

Abstract
Microbial methane oxidation is a major biofilter preventing larger emissions of this powerful greenhouse gas from marine coastal areas into the atmosphere. In these zones, various electron acceptors such as sulfate, metal oxides, nitrate, or oxygen can be used. However, the key microbial players and mechanisms of methane oxidation are poorly understood. In this study, we inoculated a bioreactor with methane‐ and iron‐rich sediments from the Bothnian Sea to investigate microbial methane and iron cycling under low oxygen concentrations. Using metagenomics, we investigated shifts in microbial community composition after approximately 2.5 years of bioreactor operation. Marker genes for methane and iron cycling, as well as respiratory and fermentative metabolism, were identified and used to infer putative microbial metabolism. Metagenome‐assembled genomes representing novel Verrucomicrobia, Bacteroidetes, and Krumholzibacteria were recovered and revealed a potential for methane oxidation, organic matter degradation, and iron cycling, respectively. This work brings new hypotheses on the identity and metabolic versatility of microorganisms that may be members of such functional guilds in coastal marine sediments and highlights that microorganisms potentially composing the methane biofilter in these sediments may be more diverse than previously appreciated.
Keywords: Bothnian Sea, coastal sediments, iron cycling, low oxygen, methane oxidation, methanotrophs
We identified novel bacteria potentially involved in methane and iron cycling in an oxygen‐limited bioreactor inoculated with methane‐ and iron‐rich Bothnian Sea sediments. Metagenomic analyses provided hypotheses about the mechanisms they may employ, such as the use of oxygen at very low concentrations. Our results imply that in more shallow sediments, where oxygen‐limited conditions are present, microorganisms potentially composing the methane biofilter may be more diverse than previously thought.

1. INTRODUCTION
Archaea and bacteria capable of methane oxidation largely prevent global emissions of methane, a greenhouse gas 28–105 times more potent than carbon dioxide for global warming, into the atmosphere (Shindell et al., 2009). In deep marine sediments, archaeal methanotrophs are predicted to consume more than 90% of the in situ generated methane in cooperation with sulfate‐reducing bacteria (Knittel & Boetius, 2009). In this way, a biofilter that prevents larger methane emissions from these ecosystems is established. Recent estimates suggest that 45–61 Tg of methane is oxidized annually in marine sediments with sulfate, the dominant terminal electron acceptor for methane oxidation in such environments (Egger et al., 2018). However, methanotrophs operating under low oxygen concentrations and using alternative electron acceptors such as iron and manganese oxides, nitrate, or even limited amounts of oxygen are poorly identified, and the mechanisms and metabolism they employ are not yet well explored. Characterizing these microorganisms and understanding their environmental functioning are fundamental to estimate the impacts of climate change and eutrophication on methane cycling in coastal sediments to design better predictive models and create possible future bioremediation and restoration strategies.
Iron oxides are globally distributed in marine coastal sediments (Aromokeye et al., 2020). Anaerobic oxidation of methane coupled to iron reduction (Fe‐AOM) is hypothesized to account for elevated dissolved iron concentrations in methanic zones, particularly in the Baltic and Bothnian Sea, North Sea, and Black Sea (Aromokeye et al., 2020). However, despite strong biogeochemical evidence for Fe‐AOM (Beal et al., 2009; Egger et al., 2015; Riedinger et al., 2014), this remains the least elucidated methane‐cycling metabolism. Archaea affiliated to the cluster ANME‐2d, of the genus Candidatus Methanoperedens, were the first identified microorganisms that show the methane oxidation activity at the expense of Fe and manganese (Mn) reduction, likely reversing the methanogenesis pathway (Ettwig et al., 2016). Candidatus Methanoperedens nitroreducens, the first archaeon described to couple methane oxidation to nitrate reduction (Haroon et al., 2013; Raghoebarsing et al., 2006), was shown to perform Fe‐ and Mn‐AOM in short‐term experiments with iron citrate, nanoparticulate ferrihydrite, and birnessite in follow‐up studies (Ettwig et al., 2016). Recent investigations (Cai et al., 2018; Leu, Cai, et al., 2020) enriched related Fe‐ and Mn‐AOM Methanoperedens species, namely Ca. Methanoperedens ferrireducens, Ca. Methanoperedens manganicus, and Ca. Methanoperedens manganireducens from organic‐rich freshwater sediments in Australia after 2 years of cultivation. The genomes of the various Ca. Methanoperendes strains encode several multiheme c‐type cytochrome proteins that are implicated in the extracellular electron transfer pathways needed to convey electrons to the metal oxides (Arshad et al., 2015; Berger et al., 2017; Leu, Cai, et al., 2020 Leu, McIlroy, et al., 2020).
Interestingly, bacteria commonly implicated in aerobic methane oxidation via particulate methane monooxygenase (PMO) have recently been suggested to be capable of Fe‐AOM via a yet unknown mechanism. Pure cultures of the gammaproteobacterial and alphaproteobacterial methanotrophs Methylomonas and Methylosinus coupled methane oxidation to ferrihydrite reduction under the availability of 0.89 mg O2 L−1 (Zheng et al., 2020). Bacterial methanotrophs were suggested to account for Fe‐AOM in oxygen‐depleted incubations with sediments from Lake Kinneret in Israel (Bar‐Or et al., 2017) and in anoxic waters of Northwestern Siberian lakes (Cabrol et al., 2020). How methane may be activated by PMO in the absence of oxygen or at nanomolar concentrations of oxygen is not yet known, but could have similarities to the mechanism employed by Ca. Methylomirabilis species, which generate oxygen intracellularly (Ettwig et al., 2010, 2012). How pure cultures of these methanotrophs could reduce iron, while their genomes lack known marker genes for iron reduction is another question that remains unanswered.
The brackish Bothnian Sea is located in the northern part of the Baltic Sea and, in contrast to the rest of the Baltic Sea basin, is an oligotrophic ecosystem. These conditions have been established due to the topography, which hinders the input of nutrient‐rich waters from the south. The Bothnian Sea is fed by several major rivers that transport freshwater, terrestrial organic carbon, and metal oxides into the ecosystem (Algesten et al., 2006). Low salinity and high sedimentation rates have enabled the establishment of a relatively shallow sulfate‐methane transition zone (SMTZ). Below the SMTZ, ferric oxides (ferrihydrite) can accumulate and act as terminal electron acceptors for AOM (Egger et al., 2015). A recent study from another area in the Baltic Sea, Pojo Bay estuary in Finland, also provided evidence for AOM below the SMTZ in Fe‐rich coastal sediments (Myllykangas et al., 2020). The microbial communities from both ecosystems exhibited significant similarities, including dominant taxa such as ANME‐2a/b, Methanomicrobia, Bathyarchaeota, Thermoplasmata, Bacteroidetes, Chloroflexi, Verrucomicrobia, and Proteobacteria (α‐, β‐, γ‐) (Myllykangas et al., 2020; Rasigraf et al., 2020). However, a direct link between particular taxa and Fe‐AOM activity in Baltic Sea sediments is still lacking.
Previous metagenomic and biogeochemical studies have indicated that various electron acceptors and different guilds of microorganisms, including various putative methanotrophs, are present in Bothnian Sea sediments (Egger et al., 2015; Rasigraf et al., 2017, 2020) and could affect carbon, sulfur, nitrogen, and iron cycling. To identify and elucidate the metabolism of organisms involved in carbon and iron cycling, we inoculated a bioreactor with oxygen‐depleted sediment from an iron‐ and methane‐rich coastal site in the Bothnian Sea. Our enrichment strategy was based on selecting sediments below the SMTZ in which previous incubation experiments (Egger et al., 2015) and modeling studies (Lenstra et al., 2018; Rooze et al., 2016) have indicated Fe‐AOM activity, as well as in which the metagenomic and 16S rRNA gene analyses evidenced candidate microorganisms (Rasigraf et al., 2017, 2020). The reactor received methane and ferric iron (both Fe(III) nitrilotriacetic acid and ferrihydrite nanoparticles) to enrich microorganisms involved in methane oxidation and iron reduction. Microbial community dynamics were followed with metagenomics after approximately 2.5 years of bioreactor operation.
2. METHODS
2.1. Bioreactor setup
The enrichment culture was operated in a sequencing batch mode in a jacketed 3L‐glass bioreactor (Applikon) at a working volume of 2 L. Medium (0.3 L day−1) was continuously supplied, except during daily settling (1 h) and effluent pumping out times (30 min). The reactor was inoculated on 20 June 2016, with 411 g of wet sediment collected on 6 August 2015, from sampling site NB8 in the Bothnian Sea (N 63°29.071, E 19°49.824) (Lenstra et al., 2018). The sediment was derived from 37 to 42 cm depth, located below the SMTZ, where the pore water sulfate concentration was below the detection limit (<75 µM), but where methane and Fe(II) concentrations were in the millimolar range, and iron oxides were abundant (Lenstra et al., 2018). Between sampling and inoculation, the sediment was stored anoxically at 4°C in the dark in a sealed aluminum bag under dinitrogen gas pressure. The medium consisted of 0.1 mM KH2PO4, 2 mM KCl, 3 mM CaCl2, 80 mM NaCl, 9.5 mM MgCl2, 0.2 mM NH4Cl, 5 mM Fe(III) nitrilotriacetic acid (NTA), and the trace element solution (2 ml/10 L) was made as previously described (van de Graaf et al., 1996) and supplemented with 0.2 mM Ce2(SO4)3. The Fe(III)‐NTA solution (200 mM) was made according to the following protocol: 57.3 g of NTA was added to 200 ml Milli‐Q water and the pH was adjusted to 7–8 with 10 M NaOH until the NTA was dissolved. After the addition of 16.4 g NaHCO3 and 27.0 g FeCl3x6H2O to the dissolved NTA, the volume was adjusted to 500 ml with Milli‐Q water. The solution was made sterile by passing it through a 0.2‐μm filter. Additionally, the reactor received 10–12 mM of Fe(OH)3 (ferrihydrite) nanoparticles, synthesized as previously described (Bosch et al., 2010), once a month from the beginning of the second year of operation.
