

Abstract
This report describes the development of an orally active analgesic that resolves inflammation and neuropathic pain without the addictive potential of opioids. EC5026 acts on the cytochrome P450 branch of the arachidonate cascade to stabilize epoxides of polyunsaturated fatty acids (EpFA), which are natural mediators that reduce pain, resolve inflammation, and maintain normal blood pressure. EC5026 is a slow-tight binding transition-state mimic that inhibits the soluble epoxide hydrolase (sEH) at picomolar concentrations. The sEH rapidly degrades EpFA; thus, inhibiting sEH increases EpFA in vivo and confers beneficial effects. This mechanism addresses disease states by shifting endoplasmic reticulum stress from promoting cellular senescence and inflammation toward cell survival and homeostasis. We describe the synthesis and optimization of EC5026 and its development through human Phase 1a trials with no drug-related adverse events. Additionally, we outline fundamental work leading to discovery of the analgesic and inflammation-resolving CYP450 branch of the arachidonate cascade.
Introduction
There is a pressing need for new pharmaceuticals to treat pain in general and neuropathic pain specifically in view of the opioid epidemic discussed below. Here we provide an overview of the discovery and development of a new class of nonopioid analgesics that appear devoid of addictive potential, resolve inflammation, and reduce endoplasmic reticulum (ER) stress. EC5026, a small molecule drug candidate disclosed in this study, is an exceptionally potent transition-state mimic inhibitor of the soluble epoxide hydrolase (sEH) enzyme which is widely distributed in mammalian tissues.1 Interestingly, the sEH was found while studying fundamental insect developmental biology where the hydration of epoxides of juvenile hormones is a key reaction in metamorphosis. This biology was exploited for the rational development of a class of green pesticides termed insect growth regulators (IGRs), and the mammalian sEH was discovered while studying the metabolism of one of these IGRs in mammals. Following the first report of the sEH in 1972, the full metabolism of the terpene epoxide developed as a vector control and agricultural chemical was described in 1974, and the properties of the sEH in mammals were investigated.2 Over the past several decades, a primary role of this enzyme was found to be the rapid conversion of anti-inflammatory, antihypertensive, and analgesic chemical mediators, the fatty acid epoxides also named epoxy-fatty acids (EpFA), into their respective 1,2-diols.3 Therefore, several classes of sEH inhibitors (sEHI) have been developed over the past several decades and were useful in establishing EpFA as key chemical mediators. In contrast to the cyclooxygenase (COX) and lipoxygenase (LOX) branches that are predominately but not exclusively pro-inflammatory, it is generally accepted that a third branch (cytochrome P450s or CYP450s) of the arachidonic acid cascade is largely anti-inflammatory and pro-resolving.3 The discovery that ureas, amides, carbamates, and various heterocycles could act, as not only epoxide mimics but also transition-state mimics, started the field down a path to several exceptionally potent sEHI including EC5026 described here.4,5 The physical, pharmacokinetic (PK), and biological properties of sEHI yielded a unique set of advantages and challenges in their development to the clinic as described below. In addition, EC5026 has been successfully moved to the clinic through a unique and largely publicly funded clinical development path.
In the following paragraphs, we cover the early chemistry and structure–activity relationship studies of sEHI, and the path to drug candidates for human, equine, and companion animals. We also present the synthetic pathway to EC5026 and its full characterization (see Supporting Information). In parallel to the medicinal chemistry, advances were made in understanding the action of sEHI with the target enzyme and on mammalian physiology. Although there are a variety of possible indications and clinical paths for sEHI, neuropathic pain has been selected for multiple reasons including lack of effective therapies for this pressing need, the reluctance of large pharmaceutical companies to enter the field, and the possibility that a small company could take a new product through trials demonstrating efficacy. We present an update on the Phase 1a clinical trial and our plans for the immediate future. We conclude with a brief discussion of moving the compounds to the clinic largely with the support of the NIH.
Chemistry
Early sEH Chemistry
Early substrate mimics and irreversible inhibitors of the sEH were useful biochemical probes but too metabolically unstable to be useful as pharmaceuticals.4 A series of fundamental studies in enzymology discovered that the sEH belongs to the α/β-fold hydrolase class of enzymes and that the key catalytic residue is a nucleophilic aspartic acid.6 This class of enzymes has been inhibited by carbodiimides, which initially appeared to be the case with sEH as well. However, Morisseau et al.7 found the actual compound that acts as a potent, time-dependent inhibitor against sEH is dicyclohexyl urea (DCU, Figure 1), a hydrated form of dicyclohexyl carbodiimide (DCC). DCU is a byproduct from amide coupling reactions with DCC. This urea was our initial lead compound, and throughout the chemical history of this series, the goal has been to maximize potency and other pharmacological properties while dealing with these high melting, poorly soluble lipophilic molecules. The redeeming features of these transition-state mimics were their exceptionally high potency on the target enzyme and their high selectivity. A variety of structures have been used as mimics of epoxides at putative receptors including ureas, amides, and carbamates, so these functional groups were examined as central pharmacophores of the inhibitors. Since epoxides of long chain polyunsaturated fatty acids were the hypothetical natural substrates of sEH, many early compounds were designed to mimic these substrates while incorporating a pharmacophore that mimics the epoxide group of the substrate or the putative transition-state(s) of the epoxide ring opening. This resulted in a series of compounds such as 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA, Figure 1), which were exceptionally potent enzyme inhibitors but also suffered from being good mimics of the biological active EpFA at their putative receptors.8 In addition, they were rapidly metabolized to inactive degradation products,9 and, as expected based on their physical properties such as high lipophilicity and high melting points as well as difficulty in formulation to obtain high bioavailability. Despite these difficulties, a number of biologists used these compounds to demonstrate multiple roles of EpFA in reducing hypertension and vascular inflammation.10 In early stages of the development of sEHI, the adamantyl group used in AUDA and AEPU (Figure 1) yielded exceptionally high detection sensitivity on an LC-MS as well as high potency on the target sEH. This detection sensitivity afforded by the adamantyl group allowed rapid PK studies of these sEHI in rodents where PK-absorption, distribution, metabolism, and excretion (ADME) could be rapidly determined using just a few microliters of blood.11 The ease of determining oral bioavailability and PK parameters as well as availability of rapid quantitative enzyme assays resulted in rapid filters to select progressively more potent orally bioavailable sEHI. Kim et al.12 found that the presence of a polar group approximately 7 Å from the carbonyl of the central pharmacophore dramatically increased solubility without loss of potency as in AEPU shown in Figure 1.
Figure 1.

Chemical structures of early stage sEHI. A commonly used four-letter abbreviation and a University of California (UC) number are provided. Our lead compound in this series was DCU. The presence of an adamantyl group made the sEHI more potent and easier to detect by LC-MS in blood. They also tended to mimic EpFA, which were the presumptive substrates.
These leads were exploited aggressively in both industrial and academic laboratories.13 Among these, an excellent compound was GSK2256294 (Figure 2).14 A major success was the discovery of ureas that are constructed with simple cyclicalkyl groups on a nitrogen atom of the urea combined with conformationally restricted substituents on the other nitrogen atom of the urea. This approach resulted in compounds such as APAU (AR9281) and t-AUCB (Figure 1) possessing drug-like properties, high potency, and improved PK profiles.15,16 These series of compounds were further optimized by replacing the metabolically unstable cyclicalkyl group such as the adamantyl group by more stable aryl groups such as the p-trifluoromethoxyphenyl group as in TPPU and t-TUCB (UC1770, EC1728, Figure 2).16,17 One compound from the conformationally restricted ureas was AR9281 (Figure 1) that was proven to be safe even at high doses in a Phase 2a clinical trial but failed to show adequate efficacy for further development. Although it is surprisingly water-soluble, it contained an adamantyl group that resulted in complex metabolites and a short half-life due to rapid hydroxylation of the adamantyl group.18 One of its human metabolites was only found in primates requiring expensive primate safety studies. This, coupled with mediocre potency on the human recombinant sEH and a short drug-target residence time on the human enzyme, probably contributed to its clinical failure. Because once or twice a day dosing is the norm for hypertension and diabetes drugs, exceptionally high levels of AR9281 were required to maintain adequate blood levels.18 Interestingly, it has never been clear why AR9281 was selected for development when the related TPPU which demonstrated much greater potency on the recombinant human enzyme and a 100-fold higher PK-AUC in mice was already reported.17,19 In pain assays in rodents, TPPU was of similar potency to AR9281 on the recombinant murine enzyme but was more potent than morphine.17 As mentioned above, AR9281 is quite potent on the recombinant mouse sEH, but it is quite a weak inhibitor with a rapid off rate on the recombinant human enzyme as discussed below. TPPU has emerged as the standard in the field used in many biological studies to inhibit the sEH due to its good PK and high potency on both primate and rodent sEH enzymes.
