

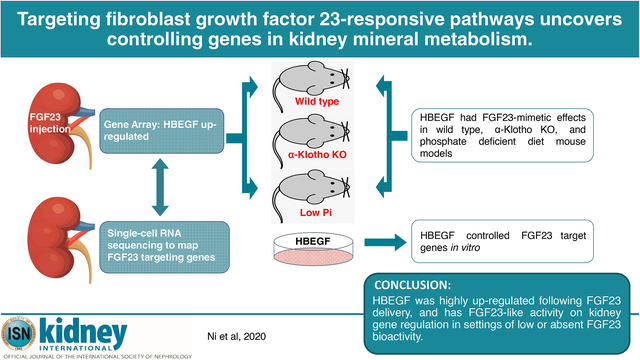
Abstract
Fibroblast Growth Factor 23 (FGF23) is a bone-derived hormone that reduces kidney phosphate reabsorption and 1,25(OH)2 vitamin D synthesis via its required co-receptor alpha-Klotho. To identify novel genes that could serve as targets to control FGF23-mediated mineral metabolism, gene array and single-cell RNA sequencing were performed in wild type mouse kidneys. Gene array demonstrated that heparin-binding EGF-like growth factor (HBEGF) was significantly up-regulated following one-hour FGF23 treatment of wild type mice. Mice injected with HBEGF had phenotypes consistent with partial FGF23-mimetic activity including robust induction of Egr1, and increased Cyp24a1 mRNAs. Single cell RNA sequencing showed overlapping HBEGF and EGF-receptor expression mostly in the proximal tubule, and alpha-Klotho expression in proximal and distal tubule segments. In alpha-Klotho-null mice devoid of canonical FGF23 signaling, HBEGF injections significantly increased Egr1 and Cyp24a1 with correction of basally elevated Cyp27b1. Additionally, mice placed on a phosphate deficient diet to suppress FGF23 had endogenously increased Cyp27b1 mRNA, which was rescued in mice receiving HBEGF. In HEK293 cells with stable alpha-Klotho expression, FGF23 and HBEGF increased CYP24A1 mRNA expression. HBEGF, but not FGF23 bioactivity was blocked with EGF-receptor inhibition. Thus, our findings support that the paracrine/autocrine factor HBEGF could play novel roles in controlling genes downstream of FGF23 via targeting common signaling pathways.
Keywords: FGF23, kidney, alpha-klotho, HBEGF, mineral metabolism
Graphical Abstract
Introduction
The hormone FGF23 was initially identified in a rare hereditary disease, autosomal dominant hypophosphatemic rickets (ADHR)1. FGF23 is produced predominantly in osteoblasts/osteocytes. After secretion, FGF23 acts on kidney to control phosphate and 1,25(OH)2 vitamin D (1,25D) metabolism through the regulation of the renal proximal tubule (PT) Type-IIa and -IIc sodium-phosphate cotransporters (NPT2a and NPT2c), and the 1,25D metabolizing enzymes 1α-hydroxylase (Cyp27b1; anabolic activity) and 24-hydroxylase (Cyp24a1; catabolic activity), respectively. With FGF23 infusion, or transgenic overexpression, NPT2a/NPT2c proteins are withdrawn from the PT apical membrane2, 3. In parallel, Cyp27b1 mRNA levels are reduced whereas Cyp24a1 is elevated4. Consistent with these findings, patients with disorders of elevated FGF23 bioactivity, including ADHR, X-linked hypophosphatemic rickets (XLH), autosomal recessive hypophosphatemic rickets (ARHR, Types 1–3), and tumor induced osteomalacia (TIO), are hypophosphatemic with inappropriately low or normal 1,25D5–10
Mouse models support that FGF23 and alpha-Klotho (KL) are requisite signaling partners, as FGF23- and KL-null mice are phenocopies with respect to mineral metabolism. Genetic ablation of these two proteins results in excessive 1,25D production contributing to hyperphosphatemia, severe growth retardation, vascular and ectopic calcifications and shortened life span11. The difference in KL-null mice is the prevailing elevated FGF23, likely due to a pathogenic feedback loop from end organ resistance to FGF235. Additionally, patients with FGF23 and KL loss of function mutations have the disorder familial hyperphosphatemic tumoral calcinosis (hfTC), and mimic the biochemical phenotypes of the respective null mouse models12–15. Under physiological conditions, FGF23 binds to FGFR-KL complexes to activate the mitogen activated protein kinase (MAPK) pathway16, supporting that high affinity interactions between FGF23 and KL are required for normal mineral metabolism.
Progress has been made in understanding FGF23 interactions with KL and FGFRs. However, whether alternative signaling pathways can be used to control genes downstream of FGF23 remains unknown, and could prove useful when FGF23 cannot be produced or during situations of ‘FGF23 resistance’ when KL is down-regulated. In the present work, a member of the autocrine epidermal growth factor (EGF) family, heparin binding EGF-like growth factor (Hbegf), was a significantly up-regulated gene in kidney following FGF23 delivery. Only the transcription factor early growth response gene-1 (Egr1), a reliable gene marker of the MAPK cascade, known to be responsive to FGFR signaling,17 was more highly induced. When delivered to mice, HBEGF possessed activities mimicking FGF23, thus our findings support that targeting pathways within the FGF23 functional axis may offer alternative means to control key renal gene expression.
Results
Kidney gene expression following FGF23 administration
To identify the factors involved in FGF23-mediated renal phosphate handling, WT mice were injected with recombinant human FGF23 (rhFGF23, 500 ng/g body weight i.p. for 1 h) and whole kidney RNA was isolated. Saline-injected WT mice were used as controls for baseline expressional activity. Gene array was then performed on the kidney RNA from the saline- and FGF23-injected WT animals. We used P<0.001 as the criteria to select significantly changed genes, and found 55 upregulated genes and 26 downregulated genes (Figure 1A, Table S1 and S2). Following array analyses, it was determined that genes previously associated with FGF23-FGFR signaling were the most up-regulated compared to saline-injected mice. These genes included Egr1, Nab2, c-Fos, and c-Jun (Table S1), supporting the validity of this approach. Among the upregulated genes, according to KEGG-pathway analysis, the most prevalent genes were associated with MAPK signaling (Ddit3, Fos, Dusp4, Dusp6 and Mapk12), proteoglycans in cancer (Ank1, Fzd5, Hbegf, Mapk12), TNF signaling (Fos, Junb and Mapk12), infection (Fos, Egr1, Fzd5 and Zfp36), as well as markers associated with cell differentiation (Fos, Junb, Mapk12). As a positive control for successful FGF23 injections, expression of the renal Cyp24a1 mRNA was tested, which showed a significant increase versus kidney expression from vehicle injected mice (Figure 1B). Interestingly, a paracrine factor, Heparin-binding EGF-like growth factor (Hbegf)18, which was not previously associated with renal FGF23 signaling, was identified as the second-most elevated transcript (Figure 1A and Table S1). The increased Hbegf mRNA detected by array was confirmed by individual qPCR using the original kidney RNA from the FGF23-injection study (Figure 1C). Modest Hbegf induction was also observed in KL-null mice (Figure 1D) and an adenine-induced chronic kidney disease (CKD) mouse model (Figure 1E).
Figure 1. Renal gene expression changes following FGF23 administration and in an FGF23 resistant state.
