

Abstract
Docking analysis of propolis's natural compound was successfully performed against SARS-CoV-2 main protease (Mpro) and spike protein subunit 2 (S2). Initially, the propolis's protein was screened using chromatography analysis and successfully identified 22 compounds in the propolis. Four compounds were further investigated, i.e., neoblavaisoflavone, methylophiopogonone A, 3′-Methoxydaidzin, and genistin. The binding affinity of 3′-Methoxydaidzin was −7.7 kcal/mol, which is similar to nelfinavir (control), while the others were −7.6 kcal/mol. However, we found the key residue of Glu A:166 in the methylophiopogonone A and genistin, even though the predicted binding energy slightly higher than nelfinavir. In contrast, the predicted binding affinity of neoblavaisoflavone, methylophiopogonone A, 3′-Methoxydaidzin, and genistin against S2 were −8.1, −8.2, −8.3, and −8.3 kcal/mol, respectively, which is far below of the control (pravastatin, −7.3 kcal/mol). Instead of conventional hydrogen bonding, the π bonding influenced the binding affinity against S2. The results reveal that this is the first report about methylophiopogonone A, 3′-Methoxydaidzin, and genistin as candidates for anti-viral agents. Those compounds can then be further explored and used as a parent backbone molecule to develop a new supplementation for preventing SARS-CoV-2 infections during COVID-19 outbreaks.
Keywords: Antiviral, Propolis compounds, Docking analysis, Binding affinity energy, SARS-CoV-2
Graphical abstract
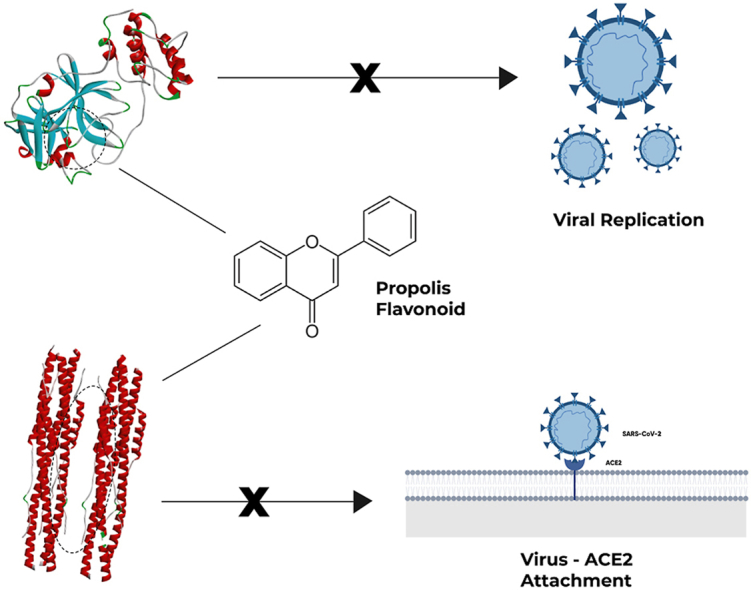
Highlights
-
•
Propolis compound consists of flavonoid, phenolic, and phenylpropanoid.
-
•
The intermolecular interaction between SARS-CoV-2 Mpro and 3′-Methoxydaidzin has the stronger binding energy among others.
-
•
Four compounds have stronger binding energy than Pravastatin against SARS-CoV-2 Spike Protein.
-
•
The candidate to inhibit the SARS-CoV-2 Mpro was methylophiopogonone A, 3'-Methoxydaidzin, and genistin.
1. Introduction
Earlier this year, a novel coronavirus (SARS-CoV 2) triggered a coronavirus disease 19 (COVID-19) outbreak in Wuhan, China, and then spreading globally. The outbreak was announced as a Public Health Emergency of International Concern in January 2020 by World Health Organization (WHO). As of May 12, 2020, there are more than 4,324,739 confirmed cases of COVID-19, and at least 297,197 deaths reported, which indicates a severe public health threat [1]. Unlike MERS-CoV but similar to SARS-CoV [2,3], SARS-CoV 2 can cause human-to-human transmission, and its intermediate host leading to the current human infection is still under investigation [4,5].
Any concrete solutions in prevention, treatment, and post-infection are still lacking. So far, several drugs have been studied as COVID-19 treatments, such as nevilnafir [6], chloroquine [7], hydrochloroquine [8] and remdesivir [9]. Several researchers identified potential anti-viral agents to prevent SARS-CoV-2 infection [10,11]. Several FDA-approved drugs were evaluated as potential anti-viral agents. They can bind to SARS-CoV-2 RdRp and blocked its active site [10]. Remdesivir and chloroquine also have an inhibitor activity to SARS-CoV-2 infection [11]. Although these drug candidates have been massively studied, its currently undergoing the clinical evaluation phase and caution must be taken to avoid any negative effects [12]. Unfortunately, there is no apparent certainty which drug is the most effective in curing the SARS-CoV-2 infection.
It has been computationally determined that the SARS-CoV-2 has a similar mechanism and receptor to SARS, angiotensin-converting enzyme 2 (ACE2) [3]. SARS-CoV-2 uses its spike glycoprotein, which has similarities with the SARS-CoV spike protein structurally, to mediate membrane fusion. The conservation is 73% with most of the variability being in the protein's host cell interaction region. SARS-CoV-2 spike protein is a class I viral fusion protein, which required a protease cleavage to activate the membrane fusion. It has two subunit monomers, S1 for mediating attachment and S2 for membrane fusion, with a similar length of around 180 kDa. Any of them has been the main target for the neutralization antibody [13].
The second in silico docking model is the 3CLPRO main protease, which is responsible for controlling several major functions of the virus and has a highly conserved catalytic domain from the SARS virus [13]. The SARS-CoV-2 main protease (Mpro) is a key CoV enzyme that plays an essential role in mediating viral replication and transcription. When viruses enter the cell, RNA viruses take over the host cell to express structural and non-structural proteins, and Mpro cleaves the large viral polyprotein into a mature viral protein [14,15]. Some of its functions include the virus's replication processes, which makes it an ideal target for drug development [6]. Therefore, Mpro is also identified as an attractive target for inhibitory viral replication [16].
Propolis is commonly used as daily consumption or supplement due to its natural compounds with good pharmacological and pharmaceutical properties [17]. It is well-known propolis has been used as a folk medicine for 300 years B.C [18]. The propolis global world production was around 100 and 200 tons a year, and China produces propolis the most than other countries [19].
The consumption of propolis continues to this day, therefore, determining the bioavailability and safety of its compounds are essential. Furthermore, propolis has been known as an immune agent, works by increasing macrophages' performance [20,21]. Most of the propolis content belongs to flavonoid and polyphenol groups [22], wherein its anti-viral potential has been the subject of several reports [[23], [24], [25], [26], [27]] [[23], [24], [25], [26], [27]] [[23], [24], [25], [26], [27]]. Propolis is reported to inhibit varicella-zoster virus infection, herpesviruses, and HIV by inhibiting viral entry [[25], [26], [27]] [[25], [26], [27]] [[25], [26], [27]].
Several reports are investigating how flavonoids inhibit the infection of SARS-CoV-2 with its host. Some of the flavonoid from propolis such as kaempferol, quercetin, rutin, and galangin has been reported to inhibit SARS-CoV-2's Mpro [11]. Besides, rutin and kaempferol are reported to possess a binding affinity more than nevilnavir against SARS-CoV 2 Mpro [28,29]. Quercetin and rutin are also reported as valuable drug candidates for medicating SARS-CoV-2 based on a molecular docking result against SARS-CoV-2 Mpro and human receptor ACE-2 [30]. Both SARS-CoV and SARS-CoV-2 use ACE2 receptors to enter their host cell via their protein spike, which has a 76% homology [13]. This attachment only occurs in ACE2 but does not involve ACE [31]. Some reports indicate an increase in the high expression of ACE2 in patients with COVID-19 in the oral cavity and lung mucosa [[32], [33], [34]] [[32], [33], [34]] [[32], [33], [34]], and the blocking of spike protein (S2) is imperative to prevent the virus from penetrating the cell membrane. Another strategy targets the main protease of SARS-CoV-2 (Mpro) to prevent structural protein cleavage during the viral formation in the host cell.
This study investigated active propolis compounds' molecular interaction against SARS-CoV-2 main protease and spike protein by molecular docking approach. The molecular interactions between targets and inhibitors were analyzed thoroughly, and the mechanism for the difference in binding ability was also investigated. Hopefully, this information can be used as a reference in building synthetic compounds with better affinity or developing drug and vaccine candidates.