Figure 2.

Chemical structures of late stage sEH inhibitors. A commonly used four-letter abbreviation and University of California numbers are provided.
Polypharmacy with sEHI
sEHI have been found to synergize with several other pharmaceuticals. In several cases, the mechanism of this synergism is quite well understood often based on monitoring the profiles of blood regulatory lipids and key regulatory pathways. The best studied to date are structures combining pharmacophores that inhibit COX-2. This synergism was predicted when lipid profiles in murine sepsis models showed that EpFA were increased and diols decreased in the blood as expected with sEHI, but the sEHI quickly and dramatically resolved the high levels of PGE2 and other inflammatory mediators. The effect on products the COX branch of the arachidonate cascade was far more dramatic than on the CYP450 branch in plasma analysis. Even in the early study, this was shown to be via transcriptional down regulation of COX-2 induced in the sepsis model.20 There is now evidence that this is due to reduction of ER stress and inflammatory pathways leading to prostaglandin synthases and COX-2.21 This synergism of COX and sEHI when combined is fortunate since in addition to synergism many of the cardiovascular and gastrointestinal side effects of COX inhibitors are blocked by sEHI.22 sEHI have been found to synergize with PPAR agonists, FLAP and FAAH inhibitors, phosphodiesterase inhibitors, and others which can be exploited by drug combinations or integrated molecules.23 A summary of this class of dual inhibitors/modulators is listed in Table S4.
SAR Leading to IND Candidate EC5026
On the basis of investigations of compounds such as TPPU, it became clear that the drug-target residence time with sEHI was a better indicator of inhibitor’s in vivo efficacy than its potency in enzyme assays (IC50 or Ki).19,24 The drug-target residence time and potency were correlated but not tightly correlated, especially for very tight-binding sEHI (IC50 or Ki ≤ 5 nM).19,24 sEHI with a urea pharmacophore like TPPU work well in vivo, but a careful formulation is critical for reproducible results, particularly for experiments requiring high doses due to the high lipophilicity, high melting point, and poor water-solubility of some sEHI. Formulation of either high melting or lipophilic compounds can normally be addressed particularly with highly potent compounds. However, high melting lipophilic compounds such as some of the potent sEHI present a challenge for the field (Figure 1). Although replacing the central urea pharmacophore by an amide group lowered the melting point and increased the water solubility in general, such replacement also reduced the potency of the inhibitors by an order of magnitude (EC5026 vs EC5049 in Table 1).1 Within EicOsis, the goal was to find sEHI that were more potent with a lower melting point, and/or more water-soluble. A new series of compounds was designed based on the scaffold shown in TPPU.5
Table 1. Physical Properties, Potency, and Target Occupancy of AR9281, TPPU, EC5023, EC5024, and EC5026.

Solubility was measured with sodium phosphate buffer (0.1 M, pH 7.4).
LogP was measured by HPLC.
Ki and tR (which is a reciprocal of koff) were determined against the affinity purified recombinant human sEH (hsEH) by a FRET-displacement assay.
Figure 3.
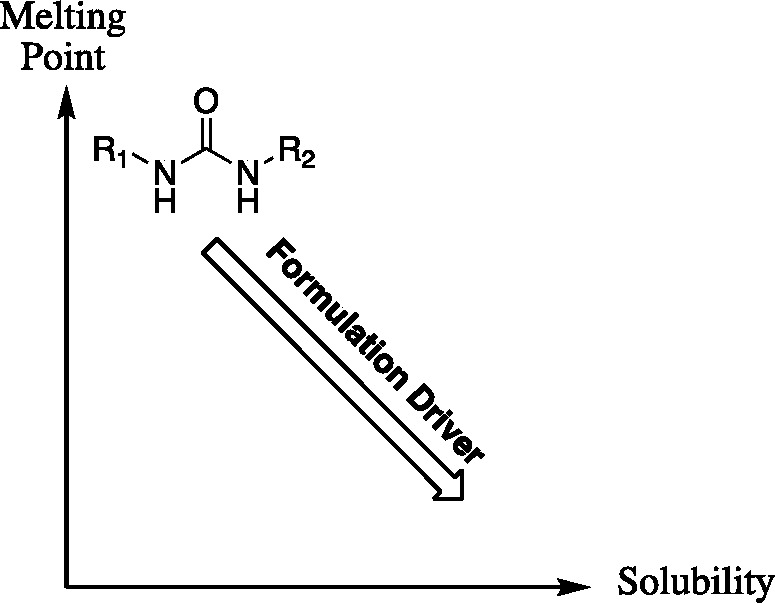
Chemical properties impact ease of formulation. Generally, the more potent the compound the more facile the formulation. There are relatively routine procedures to formulate high melting or poorly water-soluble materials, but high melting poorly soluble crystals are a challenge. This problem was addressed by synthesis to increase potency on the human target enzyme, increase water solubility, and reduce the stability of the crystal structure.
Within the series of inhibitors synthesized by EicOsis, the incorporation of a fluorine atom in the meta-position of the p-trifluoromethoxyphenyl group of sEHI (Table 1) not only increased the potency against human sEH (dramatically in some cases) but also reduced the melting point significantly while maintaining the drug-target residence time (e.g., TPPU vs EC5023 in Table 1). This came with the cost of reduced water solubility and increased LogP. It was assumed that an addition of a meta-fluoro substituent on the phenyl group of the inhibitors broke the overall symmetry of the chemical structure of the inhibitors and thus destabilized the crystalline state resulting in a drop in the melting point.5 In addition, a previous SAR study19 suggested that introduction of an extra methyl group at the α-carbon of the amide group of the inhibitor could significantly improve the potency of the inhibitor. Therefore, a significant improvement of potency was expected with EC5019 when we introduced an extra methyl group on the α-carbon of the amide group of EC5023. As expected, EC5019 improved the potency but showed poor water solubility due probably to the increased melting points. Therefore, we further modified EC5019 by performing another SAR study5 and found that introduction of another extra methyl group at the β-carbon of EC5019 could lower melting points, which in turn improve water solubility (EC5019 vs EC5026 in Table 1) and also further improve potency. To further test whether we could improve the potency and water solubility of EC5026 by reducing its size, we replaced the 4-trifluoromethoxy group on the phenyl of EC5026 by 4-trifluoromethyl group (EC5034). However, this attempt failed, resulting in lower potency and poorer water solubility compared to EC5026. Therefore, EC5026 was selected as a drug candidate for further development. The increase in potency coupled with a low melting point reduces the likelihood of off-target toxicity and makes the compounds a remarkably simple formulate for oral administration (Table 1).5
The selection of an IND candidate is based on multiple factors ranging from the potency to the cost of synthesis. Unique drivers in this series included long drug-target residence time (reciprocal of koff of the inhibitor from the enzyme),19 the moderate in vivo half-life needed for a first-in-class compound, and sometimes a compromise between reduced melting point and potency in favor of ease of formulation. With many highly potent compounds in hand, plasma protein binding was considered as an asset to facilitate a rapid drug distribution of inhibitors. Among the most potent inhibitors possessing the meta-fluoro substituent on the phenyl ring, the PK profiles in mice were found to vary dramatically. In some cases, the compounds such as EC5023 and EC5024 and even TPPU were considered too stable following oral administration in preclinical species for development for use in humans (Table 2). Thus, the slow, tight-binding EC5026 was selected as a leading candidate for the clinic because it had a significantly shorter in vivo half-life than other analogues, yet a sufficient half-life to avoid break-through pain should a patient miss a daily dose.
