

Abstract
Toxoplasma gondii is an obligate intracellular parasite and replicates inside a parasitophorous vacuole (PV) within the host cell. The membrane of the PV (PVM) contains pores that permits for equilibration of ions and small molecules between the host cytosol and the PV lumen. Ca2+ signaling is universal and both T. gondii and its mammalian host cell utilize Ca2+ signals to stimulate diverse cellular functions. Egress of T. gondii from host cells is an essential step for the infection cycle of T. gondii and a cytosolic Ca2+ increase initiates a Ca2+ signaling cascade that culminates in the stimulation of motility and egress. In this work, we demonstrate that intracellular T. gondii tachyzoites are able to take up Ca2+ from the host cytoplasm during host signaling events. Both intracellular and extracellular Ca2+ sources are important in reaching a threshold of cytosolic Ca2+ needed for successful egress. Two peaks of Ca2+ were observed in single parasites that egressed with the second peak resulting from Ca2+ entry. We patched infected host cells to allow the delivery of precise concentrations of Ca2+ for stimulation of motility and egress. Using this approach of patching infected host cells allowed to determine that increasing the host cytosolic Ca2+ to a specific concentration can trigger egress, which is further accelerated by diminishing the concentration of potassium (K+).
Keywords: Toxoplasma gondii, calcium signaling, genetically-encoded calcium indicators, GCaMP6f, RGECO, Toxoplasma egress
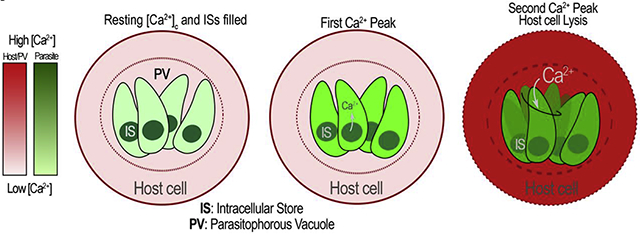
Graphical Abstract
1. Introduction
Toxoplasma gondii is an obligate intracellular parasite that infects approximately one third of the world population. T. gondii replicates inside cells and causes disease by engaging in multiple rounds of a lytic cycle, which consists of invasion of host cells, replication inside a parasitophorous vacuole (PV), egress resulting in lysis of the host cell, and invasion of a new host cell [1, 2]. Several key steps of the lytic cycle of T. gondii: motility, attachment, invasion, and egress, are regulated by fluctuations in its cytosolic Ca2+ concentration ([Ca2+]c) [3, 4].
Ca2+ signaling is universal and plays important roles in the regulation of a large number of cellular functions [5]. The [Ca2+]c is highly regulated, because prolonged high cytosolic Ca2+ levels are toxic and may result in cell death. A variety of Ca2+ pumps, channels, and transporters, located at the plasma membrane (PM) and intracellular organelles (endoplasmic reticulum (ER), acidic stores, and mitochondria) are involved in regulating cytosolic Ca2+ [6].
In T. gondii, the controlled influx of extracellular and intracellular Ca2+ into the parasite cytosol initiates a cascade of signaling pathways that promote progression through the biological steps of the parasite lytic cycle. Motile parasites loaded with fluorescent Ca2+ indicators, as well as expressing Genetically Encoded Calcium Indicators (GECIs) exhibit Ca2+ oscillations [7, 8]. Previous studies have shown that a rise in the cytosolic Ca2+ activates the motility machinery leading to egress. Blocking these cytosolic Ca2+ fluxes with BAPTA-AM (membrane permeable cytosolic Ca2+ chelator), blocks motility, conoid extrusion (apical tip of the parasite necessary for attachment), invasion, and host cell egress [9].
Measurements of T. gondii [Ca2+]c (using Fura2-AM) showed influx of Ca2+ from the extracellular environment, which did not operate as store-operated calcium entry (SOCE) as shown with experiments testing surrogate ions like Mn [10]. This result was supported by the lack of components of the SOCE pathway, STIM and ORAI from the T. gondii genome [11]. We showed that influx of Ca2+, as well as invasion-linked traits [10] and egress [8] were inhibited by nifedipine a Voltage Operated Calcium Chanel blocker (VOCC), supporting the function of this type of channel in Ca2+ influx.
Active egress of T. gondii from host cells requires rupture of the parasitophorous vacuole membrane (PVM) and the host cell membrane [12]. Egress is essential for the dissemination of the infection and it has been known for several years that Ca2+ ionophores can trigger egress [13]. The final and conclusive evidence of a cytosolic Ca2+ increase preceding egress was obtained by expressing GECIs in the cytosol of T. gondii tachyzoites [8]. Secretion of the perforin-like protein 1 (TgPLP1) from micronemes (specialized secretory organelles involved in egress, motility, and invasion by tachyzoites), assists in the permeabilization of the PVM and host cell membrane [14]. Both secretion of the microneme protein TgPLP1 and initiation of motility during egress are stimulated by an increase in cytosolic Ca2+. It has been proposed that the trigger for this cytosolic Ca2+ increase is the rupture of the host plasma membrane and the ensuing reduction in the concentration of the surrounding potassium [K+]. It was proposed that low [K+] would activate a phospholipase C activity in T. gondii that, in turn, would cause an increase in [Ca2+]c in the parasite via Ca2+ release from intracellular stores [15].
As an obligate intracellular parasite, T. gondii resides and replicates within the PV that functions as a molecular sieve and passively permits the exchange of small molecules; thus, the surrounding milieu of intracellular parasites is likely in equilibrium with the host cell cytoplasm [16]. Therefore, intracellular parasites would be exposed to the fluctuations of the host cytosolic ionic composition. The host cytosolic Ca2+ is highly regulated and the resting [Ca2+]c is maintained at ~70-100 nM, which is similar to the [Ca2+]c of replicating parasites. Ca2+ efflux from intracellular stores of the parasite must be the first step of the Ca2+ signaling pathway leading to activation of egress, so maintaining these stores replenished with Ca2+ is fundamental for continuation of the lytic cycle.
In this work, we investigated how T. gondii manages to replenish its intracellular Ca2+ stores during its intracellular replication and how the decrease of the surrounding K+ concentration impacts parasite egress. Using a variety of pharmacological tools, fluorescence microscopy, and a new approach using patched infected host cells, we show that Ca2+ signaling of the host cell influences parasite cytosolic Ca2+ and contributes to parasite egress. Rupture of the host cell during egress facilitates Ca2+ influx. The ensuing decrease in the K+ concentration modulates but does not trigger egress directly.
2. Results
2.1. Calcium influx in intracellular parasites
We previously characterized a Ca2+ influx pathway at the plasma membrane of extracellular T. gondii. Following on this finding we wanted to determine if Ca2+ influx was also operational in intracellular replicating parasites. For this, we measured cytosolic Ca2+ responses of intracellular tachyzoites and used specific natural host receptor agonists to stimulate Ca2+ signaling in the host. We expressed Genetically Encoded Ca2+ Indicators (GECIs) [17, 18] in the cytosol of HeLa cells (jRGECO1a or RGECO) and infected them with T. gondii tachyzoites expressing either cytosolic GCaMP6f or luminal PV-targeted jGCaMP7f or RGECO [18, 19] (Table S1 lists the GECIs used in this study and Table S2 lists the cell lines). PV expression of GECIs was achieved by fusing the T. gondii P30 gene to the N-terminus of the GECI, previously shown to confer PV localization [14] (See Material and Methods and Table S3). Ca2+ changes were followed via time-lapse microscopy after stimulation with agonists specific for the host cell. We used carbachol, an agonist that binds muscarinic receptors, which are present in mammalian cells but absent in the T. gondii genome [20]. Muscarinic receptors, stimulated by the neurotransmitter acetylcholine, are well-characterized G-protein coupled receptors and their activation result in an increase in cytosolic Ca2+ via activation of a phosphatidylinositol phospholipase C (PI-PLC) and generation of inositol 1,4,5-trisphosphate (IP3) [21].
To investigate the link between host and PV calcium, we infected Hela cells expressing jRGECO1a (a red GECI) with tachyzoites that express the green GECI jGCaMP7f at the PV (Fig. 1A). Treating the infected cultures with carbachol led to an almost simultaneous rise in host (red) and PV (green) Ca2+, supporting the molecular sieve model of the PV [16].
Figure 1: Ca2+ entry in intracellular T. gondii tachyzoites.
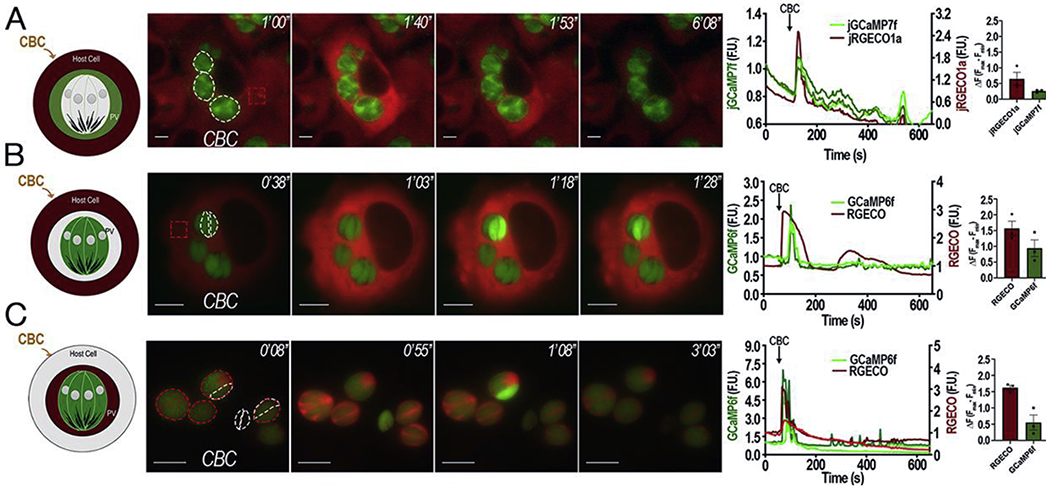
A, Representative Images of HeLa cells stably expressing jRGECO1a that were infected with tachyzoites expressing jGCaMP7f in their PVs. Dashed white outlines indicates the area used for the green fluorescence (PVs) analysis and the red square indicates the region of the host cell used for the analysis of the jRGECO1a fluorescence shown on the graphs. B, Representative Images of HeLa cells transiently expressing the red GECIRGECO infected with tachyzoites expressing cytosolic GCaMP6f. Dashed white outlines show GCaMP6f expressing parasites used for the fluorescence analysis shown in the graphs and the dashed red square shows the region of the host cell used to analyze the RGECO fluorescence. C, Representative images of HeLa cells infected with tachyzoites expressing cytoplasmic GCaMP6f and RGECO in their PVs. Dashed white outlines show GCaMP6-expressing parasites and dashed red outlines indicate the PV region used to analyze the RGECO fluorescence. For the three parts, 1 mM carbachol was added 1 min after recording started. Numbers at the upper right of each panel indicate the time frame of the video. Tracings to the right of each panel shows green fluorescence (GCaMP6f or jGCaMP7f) and red fluorescence (jRGECO1a or RGECO) fluctuations with the scales for the green fluorescence shown on the left Y axis and for the red fluorescence on the right Y scale. Ca2+ fluctuations of PVs are shown in A (green) and C (red). Single parasites fluctuations are shown in B and C (green). Host cytosolic changes are shown in A and B (red). Bar graphs represent quantification of the average ΔF values of a minimum of three independent trials (red, jRGECO1a or RGECO) (green, GCaMP6f or jGCaMP7f) with Standard Error of the Mean (S.E.M.) represented as error bars.