The medium was constantly sparged with an Ar/CO2 gas mixture (95:5), and the culture was continuously sparged with a CH4/CO2 gas mixture (95:5, the Linde Group) with a flow rate of 10 ml min−1. The liquid volume was maintained by a level‐controlled effluent pump, stirring was set at 150 r.p.m., and the reactor was kept at room temperature (21°C). The pH was monitored using an ADI 1010 Bio Controller (Applikon) and maintained at pH 7.59 by a pH controller loop using potassium hydrogen carbonate (KHCO3) as base and CO2 gas as acid. Oxygen was monitored by a Clark‐type oxygen electrode (Applikon) and measured during activity assays as described below. During the standard operation mode of the bioreactor, oxygen concentrations were below the detection limit of the electrode (0.1% of atmospheric oxygen). To prevent the growth of photosynthetic organisms and to prevent the reduction of iron by UV light, the reactor was wrapped in black foil, and black tubing with low oxygen permeability was used (Masterflex Norprene).
2.2. Activity assays
Whole reactor activity tests were conducted twice, once in the first and once in the second year of operation, as follows. Medium supply, effluent outflow, and base pump were stopped and the reactor was flushed with Ar/CO2 (95:5) while stirring. Methane concentrations were measured in the headspace, and when undetectable (below 1.8 ppm), 13CH4 was added to a final concentration of 10% in the headspace. Fe(III)‐NTA (5–10 mM) or Fe(OH)3 nanoparticles (10 mM) were tested as terminal electron acceptors. For batch activity tests, which were performed by the end of the second year of reactor operation, 12.8–15 ml of reactor biomass was placed into 30‐ml serum bottles and incubated with a combination of electron acceptors and donors (in duplicates). The following conditions were set up: All bottles were kept at 0.5 bar overpressure, electron donors were 13CH4 at 75% of the headspace or 2 mM acetate, and electron acceptors were 15 mM Fe(OH)3 nanoparticles, 15 mM Fe(III) citrate, 2 mM magnesium sulfate, or O2 at 5% of the headspace. Control bottles included biomass and only methane or oxygen.
Headspace samples (100 µl) were withdrawn with a gas‐tight glass syringe (Hamilton, Switzerland), and methane was immediately measured in technical triplicates on an HP 5890 gas chromatograph equipped with a Porapak Q column (80/100 mesh) and flame ionization detector (Hewlett‐Packard). For 13CO2 and O2 technical duplicate measurements, 50 µl of headspace was injected into an Agilent 6890 series gas chromatograph coupled to a mass spectrometer (Agilent) equipped with a Porapak Q column heated at 80°C with helium as the carrier gas. Gas concentrations were calculated using a calibration curve made with gas standards, and liquid‐dissolved concentrations were estimated with the Ostwald coefficient (Battino, 1984). Iron concentrations were measured in technical duplicates using the colorimetric ferrozine method (Lovley & Phillips, 1987). Briefly, 30 μl of a solid‐free reactor liquid sample was mixed with 30 μl 1 M HCl in an Eppendorf tube (for ferrous iron), and in another tube, 30 μl of the solid‐free liquid sample was mixed with 30 μl of saturated hydroxylamine solution in 1 M HCl (for total iron). After incubation for 1 h at room temperature, 10 μl of the solution was added to 100 μl ferrozine reagents (0.05% w/v ferrozine (PDT disulfonate: 3‐[2‐Pyridyl]‐5,6‐diphenyl‐1,2,4‐triazine‐4,4‐disulfonic acid sodium salt) in 50 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) buffer, pH 7.0) and 500 μl of Milli‐Q water. The absorbance was measured at 562 nm using a SpectraMax 190 microplate reader (Molecular Devices). The ferric iron content was calculated by subtracting the concentration of ferrous iron from the concentration of the total reduced ferrous iron. Activity data were imported into RStudio v1.2.5033 (R v3.6.3) (R Core Team, 2017), and graphs were constructed with ggplot2 v3.3.2 (Wickham, 2016).
2.3. DNA extraction, metagenomic sequencing, data processing, and analyses
DNA was extracted from original sediments (hereafter T0) and biomass after 16 months (T1) and 29 months (T2) of reactor operation. All DNA extractions were performed using the DNeasy PowerSoil DNA extraction kit (Qiagen) according to the manufacturer's instructions. One extra DNA extraction of T2 with the ammonium acetate method was conducted and used only for assembly and binning purposes. Metagenomic sequencing was performed in‐house using the Illumina Nextera® XT Library Prep Kit according to the manufacturer's instructions (Illumina). The library was normalized to 4 nM, and sequencing was performed with an Illumina MiSeq using the sequencing protocol for 300 bp paired‐end reads. The resulting reads (~4 Gbp per sample) were trimmed and quality‐controlled with BBDuk (https://sourceforge.net/projects/bbmap/), and then co‐assembled with MEGAHIT v1.2.9 (Li et al., 2016). Read mapping was performed using BBMap (https://sourceforge.net/projects/bbmap/) and mapping files were handled using samtools v1.9 (using htslib v1.9) (Li et al., 2009). Contigs were binned with four methods all using default parameters: binsanity v2.0.0 (Graham et al., 2017), concoct v1.1.0 (Alneberg et al., 2013), maxbin2 v2.2.7 (Wu et al., 2015), and metabat2 v2.15 (Kang et al., 2019). Bins were supplied to DAS tool v1.1.2 (Sieber et al., 2018) for consensus binning and to CheckM v1.1.2 (Parks et al., 2015) for quality inference of metagenome‐assembled genomes (MAGs).
Taxonomic classification of MAGs was obtained using GTDB‐Tk v0.3.2 (Chaumeil et al., 2019), and phylogenetic trees for MAG placement were constructed using UBGC v3.0 (Na et al., 2018). MAGs were gene‐called with prodigal v2.6.3 (Hyatt et al., 2010) and annotated with KEGG KAAS (Moriya et al., 2007), prokka v1.13.3 (Seemann, 2014), and DRAM v0.0.1 (Shaffer et al., 2020) in KBase (Arkin et al., 2018). Marker genes for iron metabolism were searched with FeGenie (Garber et al., 2020), while other genes of interest involved in methane, nitrogen, sulfur, and general carbon metabolism were searched using hmmsearch (HMMER 3.3 with ‐‐cut_tc) (Eddy, 2011), blastp (Altschul et al., 1990), prokka, and DRAM annotation files. The following hmms were downloaded from PFAM (https://pfam.xfam.org/) or TIGRFAMs (http://tigrfams.jcvi.org/cgi‐bin/index.cgi): PF02240.16 (MCR_gamma), PF02241.18 (MCR_beta), PF02783.15 (MCR_beta_N), PF02249.17 (MCR_alpha), PF02745.15 (MCR_alpha_N), PF14100.6 (PmoA), PF04744.12 (PmoB and AmoB), PF02461.16 (AMO), TIGR04550 (MmoD), PF02406.17 (MmoB/DmpM family), and PF02964.16 (Methane monooxygenase, hydrolase gamma chain). Average amino acid identity was calculated using the enveomics tool (Rodriguez‐R & Konstantinidis, 2016) available at http://enve‐omics.ce.gatech.edu.
Phylogenetic trees for specific genes of interest were constructed by retrieving reference sequences from NCBI (https://www.ncbi.nlm.nih.gov/protein/), aligning them with MUSCLE v3.8.31 (Edgar, 2004), stripping alignment columns with trimAl v1.4.rev22 (Capella‐Gutierrez et al., 2009) (with ‐gappyout) and calculating trees with FastTree v2.1.10 (Price et al., 2010). Trees were visualized on iTol (Letunic & Bork, 2016). Reads per kilobase per million mapped reads (RPKM) values for genes of interest were calculated by mapping reads to these genes with BBMap using the option rpkm. Data frames were imported into RStudio v1.2.5033 (R v3.6.3) (R Core Team, 2017), and heat maps were constructed using the packages vegan v2.5–6 and ggplot2 v3.3.2 (Wickham, 2016), with the function heatmap.2. All figures were edited in Adobe Illustrator CC 2018 (Adobe).
3. RESULTS AND DISCUSSION
3.1. Retrieved metagenome‐assembled genomes revealed changes in microbial community composition after 2.5 years of reactor operation
Over 29 months, the occurrence of iron reduction was obvious from the color change in the reactor and production of iron (II) in activity tests (Figure A1). Consumption of methane, especially at the expense of iron (III), was less clear (Figure A1). We did observe the production of 13CO2 from 13CH4, but could not ascertain that this was coupled to the stoichiometric iron reduction. Most surprisingly, after more than 2 years of operation under low liquid‐dissolved oxygen concentrations, generally between 4.7 and 0.27 µM, we still detected high levels of oxygen‐dependent methane oxidation (Figure A1).
To investigate changes in the microbial community, original sediments and bioreactor samples were subjected to DNA extractions and metagenomic sequencing. Illumina sequencing, co‐assembly, and binning allowed the reconstruction of 56 MAGs (Figure 1) from three time points: original sediments (T0), bioreactor biomass 16 months after reactor inoculation (T1), and 29 months after reactor inoculation (T2). Together, these 56 MAGs represented 35.8%, 81.5%, and 79.1% of the metagenome reads at T0, T1, and T2, respectively. In original sediments, three MAGs represented 6%–9% of metagenome reads each: thermoplasmata_1, aminicenantales_1, and bipolaricaulia_1. Using the percentage of reads mapped to each genome as a proxy for abundance, these organisms seemed to disappear by T2 (Table S1 at https://doi.org/10.5281/zenodo.4478410). At T1, 31% of the metagenome reads mapped to MAG desulfuromonadales_1, potentially representing the most abundant organisms in the reactor at that time, followed by bacteroidales_2, with 8.6%, and burkholderiales_1, with 6.9%. By T2, these microorganisms appeared to become rare members of the community. Finally, at T2, three MAGs accounted for 29.7% of the metagenome reads the verrucomicrobial MAG pedosphaeraceae_1, with 10.5% of reads, bacteroidales_8 with 10.1%, and krumholzibacteria_1 with 9.1% (Table S1 at https://doi.org/10.5281/zenodo.4478410).
