


Abstract
A set of >300 nonredundant high-resolution RNA–protein complexes were rigorously searched for π-contacts between an amino acid side chain (W, H, F, Y, R, E and D) and an RNA nucleobase (denoted π–π interaction) or ribose moiety (denoted sugar–π). The resulting dataset of >1500 RNA–protein π-contacts were visually inspected and classified based on the interaction type, and amino acids and RNA components involved. More than 80% of structures searched contained at least one RNA–protein π-interaction, with π–π contacts making up 59% of the identified interactions. RNA–protein π–π and sugar–π contacts exhibit a range in the RNA and protein components involved, relative monomer orientations and quantum mechanically predicted binding energies. Interestingly, π–π and sugar–π interactions occur more frequently with RNA (4.8 contacts/structure) than DNA (2.6). Moreover, the maximum stability is greater for RNA–protein contacts than DNA–protein interactions. In addition to highlighting distinct differences between RNA and DNA–protein binding, this work has generated the largest dataset of RNA–protein π-interactions to date, thereby underscoring that RNA–protein π-contacts are ubiquitous in nature, and key to the stability and function of RNA–protein complexes.
INTRODUCTION
Many different types of RNA exist that fold into diverse structural domains (such as double-stranded regions, hairpins, loops, bulges and pseudoknots) and fulfill a plethora of critical cellular functions. For example, during protein synthesis, the nucleobase sequence in messenger RNA (mRNA) dictates the amino acid sequence of the protein, transfer RNA (tRNA) brings the correct amino acid to the ribosome for protein synthesis, and ribosomal RNA (rRNA) is an essential component of the molecular machine that joins the amino acids (1,2). Similarly, several RNA types, such as microRNA (3), long-non-coding RNA (4) and small RNA (sRNA) (5), participate in the regulation of gene expression. Additionally, viral RNA can carry genetic information (6), while uniquely designed RNA aptamers have shown promise for use in diagnostics and as therapeutics (7).
To fulfill these vast functions, RNA interacts with a range of proteins from the time it is synthesized until it is degraded in the cell. RNA–protein interactions are typically noncovalent in nature in order to afford stability to the resulting complex for functional purposes, while at the same time permitting facile degradation when the task at hand has been completed. To gain a greater understanding of the nature of noncovalent interactions that govern RNA recognition, binding and processing, several previous studies have analyzed RNA–protein crystal structures published in the protein data bank (PDB) (8–20). The noncovalent contacts between RNA and proteins have traditionally been recognized to include ionic (salt-bridge or phosphate backbone), hydrophobic, hydrogen-bonding (direct or water mediated) and van der Waals interactions. Detailed structural studies have revealed that van der Waals interactions are exceptionally important, being more prevalent than hydrogen bonding (8,16) and accounting for nearly three-quarters of the interactions at RNA–protein interfaces (15).
Among van der Waals contacts, π-interactions have been proposed to play particularly central roles in RNA–protein complexes. Indeed, a variety of RNA–protein π-interactions identified in X-ray crystal structures may stabilize ribosome assembly (21). As an even more specific example, several nucleobase–amino acid (U:Y) stacking interactions have been proposed to help achieve the natural specificities and binding affinities of the PUF family of proteins (Figure 1A) (22), which regulate mRNA and play important roles in stem cell maintenance and memory (23,24). In fact, engineered PUF proteins use Y and R stacking interactions to achieve enhanced function (25). Similarly, the specificity of RNase T, an exonuclease that removes a nucleotide from the 3′-end of RNA, has been suggested to be controlled by π–π interactions between two F and the 3′ and penultimate 3′ bases (26–28). Computer modeling has hinted that G:H π–π interactions are vital for aligning and binding RNA transcripts to the b-subunit of RNA polymerases (29), while CD72 uses several Y residues to form π–π interactions with RNA nucleobases in order to act as a regulatory protein for B lymphocytes (30). In the absence of structural data, π–π stacking between the RNA-binding protein heterogeneous nuclear ribonucleoprotein H1 (HNRNPH1) and an RNA G-quadraplex has been proposed to be related to alterative splicing and the onset of cancer (31). Although the RNA and protein components are anticipated to be relatively co-planar in the previous examples, nonparallel nucleobase–amino acid binding modes also occur. For example, a C Hoogsteen binding face interacts with a W π-system when the p19 protein from tombusvirus binds with small interfering RNA (siRNA), which inhibits RNA silencing and impacts gene expression (Figure 1B) (32). These few examples highlight the structural diversity of RNA–protein π–π interactions in nature and underscore why a broader understanding of these contacts is necessary to gain a greater appreciation of essential cellular functions.
Figure 1.

Examples of (A) nucleobase–amino acid π–π stacking interaction (PDB ID: 3QGB), (B) nucleobase–amino acid T-shaped interaction (PDB ID: 1R9F), (C) ribose–amino acid sugar–π interaction (PDB ID: 2R8S) and (D) ribose–amino acid sugar–π interaction (PDB ID: 1K8W).
Despite their emerging importance, divergent conclusions regarding the nature of RNA–protein π–π interactions have arisen in the literature. For example, nucleotide–amino acid contacts between G or U and F or Y were originally determined to be prevalent at RNA binding sites based on analysis of 32 RNA–protein complexes (8). Although studies of a larger dataset that included 89 RNA–protein complexes confirmed that U:Y is one of the most favored nucleotide–amino acid pairings, A:F and G:W contacts were determined to also occur at high frequencies (16). Alternatively, interactions between W and either purine were found to be among the most prevalent contacts in 61 RNA–protein crystal structures (12). In addition to contacts with ring-containing amino acids, other studies have highlighted the abundance of π-interactions between acyclic amino acids, such as R, and RNA (11,14). Further conflicting conclusions about RNA–protein interactions have occurred in subsequent studies that specifically considered nucleobase–amino acid π–π stacking interactions (11,12,14,19).
There are likely a number of factors contributing to discrepancies in the literature on RNA–protein π–π interactions. For example, previous studies that datamined the PDB were restricted by the small number of contacts considered due to limitations in the availability of high-resolution crystal structures of RNA–protein complexes. Furthermore, most previous works typically employed search routines solely based on distance or angle cut-offs, which can erroneously lead to the inclusion of nucleic acid–protein interactions that do not truly represent π–π interactions in the datasets (see, e.g. Supplementary Figure S1) (33,34). Although a preliminary report from our group on RNA–protein π–π interactions added a visual inspection step along the analysis pathway, the study was similarly limited by the relatively small number (75) of nonredundant, high-resolution RNA–protein complexes available at the time (19). Indeed, currently available crystal structures allow for an expanded dataset, which increases the number of structures searched by a factor of ∼4 and nearly doubles the total number of RNA types represented (Supplementary Table S1). The added functional diversity in such a larger dataset will enable a more complete understanding of RNA–protein π-interactions. Thus, although previous studies collectively point to the prevalence of nucleobase–amino acid π–π interactions in nature, the true frequency, composition, structure and stability of RNA–protein π–π interactions currently remain poorly understood and warrant a more detailed investigation.
In addition to many unknowns regarding RNA–protein π–π interactions, the ribose moiety of RNA may form close contacts with amino acid π-systems (denoted sugar–π interactions). Indeed, crystal structures reveal short distances between many amino acids, including ring-containing variants (F, Y, H and W), and nucleic acids (8,10). In terms of RNA–protein complexes, close contacts have been observed between Y and the 3′/5′ ribose edge upon binding of anti-RNA antibodies to large functional RNAs (Figure 1C), which is pathologically important in autoimmune diseases (35). Alternatively, Y stacks against the 2′/3′ side of ribose in the active site of TruB (36), a pseudouridine synthase that converts uracil to pseudouridine in tRNA to modulate interactions with rRNA and mRNA (Figure 1D). These select examples illustrate the structural diversity and potential importance of ribose–amino acid contacts in nature. Furthermore, sugar–π interactions have been reported to be common in DNA–protein complexes (33,34,37) and folded RNA structures (38), while similar interactions between carbohydrates and amino acids have long been accepted to play indispensable roles in glycobiology (39–44). By analogy, sugar–π interactions could likely modulate RNA–protein binding. Indeed, the predominant single-stranded nature and additional 2′-hydroxy group may make such interactions more important, and yet distinct, for RNA compared to DNA and carbohydrates. Despite the likely prevalence and critical role of such contacts in RNA–protein systems, no previous work has characterized the occurrence, composition, structure or stability of RNA–protein sugar–π interactions.
Due to existing uncertainties surrounding the relative frequency of different RNA–protein contact classifications, the proportional involvement of different RNA and protein components, and the comparative stability and/or function the contacts can provide to RNA–protein complexes, the present study reports a detailed analysis of over 1500 RNA–protein contacts identified in a nonredundant set of >300 X-ray crystal structures of RNA–protein complexes (Supplementary Tables S3 and S4). Interactions between ring-containing (H, F, Y and W; also denoted cyclic) or acyclic (R, E and D) π–containing amino acids and the RNA nucleobases (π–π interactions) or ribose (sugar–π contacts) are considered (Supplementary Figure S2). This choice of amino acids permits the evaluation of neutral, cationic and anionic RNA–protein π-interactions, and therefore the impact of different biologically-relevant amino acid protonation states. Each RNA–protein contact identified was visually inspected to rule out close contacts due to other forces, such as hydrogen bonding, and to unambiguously categorize the interaction according to the RNA and protein components involved and their relative orientation. Quantum chemical calculations are subsequently used to determine the binding energy of each RNA–protein π–π and sugar–π interaction in order to appreciate their relative contributions to the stability of the overall RNA–protein complex.
By assembling and analyzing the largest database of RNA–protein π-interactions available to date, our work clarifies discrepancies in the previous literature by explicitly uncovering key structural and energetic features of RNA nucleobase–amino acid π–π interactions. Furthermore, for the first time, novel RNA–protein sugar–π contacts are examined using the same detailed approach, which complements existing data on sugar contacts within folded RNA (38) and DNA–protein complexes (33,34,37). The larger and more diverse data set considered in the current work leads to new conclusions regarding the dominant amino acids and nucleobases involved in RNA–protein π–π interactions, the most common nucleobase–amino acid pairings and the preferred relative monomer orientations, as well as the stability of the contacts (see Supplementary Tables S1 and S2 for a comparison to previous literature). Perhaps even more importantly, by considering the same expanded datasets for both π–π and sugar–π contacts, we uncover the relative abundance of RNA–protein π–π and sugar–π contacts, and further emphasize the broad range of π-interactions that occur in RNA biology. Finally, detailed comparison of our new dataset to previously published work on DNA–protein π-interactions (33,34) highlights both similarities and differences in noncovalent protein interactions for the two nucleic acid families. Our findings can be applied to gain a greater understanding of many critical cellular functions (e.g. protein synthesis, gene expression, and viral replication), to design improved functional RNAs (aptamers), to develop novel drugs that target RNA, and to fine-tune computational methodologies (i.e. force fields for molecular dynamic simulations and docking procedures) that can be subsequently used to further advance our understanding of RNA structure and function.