Table 2. PK Profiles of EC5023, EC5024, and EC5026 in Rats.

Sprague–Dawley rats (male, n = 6) received the compounds by oral gavage with a single dose at 0.1 mg/kg (inhibitors were dissolved in PEG 300). PK parameters of inhibitors were calculated by WinNonlin based on the best fitted one compartment model. Note that the driver was a lower AUC and shorter half-life for a first-in-class drug.
Formulation for Human Phase 1a
The EC5026 drug product was initially developed as an immediate-release oral dosage form in a gelatin capsule using liquid formulation by considering the following attributes. First, EC5026 is an uncharged molecule with a low pH-independent solubility of <0.1 mg/mL in aqueous systems. Therefore, the equilibrium solubility of EC5026 in various media was screened to aid the development of preclinical formulations and a dissolution method (Table S1). Second, the permeability (Papp) of EC5026 using a colorectal adenocarcinoma cell line (Caco-2) is 26.4 × 10–6 cm/s indicated that the compound is well absorbed (Table S2). Therefore, per the Biopharmaceutics Classification System (BCS), EC5026 is classified as a BCS Class 2 compound (high permeability and poor solubility over the physiologic pH range), and its absorption rate is limited by either solubility or dissolution or both.25
Poor solubility of BCS Class 2 compounds limits the formulation choices and drug product dosage forms that can deliver the drug to the systemic circulation. Prior experience with structurally similar compounds to EC5026 indicated the drugs need to be in solution when dosed otherwise they will not be absorbed, which is directly related to poor solubility. This directed a formulation effort seeking a fully solubilized drug substance in a dosage form for both preclinical toxicology studies and use in the clinic. If dissolution is too fast, the compounds can also be recrystallized in the stomach. Therefore, liquid-filled hard capsules were the initial target for development. Since EC5026 has good solubility in PEG 400 (Table S1), efforts focused on capsules filled with the drug substance EC5026 dissolved in PEG 400. Several different capsules were evaluated, but all of the ones acceptable to the contract laboratory were affected by long-term exposure to PEG 400. The most prevalent incompatibility was leaking, with capsule deformation as a distant second problem. Overall, the output from these studies showed that none of the capsules tested were compatible with PEG 400. To address leaking of PEG 400 from capsules, we investigated novel PEG-based formulation using combinations of various PEG compounds based our prior experience and that of others which suggested that PEG 400 in combination with even very low concentrations of PEG 3350 produces a semisolid matrix which is hard and not easily deformed.26 The semisolid matrix forms after a heated solution of the two PEGs cools to room temperature. The combination of PEG compounds in a ratio of 80% PEG 400 and 20% PEG 3350 was chosen for the semisolid matrix formulation because the ratio of the two PEG compounds provides the physical rigidity to prevent leaking, analysis showed no crystal formation, EC5026 remained in solution in the dosage form, and the process was suitable for transfer to a compounding pharmacy where the clinical drug product was to be made for Phase 1a clinical trials. Therefore, the EC5026 drug product for use in initial clinical studies was an extemporaneously compounded immediate-release capsule for oral administration. Briefly, EC5026 capsules were prepared by filling a solution heated at 70 °C consisting of EC5026 drug substance dissolved in 80% PEG 400/20% PEG 3350 (w/w) into size 0, white opaque/white opaque, hard-gelatin capsules. The capsules were hand filled by volume with the heated solution and allowed to cool prior to capping. On the basis of an in-use stability study at both accelerated (40 °C/75% RH) and long-term (25 °C/60% RH) conditions for 4 weeks, these EC5026 drug products were found to be stable, but capsules were stored at 15–30 °C at the clinical site until dosing following preparation at the clinical pharmacy in accordance with pharmacy compounding procedures. Compositions of the EC5026 capsules, as well as the function and quality standard for each component of the dosage forms, are given in Table 3 (the detailed preparation of EC5026 is described in the Supporting Information). This procedure could be considered a low technology hot melt where, even if the compound were not soluble in the PEG matrix, its glass-like consistency precludes crystal formation.
Table 3. Compositions of 0.5 mg and 8 mg EC5026 Product.
| quantity
per capsule |
||||
|---|---|---|---|---|
| component | 0.5 mg strength | 8 mg strength | function | quality standard |
| EC5026 | 0.50 mg | 8.0 mg | API | in house |
| PEG 400 | 435.6 mg | 429.6 mg | solubilizer | Ph Eur, JP |
| PEG 3350 | 108.9 mg | 107.4 mg | matrix former | USP |
| Licaps capsules white opaque, size 0a | 1 each | 1 each | dosage form encapsulation | in house |
| total capsule fill weight | 545.0 mg | 545.0 mg | ||
Manufactured by Capsugel (Greenwood, SC). Abbreviations: API = active pharmaceutical ingredient, JP = Japanese pharmacopeia; PEG = polyethylene glycol; Ph Eur = European Pharmacopeia; USP = United States Pharmacopeia
Biochemcal Mechanism of Action
Degradation of Fatty Acid Epoxides (EpFA)
By weight, well over 50% of the world’s drugs act on the arachidonic acid cascade; only the COX and LOX pathways of the cascade have been exploited, and these drugs act by blocking the production or action of predominantly inflammatory chemical mediators. Of these, the COX branch has been the most studied and predominantly exploited by NSAIDs and COXIBs. In contrast, the sEHI work on the more recently discovered and largely analgesic and inflammation-resolving CYP450 branch of the cascade. Of these, the epoxides of arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid, abbreviated EETs, EEQs, and EDPs, respectively, are generally analgesic and anti-inflammatory mediators known collectively as EpFA. One could alter the pathway by increasing precursor polyunsaturated fatty acids, by stimulating the biosynthesis of EpFA or the release of EpFA from phospholipids, by using mimic natural bioactive EpFA, or by stabilizing the biologically active EpFA as reported here with sEHI.3 The sEH is a very active enzyme with a high kcat, a low Km, and in some tissues of higher abundance than its putative EpFA substrates, and thus a powerful inhibitor with high target occupancy is needed. The sEHI discussed here fit these criteria resulting in inhibition of the target sEH and the resulting increase in EpFA (Figure 4).
Figure 4.

Roles of EETs and sEH in the arachidonic acid cascade. Similar products arise from other polyunsaturated fatty acids. EETs are epoxides of arachidonic acid, and DHETs are the corresponding diols.
Thus, the sEHI are increasing tissue levels of the natural EpFA resulting in a variety of generally positive effects. Among these positive effects are a reduction in both inflammatory and neuropathic pain.27
Structure and Inhibitor Binding to Enzyme
Several X-ray crystal structures of human sEH complexed with urea-based sEHI have been determined.19,28,29 The human sEH structures revealed that the epoxide hydrolase domain is located at C-terminus, while the phosphatase domain is located at N-terminus.
In addition, the urea of the inhibitors mimics the transition state of the epoxide hydration in sEH (Figure 5A).19 The oxygen of the urea forms two tight hydrogen bonds with tyrosine 383 and tyrosine 466. In addition, both urea hydrogens strongly interact with aspartate 336 (Figure 5A). The resulting polarization of the urea may lead to an aspartate–urea salt bridge stabilized by the two hydrogen bonds with the urea. The binding pocket of sEH resembles a long hydrophobic channel (Figure 5B); therefore, the binding pocket generally cannot tolerate the polar or charged substituent, such as a pyridyl group, on 1,3-disubstituted urea.5 Interestingly, the recent crystal structure of human sEH with TPPU also showed an extra binding pocket next to the α position of the piperidinyl amide of the TPPU as predicted earlier by Kim.12 This observation was further supported from SAR studies (EC5019 vs EC5023 in Table 1).5,19 Also, Lee et al. revealed that the binding pocket of sEH is promiscuous,30 and the left side of the pocket can tolerate chemical structures of various sizes.31 The tunnel appears to breathe or even open to accept bulky groups. Therefore, EicOsis synthesized a new series of sEHI with a methyl substituent at the alpha-carbon of the piperidinyl amide of EC5023, resulting in a very potent EC5026 (Table 1).