2. Materials and methods
2.1. Propolis screening
The propolis was supplied from PT Nano Herbaltama Internasional, South Tangerang, Indonesia. The preparation was done by adding 1 ml of propolis to 9 mL methanol in a 10 ml volumetric flask and then filtered with Minisart SRP15 Syringe Filter PTFE 0.2 μm (Sartorius, Germany). The filtered solution was added into a 2 mL vial for LC-MS/MS analysis. LC-MS/MS XEVO G2-XS Quadrupole-time-of-flight (The Waters, New Zealand) was used and separation was carried out using Acquity UPLC HSS-T3 column (100 × 2.1 mm, 1.7 μm). The column was maintained at 40 °C throughout the analysis, and the sample temperature was kept at 25 °C. The mobile phases used were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 0.6 ml/min. The elution gradient was set as follows: Starts at 1%, at 0.5 min 1% A, at 16 min 35% A, at 18 min 100% and 20 min 1%A. A total of 2 μl of the test solution was injected to screen flavonoids, and the chromatograph was recorded for 20 min. The mass scan range was set at 50–1200 Da with electrospray ionization (ESI) positive ion mode. The source temperature was 120 °C and the desolvation temperature was 550 °C. The capillary voltage was set as 2.0 kV and cone voltage was 40 V. The cone gas flow was set at 50 L/h and desolvation gas flow was 1000 L/h. The low collision energy was 6 eV and the high collision energy was 15–40 eV (ramp). 1 ng/mL solution of Leucine Enkephalin was used as external references (lock spray) for mass correction during acquisition. The Lock-Spray was infused at a flow rate of 10 μl/min and scan time was 0.1 s with an interval of 30 s.
2.2. Ligand retrieval and preparation
As much as 24 ligands were retrieved in 3D conformer with SDF formats from PubChem database (NCBI) (http://pubchem.ncbi.nlm.nih.gov). Of them, 22 ligands were the screened bioactive compound of propolis, and the other 2 were nelfinavir (C32H45N3O4S) and pravastatin (C23H36O7) with PubChem ID 64143 and 54687, respectively. Each ligand was optimized in molecular geometry, torsional barriers, and intermolecular-interaction geometry using MMFF94 partial forcefield in CHARMM and converted to a.pdb file using the Discovery Studio 2020 (BIOVIA)[35]. The ligand preparation only uses the propolis component's original ligand structure without considering the appropriate tautomeric forms of ligands, the specification of the chirality, and pH range addition.
2.3. Protein retrieval and preparation
Two proteins were used as the target for this study, SARS-CoV-2 main protease complex and SARS-CoV-2 S2 subunit protein. The crystal structures for those proteins were retrieved from Protein Data Bank (PDB) (www.rcsb.org) with the following PDB ID: 6LU7 for the complex of SARS-CoV-2 main protease (2.16 Å) and 6LXT for SARS-CoV-2 S2 subunit protein (2.90 Å). Each retrieved crystal structure was prepared using PyMOL version 2.0.4 program (The PyMOL Molecular Graphics System)[36] by removing the water molecule and ligand molecule. Prepared pdb file was then assessed for its Ramachandaran plot using RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) to check its readiness for docking analysis[37]. Binding site residues of each ligand were identified using the Computed Atlas of Surface Topography of Protein (CASTp)[38]. The binding pocket was shown as a red pocket of Mpro (Fig. 1(a)) and S2 (Fig. 1 (b)), whereas the key residue was presented in Table 1.
Fig. 1.

The binding pocket of (a) SARS-CoV-2 Main Protease (Mpro) and (b) SARS-CoV-2 Spike Protein S2 unit.
Table 1.
Grid Selection and targeted key residue.
| Protein Target | Key residue | Center | Dimension (Å) |
|---|---|---|---|
| 6LU7 | Thr24, Thr25, Thr26, Leu27, His41, Thr45, Ser46, Met49, F139, Leu140, Asn141, Gly142, Ser143, C144, His163, Met165, Glu166, His172 | X: 17.6528 Y: 23.4131 Z: 70.9264 |
X: 38.0184 Y: 34.0608 Z: 38.7272 |
| 6LXT | Leu922, Gln926, Asn928, Gly932, Lys933, Gln935, Asp936, Ser939, Ala942, Ser943, Gly946, Lys947, Gln949, Asp950, Val951, Asn953, Gln954, Asn955, Gln957, Ala958, Asn960, Thr961, Val963, Lys964, Gln965, Ser967, Ser968, Asn969, Phe970, Gly971, Ala972, Ser974, Asp1165, Leu1166, Gly1167, Asp1168, Ile1169, Ser1170, Gly1171, Ile1172, Asn1173, Ala1174, Ser1175, Val1176, Val1177, Asn1178, Ile1179, Gln1180, Lys1181, Glu1182, Ile1183, Asp1184, Arg1185, Asn1187, Glu1188, Val1189, Ala1190, Lys1191, Asn1192, Asn1194, Glu1195, Leu1197, Ile1198, Asp1199, Leu1200, Gln1201 | X: −11.7071 Y: 3.4251 Z: −10.4247 |
X: 25.0000 Y: 25.0000 Z: 99.2687 |
2.4. Docking analysis, visualization, and validation
The prepared protein and ligands were loaded to PyRx (https://pyrx.sourceforge.io/home) and then prepared to be run in the built-in AutoDock VINA in the PyRx program[39].
Site-specific docking was performed with the grid selection parameter, as shown in Table 1, and the rest of the setting was left at default. The binding result table and the best model for each protein-ligand interaction were saved to be visualized. Docking visualization was done using Discovery Studio 2020 (BIOVIA) for its 3D conformation and 2D binding diagram [35]. Docking analysis and visualization were done in Microsoft Windows PC with Intel® Core™ i7-7700H CPU @3.6 GHz, 16 GB RAM with NVIDIA Titan XP GPU.
The docking analysis validation was inspired by the Shivanika et al. (2020) study [40], which used decoy ligand as a validator of the docking parameters and accompanied with positive control. Positive controls were selected based on the published research that confirms the chemical ligand binding (Table 2). The decoy substances were obtained from the DUD-E online server (http://dude.docking.org/) [41]. The docking parameters and process were identical to the previously mentioned process.
Table 2.
Ligands for positive control.
| No | Ligands name | Pubchem CID | Reference |
|---|---|---|---|
| Mpro (6LU7) | |||
| 1 | Caulerpin | 5326018 | [42] |
| 2 | Dexamethasone | 5743 | [43] |
| 3 | Dextromethorphan | 5360696 | [44] |
| 4 | Niclosamide | 4477 | [43] |
| 5 | Nitazoxanide | 41684 | [43] |
| 6 | Prednisolone | 5755 | [44] |
| S2 Protein (6LXT) | |||
| 1 | TGG | 73178 | [45] |
| 2 | Arbidol | 131411 | [45] |
| 3 | Luteolin | 5280445 | [45] |
| 4 | Quercetin | 5280343 | [45] |
2.5. ADME and toxicity analysis
Absorption, Distribution, Metabolism, and Excretion (ADME) and Toxic analysis are important in supplement or drug design. Some properties, including molecular weight (MW), rule of five (RO5), acceptable and donatable hydrogen bonds, topological polar surface area (TPSA), polarizability, and estimated solubility (ESOL) were analyzed for compositions selected after docking. We consider the following properties for intestinal absorption, solubility levels, and Blood-Brain Barrier (BBB) penetration levels. The ADME parameters and pharmacokinetic properties' prediction was done by the SwissADME webserver (http://www.swissadme.ch/)[46]. While the toxicity information for acute effects was derived from PubChem, and some of the compounds that have no information in PubChem predicted using the consensus method in Toxicity Estimation Software Tool (T.E.S.T) was done [47].
3. Results and discussion
In this study, we successfully identified the compound of commercial propolis, which Indonesian commonly consumes. The profile of propolis compounds is shown in Table 3. A total of 22 compounds of polyphenol have been found in propolis, classified as flavonoid, phenolic acid, and phenylpropanoid. The flavonoid structure was built by fifteen carbon skeletons forming two benzene rings linked with pyrane heterocyclic ring. Moreover, three subgroups of flavonoids are known, such as flavones, flavonones, and flavonol, based on their position of hydroxyl bonds, carbonyl bonds, and hydrogen bonds [48]. 15 compounds identified as flavonoid in propolis at retention time 7.15, 7.16, 10.86, 11.2, 12.48, 12.51, 13.5, 13.55, 14.12, 14.34, 14.77, 15.08, 16.45 min were 3′-Methoxydaidzein, 3′-Methoxydaidzin, genistin, xanthomicrol, 3′,5,6,7-Tetrahydroxy-4′-methoxyisoflavone, methylophiopogonone A, 3′-Deoxysappanol, 3′4′,7-Trihydroxyflavanone, moslosooflavone, luteolin, 2′,6′-Dihydroxy-4′- methoxy-dihydrochalcone, chrysoeriol, jaceosidin, (3R)-7,2′,3′-Trihydroxy-4′- Methoxyisoflavanone and neobavaisoflavone, respectively. All flavonoid compounds found in propolis were classified into flavones group except for 3′-Deoxysappanol, which falls into the category flavonols. The rest were classified into phenolic acid, i.e., isoferulic acid, dimethylcaffeic acid, cinnamic acid, caffeic acid, benzyl caffeate phenylpropanoid (2,5- Dimethyl-7-hydroxychromone and isoaloeresin D). Isoferulic acid, cinnamic acid, caffeic acid, and dimethylcaffeic acid were classified into hydroxycinnamic acid sub-group, while benzyl caffeate was hydroxybenzoic acid [49].