To follow the Ca2+ path from the host cytosol to the parasite through the PV we first examined if the parasite cytosolic Ca2+ oscillated in response to host cytosolic Ca2+ increases. We infected HeLa cells expressing cytosolic RGECO with tachyzoites expressing cytosolic GCaMP6f and activated Ca2+ signaling with carbachol (Fig 1B, green tracing and Supporting Video S1). Carbachol caused an increase in the Hela cytosolic Ca2+, which was followed by a few parasites showing a subsequent large increase in cytosolic Ca2+ (Fig. 1B). Additionally, we infected HeLa cells (no genetic indicator) with parasites expressing both cytosolic GCaMP6f and RGECO in the lumen of the PV to follow both, PV and parasite cytosolic Ca2+. Carbachol addition caused a uniform rise in all PVs, followed by a small proportion of parasites showing increase in cytosolic Ca2+ (GCaMP6f signal) (Fig. 1C). All PV’s responded uniformly to carbachol stimulation (Fig. 1A and C). To further confirm that the increase in parasite Ca2+ is due to influx, we also stimulated the host cells with histamine which binds a different GPCR of HeLa cells and induces an IP3 mediated increase in cytosolic Ca2+ [22] (Fig S1). Histamine caused a rise in host cell Ca2+ that caused a simultaneous Ca2+ increase in the PV (Fig. S1A). Parasite Ca2+ influx trailed host cytosolic Ca2+ (Fig. S1B) and PV Ca2+ (Fig. S1C).
The % of responding parasites vs PVs was quantified for each agonist (Carbachol or Histamine) and it is depicted in the pies presented in Fig. S2. Note that exposure to either agonist causes all PVs to respond (Fig. S2A) followed by the majority of parasites responding with Ca2+ oscillations (Fig. S2B–C), with varying levels of amplitude. The reason for this could be the parasites regulation of cytosolic Ca2+ by the activity of pumps and exchangers while this is not likely to be the case for the PV. Additionally, the sensitivity of the GCaMP may not allow to see all the responding parasites. Note that once Ca2+ reaches a specific threshold, parasites exit the PV (see section 2.2).
Both agonists (Carbachol and Histamine) act specifically on host cells as T. gondii tachyzoites do not respond to either one (Fig. S3). T. gondii tachyzoites expressing GCaMP6f exposed to carbachol showed no increase in fluorescence indicating their cytosolic Ca2+ is not responsive to this agonist. These results further support that the cytosolic Ca2+ increase of intracellular tachyzoites is due to Ca2+ influx from the host. We also showed previously that histamine has no effect on T. gondii cytosolic Ca2+ in extracellular parasites loaded with Fura2-AM [8].
Overall, we demonstrate that intracellular tachyzoites are capable of taking up Ca2+ from the host cytosol, during host Ca2+ signaling events.
2.2. A threshold of Ca2+ is needed for parasite egress
Intracellular tachyzoites replicate in a PV that is able to extensively associate with host mitochondria at the PVM [23, 24] in a process that is strain specific. We took advantage of this striking feature of the T. gondii infection to visualize the path of Ca2+ from host to intracellular parasites and we transiently transfected HeLa cells with LAR-GECO1.2 (a red Ca2+ indicator optimized for expression in the host mitochondria) [25] and infected them with tachyzoites expressing cytosolic GCaMP6f. We first stimulated [Ca2+]c increase with ionomycin (IO), a Ca2+/H+ ionophore that causes Ca2+ release from all neutral stores [26]. or thapsigargin (TG), an inhibitor of the endoplasmic reticulum (ER) SERCA Ca2+-ATPase [27]. Addition of IO caused a global Ca2+ increase in both host cells and parasites, followed by rapid egress (Fig. 2A–B). Blockage of the SERCA by TG induced a rise in cytosolic Ca2+ due to uncompensated ER Ca2+ efflux [27]. This caused an increase in the fluorescence of LAR-GECO, indicating an increase of Ca2+ within the host mitochondria, most likely due to Ca2+ transfer from the ER through membrane contact sites [28]. This Ca2+ increase was trailed by Ca2+ oscillations within tachyzoites (Fig. 2C–D, green tracings, left Y axis) of smaller amplitude and parasites remained intracellular throughout all 10 min of recording (Fig. 2C and Supporting Video S2). Note that the Ca2+ oscillations triggered by Histamine or Carbachol were also not sufficient to trigger egress. This result indicates that there is a threshold for the parasite cytosolic Ca2+ increase that needs to be reached for the stimulation of motility that precedes egress. Additionally, it suggests that the host mitochondria surrounding the PV may buffer Ca2+ so tachyzoites do not egress prematurely.
Figure 2: A threshold for the tachyzoite cytosolic Ca2+:
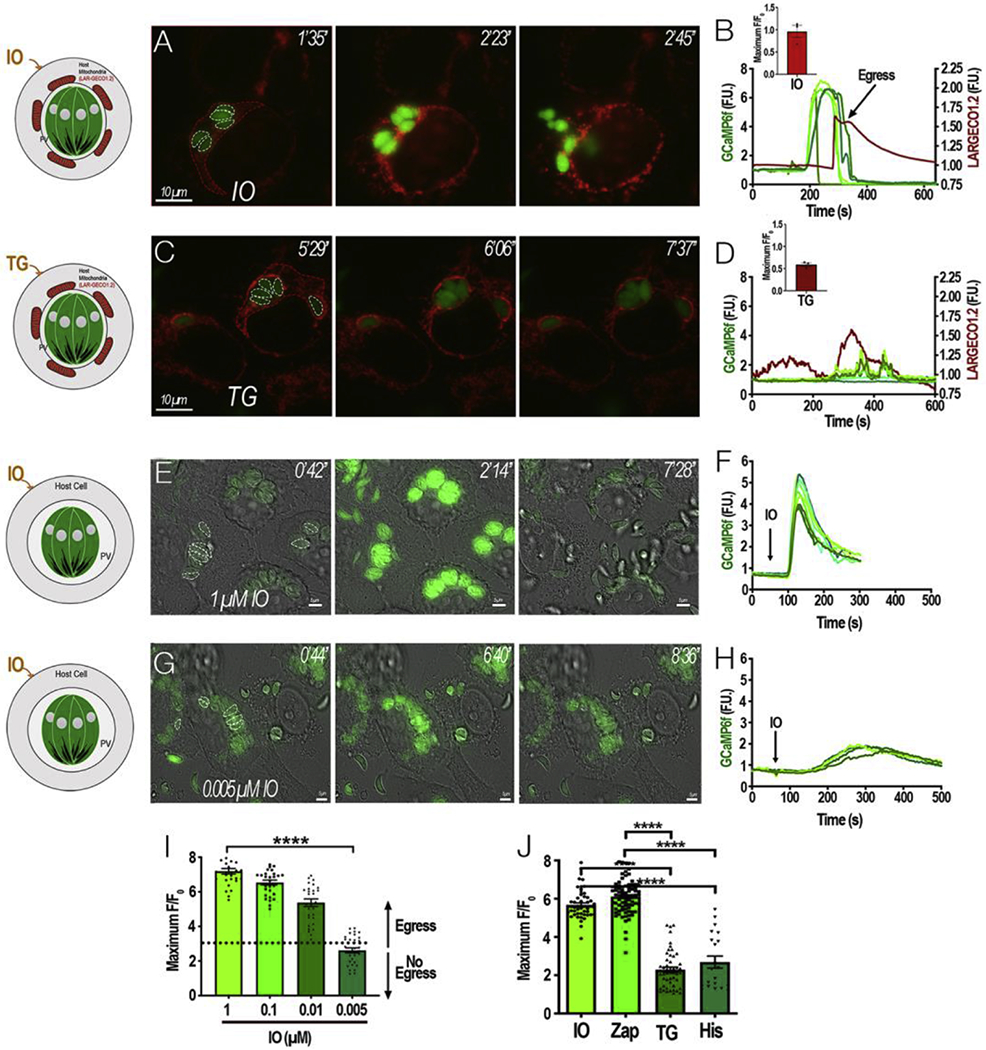
HeLa cells expressing the mitochondrial Ca2+ indicator LAR-GECO1.2 were infected with tachyzoites expressing cytosolic GCaMP6f for approximately 20 h. The host mitochondria surrounding each PV becomes labeled with LAR-GECO1.2 (red). Dashed white outlines show GCaMP6f-expressing parasites whose fluorescence were used for the analysis in B. Dashed red outlines indicate the region of the host cell that was used to analyze the LAR-GECO1.2 fluorescence in B. Numbers at the upper right of each panel indicate the time frame of the video. A, Addition of 1 μM ionomycin (IO) stimulated egress. B, Fluorescence tracings of single parasites shown in A (green) or the host cell area (red) after addition of IO. The left Y axis scale shows green fluorescence and the right Y axis scale shows red fluorescence changes. Inset bar graph represents ΔFluorescence values of IO response with S.E.M represented by error bars. C, Addition of 2 μM thapsigargin (TG) at 1 min leads to stimulation of red fluorescence (host cytosol) trailed by green fluorescence (parasite cytosol). D, Fluorescence tracings of areas indicated in B (green) or the host cell area (red) after addition of TG. Inset bar graph represents ΔFluorescence values of TG response with S.E.M represented by error bars. E and G, HeLa cells infected with GCaMP6f parasites and IO was added at the indicated concentration (1 and 0.005 μM, respectively). Dashed white outlines indicate the area used as a region of interest to analyze the fluorescence changes shown in F and H. Numbers at the upper right of each panel indicate the time frame of the video. F and H, fluorescence tracings of parasites shown in E and G respectively. I, Average ΔF of GCaMP6 fluorescence after stimulating cultures with 1, 0.1, 0.01, and 0.005 μM IO. The ΔF values of GCaMP6f expressing parasites were determined after the addition of IO. Data from 3 independent sets of experiments were combined and the average was calculated. Error bars represent the S.E.M. **** represent a p-value ≤ 0.0001 in a T-test between 1 μM and 0.005 μM IO average ΔF values. J, Comparison of ΔF values generated by different agonists.