Figure 5.
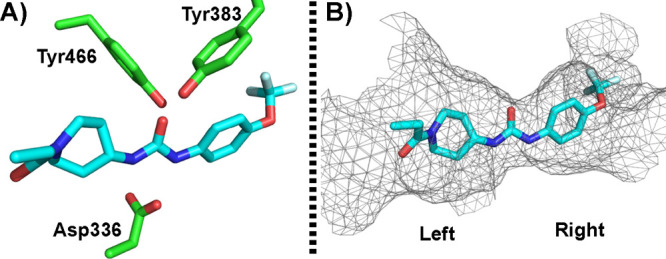
(A) The crystal structure (PDB ID: 4OD0) of human sEH bound with TPPU (cyan, stick) showed that Tyr383 and Tyr466 formed tight hydrogen bonding with the carbonyl oxygen of the urea and Asp336 formed a tight hydrogen bonding with the urea hydrogens. (B) The crystal structure revived the long hydrophobic binding channel at the C-terminus domain of human sEH.
Catalytic Enzyme Assays
Multiple assays have been developed to determine the potency of the sEHI, which includes early radiolabel assays, high-throughput colorimetric and fluorescent substrate assays, an LC-MS/MS assay with natural substrates, and FRET32 displacement assays. These and other assays have been discussed in reviews.4,33 In EicOsis, the fluorescent substrate assay was used as a primary screening assay because of the robustness and throughput of the assay. However, the fluorescent substrate screening assay has limits in distinguishing extremely potent compounds (IC50 ≤ 5 nM) effectively.32 Therefore, the potencies of the identified sEHI were further validated with a FRET displacement assay, radiometric assay, and LC-MS/MS based assay with natural substrates. Recent studies revealed that drug-target residence time is a better predictor of the in vivo activity of sEHI. This is particularly important when natural substrates have a very high apparent affinity for the enzyme. Therefore, the drug-target residence time of the sEHI which can be determined by the FRET displacement assay was also included in our inhibitor selection criteria.19,32 The availability of recombinant sEH of human and other preclinical species and the robust high-throughput assays allowed rapid SAR study of sEHI. Combining these data with a rapid determination of PK/ADME identified a promising candidate for efficacy testing in vivo.11
Physiological Mechanism of Action
History of EpFA Action
sEHI allowed for the elucidation of the biological activity of the transient EpFA, and it was discovered that EpFA are anti-inflammatory, pro-resolving, antihypertensive, and analgesic. Although the receptor for EpFA has not been identified, preclinical models of disease have identified multiple biological actions that contribute to the efficacious properties of EpFA. These actions include preventing or reversing endothelial cell dysfunction, altering the immune response, and reducing ER stress. These activities result in the regulation of cellular stress by stabilizing mitochondrial function, reducing reactive oxygen species, and shifting the ER response toward maintenance of homeostasis and away from severe activation of inflammatory pathways and cell death.
ER Stress and Mitochondrial Dysfunction as a Unifying Mechanism
This mechanism explains why there are so many possible indications for sEHI. We predict when ER stress and/or mitochondrial dysfunction is involved, sEHI administration will be beneficial. The original biological activity found for sEHI was the reduction of blood pressure.10 Considering that a search for endothelium derived hyperpolarizing factors had been in progress for years, EETs appeared to fit the criteria for an endothelium-derived hyperpolarizing factor (EDHF). However, with the ability to block the rapid hydrolysis of EpFA with sEHI, new roles for both were quickly found including attenuating inflammation, and beneficial effects in treating COPD, sepsis, stroke, cardiac contractile function, cardiac hypertrophy, and others, which is summarized in a recent review paper.1 This of course raised the concern among critics if there was any disease where the sEHI were ineffective. Since neuropathic pain remains largely an unmet medical need, and we needed to demonstrate there was a disease where the sEHI had no efficacy, we evaluated them on neuropathic pain rodent models and were surprised to find that they were exceptionally potent.34 We have since observed a lack of efficacy in preclinical models with high reactive oxygen species. Later studies resulted in the discovery that stabilization of EpFA by sEHI moves the ER stress response back toward cell survival and homeostasis and away from acute inflammation and cell death.35 Thus, a unifying mechanism was found for the combined action of EpFA and sEHI. ER stress has been found to be an underlying contributor to every disease state where EpFA are effective to date, and therefore, the presence of ER stress in a disease suggests that sEHI should be effective.
Selecting a Clinical Path
The sEHI stabilize whatever EpFA are released and biosynthesized at the time they are administered. Since various EpFA have slightly different biological action and potencies, the effects of sEHI are anticipated to vary somewhat with the individual treated, their diet, and their physiological state. However, reversing the shift of the ER stress pathway away from initiation of inflammation, pain, and other disease states and toward resolution seems a common effect of stabilizing and increasing EpFA. As indicated by Table S6 and in several review articles, this fundamental mechanism underlies many disease states ranging from hepatic steatosis to senescence and aging, and more possible clinical targets are being found as the role of ER stress in disease becomes better understood. Thus, each of the companies involved in the development of sEHI or mimics of EpFA has the “problems of the rich” in selecting a good commercial target and a viable clinical path from among many possibilities. For example, Arête Therapeutics initially selected hypertension for a short acting drug candidate, although the standard at the time in the field of hypertension was once or twice a day dosing. Numerous published studies demonstrated the efficacy of sEHI in treating hypertension driven by the renin–angiotensin system.10,36 However, for many patients, hypertension can be effectively controlled by several combination therapies delivered by drugs that appear quite safe and often inexpensive. Thus, this type of hypertension is a very hard target to address commercially with new chemistries. Since sEHI control many of the comorbidities of diabetes, Arête Therapeutics thought diabetes may be a good target and tried in their Phase 2a clinical trial to pick up efficacy on both diabetes and hypertension but failed to get clinically and economically relevant efficacy in either indication. In retrospect, we now know that ER stress is stimulated by high glucose. Diabetes is an attractive target as a growing and massive market, and certainly sEHI can address many of the comorbidities of diabetes, but diabetes itself is a difficult clinical path and comes with expensive and long-term clinical trials.
Problems caused by cardiac ischemia have long been considered potential targets. EicOsis initially considered COPD and asthma as indications as well, but GSK was already moving down this path. Atrial fibrillation also was considered since it is common, and one can envision relatively short and well-defined clinical trials.37 Atrial fibrillation also is a precursor to atrial fibrosis and cardiac hypertrophy, which are both major markets but involve expensive and long clinical trials.37 However, we were swayed toward pain by the observation that sEHI not only were powerful analgesics in inflammatory pain models where they synergized with and reduced the gastrointestinal toxicity of NSAIDs and COXIBs, but they also were analgesic in neuropathic pain models. Neuropathic pain remains an unmet medical need, and it is poorly handled by existing medications. There is a long history of pharma failures with the neuropathic pain indication. However, we were encouraged by the evidence that we could successfully treat severe equine laminitis, a neuropathic pain condition, with sEHI. This gave us confidence in moving from models into patients and from rodents into humans. Some neuropathic pain conditions also lend themselves to relatively short-term trials. With both in-house expertise and strong collaborators in the neuropathic pain area, we selected this indication. Because of the increasing appreciation that the opioid crisis in the United States is devastating both on a personal basis and economically, EicOsis was lucky in finding nondilutive resources to follow the neuropathic pain indication.
