

Abstract
Enrichment of ligands versus property-matched decoys is widely used to test and optimize docking library screens. However, the unconstrained optimization of enrichment alone can mislead, leading to false confidence in prospective performance. This can arise by over-optimizing for enrichment against property-matched decoys, without considering the full spectrum of molecules to be found in a true large library screen. Adding decoys representing charge extrema helps mitigate over-optimizing for electrostatic interactions. Adding decoys that represent the overall characteristics of the library to be docked allows one to sample molecules not represented by ligands and property-matched decoys but that one will encounter in a prospective screen. An optimized version of the DUD-E set (DUDE-Z), as well as Extrema and sets representing broad features of the library (Goldilocks), is developed here. We also explore the variability that one can encounter in enrichment calculations and how that can temper one’s confidence in small enrichment differences. The new tools and new decoy sets are freely available at http://tldr.docking.org and http://dudez.docking.org.
Graphical Abstract
INTRODUCTION
Large library docking screens seek to discover new chemotypes that are active on a target, based on molecular fit. Calculation speed has been crucial since the field’s inception,1–9 and to ensure it, several biophysical terms are either approximated or ignored entirely. While this led to programs that can screen libraries now approaching10 or exceeding11 a billion molecules and discovering novel ligands for multiple targets,10,12–24 the emphasis on throughput has forced compromises that make predicting absolute binding energies by docking or even compound rank ordering implausible.25 While molecular docking screens are thus pragmatic and while docking remains among the methods most subject to experimental testing in computational biophysics, it is also among the biophysical methods that have most surrendered “ground truth”.
Accordingly, to evaluate new docking methods or to evaluate how well docking might perform prospectively on a new target, benchmarking studies are often performed. For a new docking method, these benchmarks evaluate the key outcomes expected of a library screen: can the method reproduce the binding orientations of known ligands for a range of targets, and can it enrich known ligands from among a set of decoys over a range of disparate targets? For a particular target campaign with an established method, such benchmarks optimize for ligand pose fidelity and enrichment. This occurs by varying sampling and weighting energy terms—ideally constrained by physical reasonableness. It has been argued that careful construction of retrospective benchmarks, indeed by addressing some of the same problems that we investigate here, can lead to retrospective performance that predicts prospective success.26–28 Our own view is more conservative: given the very thin slice of top-ranking candidates from which docking predictions are drawn, it is difficult for small retrospective benchmarks to predict prospective, experimental success in docking much larger libraries. Still, without such benchmarks, the likelihood of success is reduced, as is our ability to understand failure. In docking, running detailed benchmarks for a new method or on a new target fulfills the same role as controls in experimental biology, which often also lack “ground truth”, and so must rigorously control all new experiments. However, just as in experimental biology, well-run controls simply protect against obvious failure and allow one to disentangle prospective failure when it frequently occurs; they do not predict prospective success when one is trying to discover something genuinely new.
Among the most widely used benchmarks in library docking is the enrichment of annotated ligands versus property-matched “decoy” molecules.29–31 A decoy molecule is one that is expected not to bind to a protein of interest; enrichment measures docking’s ability to highly rank (enrich) the annotated ligands vs such decoys. The idea of using decoys in benchmarks follows from analogous use in protein structure prediction32–34 and initially drew on random molecules.35–37 As is true for folding decoys, it was found that it was important that decoy molecules physically resemble the known ligands; otherwise, the docking program might be optimized to simply recognize gross physical differences, such a molecular weight, hydrophobicity, or charge.38 Property-matched decoys match ligands by physical properties but are otherwise topologically unrelated and so presumed not to bind. Enrichment of ligands against property-matched decoys, in sensible geometries, thus offers some assurance that the docking program recognizes ligands by their detailed interactions and not just gross physical differences. Several benchmarking sets of ligands and property-matched decoys have been introduced,39–46 including the DUD and DUD-E sets.29,30 The DUD-E benchmark, which covers 102 proteins, 22,886 ligands, and 1.1 million property-matched decoys, is widely used to test new methods, while its method of matching ligands to decoys is often employed to construct bespoke benchmarks as controls for individual target campaigns.
Notwithstanding its wide use, several studies have shown that DUD-E retains important liabilities. These include small differences in ligand vs decoy property matching, which can be exploited by virtual screening to falsely increase enrichment,47–49 as can self-similarity among the benchmark molecules.49 Finally, property-matched decoys do not represent the full spectrum of molecules that will be encountered in docking a diverse library, something that has become increasingly true as these have increased to 109 molecules. For instance, they will not expose one to extreme physical differences nor will they necessarily represent even the typical molecular properties of a large library;26,50,51 this argues for adding, to property-matched benchmarks, those that challenge not with property-matched decoys but decoys that represent the molecules that one might expect to encounter during the prospective screen.
Here, we investigate optimized and new benchmarks that contribute to addressing some of these pathologies. An optimized version of the DUD-E set (DUDE-Z) addresses unintended biases in the older set that others have described,47,48 allowing for false enrichment. We also investigate an extrema benchmarking set (Extrema), which seeks to address charge imbalances in docking scoring functions and by nature uses decoys that are opposite of property-matched. Finally, we investigate a benchmark that represents ligands that have average physical features of the larger library to be docked, following up on weaknesses pointed out by earlier studies,26,50,51 rather than being property-matched. Akin to the Grimm fairy tale, we call this library “Goldilocks” because its molecules are drawn at random from the middle of ZINC lead-like physical property space and are not too big, not too small, not too greasy, and not too polar. In our experience, retrospective calculations against each set help control for different pathologies in prospective docking campaigns that are chief interest in early ligand discovery research.
METHODS
DUD-E.
Three-dimensional dockable ligand and decoy files for the 41 DUD-E targets were downloaded from http://autodude.docking.org. For D4 dopamine and melatonin MT1 receptors, DUD-E decoys were generated from http://dude.docking.org/generate and built using an in-house ligand building pipeline.
Binders & Nonbinders.
Three-dimensional dockable files for binders and nonbinders for D4 dopamine and MT1 melatonin receptors were downloaded from ZINC15. This included 84 binders and 468 nonbinders and 105 binders and 65 nonbinders for D4 and MT1, respectively. Enrichment calculations were performed for all 16 scoring function coefficient combinations (see Docking Calculations).
DUDE-Z.
An initial motivation for this study was an imbalance among the charge states between ligands and decoys in DUD-E, arising from the generation of multiple protonation states for the molecules. In the DUD-E set, this had arisen because the DUD-E set was reported in 2D SMILES format with specific protonation states specified. Because our pipeline builds all molecules at all protonation states at physiological pH, the generation of new protonation states of ligands and decoys disturbs the charge balance originally controlled in the DUD-E set. To correct this in the DUDE-Z set, only prebuilt 3D decoys with specified protonation and charge states are matched to prebuilt ligands, ensuring that charge is matched exactly, and this balance is not disrupted. The DUDE-Z set is provided in both 2D and 3D formats.
As part of the current ligand building pipeline,52 ChemAxon’s CXCALC command is used on the 2D SMILES of each molecule to generate protonation and tautomer states at physiologically relevant pH.53 Each protomer is converted to 3D format using CORINA,54 and conformational ensembles of each protomer are generated using OpenEye’s Omega.55 Atomic charges and desolvation penalties are calculated using AMSOL7.1.56 Files are formatted into flexibases for docking with DOCK3.7.
Because several DUD-E systems had large numbers of ligands and decoys, we reduced the number of ligands for more rapid docking calculations. Targets with over 100 ligands had their ligands sorted by molecular weight and were clustered by an ECFP4 Tanimoto coefficient (Tc) of 0.7. The smallest ligand in each cluster served as the cluster representative for property-matched decoys, which had the added advantage of better matching the properties of the general docking library. As several of the docking targets have high molecular weight ligands and because 3D molecules in ZINC15 are biased toward lead-like properties (as of July 2020, 448 million of the 698 million 3D molecules in ZINC15 are defined by 300 ≤ MW ≤ 350 and −1 ≤ cLogP ≤ 3.5), we found that using the smallest ligand as the cluster representative had the greatest success in retrieving sufficient numbers of 3D property-matched decoys. For targets with less than 100 ligands, all ligands were retained for generating property-matched decoys.