Table 3.
The identified compounds of propolis.
| Compound | PubChem ID | Formula | Structure | Molecular ion (m/z) |
|---|---|---|---|---|
| Cinnamic acid | 444539 | C9H8O2 | 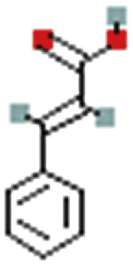 |
149.0597 |
| Caffeic acid | 689043 | C9H8O4 | 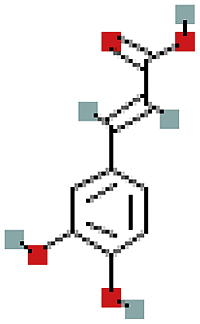 |
181.0498 |
| 2,5-Dimethyl-7-hydroxychromone | 5316891 | C11H10O3 | 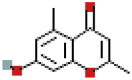 |
191.0710 |
| Isoferulic acid | 736186 | C10H10O4 | 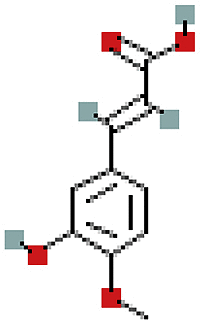 |
195.0648 |
| Dimethylcaffeic acid | 717531 | C11H12O4 | 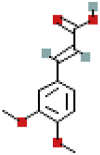 |
209.0816 |
| Benzyl caffeate | 5919576 | C16H14O4 | 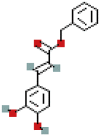 |
271.0960 |
| 3′4′,7-Trihydroxyflavanone | 3496769 | C15H12O5 | 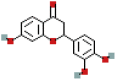 |
273.0768 |
| 2′,6′-Dihydroxy-4′ methoxydihydrochalcone | 169676 | C16H16O4 | 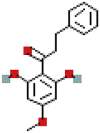 |
273.1150 |
| 3′-Methoxydaidzein | 5319422 | C16H12O5 | 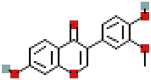 |
285.0751 |
| Luteolin | 5280445 | C15H10O6 | 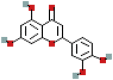 |
287.0553 |
| 3′-Deoxysappanol | 13846660 | C16H16O5 | 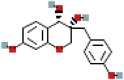 |
289.1081 |
| Moslosooflavone | 188316 | C17H14O5 |  |
299.0922 |
| Chrysoeriol | 5280666 | C16H12O6 |  |
301.0705 |
| (3R)-7,2′,3′-Trihydroxy-4′-methoxyisoflavanone | 14353662 | C16H14O6 |  |
303.0862 |
| 3′,5,6,7-Tetrahydroxy-4′-methoxyisoflavone | 10543410 | C16H12O7 |  |
317.0668 |
| Neobavaisoflavone | 5320053 | C20H18O4 | 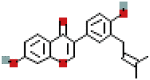 |
323.1287 |
| Jaceosidin | 5379096 | C17H14O7 | 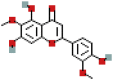 |
331.0820 |
| Methylophiopogonone_A | 10065830 | C19H16O6 | 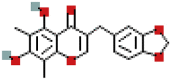 |
341.1028 |
| Xanthomicrol | 73207 | C18H16O7 | 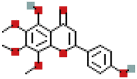 |
345.0969 |
| 3′-Methoxydaidzin | 10527347 | C22H22O10 |  |
447.1278 |
| Genistin | 5281377 | C21H20O10 |  |
455.0953 |
| Isoaloeresin D | 76332505 | C29H32O11 |  |
595.1584 |
All the identified propolis compounds were docked against two proteins target: Mpro and S2. Several positive controls were also docked to the protein target. Additionally, a subset of docking decoys was added to validate the docking parameters further. Our docking procedure was validated using decoy ligands similar to nelfinavir which were obtained from DUD-E online server.
There were 51 decoy compounds matched with nelfinavir and docked against the active site of Mpro (6LU7) and S2 protein (6LXT). This was done to enhance ligand enrichment, which is essential to assess the docking procedure and eliminate false positives. The result's binding energies remained between −8.9 kcal/mol to −5.7 kcal/mol 33 decoy compounds out of 51 showed higher binding energy compared to the Mpro-Nelfinavir complex. In addition, 27 decoy molecules showed no binding with 6LU7. Hence, this confirms the docking efficiency and protocol. When conducting this study, no other studies discussed the potential interactions between ligands and the SARS-CoV-2 S2 protein (6LXT). Therefore, we are the first to take this approach.
Previously, we have tried docking against 6LU7 for several FDA-approved anti-viral drugs as its positive controls; Caulerpin, Dexamethasone, Dextromethorphan, Niclosamide, Nitazoxanide, and Prednisolone and displayed docking scores of −8.6, −6.9, −6.2, −7.4, −6.6, and −7.1 kcal/mol, respectively. We have docked several compounds (TGG, Arbidol, Luteolin, and Quercetin) based on Xiu et al. (2020) [45] against 6LXT as its positive controls. The binding affinity is −8.3, −6.7, −7.7, and −7.6 kcal/mol for TGG, Arbidol, Luteolin, and Quercetin, respectively. However, those compounds were not the type of FDA-approved anti-viral drugs. Pravastatin belongs to statins which are lipid-lowering, anti-inflammatory, and potentially antiretroviral drugs. Furthermore, on several cellular pathways, pravastatin can block the virus-host interaction system, which can act as an effective compound as opposed to H1N1 infection [50].
Nelfinavir is identified as a potential inhibitor against COVID-19 main protease, based on binding free energy calculations using molecular mechanics with generalized born and surface area solvation (MM/GBSA) model and solvated interaction energy methods [51]. Another study stated that nelfinavir has the second-highest docking score (−7.9 kcal/mol) among favipiravir, lopinavir, simvastatin, rosuvastatin, pravastatin, pitavastatin, lovastatin, fluvastatin, and atorvastatin (−5.8, −7.9, −7.0, −7.7, −6.6, −8.2, −7.4, −7.7, and −6.8 kcal/mol) against protease enzyme of SARS-CoV-2 (PDB: 6LU7). The binding sites involved are GLN189, GLU166, and CYS145, which were available in our docking result [52].
The binding affinity of each compound towards the protein target is shown in Table 4. The free energy of binding and the binding constant (Ki) are each other related to support the docking score data. So, the more negative the free energy of binding, the more favorable the binding, whereas the lower the value of Ki, the more favorable the binding is. They showed promising binding affinities towards Mpro and S2 with variable free binding energies ranging from −5.4 to −7.7 kcal/mol, and −5.3 to −8.3 kcal/mol, respectively. The wide range of binding affinity against the targeted protein may explain the existence of hydrophobic aromatic rings and hydrophilic hydroxyl groups of flavonoids [48].
Table 4.