FIGURE 1.
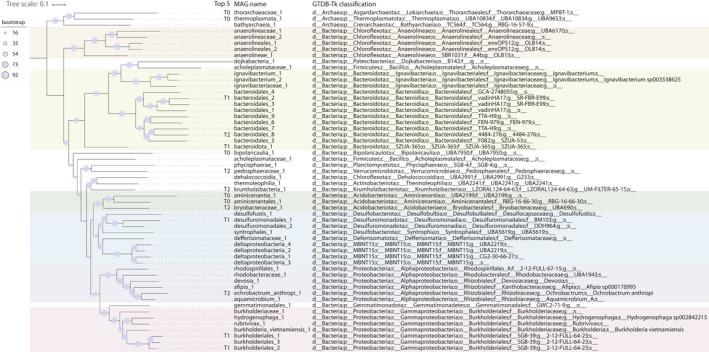
UBCG phylogenetic tree of MAGs retrieved in this study. Ninety‐two conserved genes were used for tree construction (Na et al., 2018). MAGs are named after their taxonomy, which was assigned with GTDB‐Tk (Chaumeil et al., 2019). The top five most abundant MAGs in each metagenome (T0, T1, and T2) are indicated. Colors highlight the following taxa: orange, Anaerolineae; yellow, Bacteroidota; green, Acidobacteriota; blue, Deltaproteobacteria; purple, Alphaproteobacteria; and pink, Gammaproteobacteria
The MAGs retrieved in this study reflect findings from previous metagenomic and 16S rRNA gene analyses of sediments from site NB8 (Rasigraf et al., 2020), which were used as inoculating material for the bioreactor in this study. Anaerolinea and Bacteroidetes were detected at increasing 16S rRNA gene‐based relative abundances with depth in NB8 and were hypothesized to perform fermentation. Other groups previously identified in NB8 via 16S rRNA gene analyses (Rasigraf et al., 2020) included Verrucomicrobia, Planctomycetes, Ignavibacteria, Actinobacteria, Alphaproteobacteria (particularly Rhodospirillales and Rhizobiales), Deltaproteobacteria, and Gammaproteobacteria. A previously recovered MAG classified as Aminicenantes had the potential for acetogenesis, sulfate reduction, and fermentation, while Thorarchaeota and Bathyarchaeota MAGs were hypothesized to participate in fermentative production of acetate, formate, and ethanol (Rasigraf et al., 2020). Similarly, other studies (Broman et al., 2017; Hugerth et al., 2015; Rasigraf et al., 2017; Thureborn et al., 2013) that sequenced DNA from the Baltic Sea water or sediments have identified several common groups, including Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Firmicutes, Gemmatimonadetes, Planctomycetes, Alphaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, Thermoplasmata, and Verrucomicrobia.
Shifts in microbial community composition detected in this study via several metagenomic analyses (further detailed in the next sections) highlight microbial successions over time and might be explained by a combination of factors. First, we hypothesize that, due to high terrestrial influence in the Bothnian Sea, sediments used as inoculum carried recalcitrant organic matter (Hoikkala et al., 2015) that could have been gradually degraded, providing differing pools of carbon at times and thus affecting the microbial community structure. Second, carbon fixation and biomass turnover might also have provided different pools of organic matter into the reactor. Finally, monthly shots of ferrihydrite nanoparticles in addition to the constant inflow of Fe(III)‐NTA starting approximately one year after reactor inoculation may have changed the bioreactor conditions periodically.
3.2. Marker gene analyses highlight the potential for methane and iron cycling, as well as oxygen respiration
Genes of interest were searched in MAGs to investigate the functional potential for methane and iron cycling, as well as respiratory metabolism (oxygen reduction, denitrification, and sulfate reduction) and carbon fixation pathways. The analyzed marker genes for methane cycling were subunits of the methyl‐coenzyme M reductase (mcrA, mcrB, and mcrG), soluble methane monooxygenase (mmoB and mmoX), and particulate methane monooxygenase (pmoA and pmoB). RPKM values derived from read mapping to genes were used as a proxy for gene abundance across the three metagenomes.
No canonical soluble (mmo) or particulate (pmo) methane monooxygenase genes were found in this study's entire dataset. Five mcr genes with best blastp hits to the ANME‐2 cluster archaeon assembly GCA_009649835.1 were identified in unbinned contigs (three in the same one). RPKM values indicate that the organism represented by these genes was abundant in original sediments but was selected against in the reactor (Figure A2). While it is unclear why archaea did not thrive in the reactor, we hypothesize that keeping the reactor at atmospheric pressure and room temperature (21°C) might have played a role as methane dissolves less in the liquid phase compared to a pressurized setting at colder temperatures. Alternatively, the sediment storage time until bioreactor inoculation might have been detrimental to ANME. The identification of mcr genes aligns with previous 16S rRNA gene analyses of NB8 sediments (Rasigraf et al., 2020), which revealed ANME‐2a/b dominated archaeal communities at depths 14–30, 34–45, and 50–63 cm in this site (in the SMTZ and below as well). Archaea affiliating to this group were hypothesized to mediate SO4 2−‐ and Fe‐AOM in distinct sediment zones (Rasigraf et al., 2020), directly reducing iron or, alternatively, transferring electrons to an iron‐reducing organism.
Next, we looked at C1 metabolism. Genes encoding nicotinamide adenine dinucleotide (NAD)‐dependent methanol dehydrogenases were found in the following 13 MAGs: bryobacteraceae_1, thermoleophilia_1, aminicenantales_1, burkholderiales_1, burkholderiales_2, burkholderiales_3, anaerolineaceae_1, deltaproteobacteria_1, deltaproteobacteria_2, desulfofustis_1, devosia_1, rhodospirillales_1, and pedosphaeraceae_1. Shifts in the microbial community were also clear from RPKM values of methanol dehydrogenases over time (Figure A3). By T2, MAG pedosphaeraceae_1 had the methanol dehydrogenase with the highest RPKM value, followed by rhodospirillales_1 and deltaproteobacteria_1.
Marker genes for iron oxidation and iron reduction were searched with FeGenie (Garber et al., 2020). Marker genes for iron oxidation identified in some MAGs of this study (Figure 2) encoded the following: cyc2, iron oxidases; cyc1, periplasmic cytochrome c 4, part of an iron oxidation complex; foxE, a c‐type cytochrome; and foxY, a protein containing the redox cofactor pyrroloquinoline quinone (PQQ). Other MAGs had marker genes for iron reduction that encoded several porins and outer membrane c‐type cytochromes described in various iron‐reducing microorganisms (Garber et al., 2020). RPKM values for these genes highlighted microbial community shifts over time in the reactor (Figure 2). The desulfuromonadales_1 MAG appeared to be particularly abundant in T1 when the reactor started to receive monthly inputs of ferrihydrite nanoparticles in addition to the constant inflow of Fe(III)‐NTA provided in the medium. At T2, when the supply of ferrihydrite was stopped, the functional guild of iron reduction seemed to be spread across other members represented by MAGs deltaproteobacteria_4, ignavibacteriaceae_1, deltaproteobacteria_1, and krumholzibacteria_1. As for iron oxidation, three MAGs had cyc2 genes: aminicenantales_1 and aminicenantia_1, which may represent iron‐oxidizing microorganisms abundant in original sediments, and krumholzibacteria_1, which seemed abundant at the latest enrichment culture metagenome (T2). These results are supported by whole reactor activity tests performed 6 months after reactor inoculation, which evidenced the iron reduction (Figure A1). While we did not conduct iron oxidation activity tests, we hypothesize that iron (II) oxidation by microaerophilic microorganisms could have occurred in the bioreactor. Active iron (III) reduction could have generated iron (II), which could have been oxidized with traces of oxygen detected in the bioreactor. While Deltaproteobacteria and Ignavibacteria are known iron reducers (Hedrich et al., 2011; Podosokorskaya et al., 2013), Aminicenantes have only recently been suggested to perform iron oxidation due to the identification of cyc2 gene sequences in draft genomes (Garber et al., 2020), corroborating findings from this study. Given the widespread occurrence of Aminicenantes bacteria in Bothnian Sea sediments (Rasigraf et al., 2020), their contribution to iron cycling could be significant. The identification of OmcS and Cyc2‐encoding sequences in Krumholzibacteria is described here for the first time and indicates the potential for iron reduction or oxidation in this taxonomic group.
FIGURE 2.
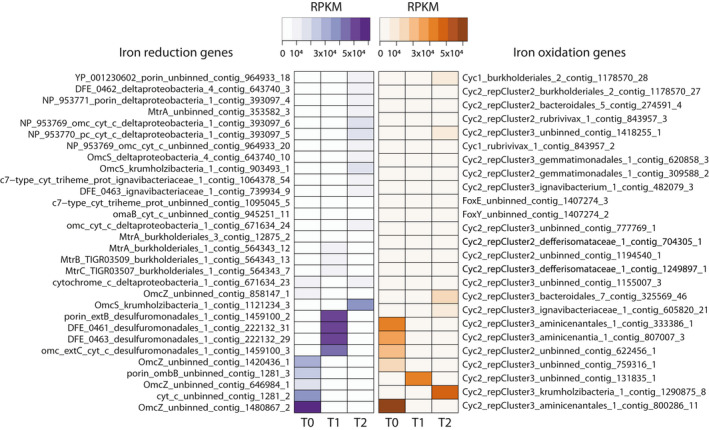
Heat maps of marker genes for iron reduction and iron oxidation in the three metagenomes (T0, T1, and T2). Marker genes were identified with FeGenie (Garber et al., 2020). RPKM values were calculated from read mapping with BBMap (https://sourceforge.net/projects/bbmap) and imported into RStudio (R Core Team, 2017) for heat map construction
MAGs were mined for the potential use of terminal electron acceptors other than iron (Table S1 at https://doi.org/10.5281/zenodo.4478410). The investigated marker genes encode enzymes involved in denitrification (nar, nir, nor, nos, and nrf), sulfate reduction (dsr), and oxygen respiration (several quinol and cytochrome c oxidases). The gene dsrA was found in 8 MAGs, although in desulfofustis_1, it likely encodes a protein subunit involved in sulfur oxidation. Marker genes for denitrification and dissimilatory nitrate reduction to ammonium (DNRA) were present in 48 MAGs, but none had the full denitrification pathway. Potential for oxygen respiration was also widespread: 37 MAGs had cox genes, 19 had cbb3‐type subunit‐encoding genes, and 32 had cyd genes. The widespread functional potential for the use of alternative terminal electron acceptors has been previously reported: Potential for dissimilatory sulfate reduction was detected in previous NB8 MAGs affiliated to Bacteroidales, Xanthomonadales/Chromatiales, Aminicenantes, Syntrophobacterales, and Gemmatimonadales (Rasigraf et al., 2020). In an investigation of Bothnian Sea site US5B, narG, napA, nirK, nirS, nor, nosZ, and nrfA were identified across the depth profile but seemed particularly abundant at shallower sediments, where nitrogen oxides would be available for respiration (Rasigraf et al., 2017).