MATERIALS AND METHODS
The careful protocol previously developed and applied by our group to understand DNA–protein π-interactions (33,34,37) was adapted in the present work. Specifically, a total of 317 crystal structures of RNA–protein complexes were chosen from the PDB for analysis based on a resolution better than 2.5 Å and redundancy of < 30% as assessed using the CD-HIT suite (45). Crystallographic copies were excluded from the dataset and the structures chosen do not involve nucleic acid oligomers that represent RNA–DNA hybrids. Although modifications to RNA are common and modified residues are included in the crystal structures searched in the present work, we did not include modifications in the data set. Specifically, modified nucleobases were not considered for π–π interactions, while modified sugar residues were not considered for sugar–π interaction. For residues consisting of multiple conformations that adopted similar orientations, only the highest occupancy conformation was considered. The lower occupancy orientation was only considered when the higher occupancy conformation did not form a π-interaction. Only contacts between fully resolved residues were included in our database. A complete list of structures analyzed is available in the Supplementary Material (Supplementary Tables S3 and S4).
For each crystal structure included in our search, pairs composed of a nucleobase or ribose and a cyclic (H, W, F and Y) or acyclic (R, E or D) π–containing amino acid that are within 5 Å were selected. This distance was chosen based on geometric characteristics known to be important for nucleic acid π–π and sugar–π contacts (33,34,37). Finally, each contact was visually inspected in order to ensure overlap between the two monomers such that each pairing identified can unambiguously be classified as a π–π or sugar–π interaction. Through this visual inspection, all hydrogen bonding dimers that satisfy the distance criteria were removed. During this analysis, the type of nucleobase, amino acid or sugar edge involved in the π-interaction was recorded, while the Mercury program (46) was used to measure the angle between the π-systems for each π–π contact. The cleaned dataset is available for download as part of the Supplementary Material.
For each RNA–protein contact identified, quantum chemical calculations were performed to estimate the stability of the interaction. Specifically, for the π–π interactions, models were used that contain only the RNA nucleobase and the amino acid π-system (i.e. the cyclic amino acids and E were truncated as Cβ, D was truncated at Cα, and R was truncated at Cγ; Supplementary Figure S2). Different protonation states were considered for the amino acid side chains, with H modeled as neutral (both δ and ϵ forms) and cationic, and D/E modeled as neutral and anionic. Furthermore, the hydroxy group of Y and of the neutral D/E models were considered in two relative orientations with respect to the nucleobase, although only the binding energy for the most stable orientation is discussed due to the observed negligible impact of hydroxy orientation on the binding strength, which parallels conclusions previously reported for DNA (33,34). The nucleobase and ring-containing amino acid models were optimized with MP2/6–31G(d) in Cs symmetry and overlaid onto the coordinates extracted from the crystal structure for each binding pair.
The same amino acid models implemented to investigate π–π contacts were used to evaluate sugar–π interactions. For the ribose moiety, the nucleotide was truncated at the nucleobase, and the 5′ and 3′ phosphorus atoms and the truncated position capped with hydrogen atoms, and the positions of all hydrogen atoms in the model were optimized with MP2/6–31G(d), while the positions of all heavy atoms were held fixed. During the optimization, the 5′ and 3′ hydroxy orientations were frozen by constraining the ∠(HO5′−O5′−C5′−C4′) and ∠(HO3′−O3′−C3′−C4′) dihedral angles to the corresponding values of the truncation points in the crystal structures. The 2′-hydroxy group was initially aligned in the minimum energy orientation for an isolated ribose according to MP2/6–31G(d) calculations, but the orientation of the substituent was not fixed during the optimizations. This approach for the sugar–π contacts circumvents issues with the range of puckering modes for ribose that are found in nature and is justified based on work on carbohydrate–π interactions that revealed small deviations in binding strengths when crystal structures versus fully optimized geometries were implemented (47,48).
For all RNA–protein π-interactions, overlays were performed using root-mean-square (RMS) fitting according to the relevant heavy atoms using HyperChem 8.0.8 (49) and PyMOL 1.8.6 (50). The geometry of monomers in the dimer are the same as the isolated monomers. The binding energy for each RNA–protein pair was evaluated using M06–2X/6–311++G(2df,2p) single-point calculations according to the following equation:
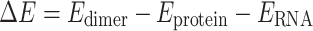 |
where 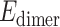 is the electronic energy of the π–π or sugar–π complex,
is the electronic energy of the π–π or sugar–π complex, 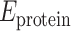 is the electronic energy of the amino acid model side chain, and
is the electronic energy of the amino acid model side chain, and 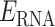 is the electronic energy of the RNA nucleobase or ribose. The reported binding energies do not contain zero-point vibrational or Gibbs energy corrections. The chosen density functional (DFT) method has proven to provide accurate data at a reduced computational cost compared to gold-standard CCSD(T) data at the complete basis set limit (CBS) for analogous DNA–protein π–π and sugar–π interactions (33). We note that the data generated using the smaller 6–31+G(d,p) basis set was used to compare RNA and DNA–protein interactions as this methodology was used in the previous work on DNA (33,34). Furthermore, this approach is justified due to the negligible difference between the small and large basis set data (<8 kJ/mol deviations, with 95% of the interactions changing by less than 4 kJ/mol, Supplementary Tables S5 and S6), as well as the maintenance of all trends in the data. All calculations were performed in the gas phase, which implies that the reported interactions represent the magnitude of the interactions in protein binding environments of low polarity. Although we acknowledge that binding free energies can be estimated from classical molecular dynamics (MD) studies in various solvents (51), the predicted binding energies reported in the present work provide upper bounds to the strength of these contacts in a variety of biological environments. Nevertheless, we acknowledge that some protein–RNA interactions may occur at more solvent exposed sites. Therefore, the impact of different environments ranging from the gas phase to water have been estimated for a selection of neutral and charged π–π and sugar–π interactions (Supplementary Tables S7 and S8), which highlights the preserved stability of such contacts in a variety of environments. Furthermore, the sample set showed the inclusion of counterpoise corrections for RNA–protein systems does not significantly change the predicted binding energy for RNA–protein systems (differences < 5 kJ/mol, Supplementary Table S9). Overall, the implemented methodology was chosen due to successes using the same approach to understand other π-interactions between nucleic acid and protein components to allow for meaningful comparisons between RNA and DNA (19,33,34,37), as well as due to computational efficiency because of the large number of contacts considered. All quantum chemical calculations were performed using Gaussian 09 (revisions A.02 and C.01).
is the electronic energy of the RNA nucleobase or ribose. The reported binding energies do not contain zero-point vibrational or Gibbs energy corrections. The chosen density functional (DFT) method has proven to provide accurate data at a reduced computational cost compared to gold-standard CCSD(T) data at the complete basis set limit (CBS) for analogous DNA–protein π–π and sugar–π interactions (33). We note that the data generated using the smaller 6–31+G(d,p) basis set was used to compare RNA and DNA–protein interactions as this methodology was used in the previous work on DNA (33,34). Furthermore, this approach is justified due to the negligible difference between the small and large basis set data (<8 kJ/mol deviations, with 95% of the interactions changing by less than 4 kJ/mol, Supplementary Tables S5 and S6), as well as the maintenance of all trends in the data. All calculations were performed in the gas phase, which implies that the reported interactions represent the magnitude of the interactions in protein binding environments of low polarity. Although we acknowledge that binding free energies can be estimated from classical molecular dynamics (MD) studies in various solvents (51), the predicted binding energies reported in the present work provide upper bounds to the strength of these contacts in a variety of biological environments. Nevertheless, we acknowledge that some protein–RNA interactions may occur at more solvent exposed sites. Therefore, the impact of different environments ranging from the gas phase to water have been estimated for a selection of neutral and charged π–π and sugar–π interactions (Supplementary Tables S7 and S8), which highlights the preserved stability of such contacts in a variety of environments. Furthermore, the sample set showed the inclusion of counterpoise corrections for RNA–protein systems does not significantly change the predicted binding energy for RNA–protein systems (differences < 5 kJ/mol, Supplementary Table S9). Overall, the implemented methodology was chosen due to successes using the same approach to understand other π-interactions between nucleic acid and protein components to allow for meaningful comparisons between RNA and DNA (19,33,34,37), as well as due to computational efficiency because of the large number of contacts considered. All quantum chemical calculations were performed using Gaussian 09 (revisions A.02 and C.01).
RESULTS
RNA–protein π–π and sugar–π interactions both commonly occur for many different RNA types, with π–π interactions being approximately 1.7 times more prevalent
In total, 1532 RNA–protein π-interactions were identified in the 317 nonredundant crystal structures of RNA–protein complexes included in our database. Among the crystal structures searched, 262 (83%) contain at least one RNA–protein π-interaction (Figure 2A). Although 32 (10%) of the structures searched contain more than six discrete contacts, each complex most commonly contains one (56 structures or 18% searched), two (48 or 15%) or three (50 or 16%) RNA–protein π-interactions. Among RNA types included in our crystal structure database, over 70% of the structures for a given RNA type contain at least one contact (Figure 2B), illustrating the abundance of π-interactions is independent of RNA type.
Figure 2.