With the aim of estimating the Ca2+ threshold needed for egress, we titrated down the concentration of IO to determine a concentration that would still cause a Ca2+ response but was insufficient to induce egress. IO at 1 μM induced a large increase in the ΔF (ΔFmax/F0: fluorescence fold change over baseline) response from GCaMP6f fluorescence, which was followed by egress (Fig. 2E–F). Titrating down the concentration of IO to 0.1 μM and 0.01 μM led to a decrease in the ΔF response and widening of the curve. Although parasite egress was still observed, it was less at lower concentrations of IO. However, no egress was observed when the concentration of IO was below 0.005 μM (Fig. 2G and H). Note that the ΔF response to 0.005 μM IO was approximately 3, while the ΔF for 1 μM IO was approximately 7 (Fig. 2I). The response to TG or histamine also resulted in a ΔF of approximately 2-3 and did not induce egress (Fig. 2J). We next evaluated the [Ca2+]cthat tachyzoites reached in response to various concentrations of IO in a separate experiment with extracellular tachyzoites loaded with the ratiometric Ca2+ indicator Fura-2-AM (Fig. S4). The cytosolic concentration reached with 5 nM IO was ~250 nM and with 1 μM was ~800 nM. We concluded that there is a cytosolic Ca2+ threshold for the stimulation of egress and it is around 300-500 nM. We tested IO in Ca2+-free conditions, and observed that even that egress still occurred, the ΔF response compared to the ΔF in Ca2+-rich media was significantly reduced and interestingly it caused some oscillations (Fig. S5).
We next tested Zaprinast, a cGMP phosphodiesterase inhibitor resulting in build-up of cGMP, which activates protein kinase G (PKG) leading to an increase of cytosolic Ca2+ and stimulation of egress [29, 30]. The increase of cytosolic Ca2+ by Zaprinast (ΔF) was comparable to the increase observed with 1 μM IO, and resulted in egress (Fig. 2J). Note that it is possible that the GCaMP6f Ca2+ indicator could be at saturation under the conditions tested, thus explaining the similarity between the ΔF responses. The ΔF responses by TG or histamine were of less amplitude ~2 fold and there was no statistically significant difference between the two responses. However, the Ca2+ response to IO or Zaprinast were significantly higher than the responses to TG or Histamine (Fig. 2J).
Our results support the concept that a threshold for cytosolic Ca2+ has to be met in order for egress to start. We also showed that stimulation of cytosolic Ca2+ by TG or Histamine do not reach the Ca2+ threshold for egress, which could be the result of the buffering effect of the host mitochondria.
2.3. Two peaks of Ca2+ lead to parasite egress
To further characterize cytosolic Ca2+ responses of intracellular parasites and ensuing egress we tested Zaprinast, which causes a cytosolic Ca2+ increase and stimulation of microneme secretion, and parasite egress. We tested first high extracellular Ca2+ (2 mM) and we noticed that egressing parasites displayed two peaks of Ca2+ (as GCaMP6 fluorescence) preceding egress (Fig 3A–C and Supporting Video S3). We repeated this experiment under low extracellular Ca2+ (100 μM EGTA) (Ca2+ free in the figure, ~50 nM) and observed that the amplitude of the second fluorescence peak was lower and wider (Fig 3D–F). Additionally, under low Ca2+, parasites took longer to egress compared to the time to egress under high Ca2+ (Fig 3G). Note that the effect of Zaprinast is specific for T. gondii Ca2+ as shown in Fig. S6 using uninfected jRGECO1a-expressing HeLa cells exposed to 100 μM Zaprinast. Under these conditions only a very small change in the fluorescence of jRGECO1a (~0.1) was observed considerably below the average fluorescence fold changes caused by carbachol or histamine. This effect of Zaprinast was insufficient to lead to the large Ca2+ increase needed to stimulate parasite egress (Fig. S6).
Figure 3: Two peaks of Ca2+ precede egress:
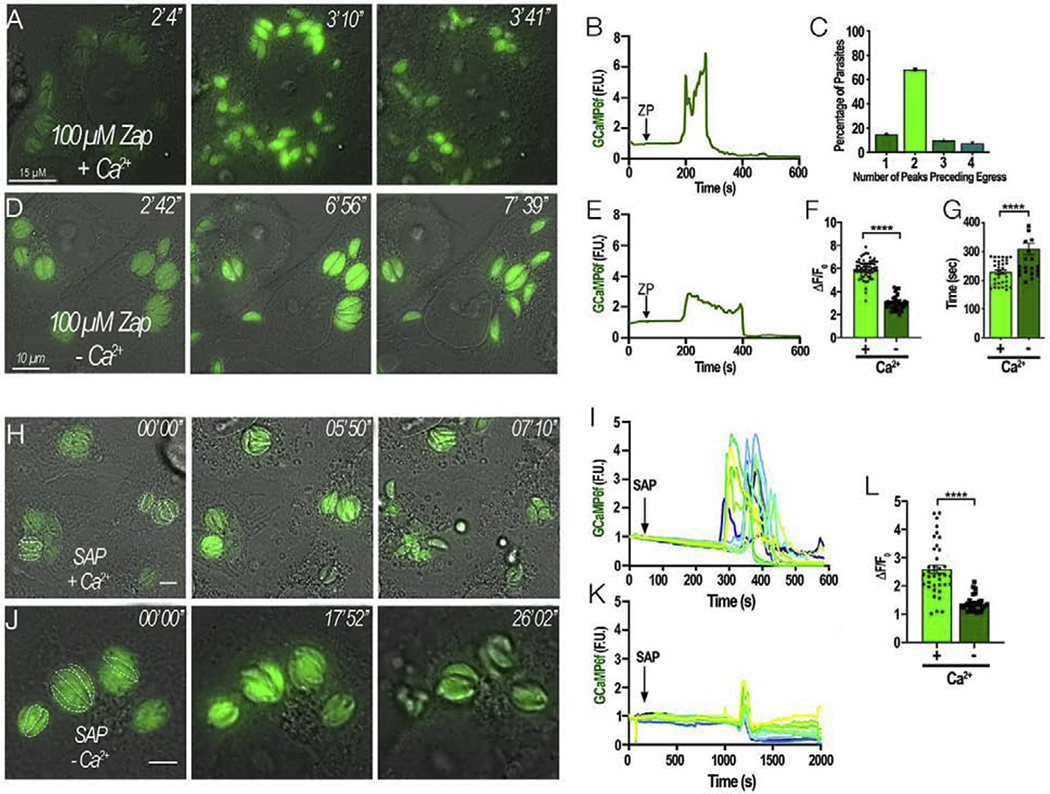
A and D, HeLa cells infected with tachyzoites expressing cytosolic GCaMP6f were stimulated to egress with 100 μM Zaprinast. They were bathed in Ringer buffer supplemented with either 2 mM Ca2+ (+ Ca2+, A) or 100 μM EGTA (− Ca2+, D). The numbers at the upper right of each panel indicate the time frame of the video. B and E, Representative fluorescence tracings of single parasites under Zaprinast simulation in the presence of high or low Ca2+, respectively. Note that under low Ca2+ conditions (100 μM EGTA) the second peak is reduced. C, Quantification of GCaMP6f peak numbers preceding egress. Graph represents the combined summation of 3 independent experiments. F, Amplitude of the second Ca2+ peak after stimulating egress with Zaprinast in the presence of high (+) and low Ca2+ (−). G, Average time of egress comparing Zaprinast induced egress from cells resuspended in extracellular media supplemented with either 2 mM Ca2+ (+) or 100 μM EGTA (−). Error bars represent the S.E.M. in which **** represents a p-value ≤ 0.0001. H, Intracellular GCaMP6f parasites were treated with 0.01% (w/v) saponin to permeabilize the host cells in Ringer buffer with 2 mM Ca2+ (SAP + Ca2+). Dashed white outlines represent the parasites that were used for the analysis. Numbers at the upper right of each panel indicate the time frame of the video. I, Representative fluorescence tracings of egressing parasites treated with saponin in +Ca2+ conditions taken from H. J, Intracellular GCaMP6f parasites were exposed to Saponin 0.01% (w/v) in Ringer buffer with 1 mM EGTA (SAP – Ca2+). Dashed white outlines represent the parasites that were used in the analysis. Numbers at the upper right of each panel indicate the time frame of the video. K, Representative fluorescence tracings of non-egressing parasites treated with saponin in Ca2+-free conditions as shown in J. L, amplitude of the ΔF changes from fluorescent parasites in H and J.
We next compared the parasite cytosolic Ca2+ fluctuations after permeabilizing the host cell with saponin under two conditions: high Ca2+ (2 mM Ca2+) or low Ca2+ (100 μM EGTA) (Fig. 3H–K). Under high extracellular Ca2+, the parasite cytosolic Ca2+ oscillated and they promptly egressed from their respective PV’s (Fig 3H–I). In the presence of extracellular Ca2+ two peaks of Ca2+ were observed (Fig. 3I). In comparison, when the extracellular Ca2+ was low, egress was rarely observed, and most parasites showed a sharp single peak, and it was difficult to observe the second peak (Fig 3J–L). The intensity of the second Ca2+ peak was significantly higher under high extracellular Ca2+ (Fig. 3L).
The microneme protein Perforin-Like Protein 1 (PLP1) functions in breaking down the host cell during egress, exposing intracellular tachyzoites to the surrounding extracellular milieu. PLP1 has been shown to be involved in the initial rupture of the PVM [14] and the release of Ca2+ from parasite intracellular stores could stimulate this release in the natural egress process. This rupture would allow the extracellular Ca2+ to influx the parasites and contribute to the second peak of Ca2+ and reaching the Ca2+ threshold for stimulation of gliding motility and egress. We used ΔPLP1 mutants, defective in their ability to breakdown the host cell, to investigate if they would display one or two Ca2+ peaks when stimulated to egress. We transfected GCaMP6f into ΔPLP1 mutants, which were used to study egress with 100 μM Zaprinast (Fig. S7). Egress of ΔPLP1 parasites, was delayed as expected [14] but the majority were able to exit host cells, due to the persistent gliding motility caused via Zaprinast stimulation. We observed that the parasites that were able to egress (~80%) still showed two peaks of cytosolic Ca2+ as the rupture of the PV and PM (in this case mechanical) would expose parasites to extracellular Ca2+. However, the ones that did not egress (~20%) displayed Ca2+ oscillations of lower amplitude and only a single broader peak, that did not reach the needed threshold for egress. We quantified the fluorescence tracings of responding parasites which are shown in Fig. S7B–C, and the data indicated that when there is rupture of the PV and PM the second peak of Ca2+ is significantly larger (Fig. S7D) supporting its extracellular source.