The persistence of oxygen respiration potential in oxygen‐depleted sediments may occur upon the rapid burial of metabolically versatile microorganisms due to high sedimentation rates, as reported for our study site (Lenstra et al., 2018). In surface sediments where oxygen is still available (here: 0–3 mm depth in summer (Hellemann et al., 2017)), oxygen respiration is expected to be the predominant metabolic mode. As microorganisms are buried and oxygen becomes depleted, alternative electron acceptors (i.e., sulfate and metal oxides) and other metabolic pathways might then be employed by these microorganisms.
Genes involved in carbon fixation pathways were investigated to determine the potential for autotrophy, which could have accounted for inputs of organic matter into the reactor (Table S1 at https://doi.org/10.5281/zenodo.4478410). Our results indicate a widespread potential for carbon fixation among MAGs: Ribulose bisphosphate carboxylase (RuBisCO) genes were used as markers for the Calvin–Benson–Bassham cycle and were identified in 7 MAGs. Genes encoding 2‐oxoglutarate synthase and pyruvate‐ferredoxin/flavodoxin oxidoreductase, markers for the reverse tricarboxylic acid (TCA) pathway, were both found in 30 MAGs. Genes for acetyl‐CoA/propionyl‐CoA carboxylase, markers of the 3‐hydroxypropionate cycle, were found in 22 MAGs. Finally, the carbon monoxide dehydrogenase/acetyl‐CoA synthase, a marker for the reductive acetyl‐CoA pathway, was encoded in 8 MAGs.
3.3. Enrichment of a novel Verrucomicrobia, Bacteroidetes, and Krumholzibacteria microorganisms
The MAG pedosphaeraceae_1 likely represented an abundant microorganism enriched after ~2.5 years of reactor operation, with the percentage of reads mapped to this genome going from 0% (T0) to 0.1% (T1) and 10.5% (T2) (Table S1 at https://doi.org/10.5281/zenodo.4478410). The estimated genome completeness was 98.6%, with 8.8% redundancy. This MAG had most genes encoding enzymes in the aerobic pathway for methane oxidation, except for a canonical methane monooxygenase. However, pedosphaeraceae_1 harbored 7 pmoA‐family sequences that, in a phylogenetic tree with verrucomicrobial reference sequences, formed a cluster (indicated by the number 2, green highlight, in Figure 3) with other hypothetical proteins and pmoA‐family sequences. Another cluster (indicated by 1, purple highlight, in Figure 3), monophyletic with cluster 2, contained mostly canonical reference pmoA sequences, while sequences not in these clusters were mostly reference pmoB.
FIGURE 3.
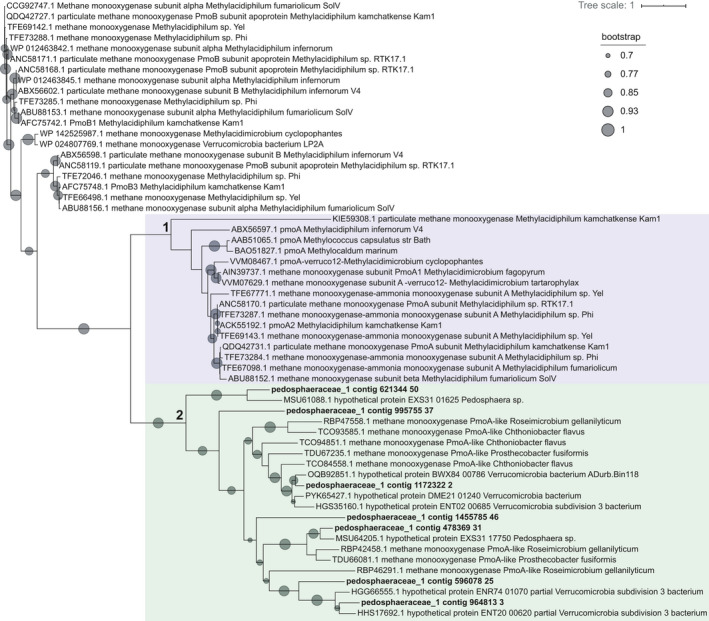
Phylogenetic tree of PmoA‐family sequences found in the MAG pedosphaeraceae_1 and reference verrucomicrobial sequences. Reference sequences were retrieved from the NCBI RefSeq database and are indicated by accession numbers. Sequences were aligned with MUSCLE (Edgar, 2004), alignment columns were stripped with trimal (Capella‐Gutierrez et al., 2009), and the tree was built with FastTree (Price et al., 2010) using the Jones–Taylor–Thornton model of amino acid evolution (Jones et al., 1992)
Other genes in the pathway for aerobic methane oxidation included a NAD‐dependent methanol dehydrogenase, the three proteins involved in the tetrahydrofolate pathway for formaldehyde oxidation to formate, and a formate dehydrogenase gamma subunit (other subunits were missing). Electron transport chain proteins for oxygen respiration were mostly present: all subunits of the NADH dehydrogenase (type I), subunits sdhABC encoding succinate dehydrogenase, oxygen reductase‐encoding genes cyoE, ctaA, coxC, cbb3‐type subunits I/II and III, and cydAB. Nitrogen dissimilatory metabolism genes narGH, napAB, norB, and nrfAH were also present in the genome, as well as all genes in glycolysis, the TCA cycle, and the pentose phosphate pathway. Marker genes for the reverse TCA cycle (korAB, por/nifJ, porB) and the 3‐hydroxypropionate cycle (accABCD), as well as the acs gene encoding acetyl‐CoA synthetase, indicated the potential for carbon fixation. The metabolic potential of pedosphaeraceae_1 is summarized in Figure 4.
FIGURE 4.
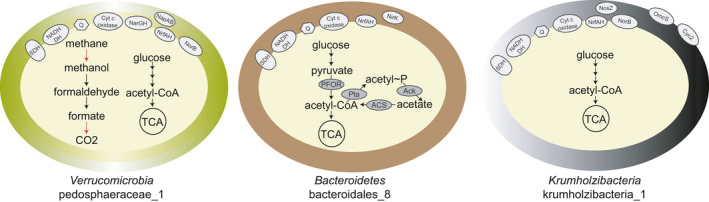
Highlights of metabolic potential found in the three enriched microorganisms after ~2.5 years of bioreactor operation. MAG name and taxonomy are indicated below each cartoon. Red arrows indicate the protein was not clearly identified or subunits are missing. SDH, succinate dehydrogenase. Ack, acetate kinase; ACS, acetyl‐CoA synthetase; Cyc2, iron oxidase; NADH DH, NADH dehydrogenase; Nap, periplasmic nitrate reductase; Nar, nitrate reductase; Nir, nitric oxide‐forming nitrite reductase; Nor, nitric oxide reductase; Nos, nitrous oxide reductase; Nrf, ammonia‐forming nitrite reductase; OmcS, outer membrane c‐type cytochrome; PFOR, pyruvate:ferredoxin oxidoreductase; Pta, phosphotransacetylase; Q, quinone; TCA, tricarboxylic acid cycle
By T2 (29 months after reactor inoculation), when pedosphaeraceae_1 was most abundant, mcr genes present in unbinned contigs had low RPKM values (~2000–2500) compared to pmoA‐family genes present in pedosphaeraceae_1 (~15,000–23,000). Activity tests suggest that the bioreactor biomass was actively oxidizing methane 6 months after reactor inoculation and that the biomass was responsive to oxygen, which promoted methane oxidation in serum bottle incubations performed in the second year of reactor operation (Figure A1). These results support the role of a methane‐oxidizing microorganism thriving under low dissolved oxygen concentrations (4.7–0.27 µM) measured in the reactor (Figure A1). Although activity data did not coincide with DNA extraction times, the presence of high‐affinity oxygen reductases in the genome indicated that MAG pedosphaeraceae_1 could have been responsible for methane oxidation potentially coupled to oxygen reduction. We hypothesize that a divergent, uncharacterized particulate methane monooxygenase in the PMO family (Figure 3) or a canonical methane monooxygenase missing from the genome could have accounted for the activity of methane oxidation.
The hypothesis that the organism represented by MAG pedosphaeraceae_1 was the main methanotroph by T2 is supported by the high estimated abundance of this organism and the absence of canonical methane monooxygenase sequences in other MAGs and unbinned contigs. However, we could not eliminate the hypothesis that an archaeon at low abundance could have performed anaerobic oxidation of methane. Methanol leakage from pedosphaeraceae_1 could have sustained other community members such as bryobacteraceae_1 and rhodospirillales_1, which also had methanol dehydrogenases and represented 4.3% and 3.5% of the metagenome reads at T2, respectively. Alternatively, pedosphaeraceae_1 might have thrived on methanol oxidation if other community members encoding methanol dehydrogenases (Figure A2) produced methanol.