As in DUD-E, decoys were matched to ligands based on the molecular weight, water–octanol partition coefficient (cLogP), number of rotatable bonds, number of hydrogen bond donors and acceptors, and net charge. We generated all protonation states for each ligand using ChemAxon’s Jchem53 at physiological pH and computed molecular properties using RDKit. Each of these protomers shares the same molecule ID; an underscore is added along with the number for each protomer; for instance, a molecule with two protomers would be designated with ZINCXXXX_0 and ZINCXXXX_1. Each protomer would be assigned up to 50 property-matched decoys, resulting in 100 property-matched decoys for this single molecule. For each protomer, the optimal goal was to find 50 property-matched decoys, but we also accepted as few as 20 if the number of decoys in ZINC15 was limited in this property space. To identify matching decoys, the ZINC15 website was queried for up to 10,000 3D molecules matching the ligand protomer for the molecular properties listed above. Once thousands of decoys for a target were retrieved, ECFP4 Tanimoto calculations were performed using in-house programs (https://github.com/docking-org/ChemInfTools) between all ligands and all potential decoys for that target. Any decoy that had greater than 0.35 ECFP4 Tc—i.e., was too similar topologically—to any ligand was discarded. The decoys were then sorted by molecular weight and clustered by an ECFP4 Tc of 0.8, with the smallest decoy being retained from each cluster. This ensured that property-matched decoys would not contain duplicates and ensured some scaffold exploration among the decoys. The remaining decoys were sorted by ECFP4 Tanimoto coefficients to all ligands and were placed such that the ligand with the least number of decoys assigned would get the decoy in an iterative procedure. If fewer than 50 decoys could be assigned to all ligands, then the highest number of decoys that could be assigned to the ligand protomers was computed. If it was difficult to find 3D decoys for a target, then an alternative approach that queries ZINC15 for molecular SMILES was used. The procedure was largely the same, except that up to 750 potential decoys were retrieved for each ligand protomer based on molecular weight and cLogP of the decoy SMILES. Then, an additional step was performed in which ChemAxon’s JChem was used to generate protonation states for these decoys’ SMILES followed by calculation of the remaining molecular properties by RDKit to determine whether they matched the ligands in property space.
Extrema.
To generate extrema sets for all 43 targets, the molecular weight and cLogP values of the DUD-E ligand set were calculated using RDKit, and the corresponding interquartile ranges were calculated. For each charge, we retrieved a minimum of 1000 “in-stock” or “make-on-demand” molecules from ZINC15, built at physiological pH of 7.4, in readily dockable format in this molecular weight and cLogP property space. Most of these molecules fall within charge ranges from −2 to +2, but there exist molecules with outlier charges as well. These dockable molecules were docked to their protein targets, and enrichment calculations were performed (see Docking Calculations).
Goldilocks.
For generating the Goldilocks decoy set, which is used for all targets, the same procedure as with Extrema was used. However, instead of matching the decoys to an input ligand set, “in-stock” 3D-built molecules for each charge ranging from −2 to +2 within the property space (300 Da ≤ MW ≤ 350 Da, 2 ≤ cLogP ≤ 3) were retrieved from ZINC15.52 For each charge, 3D-built molecules were retained until they reached half of the total number of 3D molecules with that charge and within that molecular weight and cLogP property space (on December 10, 2019). These dockable molecules were docked to their protein targets, and enrichment calculations were performed (see Docking Calculations). Of the 69,909 decoys in DUDE-Z, 5357 also appear within the 1.1 million Goldilocks set.
Docking Calculations.
DOCK3.7.257 was used for ligand docking. The orientations of candidate ligands are calculated in the site by matching ligand atoms to precalculated hot spots on the protein surface, using internal distance correspondence to ensure fidelity and to calculate a rotation-translation matrix that moves the library molecule from its initial frame-of-reference to that of the binding site.57–59 Once fit in the site, potential ligands are scored for fit based on electrostatics and van der Waals complementarity, corrected for ligand desolvation. The protein is protonated by REDUCE60 and assigned AMBER61 united atom charges. QNIFFT62 is used to calculate Poisson—Boltzmann-based electrostatic potentials, CHEM-GRID1 is used for AMBER van der Waals potentials, and SOLVMAP63 is used for calculating ligand desolvation energies. With these grids calculated, docking scores may be rapidly calculated by looking up the potentials for each ligand atom and multiplying them by the appropriate ligand property (e.g., electrostatic interactions are the partial atomic charge times the electrostatic potential at that position in space, as stored on the grid). The value of the electrostatic potential depends on where the dielectric boundary is drawn between a low-dielectric protein (ε = 2) and a high-dielectric solvent (ε = 80). This can be extended out by the addition of low-dielectric spheres in the site. To represent ligand flexibility, DOCK3.7 orients flexibases64—precomputed 3D conformer ensembles—into the binding site. After molecules are scored for complementarity with the protein, simplex minimization is performed on the top scoring pose of each molecule.
Targets chosen for docking were based on completeness of structure, no missing active site loops, diversity of protein types (enzymes, proteases, GPCRs, and kinases, among others), and diversity of ligand charge. Of the 43 targets, 41 targets were taken directly from the DUD-E set, while MT1 and DRD4 were taken from recent docking campaigns. Ligands for each of the targets were taken from the DUD-E set. As described previously,30 ligands annotated to targets with activities (EC50, IC50, Ki, Kd, and log variants thereof) of 1 μM or better were extracted from ChEMBL09.65 These are labeled as “active-s_nM_combined.ism” and can be found on the DUD-E webpage (www.dude.docking.org/targets/). Ligands that have affinities worse than 1 μM are labeled as “actives_marginal_combined.ism” on the DUD-E webpage. Except for AmpC, where we have specialist knowledge, we did not remove molecules that may be acting as colloidal aggregators nor those with PAINS functionality, hoping that the 1 μM filter will eliminate most of these. Aggregators and molecules with PAINS alerts were also not removed from the DUD-E set; other investigators may wish to filter more stringently by these criteria and may do so by building on the scripts in http://tldr.docking.org.
The PDB structures assigned to 40 DUD-E targets were retrieved and prepared in an automated fashion by in-house scripts based on the DOCK Blaster pipeline66 for generating docking grids (blastermaster.py in the DOCK3.7 distribution). The docking preparations for AmpC,10,67,68 DRD410,14 (PDB: 5WIU), and MT169 (PDB: 6ME3) adopted the parameters that had been used in published prospective docking screens against these targets, which led to experimental testing of tens to many hundreds of molecules. This allows investigators to use not only calculated decoys but also experimentally measured false positives from these prospective docking screens. Thin sphere layers were used for AmpC, DRD4, and MT1 to extend the dielectric boundary from the solute surface for Poisson–Boltzmann calculation63 radii of 2.0, 1.0, and 1.9 Å, respectively. For all other systems, the default DOCK Blaster preparation was used in which the full binding site was filled with low-dielectric spheres of radius 1.9 Å for Poisson–Boltzmann calculations, thereby modeling the full binding site as a low-dielectric solute. The magnitudes of the partial charges of five AmpC residues and two MT1 residues were increased without changing the net residue charges.68 For all DUD-E targets, their DUD-E assigned PDB ligand was used for generating up to 45 matching spheres, to which molecules are matched during docking. For DRD4 and MT1, matching spheres were generated based on the atomic coordinates of nemonapride and 2-phenylmelatonin, respectively. Ligand conformations were generated by OpenEye’s Omega.55 Ligands were only scored if the number of ligand heavy atoms contained within the ligand ranged from 4 to 100. For each ligand hierarchy (each rigid fragment contained within the ligand), the maximum number of matches generated was set to 5000. For AmpC and DRD4, the large-scale docking setup was used, in which the target number of ligand hierarchy matches was set to 1000, and up to 500 simplex minimization70 steps were performed for each top scoring pose of each docked molecule, starting with initial translations of 0.2 Å and initial rotations of 5°. For MT1, the target number of ligand hierarchy matches was set to 5000, and up to 500 simplex minimization steps were performed for each top scoring pose of each docked molecule. All other DUD-E systems did not use simplex minimization. To judge performance, the adjusted log AUC was used. The adjusted log AUC subtracts the log AUC of the random curve (14.462%) to ensure that random enrichment is 0% at any percentage of the database. For the DUD-E benchmarking calculations, the DUD-E ligands for each target are used as the ligand set for these calculations. For all plots for DUDE-Z, Extrema, and Goldilocks, the reduced ligand set after clustering by an ECFP4 Tanimoto coefficient of 0.7 is used for these calculations.
To prepare different scoring function coefficient combinations, the “electrostatic_scale” and “ligand_desolv_scale” parameters of the INDOCK files for each target were modified to be 0.3, 0.5, 0.7, or 1.0, generating 16 different combinations of DOCK scoring weights. The van der Waals scoring function coefficient was maintained at 1.0 for all docking calculations. All other parameters in the INDOCK file, docking grids, and matching spheres were kept identical.
Bootstrapping.
To add error bars to our LogAUC calculations and to compare different setups statistically, we use bootstrapping. For each bootstrap replicate (50 total for each system), ligands and decoys were chosen at random with replacement (i.e., a ligand or decoy could be chosen multiple times) until the same sample size as the original set was reached. Each new hit list was then sorted by the original docking energy, and a new adjusted log AUC is calculated. Z-tests were performed to test the significance of the difference between the means of two bootstrapped distributions. With the p-value smaller than 0.05, the null hypothesis of equal mean and distribution is rejected. The Z-test is chosen since the number of bootstrap replicates is larger than 30, and the bootstrapped distribution rapidly converges to the normal distribution with mild finite-variance assumptions.71
RESULTS
DOCK Scoring Function Optimization Using Property-Matched Decoys.