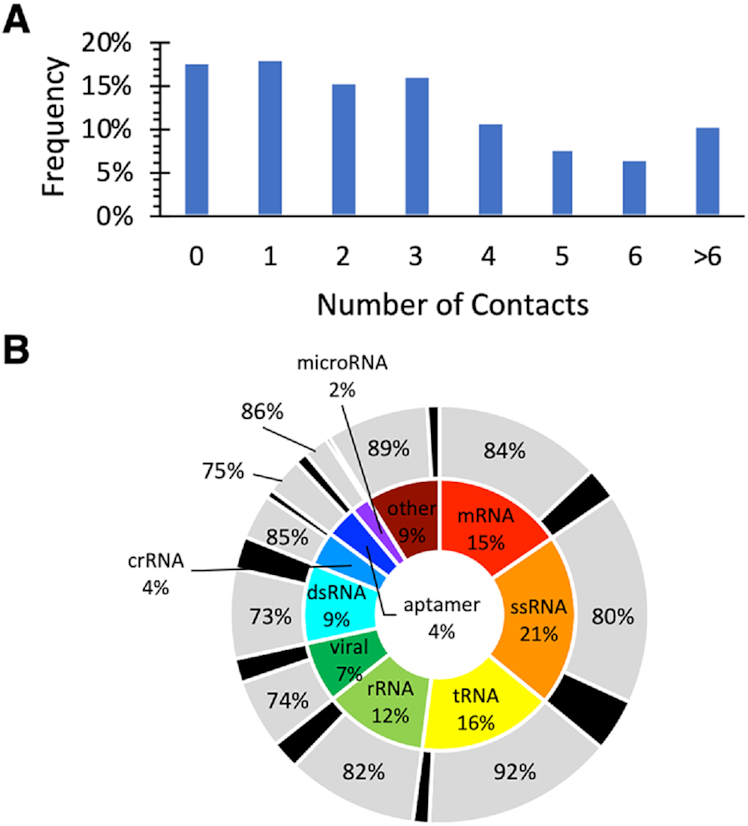
(A) Overall frequency of the number of RNA–protein π-contacts found in each crystal structure searched. (B) Distribution in the RNA types searched (inner circle) and the RNA types that form at least one π-contact (outer circle).
Among the 1532 RNA–protein π-contacts found, 897 (59%) represent π–π interactions, while 635 (41%) can be classified as sugar–π contacts. Over 73% of the structures in our database contain at least one π–π interaction, with RNA–protein complexes most commonly containing one (75 structures or 24%) or two (62 or 20%) π–π interactions (Supplementary Figure S3a). Among RNA classifications, at least half of the structures searched for a given RNA type contain one or more contact(s) (Supplementary Figure S4a). In fact, over 70% of the structures for all RNA types contain at least one π–π interaction, with the exception of only dsRNA and aptamers.
For sugar–π interactions, 53% of RNA–protein structures contain at least one sugar–π contact, with most crystal structures containing one (87 structures or 27%) or two (47 or 15%) such interactions (Supplementary Figure S3b). Three quarters of the tRNA, crRNA and microRNA structures searched contain at least one sugar–π contact. Although the percentage of structures that contain at least one sugar–π contact is smaller for each other type of RNA, a significant portion (25–54%) of the structures contains sugar–π interactions (Supplementary Figure S4b).
Y, F and R-containing π–π interactions are more frequent than anticipated based on relative natural abundances of amino acids, while R contacts dominate sugar–π interactions
Among the π–π interactions identified, the distribution as a function of the amino acid involved indicates that most contacts occur for R (34%), F (26%) and Y (21%), while few contacts exist for H, W, E and D (≤ 8% each, Figure 3A). However, the expected relative occurrence of the amino acids should be considered when analyzing this data, which can be calculated as the ratio of the frequency of the respective amino acid in proteins to the total frequency of the π-containing amino acids considered in the present study (Supplementary Tables S10 and S11). Comparison of the deviation between the expected occurrence and observed occurrence of specific amino acids reveals that only H and W are involved in π–π interactions in the expected ratio (<1.5 times more than expected). In contrast, R, F and Y interactions are more abundant than expected (∼2 times), while E and D interactions are less abundant than anticipated (by 8–10 times, Supplementary Table S10). Overall, approximately two-thirds of π–π interactions involve a ring-containing amino acid (Figure 3A).
Figure 3.
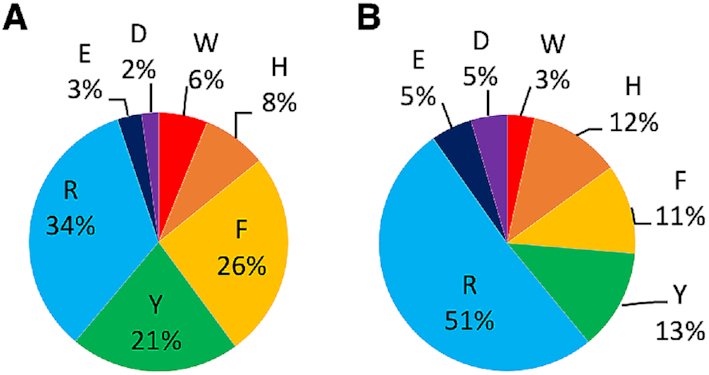
Relative composition of the RNA–protein (A) π–π and (B) sugar–π contacts identified as a function of the amino acid involved.
For the sugar–π interactions, over half of the contacts involve R (51%), while the sum of the ring-containing amino acids is 39%, with Y being the most frequently at 13%, followed by F and H (11–12% each, Figure 3B). Interestingly, unlike π–π interactions, the number of sugar–π interactions with all ring-containing amino acids is exactly as expected based on the amino acid natural abundance (<1.5 times greater or less than expected, Supplementary Tables S11 and S12). However, as discussed for π–π contacts, R sugar–π interactions occur significantly more often than expected (2.6 times), while E and D contacts are less frequent (4–5 times). As a result, in direct contrast to π–π interactions, two-thirds of the sugar–π interactions involve an acyclic π–containing amino acid.
RNA nucleobase are not equally likely to participate in π–π interactions
The estimated distribution of the four canonical nucleobases in RNA varies. For example, A has been reported to occur more frequently than the other nucleobases in rRNA (52–54). The composition of the RNA–protein π–π interactions in our structural database as a function of the nucleobase suggests that A π–π interactions occur most frequently (36%, Figure 4 and Supplementary Table S10). Indeed, A π–π contacts appear 10% more frequently and C interactions 13% less frequently than those involving G or U. As a result, purine π–π interactions (68%) are more common than pyrimidine contacts (38%). This distribution generally holds for each individual amino acid, regardless of their cyclic versus acyclic nature (Figure 4). When coupled with the frequency of interactions as a function of amino acid (Figure 3), it is therefore not surprising that the most common π–π interactions occur between A and R (13.0%), F (9.3%) or Y (7.1%), while G or U interactions with F, Y and R fall close behind (5.2–7.8%, Figure 4).
Figure 4.
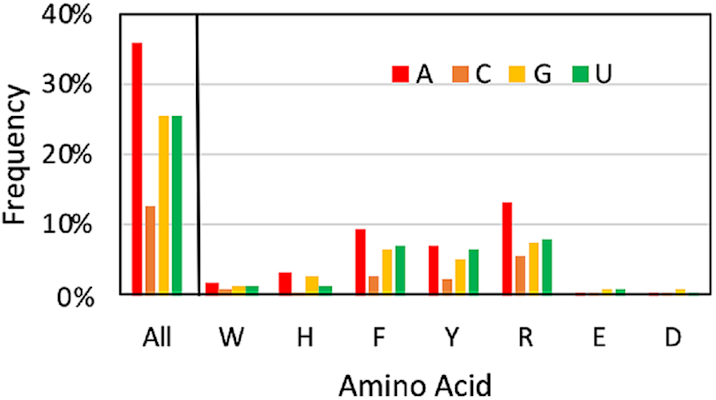
Relative composition of the RNA–protein π–π interactions identified as a function of the nucleobase (color) and amino acid involved.
RNA–protein π–π interactions exhibit a strong preference for a parallel alignment of the rings
Based on the relative arrangement of the planes of the nucleobase and amino acid π-systems (denoted tilt or ω), the RNA–protein π–π interactions were classified as stacked (ω = 0–20°), inclined (20° < ω < 70°) or T-shaped (ω = 70–90°). Approximately half of all π–π interactions identified in the present work represent a stacked or nearly parallel arrangement of the π-systems (49%, Figure 5A and Supplementary Figure S5). Although many inclined orientations (39%) also prevail, the nucleobase and amino acid π-systems are less likely to adopt a T-shaped arrangement (12%). Indeed, over three-quarters of all interactions have a tilt angle ≤30%. The T-shaped interactions can involve either a nucleobase edge directed toward the amino acid π-system or an amino acid edge directed toward the nucleobase π-system. In general, the strongest T-shaped interactions in nature typically involve the edge of the amino acid interacting with the π-system of the nucleobase, with the preference notably greater for the charged systems (Supplementary Tables S13 and S14).
Figure 5.
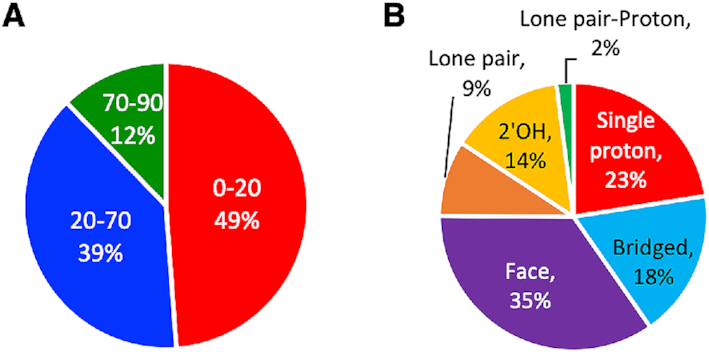
Distribution in the relative monomer orientations for RNA–protein (A) π–π (divided according to the angle between the planes of the π-systems, with 0–20° representing stacking, 20–70° representing inclined and 70–90° representing T-shaped orientations) and (B) sugar–π interactions identified.
In terms of the amino acid involved in the interaction, the stacked structures are favored for the ring-containing amino acids (48–69% of all structures), but the inclined orientation becomes preferred for acyclic R, E and D (Supplementary Figure S5a, c). Nevertheless, for R, 188 interactions (62%) fall below 30%, further emphasizing a common preference for a near parallel arrangement. Among the nucleobases, the stacked orientation is highly preferred for the purines, comprising ∼50% of the structures, while only ∼35% of the interactions adopt an inclined orientation (Supplementary Figure S5b, d). Although the stacked arrangement is still preferred for U (46%), the inclined orientation becomes slightly more important than for the purines (∼40% of all U interactions). The inclined configuration is the most common for C (51%, Supplementary Figure S5b, d).
The strong preference for nucleobase and amino acid π-systems to adopt parallel stacked arrangements is clear when all identified nucleobase–amino acid π–π interactions are overlaid using RMS fitting with respect to the nucleobase heavy atoms (see Figure 6A–D). Nevertheless, these overlays also highlight structural diversity in the nucleobase–amino acid π–π interactions, including the explicit presence of T-shaped interactions between either the nucleobase or amino acid edge and the π-system of the second monomer.
Figure 6.