The predicted binding affinity energy of propolis compound against protein targets with its Ki value.
| Compound | Sample name | Interaction with Mpro |
Interaction with S2 |
||
|---|---|---|---|---|---|
| Binding affinity energy (kcal/mol) | Ki (M) | Binding affinity energy (kcal/mol) | Ki (M) | ||
| Cinnamic acid | P1 | −5.4 | 0.9978 | −5.3 | 0.9979 |
| Caffeic acid | P2 | −5.9 | 0.9976 | −5.5 | 0.9978 |
| 2,5-Dimethyl-7-hydroxychromone | P3 | −6.2 | 0.9975 | −6.1 | 0.9975 |
| Isoferulic acid | P4 | −5.7 | 0.9977 | −5.6 | 0.9977 |
| Dimethylcaffeic acid | P5 | −5.7 | 0.9977 | −5.7 | 0.9977 |
| Benzyl caffeate | P6 | −7.1 | 0.9971 | −6.5 | 0.9974 |
| 3′4′,7-Trihydroxyflavanone | P7 | −7.5 | 0.9970 | −7.6 | 0.9969 |
| 2′,6′-Dihydroxy-4′ methoxydihydrochalcone | P8 | −6.9 | 0.9972 | −6.7 | 0.9973 |
| 3′-Methoxydaidzein | P9 | −7.3 | 0.9971 | −7.6 | 0.9969 |
| Luteolin | P10 | −7.5 | 0.9970 | −7.7 | 0.9969 |
| 3′-Deoxysappanol | P11 | −7 | 0.9972 | −7 | 0.9972 |
| Moslosooflavone | P12 | −7.4 | 0.9970 | −7.4 | 0.9970 |
| Chrysoeriol | P13 | −7.3 | 0.9971 | −7.7 | 0.9969 |
| (3R)-7,2′,3′-Trihydroxy-4′-methoxyisoflavanone | P14 | −7.1 | 0.9971 | −7.5 | 0.9970 |
| 3′,5,6,7-Tetrahydroxy-4′-methoxyisoflavone | P15 | −7.6 | 0.9969 | −7.8 | 0.9969 |
| Neobavaisoflavone | P16 | −7.6 | 0.9969 | −8.1 | 0.9967 |
| Jaceosidin | P17 | −7.2 | 0.9971 | −7.3 | 0.9971 |
| Methylophiopogonone_A | P18 | −7.6 | 0.9969 | −8.2 | 0.9967 |
| Xanthomicrol | P19 | −7.1 | 0.9971 | −7 | 0.9972 |
| 3′-Methoxydaidzin | P20 | −7.7 | 0.9969 | −8.3 | 0.9967 |
| Genistin | P21 | −7.6 | 0.9969 | −8.3 | 0.9967 |
| Isoaloeresin D | P22 | −7.4 | 0.9970 | −7.8 | 0.9969 |
| Nelfinavir | Control for Mpro | −7.7 | 0.9969 | – | |
| Pravastatin | Control for S2 | – | −7.3 | 0.9971 | |
There are no compounds found that have lower binding energy needed to bind Mpro compared to nelfinavir. The binding affinity of nelfinavir was −7.7 kcal/mol, while the other reports showed the binding energy of −7 kcal/mol [53,54]. We found the 3′-Methoxydaidzin (P20) has a similar binding affinity with nelfinavir against Mpro. In contrast, many compounds have lower binding energy compared to pravastatin (−7.3 kcal/mol) when docked against S2 except for cinnamic acid (P1), caffeic acid (P2), 2,5-Dimethyl-7- hydroxychromone (P3), isoferulic acid (P4), dimethylcaffeic acid (P5), benzyl caffeate (P6) and 2′,6′-Dihydroxy-4′ methoxydihydrochalcone (P8).
Interestingly, four compounds have binding affinity lower than −8 kcal/mol, which are neobavaisoflavone (P16, -8.1 kcal/mol), methylophiopogonone A (P18, -8.2 kcal/mol), 3′-Methoxydaidzin (P20, -8.3 kcal/mol), and genistin (P21, -8.3 kcal/mol). Those four compounds could also be embedded into the same binding pocket of Mpro with a similar orientation (Fig. 2 (a)). While S2, except for P18 and P20, shared the same binding pocket (Fig. 2 (b)). Therefore, those compounds (P16, P18, P20, and P21) were used in the molecular docking simulation against S2 to visualize the binding interaction. For comparison purposes, the same compounds were examined the binding interaction with Mpro.
Fig. 2.

Binding orientation of propolis compounds with Mpro and S2 show they could be embedded into the same binding pocket of both protein receptors. (a) Binding orientation within Mpro, nelfinavir = red; neobavaisoflavone (P16) = yellow; methylophiopogonone A (P18) = blue; 3′-Methoxydaidzin (P20) = green; and genistin (P21) = orange. (b) Binding orientation within S2, pravastatin = red; neobavaisoflavone (P16) = yellow; methylophiopogonone A (P18) = blue; 3′-Methoxydaidzin (P20) = green; and genistin (P21) = orange. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 3 (a) shows the 2D visualization of the interaction between nelfinavir against Mpro. Two primary conventional hydrogen bonds were found in the nelfinavir, i.e., Glu A:166 and Gln A:189, whereby the residue Gln A:189 shares another bond as a carbon hydrogen bond. Hydrogen bonding gives a stabilization effect; thus, it plays an important role in biomolecular structures [55]. While other key residues interact through hydrophobic bonds (π-alkyl and alkyl) and electrostatic bonds (van der Waals). Hydrophobic bonds occur due to any interaction between hydrophobic amino acids with a polar solvent, i.e., water [56]. The orientation of nelfinavir and its orientation are shown in Fig. 3 (b).
Fig. 3.

(a) Detail bonding of molecular interaction between nelfinavir and Mpro in 2D visualization, showing the presence of conventional hydrogen, carbon-hydrogen, π-Alkyl, and van der Waals bonding. (b) The 3D interaction of nelfinavir at the binding pocket of Mpro.
Glu A:166 and Gln A:189 were also found in the P18 (Fig. 4 (b)), P20 (Fig. 4 (c)), and P21 (Fig. 4 (d)) through O – H interaction, however, the hydrogen bonding only found in Glu A:166, whereas Gln A:189 forms van der Waals bond with Mpro. Contrary, only one hydrogen bonding (Tyr A:54 and hydroxyl group) was found in P16, as shown in Fig. 4 (a), without the presence of Glu A:166 and Gln A:189. It implies that there is a certain electron delocalization or charge transfer [57].
Fig. 4.

2D and 3D molecular interaction between (a) P16, (b) P18, (c) P20, and (d) P21 against Mpro.
P16 has unique interactions with amino acid residues. As shown in Fig. 4(a), the presence of π interactions can stabilize the position of the P16 in the binding site. Sulfur functional groups in CYS A: 145 (C–S–H) form π-Sulfur and π-alkyl with aromatic ring ligands. The aromatic ring has a cloud of π-electron that can interact with a lone pair electron of S–H cysteine to form π-sulfur [58]. Furthermore, the σ-bond of C–S in cysteine may also interact with the π-electron cloud to form π-alkyl. Consequently, two interactions occurred; π-sulfur and π-alkyl; at key residue CYS A:145. A pair of key residue methionine; MET A:165 and MET A:49; form π-alkyl interactions with the same aromatic ring of P16. The two π-alkyls presence helps in the intercalation of ligands at the binding site [59]. The π-π T shape interaction gives a significant effect to binding energy because there are electrostatic, dispersion, induction, and exchange-repulsion contributions, while a π-electron cloud in an aromatic ring interacts with other π key residue in T shaped position [60]. The interaction of P16-Mpro is indicated by the interaction of the π system in an aromatic ring.
Hydrogen bond becomes a priority in determining the key amino acids, therefore, Glu A:166 might play an important role in intermolecular interaction between compound and Mpro. Hence, although P18, P20, and P21 have higher binding affinity than nelfinavir, these compounds are also candidates to inhibit the SARS-CoV-2 Mpro due to the existence of Glu A:166. A report showed a decrease of binding affinity energy to ~2 kcal/mol when replacing Glu166 with an alanine residue [54], indicates the importance of Glu A:166. Moreover, the other key residues were also found to form hydrogen bonding with Mpro, such as Ser A:144 and Leu A:141 in P18, Arg A:188, Thr A:190, Thr A:26 in P20, Thr A:190, Arg A:188, Thr A:26, Gln A:192, Asn A:142 in P21 as shown in Fig. 4.
Fig. 5 (a) shows the interaction between pravastatin and S2 in 2D and its orientation (Fig. 5 (b)). It showed six hydrogen bonding of key residues, i.e., Asn F:1194, Lys B:1191, Ser F:939, Arg E:1185, and Ser F:940. Among them, only Ser F:939 were found to be conserved in P16 and P21. In general, the hydrogen bonding in the P16, P18, P20 and P21 are less than 5, as shown in Fig. 6 (a) – (d). However, interestingly, the binding affinity is lower than pravastatin, as shown in Table 4. We believe the different binding affinity between pravastatin and P16, P18, P20, and P21 is due to the presence of π bonding.
Fig. 5.

(a) Detail bonding of molecular interaction between pravastatin and S2 in 2D visualization, showing the presence of conventional hydrogen, Alkyl, and van der Waals bonding. (b) The 3D interaction of pravastatin at the binding pocket of S2.
Fig. 6.

2D and 3D molecular interaction between (a) P16, (b) P18, (c) P20, and (d) P21 against S2.