A previous investigation (Rasigraf et al., 2020) showed that verrucomicrobial 16S rRNA gene sequences dominated bacterial communities at site NB8 (and at the nearby site N10) in the top 4 cm of the sediment, but were also present throughout the depth profile of NB8 at lower abundances. In this same top 4 cm depth, few sequences of canonical methane‐oxidizing proteobacteria were identified (Rasigraf et al., 2020). Verrucomicrobia were suggested to degrade polysaccharides and algal material while performing aerobic metabolism or denitrification at shallower depths. In deeper sediments, however, oxygen and sulfate are depleted, and iron reduction is predicted to predominate (Lenstra et al., 2018). This indicates that Verrucomicrobia may be metabolically versatile and capable of surviving under oxygen‐depleted conditions and that the enrichment of such microorganisms has relevance for biogeochemical cycling in situ. We hypothesize that Verrucomicrobia potentially oxidizing methanol or methane with a distinct pmoA‐family methane monooxygenase (Figure 3) or with a canonical methane monooxygenase (missing in pedosphaeraceae_1) might thrive in a niche in shallow sediments where the transition from low to zero oxygen occurs (Figure 5). In deeper sediments, where oxygen is depleted and the redox potential is low, archaea would dominate methane oxidation (Rasigraf et al., 2020). Our results indicate that the methane biofilter in these coastal marine sediments could be more complex than previously appreciated, comprising also novel putative methanotrophs within the Verrucomicrobia adapted to low oxygen availability.
FIGURE 5.
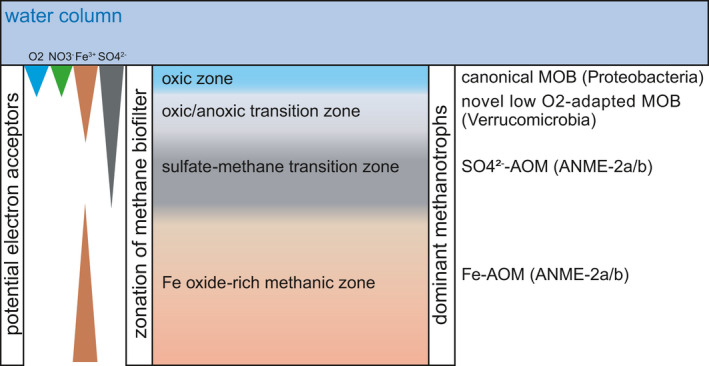
Illustration of the hypothesized biological methane filter in the coastal sediments of the Bothnian Sea. The depth of distinct zones and electron acceptor availability is not to scale. ANME, anaerobic methane‐oxidizing archaea; AOM, anaerobic oxidation of methane; MOB, methane‐oxidizing bacteria. While putative novel methanotrophic Verrucomicrobia were investigated in this study, canonical MOB and ANME sequences were previously reported (Rasigraf et al., 2020) in Bothnian Sea sediments
An important question remains regarding whether pedosphaeraceae_1 and, more generally, bacteria using particulate methane monooxygenase for methane oxidation could couple it to iron reduction. Under such a scenario, very low concentrations of oxygen could suffice for methane activation by PMO, and an unknown mechanism would allow electron transfer to iron oxides. A highly speculative hypothesis is that pedosphaeraceae_1 could have performed Fe‐AOM in the bioreactor via an unidentified pathway, and the produced Fe(II) could have served as an electron donor for microaerophilic Krumholzibacteria. This hypothesis is theoretically and solely based on a recent study, which demonstrated that two proteobacterial methanotrophs reduced ferrihydrite during growth on methane under low oxygen concentrations, in the absence of known marker genes for the iron reduction in both genomes (Zheng et al., 2020). Evidence for this phenomenon became first available from Lake Kinneret sediment incubations with 13C‐methane and different iron oxide minerals under oxygen depletion (Bar‐Or et al., 2017). In that study, pmoA copy numbers followed trends of δ13CDIC and iron (II) indicative of Fe‐AOM. More recently, Methylomonadaceae were suggested to account for methane oxidation in anoxic waters of Northwestern Siberian lakes (Cabrol et al., 2020). The authors hypothesized that iron (III) could be the electron acceptor for AOM because of total iron concentrations and the identification of taxa known to perform the iron reduction. In addition, nitrate was below their detection limit and nitrite concentrations did not exceed 0.02 mg L−1 of N‐NO2 −, which indicated the unavailability of these electron acceptors or their rapid turnover.
Although the family Pedosphaeraceae has no isolates or genomes previously implicated in methane oxidation, several known aerobic methanotrophs within the Verrucomicrobia phylum have been reported, such as Methylacidiphilum fumariolicum SolV (Pol et al., 2007) and others of the same genus, as well as some species of Methylacidimicrobium (van Teeseling et al., 2014). Our study highlights the metabolic versatility of organisms in the phylum and indicates Verrucomicrobia might play unforeseen roles in marine environments. Further investigations are needed to elucidate the metabolism of these organisms in Bothnian Sea sediments and to determine the potential for Fe‐AOM in Verrucomicrobia.
MAG bacteroidales_8 was 96.2% complete and 1.6% redundant, and the percentage of reads mapped to this genome varied from 0% (T0) to 0.01% (T1) and 10% (T2) (Table S1 at https://doi.org/10.5281/zenodo.4478410). It had the potential for oxygen respiration via low‐ and high‐affinity oxygen reductase‐encoding genes (coxBCD, cbb3‐type subunits I/II, III, and IV, and cydAB), as well as DNRA via NrfAH, and nitrite reduction to nitric oxide via NirK (Figure 4). Marker genes for the reverse TCA cycle (korAB, por/nifJ) were present. As most genes from glycolysis and the TCA cycle were found, we hypothesize that this organism could have been involved in heterotrophic respiratory metabolism. Bacteroidetes have been previously identified in water and sediment samples from the Baltic and Bothnian Sea (Broman et al., 2017; Rasigraf et al., 2017, 2020). Bacteroidetes previously detected in NB8 were hypothesized to perform fermentation, which is also supported by the presence of phosphate acetyltransferase, acetate kinase, acetyl‐CoA synthetase, pyruvate:ferredoxin oxidoreductase‐encoding gene pfor, and the NADP‐reducing hydrogenase HndABCD in bacteroidales_8. In another study, nine Bacteroidetes MAGs retrieved from water samples from the Baltic Sea also had cytochrome c oxidase genes (data retrieved from annotations on NCBI) and were hypothesized to contribute to the degradation of algal material via carbohydrate‐active enzymes (Hugerth et al., 2015), which has similarly been reported for Bacteroidetes from the North Sea (Krüger et al., 2019). Given that, as expected in the oligotrophic NB8 site, no cyanobacteria were detected in our metagenomes, it is more likely that the organism represented by MAG bacteroidales_8 could have participated in biomass turnover evidenced by the observed shifts in MAG abundances over time in the bioreactor. Whether respiratory or fermentative metabolism was employed by this microorganism remains to be elucidated.
MAG krumholzibacteria_1 was 89% complete and 2.5% redundant, and the percentage of reads mapped to these genomes varied from 0% (T0) to 1.7% (T1) and 9% (T2) (Table S1 at https://doi.org/10.5281/zenodo.4478410). It could have been involved in iron cycling, harboring OmcS and Cyc2‐encoding genes that had high RPKM values by T2 (Figures 2 and 4). Intriguingly, krumholzibacteria_1 had both iron oxidation and reduction genes. The capacity for iron oxidation and reduction in the same organism has been previously described in Geobacter sulfurreducens (Caccavo et al., 1994; Tang et al., 2019). Further studies are necessary to elucidate these potential roles of krumholzibacteria_1 in iron cycling and the metabolism of such organisms in Bothnian Sea sediments. If krumholzibacteria_1 coupled iron oxidation to oxygen reduction, it must have been able to compete for limited oxygen present in the bioreactor. Respiratory metabolism genes present in this MAG indicated this could have been possible: We identified genes encoding subunits of low‐ and high‐affinity oxygen reductases (cox, cbb3‐type, cyd, and cyo). Additionally, norB, nosZ, and nrfAH were present. Arsenic resistance genes arsC, arsB, arsM, and arsR were present, as well as complete glycolysis and most genes in the TCA pathway. Potential for carbon fixation was determined based on the presence of marker genes for the reverse TCA cycle (korAB, porABC) and the 3‐hydroxypropionate cycle (accABCD), as well as the acs gene encoding acetyl‐CoA synthetase. The genome also had a hydroxylamine dehydrogenase hao gene.
The functional potential of krumholzibacteria_1 differs from that of Candidatus Krumholzibacterium zodletonense Zgenome0171T (QTKG01.1), type material for the recently described phylum Krumholzibacteriota, recovered from the sulfur‐rich Zodletone spring in southwestern Oklahoma, USA (Youssef et al., 2019). For comparative purposes in this study, the draft genome QTKG01.1 has been annotated with DRAM (Shaffer et al., 2020). While krumholzibacteria_1 presented potential to respire iron, oxygen, nitric oxide, nitrous oxide, and nitrite, QTKG01.1 showed no clear potential to use inorganic external terminal electron acceptors. However, QTKG01.1 harbored a few subunits of the NADH:quinone oxidoreductase and complete succinate dehydrogenase, as previously described, indicating the potential for fumarate respiration. A fermentative, heterotrophic lifestyle has been previously hypothesized, with sugar catabolism genes for glucose and mannose use identified in genome QTKG01.1, as well as amino acid use via peptidases. Using the DRAM, we identified carbohydrate‐active enzyme (CAZy) sequences for the use of arabinose, chitin, pectin, mixed‐linkage glucan, and xyloglucan hemicellulose, and polyphenolics in QTKG01.1. Some functional potential is similar to krumholzibacteria_1, in which we identified CAZy sequences for the use of chitin, mixed‐linkage glucan, alpha‐mannan, and polyphenolics. These differences in functional potential between the two genomes can be expected given the degree of taxonomic novelty and low average amino acid identity (40.9%).