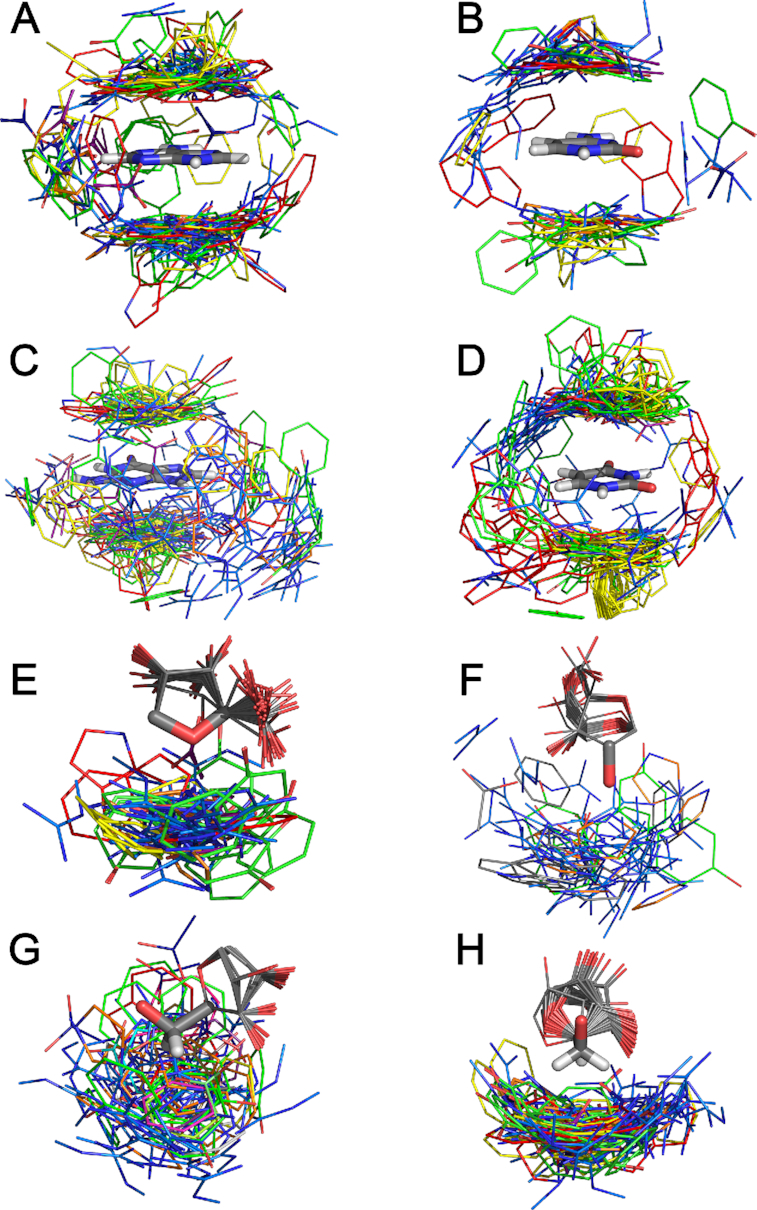
Overlay the all interactions identified between the amino acids (W (red), H (orange), F (yellow), Y (green), R (light blue), E (navy) and D (purple)) and each nucleobase [(A) adenine, (B) cytosine, (C) guanine, (D) uracil] or one of the four ribose edges [(E) O4′, (F) O2′H, (G) H4H5aH5b or (H) the H5aH5b].
Sugar–π interactions can involve ribose proton(s) and/or oxygen atom(s) directed towards the amino acid, with a critical role played by the 2′-hydroxy group
The flexible nature of single-stranded RNA allows many different faces of ribose to interact with π-systems of amino acids upon binding with proteins. In general, the sugar–π contacts can be categorized according to the identity and nature of the atom(s) in ribose directed toward the amino acid. Our classification system separates interactions that involve a sugar edge comprised of a single proton, two protons (denoted bridged), three protons (denoted face), a (O4′) lone pair, both a lone pair (lp) and a proton (lone pair-proton or lp-proton) or the 2′-hydroxyl group (2′OH, Supplementary Figure S6A). Over all amino acids, more than three-quarters of the sugar–π interactions involve a ribose proton rather than an oxygen atom (Figure 5B and Supplementary Table S11). The most common relative orientation of ribose and the amino acid π-system is a face interaction (35%), which is closely followed by contacts involving one (23%) or two (bridged, 18%) protons. Nevertheless, lone pair and lone pair-proton interactions comprise 11% of all sugar–π contacts identified. Furthermore, the 2′-OH is involved in interactions with the amino acid π-system in 14% of all contacts, being greater than all interactions with O4′ (11%). In terms of atoms involved for specific classifications of the sugar–π interactions (Figure 5 and Supplementary Figure S6B), the majority of the face interactions involve the three protons on C4′ and C5′ (26% of the total contacts identified), and the bridged interaction similarly most commonly involves the C5′ protons (15%). Single proton interactions frequently occur at many different ribose sites, including C5′, C1′, and C4′ (4–6% each). The trend of the favored classification and atoms involved in the sugar–π interactions generally holds when contacts with each amino acid are considered (Supplementary Figure S6C and S7). Nevertheless, the sugar–π interactions exhibit significant fluctuation in the position of the amino acid relative to ribose for a given binding orientation. Indeed, overlays of all contacts identified for the four most common binding configurations obtained using RMS fitting with respect to the sugar heavy atoms highlights the variation in the relative monomer orientations for a specific sugar edge (Figure 6E–H). In fact, the great fluctuation in relative monomer orientations likely leads to a continuum between sugar–π interaction classifications. These representative overlays also underscore that the sugar puckering varies across crystal structures, which adds another element of structural diversity to sugar–π contacts.
Both nucleobase–amino acid π–π and sugar–π interactions can significantly enhance the stability of RNA–protein complexes
Although the abundance and composition of RNA–protein π-interactions can be conjectured from crystal structures, the relative strength of each contact is also important to understand in order to gain a full appreciation of the role of these contacts (37). Due to the proven ability of computational methods to approximate the energetic contributions of π-interactions between nucleic acid and protein components (12,19,33,34,37,38,55–58), quantum chemical methods were used to evaluate the gas-phase binding strength for each RNA–protein π-interaction identified in the experimental crystal structures. A range in computed stabilities is found for RNA–protein π-interactions in nature (Figure 7 and Supplementary Figures S8 and S9). This variance in the binding energies reflects the differences in chemical properties of the nucleobases and amino acids involved. Furthermore, the diversity of the interaction strengths parallels the variation in the relative monomer orientations across all RNA–protein π-interactions (Figure 6A–D, and Supplementary Figures S10 and S11), including the tilt angle for the π–π interactions (Supplementary Figure S12), as well as the amino acid or nucleobase edge involved in T-shaped π–π interactions, and the identity of the sugar edge involved in the sugar–π interactions (Supplementary Figure S13). The charge of the amino acid also plays a significant role (Supplementary Figures S8 and S9).
Figure 7.
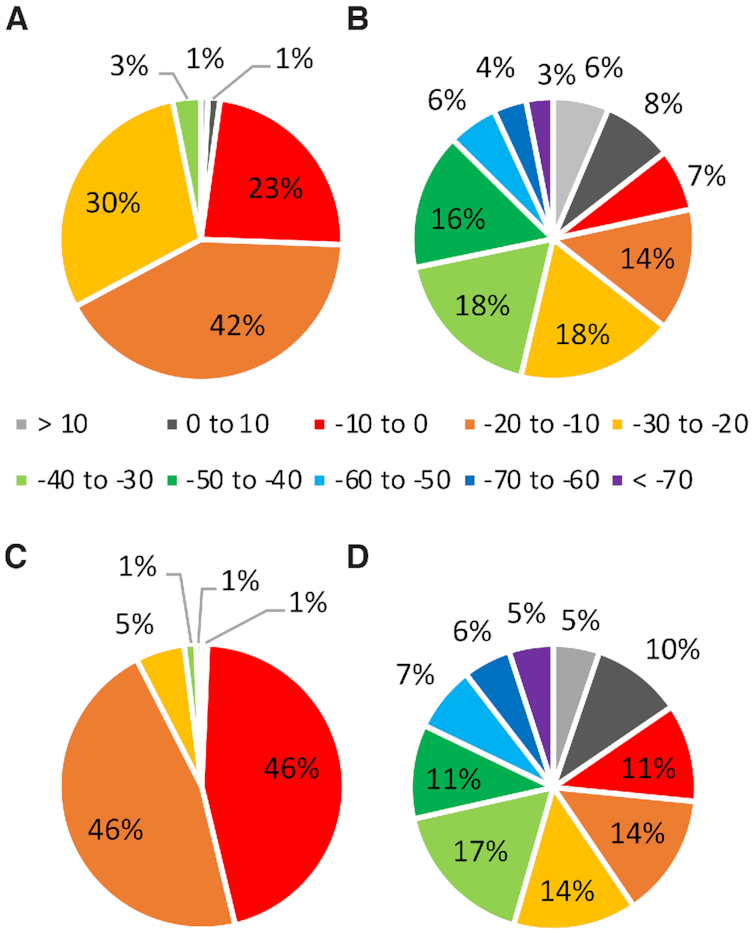
Distribution in the binding strengths (kJ/mol) for RNA–protein (A and B) π–π and (C and D) sugar–π contacts that involve a neutral (A and C) or charged (B and D) amino acid.
In terms of π–π interactions involving a neutral amino acid (W, H, F, Y and E/D), over three-quarters of all contacts have binding strengths greater than –20 kJ/mol, with 71% ranging between –10 and –30 kJ/mol (Figure 7A). The magnitude of the interaction energy more strongly depends on the amino acid (Supplementary Figure S8a) than the nucleobase (Supplementary Figure S8c). The strongest interactions involve W or Y and G or U (Supplementary Figure S8a, c). This trend parallels the near perfect parallel arrangement between W or Y and G in the most stable interactions (Supplementary Figure S10), as well as G having the largest dipole moment among the nucleobases. Indeed, the magnitude of the interaction strength is highly dependent on the tilt angle between the planes of the (neutral) π-systems, with stronger interaction energies occurring for the more predominant stacked orientations (tilt angle < 20°, Supplementary Figure S12). Furthermore, repulsive interactions rationalize any highly unstable interactions. For example, the weakest W interaction occurs with A due to close (< 2 Å) atomic contacts.