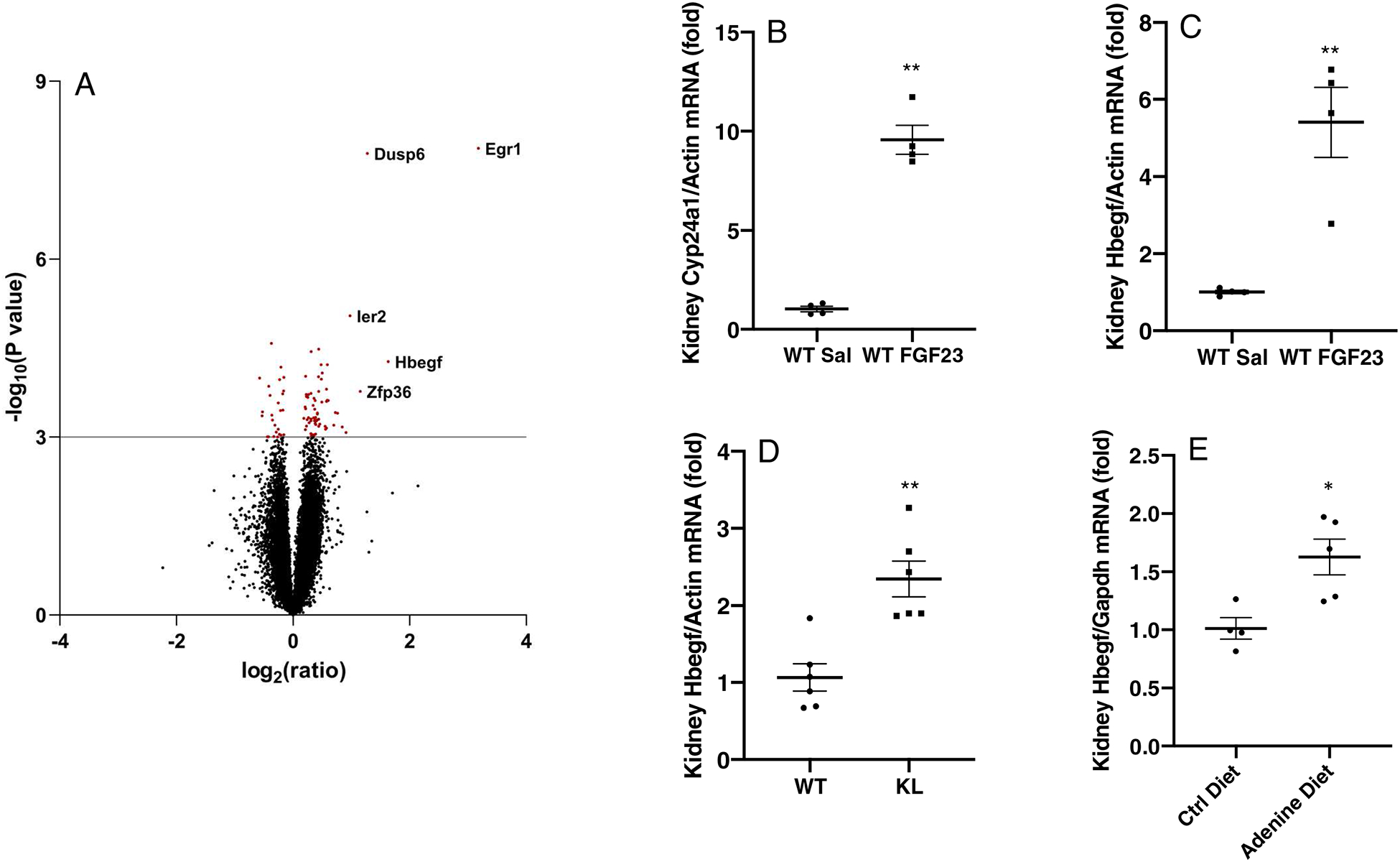
(A) Volcano-plot for microarray analysis, with Y-axis being −log10(P value) and X axis being log2(ratio). A value of P<0.001 was set as the criteria to select significantly changed genes; dots highlighted in red. The top 5 up-regulated genes were labeled with their gene names in the graph. (B-C) 1h FGF23 administration resulted in a significant increase of renal Cyp24a1 and Hbegf mRNA expression. (D) Kidney Hbegf mRNA levels were elevated in the KL knock-out mouse model and in (E) kidneys from an adenine diet induced CKD mouse model (* P<0.05, **P<0.01 versus control group. N= 4–6. ‘WT’, wild type mice. ‘KL’, KL-null).
Single cell RNA sequencing mapping in kidney
The expression of KL is essential for FGF23 to initiate its renal effects through endocrine actions on the PT-expressed genes Npt2a and Cyp27b1/Cyp24a1. Since the renal localization of KL was unclear, and to determine whether Hbegf- and FGF23-mediated signaling could potentially overlap in the nephron, single cell RNA-seq was conducted on WT mouse kidneys. The derived kidney cell clusters were plotted in a Uniform Manifold Approximation and Projection (UMAP) graph and violin plots (Figure 2). The sodium phosphate cotransporter Npt2a was used as a robust PT marker (Figure 2B; Figure S1A), and the sodium chloride cotransporter (Ncc) used as a distal tubule/connecting tubule (DT/CNT) marker (Figure 2C; Figure S1B). Analysis of the single cell sequencing data confirmed that KL mRNA was highly expressed in the DT/CNT and moderately in the PT (Figure 2D, 2G). It also demonstrated that Hbegf (Figure 2E, 2H) along with the Hbegf receptor Egfr (Figure 2F, 2I) had higher expression in PT segments compared to other renal cell clusters. Thus, these results show that KL- and Hbegf-expressing cells localize to PT nephron segments.
Figure 2. Kidney single cell RNA sequencing.
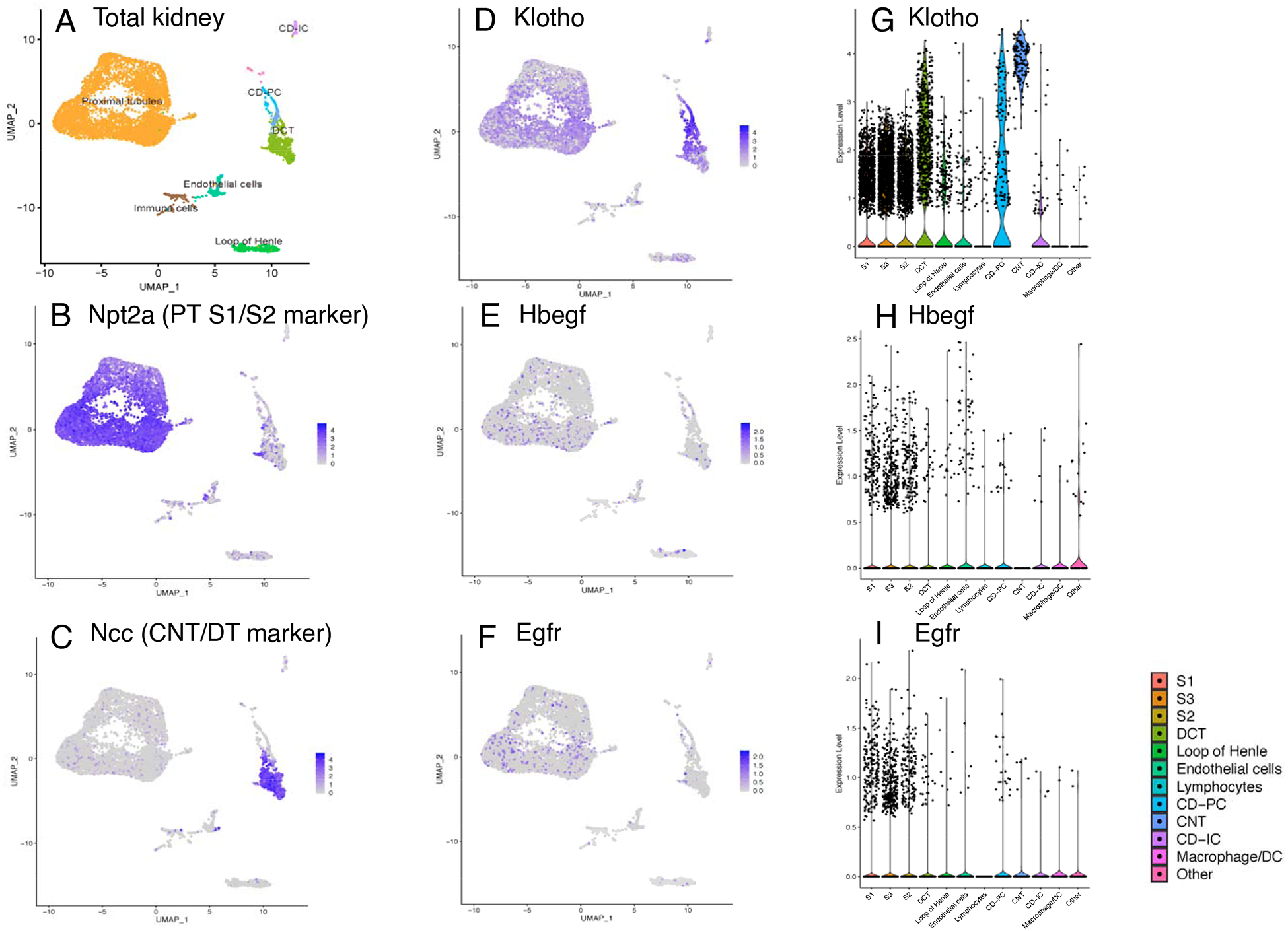
Single cell RNA-seq (scRNAseq) was conducted using WT mouse kidneys. Renal cell clusters were plotted in a UMAP graph (A) and PT and DT/CNT were marked with (B) Npt2a and (C) Ncc. (D) Klotho was expressed in both DT/CNT and PT; (E-F) Hbegf and its receptor Egfr were primarily expressed in PT but also present in other cell groups sporadically as depicted in the Klotho, Hbegf and Egfr violin plots (G-I) (PT, proximal tubule. DT/CNT, distal tubule/connecting tubule).
