Gafoor VA, Smitha B. Reversible conduction failure in acute motor axonal neuropathy. Ann Indian Acad Neurol 2014; 17: 142–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Centers for Disease Control and Prevention. Acute flaccid myelitis: clinical management. October, 2020. https://www.cdc.gov/acute-flaccid-myelitis/hcp/clinical-management.html (accessed Aug 18, 2020).
- 77.Zhang Y, Moore DD, Nix WA, Oberste MS, Weldon WC. Neutralization of enterovirus D68 isolated from the 2014 US outbreak by commercial intravenous immune globulin products. J Clin Virol 2015; 69: 172–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hixon AM, Clarke P, Tyler KL. Evaluating treatment efficacy in a mouse model of enterovirus D68-associated paralytic myelitis. J Infect Dis 2017; 216: 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rhoden E, Zhang M, Nix WA, Oberste MS. In vitro efficacy of antiviral compounds against enterovirus D68. Antimicrob Agents Chemother 2015; 59: 7779–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Messacar K, Sillau S, Hopkins SE, et al. Safety, tolerability, and efficacy of fluoxetine as an antiviral for acute flaccid myelitis. Neurology 2018; 92: e2118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vogt MR, Fu J, Kose N, et al. Human antibodies neutralize enterovirus D68 and protect against infection and paralytic disease. Sci Immunol 2020; 5: eaba4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Musharrafieh R, Ma C, Zhang J, et al. Validating enterovirus D68-2A pro as an antiviral drug target and the discovery of telaprevir as a potent D68-2A pro inhibitor. J Virol 2019; 93: e02221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Musharrafieh R, Zhang J, Tuohy P, et al. Discovery of quinoline analogues as potent antivirals against enterovirus D68 (EV-D68). J Med Chem 2019; 62: 407–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kirolos A, Mark K, Shetty J, et al. Outcome of paediatric acute flaccid myelitis associated with enterovirus D68: a case series. Dev Med Child Neurol 2019; 61: 376–80. [DOI] [PubMed] [Google Scholar]
- 85.Doi K, Sem SH, Hattori Y, et al. Surgical reconstruction for upper-extremity paralysis following acute flaccid myelitis. JB JS Open Access 2019; 4: e0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edmiston TL, Elrick MJ, Kovler ML, Jelin EB, Onders RP, Sadowsky CL. Early use of an implantable diaphragm pacing stimulator for a child with severe acute flaccid myelitis—a case report. Spinal Cord Ser Cases 2019; 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dahan V, Kimoff RJ, Petrof BJ, Benedetti A, Diorio D, Trojan DA. Sleep-disordered breathing in fatigued postpoliomyelitis clinic patients. Arch Phys Med Rehabil 2006; 87: 1352–56. [DOI] [PubMed] [Google Scholar]
- 88.Kay L, Nielsen NM, Wanscher B, Jennum P. Neurological symptoms in Danes with a history of poliomyelitis: lifelong follow-up of late symptoms, their association with initial symptoms of polio, and presence of postpolio syndrome. Eur Neurol 2018; 80: 295–303. [DOI] [PubMed] [Google Scholar]
- 89.Darras BT. Spinal muscular atrophies. Pediatr Clin North Am 2015; 62: 743–66. [DOI] [PubMed] [Google Scholar]
- 90.Haziza M, Kremer R, Benedetti A, Trojan DA. Osteoporosis in a postpolio clinic population. Arch Phys Med Rehabil 2007; 88: 1030–35. [DOI] [PubMed] [Google Scholar]
- 91.Nash MS, Bilzon JLJ. Guideline approaches for cardioendocrine disease surveillance and treatment following spinal cord injury. Curr Phys Med Rehabil Rep 2018; 6: 264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arens R, Muzumdar H. Sleep, sleep disordered breathing, and nocturnal hypoventilation in children with neuromuscular diseases. Paediatr Respir Rev 2010; 11: 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsai HC, Hung TH, Chen CC, et al. Prevalence and risk factors for upper extremity entrapment neuropathies in polio survivors. J Rehabil Med 2009; 41: 26–31. [DOI] [PubMed] [Google Scholar]
- 94.Kidd D, Howard RS, Williams AJ, Heatley FW, Panayiotopoulos CP, Spencer GT. Late functional deterioration following paralytic poliomyelitis. QJM 1997; 90: 189–96. [DOI] [PubMed] [Google Scholar]
- 95.Dalakas MC, Elder G, Hallett M, et al. A long-term follow-up study of patients with post-poliomyelitis neuromuscular symptoms. N Engl J Med 1986; 314: 959–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.