4. CONCLUSION
Biogeochemical evidence for methane and iron cycling in Bothnian Sea sediments (Lenstra et al., 2018; Rasigraf et al., 2020) highlights the importance of microbial processes in controlling greenhouse gas emissions from coastal ecosystems. Here, we enriched and identified novel taxa potentially involved in methane and iron cycling in an oxygen‐limited bioreactor inoculated with methane‐ and iron‐rich Bothnian Sea sediments. Metagenomic analyses provided hypotheses about the mechanisms that they may employ, such as the use of oxygen at very low concentrations. Our results imply that in more shallow sediments, where oxygen‐limited conditions are present, the methane biofilter is potentially composed of novel, metabolically versatile Verrucomicrobia that could contribute to mitigating methane emissions from coastal marine zones. Moreover, novel Bacteroidetes and Krumholzibacteria may contribute to carbon and iron cycling in these sediments. Therefore, this study brought new hypotheses on the identity and metabolic versatility of microorganisms potentially involved in biogeochemical cycling in Bothnian Sea sediments. Finally, our results are compared to recent evidence that bacterial methanotrophs may also be capable of anaerobic oxidation of methane. Future studies, which are needed to elucidate the in situ metabolic activity and mechanisms for methane and iron cycling in Bothnian Sea sediments, will benefit from the insights gained in this work.
5. ACKNOWLEDGMENTS
We thank Annika Vaksmaa for DNA extractions and useful discussions, Theo van Alen for sequencing the metagenomes, Jeroen Frank for binning support, and Matthias Egger for assistance with sampling. This work was funded by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) Grant ALWOP.293 [CUW], SIAM Gravitation Grant 024.002.002 [MSMJ], NESSC Gravitation Grant 024.002.001 [MSMJ, CPS], and ERC Matrix Grant 854088 [MSMJ, CPS].
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Paula Dalcin Martins: data curation (lead); formal analysis (equal); methodology (equal); visualization (lead); writing‐original draft (lead); writing‐review & editing (equal). Anniek de Jong: conceptualization (equal); formal analysis (equal); methodology (equal); writing‐review & editing (equal). Wytze K. Lenstra: conceptualization (equal); methodology (equal); writing‐review & editing (equal). Niels A. G. M. van Helmond: conceptualization (equal); methodology (equal); writing‐review & editing (equal). Caroline P. Slomp: conceptualization (equal); funding acquisition (equal); methodology (equal); writing‐review & editing (equal). Mike S. M. Jetten: conceptualization (equal); funding acquisition (equal); methodology (equal); supervision (equal); writing‐review & editing (equal). Cornelia Welte: funding acquisition (equal); methodology (equal); supervision (equal); writing‐review & editing (equal). Olivia Rasigraf: conceptualization (equal); methodology (equal); supervision (equal); writing‐review & editing (equal).
ETHICS STATEMENT
None required.
FIGURE A1.
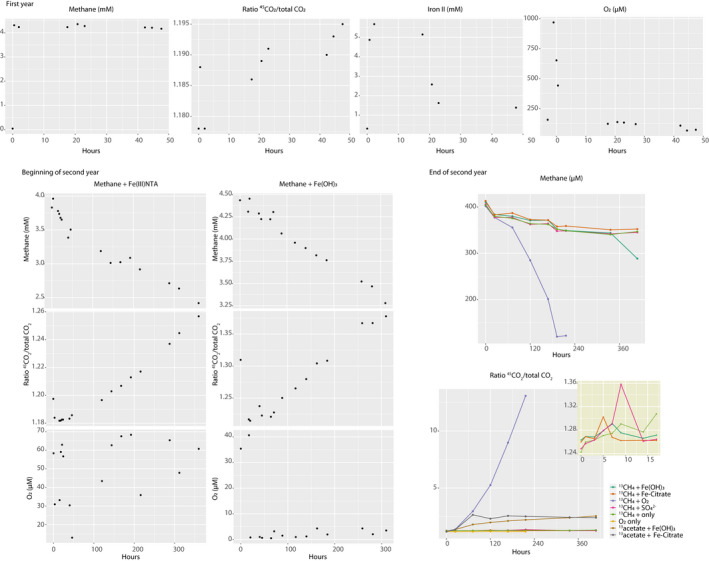
Summary of activity tests. Data are categorized based on the year when measurements were conducted. In the first year and beginning of second year of reactor operation, whole reactor experiments were performed, in which the reactor was set as a closed system and substrates were followed for several hours. In the end of the second year of reactor operation, bioreactor biomass was incubated into serum bottles with combinations of electron donors and acceptors as indicated in the figure and detailed in the methods section. The inset in yellow displays a zoom in on some incubations as specified in the legend. Oxygen measurements reflect headspace concentrations.
FIGURE A2.
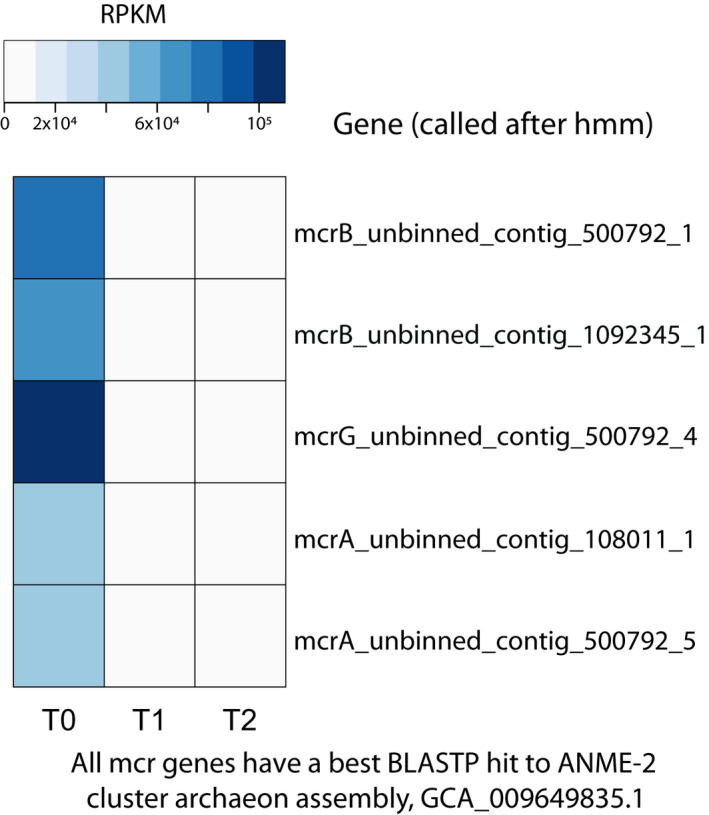
Heat map of mcr genes in the three metagenomes (T0, T1, and T2). RPKM values were calculated from read mapping with BBMap and imported into RStudio for heat map construction. Protein sequences were used for blastp against the NCBI RefSeq database
FIGURE A3.
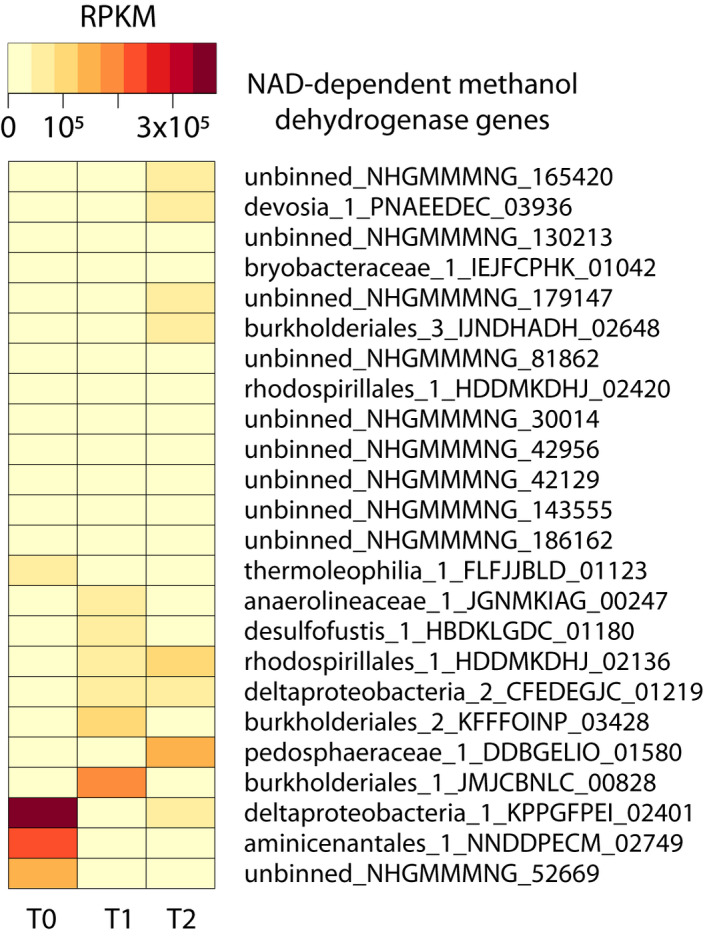
Heat map of NAD‐dependent methanol dehydrogenase genes in the three metagenomes (T0, T1, and T2). RPKM values were calculated from read mapping with BBMap and imported into RStudio for heat map construction. Protein sequences were identified via prokka annotations
Contributor Information
Paula Dalcin Martins, Email: p.dalcinmartins@science.ru.nl.
Anniek de Jong, Email: dejonganniek@gmail.com.
Wytze K. Lenstra, Email: W.K.Lenstra@uu.nl.
Niels A. G. M. van Helmond, Email: n.vanhelmond@uu.nl.
Caroline P. Slomp, Email: C.P.Slomp@uu.nl.
Mike S. M. Jetten, Email: m.jetten@science.ru.nl.
Cornelia U. Welte, Email: c.welte@science.ru.nl.
Olivia Rasigraf, Email: orasigra@gfz-potsdam.de.