Clinical Path to Treating Inflammatory and Neuropathic Pain
Importance of Pain Drugs
We chose diabetic peripheral neuropathy (DPN) for our initial neuropathic pain trials due to the prevalence and growing incidence in the U.S. population, but also because DPN is a serious condition that causes substantial disability and often leads patients to use opioid analgesics when they fail to get adequate pain relief from currently approved neuropathic pain drugs. We are following this with several other pain indications selected based on the efficacy of sEHI in rodent and nonrodent species and where the indications seem to lead to opioid dependence. Our biological data support the hypothesis that the sEHI can control both inflammatory and neuropathic pain by acting peripherally.21 Therefore, our conclusion is that central nervous system penetration is not necessary for analgesic action, which also explains why sEHI work topically. However, it does not exclude action at the dorsal root ganglia or the CNS. Recently, it was reported that TPPU and EC5026 prevent neuroinflammation in the CNS of an LPS-induced mouse model, where TPPU has shown brain-to-plasma ratio of 17.2–21.7% in mice.38
The available treatments for neuropathic pain have, at most, modest efficacy, and dose-limiting and/or serious side effects, including somnolence, decreased cognition, dizziness, and, in some cases, respiratory depression. The commonly prescribed drugs, such as gabapentin and pregabalin, can produce significant CNS side effects.39 In addition, 50% or more of patients with neuropathic pain and painful diabetic peripheral neuropathy, in particular, are prescribed opioids to manage their symptoms, with the associated risk of opioid misuse and addiction.40 Patients requiring more than one drug class are at a higher risk for these central effects and for respiratory depression, especially with opioids. CNS adverse effects are common to most currently available neuropathic pain drugs and because of their impact on the day-to-day functioning of patients often result in discontinuation of treatment. Even NSAIDs, often prescribed for inflammatory pain including osteoarthritis, present a significant risk of GI-associated adverse events. Novel drugs able to demonstrate efficacy in inflammatory and neuropathic pain without these side effects would provide an important advance for patients.
sEHI in general and EC5026, in particular, are devoid of the side effects produced by other drugs used for this therapeutic indication.41 sEHI have a unique mechanism, blocking ER stress and inflammation in the diseased state to shift responses toward resolution and repair, providing efficacy without the side effects seen with other mechanisms. On the basis of data from preclinical studies, EC5026 has the potential to provide efficacy comparable to available treatments while avoiding serious side effects observed with other pain drugs, including CNS-related adverse events (Figure 6). Previous sEHI, including AR9281 and GSK2256294, that were tested in clinical trials showed minor adverse events.14 Unlike opioid analgesics, sEH inhibition has shown no evidence thus far of tolerance, dependence, or additional liability with chronic exposure in animals, avoiding the risk of addiction. sEHI do not disturb cognition, or produce sedation preclinically, and they do not produce the GI side effects of NSAIDs, and synergize with NSAIDs to block their side effects, further supporting their unique potential as pain drugs.14
Figure 6.

(A) In a chronic constriction injury model of neuropathy in male SD rats, EC5026 blocked pain measured with a von Frey assay (mechanical withdrawal thresholds). The efficacy of EC5026 was superior to pregabalin at 10–20 fold lower doses over the time course represented as area under the response curve (AUC) over 6 h post dose. The AUC was calculated with the trapezoidal method for each animal and the average of the group reported ± SEM (n = 6–8/group). Kruskal–Wallis one way analysis of variance on ranks, H = 21.011 with 3 degrees of freedom with Dunn’s Method post hoc. EC5026 vs PEG 300 (p ≤ 0.001), EC5026 vs pregabalin (30 mg/kg) (p = 0.039). (B) The same effective dose of 3 mg/kg EC5026 was able to block withdrawal pain in morphine dependent rats (male SD rats, n = 4/group). Rats were made morphine tolerant with twice daily 10 mg/kg subcutaneous injections of morphine for 10 days. Morphine was stopped and after 18 h of withdrawal the rats were assessed and administered EC5026 or vehicle by oral gavage and tested at 30 min and 1 h post treatment. Painful withdrawal baseline scores (von Frey assay) were normalized to zero (zero is the painful state). EC5026 relieved opioid withdrawal pain compared to vehicle controls. For both graphs scores are calculated as the percent improvement over the painful baseline: the score per time point/painful baseline score *100 calculated for each animal and averaged ± SEM.
Other important aspects of drug safety include the selectivity of drugs for the target and the avoidance of drug–drug interactions. Several of the available therapies for neuropathic pain have been shown to cause drug–drug interactions, and patients with neuropathic pain often require more than one drug class.42 EC5026 has proven selective for sEH when screened against a large receptor panel in vitro, and additional in vitro data described below suggest that the potential of drug–drug interactions for EC5026 is low, further supporting the safety of EC5026 and its unique profile as a therapeutic for neuropathic pain.5
IND-Enabling Studies
Species selection for dose-range finding toxicology studies is generally determined based on species comparisons of rates of metabolism and on whether unique metabolites are generated in humans. These studies involve the use of microsomal enzymes and/or hepatocytes from rodents, canine, and primates. Rat and dog were selected as the toxicological species due to similarities in PK and metabolite formation. Studies to identify clinically relevant toxicological species based on metabolism profiles and to predict human equivalent doses based on clearance rates are described below.
In vitro metabolism studies were conducted in mouse, rat, dog, monkey, and human hepatocyte incubations at SEKISUI XenoTech and demonstrated the formation of seven putative metabolites (hydroxylated EC5026 and their corresponding dehydrated products) that were separated by HPLC and detected by mass spectrometry. All seven metabolites were detected in the mouse, rat, monkey, and human hepatocyte incubations, while only six metabolites were observed in dog hepatocytes. On the basis of the in vitro metabolite profiles, it is likely that EC5026 is mainly metabolized by CYP450 enzymes.
Considering the main route of metabolism is likely through CYP450s, inhibition and induction of CYP450s were measured after incubation of 7 μM EC5026. Of seven major CYP450 isoforms assessed for inhibition, EC5026 had little to no inhibitory effects except for CYP2C9 and CYP2C19. At 7 μM, EC5026-mediated inhibition was about 25% and 34% for CYP2C9 and CYP2C19, respectively. An in vitro study was conducted to assess the inductive effects of EC5026 on CYP1A2, CYP2B6, and CYP3A4 by using three different batches of cryopreserved human hepatocytes. CYP1A2, CYP2B6, and CYP3A4 are known to be induced via three mechanistically distinct mechanisms. Treatment of cultured human hepatocytes with EC5026 up to 30 μM had little or no effect (i.e., < 2-fold change and/or <20% of the positive control) on CYP1A2, CYP2B6, and CYP3A4 mRNA levels. These results strongly suggest that EC5026 is not a potent CYP inhibitor or inducer. Clinically effective concentrations measured at the highest dose tested (24 mg) in human clinical trials achieved 301 ng/mL, or 9.4× lower than the concentrations used in this study. Therefore, potential CYP-mediated drug interactions caused by EC5026 are not expected.
Excretion: Distribution studies were contracted with QPS (Newark, DE) to characterize the rate and extent of excretion of EC5026 after a single oral dose of [14C]EC5026 at 3 mg/kg in male and 1.5 mg/kg in female Sprague–Dawley rats. 96.5% of the dose was recovered in the urine and feces through 168 h postdose. On the basis of these data, it is believed that the elimination of EC5026 is mainly attributed to hepatic metabolism, likely through CYP450 metabolism.
Safety Studies
EC5026 has been assessed in a series of nonclinical toxicology studies consistent with ICH M3(R2): Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (January 2010) including in vitro metabolism, genotoxicity studies, CNS effects in a modified Irwin assay, and single and multiple repeat dose studies in the most sensitive species in male and female rats and dogs. Twenty-eight day repeat-dose GLP toxicity studies were also conducted to estimate safety in humans.
Three dose levels of EC5026 were tested via oral gavage were tested in GLP toxicity studies conducted in rat and dog for 28 consecutive days in independent groups of male and female Beagle dogs and Sprague–Dawley rats (6 dogs and 15 rats/sex). A control group was administered the vehicle (PEG 400) at an equivalent volume. Animals were sacrificed on Days 29 and 57 (4 dogs and 10 rats/sex each at “Main” and 2 dogs and 5 rats/sex each at “Recovery” necropsies). The following potential toxicities were evaluated: plasma drug levels and toxicokinetics, mortality/morbidity, clinical observations including electrocardiograms (dog only) and ophthalmologic exams, functional observational battery (male rats only), body weights, micronucleus evaluation for genetic toxicity (rats only), food consumption, clinical pathology (hematology, clinical chemistry, and coagulation), urinalysis, gross necropsy observations, organ weights, and microscopic evaluation of tissues.
