

Abstract
The merger of photoredox and nickel catalysis has enabled the construction of quaternary centers. However, the mechanism, role of the ligand, and effect of the spin state for this transformation, and related Ni-catalyzed cross-couplings involving tertiary alkyl radicals in combination with bipyridine and diketonate ligands, remains unknown. Several mechanisms have been proposed, all invoking a key Ni(III) species prior to undergoing irreversible inner-sphere reductive elimination. In this work, we have used open-shell dispersion-corrected DFT calculations, quasi-classical dynamics calculations, and experiments to study in detail the mechanism of carbon-carbon bond formation in Ni bipyridine- and diketonate-based catalytic systems. These calculations revealed that access to high spin states is critical for effective radical cross-coupling of tertiary alkyl radicals. Further, these calculations revealed a disparate mechanism for the C-C bond formation. Specifically, contrary to neutral Ni-bipyridyl system, diketonate ligands lead directly to the corresponding tertiary radical cross-coupling products via an outer-sphere reductive elimination step via triplet spin state from the Ni(III) intermediates. Implications to related Ni-catalyzed radical cross-couplings and the design of new transformations is discussed.
Graphical Abstract
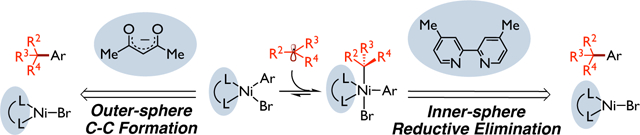
Open-shell, dispersion DFT calculations and quasi-classical dynamics calculations have been used to examine the ligand effect on the mechanism of photoredox-nickel catalyzed cross-coupling reactions with tertiary alkyl radicals. These results revealed mechanistically distinct carbon-carbon pathways for bipyridine- and diketonate-nickel based catalytic systems.
INTRODUCTION
Nickel-catalyzed cross-coupling reactions (CCRs) have emerged as powerful synthetic methods for the mild and selective construction of carbon-carbon bonds.1 Although C(sp2)–C(sp2) couplings are highly reliable and well-established,2 significant limitations are often encountered in the application of sp3-hybridized reagents, particularly in the installation of quaternary centers.3 Notable examples are from reports by the Glorius4 and Biscoe5 groups that used a Ni-based catalyst with N-heterocyclic carbene (NHC) ligands to perform C(sp2)–C(sp3) cross-couplings between aryl bromides and tertiary alkyl Grignard nucleophiles (Scheme 1A). Fu employed a nickel/bipyridine system for the cross-coupling between unactivated alkyl bromides and organoboron compounds.6 Gong used a monodentate pyridine ligand in combination with a nickel system for the reductive coupling between aryl halides and tertiary alkyl halides,7 while Watson utilized a Ni/phosphine system for the cross-coupling between tertiary benzylic acetates and organoboron compounds.8 More recently, the Molander group disclosed the first dual photoredox-Ni catalytic strategy for the cross-coupling between potassium tertiary alkyltrifluoroborates and aryl bromides (Scheme 1B).9 Notably, the bidentate bipyridyl-based ligand (4,4-di-tert-butyl-2,2-bipyridine; dtbbpy), which has proven effective in the dual photoredox-Ni catalyzed cross-coupling of secondary alkylboron reagents, was ineffective with acyclic tertiary alkyltrifluoroborates in this case. On the other hand, anionic diketonate-based bidentate ligands (e.g., 2,2,6,6-tetramethyl-3,5-heptanedionate TMHD and acetylacetonate acac) yielded the desired cross-coupling products between acyclic tertiary organoboron reagents and a wide range of electron-poor and electron-neutral aryl bromides. In this context, Baran reported the use of an anionic TMHD ligand in the nickel-catalyzed radical C(sp2)-C(sp3) cross-coupling between tertiary alkyl redox-active esters and arylzinc reagents, while Shenvi reported a dual Mn/Ni(acac)2 system to form all-carbon arylated quaternary centers from tertiary radical precursors.10
Scheme 1.
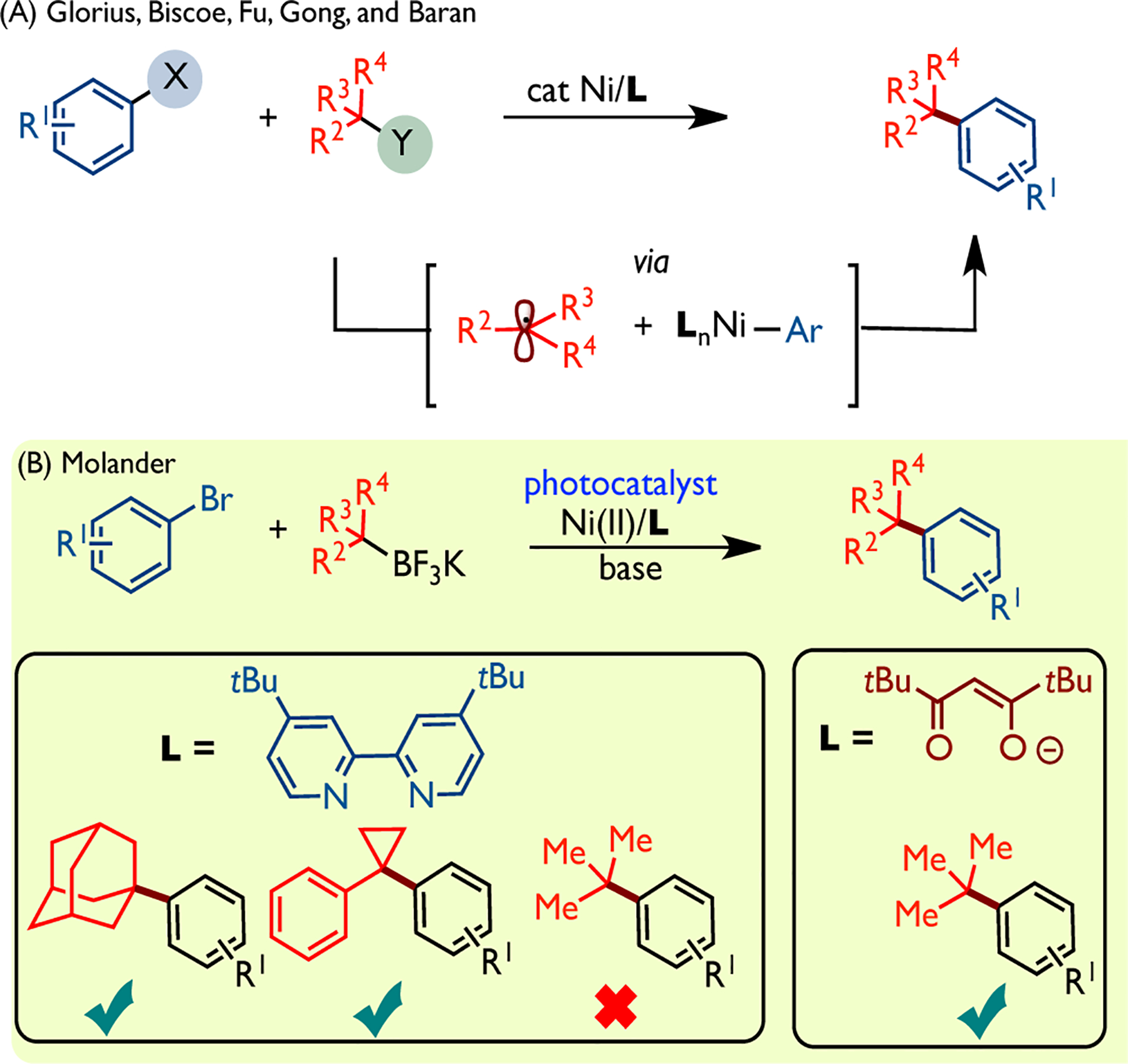
Methods for the construction of all-carbon quaternary centers via Ni-catalyzed cross-couplings between tertiaryl alkyl precursors and aryl halides.