DATA AVAILABILITY STATEMENT
Raw sequencing reads, the 56 metagenome‐assembled genomes, and unbinned contigs (SAMN16148055) have been deposited in the National Center for Biotechnology Information (NCBI) database under the BioProject accession number PRJNA663545: https://www.ncbi.nlm.nih.gov/bioproject/663545. Supporting data are available in Appendix. Table S1 is available in Zenodo at https://doi.org/10.5281/zenodo.4478410.
REFERENCES
- Algesten, G. , Brydsten, L. , Jonsson, P. , Kortelainen, P. , Löfgren, S. , Rahm, L. , Räike, A. , Sobek, S. , Tranvik, L. , Wikner, J. , & Jansson, M. (2006). Organic carbon budget for the Gulf of Bothnia. Journal of Marine Systems, 63(3–4), 155–161. 10.1016/j.jmarsys.2006.06.004 [DOI] [Google Scholar]
- Alneberg, J. , Brynjar, S.B. , de Bruijn, I. , Schirmer, M. , Quick, J. , Ijaz, U. Z. , Loman, N. J. , Andersson, A. F. , Quince, C. (2013). ‘CONCOCT: Clustering cONtigs on COverage and ComposiTion’, Arxiv preprint arXiv:1312.4038v1, 28 pp. https://arxiv.org/abs/1312.4038
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. https://arxiv.org/abs/1312.4038 [DOI] [PubMed] [Google Scholar]
- Arkin, A. P. , Cottingham, R. W. , Henry, C. S. , Harris, N. L. , Stevens, R. L. , Maslov, S. , Dehal, P. , Ware, D. , Perez, F. , Canon, S. , Sneddon, M. W. , Henderson, M. L. , Riehl, W. J. , Murphy‐Olson, D. , Chan, S. Y. , Kamimura, R. T. , Kumari, S. , Drake, M. M. , Brettin, T. S. , … Yu, D. (2018). KBase: The United States department of energy systems biology knowledgebase. Nature Biotechnology, 36(7), 566–569. 10.1038/nbt.4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromokeye, D. A. , Kulkarni, A. C. , Elvert, M. , Wegener, G. , Henkel, S. , Coffinet, S. , Eickhorst, T. , Oni, O. E. , Richter‐Heitmann, T. , Schnakenberg, A. , Taubner, H. , Wunder, L. , Yin, X. , Zhu, Q. , Hinrichs, K.‐U. , Kasten, S. , & Friedrich, M. W. (2020). Rates and microbial players of iron‐driven anaerobic oxidation of methane in methanic marine sediments. Frontiers in Microbiology, 10. 10.3389/fmicb.2019.03041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad, A. , Speth, D. R. , de Graaf, R. M. , Op den Camp, H. J. M. , Jetten, M. S. M. , & Welte, C. U. (2015). A metagenomics‐based metabolic model of nitrate‐dependent anaerobic oxidation of methane by methanoperedens‐like Archaea. Frontiers in Microbiology, 6, 1423. 10.3389/fmicb.2015.01423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Or, I. , Elvert, M. , Eckert, W. , Kushmaro, A. , Vigderovich, H. , Zhu, Q. , Ben‐Dov, E. , & Sivan, O. (2017). Iron‐coupled anaerobic oxidation of methane performed by a mixed bacterial‐archaeal community based on poorly reactive minerals. Environmental Science and Technology, 51(21), 12293–12301. 10.1021/acs.est.7b03126 [DOI] [PubMed] [Google Scholar]
- Battino, R. (1984). The Ostwald coefficient of gas solubility. Fluid Phase Equilibria, 15(3), 231–240. 10.1016/0378-3812(84)87009-0 [DOI] [Google Scholar]
- Beal, E. J. , House, C. H. , & Orphan, V. J. (2009). Manganese‐ and iron‐dependent marine methane oxidation. Science, 325(5937), 184–187. 10.1126/science.1169984 [DOI] [PubMed] [Google Scholar]
- Berger, S. , Frank, J. , Dalcin Martins, P. , Jetten, M. S. M. , & Welte, C. U. (2017). High‐quality draft genome sequence of “Candidatus Methanoperedens sp”. strain BLZ2, a nitrate‐reducing anaerobic methane‐oxidizing archaeon enriched in an anoxic bioreactor. Genome Announcements, 5(46). 10.1128/genomeA.01159-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, J. , Fritzsche, A. , Totsche, K. U. , & Meckenstock, R. U. (2010). Nanosized ferrihydrite colloids facilitate microbial iron reduction under flow conditions. Geomicrobiology Journal, 27(2), 123–129. 10.1080/01490450903456707 [DOI] [Google Scholar]
- Broman, E. , Sjöstedt, J. , Pinhassi, J. , & Dopson, M. (2017). Shifts in coastal sediment oxygenation cause pronounced changes in microbial community composition and associated metabolism. Microbiome, 5(1), 96. 10.1186/s40168-017-0311-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrol, L. , Thalasso, F. , Gandois, L. , Sepulveda‐Jauregui, A. , Martinez‐Cruz, K. , Teisserenc, R. , Tananaev, N. , Tveit, A. , Svenning, M. M. , & Barret, M. (2020). Anaerobic oxidation of methane and associated microbiome in anoxic water of Northwestern Siberian lakes. Science of the Total Environment, 736, 139588. [DOI] [PubMed] [Google Scholar]
- Caccavo, F. , Lonergan, D. J. , Lovley, D. R. , Davis, M. , Stolz, J. F. , & McInerney, M. J. (1994). Geobacter sulfurreducens sp. nov., a hydrogen‐ and acetate‐oxidizing dissimilatory metal‐reducing microorganism. Applied and Environmental Microbiology, 60(10), 3752–3759. 10.1128/AEM.60.10.3752-3759.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, C. , Leu, A. O. , Xie, G.‐J. , Guo, J. , Feng, Y. , Zhao, J.‐X. , Tyson, G. W. , Yuan, Z. , & Hu, S. (2018). A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. The ISME Journal, 12(8), 1929–1939. 10.1038/s41396-018-0109-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutierrez, S. , Silla‐Martinez, J. M. , & Gabaldon, T. (2009). trimAl: A tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25(15), 1972–1973. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil, P.‐A. , Mussig, A. J. , Hugenholtz, P. , & Parks, D. H. (2019). GTDB‐Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics, 36(6), 1925–1927. 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Computational Biology, 7(10), e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger, M. , Rasigraf, O. , Sapart, C. J. , Jilbert, T. , Jetten, M. S. M. , Röckmann, T. , van der Veen, C. , Bândă, N. , Kartal, B. , Ettwig, K. F. , & Slomp, C. P. (2015). Iron‐mediated anaerobic oxidation of methane in brackish coastal sediments. Environmental Science & Technology, 49(1), 277–283. 10.1021/es503663z [DOI] [PubMed] [Google Scholar]
- Egger, M. , Riedinger, N. , Mogollón, J. M. , & Jørgensen, B. B. (2018). Global diffusive fluxes of methane in marine sediments. Nature Geoscience, 11(6), 421–425. 10.1038/s41561-018-0122-8 [DOI] [Google Scholar]
- Ettwig, K. F. , Butler, M. K. , Le Paslier, D. , Pelletier, E. , Mangenot, S. , Kuypers, M. M. M. , Schreiber, F. , Dutilh, B. E. , Zedelius, J. , de Beer, D. , Gloerich, J. , Wessels, H. J. C. T. , van Alen, T. , Luesken, F. , Wu, M. L. , van de Pas‐Schoonen, K. T. , Op den Camp, H. J. M. , Janssen‐Megens, E. M. , Francoijs, K.‐J. , … Strous, M. (2010). Nitrite‐driven anaerobic methane oxidation by oxygenic bacteria. Nature, 464(7288), 543–548. 10.1038/nature08883 [DOI] [PubMed] [Google Scholar]
- Ettwig, K. F. , Speth, D. R. , Reimann, J. , Wu, M. L. , Jetten, M. S. M. , & Keltjens, J. T. (2012). Bacterial oxygen production in the dark. Frontiers in Microbiology, 3. 10.3389/fmicb.2012.00273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettwig, K. F. , Zhu, B. , Speth, D. , Keltjens, J. T. , Jetten, M. S. M. , & Kartal, B. (2016). Archaea catalyze iron‐dependent anaerobic oxidation of methane. Proceedings of the National Academy of Sciences of the United States of America, 113(45), 12792–12796. 10.1073/pnas.1609534113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber, A. I. , Nealson, K. H. , Okamoto, A. , McAllister, S. M. , Chan, C. S. , Barco, R. A. , & Merino, N. (2020). FeGenie: A Comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Frontiers in Microbiology, 11, 37. 10.3389/fmicb.2020.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, E. D. , Heidelberg, J. F. , & Tully, B. J. (2017). BinSanity: Unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ, 5, e3035. 10.7717/peerj.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroon, M. F. , Hu, S. , Shi, Y. , Imelfort, M. , Keller, J. , Hugenholtz, P. , Yuan, Z. , & Tyson, G. W. (2013). Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature, 500(7464), 567–570. 10.1038/nature12375 [DOI] [PubMed] [Google Scholar]
- Hedrich, S. , Schlömann, M. , & Johnson, D. B. (2011). The iron‐oxidizing proteobacteria. Microbiology, 157(6), 1551–1564. 10.1099/mic.0.045344-0 [DOI] [PubMed] [Google Scholar]
- Hellemann, D. , Tallberg, P. , Bartl, I. , Voss, M. , & Hietanen, S. (2017). Denitrification in an oligotrophic estuary: A delayed sink for riverine nitrate. Marine Ecology Progress Series, 583, 63–80. 10.3354/meps12359 [DOI] [Google Scholar]
- Hoikkala, L. , Kortelainen, P. , Soinne, H. , & Kuosa, H. (2015). Dissolved organic matter in the Baltic Sea. Journal of Marine Systems, 142, 47–61. 10.1016/j.jmarsys.2014.10.005 [DOI] [Google Scholar]
- Hugerth, L. W. , Larsson, J. , Alneberg, J. , Lindh, M. V. , Legrand, C. , Pinhassi, J. , & Andersson, A. F. (2015). Metagenome‐assembled genomes uncover a global brackish microbiome. Genome Biology, 16(1), 279. 10.1186/s13059-015-0834-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1), 119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D. T. , Taylor, W. R. , & Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Bioinformatics, 8(3), 275–282. 10.1093/bioinformatics/8.3.275 [DOI] [PubMed] [Google Scholar]