HBEGF signals in WT mouse kidney
To determine whether HBEGF could mimic the biological actions of FGF23 in vivo, WT female mice were injected with 50, 250, or 500 ng/g body weight of recombinant human HBEGF (rhHBEGF) or saline for 1h and reliable mRNA markers of kidney FGF23 bioactivity were tested. The 50 ng/g body weight rhHBEGF injections demonstrated robust induction of Egr1 mRNA (Figure 3A), a gene known to be associated with HBEGF- and FGF23-dependent MAPK stimulation17–19. Further increases were observed with larger doses (Figure 3A). Importantly, the HBEGF treated mice also had a similar pattern of increased Cyp24a1, which showed a highest induction in 500 ng/g group (Figure 3B). No significant change was observed in Cyp27b1 (Figure 3C). We next employed a longer time course for rhHBEGF to determine this factor’s effects on serum phosphate in WT mice using tail vein administration according to a previous protocol4. Renal Egr1 expression was induced 12-fold by rhHBEGF at 3 h, demonstrating delivery of rhHBEGF, and returned to baseline levels at 9 and 13 h (Figure S2A). Although this protocol showed transcriptional changes with rhHBEGF delivery, serum phosphorus concentrations remained unchanged over the time course (Figure S2B).
Figure 3. HBEGF mimicked FGF23 function on targeted gene expression in WT mice.
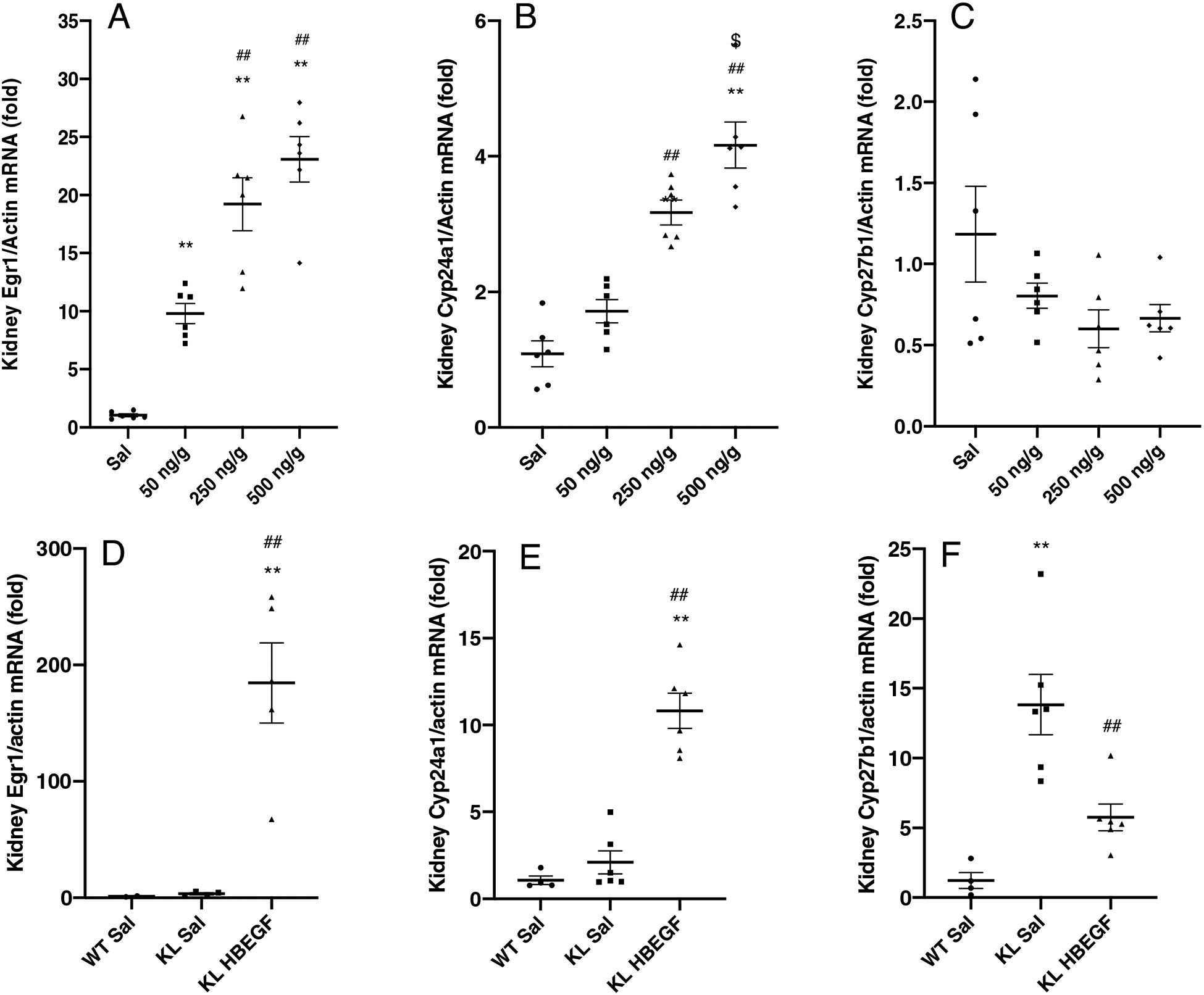
50 ng/g, 250 ng/g and 500 ng/g body weight of rhHBEGF 1 h administration in 8-week old female WT mice robustly induced kidney expression of (A) Egr1 mRNA, with a higher induction in 250 and 500 ng/g groups. (B) Cyp24a1 mRNA increased in 250 ng/g and 500 ng/g dosage groups and had a significantly increase in 500 ng/g compared to 250 ng/g. There were no significant changes in (C) Cyp27b1. rhHBEGF or saline was injected into KL-null mice and compared to WT littermates. 1-hour post-treatment, rhHBEGF administration markedly increased (D) Egr1 mRNA expression (184-fold, P<0.01). (E) Cyp24a1 mRNA, modestly elevated in KL-null mice, demonstrated a further increase in KL-null HBEGF injected mice (10-fold, P<0.01). In the KL-null group, (F) Cyp27b1 mRNA was 13-fold higher compared to WT-saline injected mice (P<0.01) and were partially corrected to 5-fold after rhHBEGF treatment (P<0.01) (*P<0.05, **P<0.01 versus saline control mice. ##P<0.01 versus 50 ng/g rhHBEGF or KL-null saline treated mice. $ P<0.05, versus 250 ng/g rhHBEGF. N=4–6. ‘Sal’, saline. ‘KL’, KL-null).
HBEGF rescues FGF23 function in signaling-deficient mice
To test HBEGF activity in a model devoid of kidney FGF23-mediated signaling capabilities, the KL-null mouse model was employed. rhHBEGF-injected (500 ng/g body weight) KL-null mice markedly increased Egr1 (Figure 3D). These findings support a distinct or downstream signaling pathway from FGF23 actions on Egr1, as FGF23 is already >1000-fold elevated in this model20. Cyp27b1 is known to be elevated at the transcriptional level in KL-null mice due to the loss of FGF23-mediated repression4. Following injection in KL-null mice, HBEGF mimicked in vivo FGF23 actions on kidney gene expression by significantly reducing the prevailing elevated Cyp27b1 mRNA (Figure 3F, already 13-fold baseline vs. WT, corrected to 5-fold, P<0.01). Interestingly, HBEGF administration further increased Cyp24a1, a known action of FGF23 (Figure 3E, 2-fold baseline vs. WT, to 10-fold; P<0.01). These results are consistent with the ability of HBEGF to control renal genes independently of FGF23.