We were confronted with the liabilities of relying on property-matched decoys in an investigation of different weighting terms in the DOCK3.7 scoring function.57,63 We initially tried to use adjusted LogAUC performance (see below) to guide the optimization of the scoring function by varying the coefficients of the electrostatics and ligand desolvation contributions to the total docking score. We scanned across electrostatics and ligand desolvation weighting for 41 DUD-E targets and for the MT1 melatonin receptor (MT1) and D4 dopamine receptor (DRD4), which have the advantage of hundreds of experimentally tested docking predictions10,69 (Figure 1). To measure performance, we used a log-weighted area under the curve approach, subtracting from this enrichment expected at random (adjusted Log AUC,63 Figure 1 and Table 1). This approach equally weights enrichment in the top 0.1 to 1% of the library with that within the top 1 to 10% and the top 10% to 100% of the library, thus up-weighting early performance. Sampling sixteen combinations of weights (four electrostatics, four ligand desolvation with constant van der Waals) revealed that performance correlated with the electrostatics and ligand desolvation terms (Figure 1a, Table 1, but see Sensitivity Analysis below for the significance of these differences). In most of the DUD-E targets, increasing the electrostatic coefficient increased enrichment of ligands among high-ranking molecules. This included systems such as GAR transformylase (PUR2), which had its best performance with weights of 1.0 for electrostatics and 0.3 for ligand desolvation (Figure 1b). These same coefficients, however, negatively impacted other systems, such as C-X-C chemokine receptor type 4 (CXCR4), where the same weights that were optimal for AmpC led to worse performance. Instead, CXCR4 had its best enrichment of ligands among high-ranking molecules with weights of 0.5 on the electrostatics and of 1.0 on the ligand desolvation terms (Figure 1c).
Figure 1.

(a) For each electrostatic coefficient (0.3, 0.5, 0.7, and 1.0), the average adjusted log AUC value and standard error, which are calculated over the four ligand desolvation coefficients (0.3, 0.5, 0.7, and 1.0), are plotted. Individual adjusted LogAUC plots for each electrostatic and ligand desolvation coefficient combination for PUR2 (b) and CXCR4 (c) are shown. Performance for PUR2 diminishes as the ligand desolvation coefficient increases, while performance for CXCR4 improves as the ligand desolvation coefficient increases.
Table 1.
Adj. LogAUC for DOCK3.7 Scoring Coefficients over 43 Targetsa
| 0.3ES | 0.5ES | 0.7ES | 1.0ES | |
|---|---|---|---|---|
| 0.3LD | 16.3 (11, 7, 25) | 13.89 (8, 10, 25) | 11.85 (6, 8, 29) | 9.95 (6, 8, 29) |
| 0.5LD | 19.3 (17, 9, 17) | 17.99 (12, 11, 20) | 15.87 (8, 14, 21) | 12.71 (4, 14, 25) |
| 0.7LD | 20.17 (17, 14, 12) | 19.88 (17, 19, 7) | 18.72 (9, 22, 12) | 16.05 (4, 17, 22) |
| 1.0LD | 19.95 (15, 15, 13) | 20.31 (17, 18, 8) | 20.1 (16, 21, 6) | 19.19 |
Values outside the parentheses are the average adjusted log AUC values, while those within the parentheses refer to those targets that improved by 1 adjusted log AUC value, stayed within ±1 log AUC, and diminished by 1 adjusted log AUC value vs the standard scoring function (1.0ES+1.0vdW+1.0LD) (see Table S1 for full results).
Closer inspection revealed that the enrichment differences and the sensitivity to scoring coefficients were often explained by different formal charge distributions between ligands and decoys. For instance, for AmpC, larger weighting of electrostatic interactions improved enrichment of high-ranking ligands because AmpC’s ligands are all anionic, whereas 35% of AmpC’s DUD-E decoys are neutral (Figure 2a). Thus, as the weight on the ligand desolvation term, which scales with net charge, decreases, AmpC’s anionic ligands are penalized less (Figure 2c). When unconstrained, as with an electrostatics weighting of 1.0 and ligand desolvation weighting of 0.5, the “optimized” scoring function, i.e., the coefficients that maximize enrichment, prioritizes charge over other molecular properties versus the unweighted, standard scoring function. Similarly, most of the PUR2 ligands are dianions, while their decoys are mainly monoanionic or neutral (Figure 2b), and docking with reduced ligand desolvation coefficients favors the ligands over the decoys (Figure 2d). Even if all our molecular properties, besides charge, are well-matched in the DUD-E benchmarking sets, altering the scoring function weights of electrostatics and ligand desolvation allows DOCK to simply recognize gross physical differences between ligands and decoys, rather than detailed molecular interactions, reflecting an imbalance in the DUD-E ligand and decoy properties.
Figure 2.

Proportion of charged ligands and decoys in the DUD-E benchmarking sets coupled with altered electrostatic and ligand desolvation weights affects the DOCK energies and thus LogAUC values. Percentage of ligands or decoys in the DUD-E set with a given charge for AmpC β-lactamase (AmpC, a) and GAR transformylase (PUR2, b). Comparison of DOCK energy and molecule charge for AmpC β-lactamase (AmpC, c) and GAR transformylase (PUR2, d) for the electrostatic coefficient of 1.0 and the four ligand desolvation weights (0.3, 0.5, 0.7, and 1.0). Central dotted lines of DOCK energies represent the medians, upper dotted lines represent the third quartiles, and lower dotted lines represent the first quartiles for both scoring functions. The lowest points represent the minimum DOCK energies, and the highest values represent the maximum DOCK energies. The AmpC ligands in DUD-E are predominantly anionic (a), and while this is also true for the decoys, the latter harbors a higher ratio of neutral molecules. Increasing the ligand desolvation coefficient ranks neutral molecules higher (as sorted by total DOCK energy), favoring decoys, and enrichment decreases (c). Conversely, increasing the electrostatic coefficient favors the anionic ligands, increasing the enrichment. The large majority of PUR2 ligands is di-anionic, while the decoys are monoanionic (b), providing an advantage to the ligands at lower ligand desolvation coefficients (as sorted by total DOCK energy) (d), as they can form more favorable electrostatic interactions with the protein without a large ligand desolvation cost.
New Property-Matched Decoy Method.
The original DUD-E benchmarking set30 was built to correct the charge imbalance in the original DUD set29 by including net charge during property matching. However, during molecular building of 3D dockable molecules, the charge populations change based on which protomers are predicted to exist at physiological pH, producing charge imbalances that were not present in the SMILES representation. For example, calculating the formal charges of the AmpC ligand and decoy SMILES contained within the DUD-E benchmarking set suggests that 60 and 38% of ligands are neutral and monoanionic, respectively, while 43 and 56% of decoys are di- and monoanionic, respectively, compared with the actual charge representation in the dockable set (Figure 2a).
To address this, we created a new decoy pipeline that better charge-matched ligands to decoys (freely available at http://tldr.docking.org), such that ligand and decoy protomers are only considered in their dockable, 3D representation. In summary, up to 50 decoys are generated for each ligand accounting for the charge, molecular weight, calculated LogP, number of rotatable bonds, and number of hydrogen bond acceptors and donors while ensuring that these decoys are structurally dissimilar to each other and to the ligands to which they are matched (Table 2 and Table S3). By default and always for proteins with more than 100 ligands, the ligands are first clustered by an ECFP4 Tc of 0.7 to reduce the dominance of narrow congeneric series. The ligand with the smallest molecular weight from each cluster is chosen for property matching. These changes improve the DUD-E design, without changing its underlying logic.
Table 2.
Ligand and Decoy Properties for 43 Protein Targets
| DUD-E | DUDE-Z | Extrema | Goldilocks | |
|---|---|---|---|---|
| # unique ligands | 8267 | 2312 | ||
| # unique decoys | 477,924 | 69,904 | 732,309 | 1,145,472 |
| # unique decoy scaffolds | 162,286 | 33,292 | 143,423 | 317,316 |
Improved Property-Matched Decoys Reduce False Enrichment.
With these changes in hand, we compared the “optimized” scoring function with a 0.5 weight on ligand desolvation to the standard, unweighted scoring function to determine whether the improved enrichments stood up to better charge matching between ligands and decoys. Competition with the better charge-matched decoys reduced the enrichment differences between the standard and the “optimized” 0.5 ligand desolvation scoring functions from 1.11 with the original DUD-E set to −0.34, supporting the hypothesis that more closely property-matched decoys would be less susceptible to imbalances in electrostatics and ligand desolvation energies (Figure 3 and see Sensitivity Analysis below for the significance of such differences). For instance, AmpC, whose enrichment was better with the optimized scoring function by 6.34 log adjusted AUC, with the new property-matched decoy background now much favors the standard scoring function, attaining an enrichment of 20.92, 12 adjusted log AUC over the “optimized” scoring function’s 8.93. Similarly, the DUD-E enrichment difference for PUR2 was 9.15 log adjusted AUC, but the difference becomes 0.35 in the new decoy set. Similar behavior where complete charge matching reduces preference for the optimized scoring function is seen in multiple systems including fatty acid binding protein 4 (FABP4), protein-tyrosine phosphatase 1 (PTN1), tryptase beta-1 (TRYB1), and trypsin I (TRY1). The opposite also occurs, where preference for the standard scoring function is diminished in the presence of better charge-matched decoys such as in rho-associated protein kinase 1 (ROCK1), C-X-C chemokine receptor type 4 (CXCR4), and epidermal growth factor receptor (EGFR). Overall, the average adjusted log AUC values for the 43 targets dropped from 19.05 and 20.2 for the standard and “optimized” scoring functions, respectively, with the original DUD-E benchmarking sets, to 14.82 and 15.17 with the new, better-matched decoy sets (Table 3). This enrichment drop reflects the better choice of decoy molecules in the new benchmarks, making the challenge harder, appropriately, for the docking program.