These experiments demonstrate that intracellular tachyzoites are capable of taking up Ca2+ from the host cytosol and also from the extracellular milieu after host cell rupture, which results in a second peak of cytosolic Ca2+ that precedes egress.
2.4. Two Ca2+ peaks precede natural egress from host cells
With the aim of investigating the presence of two cytosolic Ca2+ peaks during natural egress we synchronized intracellular parasites by pre-incubating cultures with Compound 1 (cpd1) (Fig 4 and Supplemental Video 4). Cp1 inhibits the parasite protein kinase G (PKG) [32], and it was previously used to arrest Plasmodium falciparum egress [33]. We pre-incubated HeLa cells expressing jRGECO1a infected with tachyzoites expressing cytosolic GCaMP6f, with 1 μM cpd1 for 24 h to arrest egress. Egress began approximately 2 min after washing off cp1, preceded by an increase in cytosolic Ca2+. A single “leader” tachyzoite having the largest rise in cytosolic Ca2+ egressed first (Fig 4A, stars). Egressing parasites displayed two peaks of cytosolic Ca2+ increase during egress (Fig 4 C–E). Interestingly, a rise in host Ca2+ was also evident during the natural egress process, thus highlighting the role of extracellular Ca2+ influx during natural egress (Fig. 4C and D, red tracing). Next, we tested the ΔPLP1 parasites under natural egress conditions (Fig. 4B and E). After washing off cpd1, cytosolic Ca2+ of the ΔPLP1 GCaMP6f cells oscillated randomly and nonuniformly, and parasites did not exhibit the two-peak pattern observed in wild type parasites (Fig. 4E). Some parasites displayed characteristic movement within the PV of the ΔPLP1 parasites [14] post-rise in cytosolic Ca2+ though none of these parasites displayed a second peak of higher amplitude that would be associated with Ca2+ entry, nor a rise in the jRGECO1a channel that would be indicative of rupture of the host cell and extracellular Ca2+ influx. Eventually the fluorescence of these parasites diminished indicating lower cytosolic Ca2+, and the parasites stopped moving. We conclude that two-peaks of Ca2+ are part of the natural egress progression of T. gondii, and the second peak is due to Ca2+ influx from the extracellular milieu.
Figure 4: Cytosolic Ca2+ during natural egress:
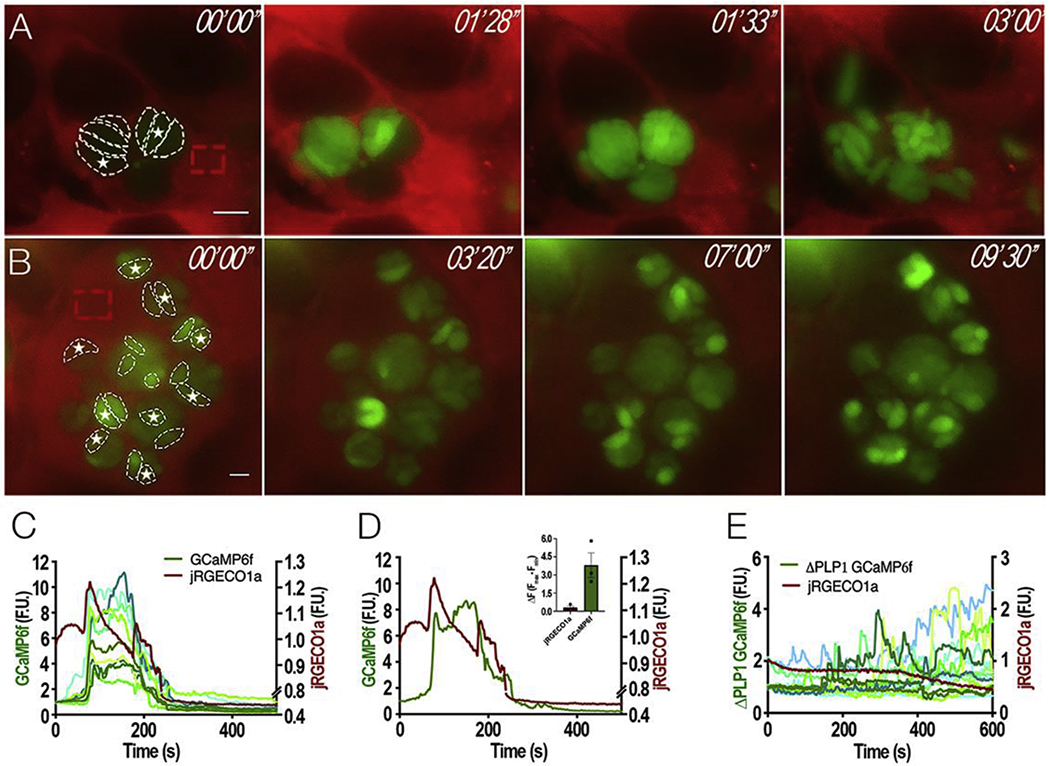
Intracellular parasites expressing cytosolic GCaMP6f were synchronized for natural egress with Compound 1 (cpd1). A, still images of HeLa cells expressing jRGECO1a infected with tachyzoites expressing GCaMP6f and treated with 1 μM cpd1 for 24 hours. After washing off cpd1, parasites egressed within 3-5 min. Dashed white outlines show fluorescent parasites used for the analyses in C and D. Dashed red outlines indicate the region of the host cell used to analyze the jRGECO1a fluorescence. Numbers at the upper right of each panel indicate the time frame of the video. White stars indicate “leader” parasites whose fluorescence begins the egress process. B, still images of HeLa cells expressing jRGECO1a infected with ΔPLP1 tachyzoites expressing cytosolic GCaMP6f and treated with 1 μM cpd1 for 24 hours. Washing off cpd1, results in egress within 3–5 mins. Dashed regions show the ΔPLP1 fluorescent parasites used for the analysis shown in E. Dashed red outlines indicate the region of the host cell that was used to analyze the jRGECO1a fluorescence. Numbers at the upper right of each panel indicate the time frame of the video. White stars indicate “leader” parasites, which shows the highest increase in cytosolic Ca2+. C, Fluorescence tracings of host cell jREGO1a (red tracing, scale shown on the right Y axis) and GCaMP6f parasites (green tracings, left Y axis); D, Fluorescence tracings of host cell jREGO1a (red tracing) and a single parasite expressing GCaMP6f (green tracing). Note that parasites that egress show two fluorescence peaks; Inset, Quantification of 3 independent trials of ΔF of jRGECO1a (red bar) and GCaMP6f (green bar) after cpd1 washout. Error bars represent the S.E.M. E, Fluorescence tracings of host cell jREGO1a (red tracing, right Y axis) and ΔPLP1 GCaMP6f parasites (green tracings, Y left axis). Size bars are 5 μm.
2.5. Blocking Ca2+ influx using pharmacological agents blocks parasite egress
To characterize further the source of the two peaks of Ca2+ we tested nifedipine (NIF), a voltage-operated Ca2+ channel inhibitor, that we previously showed to inhibit Ca2+ influx in tachyzoites [10], and pretreated intracellular tachyzoites expressing cytosolic GCaMP6f with the drug to compare egress with that of control parasites (Fig. 5A). The surrounding extracellular buffer was supplemented with 2 mM Ca2+, and the host cells were permeabilized with saponin. A sharp rise in cytosolic Ca2+ trailed by smaller Ca2+ oscillations was observed followed by egress. Pretreatment with NIF resulted in parasites that did not egress, and showed only modest Ca2+ oscillations of approximately two-fold range (Fig. 5C and D), which were below the threshold needed for egress (Fig. 5E). We next tested cpd1 to inhibit PKG and induced egress with Zaprinast. While control parasites egressed following a fast rise in cytosolic Ca2+ with two peaks (Fig. 5F and G), pretreatment with cpd1 abolished egress, and the parasites’ cytosolic Ca2+ only raised about 3-fold (Fig. 5G–J). The two peaks were not clearly evident. These results showed that blocking extracellular Ca2+ influx with NIF or cytosolic Ca2+ increase with cpd1, resulted in intracellular parasites unable to reach the threshold needed for egress, stressing the role of both intracellular and extracellular sources of Ca2+ for egress.