Effects of HBEGF during suppressed FGF23
To determine if HBEGF could function under conditions of reduced FGF23 production, a model of clamped FGF23 expression was used. In this regard, WT mice were placed on a phosphate (Pi) deficient diet (0.02% Pi) for 7 days to suppress intact FGF23 as previously reported21. The low-Pi diet mice had suppressed intact FGF23 (bioactive form) of approximately 95% as measured by ELISA (Figure 4A, P<0.01) compared to mice provided the control diet. Following the diets, rhHBEGF delivery (500 ng/g body weight, one i.p. injection for 1 h) was confirmed by highly increased Egr1 mRNA expression (Figure 4B). In the saline-injected low-Pi diet mice, although intact bioactive FGF23 was significantly suppressed, Cyp24a1 was not different compared to those on the control diet (Figure 4C). In contrast, following rhHBEGF injection, Cyp24a1 mRNA increased by ~5-fold in the low-Pi diet group (Figure 4C). The prevailing elevated Cyp27b1 mRNA during low-Pi diet was reduced from 3-fold to ~2-fold after rhHBEGF administration (Figure 4D). These results support that during marked suppression of FGF23, HBEGF delivery can independently affect genes associated with renal mineral handling.
Figure 4. HBEGF has FGF23-like activity during low phosphate diet.
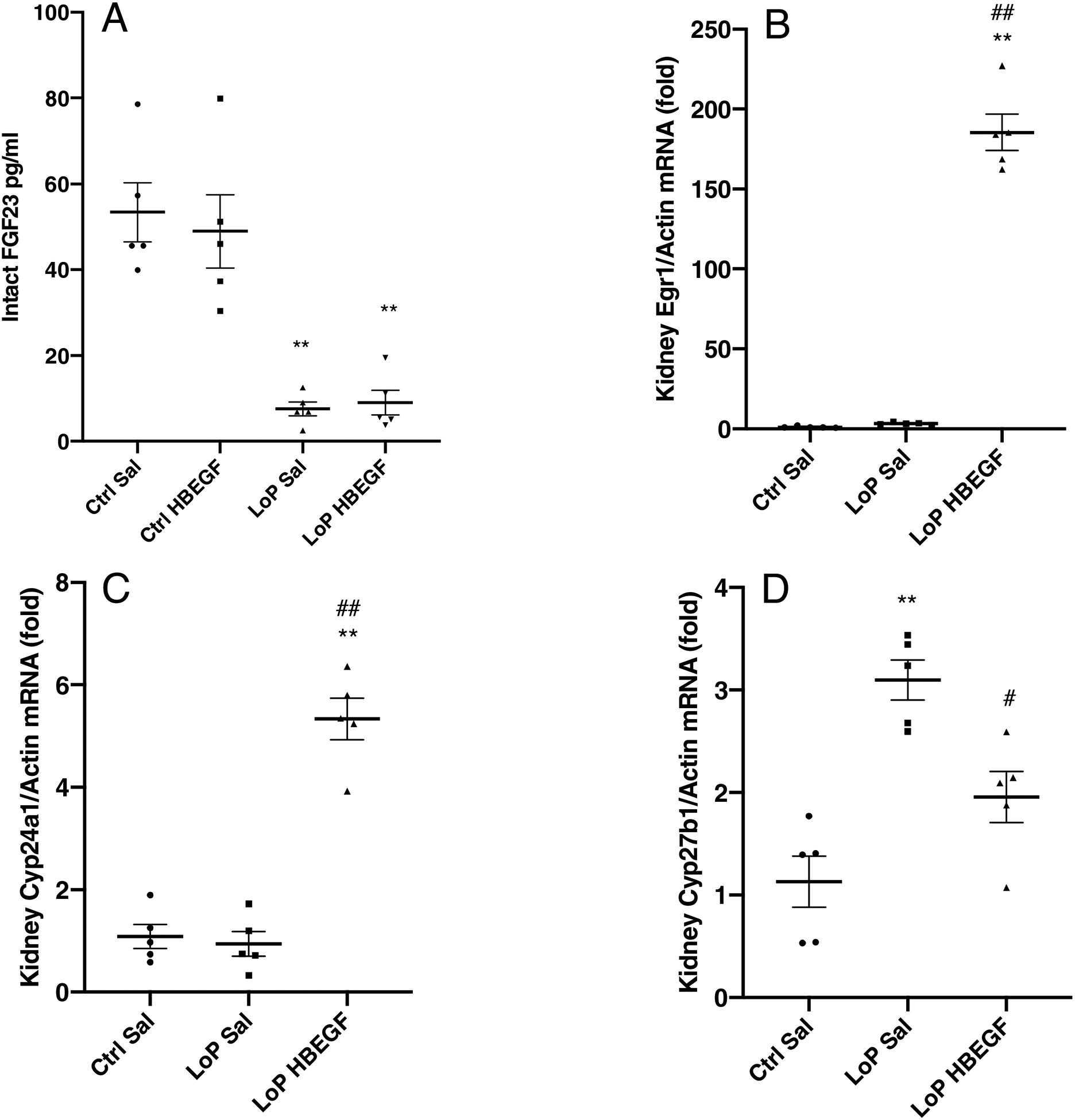
Six-week old WT female mice were placed on a phosphate (Pi) deficient diet (0.02% phosphorus, ‘LoP’) for 1-week and given one dose of rhHBEGF or saline for 1 hour. Low-Pi mice had significantly lowered circulating (A) iFGF23. With rhHBEGF administration, (B) Egr1 increased (180-fold, P<0.01), (C) Cyp24a1 mRNA was elevated 5-fold (P<0.01) and (D) baseline elevated Cyp27b1 mRNA was reduced to 63% compared to saline-injected mice (P<0.05) (**P<0.01 versus control diet saline injection mice. #P<0.05, ##P<0.01 versus HBEGF treatment within the same diet group. N=5. ‘Sal’, saline. ‘LoP’, low phosphate diet).
HBEGF effects on 1,25D metabolizing enzymes in vitro
To examine HBEGF and FGF23 activity in vitro, the native human renal epithelial cell line HEK293, as well as HEK293 cells stably transfected with membrane KL (‘HEK-mKL’ cells), were utilized. The HEK-mKL cells were previously shown to elicit MAPK signaling with FGF23 stimulation, and are known to also express the EGFR16. As examined by immunoblot, HEK-mKL cells receiving rhFGF23 or HEK293 with rhHBEGF treatment for 15 minutes showed robust p-ERK1/2 induction, confirming HBEGF activated the MAPK pathway, similar to FGF23 in the presence of mKL (Figure 5A). HBEGF-treated, but not FGF23-treated cells, showed p-EGFR immunoreactivity (Figure 5A). To test whether HBEGF could target FGF23-controlled genes, HEK293 cells were treated with 500 ng/ml rhHBEGF for 24 hours. A 25-fold increase of EGR1 gene expression was observed (Figure 5B) in concert with a significant increase in CYP24A1 expression (Figure 5C), supporting that in vitro HBEGF could mimic FGF23 function. No significant changes were observed in CYP27B1 (Figure 5D). Additional incremental doses were also tested and were associated with increased CYP24A1 (Figure S3). In parallel, HEK-mKL cells were treated with 500 ng/ml rhFGF23 for 24 hours and showed a 47-fold increase of EGR1 expression (Figure 5E), a 3-fold induction of CYP24A1 (Figure 5F), and a significant decrease in CYP27B1 (Figure 5G). Also, confirming our in vivo results, HBEGF expression increased with FGF23 administration (Figure 5H). These findings support the concept that HBEGF can stimulate similar renal pathways to FGF23, controlling related gene expression.
Figure 5. HBEGF activity on 1,25D processing enzymes in vitro.