With the available animal data via the 28 Day GLP toxicity studies and efficacy studies in preclinical species, a therapeutic index (TI) was estimated based blood level exposures to EC5026. For this estimation there was no toxic dose in 50% of the population (TD50) observed so the dose for the No Observed Adverse Effect Level (NOAEL) was used. To correlate to this level the minimum dose with observed efficacy was used rather than the efficacious dose in 50% of subjects (ED50). The Area Under the Curve after the last dose (AUClast) in ng*h/mL exposure for the NOAEL was divided by the AUClast of the minimum efficacious dose previously determined in a rat chronic constriction injury model of neuropathic pain.
This resulted in a 37–111× therapeutic index based on rat and dog exposures, respectively.
In addition, Panigrahy et al. reported that EETs promote tumor growth. In particular, EETs can stimulate multiorgan metastasis and escape from tumor dormancy in several tumor mouse models.43 EETs promoted metastasis by triggering secretion of VEGF by the endothelium, which was critical for EETs’ cancer-stimulating activity. The stimulation of metastasis by EETs is due to their action at the secondary (metastasis) site and not due to their effect on the cells of the primary tumor. The authors failed to show EETs-promoted growth of the primary tumor and at the site of metastasis using sEH-null mice while exogenously administrated EETs promote them where systemic EET level is twice than the sEH-null mice. However, such high EET level in the sEH-null mice cannot be achieved by sEHI.
More importantly, GSK was also aware of this issue and concerned about the role of EETs and sEH in VEGF signaling and expression. Therefore, GSK measured VEGF concentrations to ensure human safety in their Phase 1 clinical trial and found no significant changes in VEGF concentrations from baseline to day 15 in the repeat dose subjects. Overall, serum VEGF concentrations were not increased and actually trended lower in subjects who received the higher dose. Meanwhile, near maximal sEH inhibition (98–99%) was observed for the doses following two-week repeated dosing.14b Considering the reasons listed above, the results from using mouse models of cancer are not likely to translate to human cancers, and no such concern was raised by the FDA when EC5026 moved on in the IND application process to Phase 1a clinical trial. Despite this, long-term studies are still necessary to fully assess the potential effects.
Fast Track
In April 2020, the FDA granted Fast Track designation to EC5026 for the treatment of neuropathic pain. The FDA’s Fast Track process is intended to facilitate the development and expedite the review of new therapies to address an unmet medical need in the treatment of a serious condition. Neuropathic pain is estimated to affect 7–10% of the general population, substantially affecting day-to-day functioning and quality of life of patients. Currently available treatments for neuropathic pain remain ineffective and are associated with frequent adverse effects and poor tolerability. The FDA’s Fast Track designation recognizes the potential of EC5026 to address the pressing need for effective, nonaddictive alternative drugs for pain management, decrease the use of prescription opioids, and reduce the opioid abuse epidemic. The Fast Track program enables drug companies to have early and frequent communication with the FDA throughout the drug development and review process, and confers important benefits, including the potential eligibility for Priority Review of a New Drug Application.
Phase 1a Clinical Trial
A Phase 1a single ascending dose study to investigate the safety, tolerability, and PK of EC5026 in healthy volunteers was recently completed at PPD in Austin, TX (ClinicalTrials.gov Identifier: NCT04228302). Oral EC5026 was administered in five escalating single dose regimens (0.5–24 mg) in 40 healthy male and female subjects, eight subjects in each cohort, equal male and female with six subjects randomly assigned to receive EC5026 and two subjects the placebo. Dose escalation was done in a stepwise fashion following acceptable safety and tolerability of the preceding dose(s), as determined by the safety review committee. EC5026 was monitored in urine and plasma over 14 days. The study was completed with no drug-related safety concerns or adverse effects, and all doses were very well tolerated. EC5026 was well absorbed with linear dose-proportionality (Figure 7). Safety was demonstrated at ∼12× the predicted efficacious dose, and concentrations were 600× higher than the predicted efficacious concentrations determined from preclinical pain studies. The mean terminal half-life of EC5026 in plasma was between 42 and 59 h at doses of 8–24 mg, consistent with predictions based on IND-enabling toxicity studies, and suitable for once daily dosing. These results are reassuring for the advancement of EC5026 into multiple ascending dose (MAD) Phase 1b studies. EicOsis is planning to start a Phase 1b MAD clinical trial in healthy volunteers in the second half of 2021, followed by two nested Phase 1b MAD studies in patients with two chronic pain conditions that are anticipated to start in 2021. These MAD Phase 1b studies will evaluate the safety and tolerability of three escalating dose regimens of EC5026, administered as a single oral dose daily, for 7 consecutive days. Each dose regimen will be tested first in the healthy volunteer population before being tested in the two chronic pain populations.
Figure 7.

(A) Cmax and (B) area under the curve (AUC) of EC5026 after single oral dose administration in humans. Following single oral administration of EC5026 in healthy male and female subjects, plasma concentrations peaked rapidly with Tmax observed at 1.25 h across all dose groups and declined slowly through the end of sampling (14 days, data not shown). Plasma concentrations following 0.5 mg EC5026 were below the limit of quantification for all subjects at all time. Linear exposure was observed in the highest concentration measured at a specific time.
Rationale for the Anticipated Therapeutic Index
With the assumption that the in vivo IC90 of EC5026 in humans is similar to the in vitro IC90 (0.62 nM), the effective plasma AUC required to maintain target inhibition over 24 h is estimated to be ≥892 nM·min (362 ng·min/mL). For a daily dosing regimen, the effective total AUC of EC5026 can then be calculated to be 6 ng h/mL. After a single oral dose, administration of 2 mg EC5026 exceeded these concentrations; therefore, we predict that the efficacious dose level will be <2 mg administered once daily and the therapeutic index >12x based on the highest concentration tested to date in human, 24 mg.
Metabolism
The abundant metabolites found in human blood after a single oral dose of EC5026 in Phase 1a trial were hydroxylated products on carbons that are susceptible to oxidation by CYPs as predicted from a previous metabolism study of the related sEHI compound TPPU44 and previous microsomal and hepatocyte metabolism studies prepared from rat, dog and monkey with EC5026 (Figure 8). Of note, there were no detectable cleavage products of the urea moiety or deacylation products of the hindered amide. Neither glucuronides formed from conjugation of the oxidative metabolites nor the N,N’, or diglucuronides formed from N-glucuronidation of the ureas as seen with the urea triclocarban45 were detected in human blood.44 These glucuronide metabolites would be predicted to be rapidly excreted into the urine where they are found.
Figure 8.

Chemical structures of metabolites of EC5026 identified from human blood in Phase 1a clinical trial. Amide or urea cleavage was not observed.
Future Plans
As a small company, EicOsis must focus on a path to the clinic targeting neuropathic and inflammatory pain. However, stabilization of EpFA regulates a homeostatic mechanism of ER stress and has the potential for treating multiple disease states as mentioned above. Hopefully, given the extensive library of highly potent compounds held by EicOsis, other promising indications can be followed. In addition to the conditions discussed previously, additional indications could include reduction of postsurgical pain, chemotherapy, and CAR-T induced tumor growth and metastasis due to cell debris,46 modulation of cytokine storms associated with sepsis and COVID-19,47 and inflammation of the central nervous system.48
Equine laminitis and Treating Companion Animals
The purpose of the ACS symposium this report is associated with was to highlight “First-Time Disclosures of Clinical Candidates” specifically for new human health drugs; however, EicOsis is also pursuing a second sEHI in a different patent class, t-TUCB or EC1728 (Figure 2), for efficacy in horses and companion animals with neuropathic and arthritic pain. Animal health represents a major market, but more relevant to human health, demonstration of efficacy in these studies validates the ability to translate preclinical efficacy studies to a heterogeneous human patient population with diverse disease ideologies. Translating success in rodent pain models to humans has proven difficult in the past. This may be due to vast genetic differences in species, or possibly the use of models where pain is generated mechanically, genetically, or clinically. At EicOsis, we have had the advantage of close collaboration with a veterinary school where we can evaluate sEHI on veterinary patients with real disease states rather than models, patients presenting with complex comorbidities, and patients of four significantly different mammalian orders. The sEHI successfully relieving neuropathic and inflammatory pain certainly raises confidence that translation of this work to pain patients in another mammalian orders will be successful.