Figure 5: Pharmacological inhibition of Ca2+ influx leads to inhibition of egress:
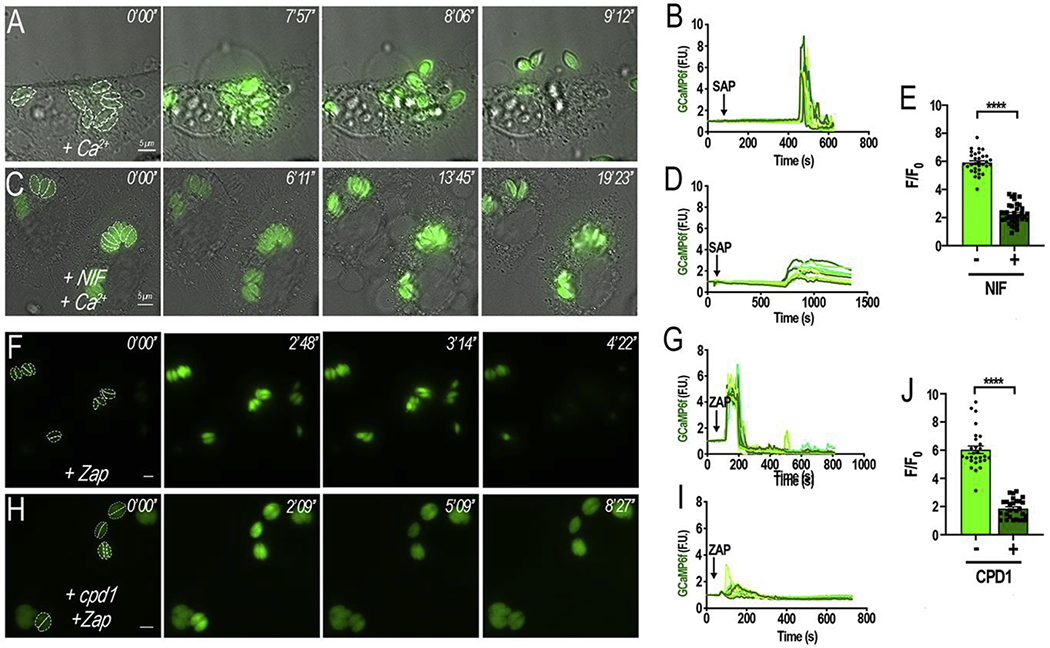
A, HeLa cells infected with tachyzoites expressing cytosolic GCaMP6f were exposed to 2 mM Ca2+ (+ Ca2+) containing 0.01% (w/v) saponin. B, Fluorescence tracings of egressing parasites (dashed outlines in A) after the addition of saponin in media with 2 mM Ca2+; C, HeLa cells infected with tachyzoites expressing GCaMP6f were pretreated for 5 min with 10 μM nifedipine (NIF) in the presence of 2 mM Ca2+ (+NIF +Ca2+) containing 0.01% (w/v) saponin. Note that parasites did not egress under these conditions; D, Tracings obtained following the fluorescence signal from the parasites highlighted in C. E, quantification and statistical analysis of the ΔF + and − NIF calculated from three independent experiments (p< 0.001). F, HeLa cells infected with tachyzoites expressing cytosolic GCaMP6f were stimulated with 100 μM Zaprinast; G, Tracings obtained following the fluorescence signal from the parasites highlighted in F. G, HeLa cells infected with tachyzoites expressing cytosolic GCaMP6f were pretreated with 1 μM of cpd1 for 5 mins prior to stimulation with 100 μM Zaprinast. Cytosolic Ca2+ slightly and briefly oscillated but no egress was evident; I, Tracings obtained following the fluorescence signal from the parasites highlighted in H. Dashed outlines indicate the area used as a region of interest for the analysis of the fluorescence changes shown in the tracings. Numbers at the upper right of each panel indicate the time frame of the video. J, quantification and statistical analysis of the ΔF with and without cpd1 calculated from three independent experiments (p< 0.001). Size bars are 5 μm.
2.6. Role of K+ and Ca2+ in parasite egress
We next investigated the impact of defined host cytosolic Ca2+ concentrations on parasite egress. With this aim we patched infected HeLa cells as shown in Fig 6A forming a seal between the plasma membrane and the pipette, and broke the membrane within the pipet, thus equilibrating the solution in the patch pipette with the cytosol of the host cell [34, 35]. This technique allowed us to control the composition of the buffer surrounding the PVs by modifying the pipette buffer (Fig 6A). The patch pipette contained 2 mM ATP to compensate for ATP loss and we kept the pH constant to prevent acidification as a potential variable [36]. We tested increasing concentrations of cytosolic free Ca2+ (0.1, 0.5, 1, 5, and 10 μM) in a 140 mM K+ solution (Fig. 6B). No egress was observed at 0.1 or 0.5 μM Ca2+, and a small percentage of parasites egressed at 1 μM Ca2+. Approximately one third of parasites egressed when exposed to 5 μM Ca2+ and all parasites egressed at 10 μM Ca2+ (Supplemental Video 5) (Fig. 6C, purple pies). Previous literature has stated that the high K+ concentration of the host cytosol prevented parasite egress. It was postulated that a decrease in the concentration of K+ would activate a Ca2+ signal, inducing microneme secretion and subsequent egress [15]. To test the role of K+ in parasite egress, and considering that upon host cell lysis its concentration would decrease, we repeated the whole-cell patch at lower K+ concentrations within the patch pipette (Fig 6D). Choline chloride was added to maintain the same osmolarity and anionic composition. Under these conditions, 0.5 μM Ca2+ did not trigger egress and two thirds of the parasites of the patched cells egressed at 1 μM Ca2+ (Fig. 6C, pink pies). Approximately 80% of parasites egressed at 5 μM Ca2+, which occurred in approximately 3 min compared to 5 min in high K+. Similar to the high K+ conditions, 10 μM Ca2+ caused 100% egress, but at a much faster rate. Parasites egressed faster at low compared to high K+. Interestingly, no egress was observed at 0.1 or 0.5 μM Ca2+, for both concentrations of K+. As we increased the host cytosolic Ca2+ concentration (2-20 μM), the percentage of egressing parasites increased under both K+ concentrations. Under high K+ conditions the percentage of egressing parasites increased quasi-linearly from 1-5 μM Ca2+, and 10 μM Ca2+ was sufficient to induce 100% egress (Supplemental Video 6). At 1 μM Ca2+, parasites egressed in approximately 550 and 320 sec for high and low K+, respectively. Under lower K+ conditions parasites egressed faster at 5 μM Ca2+ (200 sec) than at 2 μM Ca2+ (~300 seconds). Though 100% egress was observed at 10 μM Ca2+ for both conditions, in high K+ parasites egressed in ~250 seconds, versus ~50 seconds in low K+ conditions. In summary, according to these results, Ca2+ is essential for egress but a decrease in the concentration of K+ results in acceleration but it is not the trigger for egress.
Figure 6: Role of host cytosolic Ca2+ and K+ studied by patching the host plasma membrane.
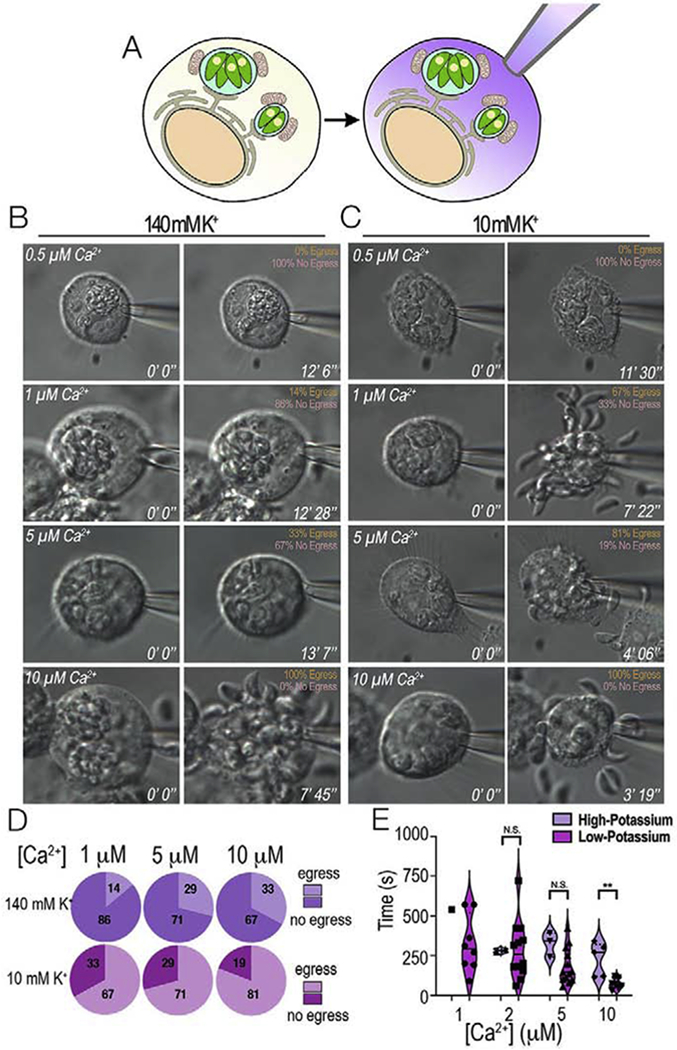
HeLa cells infected with T. gondii tachyzoites were whole-cell patched and egress was monitored. A, whole-cell patch allowed the exposure of PVs to defined Ca2+ concentrations by exchanging the cytosol of the host cell with the composition of the buffer inside the patch pipette. B, Representative still images of infected host cells patched under high potassium conditions (140 mM K+). Various concentrations of free Ca2+ were tested to monitor egress. The percentage of egressing vs non-egressing parasites is shown in the upper left-hand corner. C, Representative still images of infected hosts cells patched under low potassium conditions (10 mM K+ and 130 mM choline chloride) and egress monitored under the same experimental conditions as in A. D, Percentage of egressing parasites presented as pie charts of increasing Ca2+ concentration. Purple, 140 mM K+, pink, 10 mM K+. E, Violin Plots of the average time to egress under high (140 mM K+) and low potassium conditions (10 mM K+). Note that under low K+ conditions the percentage of egressing parasites increases, and parasites egress faster. N.S. was used to represent non-significant results and ** was used to represent p-values ≤ 0.01 of T-tests between the different Ca2+ concentrations.
3. Discussion
In this work we showed that intracellular replicating Toxoplasma gondii take up Ca2+ through their plasma membrane. The host cytosolic Ca2+ is likely tightly regulated and kept low although physiological Ca2+ signaling events will result in Ca2+ increase. In our experimental set-up we stimulated signaling in host cells with specific agonists that had no direct effect on the parasite Ca2+. Host cytosol Ca2+ did respond to these agonists and was followed by a simultaneous increase in the PV Ca2+ trailed by parasite cytosolic Ca2+ oscillations. We also showed that two peaks of cytosolic Ca2+ increase occurred in egressing tachyzoites, the first peak probably of intracellular origin and the second peak associated with Ca2+ influx after host cell lysis. Inhibition of Ca2+ influx with the voltage operated Ca2+ channel blocker nifedipine blocked the second peak. We tested ΔPLP1 parasites, which are defective in egress because they do not secrete the microneme protein PLP1, which lyses the PV and host PM. The second cytosolic Ca2+ peak was absent in the ΔPLP1 parasites that did not egress. A threshold for the tachyzoite cytosolic Ca2+ increase leading to egress was calculated to be around 300-500 nM [Ca2+]i. Patching of the plasma membrane of infected HeLa cells showed that increasing the host cytosolic Ca2+ alone was sufficient to stimulate egress, which was accelerated by decreasing the concentration of K+.
Keeping intracellular Ca2+ stores replenished in replicating T. gondii is essential for the continuation of its lytic cycle as exit from the host cell is preceded by a rapid, required spike in cytosolic Ca2+ [8]. It is puzzling that intracellular tachyzoites replicating inside the low Ca2+ environment of the host cytosol, are still able to do this. Our experiments showed that stimulation of Ca2+ signaling in the infected host cell led to Ca2+ influx into intracellular parasites resulting in parasite Ca2+ oscillations, though it was insufficient to induce egress.