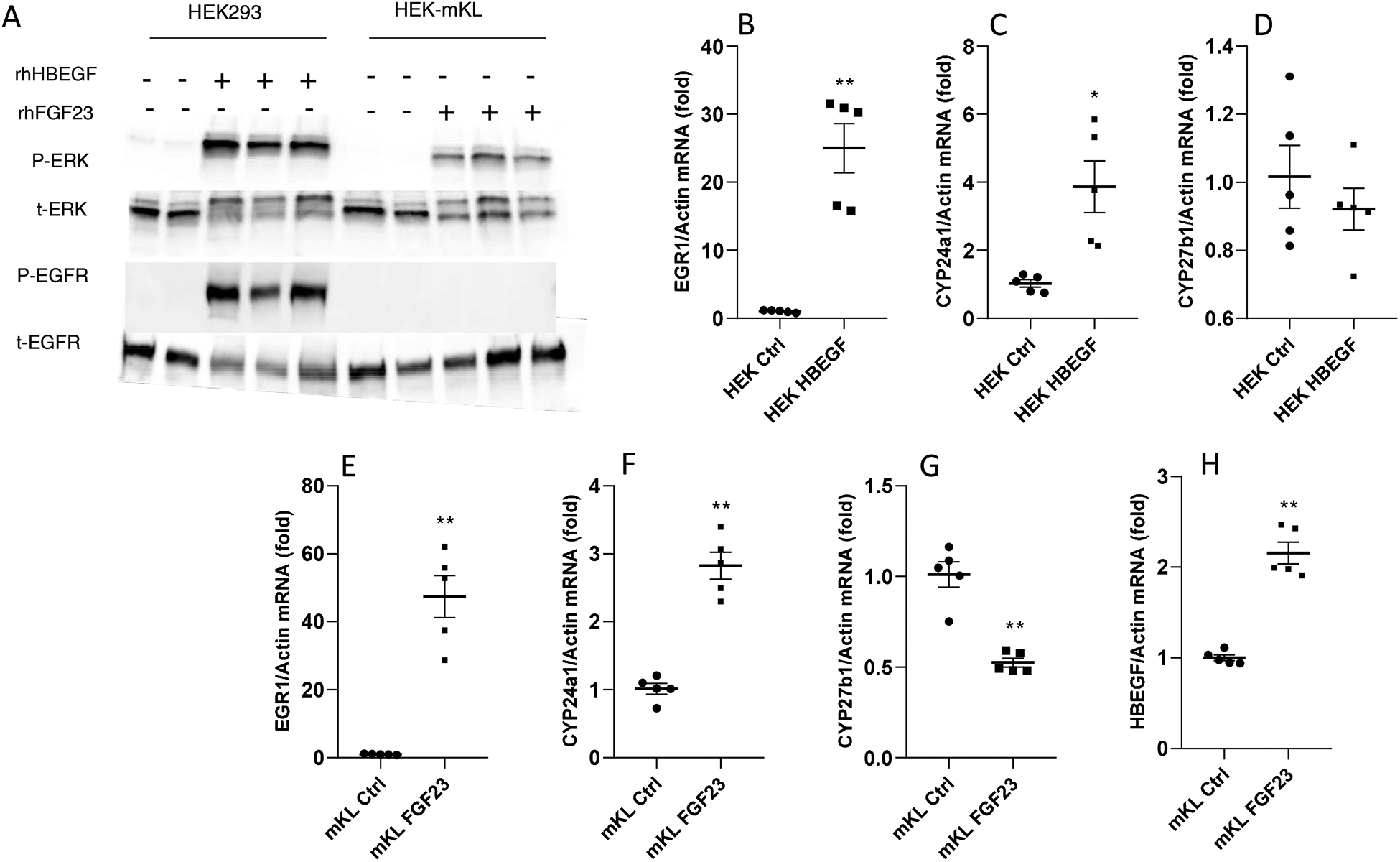
(A) HEK-mKL cells and HEK293 cells were treated with 500 ng/ml rhFGF23 or rhHBEGF respectively for 15-min and then collected for immunoblot analysis, and probed with phospho-ERK1/2, total ERK, phospho-EGFR and total EGFR. HEK293 cells treated with 500 ng/ml rhHBEGF for 24 h had a 25-fold increase of (B) EGR1 mRNA in concert with significantly raised (C) CYP24A1 mRNA, and no changes in CYP27B1 mRNA compared to the saline control (D). HEK-mKL cells were treated with 500 ng/ml rhFGF23 for 24 h and showed significantly increased EGR1 (E) and CYP24A1(F), and a decreased CYP27B1 mRNA level (G). HBEGF mRNA expression was significantly elevated after rhFGF23 treatment (H) (*P<0.05, **P<0.01 versus control group. N=5. ‘Ctrl’, control. ‘mKL’, HEK-mKL).
Function of FGF23 after blocking EGFR in vitro
To determine whether activation of HBEGF is required for FGF23 bioactivity, HEK293 and HEK-mKL cells were treated with AG1478, a specific EGFR tyrosine kinase inhibitor. Treatment with rhHBEGF increased EGR1 mRNA, but AG1478 completely blocked this effect (Figure 6A). In addition, CYP24A1 expression was also completely inhibited by AG1478, (Figure 6B), whereas there was no significant change in CYP27B1 (Figure 6C). In a parallel study in HEK-mKL cells, following FGF23 treatment, EGR1 mRNA expression was slightly reduced with AG1478 pre-treatment (Figure 6D), whereas there was no effect on the increased CYP24A1 mRNA levels (Figure 6E). Therefore, inhibiting EGFR blocked the activation of MAPK by HBEGF and its regulation of vitamin D metabolic genes, however it did not affect FGF23-mediated control of downstream targets.
Figure 6. EGFR inhibition on HBEGF and FGF23 activity in vitro.
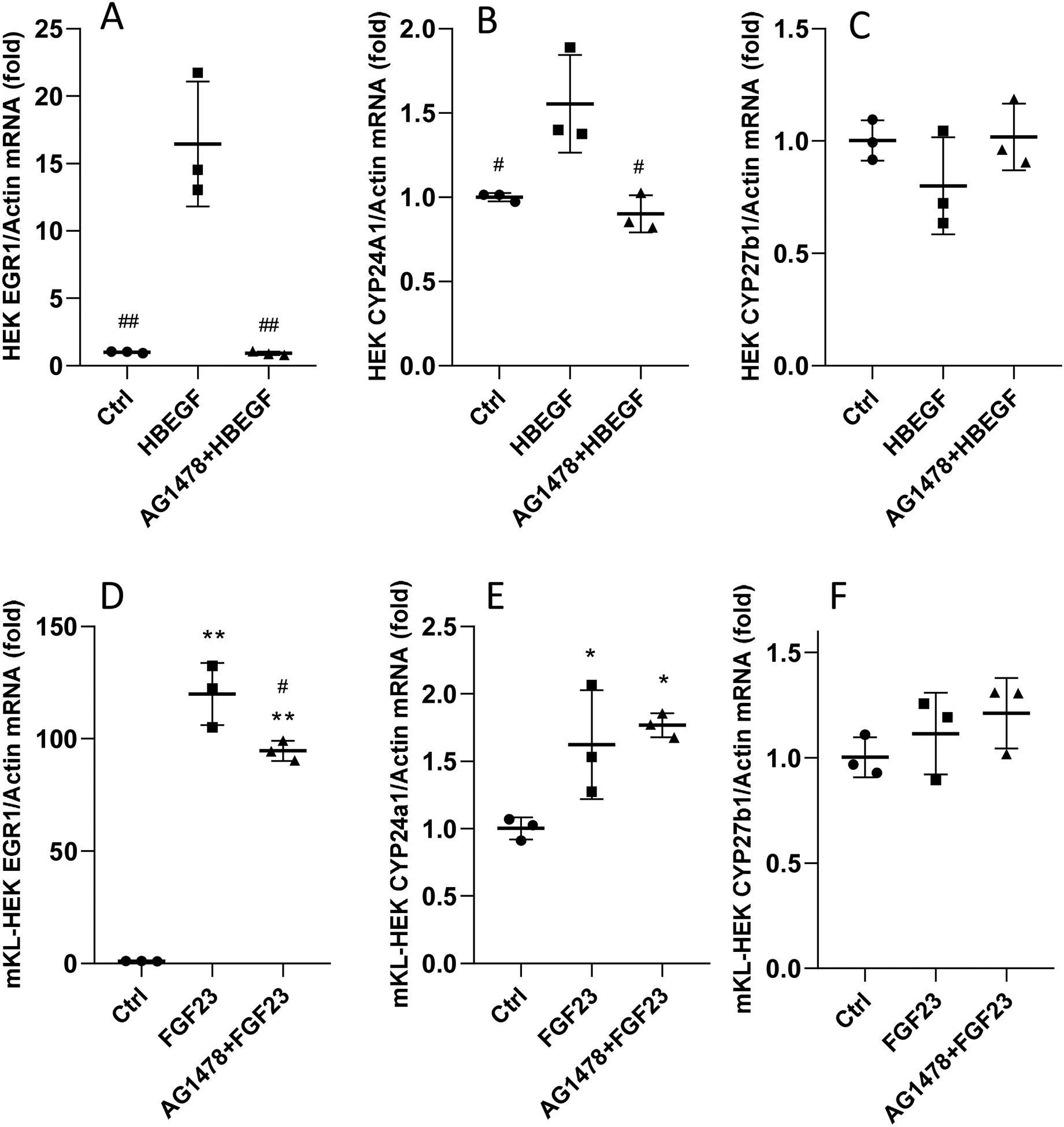
HEK293 cells and HEK-mKL cells were pre-treated with 5 uM AG1478 for 30 minutes, followed by 8 h 500 ng/ml rhHBEGF or rhFGF23; the control cells were treated with 10 ul of DMSO. In HEK293 cells, EGR1 (A) and CYP24A1 (B) mRNAs were induced with rhHBEGF treatment; this increase was blocked by AG1478 and there were no changes in CYP27B1 (C). In HEK-mKL cells, the increased EGR1 was modestly suppressed by AG1478 (D), however CYP24A1 expression remained elevated (E); there were no changes in CYP27B1 (F) (# P<0.05, ##P<0.01 versus HBEGF or FGF23 group. *P<0.05, **P<0.01 versus control group. N=3. ‘Ctrl’, control).