Figure 3.

Adjusted LogAUC differences between the standard, unweighted scoring function, and the optimized scoring function (1.0ES+1.0vdW+0.5LD), comparing the original DUD-E decoys (blue bars) and decoys prepared with the new DUDE-Z pipeline (orange bars), in which decoys are better charge-matched. Apparent advantages for the weighted scoring function dissipate on better charge matching. Average adjusted log AUC differences of −1.11 (DUD-E) and −0.34 (DUDE-Z).
Table 3.
Average Adusted logAUC Values for Different Decoy Sets
| Extrema |
Goldilocks |
|||||
|---|---|---|---|---|---|---|
| DUD-E | DUDE-Z | DUD-E ligands | DUDE-Z ligands | DUD-E ligands | DUDE-Z ligands | |
| optimized (1.0ES+1.0vdW+0.5LD) | 20.31 | 15.26 | 26.05 | 16.24 | 42.18 | 28.68 |
| standard (1.0ES+1.0vdW+1.0LD) | 19.2 | 14.92 | 26.16 | 16.02 | 41.71 | 28.13 |
| difference | −1.11 | −0.34 | 0.11 | −0.22 | −0.47 | −0.55 |
To ensure that these differences were not due to the reduced ligand set used in DUDE-Z vs the larger ligand sets in DUD-E, we generated charge-matched decoys for the 43 targets using the full ligand set from DUD-E (Table S6). Preparing the original DUD-E set using the protocols on the DUD-E site, the “optimized” scoring function performs better than the standard one by 3.4 units of adjusted logAUC. When this full DUD-E set is now optimized for charge matching, using the DUDE-Z procedures, the difference between the adjusted logAUC drops to 0.82 units between the two scoring functions. With both the reduced ligand set and the charge matching, the difference between the two scoring functions falls to 0.33 adjusted LogAUC. This supports the idea that the difference between the two scoring functions with DUD-E largely reflects a charge mismatch between ligands and decoys within that set. We note that for AmpC β-lactamase, the dopamine D4 and the s melatonin receptors, hand-optimized docking parameters, used in past prospective campaigns against these targets,10,69 were employed. We therefore compared performance with the DUD-E and DUDE-Z benchmarks with and without optimized parameters in these three systems. The DUDE-Z benchmark was typically more stringent, though the opposite was true for the melatonin receptor, on whose neutral-dominated ligands and decoys the optimization in DUDE-Z will have less impact (Table S7). It is also interesting that retrospective enrichment did not always improve with the hand-optimized parameters used in the actual prospective campaigns. For instance, those optimized parameters reduced enrichment for the DUDE-Z and Goldilocks sets for the dopamine D4 receptor versus unoptimized parameters. However, enrichment vs the extrema set was improved, reflecting better charge matching with the optimized parameters, as did geometric fidelity to competent ligand poses. These observations emphasize that multiple criteria may be considered in optimizing docking parameters, not simply enrichment against a decoy set (we do not discount the possibility of improving benchmarks to make this more s automatic; we would note that the prospective campaigns against the D4 dopamine and MT1 receptors and against AmpC revealed novel, potent ligands with high hit rates10,6).
Beyond Property-Matched Decoys: Charge Extrema.
Given the sensitivity to even small differences in charge matching between ligands and decoys, we thought it worthwhile to investigate how sensitive the docking was not only to property matching but to extremes intentionally outside the property range of the ligands. We reasoned that docking parameters might be unintentionally optimized to weight particular energetic terms at the expense of others. Such blind spots might only be illuminated when comparing the performance of physically extreme molecules.
Based on our experience with the impact of electrostatic and desolvation weighting above, we focused on ligands representing charge extremes, probing for overweighted electrostatic interactions or underweighted desolvation penalties in our scoring function. These charge-extrema sets were populated with decoys that have similar physical properties (molecular weight, cLogP) to the ligands queried but include all charges from −2 to +2, taken from “in-stock” and “make-on-demand” libraries in ZINC15.52 If many molecules bearing a net charge of −2 score better than AmpC’s monoanions, for instance, then this would indicate a bias in the scoring that would have been concealed by the charge-matched decoys. We generated sets of property-matched charge-extreme decoys for 43 targets (Table S4). These charge outlier decoys (≤ −2 and ≥ +2) comprised on average 37% (272K of 732K molecules) of benchmarks, ranging from 15% (tryptase beta-1, TRYB1) to 57% (neuraminidase, NRAM). For a well-balanced scoring function, which properly captures molecular interactions, including charge extrema should improve ligand enrichment since decoys bearing unreasonable charges should be readily recognized, which is indeed what we see, though performance improves only slightly (Figure 4, Table 3, and see Sensitivity Analysis below for the significance of such differences), with systems with charged ligands being affected significantly. For example, GAR transformylase (PUR2, Figure 4b) recognizes tri- and dianionic ligands. When screened against a large extrema set with down-weighted desolvation, cations begin to dominate, behavior that the standard scoring function is at least partially able to combat (Figure 4b). Similar behavior is seen with protein-tyrosine phosphatase 1b (PTN1), which predominantly binds mono- and dianions in the standard scoring function but begins to prioritize tri- and tetra-anions when the optimized scoring function is utilized. As with GAR transformylase, the increased desolvation cost in the standard scoring function actually diminishes performance relative to the “optimized” scoring function as it penalizes both extreme-charged ligands and decoys. On the other hand, epidermal growth factor receptor (EGFR) and macrophage colony stimulating factor (CSF1R, Figure 4c), which perform better with the standard scoring function over the optimized scoring function with extrema, both recognize neutral ligands. When these two targets are screened with charge extrema, the standard scoring function is more equipped to penalize inappropriate charges over the optimized scoring function, which in the presence of charge extrema is flooded with anions and cations. Each of these cases can be explained by the underweighting of the ligand desolvation penalty in a scoring function optimized against the DUD-E set that both had a discrepancy between ligand and decoy charges and were not challenged with charged extrema, as we show here.
Figure 4.

(a) Adjusted LogAUC differences between the standard scoring function and the weighted scoring function using the new DUDE-Z decoy pipeline and the charge extrema decoys. (b,c) through (e). Comparing DOCK energy and molecule charge of the standard and optimized scoring functions using DUDE-Z ligands and using charge extrema decoys for (b) protein-tyrosine phosphatase 1 (PTN1) and (c) macrophage colony stimulating factor receptor (CSF1R). Central dotted lines of DOCK energies represent the medians, upper dotted lines represent the third quartiles, and lower dotted lines represent the first quartiles. The lowest points represent the minimum DOCK energies, and the highest values represent the maximum DOCK energies for both scoring functions. As ligand desolvation is down-weighted in the optimized scoring function, more extreme charges score better, which is advantageous for targets that have extreme charged ligands like PUR2 and PTN1. However, this becomes problematic and decreases enrichment for systems whose ligands are less extreme like EGFR and CSF1R.
If charge extrema can reveal cryptic pathologies in docking scoring, then so too can testing against molecules that are intentionally unmatched from the physical properties of the ligands but instead reflect the molecules of the overall library itself. Since each receptor will have its own ligand preferences, certainly with the biases from the medicinal chemistry literature, for any given receptor, the average library molecule may well-represent a physical property outside those of the receptor’s ligands, exposing the docking screen to new, previously unsampled physical properties. Thus, we investigated control calculations with a set of 1.1 million ZINC molecules. These comprised over 300,000 Bemis–Murcko scaffolds72 representing the middle of the range of physical parameters of the library: not too big, not too small, not too polar, and not too greasy (Goldilocks, Table S5). Whereas this benchmark was meant to represent the middle range of properties of a much larger library to be docked prospectively, we also compared it to the physical properties of a large high-throughput screening deck, the 400,000 molecule Molecular Libraries Small Molecule Repository (MLSMR), and to hits from screening and other techniques that have been advanced to candidacy.73 Gratifyingly, the molecules in Goldilocks overlapped both of these sets in key physical properties including MWT, cLogP, the number of rotatable bonds, and the number of hydrogen bond acceptors and donors (Figure S6). Docking the Goldilocks benchmark against the 43 targets resulted in log adjusted AUC values of 28.13 and 28.68 for the s standard and “optimized” scoring functions, respectively (Table 3). These are higher than the enrichments with the property-matched sets, as expected owing to its non-property-matched nature; the differences between the two scoring functions against the Goldilocks set are small (see Sensitivity Analysis below).
As an aside, the Goldilocks set also allowed us to return to one of the earliest motivations for property-matched benchmarks,38 the idea that they would prevent docking scoring functions from cheating by optimizing against a particular physical property, such as molecular weight. Thus, property-matched sets are meant to be and widely thought to be harder for docking than random sets of molecules. The 1.1 million molecule size of the Goldilocks set allows us to return to this point quantifiably, comparing performance against this benchmark vs DUDE-Z across 43 receptor systems. A random set of Goldilocks decoys was chosen for a fixed set of ligands (common to both benchmarks), with a ratio of 50:1 decoys to ligands; this was repeated 100 times with different random Goldilocks decoys, and a distribution of LogAUC values was calculated (Figure S5). In 39 of 43 systems, the LogAUC of the Goldilocks scores was compelling and certainly statistically larger than the DUDE-Z LogAUC values, with z-scores for the average difference in LogAUC typically exceeding 100. This supports the longstanding idea that property-matched decoys provide harder tests for docking than random collections of ligands.