Although the neutral π–π interactions reach a maximum of –38.3 kJ/mol, previous literature has highlighted the significant strength of nucleobase–amino acid π-interactions involving one cationic (59–61) or anionic (62) π-system. Therefore, the corresponding cationic π–π interactions were considered for H and R and anionic interactions for E and D. The π–π interactions involving charged amino acids (H, R, E/D) are even stronger, being stable by over 70 kJ/mol in the extreme cases (Figure 7B). Although more than 66% of the contacts have binding strengths that fall between –10 and –50 kJ/mol, a significant portion (13%) also have binding strengths stronger than –50 kJ/mol. The most stable charged interactions typically occur for (cationic) R and G, up to –95.7 kJ/mol (Supplementary Figure S8b and d). Nevertheless, contacts with cationic H are very strong (up to –70.3 kJ/mol). Furthermore, the anionic interactions can be extremely stable, with E interactions being up to –135.3 kJ/mol and D contacts up to –93.0 kJ/mol. The significantly more stable anionic compared to neutral π–π interactions for E/D occur at least in part due to an increase in the size (number of atoms) involved in the amino acid π-system. It should be noted that a percentage (6%) of all contacts are unstable (positive binding strengths) due to repulsive interactions between the charged π-system and acid/basic atoms of the nucleobase. For example, a repulsive interaction occurs between the amino group of G and cationic H or the electronegative carbonyl of C and the anionic π-system of E in the least stable monomer orientations (Supplementary Figure S10). Since previous studies have highlighted the great potential stability of cationic and anionic nucleobase–amino acid π–π interactions (59–62), this repulsive nature suggests that the corresponding interactions in the crystal structures likely involve a neutral amino acid.
Similar to the nucleobase–amino acid π–π interactions, the sugar–π contacts exhibit a range of binding strengths (Figure 7C, D and Supplementary Figure S9). For sugar–π interactions involving neutral amino acids (W, H, F, Y and E/D), 51% have binding strengths ranging between –10 and –30 kJ/mol, with the stability reaching a maximum of –58.9 kJ/mol. Although many sugar edges are involved in these interactions, a range of binding strengths is found for each interaction type (i.e. single proton, bridge, face, lone pair, lone pair-proton and 2′OH, Supplementary Figure S13) and the interaction strength depends more strongly on the nature of the amino acid involved (Supplementary Figure S9a). In general, the most stable neutral sugar–π interactions involve Y, as well as H to a lesser degree. Nevertheless, a range of binding strengths are seen for each amino acid due to variations in their arrangement with respect to ribose (Figure 6E–H). For example, the strongest Y interaction occurs at least in part due to hydrogen bonding between the hydroxyl group of Y and O5′, while repulsive interactions lead to an unstable Y–ribose binding arrangement (e.g. <2 Å between a carbon atom of Y and H5′, Supplementary Figure S11).
Similar to the π–π interactions, the sugar–π contacts may involve cationic H/R or anionic E/D. The sugar–π interactions involving charged amino acids can be significantly more stable than the neutral counterparts, with 56% of the charged contacts having binding strengths between –10 and –50 kJ/mol, and 18% of contacts having interaction energies stronger than –50 kJ/mol. The strongest interaction with a cationic amino acid (–105.1 kJ/mol) occurs for R, while the most stable sugar–π interaction with an anionic amino acid (–91.5 kJ/mol) occurs with D (Supplementary Figures S9 and S11). There is not a strict dependence of the strength of the interaction on the classification of the sugar–π contact, with all types of sugar–π interactions adopting the full spectrum of predicted binding strengths for both neutral and charged amino acids (Supplementary Figure S13). Nevertheless, the range in the interaction energies is greater for the charged than neutral amino acids (Supplementary Figure S13). As discussed for the π–π contacts, this reflects repulsive interactions for some systems (e.g., repulsion between R and O3′–H in the least stable R–ribose contact, Supplementary Figure S11). This may indicate that such interactions involve a neutral amino acid. Overall, the quantum mechanical data emphasizes the great stability both nucleobase π–π and sugar–π interactions can bring to RNA–protein complexes.
DISCUSSION
A variety of RNA–protein π-interactions occur in nature that can stabilize and impart function to RNA–protein complexes
Amongst the 317 nonredundant crystal structures considered, 1532 RNA–protein π-interactions were identified in a range of RNA types, with 59% of the contacts involving a nucleobase and 41% involving ribose. Approximately 70% of the π–π interactions identified occur between F, Y or R and A, U or G. In fact, the interactions with F, Y and R are more abundant in our π–π interaction data set than anticipated based on the relative natural occurrences of the amino acids. In contrast, over half of the sugar–π interactions involve R, while the ring-containing (cyclic) amino acids participation in sugar–π contacts correlates with their relative natural abundances. The overall more dominant involvement of R in RNA–protein π-interactions stands to reason based on the negative charge of the backbone. These results are consistent with the previously reported prevalence of Y, F and R at RNA binding sites (8,11,14,16), as well as suggestions that W is the least likely ring-containing amino acid to participate in RNA–protein π–π stacking interactions (14). However, our data contradicts predictions that G:W pairings are the most prevalent at RNA-binding sites (16), potentially due to W being the most common ring-containing amino acid involved in RNA recognition (12). In terms of nucleobase, interactions with A are more predominant, which contrasts previous conclusions based on much smaller datasets that U and G contacts (8), or both purines (16) most frequently participate, and underscores the importance of the expanded dataset considered in the present work. Nevertheless, π-interactions can involve any of the four canonical RNA bases or ribose and cyclic (W, H, F and Y) or acyclic (R, E and D) π–containing amino acids, which highlights the great diversity in the composition of RNA–protein π-interactions in nature.
In terms of structure, the nucleobase–amino acid interactions with parallel arrangements of the π–rings comprise over 50% of all interactions, with 39% of the contacts adopting inclined ring orientations and very few T-shaped arrangements being assumed. Nevertheless, we have imposed an arbitrary cut-off between stacked and inclined structures, and overall >75% of all interactions have a tilt angle between the rings of <30°. The strongly preferred parallel, stacked arrangement of RNA–protein π–π interactions likely stems from the structurally flexible nature of RNA, which mostly occurs in a single-stranded form. Interestingly, the stacked arrangement is even more prevalent for the purines compared to pyrimidines, which likely simply reflects the larger π-system of the purines.
The RNA ribose moiety uses a variety of orientations to participate in sugar–π contacts. Most often one or three hydrogen atom(s) bound to carbon is (are) directed toward the π-system (58% of contacts). The favored edge of ribose involved in sugar–π interactions is the C4′ proton and C5′-C4′ face, which would be the least disruptive for nucleobase binding, potentially allowing for multiple RNA–protein noncovalent interactions to simultaneously be involved in RNA binding and/or function. Nevertheless, the 2′-hydroxy group is involved in ∼14% of all sugar–π interactions, illustrating the importance of this structural feature that at least in part distinguishes RNA from DNA. Indeed, it has been previously estimated that the 2′-hydroxy group is involved in over one quarter of all RNA–protein hydrogen bonds (15) and has important functions such as aiding RNA recognition (13,14). Nevertheless, many edges can be involved in RNA–protein binding, which highlights the critical role played by many sugar faces despite an overall preference for interactions with the C5′ side.
Regardless of whether an RNA nucleobase or ribose interacts with a π-containing amino acid, the π-interaction can impart significant stability to the RNA–protein complex. Indeed, both types of interactions between neutral nucleic acid and protein components can approach binding strengths of up to –40 (π–π) or –60 (sugar–π) kJ/mol. Furthermore, when charged amino acid π-systems are considered, the interactions become even more stable, with binding strengths up to –105 (π–π) or –135 (sugar–π) kJ/mol. Since the binding strength of a canonical A:T hydrogen-bonded pair is ∼–70 kJ/mol, it is clear that nucleobase/ribose–amino acid π-interactions can provide similar, and even greater, stability to RNA–protein complexes as commonly accepted biologically-relevant noncovalent interactions.
Based on the abundance and strength of RNA–protein π-interactions identified in the present work, it is not surprising that such contacts have been proposed to play a number of roles in RNA binding and processing. For example, two conserved F residues of the human RNA-binding motif protein 38 (RBM38) were shown to stack with two Us in the 30 untranslated region (30UTR) of mRNAs corresponding to several cancer-related proteins (Supplementary Figure S14a). Notably, mutation of either F residue results in a >100-fold decrease in binding (63). Another example is the use of π–π interactions with W to distinguish between modified and unmodified RNA nucleobases during the maternal-to-zygotic transition in early embryogenesis (64). Specifically, a W residue of the Y-box binding protein 1 (Ybx1) (65,66) has been determined to be integral in the identification of epigenetically-modified bases that need to be preserved during this process (Supplementary Figure S14b).
In addition to furthering our understanding of possible functions of RNA–protein π-interactions in biology, the present work paves the way to better understand new roles of noncovalent interactions in proteins. As highlighted by the example of modified PUF enzymes in the introduction (25), an improved working knowledge of the ways amino acids and nucleic acids can interact will permit the design of proteins with specific functions, such as the regulation of RNA sequences (67) and the controlled modification of nucleic acids (68). Beyond interactions between canonical amino acids and nucleosides, the greater insight into π–π interactions obtained from this study can be applied to better comprehend and develop ligands and drugs that bind to nucleic acids and/or proteins (69,70). Furthermore, understanding the complex π-interactions that occur between biomolecules can provide greater insight into the operation, and possibly the design, of unique ligands for inorganic complexes that can be used as catalysts and materials (71–73).
RNA–protein π-interactions exhibit both key similarities and differences compared to DNA–protein π-interactions
In direct complement to the biological importance of RNA–protein interactions, a diverse array of proteins must interact with DNA to replicate and preserve our genetic information. Indeed, DNA–protein complexes form as a critical part of DNA replication during cell growth and division, epigenetic regulation and the repair of DNA damage arising from exposure to many harmful agents in the environment. Nevertheless, the interactions between proteins and each nucleic acid type can be very different due to their unique functions and structures. Indeed, the predominant double helical structure adopted by DNA in which the nucleotides interact through Watson-Crick hydrogen bonding changes the availability of the nucleobases and the conformation of the sugar–phosphate backbone for interactions with proteins compared to the widely diverse structures adopted by single-stranded RNA. It is therefore not surprising that key differences between the recognition of RNA and DNA have been reported (8–14). Furthermore, the distinguishing 2′-hydroxy group of RNA expands the types of interactions with proteins that are possible compared to DNA. Indeed, the 2′-hydroxy group has been previously reported to participate in one quarter of RNA–protein hydrogen bonds (15) and the present work highlights that a significant percentage of all sugar–π interactions involve the 2′-hydroxy group. Therefore, it is interesting to compare the abundance, composition and strength of RNA and DNA–protein π-interactions.