In sum, Hbegf was significantly up-regulated in the kidney following FGF23 injection. When delivered to WT mice and cells, as well as mice with diet-induced suppression of FGF23 or loss of canonical FGF23 signaling (KL-null mice), HBEGF possessed activity mimicking FGF23 on genes controlling MAPK signaling and renal 1,25D metabolism. Our findings support that targeting shared pathways within the FGF23 functional axis may offer alternative means to circumvent situations of altered FGF23 bioactivity.
Discussion
FGF23 over- and under-expression is the cause of multiple disorders of aberrant mineral metabolism; however, the molecular mechanisms directing FGF23 bioactivity in the kidney remain poorly understood. HBEGF is a member of the EGF-ligand family, which constitutes a group of membrane-bound proteins that are proteolytically cleaved to produce paracrine and autocrine factors, and are best known as being associated with cell proliferation22. HBEGF is synthesized as a transmembrane precursor then matured through proteolytic processing by ADAM proteases at the cellular surface causing extracellular shedding22, 23. Soluble HBEGF, the bioactive form of HBEGF, likely acts to activate the MAPK pathway through EGFR (ErbB1) and Her4 (ErbB4)18. Our data supports that associated MAPK activation by HBEGF can result in FGF23-mimetic activity, and in agreement with our studies, Perwad and colleagues showed that HBEGF was an EGR1-target gene using a combination of ChIP-seq and microarray24. Although the previous array data across platforms generally agree with ours, slight discrepancies in changed genes exist24–26. These differences may be explained by experimental design, as some groups injected rat versus human recombinant FGF23, and alternative gene chip manufacturers were used. Even with these differences, FGF23-mediated Hbegf induction was consistently observed across studies. Herein, we used several approaches to test HBEGF’s potential activity in controlling FGF23-related genes. In vivo studies focused on acute time course were conducted first to maximize the direct effects of FGF23 and HBEGF on kidney and reduce compensatory hormone induction. In addition, a longer time course was employed in WT mice to test HBEGF’s effects on serum phosphate, however with direct HBEGF delivery, no change was detected. Although we determined that HBEGF could initiate signaling in the kidney with i.v. delivery, it is possible that in a normal physiological state in wild type mice, HBEGF plays a more direct role in regulating 1,25D metabolic enzymes downstream of MAPK activity versus genes involved in renal phosphate handling. Whether a role for HBEGF in renal phosphate reabsorption could be revealed in animal models with altered FGF23 expression will require further investigation. Our results provided in vivo and in vitro were consistent with the idea that HBEGF targeted known FGF23 downstream transcripts, including 1,25D-metabolizing enzymes, and HBEGF exhibited FGF23-like activity in mice during low-Pi diet administration. Importantly, in the phosphate deficient diet model21, bioactive FGF23 levels were suppressed, and did not change with HBEGF delivery, supporting autonomous HBEGF kidney gene regulation. At this time, however, secondary effects of HBEGF on 1,25D production cannot be fully ruled out. Additionally, in the KL-null mouse model, which lacks high affinity FGF23-mediated bioactivity, but has markedly elevated circulating FGF23, HBEGF administration partially rescued renal Cyp24a1 and Cyp27b1 mRNA expression. Collectively, these studies support that HBEGF can independently activate pathways central to FGF23 function.
The control of genes critical for homeostatic events in the kidney can be regulated through other autocrine ligands of the EGF superfamily. In this regard, it was shown that inactivating mutations in EGF leads to a renal magnesium (Mg2+) wasting syndrome27. Molecular studies supported that the EGF mutations lead to improper cell sorting of the membrane-bound pro-EGF isoform, thus the magnesium wasting is likely due to insufficient Mg2+ reabsorption via the DT apical membrane Mg2+ transporter TRPM6 (transient receptor potential melastatin)27, 28. Additionally, clinical studies demonstrated that patients receiving anti-epidermal growth factor receptor (EGFR) antibody therapy in trials for colorectal and other forms of cancer also demonstrate kidney magnesium wasting29. Previous studies showed when rodents were treated with an anti-EGFR monoclonal antibody (ME-1), they had mild, yet significantly decreased phosphate excretion, increased serum phosphate, as well as decreased serum magnesium30. Taken together, these results potentially support the roles of EGFR acting as the receptor for EGF to control Mg2+ handling, and HBEGF acting in FGF23-regulatory systems.
It was previously demonstrated that when injected acutely, FGF23 activates detectable phosphorylation of ERK1/2 that was originally localized within the renal DT with KL, supporting high KL expression in the DT2,16. Further, it was shown that conditional deletion of KL alleles in DT epithelial cells using the Ksp-cadherin-Cre results in hyperphosphatemia with increased NPT2a that positively correlates with the degree of KL knockdown31. KL has also been reported to be detected in PT, although to a lower extent than DT32. Some have found varying phenotypes with conditional PT-deletion of KL33. One group bred flox-KL mice with the Ndrg1-Cre-ERT2 mice (Ndrg: N-myc downstream-regulated gene-1) to target KL in the PT34. Interestingly this model had increased FGF23, hyperphosphatemia and 1,25D dysregulation, similar to KL-null mice35, 36, consistent with renal PT KL localization and bioactivity. Since the specific targets for these Cres and their expression levels in the nephron segments vary, our group applied single cell sequencing to map KL expression across renal compartments. Abundant KL mRNA was confirmed in the DT/CNT, with modest but widespread expression across the renal PT S1–S3 segments. It is plausible that previous results solely localizing KL to the renal DT were due to relatively low antibody affinity, or alternative KL processing within the PT.
Deriving alternative pathways to potentially modify FGF23 signaling could be relevant for several diseases, as elevated circulating FGF23 in the context of KL deficiency in mice and humans leads to marked disturbances in mineral metabolism14, 37–39. Indeed, our previous studies demonstrated that conditional inhibition of FGF23 expression during CKD exacerbated disease progression5. Herein, it was shown that renal Hbegf mRNA expression was slightly increased in KL-null mice, as well as in a CKD mouse model. In other kidney microarray studies (GSE66494), HBEGF also had a statistically significant but modest increase in CKD patients, and a data set investigating human diabetic nephropathy (GSE131882) showed a possible increase of HBEGF in kidney parietal epithelial cells (PECs). However, additional studies will be needed to determine whether these changes were clinically significant.
In summary, HBEGF is a member of a gene family known to have renal paracrine and autocrine effects. In the kidney, this factor is primarily expressed in the PT under normal physiological conditions, and up-regulated following FGF23 delivery. HBEGF possessed FGF23-like activity in the setting of loss of KL-mediated bioactivity (KL-null mice) and during suppressed circulating FGF23 (phosphate deficient diet). Thus, understanding FGF23-mediated signaling pathways may lead to new targets in diseases of altered FGF23 expression.