However, although the evidence for the presence of radical intermediates in these systems is strong,11 the mechanism, effect of ligand (neutral versus anionic), and molecular-level understanding of the key C(sp2)-C(sp3) bond-forming step remain poorly understood. The use of quantum mechanical calculations to investigate the mechanisms, electronic properties, and dynamics of transition metal complexes has led to a deeper understanding of the molecular-level interactions controlling reactivity and selectivity in complex catalytic cycles.12,16,17a,18,19 Herein, quantum mechanical calculations were used to address the following questions: (1) What is the effect of anionic diketonate-based ligands versus neutral bipyridine ligands in the commonly proposed Ni(0)/Ni(I)/Ni(III) and Ni(0)/Ni(II)/Ni(III) pathways?14 (2) What are the factors responsible for the distinct reactivity of 2° vs. 3° alkyl radicals in the Ni-bipyridine system? (3) What is the role of spin state of the purported Ni complexes and the nature of C(sp2)-C(sp3) bond formation in the nickel radical cross-couplings? (4) What is the lifetime of the Ni(III) intermediate prior to undergoing inner-sphere reductive elimination and dynamic effects on radical dissociation/rebound? In this article, we report a comprehensive computational study of the mechanism of Ni-catalyzed CCRs between aryl halides and tertiary alkyl radicals using both neutral (dtbbpy) and anionic (TMHD) ligands with broad implications to related Ni-catalyzed radical cross-couplings involving tertiary alkyl radicals as reported by Fu, Baran, and others.6,7,9,13
RESULTS AND DISCUSSION
Methods.
All optimizations were performed without restrictions using open-shell, dispersion-corrected DFT (with guess=mix keyword) using THF with CPCM implicit solvation model [noted as UB3LYP-D3/def2-SVP-CPCM(THF)] as implemented in Gaussian 09 (see Supporting Information for complete reference). Dispersion correction with Becke-Johnson damping (with EmpiricalDispersion=GD3BJ keyword) was also used to calculate the key pathway in the Ni-bipyridine system, and the results were consistent with those calculated with zero-damping (see Figure S9 in Supporting Information for comparison of calculated results). Stability tests were conducted (with stable keyword) on all singlet species to confirm that the wavefunctions are stable as implemented in Gaussian 09, and all singlet species were identified as closed-shell except 1F’ and 1G’ based on S2 values (shown in Coordinates and Energetics section in the Supporting Information). Further, for comparison and to refine energetics, we also performed single point energy calculations using a larger basis set (def2-TZVPP), different solvents used experimentally (DMA and THF), other dispersion-corrected DFT functionals (e.g., UM06), and DLPNO-CCSD(T) method (in ORCA; see Supporting Information for complete reference). These methods have been used extensively to rationalize and predict reactivity and selectivity in transition metal-catalyzed transformations, including Ni-catalyzed cross-couplings.16 Overall, all methods lead to the same conclusions (see Supporting Information for details). For simplicity, only the energies obtained from UB3LYP-D3/def2-TZVPP-CPCM (DMA or THF) will be discussed in the manuscript (See Figure S1 in the Supporting Information for energetics using other methods of Ni-TMHD system). For all purported intermediates, we considered both low and high spin states (i.e., singlet/doublet and triplet/quartet) and performed a manual conformational search to identify the lowest energy structure (See Figures S2 in the Supporting Information). Initially, to reduce the computational cost, we modeled the 2,2,6,6-tetramethyl-3,5-heptanedionate TMHD ligand used experimentally (red; Scheme 1B) as acetylacetonate (acac). Notatbly, the acac ligand has also enabled nickel-catalyzed cross-couplings with tertiary radicals, albeit with lower yields as demonstrated by us and Baran.9,10 Nonetheless, to validate our model, we also compared the lowest energy pathways using the much bulkier TMHD ligand. The calculated energy values and the obtained structures with TMHD ligand are similar to those using the acac ligand (see Figure S15 in the Supporting Information).
Previously, we used open-shell DFT to investigate the mechanism of the dual photoredox/Ni-catalyzed cross-coupling between aryl bromides and benzyl radicals.14 Therein, we found that the energetically favored pathway, using neutral bipyridine and bisoxazoline ligands, proceeds via facile radical addition to Ni(0) followed by oxidative addition to the aryl halide to form a Ni(III) intermediate [i.e., Ni(0)/Ni(I)/Ni(III) mechanism]. Importantly, although based on the energetics, we could not rule out the Ni(0)/Ni(II)/Ni(III) pathway; we found that both pathways converged to a Ni(III) intermediate, which could undergo reversible radical dissociation/addition prior to undergoing irreversible (and stereodetermining) inner-sphere C(sp2)-C(sp3) bond formation. Based on these studies, we first explored the effect of an anionic ligand on the purported mechanisms for comparison (Figure 1).
Figure 1.
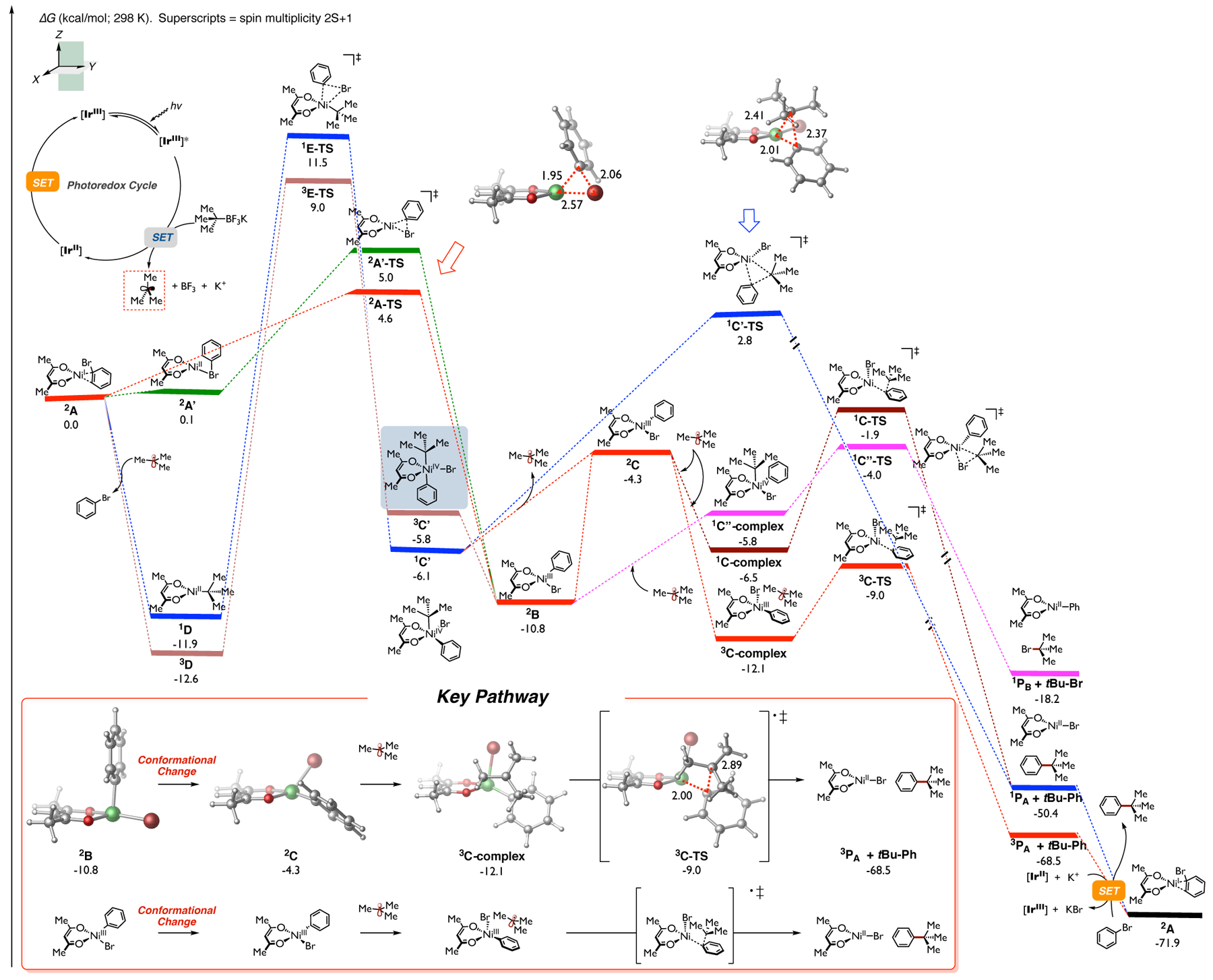
Calculated energetics of the Ni-catalyzed cross-coupling between tert-butyl radical and bromobenzene using anionic TMHD as ligand. Relative Gibbs free energy values were computed at the UB3LYP-D3/def2-TZVPP-CPCM(DMA)//UB3LYP-D3/def2-SVP-CPCM(THF).