Cytosolic Ca2+ oscillations in T. gondii tachyzoites were previously observed [7, 37], a fascinating phenomenon for which there is no molecular explanation. Ca2+ oscillations arise from cyclical release and re-uptake of intracellularly stored Ca2+ [38], and a role for influx through plasma membrane channels has also been demonstrated to be important for the maintenance and delivery of Ca2+ into the cytosol. Because of the apparent digital nature of these Ca2+ oscillations [39] they would be perfectly suited for signaling specific biological responses like secretion of micronemes, stimulation of motility and egress. It was shown that Ca2+ oscillations in extracellular tachyzoites loaded with Ca2+ dyes were associated with microneme discharge and bursts of motility [7]. In intracellular tachyzoites, we believe that Ca2+ oscillations, serve to ensure the filling of intracellular stores. These oscillations may initiate from Ca2+ influx, the result of uptake from the host through a plasma membrane mechanism, as we demonstrated in this work, followed by pumping into organelles like the ER via the SERCA-Ca2+ ATPase [37]. This initial Ca2+ increase would stimulate the activity of the PI-PLC at the plasma membrane [40, 41] to synthesize IP3, which would open an unknown ER channel and release Ca2+ into the cytosol potentiating the Ca2+ signals and leading to additional downstream oscillations.
Within host cells parasites are stationary, non-motile, and surrounded by low Ca2+, yet activation by an intrinsic signal like phosphatidic acid, as recently proposed [42], would start a signaling cascade leading to a rise in cytosolic Ca2+ followed by stimulation of motility [43]. It has been proposed that the high K+ content of the host cytosol blocks parasite egress [15]. When the integrity of the host cell becomes compromised, the K+ concentration drops due to its dilution into the extracellular media. We propose a model that would involve the participation of a Ca2+-activated K+ channel(s) for which two candidate genes are annotated in the T. gondii database (TGME49_238995 and TGME49_273380) [44]. During intracellular growth, these channels could activate/open in response to Ca2+ release from intracellular stores. Initially, no conductance would occur because the K+ concentration of both parasite and host cytosol would be similar. Lysis of the host cell would result in decrease of the concentration of K+, generating a gradient, and conductance of the ion with loss of K+ from the parasite, leading to an unbalance in the intracellular concentration of K+ that would be counteracted by a plasma membrane mechanism such as a K+/H+ exchanger that would exchange K+ for H+ leading to acidification of the PV, which has been shown to lead to egress [36]. T. gondii expresses four predicted sodium proton exchangers and one of them localized to the plasma membrane of T. gondii (TgNHE1) (TGME49_259200) [45]. It is possible that one of these exchangers could use K+ instead of Na+ and be responsible for the exchange activity.
In summary, we propose that as the parasites grow and replicate intracellularly, host Ca2+ derived from Ca2+ signaling is taken up by parasites through their plasma membrane and pumped into intracellular stores in order to keep them filled during repetitive rounds of replication (Fig. 7). Egress initiates via an unknown signal that induces release of Ca2+ from intracellular stores. Within the PV a leader parasite would respond and release PLP1 among other microneme proteins contributing to the partial breakdown of the host cell [14]. Lysis of the host cell would permit for extracellular Ca2+ influx and a drop of host K+, two factors that would contribute to egress of the remaining parasites. We found that the decrease in the concentration of K+ plays a role in timing parasite egress, and Ca2+ influx from the host cell or the extracellular milieu would be essential for parasite egress. Our data demonstrates that Ca2+ influx, through an unknown channel is still functional in the low Ca2+ environment of the host cytosol. The use of whole-cell patch presents a new methodology to study the role of ions involved in parasite egress, an essential component of the lytic cycle.
Figure 7: Model of host Ca2+ influx during intracellular growth.
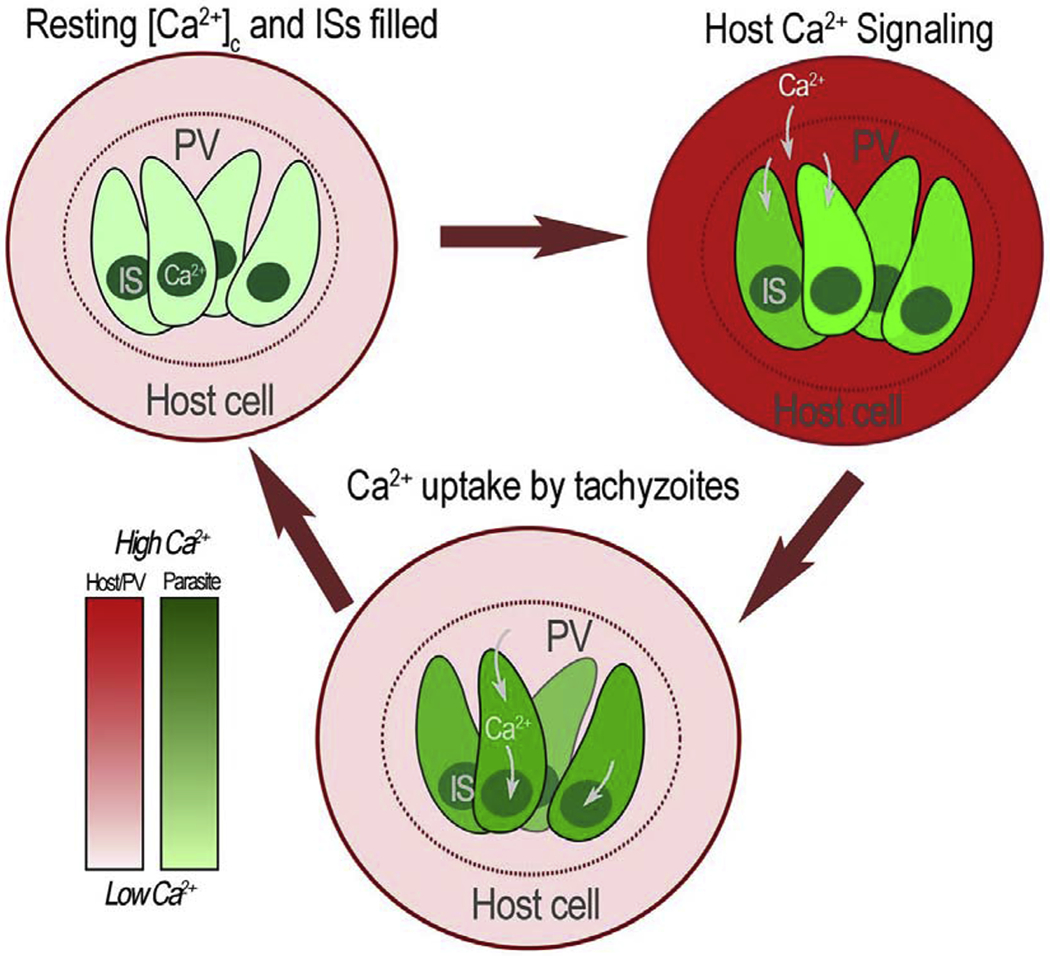
Top left: An infected host cell with tachyzoites both with cytosolic Ca2+ at resting levels. Intracellular Stores (IS) are filled. Top right: A host cell Ca2+ signaling event triggers an increase in cytosolic Ca2+. Given that the PV is in equilibrium with the host cell cytosol, PV Ca2+ rises simultaneously. Bottom: Rise in PV Ca2+ is followed by Ca2+ influx into the parasite via a plasma membrane Ca2+ channel causing a rise in the cytosolic Ca2+ of the parasite. Host cytosolic Ca2+ returns to resting level and the tachyzoite cytosolic Ca2+ is pumped into intracellular stores (IS). Parasites will continue replicating within the host cell while utilizing the Ca2+ influx from the host cell to maintain IS Ca2+ filled to be eventually utilized and released during egress. The red scale estimates the concentration of Ca2+ in the host cell and PV and the green scale in the parasite cytosol.
4. Materials and Methods
4.1. Cell culture
T. gondii tachyzoites (RH strain) were maintained in hTERT human fibroblasts (BD Biosciences) using Dulbeco’s modified essential media (DMEM) with 1% fetal bovine serum (FBS), as described previously [46]. GCaMP6f expressing tachyzoites were maintained under similar conditions, in the presence of 20 μM chloramphenicol. The selection-less strain of GCaMP6f was grown under the same conditions as the RH strain. hTERT cells were maintained in high glucose DMEM with 10% calf serum. HeLa cells (ATCC) were used for egress and whole-cell patch experiments and were maintained in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, and 2 mM L-glutamine. Cell cultures were grown at 37°C with 5% CO2. Parasites were purified by centrifugation and filtration through a Whatman 8 μM nuclepore membrane (GE Healthcare) followed by a second filtration step through a 5 μM nuclepore membrane. Filtered parasites were counted and centrifuged following the protocols specific for each experiment. T. gondii lines created in this work are described in Table S2.
4.2. Chemicals and Reagents
Transient transfections of HeLa cells were performed using PolyJet purchased from SignaGen (http://signagen.com/). Plasmids for GCaMP6f (fast version of GCaMP6), R-GECO1.2, jGCaMP7f, and LAR-GECO1.2 were obtained from Addgene, and the plasmids were used for transient transfection in HeLa cells. The respective genes were cloned into the T. gondii expression vector pCTH3 and pDHFRTubGFP for chloramphenicol and selection-less stable expression of GCaMP6f in tachyzoites, respectively. Thapsigargin, ionomycin, saponin, histamine, Zaprinast, and all other chemicals were obtained from Sigma. All plasmids used in this work are shown in Table S3.
4.3. Preparation of GECI-expressing tachyzoites and HeLa cells
GCaMP6f expressing parasites were obtained as described previously [8]. Briefly, plasmids for expressing GCaMP6f in T. gondii were a gift from Kevin Brown and David Sibley. The coding DNA sequence for GCaMP6f was amplified by PCR and cloned into a T. gondii vector for expression downstream the tubulin promoter (pCTH3 and pDTGCaMP6f), using the BglII and AvrII restriction sites and adding a stop codon in front of the GFP sequence. The primers used were forward 5′-AGGCGTGTACGGTGGGAGGTC-3′ and reverse 5′CTTCCTAGGTTACTTCGCTGTCATCATTTG-3′ (Table S4). The plasmids were electroporated into the RH strain parasites and clones were selected with chloramphenicol. Parasites with low fluorescence were isolated by cell sorting to eliminate those highly fluorescent cells in which the GCaMP6f could be buffering Ca2+ [47].