Methods
Animal studies
Animal studies were conducted according to the Institutional Animal Care and Use Committee for Indiana University School of Medicine, and comply with the NIH guidelines. Female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). At 8 weeks of age, 50 ng/g, 250 ng/g or 500 ng/g body weight of recombinant human HBEGF (rhHBEGF; R&D System, Minneapolis, MN), 500 ng/g body weight of recombinant human FGF23 (rhFGF23; R&D System), or 100 μl saline vehicle were given intraperitoneally (i.p.) (N=4–6). Mice were euthanized after one hour and tissues were harvested. 9–10 weeks of age female C57BL/6 mice were tail vein injected with 5 ug rhHBEGF or saline vehicle, and were euthanized after 3 h, 9 h and 13 h (N=4–6). Sets of KL-null mice37 and wild type littermates (N=4–6) were maintained on standard diet for 5–8 weeks and given one i.p. injection of 500 ng/g body weight recombinant of rhHBEGF, 500 ng/g body weight of rhFGF23 or 100 μl saline vehicle i.p., then euthanized after one hour. For the diet study, 6-week-old wild type male and female C57BL/6 mice (N=4–6) were placed on a phosphate deficient diet (0.02% phosphorus, Envigo, Indianapolis, IN) for one week, then administrated one dose of 500 ng/g body weight of rhHBEGF or 100 μl saline i.p.. Mice were euthanized after one hour. To induce CKD, 8 weeks old female WT mice were fed a 0.2% adenine-containing rodent diet (Envigo) for 4 weeks, then switched to a 0.15% adenine containing diet (Envigo) for 5 weeks. Casein diet (0.9% phosphate and 0.6% calcium, Envigo) was used as the control diet5.
Gene array
Kidney RNA was labeled and hybridized to Affymetrix Mouse Gene 1.0 ST arrays following the standard WT protocol (GeneChip® Whole Transcript (WT) Sense Target Labeling Assay, rev. 5, www.affymetrix.com). Arrays were scanned and data were imported into Partek Genomics Suite version 6.2 (Partek, Inc., St. Louis, MO). Robust Multichip Average signals (RMA)40 were generated for the core probe sets using the RMA background correction. The log2 transformed signals were used for principal components analysis, hierarchical clustering and signal histograms to determine if there were any outlier arrays41; none were found. The probe sets were analyzed using a 1-way ANOVA by treatment (WT with saline, WT with 1h FGF23 injection). False discovery rates (FDR) were calculated using q-value42. The gene array data was deposited with GEO accession number GSE138992.
Single cell sequencing of kidney
WT kidney single cell samples were prepared per the protocol “Dissociation of mouse kidney using the Multi Tissue Dissociation Kit 2” (https://www.miltenyibiotec.com/upload/assets/IM0015569.PDF) with the following modifications: After termination of the program “Multi_E_2”, 10 mL RPMI1640/10% BSA was added, and homogenate centrifuged (300 rcf for 5 minutes at 4°C). Annexin V dead cell removal (StemCell, Vancouver, Canada) was then performed. The sample was targeted to 10,000 cell recovery and applied to a single cell master mix with lysis buffer and reverse transcription reagents, following the Chromium Single Cell 3’ Reagent Kits V3 User Guide, CG000183 Rev A (10X Genomics Inc., Pleasanton, CA). The resulting library was sequenced on an Illumina NovaSeq 6000; 50K reads per cell were generated and 91% of the sequencing reads reached Q30 (99.9% base call accuracy).
Single cell data analysis
The CellRanger (10x Genomics) pipeline was utilized to align data from sequencing to mm10 genome. CellRanger.mtx and .tsv files were then processed via R software (v.3.5.0), using Seurat (v. 3.0). Cells were filtered for downstream analysis using gene counts between 200–3000, with mitochondrial percentages less than 50. Gene counts were log normalized and scaled to 104. Unsupervised clustering analysis was performed using the Seurat single cell pipeline. Data was deposited as GSE142113.
Cell culture
HEK293 cells and HEK293 stably transfected with a full-length Klotho cDNA (‘HEK-mKL’ cells) were cultured in DMEM/F-12 (Hyclone, Chicago, IL) supplemented with 10% fetal bovine serum, exactly as described16. 2.5 × 105 cells were plated on 12-well plates or 1×105 cells on 24-well plates (Midwest Scientific, Valley Park, MO) for one day, then treated with 500 ng/ml rhFGF23 or 500 ng/ml rhHBEGF, with or without 30 minutes 5 uM AG1478 (EGFR inhibitor; Calbiochem, San Diego, CA) pretreatment. The rhFGF23 and rhHBEGF were added for various treatment times as described in Results. Cell protein lysates were collected with 200 μl 1X cell lysis buffer (Cell Signaling, Danvers, MA) with 0.1mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, Santa Cruz, Dallas, TX), and cell RNAs were collected using ISOLATE II RNA mini kit (Bioline, Boston, MA).
RNA isolation and qPCR
Kidney and cell RNAs were extracted using Trizol (Life Technologies, Carlsbad, CA) or Lysis buffer (Bioline) for in vivo and in vitro isolation, respectively, according to the manufacturer’s protocols. Samples were tested with primers purchased from Applied Biosystems/Life Technologies, Inc., specific for human or mouse EGR1, CYP27B1, CYP24A1, KL, Hbegf, β-actin and Gapdh. The TaqMan One-Step RT-PCR kit (Life Technologies) was used for all analyses, and data were collected with the 7500 Real Time PCR/StepOne Plus system and software (Life Technologies). The qPCR data was analyzed using the 2−ΔΔCT method according to Livac43.
Immunoblotting
Homogenized cell samples were measured for protein concentrations using the Coomassie Plus (Bradford) Assay kit (ThermoFisher Scientific) 35 μg lysates per sample were electrophoresed on AnyKD or 7.5% Mini-Protean TGX Gels (Bio-Rad, Hercules, CA), and transferred to a PVDF membrane (Bio-Rad). Blots were probed with primary antibodies phospho-EGF Receptor (Try1068; Cell Signaling) and phospho-ERK1/2 (Cell Signaling) first. After being stripped, the same membranes were re-probed with total EGF receptor (D38B1; Cell Signaling) and total ERK1/2 (Promega, Madison, WI). Enhanced chemiluminescence (GE Healthcare, Chicago, IL) was used to detect conjugated horseradish peroxidase (HRP) activity.
Serum biochemistries
Serum samples were tested using a COBAS MIRA Plus Chemistry Analyzer (Roche Diagnostics, Basel, Switzerland). Plasma FGF23 was tested using a commercialized Intact FGF23 (‘iFGF23’) ELISA kit (Kainos Laboratories, Tokyo, Japan).
Statistical analysis
Statistical analyses of the data were performed by two-tailed Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test. Means and standard errors of the mean were used in bar graphs. Significant changes were considered when P<0.05.
Supplementary Material
Supplemental Material
Table S1. The top 15 up-regulated genes after acute FGF23 administration
Table S2. The 15 most down-regulated genes after acute FGF23 administration
Figure S1. Violin plots of nephron segment gene markers
Figure S2. Long term effect of HBEGF on WT renal genes
Figure S3. Incremental doses of HBEGF in vitro
Supplementary information is available at Kidney International’s website.
Translational Statement.
FGF23 is a hormone produced in bone, and is increased or decreased in diseases of altered mineral metabolism. The ability to influence pathways associated with FGF23 signaling in the kidney may provide patient benefit by controlling genes independently of FGF23. Studies are needed to determine whether direct targeting of renal pathways could modify expression of FGF23-regulated genes. This information may provide insight into FGF23 downstream signaling, which is not well understood.
Acknowledgements
The authors would like to acknowledge the support by NIH grants R21-AR059278, DK112958-01A1, R01-HL145528 (KEW); and by T32-HL007910 and F31- DK122679 (MLN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
KEW receives royalties for licensing the FGF23 gene to Kyowa Hakko Kirin, Ltd.
Conflict of interest statement: KEW receives royalties for licensing the FGF23 gene to Kyowa Hakko Kirin, Ltd.