- Kang, D. D. , Li, F. , Kirton, E. , Thomas, A. , Egan, R. , An, H. , & Wang, Z. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ, 7, e7359. 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittel, K. , & Boetius, A. (2009). Anaerobic oxidation of methane: Progress with an unknown process. Annual Review of Microbiology, 63(1), 311–334. 10.1146/annurev.micro.61.080706.093130 [DOI] [PubMed] [Google Scholar]
- Krüger, K. , Chafee, M. , Ben Francis, T. , Glavina del Rio, T. , Becher, D. , Schweder, T. , Amann, R. I. , & Teeling, H. (2019). In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. The ISME Journal, 13(11), 2800–2816. 10.1038/s41396-019-0476-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra, W. K. , Egger, M. , van Helmond, N. A. G. M. , Kritzberg, E. , Conley, D. J. , & Slomp, C. P. (2018). Large variations in iron input to an oligotrophic Baltic Sea estuary: Impact on sedimentary phosphorus burial. Biogeosciences, 15(22), 6979–6996. 10.5194/bg-15-6979-2018 [DOI] [Google Scholar]
- Letunic, I. , & Bork, P. (2016). Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research, 44(W1), W242–W245. 10.1093/nar/gkw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu, A. O. , Cai, C. , McIlroy, S. J. , Southam, G. , Orphan, V. J. , Yuan, Z. , Hu, S. , & Tyson, G. W. (2020). Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. The ISME Journal, 14(4), 1030–1041. 10.1038/s41396-020-0590-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu, A. O. , McIlroy, S. J. , et al. (2020b). Lateral gene transfer drives metabolic flexibility in the anaerobic methane‐oxidizing archaeal family methanoperedenaceae. mBio, 11(3). 10.1128/mBio.01325-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Luo, R. , Liu, C.‐M. , Leung, C.‐M. , Ting, H.‐F. , Sadakane, K. , Yamashita, H. , & Lam, T.‐W. (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods, 102, 3–11. 10.1016/j.ymeth.2016.02.020 [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley, D. R. , & Phillips, E. J. P. (1987). Rapid assay for microbially reducible ferric iron in aquatic sediments. Applied and Environmental Microbiology, 53(7), 1536–1540. 10.1007/BF01611203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya, Y. , Itoh, M. , Okuda, S. , Yoshizawa, A. C. , & Kanehisa, M. (2007). KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Research, 35(Web Server), W182–W185. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllykangas, J.‐P. , Rissanen, A. J. , Hietanen, S. , & Jilbert, T. (2020). Influence of electron acceptor availability and microbial community structure on sedimentary methane oxidation in a boreal estuary. Biogeochemistry, 148(3), 291–309. 10.1007/s10533-020-00660-z [DOI] [Google Scholar]
- Na, S.‐I. , Kim, Y. O. , Yoon, S.‐H. , Ha, S.‐M. , Baek, I. , & Chun, J. (2018). UBCG: Up‐to‐date bacterial core gene set and pipeline for phylogenomic tree reconstruction. Journal of Microbiology, 56(4), 280–285. 10.1007/s12275-018-8014-6 [DOI] [PubMed] [Google Scholar]
- Parks, D. H. , Imelfort, M. , Skennerton, C. T. , Hugenholtz, P. , & Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research, 25(7), 1043–1055. 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podosokorskaya, O. A. , et al. (2013). Characterization of Melioribacter roseus gen. nov., sp. nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae . Environmental Microbiology, 15(6), 1759–1771. 10.1111/1462-2920.12067 [DOI] [PubMed] [Google Scholar]
- Pol, A. , et al. (2007). Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature, 450(7171), 874–878. 10.1038/nature06222 [DOI] [PubMed] [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2010). FastTree 2 – Approximately maximum‐likelihood trees for large alignments. PLoS One, 5(3), 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2017). R: A language and environment for statistical computing, Vienna, Austria: R Foundation for Statistical Computing. Retrieved from https://www.r‐project.org [Google Scholar]
- Raghoebarsing, A. A. , Pol, A. , van de Pas‐Schoonen, K. T. , Smolders, A. J. P. , Ettwig, K. F. , Rijpstra, W. I. C. , Schouten, S. , Damsté, J. S. S. , Op den Camp, H. J. M. , Jetten, M. S. M. , & Strous, M. (2006). A microbial consortium couples anaerobic methane oxidation to denitrification. Nature, 440(7086), 918–921. 10.1038/nature04617 [DOI] [PubMed] [Google Scholar]
- Rasigraf, O. , Helmond, N. A. G. M. , Frank, J. , Lenstra, W. K. , Egger, M. , Slomp, C. P. , & Jetten, M. S. M. (2020). Microbial community composition and functional potential in Bothnian Sea sediments is linked to Fe and S dynamics and the quality of organic matter. Limnology and Oceanography, 65(S1), S113–S133. 10.1002/lno.11371 [DOI] [Google Scholar]
- Rasigraf, O. , Schmitt, J. , Jetten, M. S. M. , & Lüke, C. (2017). Metagenomic potential for and diversity of N‐cycle driving microorganisms in the Bothnian Sea sediment. MicrobiologyOpen, 6(4), e00475. 10.1002/mbo3.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedinger, N. , Formolo, M. J. , Lyons, T. W. , Henkel, S. , Beck, A. , & Kasten, S. (2014). An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments. Geobiology, 12(2), 172–181. 10.1111/gbi.12077 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐R, L. M. , & Konstantinidis, K. T. (2016). The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints, 4, e1900v1. 10.7287/peerj.preprints.1900v1 [DOI] [Google Scholar]
- Rooze, J. , et al. (2016). Iron‐dependent anaerobic oxidation of methane in coastal surface sediments: Potential controls and impact. Limnology and Oceanography, 61(S1), S267–S282. 10.1002/lno.10275 [DOI] [Google Scholar]
- Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics, 30(14), 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Shaffer, M. , et al. (2020). DRAM for distilling microbial metabolism to automate the curation of microbiome function. bioRxiv, p. 2020.06.29.177501. 10.1101/2020.06.29.177501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindell, D. T. , Faluvegi, G. , Koch, D. M. , Schmidt, G. A. , Unger, N. , & Bauer, S. E. (2009). Improved attribution of climate forcing to emissions. Science, 326(5953), 716–718. 10.1126/science.1174760 [DOI] [PubMed] [Google Scholar]
- Sieber, C. M. K. , Probst, A. J. , Sharrar, A. , Thomas, B. C. , Hess, M. , Tringe, S. G. , & Banfield, J. F. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nature Microbiology, 3(7), 836–843. 10.1038/s41564-018-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H.‐Y. , Holmes, D. E. , Ueki, T. , Palacios, P. A. , & Lovley, D. R. (2019). Iron corrosion via direct metal‐microbe electron transfer. mBio, 10(3), 10.1128/mBio.00303-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thureborn, P. , Lundin, D. , Plathan, J. , Poole, A. M. , Sjöberg, B.‐M. , & Sjöling, S. (2013). A metagenomics transect into the deepest point of the baltic sea reveals clear stratification of microbial functional capacities. PLoS One, 8(9), e74983. 10.1371/journal.pone.0074983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Graaf, A. A. , de Bruijn, P. , Robertson, L. A. , Jetten, M. S. M. , & Kuenen, J. G. (1996). Autotrophic growth of anaerobic ammonium‐oxidizing micro‐organisms in a fluidized bed reactor. Microbiology, 142(8), 2187–2196. 10.1099/13500872-142-8-2187 [DOI] [Google Scholar]
- van Teeseling, M. C. F. , Pol, A. , Harhangi, H. R. , van der Zwart, S. , Jetten, M. S. M. , Op den Camp, H. J. M. , & van Niftrik, L. (2014). Expanding the verrucomicrobial methanotrophic world: Description of three novel species of Methylacidimicrobium gen. nov. Applied and Environmental Microbiology, 80(21), 6782–6791. 10.1128/AEM.01838-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. , & (2016). ggplot2: Elegant graphics for data analysis. Springer‐Verlag. Retrieved from 10.1007/978-3-319-24277-4 [DOI] [Google Scholar]
- Wu, Y. W. , Simmons, B. A. , & Singer, S. W. (2015). MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics, 32(4), 605–607. 10.1093/bioinformatics/btv638 [DOI] [PubMed] [Google Scholar]
- Youssef, N. H. , Farag, I. F. , Hahn, C. R. , Premathilake, H. , Fry, E. , Hart, M. , Huffaker, K. , Bird, E. , Hambright, J. , Hoff, W. D. , & Elshahed, M. S. (2019). Candidatus Krumholzibacterium zodletonense gen. nov., sp nov, the first representative of the candidate phylum Krumholzibacteriota phyl. nov. recovered from an anoxic sulfidic spring using genome resolved metagenomics. Systematic and Applied Microbiology, 42(1), 85–93. 10.1016/j.syapm.2018.11.002 [DOI] [PubMed] [Google Scholar]
- Zheng, Y. , Wang, H. , Liu, Y. , Zhu, B. , Li, J. , Yang, Y. , Qin, W. , Chen, L. , Wu, X. , Chistoserdova, L. , & Zhao, F. (2020). Methane‐dependent mineral reduction by aerobic methanotrophs under hypoxia. Environmental Science & Technology Letters, 7(8), 606–612. 10.1021/acs.estlett.0c00436 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequencing reads, the 56 metagenome‐assembled genomes, and unbinned contigs (SAMN16148055) have been deposited in the National Center for Biotechnology Information (NCBI) database under the BioProject accession number PRJNA663545: https://www.ncbi.nlm.nih.gov/bioproject/663545. Supporting data are available in Appendix. Table S1 is available in Zenodo at https://doi.org/10.5281/zenodo.4478410.