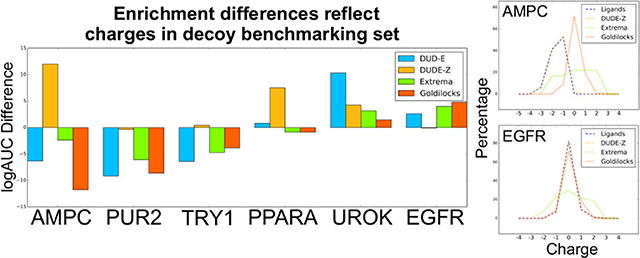
Even against a background of high enrichment, there are targets for which performance varies between the two scoring functions. Here, we focus on illustrative targets where the differences are substantial and significant (see Sensitivity Analysis below). In AmpC β-lactamase, tests against the DUDE-Z set suggest that the standard, unweighted scoring function led to better enrichments than the putatively optimized one where ligand desolvation was down-weighted by 0.5 (Figure 3), in contrast to the DUD-E benchmark test that had led to this new weighting. Against the Goldilocks benchmark, however, the situation reverts, with the optimized scoring function performing better than the standard scoring function, with an enrichment difference over 11 in adjusted log AUC (Figure 5). This difference is only partly captured by the extrema set, where the difference is only slightly larger than 2 adjusted log AUC. Similarly, GAR transformylase (PUR2) sees the relative enrichment of the optimized scoring function rise by almost 10 units of adjusted log AUC versus the standard scoring function with the Goldilocks set vs DUDE-Z, while with trypsin I (TRY1), ligands favor the optimized scoring function using the Goldilocks benchmark by almost 4 adjusted log AUC units versus the less than 1 unit difference using the DUDE-Z set. A few targets, such as FK506-binding protein 1A (FKB1A) and polo-like kinase 1 (PLK1), see the opposite effect—the optimized scoring function performs noticeably worse with the Goldilocks benchmark versus DUDE-Z. These differences are explained by differences in the properties of the decoys in the different benchmarks. In DUDE-Z, the decoy physical properties are tightly calibrated to those of the ligands. Conversely, Goldilocks represents the physical properties of the library to be docked. For targets recognizing ligands with physical properties much different from “lead-like”74 molecules, which dominate the Goldilocks benchmark and the library it represents, such as AmpC, GAR transformylase (PUR2), and trypsin I (TRY1), the DUDE-Z set will be a more stringent test (Figure 5b). However, scoring term weights that optimize performance against it will not always translate to a lead-like benchmark like Goldilocks. For these systems, the key differences are in the distribution of charge states of the ligands and the decoys: in DUDE-Z, these are well-matched, while in Goldilocks and the ultra-large library that it represents, mono-, di-, and trianions, as well as dications, are far less common than among the known inhibitors of these targets (Figure 5c–e), providing opportunities for these ligands to exploit the optimized scoring function with its down-weighted ligand desolvation term and score well. For systems that bind molecules within lead-like space, such as peroxisome proliferator-activated receptor alpha (PPARA), urokinase-type plasminogen activator (UROK), and epidermal growth factor receptor (EGFR), the enrichment differences between the standard and optimized scoring functions diminish and even begin to favor the standard scoring function (Figure 5b,f–h), as outlier charges are unable to exploit liabilities within the optimized scoring function.
Figure 5.

(a) Enrichment differences between the standard scoring function and optimized scoring function comparing the new DUDE-Z benchmarks, charge extrema decoys, and the Goldilocks benchmarks, with a focus on the enrichment changes in specific targets (b). Comparison of net charge of ligands and benchmark decoys for AmpC β-lactamase (AmpC, c), GAR transformylase (PUR2, d), trypsin I (TRY1, e), peroxisome proliferator-activated receptor alpha (PPARA, f), urokinase-type plasminogen activator (UROK, g), and epidermal growth factor receptor (EGFR, h). For systems whose ligands have more extreme charges, there is a typically small overlap in ligand charges and decoy charges, providing an advantage to the extreme charged ligands with the optimized scoring function. However, in systems where the ligand charges overlap more significantly with the decoy charges, the standard scoring function begins to perform better as there are no extreme charged ligands to exploit the lower desolvation cost and rank more favorably.
Up until now, we have seen results shift as we change the benchmark from DUD-E to the optimized DUDE-Z to Extrema to Goldilocks. A natural reaction might be to despair benchmarking entirely. Our own view is that each of these benchmarks is useful (we suggest the optimizations in DUDE-Z over DUD-E), and together can inure developers and users from false conclusions around the scoring function and docking parameter optimization. The different lessons that each benchmark teaches reflect weaknesses of enrichment as a metric; it nevertheless remains a crucial criterion for docking performance. These are points to which we will return.
Sensitivity Analysis & Statistical Significance.
The area under the curve (AUC) and its variants are widely used as a single value measure of docking performance.57,63,75–80 In comparing an innovation with the current best practice, it is common to see improvements in enrichment across a benchmarking set. It is important to understand when such improvements are significant beyond the variation one might see with small changes to docking parameters. To assess confidence intervals on enrichment plots, we turned to an empirical bootstrapping approach. In this method, we calculate enrichments multiple times for any given benchmark, each time picking a random subset of the ligands and decoys in the set, retaining the same sample size as the original set. For many of the DUDE-Z targets, this is readily done, as only a subset of the possible ligands is typically represented, and many more property-matched decoys are typically available from ZINC. With the new benchmark, whose ligands closely resemble the canonical ones and whose decoys reflect the same property matching, a new enrichment is calculated.
Repeated for 50 random subsets of ligands and decoys for each target, this approach allows one to calculate confidence intervals of enrichment (adjusted log AUC). We did so for the same 43 targets, recording the variance of the enrichments. Based on these bootstrapping calculations, we find that the average 95 and 75% confidence interval over the 43 systems is about 9.4 and 5.8 adjusted log AUC units, respectively. Naturally, individual systems varied in their confidence levels: from a relatively tight distribution for androgen receptor (ANDR, 95% CI of 3.0) to a much wider distribution for fatty acid binding protein-4 (FABP4, 95% CI of 15.6) (Figure S1). Bootstrapping can also be used to compare the performance of two docking methods or two scoring functions. The Z-test and corresponding p-values are used here since the number of bootstrap replicates is over 30, and the bootstrapped distribution follows the normal distribution.
Figure 6 shows the bootstrapped distribution comparison between the standard (STD) and “optimized” (0.5LD) scoring functions with DUD-E, DUDE-Z, Extrema, and Goldilocks as decoy sets on 41 DUD-E targets, as well as the melatonin MT1 receptor and the dopamine D4 receptor where we have not only experimentally measured docking true but also docking false positives (Figure S2). Here, for the combined sets, the change in the mean adjusted log AUC between the standard and optimized scoring functions is 0.49, 0.36, −0.05, and 0.23 for the DUD-E, DUDE-Z, Extrema, and Goldilocks backgrounds, respectively (Figure 6a). For the aggregate, only the DUD-E background difference is significant with a p-value less than 0.05, likely reflecting its flawed charge matching between ligands and decoys, while all other decoy backgrounds are not. Innovations that we might have otherwise considered successful are often found to be statistically indistinguishable or to be significant against one background but not on another. Screening poly-ADP-ribose polymerase 1 (PARP1) with DUD-E, DUDE-Z, and Goldilocks decoy sets shows significant improvement with the optimized scoring function over the standard scoring function, whereas performance is significantly worse with Extrema (Figure 6b). In the adenosine 2A receptor (AA2AR, Figure 6c), ligands in the presence of DUD-E and DUDE-Z decoy sets significantly favor the optimized scoring function but flip to favoring the standard scoring function in the presence of Extrema and Goldilocks sets, versus in coagulation factor VII (FA7, Figure 6d); ligands always significantly favor the optimized scoring function regardless of the decoy background (see Figure S3 for difference distributions and Figure S4 for bootstrapping plots of all 43 systems). However, we note that only when screened with the DUD-E decoys are the enrichment differences in these scoring functions significantly different (Figure 6a), showing for all other decoy sets insignificant differences. When all decoy sets are combined, the bootstrapping enrichment differences remain insignificant.
Figure 6.

Applying bootstrapping to the different decoy backgrounds demonstrates that while there may be statistically significant differences in terms of performance between the scoring functions for particular systems, if all bootstrapping enrichments are combined for all decoy sets over all 43 systems, then there is no statistically significant difference between the standard and optimized scoring functions, demonstrating that one can be deceived by significant differences between the two scoring functions when only considering one decoy background. Average bootstrapping statistics on the enrichments for DUD-E, DUDE-Z, Extrema, Goldilocks, and all decoy sets (Combined) for all 43 systems (a). Individual bootstrapping statistics (50 for each) on the enrichments (adjusted log AUC values) for DUD-E, DUDE-Z, Extrema, and Goldilocks decoy backgrounds for poly-ADP-ribose polymerase I (PARP1, b), adenosine 2A receptor (AA2AR, c), and coagulation factor VII (FA7, d). From the 50 bootstrapped adjusted log AUC values generated, central dotted lines represent the medians, upper dotted lines represent the third quartiles, and lower dotted lines represent the first quartiles. The lowest points represent the minimum adjusted log AUC values, and the highest points represent the maximum adjusted log AUC values generated from bootstrapping. See Figure S3 for difference distributions and Figure S4 for bootstrapping plots for all 43 systems.