Possibly the most visual demonstration of the use of sEHI in animals resulted from research by Guedes49 in horses with laminitis. Laminitis is a painful inflammatory condition that shifts to a neuropathic pain resulting from initial inflammation of the hoof laminae. It is often of idiopathic origin and presents as both chronic and acute duration, either of which often ends in euthanasia of the horse due to pain and inability to stand. After discovering that sEH is upregulated in laminitic joints, Guedes50 implemented emergency measures to treat a laminitic horse scheduled for euthanasia with t-TUCB after all available treatments failed. After 5 days of treatment, this horse made a complete recovery with no recurrent laminitis (Figure 9).49 Since this initial case, seven other laminitic horses showed improvement in disease after having been treated with t-TUCB when all other treatments failed.50
Figure 9.

Hulahalla (4-year old female thoroughbred), shown above, before (left) and after (right) a five-day treatment of once daily t-TUCB at 0.1 mg/kg. Hulahalla presented with acute laminitis and was treated with standard of care for 3 days with little effect. Her condition became extremely painful, and given the lack of response to the medications and worsening of the condition, humane euthanasia was being considered as the last resort to relieve pain and suffering. At this point, daily t-TUCB was administered in addition to her standard of care treatment. Over the course of the five-day treatment, her pain level decreased, and no signs of adverse effects were observed both from clinical exams and evaluation of blood work. Hulahalla continued to do well, and laminitis has not reoccurred.
In another example, dogs with naturally occurring osteoarthritis were treated with sEHI for 5 days and had significant reduction in pain, as determined by a questionnaire quantifying pain behaviors and recorded by a blinded technician, compared to placebo-treated animals.51t-TUCB was selected for use in animals over EC5026 due to potency on companion animal enzymes.
Fundamental Concepts Advanced by This sEHI Project
The mammalian sEH was discovered investigating the rodent metabolism chemistry of an agricultural chemical. Although it was discovered in an applied project, the enzyme research was driven by fundamental curiosity primarily in the academic sector with inhibitors of sEH and mimics of EpFA initially prepared as research tools. However, with translating the sEHI to the clinic, the scientists involved have made a number of fundamental advances. Since the sEH is so efficient at hydrolyzing most EpFA, the first application of this work was in allowing researchers to evaluate the role of EpFA in a number of physiological systems where previously the metabolism was so fast the biological results were not reproducible. As the sEHI were increasingly used in vivo, they illustrated new and often unexpected involvement of EpFA in biological systems. These discoveries continue a broad front. For example, in nutrition they illustrate that many of the attributes of dietary omega-3 fatty acids may be through their epoxide metabolites. The most recent example of unexpected activities comes from the observation that sEHI prevent and ameliorate several CNS diseases in rodent models. They were also valuable in dissection of the crosstalk between predominantly inflammatory pathways such as those driven by the COX and the LOX enzymes. Using dual inhibitors and inhibitor combinations, this has been extended to the interaction of sEHI with the PPARs. The sEHI are an excellent example of mimicking the transition state(s) along reaction coordinates to design inhibitors and the value of kinetic treatments to describe this interaction. The slow-tight-binding nature of sEHI and often their slow kinetic off rates illustrated the value of target occupancy in describing potent inhibitors. This slow off rate of sEHI from the target enzyme has been used to illustrate the role of target mediated drug disposition (TMDD) in explaining both efficacy and PK.52 Recent work illustrates a key role of lipid chemical mediators in regulating endoplasmic reticulum stress, resolving inflammation and controlling pain in general. Hopefully, there will be more fundamental insights as EC5026 and other sEHI move toward the clinic.
Conclusion
Research by numerous academic scientists around the world discovered a plethora of biological effects mediated by the CYP450 branch of the arachidonate cascade due, in part, to the availability of high-quality EpFA and sEH inhibitors to stabilize them. A variety of technologies ranging from cloning and expressing the relevant enzymes to antibodies and enzyme assays for monitoring them supported this biological work. Demonstration of the in vivo efficacy of sEHI by a variety of international scientists demonstrated their potential roles as pharmaceuticals. As indicated by the patent literature, numerous companies followed these leads with both classical SAR driven and very innovative high-throughput approaches, leading to potent sEHI. The extensive information on the biology and chemistry of the EpFA came largely from government support through competitive funding and a spirit of openness and collaboration among academic and government scientists.
The development of the sEHI class was based on knowledge of enzymology and a methodical stepwise synthesis and screening approach over many years enabling the discovery of the initial lead sEHI. Parallel tracks were taken in the academic and industrial paths toward structural optimization. Key to the success of the academic path in medicinal chemistry was simple synthesis, rapid feedback from quantitative enzyme assays, rapid analysis of parent molecules in the blood following oral administration, technology to monitor blood eicosanoids as indicators of efficacy and target engagement, and, of course, a rigorous filter to only move the most promising compounds forward into animal pain models. The resulting university use patents were supplemented by later composition of matter patents allowing EicOsis to spin out of the University of California. With several SBIR grants from NIEHS, then the NINDS Blueprint Development Grant, and now a NIDA Heal Grant and NCI SBIR, EicOsis was able to develop the independent composition of matter IP described here, obtain an IND and FDA Fast Track Status, and complete human Phase 1a human trials. This example of sEHI development is among the first successful examples to date of the NIH-academic collaborative approach to drug development. The NIH-academic-industry collaboration illustrated here will add to the range of current approaches in drug development and to the diversity of new therapeutics that are so desperately needed.
Experimental Section
General Synthetic Procedures
This synthesis was performed by Adesis, Inc. All reagents and solvents were obtained from commercial suppliers and were used without further purification. All reactions, unless otherwise described, were performed under an inert atmosphere of dry nitrogen. Melting points were determined on an OptiMelt melting point apparatus and are uncorrected. 1H NMR, 13C NMR, and 19F NMR spectra were recorded at 600 or 400, 150, and 376 MHz, respectively. 1H–1H2D NMR was recorded at 400 MHz. ATR FT-IR, UV–vis, and elemental analyses were determined at Robertson Microlit laboratories, Ledgewood, NJ. Ultraviolet absorbance spectrum was obtained on a PerkinElmer Lambda 35 UV–vis spectrometer. Mass spectra were measured by an Agilent Infinity 1290 LC with Agilent 6150 Quadrupole MS using electrospray (+) ionization. All final products synthesized to >93% purity as determined by HPLC or LC/MS. HPLC analysis was performed using Agilent 1100 LC. HPLC conditions; wavelength 210 nm, bandwidth 4, column: SorbTech C18AQ, 2.1 × 50 mm, 3 μm; gradient method from 95:5 to 5:95 water/MeCN (0.1% formic acid in both) in 14 min with a 4 min hold at 95% acetonitrile, 0.7 mL/min (retention time 6.81 min). LC-MS analysis was performed using Agilent 1260 with Bruker timeTOF instrument. HPLC conditions; column: Phenomenex Kinetex C18, 21. X150 m, 1.7 μm; gradient method from 98:2 to2:98 water/MeCN (0.1% formic acid in both) in 10 min. EC5026 was synthesized to >99% ee as determined by chiral HPLC Analysis (Agilent 1100 LC with a Chiralpak OD column, 25 cm x 4.6 mm, 10 μm). Optical rotation was measured using PerkinElmer polarimeter 343. Experimental details for the synthesis and characterization of standards of EC5026 metabolites 1 and 2 are available in the Supporting Information.
Scale-Up Synthesis of EC5026
The scale-up non-GMP synthesis of EC5026 is shown in Scheme 1
Scheme 1. Scale-up Non-GMP Synthesis of EC5026.