π bond is non-covalent chemical bonds. For instance, π-alkyl (Lys B:1191) and π-cation (Arg E:1185) were found in P16, in addition to the hydrogen bonding of Asp B:1184 (Fig. 6 (a)). The aromatic ring of P16 binds to arginine in its position and forms of π-cation between the nitrogen (of the positively charged guanidine) and the π electron cloud of its aromatic ring. Electrostatic interactions dominate these bonds. Arginine itself is an amino acid with a positively charged guanidine side chain [61]. A report has reported that the non-covalent interaction of guanidine (arginine) -benzene iso-surface showed that the region of π-cation attraction is due to NH polarity, and the average π-cation strength of arginine has been reported to be −2.9 ± 1.4 kcal/mol [62]. The π-alkyl interactions are dominated by the dispersion strength between π and –CH groups of the amino acids, but these bonds are weaker than π-cations. It has been reported that CH4-π has interaction energy of-1.3 kcal/mol [60]. The hydrogen carbon bond is also observed in Fig. 6 (a). The carbon-hydrogen bond (CH ⋯ O) is weaker than the conventional hydrogen bond because CH O's average distance is longer than the conventional hydrogen bond (N–H ⋯ O, N–H ⋯ N, O–H ⋯ O). However, it is known that the carbon-hydrogen C–H O bond has an important role in the molecular recognition process [61,62]. The π-donor H is the interaction between the hydrogen bond and the π aromatic ring ligand system and has a significant effect on stabilizing the ligands' position at the binding site. All P16 aromatic rings were observed to form π interactions with residual amino acids, where these interactions have an important contribution to the binding energy.
Asp B:1199, Asn F:928, Gln F:935 formed hydrogen bonding with S2, while Glu B:1195 and Leu F:1197 was π-anion and π-σ bonding, respectively, as shown in Fig. 6 (b). The π-anion is defined as the electrostatic interaction between anions positioned above the aromatic ring where the binding energy is dominated by the electrostatic anion-induced polarity contribution [63,64]. Therefore, the side-chain carboxylic acid functional groups of glutamic acid interact with the π-electron cloud on the ligands' aromatic ring. The same aromatic ring is attached to the σ system in the hydrophobic side chain of leucine to form π-σ. It is thought that the two π interactions stabilize the conformational position of the ligands, while the other ligand side chains bond to the hydrogen at the binding site.
P20 has quite a lot of hydrogen bonding among the other compounds, which are Leu F:1200, Ile F:1198, Asn F:928, and Glu B:1195, as shown in Fig. 6 (c). Furthermore, the presence of π-bindings was also observed, such as π-cation, π-anion, π-σ, and π-alkyl. The lower binding affinity of P20 more likely due to the double bonding of key residue, for instance, Glu B:1195 interacts in hydrogen and π-anion bonding, while Asn F:928 forms hydrogen and π-σ bonding. The π-anion has binding energy that is proportional to π-cation, besides π-σ and π-alkyl have strong dispersion energy [60,65]. It is predicted that the π-interactions present a significant contribution to the ligands' binding energy to their receptors. Therefore, the binding affinity of P20 is −8.3 kcal/mol, which is less than 1 kcal/mol compared to pravastatin.
It should be noted that P21 did not have any hydrogen bonding, as shown in Fig. 6 (d), however, the binding affinity of P21 is similar to P20, i.e., −8.3 kcal/mol. It believes the presence of amide-π stacked bonding (Asn B:1187) and π-alkyl bonding (Lys B:1191 and Ala F:1190) greatly influence the binding affinity of P21. An amide-π bond is formed between π surface of the amide bond, which overlaps with the π on the surface of the aromatic ring ligand [62]. This interaction is proportional to the hydrogen bond's strength based on the contribution of electrostatic, induction energy, and dispersion energy to the bond energy. It has been reported that almost all amide-π-accumulation interactions occur between the protein amide backbone group and the ligand aromatic ring [66]. The π-alkyls also contribute to energy binding through their dispersion forces. It has been reported that CH4-π has an interaction energy of −1.3 kcal/mol [60].
The distribution of π -electron of ligands can interact with positive and negative charges of the amino acid side chain, also hydrophobic side chains, the π system as well as amida bonds, therefore forming π -cation, π -anion, π -σ, π- π interactions, and π -amide respectively. This bonding occurred when the ligand is face-to-face or face to edge position with key residue at the binding site. Apart from hydrogen, pi-bonds are important in biological structure [59,62,67].
Several studies have been reported that the amide-π interaction is important in structural biology [[68], [69], [70], [71], [72], [73]] [[68], [69], [70], [71], [72], [73]] [[68], [69], [70], [71], [72], [73]]. Such as in acetylated lysine (KAc), which showed highly conserved aromatic residues in the bromodomain [72], while bromodomain is a protein domain in most acyltransferases and associated with chromatin structure regulation. Also, the amide-π interaction exists in human deoxyhemoglobin bind to its drug [74], also for the binding of other ligands to enzymes [75], furthermore, to be a selective binding for an organic solution model system.
The occurrence of π-bonding may cause its enthalpy/entropy on free binding energy be minimal. This interaction is unaffected by the solvation/desolvation process, which breaks water-mediated bonds and residue position rearrangement. π interaction has various binding confirmation which explains why there is various type of π interaction was detected between propolis' compound and the targeted protein (Mpro and S2), such as face-to-face (π-σ, π-cation, and π-π), parallel-displace and edge-to face (both occurring in π-π) [76].
Understanding hydrophobic interaction at molecular length scales may support a better understanding of protein folding, which remains unclear [77,78]. Hydrophobic amino acids are including Ala, Gly, Ile, Leu, Pro, Val, and Phe, which have hydrocarbon sidechains due to their non-polar chemistry [[79], [80], [81]] [[79], [80], [81]] [[79], [80], [81]]. In general, nelfinavir – Mpro interaction involves more hydrophilic amino acids (Glu, Gln, Cys, His and Asp) rather than hydrophobic amino acids (Leu and Val) also amphipathic amino acid (Met), which makes this bonding more hydrophilic than hydrophobic. The four potential inhibitors (P16, P18, P20, and P21) had a similar hydrophilic profile as nelfinavir - Mpro. Whereas pravastatin – S2 also interaction involves hydrophilic amino acids (Glu, Gln, Asp, Asn, Ser, and Arg) rather than hydrophobic amino acids (Ala) also amphipathic amino acid (Lys). This bonding is more hydrophilic rather than hydrophobic, also then nelfinavir – Mpro hydrophilic interaction. Among the four potential compounds (P16, P18, P20, and P21), P21 has the highest hydrophilic profile similarity with pravastatin – S2 interaction. These profiles crucially may affect herbal compound's ability to dissolve in the gastrointestinal fluids [82]. It is further explained in Table 5, which shows each compound's lipophilicity was according to XLOGP3. However, how the hydrogen bond at the identified ligand-protein interface relates to the hydrophobic interactions, which may increase the binding affinity has not been properly elucidated.
Table 5.
ADME properties prediction of all propolis’ compounds.
| Sample | MWa (g/mol) |
RO5b | #H-bond acceptorsc |
#H-bond donorsd |
TPSAe (Åb) |
XLOGP3f | ESOL (mol/l)g |
log Kp (cm/s)h |
LD50 (mg/kg)i |
|---|---|---|---|---|---|---|---|---|---|
| P16 | 322.35 | 0 | 4 | 2 | 70.67 | 4.40 | 1.24E-05 | −5.14 | 768.89 |
| P18 | 340.33 | 0 | 6 | 2 | 89.13 | 3.86 | 1.89E-05 | −5.64 | 206 |
| P20 | 446.40 | 0 | 10 | 5 | 159.05 | 0.64 | 6,98E-04 | −7,05 | 934.65 |
| P21 | 432.38 | 1 | 10 | 6 | 170.05 | 0.86 | 6.60E-04 | −8.33 | 2000 |
Molecular Weight (acceptable range 130–725 gm/mol).
Rule of five Lipinski rules.
Acceptable H-bonds (acceptable range 2–20).
Donatable H-bonds (acceptable range 0–6).
Topological Polar Surface Area (TPSA < 140 Åb good intestinal absorption) (TPSA < 70 Åb good brain penetration).
Lipophilicity descriptor (Acceptable range -2 – 6.5).
Estimated solubility in water.
Skin permeant (Acceptable range (−8) - (−1)).
Predicted Oral Rat LD50 (Acute Effects).