Ni-TMHD Catalytic System with tert-Butyl Radical as Substrate.
As shown in Figure 1 (red), in the absence (or low concentration) of alkyl radical, (acac)Ni(I)•••substrate complex 2A could undergo facile oxidative addition via 2A-TS (overall barrier is only 4.6 kcal/mol) leading to Ni(III) 2B intermediate (~11 kcal/mol downhill in energy). We also located a different, slightly higher energy conformation (2A’-TS) that permits direct oxidative addition from 2A’, after isomerization from 2A to complex 2A’, to form Ni(III) 2B (green-red). In turn, Ni(III) 2B will then undergo an internal reorganization from distorted tetrahedral to distorted square planar conformation, leading to Ni(III) 2C (uphill in energy by ~6 kcal/mol from 2B).15 The distorted square planar 2C is now poised to undergo facile radical “outer-sphere” cross-coupling with the tertiary alkyl radical. Specifically, in this geometry, the tertiary alkyl radical is energetically favored to complex, but not add, to the Ni center, leading to 3C-complex. Finally, this radical will then undergo a spin-selective (barrier is only 3.1 kcal/mol from 3C-complex) outer-sphere C(sp2)-C(sp3) bond formation (via triplet spin state 3C-TS), leading to the desired product and bromo Ni(acac) products (3PA + t-Bu-Ph) downhill in energy by ~69 kcal/mol. 3PA will then undergo an energetically favorable single electron transfer (SET) with the photocatalyst ([IrIII]=[Ir{dFCF3ppy}2(bpy)]+) to generate the nickel catalyst 2A with concomitant formation of KBr complex to restart the transition metal catalytic cycle (See Figure S17 in Supporting Information for details).
Notably, the outer-sphere radical coupling via the singlet spin state (1C-TS) is found to be ~7 kcal/mol higher in energy than the calculated 3C-TS and is thus not productive.17 We also explored alternative pathways initiated from radical addition to the bromobenzene Ni(acac) complex 2A, but all were found higher in energy. Specifically, as shown in Figure 1 (light-brown), in the presence of alkyl radical, radical addition to 2A oxidative addition via triplet spin state (3E-TS), leading to 3C’. Unexpectedly, this 3C’ intermediate will then undergo barrierless tertiary radical dissociation, leading back to distorted tetrahedral 2B, merging both pathways. Alternatively, 3C’ could undergo an intersystem crossing (3C’ −> 1C’; not calculated) followed by radical dissociation leading back to distorted square planar 2C intermediate. Ultimately, both of these pathways will lead to triplet spin, outer-sphere C(sp2)-C(sp3) bond formation (via 3C-TS). Based on prior computational studies by us and others,14,16,17a,18,19 we also considered the possibility of the commonly proposed singlet spin state, inner-sphere reductive elimination (1C’-TS) that can be directly accessed from 1C’. However, this pathway was found much higher in energy (~12 kcal/mol) than the triplet spin, outer-sphere C(sp2)-C(sp3) bond formation, and thus this pathway, with an anionic diketonate ligand, can be ruled out. In addition, based on our prior work in Fe-catalyzed radical cross-couplings,20 we also located (pink) the reductive elimination transition state (1C”-TS) and the and concomitant displacement of bromobenzene could lead to singlet 1D which is exergonic by ~12 kcal/mol. The triplet spin state (3D) is also favored by more than 12 kcal/mol. From 3D, calculations predict a high-barrier (~22 kcal/mol) and rate-determining, corresponding Ni-tertiary alkyl intermediate 1C”-complex that leads to the formation of tertiary alkyl bromide (t-Bu-Br) and phenyl Ni(acac) (1PB). However, this pathway was also found to be higher in energy and thus unproductive. Nonetheless, akin to Ni(I)-Ar intermediates invoked in related cross-coupling reactions,6 in principle, LNi-Ph (1PB) could undergo oxidative addition to the tertiary alkyl bromide (reverse reaction, the barrier is only ~14 kcal/mol), leading 1C”-complex. In turn, these complex computational results also predict radical dissociation to form distorted 2B, and, ultimately, lead to outer-sphere C(sp2)-C(sp3) bond formation. Thus, independent of active nickel species (LNi, LNi-alkyl, or LNi-aryl; L = acac), these calculations revealed a new mechanistic manifold in which tertiary alkyl radicals involved in Ni-diketonate systems are predicted to proceed via outer-sphere C(sp2)-C(sp3) bond formation via triplet spin state. We also considered the likelihood of explicit DMA solvent coordinating to the key nickel species 2A or 2C and promoting ligand dissociation. However, calculations showed that the DMA solvent is unlikely to cause ligand dissociation during the transition metal catalytic cycle (See Figure S14 in the Supporting Information). Moreover, we also carried out experiments using excess TMHD anionic ligand (See Supporting Information). These results showed that excess TMHD ligand (50 and 100 mol %), which would coordinatively saturate the nickel intermediates, have a minor effect on the efficiency of the system (vide infra). Finally, the electronic properties of the aryl bromide do not change the overall mechanistic conclusions (See Figure S3 in the Supporting Information).
Overall, as shown in Figure 1 (inset), the key findings from calculations are summarized as follows: For anionic diketonate-Ni systems involving tertiary alkyl radicals (i.e., as reported by Molander9, Baran10a, and Shenvi10b) after oxidative addition from Ni(I) complex 2A, distorted tetrahedral Ni(III) species 2B can be formed, which requires a conformational change to form the distorted square planar 2C. Subsequently, 2C can form the triplet spin 3C-complex with tertiary alkyl radical, followed by triplet spin, outer-sphere C(sp2)-C(sp3) bond-forming pathway via 3C-TS to generate the desired product and bromo Ni(acac) (3PA + t-Bu-Ph). In sum, independent of mechanism, in the presence of tertiary alkyl radicals, these results strongly support a novel, low barrier, spin-selective, “outer-sphere” radical cross-coupling pathway via triplet spin state and, contrary to commonly proposed pathways, disfavor the formation of sterically hindered halo-aryl-alkyl-Ni intermediates using anionic ligands.21 These results could be applied to a wide range of experimental observations in which privileged anionic ligands, in combination with nickel complexes, allow radical cross-coupling with tertiary radical precursors.9,10
Ni-Bipyridine Catalytic System with t-Bu Radical Substrate.
Previously, neutral bipyridine ligands in combination with nickel as catalyst failed to undergo C(sp2)-C(sp3) cross-couplings with acyclic tertiary radical under dual photoredox/nickel catalysis (Scheme 1B). Nonetheless, this bipyridyl-nickel system has found success for a wide range of Ni-catalyzed radical C(sp2)-C(sp3) cross-couplings with secondary and cyclic tertiary alkyl radical precursors, and many groups, including ours,14,16c,22c have taken advantage of this reactivity to promote selective dicarbofunctionalization of olefins.19,22 However, despite the widespread utility of bipyridyl-nickel system in organic synthesis with tertiary alkyl radicals, the mechanism and, in particular, the nature of the critical C-C bond formation is not known. To gain insights from this divergent reactivity with tertiary alkyl radicals, we examined the competing reaction pathways (Figure 2, see Figure S4 in the Supporting Information for energetics using other methods).
Figure 2.