HeLa cells (5 x 105) were grown on coverslips in high-glucose DMEM with 10% FBS. After 24 hours, cells were transfected with 1 μg of plasmid DNA encoding RGECO or LAR-GECO1.2 using PolyJet following the instructions of the manufacturer. 6-8 hours later, HeLa cells were infected with 1 x 106 tachyzoites expressing GCaMP6f and were grown for 15-20 hours. Rosettes containing 4-8 parasites were used in all experiments. To construct a cell line of HeLa cells stably expressing the sensitive red GECI, jRGECO1a, the coding sequence of jRGECO1a [17] was PCR amplified with primers Fwd 5’- ACCGGTATGCTGCAGAACGAGCTTGCTCTTA -3’ and Rev5’ -GAATTCGCCTACTTCGCTGTCATCATTTGTACA-3’ and cloned into the TOPO vector sequence. The resulting product was digested with Agel and EcoRI restriction enzymes and cloned into the 2nd generation lentivirus expression plasmid pUltra (Table S3). Transfection and cloning of a stable cell line of jRGECO1a was performed using previously established protocols [48]. Briefly, 2nd generation viral particles were produced in HEK293T cells, and the viral supernatant was overlaid on Hela cells for spinfection for 2 hr. Stable cell lines were enriched and selected by FAC’s sorting.
4.4. Cytosolic Ca2+ measurements
Extracellular tachyzoites of the RH strain were loaded with fura2-AM as previously described [10]. The parasites were washed twice in Ringer buffer (155 mM NaCl, 3 mM KCL, 2 mM CaCl2, 1 mM MgCl2, 3 mM NaH2PO4, 10 mM Hepes, pH 7.3, and 5 mM glucose), resuspended in the same buffer to a final density of 1 x 109 cells/mL, and kept on ice. For fluorescence measurements, 50 μL-portions of the cell suspension were diluted in 2.5 mL of Ringer (2 x 107 cells/mL final density) in a cuvette placed in a thermostatically controlled Hitachi 7000 fluorescence spectrometer. Traces shown are representative of three independent experiments conducted on separate cell preparations unless indicated differently. Calcium-defined conditions were determined by using EGTA or 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) and calcium chloride to reach specific concentrations of free calcium. Calcium-EGTA combinations were determined using Maxchelator software (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/downloads.htm).
4.5. Egress assays
Egress assays were done as described previously [47]. HeLa cells were grown in high glucose DMEM with 10% FBS in 35 mm glass bottom dishes (MatTek) until confluency. 8-12 hours after transfection, HeLa cells were infected with 1 x 106 GCaMP6f-expressing tachyzoites, replacing the media with high glucose DMEM with 1% FBS. Thirty hours after infection, parasitophorous vacuoles containing 4-8 parasites were observed by microscopy after washing them in the specific buffer for each experiment. Drugs were added as indicated in the figures at the concentrations indicated: ionomycin (0.005-1 μM), histamine (100 μM), thapsigargin (1 μM), saponin (0.01%), nifedipine (10 μM), and Zaprinast (100 μM). Ringer buffer was used as extracellular buffer (EB). CaCl2 was omitted in the experiments without extracellular Ca2+, and the media was supplemented with either 100 μM EGTA, 1 mM EGTA or 1 mM BAPTA. The composition of the intracellular buffer (IB) is: 140 mM potassium gluconate, 10 mM NaCl, 2.7 mM MgSO4, 2 mM ATP (sodium salt), 1 mM glucose, 200 μM EGTA, 65 μM CaCl2 (90 nM free Ca2+), and 10 mM Tris/Hepes, pH 7.3. The parasites were imaged at 37°C. Fluorescence images were captured using an Olympus IX-71 inverted fluorescence microscope with a Photometrix CoolSnapHQ charge-coupled device (CCD) camera driven by DeltaVision software (Applied Precision). Images were collected using time-lapse mode with an acquisition rate of at least 2-3 seconds during 10-20 minutes. Images were converted into videos using SoftWorx suite 2.0 software from Applied Precision. Fiji was used for the analysis of the video data. Fluorescence tracings were produced by drawing a region of interest (ROI) around the host or parasite of interest and measuring the mean fluorescence. Prism was used for statistical analysis.
For natural egress, 3.5 x 105 HeLa cells stably expressing the red GECI jRGECO1a [17] were plated on 35 mm glass bottom MatTek dishes in DMEM-HG with 10% FBS. 24 h later cells were infected with 2.75 x 106 parasites, and on day three the parasites were synchronized using 1 μM cpd1, and one dish was treated with 2 μL of DMSO as a vehicle control. On day four lysis of the control dish was examined for 70-80% lysis, and the synchronized dishes were used for video microscopy. The dishes were washed once with pre-warmed Ringer buffer, then again with pre-warmed Ringer buffer for 1 min and finally a solution of Ringer buffer supplemented with 2 mM CaCl2 was added before commencing imaging.
4.6. Whole cell patch
Whole cell patch recording was applied according to the method previously described [49, 50]. Briefly, coverslips with infected HeLa cells were transferred into a perfusion chamber (RC-26GLP, Warner Instruments, USA) on a stage of an inverted IX51 Olympus microscope. Growth media was immediately replaced with an extracellular buffer prior to the start of the recordings, and cells were kept at room temperature for no more than 2 hours. Coverslips were replaced every 2 hours. Intracellular thin-walled recording capillars (1.5-mm outer diameter, 1.17-mm inner diameter) made of borosilicate glass (World Precision Instruments, Sarasota, FL) were used to pull patch pipettes (3–4 MΩ) on a horizontal Flaming/Brown micropipette puller (P-97, Sutter Instruments) and then were backfilled with intracellular solution. Recordings from HeLa cells were obtained with an Axopatch 200B amplifier (Molecular Devices) using high-resolution videomicroscopy (Ameriscope). The voltage output was digitized at 16-bit resolution, 20 kHz, and filtered at 1 kHz (Digidata 1550A, Molecular Devices). All experiments were performed at −60 mV holding membrane potential (Vh) which is close to values reported as physiological for HeLa cells [51] to prevent activation of voltagegated calcium channels [52]. After establishing a “seal” (>1 MΩ) slight negative pressure was applied together with “ZAP” electrical pulse (0.5-5 ms, +1.3V) to break cell membrane. The time from membrane opening (start of pipette/cytosol solution exchange) to egress was visually recorded. All statistical analyses were conducted using GraphPad Prism 8 (GraphPad Software, San Diego, CA). All values are expressed as means ± SEM. Graphs used for quantification illustrate the results across three independent trials with a total of 10 cells patched per each condition.
HeLa cells grown in high glucose DMEM with 10% FBS on 22 x 40 mm glass coverslips until 70% confluency were infected with 1 x 106 GCaMP6f-expressing tachyzoites. 24 h post infection coverslips were placed in an electrophysiology recording chamber (~1 ml) and bathed in extracellular solution (mM): 140 NaCl, 5 KCl, 1 MgSO4, 1.8 CaCl2, 10 Hepes, pH 7.5 adjusted with NaOH/HCl. Composition of “high potassium” intracellular pipette solution (mM): 140 KC1, 5 NaCl, 2 MgCl2, 1 Glucose, 10 Hepes, 1 BAPTA, pH 7.3 adjusted with KOH/HC1. “Low potassium” solution was prepared as described above with equimolar substitution of 130 mM KC1 with choline chloride (ChCl). Disodium ATP (2 mM) was added to all intracellular solutions to maintain cell energy supply. Solutions with different free Ca2+ concentrations were prepared by adding Ca2+ and BAPTA at proportions calculated with Webmaxc software (Stanford University, USA). For control experiments in order to exclude the role of host cell extracellular Ca2+ entry that may contribute to T. gondii egress, we used a modified extracellular solution with MgCl2 and CaCl2 adjusted to 5 mM and 0.1 mM, respectively.
4.7. Quantification and Statistical Analysis
Statistical analysis of fluorescence images was performed using FIJI/ImageJ [53]. Briefly, images were background subtracted and normalized using an average of the first 5 frames of imaging. ΔF (Fmax/Fmin) represents the highest fold change over baseline in fluorescence after addition of stimuli or reagent. Figures were constructed using Prism analysis suite and error bars represent the standard error of the mean S.E.M. of three independent experiments. Significant differences were only considered if P values were < 0.05, where * p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. NS designates when the comparison is not statistically significant. Experiment-specific statistical information is provided in the figure legends or associated method details including trials (n), standard error of the mean SEM, and statistical test performed.
Supplementary Material
Highlights.
Toxoplasma gondii replicates inside host cells and takes up Ca2+ from the host cytosol
T. gondii uses extracellular Ca2+ which contributes to reach a threshold needed for egress
Two peaks of Ca2+ precede parasite egress. Intracellular and extracellular stores contribute.
It is possible to patch infected cells to deliver defined Ca2+ concentrations to trigger egress
Reduction of the surrounding potassium concentration modulates the rate of egress
Acknowledgements
We would like to thank Alex W. Chan and Dr. Sebastian Lourido for the protocol of natural egress using Compound 1; Dr. Vern Carruthers for the ΔPLP1 mutant parasites; Dr. Diego Huet for reading the manuscript; Daniel Williamson for assisting on the egress of the ΔPLP1 mutant; Beejan Asady for technical assistance; Julie Nelson for the FAC’s sorting assistance; Dr. Kandasamy for technical assistance on the use of the microscopes at the Biomedical Microscopy Core.