Those potential compounds of the propolis (P16, P18, P20, and P21) were evaluated for their ADMET physicochemical properties prediction using SwissADME and toxicity using T.E.S.T, as shown in Table 5. P21 has the highest MW among the compounds, while P16 is the lowest. Indeed, the MW of all compounds within the acceptable range. All of these compounds obey Lipinski rule, except for P21, as indicated by the RO5 value higher than 0. For the H-bond acceptor and donor, all compounds showed the value in the acceptable range, even though H-bond donor P21 is in the edge, i.e., 6. Each compound's pharmacokinetics was also evaluated, which reveals P16 and P18 may be absorbed by gastrointestinal organ due to topological polar surface area (TPSA) is less than 140 Å2. The lipophilicity of compound was according to XLOGP3 value and showing its polarity. The lower the value, the higher the polarity. Only P21 was out of the acceptable range of skin permeant (log Kp) with a value of −8.33 cm/s. Toxicity analysis was done to predict each compound's LD50 based on Hodge and Terner (1995). P16, P20, and P21 belong to the slightly toxic drug category (500–5000 mg/kg) with LD50 of 768.89, 934.65 and 2000 mg/kg, respectively. Whereby P18 is in the moderately toxic drug category (50–500 mg/kg) with LD50 of 206 mg/kg.
To the best of the authors' knowledge, this study is the first to report the potential of 3′-Methoxydaidzin, genistin, methylophiopogonone A, and neobavaisoflavone to inhibit virus infection. 3′-Methoxydaidzin of propolis as the best binding ligand predicted for both protein targets; SARS-CoV-2 Mpro and S2. Blocking its spike protein may prevent SARS-CoV-2 bonding with the ACE2 receptor. Moreover, 3′-Methoxydaidzin can inhibit the extensive proteolytic activity of viral polyproteins [[83], [84], [85]] [[83], [84], [85]] [[83], [84], [85]]. 3′-Methoxydaidzin has no profound information except for its neuroprotective effect [86]. Interestingly, this chemical compound is extracted from Radix puerariae or kudzu plant commonly used as traditional Chinese medicine to treat diarrhea, acute dysentery, deafness, and cardiovascular diseases [87]. Genistin has evaluated to be anti-microbial, anti-cancer, anti-oxidative, cardioprotector, anti-apoptotic, neuroprotector, hepatoprotective [88], and anti-HIV [89]. Genistin is also found in kudzu plants, and a previous study showed that this compound could block the tyrosinase enzyme in its active site [[87], [88], [89], [90]] [[87], [88], [89], [90]] [[87], [88], [89], [90]].
On the other hand, methylophiopogonone A presents an anti-inflammatory activity [91,92]. Some of the publications about methylophiopogonone A are related to antioxidant activity in the form of herbal medicine supplementation [[87], [88], [89], [90]] [[87], [88], [89], [90]] [[87], [88], [89], [90]]. Lipinski [93] indicates a higher bioavailability is the compound's character related to lower molecular weight, lower H-bond capacity, and low lipophilicity. Furthermore, neoblavaisoflavone, methylophiopogonone A, 3′-Methoxydaidzin, and genistin exhibit the potential to be good for oral bioavailability. The median lethal dose (LD50) is a standard measurement for acute toxicity that predicts the adverse effect of substances in a short period after exposure [94]. No compounds of propolis in our study indicate highly toxic or extremely toxic. Propolis and its constituent are well tolerated nontoxic based on clinical and research reports [17,95,96].
4. Conclusion
In this study, we examined the propolis compounds to inhibit the SARS-CoV-2 Mpro and S2 using a molecular docking simulation. The propolis examined in this study has 22 compounds, which can be categorized as flavonoid, phenolic acid, and phenylpropanoid. The candidate to inhibit the SARS-CoV-2 Mpro was methylophiopogonone A, 3′-Methoxydaidzin, and genistin. Although the binding affinity energy of those compounds is not lower than nelfinavir (control), the existence of GLU A:166 in those compounds is essential to inhibit SARS-CoV-2 Mpro. In addition to GLU A:166, many other hydrogen bonds were observed in those compounds. For the blocking of SARS-CoV-2 S2, four propolis compounds (neoblavaisoflavone, methylophiopogonone A, 3′-Methoxydaidzin, and genistin) have binding affinity energy lower than the pravastatin (control), makes those compounds a candidate to inhibit SARS-CoV-2 S2. Although only neoblavaisoflavone and methylophiopogonone A has the ability to absorbed by gastrointestinal organ, the main purpose of propolis is used as a supplement instead of a drug. Moreover, the finding on methylophiopogonone A, 3′-Methoxydaidzin, and genistin as a candidate for anti-viral reported for first time in this study. Thus, those compounds can be further explored and could be used as a parent backbone molecule to develop new supplementation for preventing SARS-CoV-2 infections during COVID-19 outbreaks.
CRediT author statement
Azza Hanif Harisna: Conceptualization, Methodology, Software, Writing - Original Draft. Rizky Nurdiansyah: Conceptualization, Methodology, Software, Writing - Original Draft. Putri Hawa Syaifie: Formal analysis, Investigation, Writing - Original Draft, Project administration. Dwi Wahyu Nugroho: Validation, Formal analysis, Investigation, Writing - Original Draft. Kurniawan Eko Saputro: Formal analysis, Investigation, Data Curation, Writing - Original Draft. Firdayani: Formal analysis. Chandra Dwi Prakoso: Formal analysis. Nurul Taufiqu Rochman: Resources. Nurwenda Novan Maulana: Resources. Alfian Noviyanto: Visualization, Supervision, Writing - Review & Editing. Etik Mardliyati: Resources.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
No funding used to support the research. We thank to PT Nano Herbaltama Internasional for providing the sample of propolis.
References
- 1.Livingston E., Bucher K., Rekito A. Coronavirus disease 2019 and influenza 2019-2020. JAMA, J. Am. Med. Assoc. 2020;323:1122. doi: 10.1001/jama.2020.2633. [DOI] [PubMed] [Google Scholar]
- 2.Zhou P., Lou Yang X., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Di Jiang R., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020;94:1–9. doi: 10.1128/jvi.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E.C., Zhang Y.-Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 7.Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. Chemrxiv.Org. 2020:1–8. doi: 10.26434/chemrxiv.11860011.v2. [DOI] [Google Scholar]
- 8.Andrade B., Ghosh P., Barth D., Tiwari S., Silva R.J.S., de A Soares W.R., Melo T.S., Freitas A. dos S., González-Grande P., Palmeira L.S., Alcantara L.C.J., Giovanetti M., Góes-Neto A., de C Azevedo V.A. 2020. Computational Screening for Potential Drug Candidates against SARS-CoV-2 Main Protease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez M.A. Compounds with therapeutic potential against novel respiratory 2019 coronavirus. Antimicrob. Agents Chemother. 2020;64:1–7. doi: 10.1128/AAC.00399-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castaldo S., Capasso F. Propolis, an old remedy used in modern medicine. Fitoterapia. 2002;73:1–6. doi: 10.1016/S0367-326X(02)00185-5. [DOI] [PubMed] [Google Scholar]
- 11.Zheng Y., Wu Y., Chen X., Jiang X., Wang K., Hu F. Chinese propolis exerts anti-proliferation effects in human melanoma cells by targeting NLRP1 inflammatory pathway, inducing apoptosis, cell cycle arrest, and autophagy. Nutrients. 2018;10 doi: 10.3390/nu10091170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer G., Conceição F.R., Leite F.P.L., Dummer L.A., Vargas G.D.A., de O Hübner S., Dellagostin O.A., Paulino N., Paulino A.S., Vidor T. Immunomodulation produced by a green propolis extract on humoral and cellular responses of mice immunized with SuHV-1. Vaccine. 2007;25:1250–1256. doi: 10.1016/j.vaccine.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan G.C.F., Cheung K.W., Sze D.M.Y. The immunomodulatory and anticancer properties of propolis. Clin. Rev. Allergy Immunol. 2013;44:262–273. doi: 10.1007/s12016-012-8322-2. [DOI] [PubMed] [Google Scholar]
- 15.Monzote L., Cuesta-Rubio O., Fernandez M.C., Hernandez I.M., Fraga J., Pérez K., Kerstens M., Maes L., Cos P. In vitro antimicrobial assessment of Cuban propolis extracts. Mem. Inst. Oswaldo Cruz. 2012;107:978–984. doi: 10.1590/S0074-02762012000800003. [DOI] [PubMed] [Google Scholar]