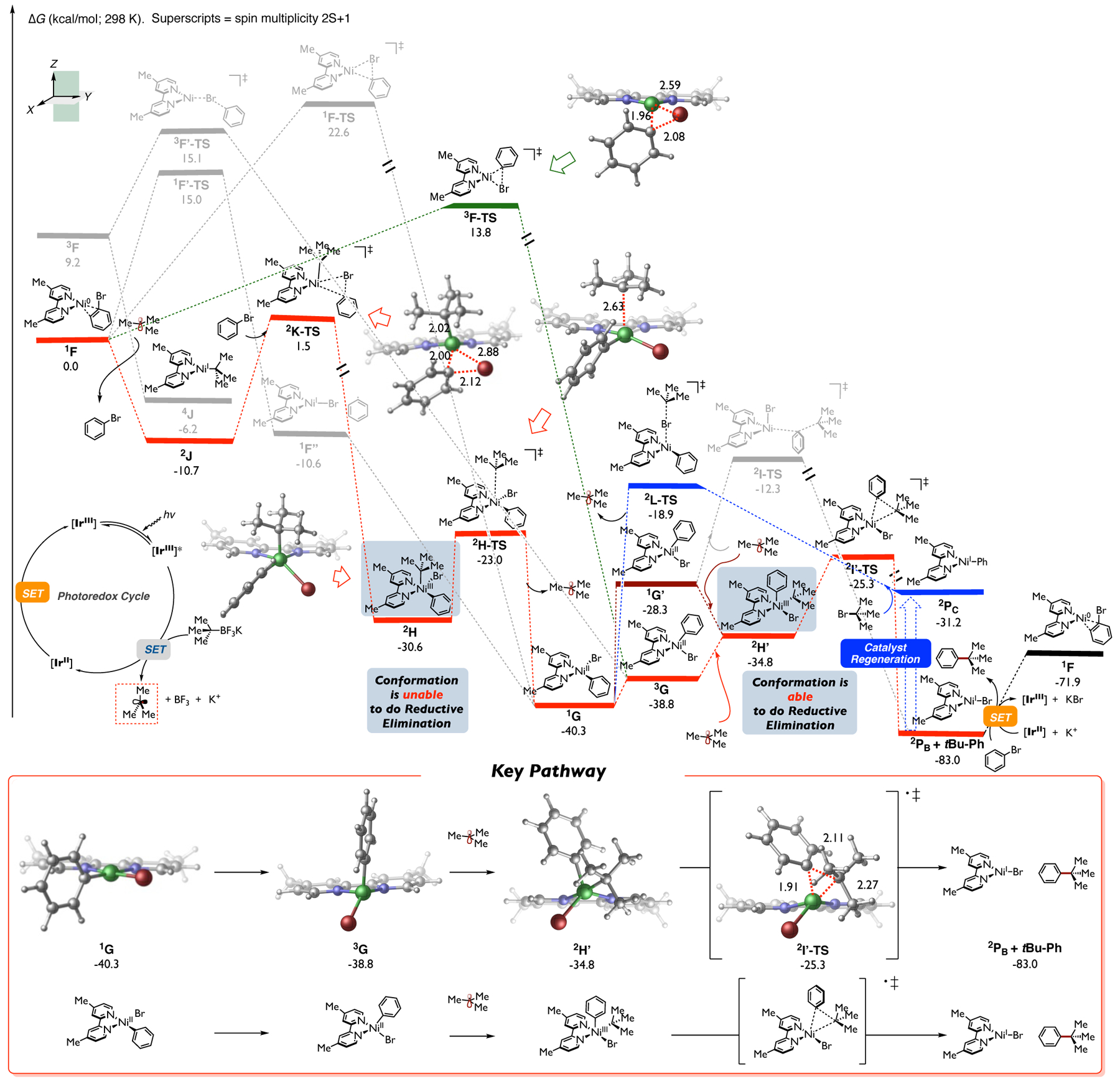
Calculated energetics of the Ni-catalyzed cross-coupling between tert-butyl radical and bromobenzene using neutral bipyridine as ligand. Relative Gibbs free energy values were computed at the UB3LYP-D3/def2-TZVPP-CPCM(THF)//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory.
Ni(0)/Ni(I)/Ni(III) Pathway:
As with previous calculations, in the presence of alkyl radical, overall the lowest energy pathway (red) proceeds via barrierless tert-butyl radical addition to Ni(0), leading to Ni(I) intermediate 2J. In turn, Ni(I) 2J undergoes oxidative addition to aryl halide via 2K-TS – the overall barrier is ~12 kcal/mol from 2J, leading to Ni(III) intermediate 2H. However, despite numerous attempts, we were unable to locate the direct, inner-sphere reductive elimination from this isomer. Instead, we found a very low barrier tertiary alkyl radical dissociation (via 2H-TS) that leads to the formation of square planar Ni(II) intermediate 1G. This square planar aryl halo Ni(II) intermediate is favored to undergo an intersystem crossing/conformational change to form the tetrahedral, triplet spin state Ni(II) intermediate 3G. In turn, 3G is now poised to undergo radical addition to form Ni(III) intermediate 2H’ followed by inner-sphere reductive elimination (via 2I’-TS), leading to the desired products 2PB and t-Bu-Ph, which is exergonic by 83 kcal/mol. After the C(sp2)-C(sp3) bond-forming step, SET can occur between the bromo Ni species and the reduced Ir photocatalyst to regenerate the Ni(0) catalytic species 1F with concomitant formation of KBr complex and restart the transition-metal catalytic cycle (See Figure S17 in Supporting Information for details). Overall, catalyst regeneration is ~11 kcal/mol uphill in energy but, based on the free energy span, energetically favorable. Given the prevalence of proposed Ni(I)/Ni(III)/Ni(I) pathways by the community and the lack of understanding of the dynamic effects in nickel-catalyzed radical cross-coupling reactions, we conducted preliminary quasi-classical dynamics simulations on the transition state of oxidative addition 2K-TS and that of reductive elimination 2I’-TS, respectively (see Figure S13 in the Supporting Information). Simulated results suggest that starting from 2K-TS in the forward direction, 60% of the trajectories resulted in the formation of a Ni(III) intermediate, while 40% formed the square planar Ni(II) intermediate and concomitant dissociation of tert-butyl radical. No cross-coupling product (t-Bu-Ph) was observed in any simulated trajectory (see Figure S13a in Supporting Information for details). These observations support our static DFT calculations that the Ni(III) intermediate obtained from direct oxidative addition on the alkyl Ni(I) intermediate cannot undergo reductive elimination, but rather undergoes radical dissociation to generate the square planar Ni(II) intermediate 1G. On the other hand, within dynamics calculations propagated from 2I’-TS in the reverse direction, all trajectories ended up in Ni(III) intermediate without radical dissociation from the Ni center or structural rearrangement (see Figure S13b in Supporting Information for details). This is also consistent with our assumption concerning the overall process because the radical addition to Ni involves a change of spin state of the Ni(II) species, while quasi-classical dynamics simulations only operate in a single spin state.24 From a broader perspective, these results suggest a novel Ni(0)/Ni(I)/Ni(III)/Ni(II)/Ni(III)/Ni(I) pathway in bipyridine-nickel radical cross-couplings with tertiary alkyl radicals.
Ni(0)/Ni(II)/Ni(III) Pathway:
In the absence (or low concentration) of tert-butyl radical, Ni(0) could undergo oxidative addition via triplet spin state (3F-TS; green) leading directly to the productive tetrahedral, triplet spin state Ni(II) 3G intermediate, thus merging both Ni(0)/Ni(I)/Ni(III)/Ni(II) (red) and Ni(0)/Ni(II) (green) pathways. We also considered the possible outer-sphere C-C bond formation pathway between tert-butyl radical and Ni(II) intermediate, but the transition states of both of the doublet (2I-TS) and quartet spin state pathways (4I-TS) are higher in energy compared to 2I’-TS (see Figure S4 in the Supporting Information for details). Therefore, inner-sphere reductive elimination via Ni(III) intermediate 2H’ is preferred to that of the outer-sphere pathway from Ni(II) intermediate in Ni-bipyridine catalytic systems.
Ni(I)/Ni(II)/Ni(III) Pathway:
Previously, alkyl halide activation by Ni(I) species (akin to 2Pc) have been proposed in a range of Ni-catalyzed cross-couplings.25 As shown in Figure 2 (blue), in these cases, aryl bipyridine-Ni(I) could undergo halogen abstraction (via 2L-TS; barrier ~12 kcal/mol from 2Pc) leading to the Ni(II) 1G, thus also merging the cross-coupling cycles. Finally, Ni(II) 1G will need to undergo an isomerization/spin-crossing to form the productive tetrahedral Ni(II) 3G which, after barrierless radical addition, will form the productive Ni(III) 2H’. Finally, inner-sphere reductive elimination (via 2I’-TS) will lead to the desired product and bromo Ni(I). From a broader perspective, although the pathway for catalyst regeneration from the bromo Ni(I) varies among nickel-catalyzed cross-coupling methods (e.g., it could undergo SET by photocatalyst or external reductant to form a Ni(0) 1F or, alternatively, could undergo transmetalation to form the Ni(I) 2PC 6), calculations show that these systems involving tertiary alkyl radicals share the same critical C(sp2)-C(sp3) bond formation step! As an example, the phenyl bpy-Ni(I) species was proposed to be obtained via transmetalation in Fu’s experiments.6 The overall key findings of these calculations are summarized as follows (Figure 2; inset): after singlet spin square planar Ni(II) species 1G forms, this species needs to undergo intersystem crossing/conformational change to generate the triplet spin tetrahedral 3G complex followed by tertiary alkyl radical addition to form Ni(III) intermediate 2H’ (see Figure S6 in the Supporting Information for more detailed analysis). The requirement for the tetrahedral Ni(II) conformation to undergo Ni(II)/Ni(III) radical addition was also reported in a recent paper with neutral bis(oxazoline) ligand, in which phenyl bromo Ni(II)-bis(oxazoline) is in a square planar geometry, while the radical adds to a tetrahedral phenyl bromo Ni(II) species.16e
Moreover, the results of our preliminary quasi-classical dynamics calculation are consistent with this suggested pathway (see Figure S7 and Figure S8 in the Supporting Information). Finally, 2H’ will undergo doublet spin, inner-sphere reductive elimination pathway via 2I’-TS to generate the desired C(sp2)-C(sp3) cross-coupling product and bromo Ni(bipyridine) (2PB + t-Bu-Ph).