DISCUSSION
Four themes emerge from this work. First, for all their strengths, property-matched decoys alone can mislead in evaluating docking performance. Scoring functions can exploit physical property differences between ligands and decoys even in relatively well-balanced sets, as we see by comparing the original DUD-E and the refined DUDE-Z sets. Decoys that are intentionally non-property-matched, such as the Extrema set that explores ligands with high molecular charges and the Goldilocks set, whose decoys can be far different from the known ligands but which represent the properties of the ultra-large database to be docked, reveal liabilities that are hidden by the property-matched sets. Second, enrichment, which is perhaps the key criterion for library docking assessment, remains a weak metric, ungrounded in physical theory or observables. Third, our understanding of this metric can be strengthened with confidence intervals, which can be readily estimated. These confidence margins are often surprisingly large, and apparently different enrichments are often statistically indistinguishable. Finally, we make the new tools developed here, including generation of better property-matched decoys (DUDE-Z), charge Extrema, Goldilocks, and bootstrapping adjusted log AUC ranges, available and free to use for the community.
Property-matched decoys remain crucial for docking evaluation,29,30,38 reducing the ability of scoring functions to exploit gross physical property differences between ligands and the random molecules that had initially been used in the field.35 However, property matching has its own liabilities, revealed by other backgrounds. For instance, property matching decoys to the GAR transformylase, AmpC β-lactamase, or trypsin I receptor ligands will result in decoys that have charge ranges tightly distributed around −2, −1, and +1 to +2 formal charges, respectively. A scoring function that overweights electrostatic interaction energies or underweights desolvation energies may not be revealed by such property-matched decoys. This is what we observed with what appeared to be an “optimized” function that down-weighted ligand desolvation, improving average enrichment over 43 systems. This apparent improvement was eliminated not only by better charge matching in the optimized DUDE-Z set, but its basis in overweighted electrostatic interactions was illuminated by a charge Extrema set (Figure 4). Similarly, benchmarks that are well-matched around ligands with unusual physical properties—in this study, highly charged ligands—will not reveal liabilities that a background representing the properties of the overall library can illuminate. This is what we observe for the Goldilocks benchmark (Figure 5).
Enrichment of ligands over property-matched decoys30,75,76,81–85 is widely used for parameter optimization and scoring function development.47,50,51,63,86–93 Because enrichment is ungrounded in physical theory, it is sensitive both to changes in the decoy background,49 which are usually only reasonable guesses, and to the ligands, which represent experimental observables, flawed though these too can be. In principle, development of decoy sets and new sampling and scoring functions would be matched with carefully controlled wet experiments to test them. While there are several model87,94–97and biological systems10,26 that support doing so and that allow for comparisons among docking programs, purely computational controls will continue to play a key role in benchmarking docking performance (as an aside, the advent of ultra-large docking libraries and the experimental testing of large numbers of docking hits that flow from them10,69 will reveal experimental decoys that will complement what have been, until now, only presumed decoy sets). As such, we do not wish to undercut enrichment as a metric of docking—weak as it is, it remains crucial to progress in the field. What this study teaches is that our confidence in enrichment can be much strengthened by using multiple decoy backgrounds. Correspondingly, the significance of enrichment differences with different docking parameterization and with different scoring functions should be controlled for. One way to do so is via the bootstrapping method that we outline here (Figure 6), which can insulate one from false conclusions about differences that fall within the variation expected from small changes in the ligands and decoys used (scripts to implement this are available at http://tldr.docking.org).
Confronted with ever more decoy benchmarks and the time it takes to run a full set of controls, it is natural to wonder if there is no end to the cottage industry of new benchmarks. One can imagine spending too much time on these sanity checks and too little on the actual prediction of new chemical matter with prospective docking. Nevertheless, the time and expense of sourcing and physically testing new chemical matter and of eliminating experimental artifacts52,98,99 still far exceed the cost of running these computational controls. Property-matched benchmarks are rarely composed of more than a few thousand molecules for a given target, and even the Goldilocks set comprises less than 2 million molecules, less than 1% the size of the ultra-large libraries now being prosecuted.10,11,69 To make these controls accessible to the community, we provide the optimized DUDE-Z benchmarks at http://dudez.docking.org. We also provide a web service that allows investigators to create bespoke Extrema and Goldilocks sets and enables bootstrapping tests for statistical significance—freely available at http://tldr.docking.org.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by US National Institutes of Health grants GM71896 (to J.J.I.) and R35GM122481 (to B.K.S.). We are grateful to OpenEye Scientific Software for an academic license for Omega, OEChem, and other tools, to ChemAxon for an academic license for JChem, Marvin, and other software, and to Schrodinger for access to their software platform. We thank the providers of public databases and free software from which ZINC has benefitted: RDKit, DrugBank, HMDB, ChEBI, and ChEMBL. We thank members of the Shoichet Lab for testing software and for timely feedback and thank Roger Sayle and John Mayfield at Nextmove Software for access to Arthor and SmallWorld and for discussions.
ABBREVIATIONS
- AA2AR
adenosine A2a receptor
- ABL1
tyrosine-protein kinase ABL
- ACES
acetylcholinesterase
- ADA
adenosine deaminase
- ADRB2
beta-2 adrenergic receptor
- AMPC
beta-lactamase
- ANDR
androgen receptor
- CSF1R
macrophage colony stimulating factor receptor
- CXCR4
C-X-C chemokine receptor type 4
- DEF
peptide deformylase
- DRD4
D4 dopamine receptor
- EGFR
epidermal growth factor receptor erbB1
- FA10
coagulation factor X
- FA7
coagulation factor VII
- FABP4
fatty acid binding protein adipocyte
- FGFR1
fibroblast growth factor receptor 1
- FKB1A
FK506-binding protein 1A
- GLCM
beta-glucocerebrosidase
- HDAC8
histone deacetylase 8
- HIVPR
human immunodeficiency virus type 1 protease
- HMDH
HMG-CoA reductase
- HS90A
heat shock protein HSP 90-alpha
- ITAL
leukocyte adhesion glycoprotein LFA-1 alpha
- KIT
stem cell growth factor receptor
- KITH
thymidine kinase
- LCK
tyrosine-protein kinase LCK
- MAPK2
MAP kinase-activated protein kinase 2
- MK01
MAP kinase ERK2
- MT1
melatonin MT1 receptor
- NRAM
neuraminidase
- PARP1
poly-ADP-ribose polymerase 1
- PLK1
serine/threonine-protein kinase PLK1
- PPARA
peroxisome proliferator-activated receptor alpha
- PTN1
protein-tyrosine phosphatase 1B
- PUR2
GAR transformylase
- RENI
renin
- ROCK1
rho-associated protein kinase 1
- SRC
tyrosine-protein kinase SRC
- THRB
thrombin
- TRY1
trypsin I
- TRYB1
tryptase beta-1
- UROK
urokinase-type plasminogen activator
- XIAP
inhibitor of apoptosis protein 3
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00598.
(Figure S1) Example bootstrapping results, (Figure S2) bootstrapping on binders and nonbinders for DRD4 and MT1, (Figure S3) bootstrapping logAUC differences using different decoy backgrounds, (Figure S4) bootstrapping statistics for all 43 targets, (Figure S5) comparison of logAUC between random Goldilocks decoys and DUDE-Z decoys, (Figure S6) comparison of molecular properties between Goldilocks and screening libraries (PDF)
(Methods S1) Code implemented for calculating the adjusted logAUC (PDF)
(Table S1) logAUC values for all scoring function coefficients (XLSX)
(Table S2) logAUC values for standard and optimized scoring functions for DUD-E, DUDE-Z, Extrema, and Goldilocks (XLSX)
(Table S3) Properties of the DUDE-Z set (XLSX)
(Table S4) Properties of Extrema decoys (XLSX)
(Table S5) Properties of Goldilocks decoys (XLSX)
(Table S6) Comparison of logAUC values for all 43 targets using original DUD-E, DUDE-Z, and new charge-matched DUD-E with the full ligand set (XLSX)
(Table S7) Comparison of unoptimized versus optimized docking setups for AmpC, DRD4, and MT1 for DUD-E, DUD-E, Extrema, and Goldilocks (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.0c00598
Contributor Information
Reed M. Stein, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States
Ying Yang, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
Trent E. Balius, Cancer Research Technology Program, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, Maryland 21702, United States
Matt J. O’Meara, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, United States
Jiankun Lyu, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
Jennifer Young, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
Khanh Tang, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
Brian K. Shoichet, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States.