Funding
This work was supported by an NIH grant R01AI128356 to SNJM. SAV and EP were partially supported by fellowships (pre-doc and post-doc respectively) through a Training Grant in Tropical and Emerging Global Diseases (T32AI060546).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Black MW, Boothroyd JC, Lytic cycle of Toxoplasma gondii, Microbiol Mol Biol Rev, 64 (2000) 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blader IJ, Coleman BI, Chen CT, Gubbels MJ, Lytic Cycle of Toxoplasma gondii: 15 Years Later, Annu Rev Microbiol, 69 (2015) 463–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arrizabalaga G, Boothroyd JC, Role of calcium during Toxoplasma gondii invasion and egress, Int J Parasitol, 34 (2004) 361–368. [DOI] [PubMed] [Google Scholar]
- [4].Hortua Triana MA, Marquez-Nogueras KM, Vella SA, Moreno SNJ, Calcium signaling and the lytic cycle of the Apicomplexan parasite Toxoplasma gondii, Biochim Biophys Acta Mol Cell Res, 1865 (2018) 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Clapham DE, Calcium signaling, Cell, 131 (2007) 1047–1058. [DOI] [PubMed] [Google Scholar]
- [6].Bootman MD, Bultynck G, Fundamentals of Cellular Calcium Signaling: A Primer, Cold Spring Harb Perspect Biol, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lovett JL, Sibley LD, Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells, J Cell Sci, 116 (2003) 3009–3016. [DOI] [PubMed] [Google Scholar]
- [8].Borges-Pereira L, Budu A, McKnight CA, Moore CA, Vella SA, Hortua Triana MA, Liu J, Garcia CR, Pace DA, Moreno SN, Calcium Signaling throughout the Toxoplasma gondii Lytic Cycle: A STUDY USING GENETICALLY ENCODED CALCIUM INDICATORS, J Biol Chem, 290 (2015)26914–26926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lourido S, Moreno SN, The calcium signaling toolkit of the Apicomplexan parasites Toxoplasma gondii and Plasmodium spp, Cell Calcium, 57 (2015) 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pace DA, McKnight CA, Liu J, Jimenez V, Moreno SN, Calcium entry in Toxoplasma gondii and its enhancing effect of invasion-linked traits, J Biol Chem, 289 (2014) 19637–19647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Prole DL, Taylor CW, Identification of intracellular and plasma membrane calcium channel homologues in pathogenic parasites, PloS one, 6 (2011) e26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D, Functional dissection of the apicomplexan glideosome molecular architecture, Cell Host Microbe, 8 (2010) 343–357. [DOI] [PubMed] [Google Scholar]
- [13].Endo T, Sethi KK, Piekarski G, Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages, Exp Parasitol, 53 (1982) 179–188. [DOI] [PubMed] [Google Scholar]
- [14].Kafsack BF, Pena JD, Coppens I, Ravindran S, Boothroyd JC, Carruthers VB, Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells, Science, 323 (2009)530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moudy R, Manning TJ, Beckers CJ, The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii, J Biol Chem, 276 (2001) 41492–41501. [DOI] [PubMed] [Google Scholar]
- [16].Schwab JC, Beckers CJ, Joiner KA, The parasitophorous vacuole membrane surrounding intracellular Toxoplasma gondii functions as a molecular sieve, Proc Natl Acad Sci U S A, 91 (1994) 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, Patel R, Macklin JJ, Bargmann CI, Ahrens MB, Schreiter ER, Jayaraman V, Looger LL, Svoboda K, Kim DS, Sensitive red protein calcium indicators for imaging neural activity, Elife, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE, An expanded palette of genetically encoded Ca(2)(+) indicators, Science, 333 (2011) 1888–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, Macklin JJ, Chen Y, Konnerth A, Jayaraman V, Looger LL, Schreiter ER, Svoboda K, Kim DS, High-performance calcium sensors for imaging activity in neuronal populations and microcompartments, Nat Methods, 16 (2019) 649–657. [DOI] [PubMed] [Google Scholar]
- [20].Walker JK, Gainetdinov RR, Feldman DS, McFawn PK, Caron MG, Lefkowitz RJ, Premont RT, Fisher JT, G protein-coupled receptor kinase 5 regulates airway responses induced by muscarinic receptor activation, Am J Physiol Lung Cell Mol Physiol, 286 (2004) L312–319. [DOI] [PubMed] [Google Scholar]
- [21].Ockenga W, Kuhne S, Bocksberger S, Banning A, Tikkanen R, Non-neuronal functions of the m2 muscarinic acetylcholine receptor, Genes (Basel), 4 (2013) 171–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tasaka K, Mio M, Okamoto M, Intracellular calcium release induced by histamine releasers and its inhibition by some antiallergic drugs, Ann Allergy, 56 (1986) 464–469. [PubMed] [Google Scholar]
- [23].Jones TC, Hirsch JG, The interaction between Toxoplasma gondii and mammalian cells. II. The absence of lysosomal fusion with phagocytic vacuoles containing living parasites, J Exp Med, 136 (1972) 1173–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Melo EJ, de Carvalho TU, de Souza W, Penetration of Toxoplasma gondii into host cells induces changes in the distribution of the mitochondria and the endoplasmic reticulum, Cell Struct Funct, 17 (1992) 311–317. [DOI] [PubMed] [Google Scholar]
- [25].Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SR, Usachev YM, Taylor CW, Campbell RE, Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum, Biochem J, 464 (2014) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu C, Hermann TE, Characterization of ionomycin as a calcium ionophore, The Journal of biological chemistry, 253 (1978) 5892–5894. [PubMed] [Google Scholar]
- [27].Berman MC, Characterisation of thapsigargin-releasable Ca(2+) from the Ca(2+)-ATPase of sarcoplasmic reticulum at limiting [Ca(2+)], Biochim Biophys Acta, 1509 (2000) 42–54. [DOI] [PubMed] [Google Scholar]
- [28].Tepikin AV, Mitochondrial junctions with cellular organelles: Ca(2+) signalling perspective, Pflugers Arch, 470 (2018) 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sidik SM, Hortua Triana MA, Paul AS, El Bakkouri M, Hackett CG, Tran F, Westwood NJ, Hui R, Zuercher WJ, Duraisingh MT, Moreno SN, Lourido S, Using a Genetically Encoded Sensor to Identify Inhibitors of Toxoplasma gondii Ca2+ Signaling, J Biol Chem, 291 (2016) 9566–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lourido S, Tang K, Sibley LD, Distinct signalling pathways control Toxoplasma egress and host-cell invasion, EMBO J, 31 (2012) 4524–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brown KM, Lourido S, Sibley LD, Serum Albumin Stimulates Protein Kinase G-dependent Microneme Secretion in Toxoplasma gondii, J Biol Chem, 291 (2016) 9554–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, Wiltsie J, Diaz CA, Harris G, Chang B, Darkin-Rattray SJ, Nare B, Crumley T, Blum PS, Misura AS, Tamas T, Sardana MK, Yuan J, Biftu T, Schmatz DM, Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target, J Biol Chem, 277 (2002) 15913–15922. [DOI] [PubMed] [Google Scholar]
- [33].Collins CR, Hackett F, Strath M, Penzo M, Withers-Martinez C, Baker DA, Blackman MJ, Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress, PLoS Pathog, 9 (2013) e1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Takahashi A, Yamaguchi H, Miyamoto H, Change in K+ current of HeLa cells with progression of the cell cycle studied by patch-clamp technique, Am J Physiol, 265 (1993) C328–336. [DOI] [PubMed] [Google Scholar]
- [35].Fertig N, Blick RH, Behrends JC, Whole cell patch clamp recording performed on a planar glass chip, Biophys J, 82 (2002) 3056–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roiko MS, Svezhova N, Carruthers VB, Acidification Activates Toxoplasma gondii Motility and Egress by Enhancing Protein Secretion and Cytolytic Activity, PLoS Pathog, 10 (2014) e1004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nagamune K, Beatty WL, Sibley LD, Artemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii, Eukaryotic cell, 6 (2007) 2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Putney JW, Bird GS, Cytoplasmic calcium oscillations and store-operated calcium influx, J Physiol, 586 (2008) 3055–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Berridge MJ, Galione A, Cytosolic calcium oscillators, FASEB J, 2 (1988) 3074–3082. [DOI] [PubMed] [Google Scholar]
- [40].Fang J, Marchesini N, Moreno SN, A Toxoplasma gondii phosphoinositide phospholipase C (TgPI-PLC) with high affinity for phosphatidylinositol, Biochem J, 394 (2006) 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bullen HE, Jia Y, Yamaryo-Botte Y, Bisio H, Zhang O, Jemelin NK, Marq JB, Carruthers V, Botte CY, Soldati-Favre D, Phosphatidic Acid-Mediated Signaling Regulates Microneme Secretion in Toxoplasma, Cell Host Microbe, 19 (2016) 349–360. [DOI] [PubMed] [Google Scholar]
- [42].Bisio H, Lunghi M, Brochet M, Soldati-Favre D, Phosphatidic acid governs natural egress in Toxoplasma gondii via a guanylate cyclase receptor platform, Nat Microbiol, 4 (2019) 420–428. [DOI] [PubMed] [Google Scholar]
- [43].Williams MJ, Alonso H, Enciso M, Egarter S, Sheiner L, Meissner M, Striepen B, Smith BJ, Tonkin CJ, Two Essential Light Chains Regulate the MyoA Lever Arm To Promote Toxoplasma Gliding Motility, MBio, 6 (2015) e00845–00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Prole DL, Marrion NV, Identification of putative potassium channel homologues in pathogenic protozoa, PLoS One, 7 (2012) e32264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Arrizabalaga G, Ruiz F, Moreno S, Boothroyd JC, Ionophore-resistant mutant of Toxoplasma gondii reveals involvement of a sodium/hydrogen exchanger in calcium regulation, J Cell Biol, 165 (2004) 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Miranda K, Pace DA, Cintron R, Rodrigues JC, Fang J, Smith A, Rohloff P, Coelho E, de Haas F, de Souza W, Coppens I, Sibley LD, Moreno SN, Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole, Molecular microbiology, 76 (2010) 1358–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vella SA, Calixto A, Asady B, Li ZH, Moreno SNJ, Genetic Indicators for Calcium Signaling Studies in Toxoplasma gondii, Methods Mol Biol, 2071 (2020) 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lou YL, Guo F, Liu F, Gao FL, Zhang PQ, Niu X, Guo SC, Yin JH, Wang Y, Deng ZF, miR-210 activates notch signaling pathway in angiogenesis induced by cerebral ischemia, Mol Cell Biochem, 370 (2012) 45–51. [DOI] [PubMed] [Google Scholar]
- [49].Potapenko ES, Biancardi VC, Florschutz RM, Ryu PD, Stern JE, Inhibitory-excitatory synaptic balance is shifted toward increased excitation in magnocellular neurosecretory cells of heart failure rats, J Neurophysiol, 106 (2011) 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Potapenko ES, Biancardi VC, Zhou Y, Stern JE, Astrocytes modulate a postsynaptic NMDA-GABAA-receptor crosstalk in hypothalamic neurosecretory neurons, J Neurosci, 33 (2013) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chemin J, Monteil A, Briquaire C, Richard S, Perez-Reyes E, Nargeot J, Lory P, Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation, FEBS Lett, 478 (2000) 166–172. [DOI] [PubMed] [Google Scholar]
- [52].Inayat S, Pinto LH, Troy JB, Minimizing cytosol dilution in whole-cell patch-clamp experiments, IEEE Trans Biomed Eng, 60 (2013) 2042–2051. [DOI] [PubMed] [Google Scholar]
- [53].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis, Nat Methods, 9 (2012) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.