References Cited
- 1.Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature genetics. November 2000;26(3):345–8. doi: 10.1038/81664 [DOI] [PubMed] [Google Scholar]
- 2.Farrow EG, Summers LJ, Schiavi SC, McCormick JA, Ellison DH, White KE. Altered renal FGF23-mediated activity involving MAPK and Wnt: effects of the Hyp mutation. The Journal of endocrinology. October 2010;207(1):67–75. doi: 10.1677/joe-10-0181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. July 2004;145(7):3087–94. doi: 10.1210/en.2003-1768 [DOI] [PubMed] [Google Scholar]
- 4.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. March 2004;19(3):429–35. doi: 10.1359/jbmr.0301264 [DOI] [PubMed] [Google Scholar]
- 5.Clinkenbeard EL, Noonan ML, Thomas JC, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI insight. February 21 2019;4(4)doi: 10.1172/jci.insight.123817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. The Journal of clinical endocrinology and metabolism. February 1997;82(2):674–81. doi: 10.1210/jcem.82.2.3765 [DOI] [PubMed] [Google Scholar]
- 7.Ruppe MD. X-Linked Hypophosphatemia. In: Pagon RA, Adam MP, Ardinger HH, et al. , eds. GeneReviews(R). University of Washington, Seattle University of Washington, Seattle. All rights reserved.; 1993. [PubMed] [Google Scholar]
- 8.Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nature genetics. November 2006;38(11):1310–5. doi: 10.1038/ng1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proceedings of the National Academy of Sciences of the United States of America. May 22 2001;98(11):6500–5. doi: 10.1073/pnas.101545198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White KE, Bringhurst FR, Econs MJ. Genetic Disorders of Phosphate Homeostasis. In: Potts John T. J, Jameson JL, Groot LJD, eds. Endocrinology Adult and Pediatric: The Parathyroid Gland and Bone Metabolism. 2010. [Google Scholar]
- 11.Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. The Journal of biological chemistry. March 10 2006;281(10):6120–3. doi: 10.1074/jbc.C500457200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garringer HJ, Fisher C, Larsson TE, et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. The Journal of clinical endocrinology and metabolism. October 2006;91(10):4037–42. doi: 10.1210/jc.2006-0305 [DOI] [PubMed] [Google Scholar]
- 13.Larsson T, Yu X, Davis SI, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. The Journal of clinical endocrinology and metabolism. April 2005;90(4):2424–7. doi: 10.1210/jc.2004-2238 [DOI] [PubMed] [Google Scholar]
- 14.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Musculoskelet Neuronal Interact. Oct-Dec 2007;7(4):318–9. [PubMed] [Google Scholar]
- 15.Nabeshima Y, Imura H. alpha-Klotho: a regulator that integrates calcium homeostasis. Am J Nephrol. 2008;28(3):455–64. doi: 10.1159/000112824 [DOI] [PubMed] [Google Scholar]
- 16.Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. Journal of the American Society of Nephrology : JASN. May 2009;20(5):955–60. doi: 10.1681/asn.2008070783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guha M, O’Connell MA, Pawlinski R, et al. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. September 1 2001;98(5):1429–39. [DOI] [PubMed] [Google Scholar]
- 18.Saito T, Okada S, Ohshima K, et al. Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by betacellulin and EGF. Endocrinology. September 2004;145(9):4232–43. doi: 10.1210/en.2004-0401 [DOI] [PubMed] [Google Scholar]
- 19.Mehta VB, Besner GE. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth factors (Chur, Switzerland). August 2007;25(4):253–63. doi: 10.1080/08977190701773070 [DOI] [PubMed] [Google Scholar]
- 20.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. December 7 2006;444(7120):770–4. doi: 10.1038/nature05315 [DOI] [PubMed] [Google Scholar]
- 21.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. December 2005;146(12):5358–64. doi: 10.1210/en.2005-0777 [DOI] [PubMed] [Google Scholar]
- 22.Vinante F, Rigo A. Heparin-binding epidermal growth factor-like growth factor/diphtheria toxin receptor in normal and neoplastic hematopoiesis. Toxins. June 2013;5(6):1180–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahin U, Weskamp G, Kelly K, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. The Journal of cell biology. March 1 2004;164(5):769–79. doi: 10.1083/jcb.200307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Portale AA, Zhang MY, David V, et al. Characterization of FGF23-Dependent Egr-1 Cistrome in the Mouse Renal Proximal Tubule. PloS one. 2015;10(11):e0142924. doi: 10.1371/journal.pone.0142924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsell R, Krajisnik T, Goransson H, et al. Gene expression analysis of kidneys from transgenic mice expressing fibroblast growth factor-23. Nephrol Dial Transplant. March 2008;23(3):827–33. doi: 10.1093/ndt/gfm672 [DOI] [PubMed] [Google Scholar]
- 26.Dai B, David V, Martin A, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PloS one. 2012;7(9):e44161. doi: 10.1371/journal.pone.0044161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groenestege WM, Thebault S, van der Wijst J, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. The Journal of clinical investigation. August 2007;117(8):2260–7. doi: 10.1172/jci31680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voets T, Nilius B, Hoefs S, et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. The Journal of biological chemistry. January 2 2004;279(1):19–25. doi: 10.1074/jbc.M311201200 [DOI] [PubMed] [Google Scholar]
- 29.Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S. Risk of anti-EGFR monoclonal antibody-related hypomagnesemia: systematic review and pooled analysis of randomized studies. Expert Opin Drug Saf. May 2012;11 Suppl 1:S9–19. doi: 10.1517/14740338.2011.606213 [DOI] [PubMed] [Google Scholar]
- 30.Ortega B, Dey JM, Gardella AR, Proano J, Vaneerde D. Antibody-mediated inhibition of EGFR reduces phosphate excretion and induces hyperphosphatemia and mild hypomagnesemia in mice. Physiol Rep. March 2017;5(5)doi: 10.14814/phy2.13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao X, Johnson JE, Richardson JA, Hiesberger T, Igarashi P. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. Journal of the American Society of Nephrology : JASN. July 2002;13(7):1824–36. [DOI] [PubMed] [Google Scholar]
- 32.Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. September 2010;24(9):3438–50. doi: 10.1096/fj.10-154765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ide N, Olauson H, Sato T, et al. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int. August 2016;90(2):348–362. doi: 10.1016/j.kint.2016.04.009 [DOI] [PubMed] [Google Scholar]
- 34.Endo T, Nakamura J, Sato Y, et al. Exploring the origin and limitations of kidney regeneration. J Pathol June 2015;236(2):251–63. doi: 10.1002/path.4514 [DOI] [PubMed] [Google Scholar]
- 35.Takeshita A, Kawakami K, Furushima K, Miyajima M, Sakaguchi K. Central role of the proximal tubular alphaKlotho/FGF receptor complex in FGF23-regulated phosphate and vitamin D metabolism. Sci Rep. May 2 2018;8(1):6917. doi: 10.1038/s41598-018-25087-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noonan ML, White KE. FGF23 Synthesis and Activity. Curr Mol Biol Rep. March 2019;5(1):18–25. doi: 10.1007/s40610-019-0111-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. November 6 1997;390(6655):45–51. doi: 10.1038/36285 [DOI] [PubMed] [Google Scholar]
- 38.Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. December 2003;17(12):2393–403. doi: 10.1210/me.2003-0048 [DOI] [PubMed] [Google Scholar]
- 39.Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. Journal of the American Society of Nephrology : JASN. January 2011;22(1):124–36. doi: 10.1681/asn.2009121311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. January 22 2003;19(2):185–93. doi: 10.1093/bioinformatics/19.2.185 [DOI] [PubMed] [Google Scholar]
- 41.McClintick JN, Edenberg HJ. Effects of filtering by Present call on analysis of microarray experiments. BMC Bioinformatics. January 31 2006;7:49. doi: 10.1186/1471-2105-7-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America. August 5 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. December 2001;25(4):402–8. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material
Table S1. The top 15 up-regulated genes after acute FGF23 administration
Table S2. The 15 most down-regulated genes after acute FGF23 administration
Figure S1. Violin plots of nephron segment gene markers
Figure S2. Long term effect of HBEGF on WT renal genes
Figure S3. Incremental doses of HBEGF in vitro
Supplementary information is available at Kidney International’s website.