John J. Irwin, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94158, United States
REFERENCES
- (1).Meng EC; Shoichet BK; Kuntz ID Automated docking with grid-based energy evaluation. J. Comput. Chem 1992, 13, 505–524. [Google Scholar]
- (2).Shoichet BK; Kuntz ID Matching chemistry and shape in molecular docking. Protein Eng. 1993, 6, 723–732. [DOI] [PubMed] [Google Scholar]
- (3).Goodsell DS; Morris GM; Olson AJ Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit 1996, 9, 1–5. [DOI] [PubMed] [Google Scholar]
- (4).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (5).Lemmen C; Lengauer T Time-efficient flexible superposition of medium-sized molecules. J. Comput.-Aided Mol. Des 1997, 11, 357–368. [DOI] [PubMed] [Google Scholar]
- (6).Eldridge MD; Murray CW; Auton TR; Paolini GV; Mee RP Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des 1997, 11, 425–445. [DOI] [PubMed] [Google Scholar]
- (7).Rarey M; Kramer B; Lengauer T; Klebe G A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol 1996, 261, 470–489. [DOI] [PubMed] [Google Scholar]
- (8).Welch W; Ruppert J; Jain AN Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol 1996, 3, 449–462. [DOI] [PubMed] [Google Scholar]
- (9).Abagyan R; Totrov M; Kuznetsov D ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem 1994, 15, 488–506. [Google Scholar]
- (10).Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; L O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gorgulla C; Boeszoermenyi A; Wang Z-F; Fischer PD; Coote PW; Das KMP; Malets YS; Radchenko DS; Moroz YS; Scott DA; Fackeldey K; Hoffmann M; Iavniuk I; Wagner G; Arthanari H An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Manglik A; Lin H; Aryal DK; McCorvy JD; Dengler D; Corder G; Levit A; Kling RC; Bernat V; Hübner H; Huang X-P; Sassano MF; Giguére PM; Löber S; Da D; Scherrer G; Kobilka BK; Gmeiner P; Roth BL; Shoichet BK Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lansu K; Karpiak J; Liu J; Huang XP; McCorvy JD; Kroeze WK; Che T; Nagase H; Carroll FI; Jin J; Shoichet BK; Roth BL In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol 2017, 13, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang S; Wacker D; Levit A; Che T; Betz RM; McCorvy JD; Venkatakrishnan AJ; Huang XP; Dror RO; Shoichet BK; Roth BL D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Korczynska M; Clark MJ; Valant C; Xu J; Moo EV; Albold S; Weiss DR; Torosyan H; Huang W; Kruse AC; Lyda BR; May LT; Baltos JA; Sexton PM; Kobilka BK; Christopoulos A; Shoichet BK; Sunahara RK Structure-based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E2419–E2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Huang XP; Karpiak J; Kroeze WK; Zhu H; Chen X; Moy SS; Saddoris KA; Nikolova VD; Farrell MS; Wang S; Mangano TJ; Deshpande DA; Jiang A; Penn RB; Jin J; Koller BH; Kenakin T; Shoichet BK; Roth BL Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Irwin JJ; Shoichet BK Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem 2016, 59, 4103–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ballante F; Rudling A; Zeifman A; Luttens A; Vo DD; Irwin JJ; Kihlberg J; Brea J; Loza MI; Carlsson J Docking Finds GPCR Ligands in Dark Chemical Matter. J. Med. Chem 2020, 63, 613–620. [DOI] [PubMed] [Google Scholar]
- (19).Patel N; Huang XP; Grandner JM; Johansson LC; Stauch B; McCorvy JD; Liu Y; Roth B; Katritch V Structure-based discovery of potent and selective melatonin receptor agonists. eLife 2020, 9, No. e53779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kiss R; Kiss B; Könczöl A; Szalai F; Jelinek I; László V; Noszál B; Falus A; Keserű GM Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J. Med. Chem 2008, 51, 3145–3153. [DOI] [PubMed] [Google Scholar]
- (21).Männel B; Jaiteh M; Zeifman A; Randakova A; Möller D; Hübner H; Gmeiner P; Carlsson J Structure-Guided Screening for Functionally Selective D2 Dopamine Receptor Ligands from a Virtual Chemical Library. ACS Chem. Biol 2017, 12, 2652–2661. [DOI] [PubMed] [Google Scholar]
- (22).Scharf MM; Bünemann M; Baker JG; Kolb P Comparative Docking to Distinct G Protein-Coupled Receptor Conformations Exclusively Yields Ligands with Agonist Efficacy. Mol. Pharmacol 2019, 96, 851–861. [DOI] [PubMed] [Google Scholar]
- (23).Dahlgren MK; Garcia AB; Hare AA; Tirado-Rives J; Leng L; Bucala R; Jorgensen WL Virtual screening and optimization yield low-nanomolar inhibitors of the tautomerase activity of Plasmodium falciparum macrophage migration inhibitory factor. J. Med. Chem 2012, 55, 10148–10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhou Y; Ma J; Lin X; Huang X-P; Wu K; Huang N Structure-Based Discovery of Novel and Selective 5-Hydroxytryptamine 2B Receptor Antagonists for the Treatment of Irritable Bowel Syndrome. J. Med. Chem 2016, 59, 707–720. [DOI] [PubMed] [Google Scholar]
- (25).Jorgensen WL Efficient drug lead discovery and optimization. Acc. Chem. Res 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Adeshina YO; Deeds EJ; Karanicolas J Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 18477–18488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sun H; Pan P; Tian S; Xu L; Kong X; Li Y; Li D; Hou T Constructing and Validating High-Performance MIEC-SVM Models in Virtual Screening for Kinases: A Better Way for Actives Discovery. Sci. Rep 2016, 6, 24817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liu S; Alnammi M; Ericksen SS; Voter AF; Ananiev GE; Keck JL; Hoffmann FM; Wildman SA; Gitter A Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model 2019, 59, 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Huang N; Shoichet BK; Irwin JJ Benchmarking sets for molecular docking. J. Med. Chem 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mysinger MM; Carchia M; Irwin JJ; Shoichet BK Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Réau M; Langenfeld F; Zagury JF; Lagarde N; Montes M Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Front. Pharmacol 2018, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Novotný J; Bruccoleri R; Karplus M An analysis of incorrectly folded protein models: Implications for structure predictions. J. Mol. Biol 1984, 177, 787–818. [DOI] [PubMed] [Google Scholar]
- (33).Park B; Levitt M Energy functions that discriminate X-ray and near native folds from well-constructed decoys. J. Mol. Biol 1996, 258, 367–392. [DOI] [PubMed] [Google Scholar]
- (34).Samudrala R; Levitt M Decoys ‘R’ Us: a database of incorrect conformations to improve protein structure prediction. Protein Sci. 2000, 9, 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pham TA; Jain AN Parameter estimation for scoring protein-ligand interactions using negative training data. J. Med. Chem 2006, 49, 5856–5868.17004701 [Google Scholar]
- (36).Bissantz C; Folkers G; Rognan D Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem 2000, 43, 4759–4767. [DOI] [PubMed] [Google Scholar]
- (37).Kellenberger E; Rodrigo J; Muller P; Rognan D Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [DOI] [PubMed] [Google Scholar]
- (38).Verdonk ML; Berdini V; Hartshorn MJ; Mooij WTM; Murray CW; Taylor RD; Watson P Virtual screening using protein-ligand docking: avoiding artificial enrichment. J. Chem. Inf. Comput. Sci 2004, 44, 793–806. [DOI] [PubMed] [Google Scholar]
- (39).Gatica EA; Cavasotto CN Ligand and decoy sets for docking to G protein-coupled receptors. J. Chem. Inf. Model 2012, 52, 1–6. [DOI] [PubMed] [Google Scholar]
- (40).Weiss DR; Bortolato A; Tehan B; Mason JS GPCR-Bench: A Benchmarking Set and Practitioners’ Guide for G Protein-Coupled Receptor Docking. J. Chem. Inf. Model 2016, 56, 642–651. [DOI] [PubMed] [Google Scholar]
- (41).Wallach I; Lilien R Virtual decoy sets for molecular docking benchmarks. J. Chem. Inf. Model 2011, 51, 196–202. [DOI] [PubMed] [Google Scholar]
- (42).Wang L; Pang X; Li Y; Zhang Z; Tan W RADER: a RApid DEcoy Retriever to facilitate decoy based assessment of virtual screening. Bioinformatics 2017, 33, 1235–1237. [DOI] [PubMed] [Google Scholar]
- (43).Cleves AE; Jain AN Structure- and Ligand-Based Virtual Screening on DUD-E(+): Performance Dependence on Approximations to the Binding Pocket. J. Chem. Inf. Model 2020, 4296. [DOI] [PubMed] [Google Scholar]
- (44).Bauer MR; Ibrahim TM; Vogel SM; Boeckler FM Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets. J. Chem. Inf. Model 2013, 53, 1447–1462. [DOI] [PubMed] [Google Scholar]
- (45).Xia J; Jin H; Liu Z; Zhang L; Wang XS An unbiased method to build benchmarking sets for ligand-based virtual screening and its application to GPCRs. J. Chem. Inf. Model 2014, 54, 1433–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Fine J; Muhoberac M; Fraux G; Chopra G, DUBS: A Framework for Developing Directory of Useful Benchmarking Sets for Virtual Screening. bioRxiv 2020, 2020.01.31.929679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chaput L; Martinez-Sanz J; Saettel N; Mouawad L Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. Aust. J. Chem 2016, 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chen L; Cruz A; Ramsey S; Dickson CJ; Duca JS; Hornak V; Koes DR; Kurtzman T Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS One 2019, 14, No. e0220113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wallach I; Heifets A Most Ligand-Based Classification Benchmarks Reward Memorization Rather than Generalization. J. Chem. Inf. Model 2018, 58, 916–932. [DOI] [PubMed] [Google Scholar]