Previous work by our group has analyzed 672 crystal structures of DNA–protein complexes published in the PDB (34). In total, 1765 nucleobase or sugar–amino acid π-interactions were identified and rigorously analyzed. Comparison of the previous DNA dataset with that compiled in the present work for RNA shows many general conclusions hold across both sets of nucleic acid π-interactions, although there are also some distinguishing differences. Overall, nucleic acid–protein complexes are highly likely to contain π-interactions regardless of whether DNA or RNA is involved. Although complexes involving DNA are more likely to contain at least one π-interaction (79% of structures searched) compared to those with RNA (73%, Figure 8A), the RNA–protein complexes generally contain a greater number of π-contacts. Specifically, when the ratio of the number of interactions found to the number of crystal structures searched is considered, DNA–protein complexes contain on average 2.6 contacts/structure, while RNA–protein complexes contain 4.8 contacts/structure.
Figure 8.
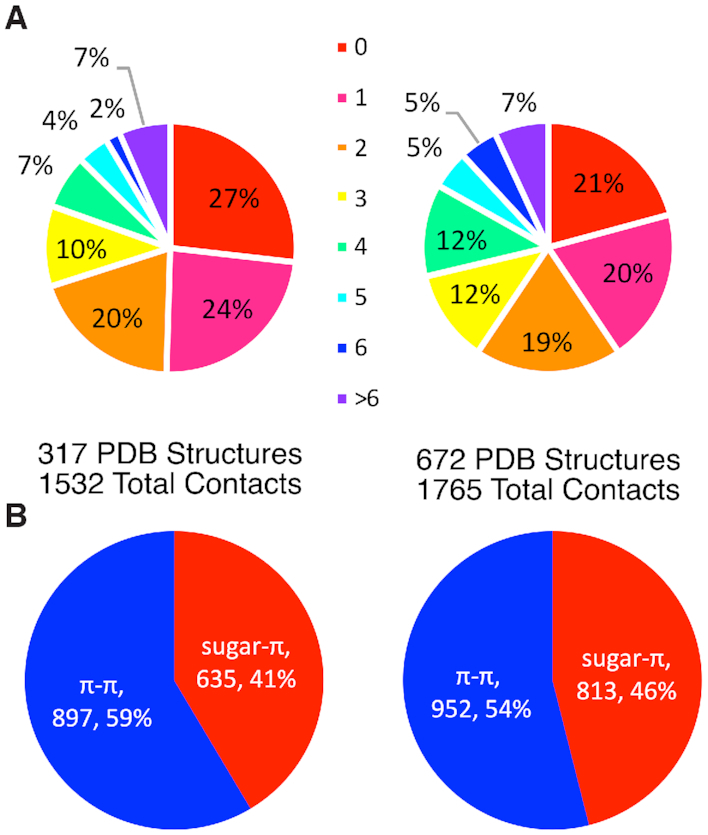
Comparison of (A) the overall frequency of the number of nucleic acid–protein π-interactions and (B) the ratio of π–π versus sugar–π interactions for RNA (left) and DNA (right).
Although both DNA and RNA–protein complexes involve a significant number of nucleobase and sugar–amino acid contacts, RNA more often forms interactions with π–containing amino acids through a nucleobase than the sugar moiety compared to DNA (Figure 8B). This correlates with structural differences between the nucleic acids, with the DNA double helix forcing many contacts with proteins through the phosphate backbone, while the single-stranded form of RNA making the nucleobase more readily available for π-interactions. Indeed, proteins often interact with RNA nucleobases in structural domains containing loops, kinks or bulges (9,10,13).
All four canonical DNA or RNA nucleobases are involved in π–π interactions with both cyclic and acyclic π–containing amino acids (Table 1). Nevertheless, although the composition is relatively equally distributed among the four DNA nucleobases, A interactions are greater than average and C interactions less than average for RNA. Furthermore, more RNA–protein π–π interactions involve acyclic protein residues (39%) than the DNA–protein counterparts (25%). Among the ring-containing amino acids, F and Y contacts dominate for both RNA and DNA, comprising 46–54% of the total π–π contacts. Similarly, acyclic amino acids are more prevalent in RNA sugar–π interactions (61%) than DNA sugar–π contacts (46%, Table 2). Additionally, although Y and F participate in a large number of DNA sugar–π interactions (45%), Y, F and H sugar–π contacts are more equally distributed for RNA (∼11–12% each). Despite interactions involving one, two or three sugar hydrogen atoms being the most populated sugar–π interactions, the number is reduced for RNA (76%) compared to DNA (85%). This difference at least in part arises due to the additional 2′-hydroxy group in RNA, which is involved in a significant portion of all RNA–protein sugar–π contacts.
Table 1.
Comparison of the relative abundance of nucleobase–amino acid pairings in RNA or DNA–protein π–π interactions found in nucleic acid–protein complexes
| RNAa | DNAb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | U | G | C | Total | A | T | G | C | Total | |
| W | 2.0% | 1.4% | 1.6% | 1.1% | 6.1% | 3.2% | 3.6% | 2.8% | 1.7% | 11.3% |
| H | 3.3% | 1.6% | 2.7% | 0.4% | 8.0% | 1.8% | 2.4% | 0.3% | 4.4% | 8.9% |
| F | 9.3% | 7.2% | 6.6% | 2.7% | 25.8% | 9.6% | 10.2% | 5.6% | 7.6% | 33.0% |
| Y | 7.1% | 6.6% | 5.2% | 2.2% | 21.2% | 2.6% | 9.2% | 5.4% | 4.5% | 21.7% |
| R | 13.0% | 7.8% | 7.4% | 5.6% | 33.8% | 6.0% | 3.8% | 6.9% | 4.0% | 20.7% |
| E | 0.7% | 0.8% | 1.1% | 0.4% | 3.0% | 0.3% | 0.3% | 0.0% | 0.4% | 1.0% |
| D | 0.4% | 0.2% | 1.0% | 0.4% | 2.1% | 0.9% | 0.3% | 2.1% | 0.1% | 3.4% |
| Total | 35.9% | 25.5% | 25.6% | 12.9% | 24.4% | 29.8% | 23.1% | 22.7% | ||
Table 2.
Comparison of the relative abundance of sugar edge–amino acid pairings in RNA or DNA–protein sugar–π interactions found in nucleic acid–protein complexes
| RNAa | DNAb | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Single H | lp | 2′OH | lp-H | Bridged | Face | Total | Single H | lp | lp-H | Bridged | Face | Total | |
| W | 0.3% | 0.2% | 0.3% | 0.2% | 0.6% | 1.7% | 3.3% | 0.7% | 0.6% | 0.5% | 0.5% | 3.7% | 6.0% |
| H | 3.8% | 1.4% | 1.3% | 0.2% | 2.4% | 2.7% | 11.8% | 1.0% | 0.9% | 0.5% | 0.7% | 0.4% | 3.5% |
| F | 2.7% | 0.6% | 0.9% | 0.2% | 1.9% | 5.0% | 11.3% | 2.7% | 1.4% | 0.5% | 6.8% | 5.4% | 16.8% |
| Y | 2.4% | 1.7% | 1.4% | 0.2% | 1.7% | 5.4% | 12.8% | 5.8% | 1.7% | 0.1% | 8.9% | 11.2% | 27.7% |
| R | 10.6% | 4.3% | 8.5% | 0.9% | 8.8% | 18.1% | 51.2% | 12.6% | 7.3% | 1.0% | 5.9% | 7.2% | 34.0% |
| D/E | 2.8% | 0.9% | 1.3% | 0.5% | 2.4% | 1.9% | 9.8% | 2.5% | 0.5% | 0.0% | 7.0% | 2.1% | 12.1% |
| Total | 22.5% | 9.1% | 13.7% | 2.0% | 17.8% | 34.8% | 25.3% | 12.3% | 2.6% | 29.8% | 30.0% | ||
The strengths of the DNA/RNA–protein intramolecular forces span a wide range of stabilities regardless of the nucleic acid component or amino acid side chain involved (Tables 3 and 4). There is no clear correlation between the stability of the interactions and the relative population of different DNA/RNA–protein pairings in nature. The RNA–protein π–π and sugar–π interactions generally access a wider range of binding strengths compared to the corresponding type of DNA contact. Indeed, the strongest π–π and sugar–π interactions for RNA are ∼40–45 kJ/mol more stable than the corresponding DNA contact. Nevertheless, the mean interaction energies for a given RNA and DNA interaction type are generally within 5 kJ/mol. On the other hand, π-interactions involving a cationic side chain are on average consistently stronger for RNA than DNA (by up to 20 kJ/mol), while contacts involving an anionic side chain are weaker for RNA than DNA (by up to 13 kJ/mol).
Table 3.
Comparison of the stability of nucleobase–amino acid π–π interactions in RNA or DNA–protein complexes
| RNAa | DNAb | |||||
|---|---|---|---|---|---|---|
| Range | Most common | Meanc | Range | Most common | Meanc | |
| W | 19.8 to –38.3 | –20 to –25 (22%) | –16.8±11.9 | 4.7 to –39.4 | –20 to –25 (44%) | –22.6±6.0 |
| H | –3.3 to –33.0 | –20 to –25 (31%) | –19.2±7.2 | 16.1 to –28.8 | –20 to –25 (40%) | –15.2±8.5 |
| F | 7.4 to –27.3 | –20 to –25 (26%) | –14.5±7.0 | 3.1 to –26.9 | –5 to –10 (25%) | –13.3±6.5 |
| Y | 23.7 to –37.9 | –20 to –25 (25%) | –18.8±8.6 | –0.4 to –33.1 | –15 to –20 (23%) | –17.7±6.9 |
| E | 12.0 to –21.9 | –5 to –10 (30%) | –10.2±7.9 | 4.2 to –16.3 | 0 to –5 (50%) | –6.1±5.7 |
| –15 to –20 (30%) | ||||||
| D | 29.1 to –25.7 | –10 to –15 (26%) | –10.1±11.5 | 8.7 to –40.1 | –10 to –15 (28%) | –15.5±10.3 |
| H+ | 30.0 to –70.3 | –40 to –45 (13%) | –36.5±19.2 | 39.9 to –49.6 | –20 to –25 (19%) | –17.1±17.1 |
| R | 96.6 to –95.7 | –25 to –30 (11%) | –24.9±24.8 | 34.6 to –96.5 | 0 to –5 (12%) | –19.6±24.9 |
| E– | 35.2 to –135.3 | 0 to 5 (26%) | –26.8±37.1 | –5.4 to –95.5 | –5 to –10 (20%) | –33.5±25.9 |
| –25 to –30 (20%) | ||||||
| D– | 5.8 to –93.0 | 0 to 5 (15%) | –25.4±34.7 | 36.2 to –87.5 | 30 to 35 (19%) | –28.0±43.3 |
aThe present work as determined using 6–31+G(d) to permit direct comparison, which yields comparable results to the 6–311++G(2df,2p) basis set (Supplementary Table S5) and permits comparison to previous work on DNA interactions.
cAveraged over all nucleobases for a given amino acid.