Origin of the Difference in C(sp2)-C(sp3) Bond-Forming Step in Ni-TMHD and Ni-Bipyridine Catalytic Systems:
As shown above, independent of the operative catalytic active species, all pathways converge at the Ni(II) and tertiary radical. Thus, assuming facile intersystem crossing/conformational change, the barrier for inner-sphere tertiary radical C(sp2)-C(sp3) cross-coupling using neutral bipyridine nickel-system (~15 kcal/mol from 1G) is much higher in energy than using anionic diketonate nickel-system (~4 kcal/mol). These relative barriers are consistent with the experiments that prevented the formation of the desired C(sp3)-C(sp2) bond using acyclic tertiary radicals under the Ni-bipyridyl/PC system.9 Moreover, the higher barrier could lead to unwanted side reactions with tertiary radicals, such as H-atom abstraction from the solvent, as observed by us.9, 23 Further, in accord with experiments by Fu,6 switching to solvent systems with high bond dissociation energies (BDE) (e.g., benzene) likely increases the barrier for H-abstraction and, in turn, allows favorable kinetics to promote C(sp3)-C(sp2) bond formation with tertiary alkyl radicals. The competence of alkyl radical undergoing H-abstraction (e.g., with solvent or other reagents) or C(sp3)-C(sp2) bond formation is not only seen in Ni-bipyridine system in Fu’s report,6 but also observed in Ni-TMHD(acac) system,10 suggesting that the H-abstraction reactions with alkyl radical are competing with C-C bond formation in related cross-coupling reactions and the efficiency of C-C bond formation (e.g., H-abstraction versus C-C cross-coupling) is likely system dependent.
On the other hand, the cross-coupling barrier for the diketonate-nickel system is only ~3 kcal/mol from the respective nickel complex and tertiary alkyl radical. Thus, we attribute the efficiency of Ni-diketonate systems with tertiary alkyl radicals under photocatalytic conditions9,10 to the nature of the outer-sphere C-C bond formation that avoids the formation of a high energy, sterically congested aryl-alkyl-halo-Ni intermediate. Experiments using less sterically hindered secondary alkyltrifluoroborates also led to the desired product in 95% yield (Scheme 2). However, in contrast to tertiary alkyl radicals, through DFT calculations we found that the favored pathway for less sterically secondary alkyl radicals is via inner-sphere C-C bond formation with overall barriers for C-C bond formation of ~9 kcal/mol (see Figure S21 in the Supporting Information). Presumably, the less sterically hindered secondary alkyl radical (in contrast to the tertiary alkyl radical) does not pay a penalty to form the aryl-alkyl-halo-Ni species and can quickly undergo inner sphere C-C bond formation. In contrast to bipyridyl-Ni system, these results suggest that for acac/TMHD-Ni systems, the nature of the C-C bond formation and whether inner- or outer-sphere C(sp2)-C(sp3) is operative is dependent on the steric properties of the alkyl radical. Surprisingly, for cyclic tertiary systems, we found that this system did not undergo aryl substrate activation in the experiment, leading, after 24 hours, to the recovered starting material (see Supporting Information). Although the reasons for lack of reactivity are not completely understood, computations showed that, if the cyclic radical were to engage with the nickel in the cross-coupling cycle, the formation of 1-bromoadamantane via inner-sphere C-Br bond formation is favored over C-C bond formation. (See Figure S22 in the Supporting Information). However, because substoichiometric additives (e.g., LiBr, ZnBr2, ZnCl2, etc.) were found to influence the reactivity of substrates in the Ni-TMHD system where bicyclic substrate (adamantyl-like) also failed,10 the exact reason for the failure of adamantyl substrate remains unknown and is under exploration in our group.
Scheme 2. Cross-coupling of secondary alkyltrifluoroborates with Ni-TMHD.
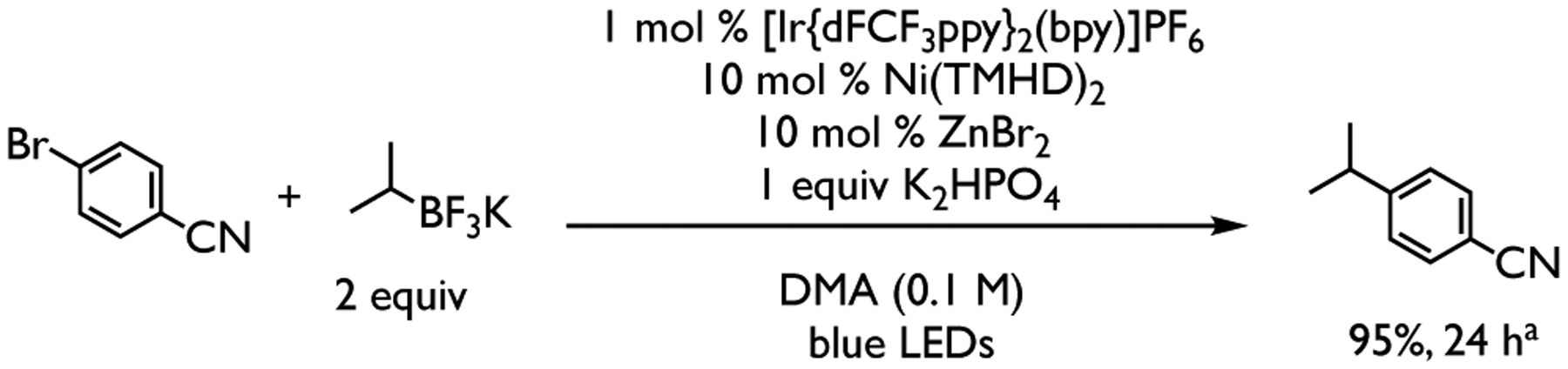
aNMR yield was calculated using 1,3,5-trimethoxybenzene as the internal standard.
To understand more fully the origins of the distinct reactivity (i.e., ligand effect on C-C bond formation), we conducted an activation strain-distortion/interaction analysis.26 As shown in Table 1, comparing inner-sphere and outer-sphere C-C bond formation transition states in the Ni-bipyridine catalytic system (green background), the relative activation electronic energy of inner-sphere transfer is negative (−3.9 kcal/mol) while that of outer-sphere transfer is positive, meaning that the C-C bond formation is favorable via inner-sphere pathway. (See Table S1 in the Supporting Information for energetics using other methods). Despite the larger distortion energy of the inner-sphere compared to the outer-sphere pathway, its interaction energy compensates for the larger distortion energy and makes the overall process favorable. On the other hand, for the Ni-TMHD system (blue background) both inner-sphere and outer-sphere transition states possess negative activation electronic energies, and both of their distortion energy and interaction energy are close to each other. Although the inner-sphere and outer-sphere pathways of the Ni-TMHD system do not show as large a difference as those of the Ni-bipyridine system, the triplet spin state outer-sphere pathway is still found most favorable (−13.3 kcal/mol) because of its lower distortion energy (18.4 kcal/mol) and larger interaction energy (−31.7 kcal/mol).
Table 1.