- (50).Kurczab R; Smusz S; Bojarski AJ The influence of negative training set size on machine learning-based virtual screening. Aust. J. Chem 2014, 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Li H; Sze K-H; Lu G; Ballester PJ Machine-learning scoring functions for structure-based virtual screening. WIREs Comput. Mol. Sci 2020, 11, e1478. [Google Scholar]
- (52).Sterling T; Irwin JJ ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Csizmadia F JChem: Java applets and modules supporting chemical database handling from web browsers. J. Chem. Inf. Comput. Sci 2000, 40, 323–324. [DOI] [PubMed] [Google Scholar]
- (54).Gasteiger J; Rudolph C; Sadowski J Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comput. Methodol 1990, 3, 537–547. [Google Scholar]
- (55).Hawkins PC; Skillman AG; Warren GL; Ellingson BA; Stahl MT Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hawkins G; Giesen D; Lynch G; Chambers C; Rossi I; Storer J; Li J; Thompson J; Winget P; Lynch BJ AMSOL-version 7.1; University of Minnesota: Minneapolis, 2004. [Google Scholar]
- (57).Coleman RG; Carchia M; Sterling T; Irwin JJ; Shoichet BK Ligand pose and orientational sampling in molecular docking. PLoS One 2013, 8, No. e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kuntz ID; Blaney JM; Oatley SJ; Langridge R; Ferrin TE A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [DOI] [PubMed] [Google Scholar]
- (59).DesJarlais RL; Shoichet B; Seibel G; Kuntz ID Inhibitor Design from Known Structures. In Crystallographic and Modeling Methods in Molecular Design; Bugg CE; Ealick SE, Eds.; Springer New York: New York, NY, 1990, pp 200–210. [Google Scholar]
- (60).Word JM; Lovell SC; Richardson JS; Richardson DC Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol 1999, 285, 1735–1747. [DOI] [PubMed] [Google Scholar]
- (61).Weiner PK; Kollman PA AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem 1981, 2, 287–303. [Google Scholar]
- (62).Gallagher K; Sharp K Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J 1998, 75, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mysinger MM; Shoichet BK Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model 2010, 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- (64).Lorber DM; Shoichet BK Flexible ligand docking using conformational ensembles. Protein Sci. 1998, 7, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bento AP; Gaulton A; Hersey A; Bellis LJ; Chambers J; Davies M; Krüger FA; Light Y; Mak L; McGlinchey S; Nowotka M; Papadatos G; Santos R; Overington JP The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014, 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Irwin JJ; Shoichet BK; Mysinger MM; Huang N; Colizzi F; Wassam P; Cao Y Automated docking screens: a feasibility study. J. Med. Chem 2009, 52, 5712–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Powers RA; Morandi F; Shoichet BK Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure 2002, 10, 1013–1023. [DOI] [PubMed] [Google Scholar]
- (68).Eidam O; Romagnoli C; Dalmasso G; Barelier S; Caselli E; Bonnet R; Shoichet BK; Prati F Fragment-guided design of subnanomolar beta-lactamase inhibitors active in vivo. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 17448–17453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Stein RM; Kang HJ; McCorvy JD; Glatfelter GC; Jones AJ; Che T; Slocum S; Huang XP; Savych O; Moroz YS; Stauch B; Johansson LC; Cherezov V; Kenakin T; Irwin JJ; Shoichet BK; Roth BL; Dubocovich ML Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Gschwend DA; Kuntz ID Orientational sampling and rigid-body minimization in molecular docking revisited: on-the-fly optimization and degeneracy removal. J. Comput.-Aided Mol. Des 1996, 10, 123–132. [DOI] [PubMed] [Google Scholar]
- (71).Efron B Bootstrap Methods: Another Look at the Jackknife. Ann. Statist 1979, 7, 1–26. [Google Scholar]
- (72).Bemis GW; Murcko MA The properties of known drugs. 1. Molecular frameworks. J. Med. Chem 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- (73).Keserü GM; Makara GM The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug. Discov 2009, 8, 203–212. [DOI] [PubMed] [Google Scholar]
- (74).Oprea TI; Davis AM; Teague SJ; Leeson PD Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci 2001, 41, 1308–1315. [DOI] [PubMed] [Google Scholar]
- (75).Neves MAC; Totrov M; Abagyan R Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput.-Aided Mol. Des 2012, 26, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Repasky MP; Murphy RB; Banks JL; Greenwood JR; Tubert-Brohman I; Bhat S; Friesner RA Docking performance of the glide program as evaluated on the Astex and DUD datasets: a complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide. J. Comput.-Aided Mol. Des 2012, 26, 787–799. [DOI] [PubMed] [Google Scholar]
- (77).Perryman AL; Santiago DN; Forli S; Santos-Martins D; Olson AJ Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: participation in the SAMPL4 protein-ligand binding challenge. J. Comput.-Aided Mol. Des 2014, 28, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Lätti S; Niinivehmas S; Pentikäinen OT Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. Aust. J. Chem 2016, 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Swift RV; Jusoh SA; Offutt TL; Li ES; Amaro RE Knowledge-Based Methods To Train and Optimize Virtual Screening Ensembles. J. Chem. Inf. Model 2016, 56, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Nicholls A What do we know and when do we know it? J. Comput.-Aided Mol. Des 2008, 22, 239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Zhou Z; Felts AK; Friesner RA; Levy RM Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J. Chem. Inf. Model 2007, 47, 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Brozell SR; Mukherjee S; Balius TE; Roe DR; Case DA; Rizzo RC Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput.-Aided Mol. Des 2012, 26, 749–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).McGann M FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des 2012, 26, 897–906. [DOI] [PubMed] [Google Scholar]
- (84).Spitzer R; Jain AN Surflex-Dock: Docking benchmarks and real-world application. J. Comput.-Aided Mol. Des 2012, 26, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ashtawy HM; Mahapatra NR Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model 2018, 58, 119–133. [DOI] [PubMed] [Google Scholar]
- (86).Wei BQ; Baase WA; Weaver LH; Matthews BW; Shoichet BK A model binding site for testing scoring functions in molecular docking. J. Mol. Biol 2002, 322, 339–355. [DOI] [PubMed] [Google Scholar]
- (87).Balius TE; Fischer M; Stein RM; Adler TB; Nguyen CN; Cruz A; Gilson MK; Kurtzman T; Shoichet BK Testing inhomogeneous solvation theory in structure-based ligand discovery. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E6839–E6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Murphy RB; Repasky MP; Greenwood JR; Tubert-Brohman I; Jerome S; Annabhimoju R; Boyles NA; Schmitz CD; Abel R; Farid R; Friesner RA WScore: A Flexible and Accurate Treatment of Explicit Water Molecules in Ligand-Receptor Docking. J. Med. Chem 2016, 59, 4364–4384. [DOI] [PubMed] [Google Scholar]
- (89).Good AC; Oprea TI Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection? J. Comput.-Aided Mol. Des 2008, 22, 169–178. [DOI] [PubMed] [Google Scholar]
- (90).Moffat K; Gillet VJ; Whittle M; Bravi G; Leach AR A comparison of field-based similarity searching methods: CatShape, FBSS, and ROCS. J. Chem. Inf. Model 2008, 48, 719–729. [DOI] [PubMed] [Google Scholar]
- (91).Yang J; Shen C; Huang N Predicting or Pretending: Artificial Intelligence for Protein-Ligand Interactions Lack of Sufficiently Large and Unbiased Datasets. Front. Pharmacol 2020, 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Goldfeld DA; Murphy R; Kim B; Wang L; Beuming T; Abel R; Friesner RA Docking and free energy perturbation studies of ligand binding in the kappa opioid receptor. J. Phys. Chem. B 2015, 119, 824–835. [DOI] [PubMed] [Google Scholar]
- (93).Damm-Ganamet KL; Smith RD; Dunbar JB Jr.; Stuckey JA; Carlson HA CSAR benchmark exercise 2011–2012: evaluation of results from docking and relative ranking of blinded congeneric series. J. Chem. Inf. Model. 2013, 53, 1853–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Teotico DG; Babaoglu K; Rocklin GJ; Ferreira RS; Giannetti AM; Shoichet BK Docking for fragment inhibitors of AmpC beta-lactamase. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).London N; Miller RM; Krishnan S; Uchida K; Irwin JJ; Eidam O; Gibold L; Cimermančič P; Bonnet R; Shoichet BK; Taunton J Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Fischer M; Coleman RG; Fraser JS; Shoichet BK Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem 2014, 6, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Merski M; Fischer M; Balius TE; Eidam O; Shoichet BK Homologous ligands accommodated by discrete conformations of a buried cavity. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 5039–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Shoichet BK Screening in a spirit haunted world. Drug Discovery Today 2006, 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.