Table 4.
Comparison of the stability (kJ/mol) of nucleic acid–protein sugar–π interactions in RNA or DNA–protein complexes
| RNAa | DNAb | |||||
|---|---|---|---|---|---|---|
| Range | Most common | Meanc | Range | Most common | Meanc | |
| W | –1.2 to –25.4 | –15 to –20 (41%) | –15.7±6.2 | –0.7 to –29.3 | –20 to –25 (31%) | –18.6±7.2 |
| H | 16.5 to –46.6 | –10 to –15 (26%) | –12.6±8.0 | 2.6 to –19.9 | –5 to –10 (40%) | –10.8±3.9 |
| F | –0.8 to –19.4 | –5 to –10 (47%) | –8.9±4.3 | 8.0 to –37.4 | –10 to –15 (39%) | –10.7±5.0 |
| Y | 12.8 to –58.9 | –10 to –15 (30%) | –14.9±10.9 | 5.2 to –16.1 | –5 to –15 (72%) | –6.4±4.2 |
| E | 8.3 to –30.3 | –10 to –15 (32%) | –10.5±7.4 | 58.7 to –23.1 | –5 to –10 (67%) | –4.1±7.9 |
| D | –0.3 to –38.3 | –5 to –10 (36%) | –12.6±8.4 | 5.2 to –68.2 | –5 to –15 (22%) | –25.3±21.6 |
| H+ | 27.9 to –86.7 | –25 to –20 (12%) | –28.4±23.8 | 95.1 to –75.1 | –25 to –30 (11%) | –22.8±21.4 |
| R | 45.9 to –105.1 | –40 to –35 (8%) | –29.2±26.5 | –9.3 to –59.1 | –15 to –20 (36%) | –27.3±13.2 |
| E– | 30.2 to –66.1 | –5 to 0 (18%) | –24.0±22.1 | 37.2 to –89.5 | –50 to –55 (23%) | –37.1±19.2 |
| D– | 30.2 to –91.5 | –25 to –20 (24%) | –13.0±24.5 | –0.7 to –29.3 | –20 to –25 (31%) | –18.6±7.2 |
aThe present work as determined using 6–31+G(d) to permit direct comparison, which yields comparable results to the 6–311++G(2df,2p) basis set (Supplementary Table S6) and permits comparison to previous work on DNA interactions.
cAveraged over all sugar edges for a given amino acid.
The differences in the stabilities of individual π-interactions for RNA and DNA is reflected in the preferred relative monomer orientations for each type of contact (Supplementary Figures S10–S11). This variation arises at least in part due to the enhanced flexibility of single-stranded RNA. Indeed, the major difference in the nucleobase–amino acid π–π interactions between RNA and DNA is the dimer geometry, as well as slight variations arising from C–H···π interactions associated with the methyl group of T in DNA. Although the sugar–π interactions are also highly similar for RNA and DNA, the 2′–OH in RNA can introduce new and influence the strength of these contacts. Regardless, the π-interactions between different nucleic acid and protein components are overall extremely stable for both RNA and DNA, which further emphasizes the importance of such noncovalent interactions for the structure and function of biomolecular complexes.
DATA AVAILABILITY
The full dataset generated and analyzed during the current study is available as part of the Supplementary Data or from the corresponding author on reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Devany Holland, Luciana Prada and Rochelle Caruso for help with preliminary analysis and/or calculations. Computational resources provided by Westgrid and Compute/Calcul Canada are greatly appreciated.
Contributor Information
Katie A Wilson, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta T1K 3M4, Canada.
Ryan W Kung, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta T1K 3M4, Canada.
Simmone D’souza, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta T1K 3M4, Canada.
Stacey D Wetmore, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive West, Lethbridge, Alberta T1K 3M4, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council of Canada (NSERC) [2016-04568]; S.D.W. also thanks the Board of Governors Research Chair program of the University of Lethbridge; K.A.W. and R.W.K. also thank NSERC, Alberta Innovates-Technology Futures and the University of Lethbridge for student scholarships. Funding for open access charge: Natural Sciences and Engineering Research Council of Canada and the University of Lethbridge.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chapeville F., Lipmann F., Von Ehrenstein G., Weisblum B., Ray W.J. Jr., Benzer S.. On the role of soluble ribonucleic acid in coding for amino acids. Proc. Natl. Acad. Sci. U.S.A. 1962; 48:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shine J., Dalgarno L.. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975; 254:34–38. [DOI] [PubMed] [Google Scholar]
- 3. He L., Hannon G.J.. Micrornas: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004; 5:522–531. [DOI] [PubMed] [Google Scholar]
- 4. Ponting C.P., Oliver P.L., Reik W.. Evolution and functions of long noncoding RNAs. Cell. 2009; 136:629–641. [DOI] [PubMed] [Google Scholar]
- 5. Storz G., Vogel J., Wassarman K.M.. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011; 43:880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drake J.W., Holland J.J.. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hermann T., Patel D.J.. Biochemistry - adaptive recognition by nucleic acid aptamers. Science. 2000; 287:820–825. [DOI] [PubMed] [Google Scholar]
- 8. Jones S., Daley D.T., Luscombe N.M., Berman H.M., Thornton J.M.. Protein-RNA interactions: a structural analysis. Nucleic. Acids. Res. 2001; 29:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allers J., Shamoo Y.. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J. Mol. Biol. 2001; 311:75–86. [DOI] [PubMed] [Google Scholar]
- 10. Lejeune D., Delsaux N., Charloteaux B., Thomas A., Brasseur R.. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins. 2005; 61:258–271. [DOI] [PubMed] [Google Scholar]
- 11. Morozova N., Allers J., Myers J., Shamoo Y.. Protein-RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics. 2006; 22:2746–2752. [DOI] [PubMed] [Google Scholar]
- 12. Baker C.M., Grant G.H.. Role of aromatic amino acids in protein-nucleic acid recognition. Biopolymers. 2007; 85:456–470. [DOI] [PubMed] [Google Scholar]
- 13. Bahadur R.P., Zacharias M., Janin J.. Dissecting protein-RNA recognition sites. Nucleic. Acids. Res. 2008; 36:2705–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barik A., C N., Pilla S.P., Bahadur R.P. Molecular architecture of protein-RNA recognition sites. J. Biomol. Struct. Dyn. 2015; 33:2738–2751. [DOI] [PubMed] [Google Scholar]
- 15. Treger M., Westhof E.. Statistical analysis of atomic contacts at RNA-protein interfaces. J. Mol. Recognit. 2001; 14:199–214. [DOI] [PubMed] [Google Scholar]
- 16. Ellis J.J., Broom M., Jones S.. Protein-RNA interactions: structural analysis and functional classes. Proteins. 2007; 66:903–911. [DOI] [PubMed] [Google Scholar]
- 17. Jeong E., Kim H., Lee S.W., Han K.. Discovering the interaction propensities of amino acids and nucleotides from protein-RNA complexes. Mol. Cells. 2003; 16:161–167. [PubMed] [Google Scholar]
- 18. Cheng A.C., Chen W.W., Fuhrmann C.N., Frankel A.D.. Recognition of nucleic acid bases and base-pairs by hydrogen bonding to amino acid side-chains. J. Mol. Biol. 2003; 327:781–796. [DOI] [PubMed] [Google Scholar]
- 19. Wilson K.A., Holland D.J., Wetmore S.D.. Topology of RNA-protein nucleobase-amino acid π-π interactions and comparison to analogous DNA-protein π-π contacts. RNA. 2016; 22:696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chavali S.S., Cavender C.E., Mathews D.H., Wedekind J.E.. Arginine forks are a Widespread motif to recognize phosphate backbones and guanine nucleobases in the RNA major Ggoove. J. Am. Chem. Soc. 2020; 142:19835–19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pilla S.P., Thomas A., Bahadur R.P.. Dissecting macromolecular recognition sites in ribosome: implication to its self-assembly. RNA Biol. 2019; 16:1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu C., Dutcher R.C., Porter D.F., Arava Y., Wickens M., Hall T.M.T.. Distinct RNA-binding modules in a single PUF protein cooperate to determine RNA specificity. Nucleic. Acids. Res. 2019; 47:8770–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morrison S.J., Kimble J.. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006; 441:1068–1074. [DOI] [PubMed] [Google Scholar]
- 24. Nishanth M.J., Simon B.. Functions, mechanisms and regulation of Pumilio/Puf family RNA binding proteins: a comprehensive review. Mol. Biol. Rep. 2020; 47:785–807. [DOI] [PubMed] [Google Scholar]
- 25. Zhao Y.-Y., Mao M.-W., Zhang W.-J., Wang J., Li H.-T., Yang Y., Wang Z., Wu J.-W.. Expanding RNA binding specificity and affinity of engineered PUF domains. Nucleic. Acids. Res. 2018; 46:4771–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsiao Y.Y., Yang C.C., Lin C.L., Lin J.L., Duh Y., Yuan H.S.. Structural basis for RNA trimming by RNase T in stable RNA 3′-end maturation. Nat. Chem. Biol. 2011; 7:236–243. [DOI] [PubMed] [Google Scholar]
- 27. Li Z., Deutscher M.P.. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA. 2002; 8:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsiao Y.Y., Duh Y., Chen Y.P., Wang Y.T., Yuan H.S.. How an exonuclease decides where to stop in trimming of nucleic acids: crystal structures of RNase T-product complexes. Nucleic Acids Res. 2012; 40:8144–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vedithi S.C., Rodrigues C.H.M., Portelli S., Skwark M.J., Das M., Ascher D.B., Blundell T.L., Malhotra S.. Computational saturation mutagenesis to predict structural consequences of systematic mutations in the beta subunit of RNA polymerase in Mycobacterium leprae. Comput. Struct. Biotechnol. J. 2020; 18:271–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akatsu C., Shinagawa K., Numoto N., Liu Z., Ucar A.K., Aslam M., Phoon S., Adachi T., Furukawa K., Ito N.. CD72 negatively regulates B lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand Sm/RNP. J. Exp. Med. 2016; 213:2691–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neckles C., Boer R.E., Aboreden N., Cross A.M., Walker R.L., Kim B.-H., Kim S., Schneekloth J.S., Caplen N.J.. HNRNPH1-dependent splicing of a fusion oncogene reveals a targetable RNA G-quadruplex interaction. RNA. 2019; 25:1731–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ye K., Malinina L., Patel D.J.. Recognition of small interfering RNA by a viral suppressor of RNA silencing. Nature. 2003; 426:874–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilson K.A., Kellie J.L., Wetmore S.D.. DNA-protein π-interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res. 2014; 42:6726–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson K.A., Wells R.A., Abendong M.N., Anderson C.B., Kung R.W., Wetmore S.D.. Landscape of π-π and sugar-π contacts in DNA-protein interactions. J. Biomol. Struct. Dyn. 2016; 34:184–200. [DOI] [PubMed] [Google Scholar]