Activation Strain-Distortion/Interaction analysis of C(sp2)-C(sp3) bond formation step.a
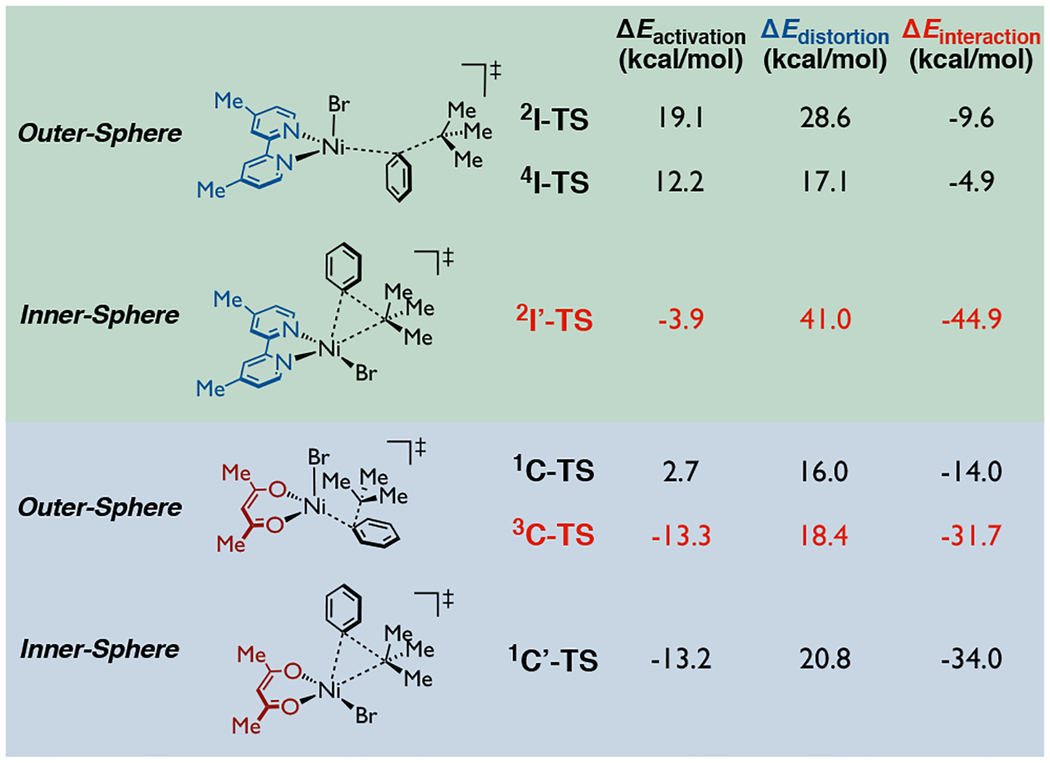
|
Relative electronic energy values were computed with respect to the separate corresponding phenyl-bromo-Ni-ligand species and tert-butyl radical at the UB3LYP-D3/def2-TZVPP//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory.
Origin of Distinct Alkyl Reactivity in the Ni-Bipyridine System:
Because of the observed experimental differences of structurally different alkyl radicals in Ni-bipyridine systems,9 we became interested in exploring the potential reactivity of different alkyl radicals in the above key pathways (Table 2). Specifically, in the dual photoredox-Ni catalytic system, C(sp2)-C(sp3) cross-coupling succeeded with aryl bromides as the electrophile and potassium 1-adamantyltrifluoroborate and potassium 1-phenylcyclopropyltrifluoroborate as the nucleophile (Scheme 1B).9 Therefore, we compare here the reactivity of tert-butyl radical, isopropyl radical, 1-phenylcyclopropyl radical, and 1-adamantyl radical in the key inner-sphere reductive elimination step (see Figure S6 in the Supporting Information for the comparison of different radicals in other steps). As shown in Table 2 (gray), using distinct Ni(II) species 1G and the purported alkyl radicals as a reference, we observed that the relative energy values of Ni(III) 2H’ intermediates, which are reactive toward inner-sphere reductive elimination, vary for different alkyl radicals. First, we observed that the Ni(III) intermediate with tert-butyl radical (black) is endergonic by 5.6 kcal/mol, while all other Ni(III) intermediates with tertiary and secondary alkyl radicals (red, blue, and green) are exergonic (by 2–3 kcal/mol). These results suggest that tert-butyl radical is more favorable to dissociate from Ni(III) center to form Ni(II) and tert-butyl radical, while for the other tertiary and secondary alkyl radicals the energetics favor the formation of Ni(III) complex. This trend is also observable in the barriers for reductive elimination. Specifically, the barriers for inner-sphere reductive elimination of cyclic and secondary alkyl radicals are significantly lower (by 7–12 kcal/mol) than with tert-butyl radical. This observation is consistent with the results in experiments by Fu, where 1-iodoadamantane, with the assumption of 1-adamantyl radical being generated in situ, can also be cross-coupled under non-photocatalytic conditions despite their slightly higher barrier of reductive elimination (7.3 kcal/mol) compared to a secondary isopropyl radical.6
Table 2.
Analysis of distinct alkyl radical reactivity in Ni-bipyridine system.a
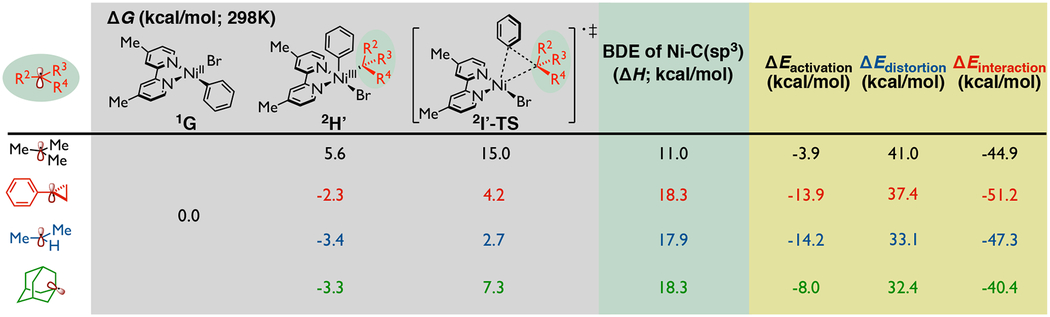
|
Gray: calculated energetics on inner-sphere reductive elimination step with different alkyl radicals; Green: related analysis on bond dissociation energy of Ni(III) intermediate; Yellow: activation strain-distortion/interaction analysis on reductive elimination transition state [relative electronic energy values were calculated with respect to the separate corresponding Ni(II) species and tert-butyl radical]. Relative free energy values were computed concerning the Ni(II) species at the UB3LYP-D3/def2-TZVPP-CPCM(THF)//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory, bond dissociation energy and relative electronic energy values were calculated at the UB3LYP-D3/def2-TZVPP//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory.
Further, to gain insight into the observed trends, we computed the bond dissociation energy (BDE) of Ni-C(sp3) bond with different alkyl radicals. As shown in Table 2 (green), we found that Ni-C(tert-butyl) bond has the lowest BDE (11.0 kcal/mol), which indicates that the dissociation of tert-butyl radical from the Ni(III) center is easier than the dissociation of secondary and cyclic tertiary alkyl radicals (~18 kcal/mol). Also, from an activation strain-distortion/interaction analysis on the reductive elimination step (yellow), we found that the transition state with tert-butyl radical shows a less negative activation energy and larger distortion energy compared to the rest of the systems. This suggests that reductive elimination with tert-butyl radical is less favorable because it needs to pay a higher energy penalty for distortion to reach the geometry in the C(sp2)-C(sp3) bond-forming step. Although the exact mechanism of bipyridyl-nickel-involved radical cross-coupling reactions could be significantly different depending on the experimental condition and applied substrates/additives, our findings on the nature of C(sp2)-C(sp3) bond formation can be used to explain some of the observed selectivity in the selective dicarbofunctionalization of olefins with a broad variety of secondary and tertiary alkyl radical precursors.14,16c,19,22
CONCLUDING REMARKS
In summary, quantum mechanical calculations and quasi-classical direct dynamics simulations have been used to investigate the mechanism of dual photoredox-Ni-catalyzed C(sp2)-C(sp3) cross-coupling reactions between tertiary alkyltrifluoroborates and aryl halides with Ni bipyridine- and diketonate-based catalytic systems. Calculations showed the prominent effect of the charge on the ligand on key catalytic intermediates and the C(sp2)-C(sp3) bond-forming step in such transformations. Specifically, as outlined in Scheme 3, in the Ni-TMHD (anionic ligand) system, the tertiary alkyl radical can directly interact with the aryl-bromo-Ni species to obtain the desired product via high-spin, outer-sphere reductive elimination pathway without engaging the Ni center first. In the Ni-bipyridine (neutral ligand) system, as shown by transition state calculations and supported by quasi-classical dynamics, the Ni(III) intermediate directly obtained from oxidative addition on alkyl Ni(I) species needs to undergo radical dissociation to form the singlet-spin square planar Ni(II) intermediate. Then, it needs to undergo a conformational change/intersystem crossing followed by radical addition to gain access to the productive Ni(III) intermediate, followed by subsequent low-spin, inner-sphere reductive elimination to generate the desired product and halo Ni(I). From a broader perspective, although the pathway for catalyst regeneration from the halo Ni(I) varies among different nickel-catalyzed cross-coupling methods (e.g., it could undergo SET by photocatalyst or external reductant to form a Ni(0) or, alternatively, could undergo transmetalation to form an aryl Ni(I)), calculations show that these systems share the same critical C(sp2)-C(sp3) formation step!