- 16.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braakhuis A. Evidence on the health benefits of supplemental propolis. Nutrients. 2019;11 doi: 10.3390/nu11112705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bankova V. Recent trends and important developments in propolis research, Evidence-Based Complement. Alternative Med. 2005;2:29–32. doi: 10.1093/ecam/neh059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crane E. second ed. 2009. Encyclopedia of Insects. [Google Scholar]
- 20.Sforcin J.M. Propolis and the immune system: a review. J. Ethnopharmacol. 2007;113:1–14. doi: 10.1016/j.jep.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Orsatti C.L., Missima F., Pagliarone A.C., Bachiega T.F., Búfalo M.C., A J.P., Jr., Sforcin J.M. Propolis immunomodulatory action in Vivo on toll-like receptors 2 and 4 expression and on pro-inflammatory cytokines production in mice. Phyther. Res. 2010:1141–1146. doi: 10.1002/ptr.3086. [DOI] [PubMed] [Google Scholar]
- 22.Sforcin J.M. Biological properties and therapeutic applications of propolis. Phyther. Res. 2016;30:894–905. doi: 10.1002/ptr.5605. [DOI] [PubMed] [Google Scholar]
- 23.Vynograd N., Vynograd I., Sosnowski Z. A comparative multi-centre study of the efficacy of propolis, acyclovir and placebo in the treatment of genital herpes (HSV) Phytomedicine. 2000;7:1–6. doi: 10.1016/S0944-7113(00)80014-8. [DOI] [PubMed] [Google Scholar]
- 24.Ito J., Chang F.R., Wang H.K., Park Y.K., Ikegaki M., Kilgore N., Lee K.H. Anti-AIDS agents. 48. Anti-HIV activity of moronic acid derivatives and the new melliferone-related triterpenoid isolated from Brazilian propolis. J. Nat. Prod. 2001;64:1278–1281. doi: 10.1021/np010211x. [DOI] [PubMed] [Google Scholar]
- 25.Nolkemper S., Reichling J., Sensch K.H., Schnitzler P. Mechanism of herpes simplex virus type 2 suppression by propolis extracts. Phytomedicine. 2010;17:132–138. doi: 10.1016/j.phymed.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Gekker G., Hu S., Spivak M., Lokensgard J.R., Peterson P.K. Anti-HIV-1 activity of propolis in CD4+ lymphocyte and microglial cell cultures. J. Ethnopharmacol. 2005;102:158–163. doi: 10.1016/j.jep.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 27.Búfalo M.C., Figueiredo A.S., De Sousa J.P.B., Candeias J.M.G., Bastos J.K., Sforcin J.M. Anti-poliovirus activity of Baccharis dracunculifolia and propolis by cell viability determination and real-time PCR. J. Appl. Microbiol. 2009;107:1669–1680. doi: 10.1111/j.1365-2672.2009.04354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adem S., Eyupoglu V., Sarfraz I., Rasul A., Ali M. 2020. Identification of Potent COVID-19 Main Protease (Mpro) Inhibitors from Natural Polyphenols: an in Silico Strategy Unveils a Hope against CORONA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khaerunnisa S., Kurniawan H., Awaluddin R., Suhartati S. Potential inhibitor of COVID-19 main protease ( M pro ) from several medicinal plant compounds by molecular docking study. Preprints. 2020:1–14. doi: 10.20944/preprints202003.0226.v1. [DOI] [Google Scholar]
- 30.Yan Y.-M., Shen X., Cao Y.-K., Zhang J.-J., Wang Y., Cheng Y.-X. Discovery of anti-2019-nCoV agents from 38 Chinese patent drugs toward respiratory diseases via docking screening. Preprints. 2020 doi: 10.20944/preprints202002.0254.v2. [DOI] [Google Scholar]
- 31.Zhang H., Penninger J.M., Li Y., Zhong N., Slutsky A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y., Zhao Y., Li Y., Wang X., Peng Z. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA, J. Am. Med. Assoc. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan W., Ni Z., Hu Y., Liang W., Ou C., He J., Liu L., Shan H., Lei C., Hui D.S.C., Du B., jua Lan, Zhong N. 2020. Clinical Characteristics of 2019 Novel Coronavirus Infection in China. [Google Scholar]
- 35.Dassault Systèmes BIOVIA . 2016. BIOVIA Discovery Studio Visualizer. [Google Scholar]
- 36.The PyMOL Molecular Graphics System, The PyMOL Molecular Graphics System Version 2.0 Schrödinger, (n.d.).
- 37.Lovell S.C., Davis I.W., Adrendall W.B., de Bakker P.I.W., Word J.M., Prisant M.G., Richardson J.S., Richardson D.C. Structure validation by C alpha geomF. In: Altschul S., Gish W., Miller W.W., Myers E., Lipman D.J., editors. vol. 50. 2003. pp. 437–450. ((1990). Basic Local Alignment Search Tool. Journal of Molecular Biology.Etry: Phi,psi and C Beta Deviation, Proteins-Structure Funct. Genet). [DOI] [PubMed] [Google Scholar]
- 38.Tian W., Chen C., Lei X., Zhao J., Liang J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trott A.J., Olson O. Autodock vina: improving the speed and accuracy of docking. J. Comput. Chem. 2019;31:455–461. doi: 10.1002/jcc.21334.AutoDock. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.C S., S D.K., Ragunathan V., Tiwari P., A S., Brindha Devi B.D. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020:1–27. doi: 10.1080/07391102.2020.1815584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmed S.A., Abdelrheem D.A., El-Mageed H.R.A., Mohamed H.S., Rahman A.A., Elsayed K.N.M., Ahmed S.A. Destabilizing the structural integrity of COVID-19 by caulerpin and its derivatives along with some anti-viral drugs: an in silico approaches for a combination therapy. Struct. Chem. 2020;31:2391–2412. doi: 10.1007/s11224-020-01586-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mostafa A., Kandeil A., Elshaier Y.A.M.M., Kutkat O., Moatasim Y., Rashad A.A., Shehata M., Gomaa M.R., Mahrous N., Mahmoud S.H., Gaballah M., Abbas H., El Taweel A., Kayed A.E., Kamel M.N., El Sayes M., Mahmoud D.B., El-Shesheny R., Kayali G., Ali M.A. Fda-approved drugs with potent in vitro anti-viral activity against severe acute respiratory syndrome coronavirus 2. Pharmaceuticals. 2020;13:1–24. doi: 10.3390/ph13120443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar I., Sen A. In silico screening predicts common cold drug Dextromethorphan along with Prednisolone and Dexamethasone can be effective against novel Coronavirus disease (COVID-19) J. Biomol. Struct. Dyn. 2020:1–5. doi: 10.1080/07391102.2020.1850528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J. Med. Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin T. 2016. Toxicity Estimation Software Tool (TEST) [Google Scholar]
- 48.Kumar S., Pandey A.K. Chemistry and biological activities of flavonoids: an overview. Sci. World J. 2013 doi: 10.1155/2013/162750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J., Du C., Beaman H.T., Monroe M.B.B. Characterization of phenolic acid antimicrobial and antioxidant structure–property relationships. Pharmaceutics. 2020;12 doi: 10.3390/pharmaceutics12050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehrbod P., Omar A.R., Hair-Bejo M., Haghani A., Ideris A. Mechanisms of action and efficacy of statins against influenza. BioMed Res. Int. 2014:11–14. doi: 10.1155/2014/872370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mothay D., Ramesh K.V. Binding site analysis of potential protease inhibitors of COVID-19 using AutoDock. VirusDisease. 2020;31:194–199. doi: 10.1007/s13337-020-00585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiner Ž., Hatamipour M., Banach M., Pirro M., Al-Rasadi K., Jamialahmadi T., Radenkovic D., Montecucco F., Sahebkar A. Statins and the Covid-19 main protease: in silico evidence on direct interaction. Arch. Med. Sci. 2020;16:490–496. doi: 10.5114/aoms.2020.94655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huynh T., Wang H., Luan B. In silico exploration of the molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2’s main protease. J. Phys. Chem. Lett. 2020;11:4413–4420. doi: 10.1021/acs.jpclett.0c00994. [DOI] [PubMed] [Google Scholar]
- 54.Ngo S.T., Quynh Anh Pham N., Thi Le L., Pham D.-H., Vu V.V. Computational determination of potential inhibitors of SARS-CoV-2 main protease. J. Chem. Inf. Model. 2020 doi: 10.1021/acs.jcim.0c00491. [DOI] [PubMed] [Google Scholar]
- 55.Jeffrey G., Saenger W. Springer-Verlag; Berlin: 1991. Hydrogen Bonding in Biological Structures. [Google Scholar]