- 35. Ye J.-D., Tereshko V., Frederiksen J.K., Koide A., Fellouse F.A., Sidhu S.S., Koide S., Kossiakoff A.A., Piccirilli J.A.. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoang C., Ferre-D’Amare A.R.. Cocrystal structure of a tRNA Psi55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001; 107:929–939. [DOI] [PubMed] [Google Scholar]
- 37. Wilson K.A., Wetmore S.D.. Combining crystallographic and quantum chemical data to understand DNA-protein π-interactions in nature. Struct. Chem. 2017; 28:1487–1500. [Google Scholar]
- 38. Chawla M., Chermak E., Zhang Q., Bujnicki J.M., Oliva R., Cavallo L.. Occurrence and stability of lone pair–π stacking interactions between ribose and nucleobases in functional RNAs. Nucleic Acids Res. 2017; 45:11019–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ren W., Pengelly R., Farren-Dai M., Shamsi Kazem Abadi S., Oehler V., Akintola O., Draper J., Meanwell M., Chakladar S., Świderek K.et al.. Revealing the mechanism for covalent inhibition of glycoside hydrolases by carbasugars at an atomic level. Nat. Commun. 2018; 9:3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paulsen P.A., Custódio T.F., Pedersen B.P.. Crystal structure of the plant symporter STP10 illuminates sugar uptake mechanism in monosaccharide transporter superfamily. Nat. Commun. 2019; 10:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hudson K.L., Bartlett G.J., Diehl R.C., Agirre J., Gallagher T., Kiessling L.L., Woolfson D.N.. Carbohydrate–aromatic interactions in proteins. J. Am. Chem. Soc. 2015; 137:15152–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montalvillo-Jiménez L., Santana A.G., Corzana F., Jiménez-Osés G., Jiménez-Barbero J., Gómez A.M., Asensio J.L.. Impact of aromatic stacking on glycoside reactivity: balancing CH/π and cation/π Interactions for the stabilization of glycosyl-oxocarbenium ions. J. Am. Chem. Soc. 2019; 141:13372–13384. [DOI] [PubMed] [Google Scholar]
- 43. Cuétara-Guadarrama F., Hernández-Huerta E., Rojo-Portillo T., Reyes-López E., Jiménez-Barbero J., Cuevas G.. Experimental and theoretical study of the role of CH/π interactions in the aminolysis reaction of acetyl galactoside. Carbohydr. Res. 2019; 486:107821. [DOI] [PubMed] [Google Scholar]
- 44. Houser J., Kozmon S., Mishra D., Hammerová Z., Wimmerová M., Koča J.. The CH–π interaction in protein–carbohydrate binding: bioinformatics and in vitro quantification. Chem. Eur. J. 2020; 26:10769–10780. [DOI] [PubMed] [Google Scholar]
- 45. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Macrae C.F., Bruno I.J., Chisholm J.A., Edgington P.R., McCabe P., Pidcock E., Rodriguez-Monge L., Taylor R., van de Streek J., Wood P.A.. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008; 41:466–470. [Google Scholar]
- 47. Spiwok V., Lipovová P., Skálová T., Buchtelová E., Hasek J., Králová B.. Role of CH/π interactions in substrate binding by Escherichia coli beta-galactosidase. Carbohydr. Res. 2004; 339:2275–2280. [DOI] [PubMed] [Google Scholar]
- 48. Sujatha M.S., Sasidhar Y.U., Balaji P.V.. Insights into the role of the aromatic residue in galactose-binding sites: MP2/6-311G++** study on galactose− and glucose−aromatic residue analogue complexes. Biochemistry. 2005; 44:8554–8562. [DOI] [PubMed] [Google Scholar]
- 49. HyperChem(TM) Professional 7.51 Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA.
- 50. Schrodinger, LLC The PyMOL Molecular Graphics System. 2015; Version 2.0.
- 51. de Ruiter A., Zagrovic B.. Absolute binding-free energies between standard RNA/DNA nucleobases and amino-acid sidechain analogs in different environments. Nucleic. Acids. Res. 2014; 43:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nissen P., Ippolito J.A., Ban N., Moore P.B., Steitz T.A.. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gutell R.R., Gray M.W., Schnare M.N.. A compilation of large subunit (23S and 23S-like) ribosomal RNA structures: 1993. Nucleic Acids Res. 1993; 21:3055–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ware V.C., Tague B.W., Clark C.G., Gourse R.L., Brand R.C., Gerbi S.A.. Sequence analysis of 28S ribosomal DNA from the amphibian Xenopus laevis. Nucleic Acids Res. 1983; 11:7795–7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mao L., Wang Y., Liu Y., Hu X.. Molecular determinants for ATP-binding in proteins: a data mining and quantum chemical analysis. J. Mol. Biol. 2004; 336:787–807. [DOI] [PubMed] [Google Scholar]
- 56. Cauët E., Rooman M., Wintjens R., Liévin J., Biot C.. Histidine−aromatic interactions in proteins and protein−ligand complexes: quantum chemical study of X-ray and model structures. J. Chem. Theory Comput. 2005; 1:472–483. [DOI] [PubMed] [Google Scholar]
- 57. Copeland K.L., Anderson J.A., Farley A.R., Cox J.R., Tschumper G.S.. Probing phenylalanine/adenine π-stacking interactions in protein complexes with explicitly correlated and CCSD(T) computations. J. Phys. Chem. B. 2008; 112:14291–14295. [DOI] [PubMed] [Google Scholar]
- 58. Copeland K.L., Pellock S.J., Cox J.R., Cafiero M.L., Tschumper G.S.. Examination of tyrosine/adenine stacking interactions in protein complexes. J. Phys. Chem. B. 2013; 117:14001–14008. [DOI] [PubMed] [Google Scholar]
- 59. Churchill C.D.M., Wetmore S.D. Noncovalent interactions involving histidine: the effect of charge on π−π stacking and T-shaped interactions with the DNA nucleobases. J. Phys. Chem. B. 2009; 113:16046–16058. [DOI] [PubMed] [Google Scholar]
- 60. Leavens F.M.V., Churchill C.D.M., Wang S., Wetmore S.D. Evaluating how discrete water molecules affect protein–DNA π–π and π+–π stacking and T-shaped interactions: The case of histidine-adenine dimers. J. Phys. Chem. B. 2011; 115:10990–11003. [DOI] [PubMed] [Google Scholar]
- 61. Rutledge L.R., Churchill C.D.M., Wetmore S.D. A preliminary investigation of the additivity of π−π or π+−π stacking and T-shaped interactions between natural or damaged DNA nucleobases and histidine. J. Phys. Chem. B. 2010; 114:3355–3367. [DOI] [PubMed] [Google Scholar]
- 62. Wells R.A., Kellie J.L., Wetmore S.D.. Significant strength of charged DNA-protein π-π interactions: a preliminary study of cytosine. J. Phys. Chem. B. 2013; 117:10462–10474. [DOI] [PubMed] [Google Scholar]
- 63. Qian K., Li M., Wang J., Zhang M., Wang M.. Structural basis for mRNA recognition by human RBM38. Biochem. J. 2020; 477:161–172. [DOI] [PubMed] [Google Scholar]
- 64. Yang X.J., Zhu H., Mu S.R., Wei W.J., Yuan X., Wang M., Liu Y., Hui J., Huang Y.. Crystal structure of a Y-box binding protein 1 (YB-1)-RNA complex reveals key features and residues interacting with RNA. J. Biol. Chem. 2019; 294:10998–11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kretov D.A., Clément M.-J., Lambert G., Durand D., Lyabin D.N., Bollot G., Bauvais C., Samsonova A., Budkina K., Maroun R.C.et al.. YB-1, an abundant core mRNA-binding protein, has the capacity to form an RNA nucleoprotein filament: a structural analysis. Nucleic. Acids. Res. 2019; 47:3127–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang Y., Wang L., Han X., Yang W.L., Zhang M., Ma H.L., Sun B.F., Li A., Xia J., Chen J.et al.. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol. Cell. 2019; 75:1188–1202. [DOI] [PubMed] [Google Scholar]
- 67. Chen Y., Varani G.. Engineering RNA-binding proteins for biology. FEBS J. 2013; 280:3734–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xiong T., Rohm D., Workman R.E., Roundtree L., Novina C.D., Timp W., Ostermeier M.. Protein engineering strategies for improving the selective methylation of target CpG sites by a dCas9-directed cytosine methyltransferase in bacteria. PLoS One. 2018; 13:e0209408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ellenbarger J.F., Krieger I.V., Huang H.L., Gomez-Coca S., Ioerger T.R., Sacchettini J.C., Wheeler S.E., Dunbar K.R.. Anion-π interactions in computer-aided drug design: modeling the inhibition of malate synthase by phenyl-diketo acids. J. Chem. Inf. Model. 2018; 58:2085–2091. [DOI] [PubMed] [Google Scholar]
- 70. Li F., Wang H., Wang Y., Feng S., Hu B., Zhang X., Wang J., Li W., Cheng M.. Computational investigation reveals Picrasidine C as selective PPARalpha lead: binding pattern, selectivity mechanism and ADME/tox profile. J. Biomol. Struct. Dyn. 2019; 38:5401–5418. [DOI] [PubMed] [Google Scholar]
- 71. Durán-Solares G., Fugarolas-Gómez W., Ortíz-Pastrana N., López-Sandoval H., Villaseñor-Granados T.O., Flores-Parra A., Altmann P.J., Barba-Behrens N.. Lone pair···π interactions on the stabilization of intra and intermolecular arrangements of coordination compounds with 2-methyl imidazole and benzimidazole derivatives. J. Coord. Chem. 2018; 71:1935–1958. [Google Scholar]
- 72. Tocana E., Siminel A., Croitor L.. Synthesis and crystal structures of luminescent mononuclear Ni(II) and Cd(II) complexes with 1,10-phenanthroline. Chem. J. Mold. 2017; 12:102–108. [Google Scholar]
- 73. Janiak C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc., Dalton Trans. 2000; 21:3885–3896. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full dataset generated and analyzed during the current study is available as part of the Supplementary Data or from the corresponding author on reasonable request.