Scheme 3.
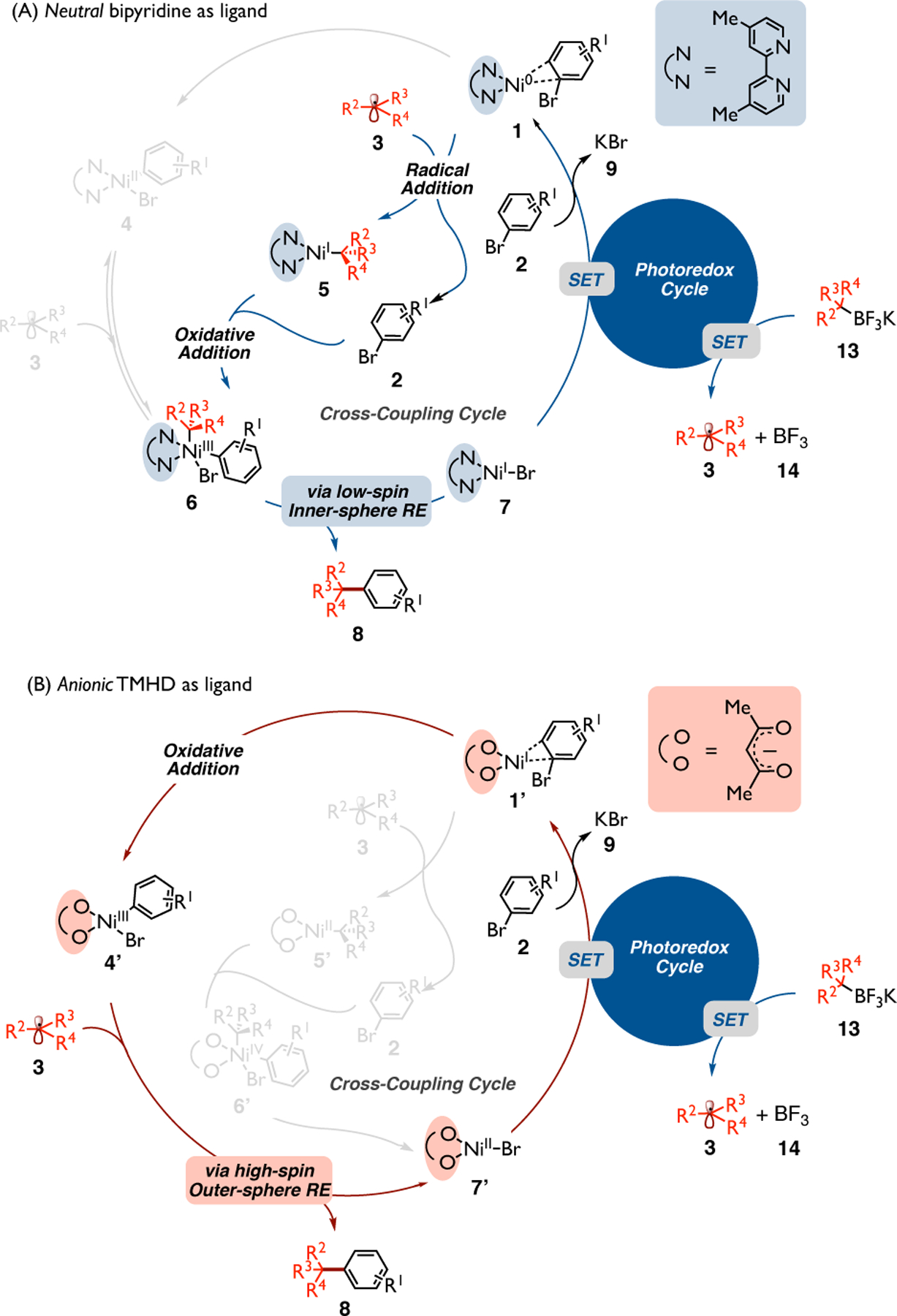
Catalytic cycle for photoredox/Ni dual catalytic system using (A) neutral (blue) and (B) anionic (red) ligands.
The reactivity of different alkyl radicals was also compared in the Ni-bipyridine system. This information suggests that relatively subtle changes in the alkyl radical precursors and ligands have a dramatic effect on the mechanism of the reactions. Nonetheless, this investigation sheds light on mechanistic possibilities that were never considered previously, and inspires the design of catalytic systems and modification of substrates for Ni-catalyzed C(sp2)-C(sp3) cross-coupling reactions. Further exploration will be focused on the comparison of more neutral and anionic ligand systems and the behavior of different alkyl radicals based on the mechanistic models proposed here.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for the financial support by the NSF (CAREER 1751568). O.G. is grateful to the University of Maryland College Park for start‐up funds and computational resources from UMD Deepthought2 and MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053). G. M. thanks the National Institutes of General Medical Sciences for support (R35 GM 131680.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental and computational details, coordinates, and spectral data (PDF)
REFERENCES
- 1.de Meijere A, Bras e S, Oestreich M, Metal-Catalyzed Cross-Coupling Reactions and More; Eds.; Wiley-VCH Ver-lag GmbH: Weinheim, 2014. [Google Scholar]
- 2.(a) Cho CH; Yun HS; Park K Nickel(0)-Catalyzed Cross-Coupling of Alkyl Arenesulfonates with Aryl Grignard Reagents. J. Org. Chem 2003, 68, 3017–3025; [DOI] [PubMed] [Google Scholar]; (b) Cheng Y; Wu Y; Tan G; You J Nickel Catalysis Enables Oxidative C(sp2)-H/C(sp2)-H Cross-Coupling Reactions between Two Heteroarenes. Angew. Chem. Int. Ed 2016, 55, 12275–12279. [DOI] [PubMed] [Google Scholar]
- 3.(a) Quasdorf KW; Overman LE Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Das JP; Marek I Enantioselective synthesis of all-carbon quaternary stereogenic centers in acyclic systems. Chem. Commun 2011, 47, 4593–4623. [DOI] [PubMed] [Google Scholar]; (c) Christoffers J; Baro A Stereoselective Construction of Quaternary Stereocenters. Adv. Synth. Catal 2005, 347, 1473–1482. [Google Scholar]
- 4.Lohre C; Dröge T; Wang C; Glorius F Nickel-Catalyzed Cross-Coupling of Aryl Bromides with Tertiary Grignard Reagents Utilizing Donor-Functionalized N-Heterocyclic Carbenes (NHCs). Chem. Eur. J 2011, 17, 6052–6055. [DOI] [PubMed] [Google Scholar]
- 5.Joshi-Pangu A; Wang C-Y; Biscoe MR Nickel-Catalyzed Kumada Cross-Coupling Reactions of Tertiary Alkylmagnesium Halides and Aryl Bromides/Triflates. J. Am. Chem. Soc 2011, 133, 8478–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zultanski SL; Fu GC Nickel-Catalyzed Carbon–Carbon Bond-Forming Reactions of Unactivated Tertiary Alkyl Halides: Suzuki Arylations. J. Am. Chem. Soc 2013, 135, 624–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wang X; Wang S; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Aryl Bromides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2015, 137, 11562–11565. [DOI] [PubMed] [Google Scholar]; (b) Wang X; Ma G; Peng Y; Pitsch CE; Moll BJ; Ly TD; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Electron-Rich Aryl Iodides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2018, 140, 14490–14497. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Q; Cobb KM; Tan T; Watson MP Stereospecific Cross Couplings To Set Benzylic, All-Carbon Quaternary Stereocenters in High Enantiopurity. J. Am. Chem. Soc 2016, 138, 12057–12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Chen T; Zhang H; Mykhailiuk PK; Merchant RR; Smith CA; Qin T; Baran PS Quaternary Centers by Nickel-Catalyzed Cross-Coupling of Tertiary Carboxylic Acids and (Hetero)Aryl Zinc Reagents. Angew. Chem., Int. Ed 2019, 58, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green SA; Huffman TR; McCourt RO; Puyl V; Shenvi RA Hydroalkylation of Olefins To Form Quaternary Carbons. J. Am. Chem. Soc 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Tellis JC; Primer DN; Molander GA Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Single-Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3–sp2 Cross-Coupling Acc. Chem. Res 2016, 49 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Primer DN; Karakaya I; Tellis JC; Molander GA Single-Electron Transmetalation: An Enabling Technology for Secondary Alkylboron Cross-Coupling J. Am. Chem. Soc 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For recent reviews, see:; (a) Sperger T; Sanhueza IA; Kalvet I; Schoenebeck F Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights. Chem. Rev 2015, 115, 9532–9586. [DOI] [PubMed] [Google Scholar]; (b) Vogiatzis KD; Polynski MV; Kirkland JK; Townsend J; Hashemi A; Liu C; Pidko EA Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem.Rev 2019, 119, 2453–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green SA; Vaśquez-Ceśpedes S; Shenvi R Iron–Nickel Dual-Catalysis: A New Engine for Olefin Functionalization and the Formation of Quaternary Centers J. Am. Chem. Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Identification of geometry is based on the average angle of the corresponding Ni species, see Figure S10 in Supporting Information for details.