- 56.Meyer E.E., Rosenberg K.J., Israelachvili J. Recent progress in understanding hydrophobic interactions. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15739–15746. doi: 10.1073/pnas.0606422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakhaei E., Nowroozi A., Ravari F. The hydrogen-bonded complexes of the 5-fluorouracil with the DNA purine bases: a comprehensive quantum chemical study. Struct. Chem. 2018;29:69–80. doi: 10.1007/s11224-017-1001-4. [DOI] [Google Scholar]
- 58.Daeffler K.N.M., Lester H.A., Dougherty D.A. Functionally important aromatic-aromatic and sulfur-π interactions in the D2 dopamine receptor. J. Am. Chem. Soc. 2012;134:14890–14896. doi: 10.1021/ja304560x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arthur D.E., Uzairu A. Molecular docking studies on the interaction of NCI anticancer analogues with human Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit. J. King Saud Univ. Sci. 2019;31:1151–1166. doi: 10.1016/j.jksus.2019.01.011. [DOI] [Google Scholar]
- 60.Sinnokrot M.O., Sherrill C.D. Substituent effects in π-π interactions: sandwich and t-shaped configurations. J. Am. Chem. Soc. 2004;126:7690–7697. doi: 10.1021/ja049434a. [DOI] [PubMed] [Google Scholar]
- 61.Kumar K., Woo S.M., Siu T., Cortopassi W.A., Duarte F., Paton R.S. Cation-π interactions in protein-ligand binding: theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018;9:2655–2665. doi: 10.1039/c7sc04905f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferreira De Freitas R., Schapira M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm. 2017;8:1970–1981. doi: 10.1039/c7md00381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watt M.M., Collins M.S., Johnson D.W. Ion-pi interactions in ligand design for anions and main group cations. Acc. Chem. Res. 2013;46:955–966. doi: 10.1021/ar300100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schottel B.L., Chifotides H.T., Dunbar K.R. Anion-Π interactions. Chem. Soc. Rev. 2008;37:68–83. doi: 10.1039/b614208g. [DOI] [PubMed] [Google Scholar]
- 65.Quiñonero D., Garau C., Frontera A., Ballester P., Costa A., Deyà P.M. Structure and binding energy of anion-π and cation-π complexes: a comparison of MP2, RI-MP2, DFT, and DF-DFT methods. J. Phys. Chem. 2005;109:4632–4637. doi: 10.1021/jp044616c. [DOI] [PubMed] [Google Scholar]
- 66.Ottiger P., Pfaffen C., Leist R., Leutwyler S., Bachorz R.A., Klopper W. Strong N-H. . .π hydrogen bonding in amide-benzene interactions. J. Phys. Chem. B. 2009;113:2937–2943. doi: 10.1021/jp8110474. [DOI] [PubMed] [Google Scholar]
- 67.Gallivan J.P., Dougherty D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U.S.A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meyer E.A., Castellano R.K., Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem. Int. Ed. Engl. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 69.Burley S.K., Petsko G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 70.Steiner T., Koellner G. Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J. Mol. Biol. 2001;305:535–557. doi: 10.1006/jmbi.2000.4301. [DOI] [PubMed] [Google Scholar]
- 71.Duan G., Smith V., Weaver D. Characteri- zation of aromatic- amide (side-chain) interactions in proteins through systematic ab initio calcula- tions and data mining analyses. J. Phys. Chem. 2000;104:4521–4532. [Google Scholar]
- 72.Hughes R.M., Waters M.L. Effects of lysine acetylation in a β-hairpin peptide: comparison of an Amide−π and a Cation−π interaction. J. Am. Chem. Soc. 2006;128:13586–13591. doi: 10.1021/ja0648460. [DOI] [PubMed] [Google Scholar]
- 73.Tóth G., Murphy R.F., Lovas S. Investigation of aromatic-backbone amide interactions in the model peptide Acetyl-Phe-Gly-Gly-N-Methyl amide using molecular dynamics simulations and protein database search. J. Am. Chem. Soc. 2001;123:11782–11790. doi: 10.1021/ja011245u. [DOI] [PubMed] [Google Scholar]
- 74.MichaelLevitt M. F.Perutz. Aromatic rings act as hydrogen bond acceptors. J. Mol. Biol. 1988;201:751–754. doi: 10.1016/0022-2836(88)90471-8. [DOI] [PubMed] [Google Scholar]
- 75.Perutz M.F. The role of aromatic rings as hydrogen-bond acceptors in molecular recognition. Phil. Trans. Phys. Sci. Eng. 1993;345:105–112. [Google Scholar]
- 76.Bernaldez M.J.A., Billones J.B., Magpantay A. In silico analysis of binding interactions between GSK983 and human DHODH through docking and molecular dynamics. AIP Conf. Proc. 2018;2045 doi: 10.1063/1.5080886. [DOI] [Google Scholar]
- 77.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 78.Tanford C. How protein chemists learned about the hydrophobic factor. Protein Sci. 1997;6:1358–1366. doi: 10.1002/pro.5560060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dill K. The meaning of hydrophobicity. Science (80-. ) 1990;250:297–298. doi: 10.1126/science.2218535. [DOI] [PubMed] [Google Scholar]
- 80.Dill K.A., Ozkan S.B., Shell M.S., Weikl T.R. The protein holding problem. Annu. Rev. Biophys. 2008:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Honig B., Yang A.S. Free energy balance in protein folding. Adv. Protein Chem. 1995;46:27–58. doi: 10.1016/S0065-3233(08)60331-9. [DOI] [PubMed] [Google Scholar]
- 82.Wenlock M.C., Austin R.P., Barton P., Davis A.M., Leeson P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003;46:1250–1256. doi: 10.1021/jm021053p. [DOI] [PubMed] [Google Scholar]
- 83.Ramajayam R., Tan K.P., Liang P.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011;39:1371–1375. doi: 10.1042/BST0391371. [DOI] [PubMed] [Google Scholar]
- 84.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles'’ heel": current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dai W., Zhang B., Su H., Li J., Zhao Y., Xie X., Jin Z., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.-K., Xu Y., Yang H., Liu H. Structure-based design of anti-viral drug candidates targeting the SARS-CoV-2 main protease. Science (80-. ) 2020;4489:eabb4489. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo M., Zhang Y.F., Wang Y., Zhao X.-P. Development of a rapid screening method for discovering neuroprotective components from traditional Chinese medicine. Zhongguo Zhongyao Zazhi. 2013;38:1581–1584. [PubMed] [Google Scholar]
- 87.Zhang Z., Lam T., Zuo Z. 2013. Edible Medicinal and Non-medicinal Plants: Volume 10, Modified Stems, Roots, Bulbs, Zhen Zhang, MSc1, Tai‐Ning Lam, PharmD, PhD1, Zhong Zuo, PhD1. [Google Scholar]
- 88.Islam A., Islam S., Uddin N., Hasan I., Akanda R. The potential health benefits of the isoflavone glycoside genistin. Arch Pharm. Res. (Seoul) 2020;Vol. 43:395–408. doi: 10.1007/s12272-020-01233-2. [DOI] [PubMed] [Google Scholar]
- 89.Sauter D., Schwarz S., Wang K., Zhang R., Sun B., Schwarz W. Genistein as antiviral drug against HIV ion channel. Planta Med. 2014;80:682–687. doi: 10.1055/s-0034-1368583. [DOI] [PubMed] [Google Scholar]
- 90.Wu Q.-L., Yang Y.-H., Simon J. Chemical profiling and quantification of isoflavone phytoestrogens in kudzu using LC/UV/MSD. Am. J. Anal. Chem. 2011;2:665–674. doi: 10.4236/ajac.2011.26076. [DOI] [Google Scholar]
- 91.Zhao J.W., Chen D.S., Deng C.S., Wang Q., Zhu W., Lin L. Evaluation of anti-inflammatory activity of compounds isolated from the rhizome of Ophiopogon japonicas. BMC Compl. Alternative Med. 2017;17:1–12. doi: 10.1186/s12906-016-1539-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li N., Zhang J.Y., Zeng K.W., Zhang L., Che Y.Y., Tu P.F. Anti-inflammatory homoisoflavonoids from the tuberous roots of Ophiopogon japonicus. Fitoterapia. 2012;83:1042–1045. doi: 10.1016/j.fitote.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 93.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 94.Erhirhie E.O., Ihekwereme C.P., Ilodigwe E.E. Advances in acute toxicity testing: strengths, weaknesses and regulatory acceptance. Interdiscipl. Toxicol. 2018;11:5–12. doi: 10.2478/intox-2018-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bazmandegan G., Boroushaki M.T., Shamsizadeh A., Ayoobi F., Hakimizadeh E., Allahtavakoli M. Brown propolis attenuates cerebral ischemia-induced oxidative damage via affecting antioxidant enzyme system in mice. Biomed. Pharmacother. 2017;85:503–510. doi: 10.1016/j.biopha.2016.11.057. [DOI] [PubMed] [Google Scholar]
- 96.Cornara L., Biagi M., Xiao J., Burlando B. Therapeutic properties of bioactive compounds from different honeybee products. Front. Pharmacol. 2017;8:1–20. doi: 10.3389/fphar.2017.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]