- 16.For selected examples on computational analysis of nickel-catalyzed reactions, see:; (a) Oshita H; Suzuki T; Kawashima K; Abe H; Tani F; Mori S; Yajima T; Shimazaki Y The effect of π–π stacking interaction of the indole ring with the coordinated phenoxyl radical in a nickel(II)-salen type complex. Comparison with the corresponding Cu(II) complex. Dalton Trans. 2019, 48, 12060–12069. [DOI] [PubMed] [Google Scholar]; (b) Slater JW; Marguet SC; Cirino SL; Maugeri PT; Shafaat HS Experimental and DFT Investigations Reveal the Influence of the Outer Coordination Sphere on the Vibrational Spectra of Nickel- Substituted Rubredoxin, a Model Hydrogenase Enzyme. Inorg. Chem 2017, 56, 3926–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matsui JK; Gutierrez-Bonet A; Rotella M; Alam R; Gutierrez O; Molander GA Photoredox/Nickel-Catalyzed Single-Electron Tsuji–Trost Reaction: Development and Mechanistic Insights. Angew. Chem 2018, 130, 16073–16077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dohm S; Hansen A; Steinmetz M; Grimme S; Checinski MP Comprehensive Thermochemical Benchmark Set of Realistic Closed-Shell Metal Organic Reactions. J. Chem. Theory. Comput 2018, 14, 2596–2608. [DOI] [PubMed] [Google Scholar]; (e) Yin H; Fu GC Mechanistic Investigation of Enantioconvergent Kumada Reactions of Racemic α-Bromoketones Catalyzed by a Nickel/Bix(oxazoline) Complex. J. Am. Chem. Soc 2019, 141, 15433–15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For examples of nickel-catalyzed transformations via outer-sphere pathway, see:; (a) Jiang J; Fu M; Li C; Shang R; Fu Y Theoretical Investigation on Nickel-Catalyzed Hydrocarboxylation of Alkynes Employing Formic Acid. Organometallics 2017, 36, 2818–2825. [Google Scholar]; (b) Brintzinger H; Hammes GG Inner- and Outer-Sphere Complex Formation in Aqueous Solutions of Nickel(II)-Methyl Phosphate. Inorg. Chem 1966, 5, 1286–1287. [Google Scholar]
- 18.(a) Luo Y; Gutierrez-Bonet A; Matsui JK; Rotella ME; Dykstra R; Gutierrez O; Molander GA Oxa- and Azabenzonorbornadienes as Electrophilic Partners under Photoredox/Nickel Dual Catalysis. ACS Catal. 2019, 9, 8835–8842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Desrosiers JN; Wei X; Gutierrez O; Savoie J; Qu B; Zeng X; Lee H; Grinberg N; Haddad N; Yee NK; Roschangar F; Song JJ; Kozlowski MC; Senanayake CH Nickel-catalyzed C-3 direct arylation of pyridinium ions for the synthesis of 1-azafluorenes. Chem. Sci 2016, 7, 5581–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu W; García-Domínguez A; Quiroś MT; Mondal R; Caŕdenas DJ; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc 2019, 141, 13812–13821. [DOI] [PubMed] [Google Scholar]
- 20.(a) Liu L; Lee W; Zhou J; Bandyopadhyay S; Gutierrez O Radical-clock a-halo-esters as mechanistic probes for bisphosphineiron-catalyzed cross-coupling reactions. Tetrahedron. 2019, 75, 129–136. [Google Scholar]; (b) Liu L; Lee W; Yuan M; Gutierrez O Mechanisms of Bisphosphine Iron-Catalyzed C(sp2)-C(sp3) Cross-Coupling Reactions: Inner-Sphere or Outer-Sphere Arylation? Comment. Inorg. Chem 2018, 38, 210–237. [Google Scholar]; (c) Lee W; Zhou J; Gutierrez O Mechanism of Nakamura’s Bisphosphine Iron-Catalyzed Asymmetric C(sp2)-C(sp3) Cross-Coupling Reaction: The Role of Spin in Controlling Arylation Pathway. J. Am. Chem. Soc 2017, 139, 16126–16133. [DOI] [PubMed] [Google Scholar]
- 21.Calculations using other substrates and different methods led to the same conclusions (see Figure S3 in the Supporting Information).
- 22.For selected examples, see:; (a) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-active Esters and Alkylzinc Reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gu J-W; Min Q-Q; Yu L-C; Zhang X Tandem Difluoroalkylation-Arylation of Enamides Catalyzed by Nickel. Angew. Chem., Int. Ed 2016, 55, 12270–12274. [DOI] [PubMed] [Google Scholar]; (c) Campbell MW; Compton JS; Kelly CB; Molander GA J. Am. Chem. Soc 2019, 141, 20069–20078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Further calculations suggest that these side reactions are feasible in the reaction system, which could explain the observation of THF-Ar adduct and Ar-H in the stoichiometric studies of aryl bromo Ni(II) with tBu-BF3K in experiment (see Figure S11 and S12 in the Supporting Information for the calculation of possible side reaction pathways).
- 24.We investigated the possibility of ligand dissociation from Ni(III) intermediate, which might invoke structural rearrangement of Ni(III) intermediates. However, based on our calculations the complete ligand dissociation is unlikely to occur on neither 2H nor 2H’ since these processes are both uphill in energy (see Figure S19 for energetic details in the Supporting Information). In addition, although transient decoordination of one of the pyridine ligands was observed in quasi-classical dynamics simulations on 2K-TS in forward direction (see Figure S20 for an example trajectory), the Ni(III) intermediates didn’t undergo any structural rearrangement, but rather stayed the same conformation until the end of simulation (60% of trajectories) or underwent radical dissociation of tBu radical from axial position of Ni center (40% of trajectories).
- 25.For selected reviews, see:; (a) Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends in Chemistry. 2019, 1, 830–844. [Google Scholar]; (b) Shi R; Zhang Z; Hu X Nickamine and Analogous Nickel Pincer Catalysts for Cross-Coupling of Alkyl Halides and Hydrosilylation of Alkenes. Acc. Chem. Res 2019, 52, 1471–1483. [DOI] [PubMed] [Google Scholar]; (c) Feng Z; Xiao Y; Zhang X Transition-Metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-Coupling with Difluoroalkyl Halides. Acc. Chem. Res 2018, 51, 2264–2278. [DOI] [PubMed] [Google Scholar]; (d) Gu J; Wang X; Xue W; Gong H Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front, 2015, 2, 1411–1421. [Google Scholar]; (e) Hu X Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci, 2011, 2, 1867–1886. [Google Scholar]
- 26.For reviews, see:; (a) Ess DH; Houk KN Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc 2007, 129, 10646–10647. [DOI] [PubMed] [Google Scholar]; (b) Ess DH; Houk KN Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models. J. Am. Chem. Soc 2008, 130, 10187–10198. [DOI] [PubMed] [Google Scholar]; (c) Fernandez I; Bickelhaupt FM The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev 2014, 43, 4953–4967. [DOI] [PubMed] [Google Scholar]; (d) Wolters LP; Bickelhaupt FM The activation strain model and molecular orbital theory. Wiley Interdisciplinary Reviews: Computational Molecular Science 2015, 5, 324–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.