Reagents and conditions: (a) triphosgene, tert-butyl 4-amino-piperidine-1-carboxylate, Et3N, DCM, −78 °C to rt, overnight; (b) 4 N HCl in MeOH, rt, overnight; (c) 7, Jones reagent, acetone, 0 °C to rt, 2.4 h; (d) EDCI, Et3N, DCM, rt, overnight.
tert-Butyl 4-(3-(3-fluoro-4-(trifluoromethoxy)phenyl)ureido)piperidine-1-carboxylate (4)
A solution of 3-fluoro-4-(trifluoromethoxy)aniline (3) (250 g, 1.28 mol) and Et3N (260 mL, 1.86 mol) in DCM (500 mL) was added dropwise over 4 h to a thick slurry of triphosgene (171 g, 0.576 mol) in DCM (500 mL) in a dry ice/acetone bath while maintaining the temperature below −64 °C. The cold bath was removed, and the mixture was warmed to rt and stirred for 30 min. The reaction was cooled to −78 °C, and a slurry of tert-butyl 4-amino-piperidine-1-carboxylate (386.4 g, 1.92 mol) and Et3N (260 mL, 1.86 mol) in DCM (1.2 L) was added in small portions over 3 h while maintaining the temperature below −65 °C. The reaction was warmed to rt and stirred overnight, at which point LCMS indicated a mixture of compound 3 (major), as well as both symmetrical ureas. The reaction mixture was washed with 10% aq HCl (4 × 1 L) The combined organic layer was dried over Na2SO4 and concentrated under reduced pressure to give a crude compound 4 as a viscous yellow oil (800 g) which was used in the next step without further purification.
1-(3-Fluoro-4-(trifluoromethoxy)phenyl)-3-(piperidin-4-yl)urea (5)
to a solution crude compound 4 (800 g) in MeOH (1.5 L) was added a solution of 4 N HCl in MeOH (1.8 L), which was prepared with 600 mL of conc. HCl and 1.2 L of MeOH. The reaction mixture was stirred at room temperature overnight at which point LC-MS indicated the reaction was complete. The MeOH was removed under reduced pressure and the residue was slurried in a mixture of water (500 mL) and DCM (250 mL) and filtered. The filtrate was transferred to a separatory funnel and the layers were separated. The aqueous layer was transferred to a 5 L 4-neck flask and basified to pH 12 by adding 300 mL of 50% aq NaOH solution dropwise. After stirring for 10 min, the precipitates were filtered and washed with water (500 mL) followed by heptanes (500 mL). The product was initially dried in a convection oven overnight at 40 °C. The material from two reactions of similar size was combined and triturated with 15% DCM in heptanes (2 L). The solid was dried overnight in a convection oven at 40 °C to give compound 5 (778 g, 96% yield over 2 steps) as a white powder. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 12.1, 2.6 Hz, 1H), 7.17 (t, J = 8.6 Hz, 1H), 6.97 (ddd, J = 8.8, 2.5, 1.5 Hz, 1H), 6.69 (br s, 1H), 4.77 (br s, 1H), 3.81–3.70 (m, 1H), 3.07 (td, J = 12.6, 3.3 Hz, 2H), 2.73–2.65 (m, 2H), 1.98 (br dd, J = 12.3, 2.8 Hz, 2H), 1.38–1.28 (m, 2H).
(S)-2-Methylbutanoic acid (7)
To a solution of (S)-2-methylbutan-1-ol 6 (140 g, Sigma 65980-100 ML, ≥ 95%ee, 1.59 mol, 1 equiv) in 1.4 L of acetone was added dropwise Jones reagent (a solution of CrO3 (263 g, 2.63 mol) in 500 mL of water and 250 mL of conc. H2SO4) while maintaining the temperature below 5 °C. During the addition, chromium salts began to precipitate out, either forming a gum on the bottom of the flask of a golf size ball. The addition was considered complete when an orange/brown color persisted (620 mL of Jones reagent)). After removal of the cooling bath, the reaction mixture was stirred for additional 2.5 h at ambient temperature. The reaction was quenched by adding 50 mL of IPA (50 mL), and the reaction mixture was stirred overnight. The solution was decanted and concentrated under reduced pressure. All solids were dissolved in water (1.3 L) and transferred to a separatory funnel. After tert-butyl methyl ether (750 mL) was added, the layers were separated. The remained organic layer was further washed with water (250 mL). The combined aqueous layer was extracted with tert-butyl methyl ether (750 mL), and the organic layer was washed with water (200 mL). All combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The material from two reactions of equal size was combined and distilled under reduced pressure (Teflon pump, 20 Torr, 80–85 °C) to give (S)-2-methylbutanoic acid (7, 234 g, 72% yield) as a colorless liquid, [∝]D20= +19.2 (c = 1.671 g/100 mL chloroform).
(S)-1-(3-Fluoro-4-(trifluoromethoxy)phenyl)-3-(1-(2-methylbutanoyl)piperidin-4-yl)urea (EC5026)
A mixture of (S)-2-methylbutanoic acid 7 (175 g, 1.71 mol, 1.3 equiv). EDCI (327 g, 1.71 mol, 1.3 equiv), Et3N (260 mL, 1.86 mol, 1.4 equiv), and compound 5 (425 g, 1.32 mol, 1 equiv) in DCM (6 L) was stirred overnight at room temperature, at which point LC-MS indicated ∼75% conversion to the desired product. 10% aqueous HCl solution (2 L) was added to the reaction flask, and stirring was continued for 5 min. The mixture was transferred to a 22 L separatory funnel containing 4 L of 10% aq HCl solution. After stirring for 10 min, the layers were separated. The organic layer was washed with 10% HCl (4 L) followed by saturated aq. Na2CO3 solution (2 × 6 L). The organic layer from two reactions of the same scale were combined, dried over anhydrous Na2SO4, and concentrated to dryness under reduced pressure. The residue was slurried in EtOAc (2 L) and reconcentrated. EtOAc (2 L) was added to the residue, the vacuum was stopped, and the bath temperature was raised to 60 °C. A clear yellow solution was obtained. The heat was turned off, and the solution was cooled with rotation to ambient temperature. After no visible solids were confirmed, EtOAc was removed in vacuo until solids began to form. The vacuum was terminated, and the mixture was stirred overnight at ambient temperature and pressure. The solids were collected by filtration and dried in a convection oven at 40 °C overnight to give EC5026 (526.6 g, 49% yield, > 99% purity) as a white solid. The filtrate was concentrated under reduced pressure to give a yellow waxy solid. After triturating with heptanes (400 mL), the resulting solid was recrystallized twice with EtOAc (250 and 150 mL) to give additional EC5026 (63.6 g, > 99% purity) for an overall yield of 55%.
Final Purification
A 12 L 4-neck flask was rinsed with absolute EtOH before use. Absolute EtOH (1 L) was added to the flask, the heat was turned on, and EC5026 (1034 g) was added in portions with stirring. Once all the solid had been added, additional EtOH was added in portions until a clear solution was obtained (1.45 L). The heat was turned off and DIUF water was added dropwise until a cloudy solution was obtained (1.38 L). The mixture was then reheated to obtain a clear solution and left to cool gradually with stirring. Once the solution became cloudy, the mixture was seeded and stirring was continued for 2 h. The resulting solids were filtered and washed with water (500 mL). The solids were dried in a convection oven at 40 °C overnight to give EC5026 (854 g, 82.6% recovery, 99.7% purity, 97.5% ee) as a white solid. mp 148.8–150.0 °C, 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 38.3 Hz, 1H), 7.53 (ddd, J = 12.4, 6.0, 2.5 Hz, 1H), 7.15 (t, J = 8.6 Hz, 1H), 7.04–7.00 (m, 1H), 5.42 (d, J = 8.1 Hz, 1H), 4.66–4.46 (m, 1H), 4.05–3.85 (m, 2H), 3.21 (td, J = 12.1, 2.7 Hz, 1H), 2.90–2.73 (m, 1H), 2.68 (p, J = 6.8 Hz, 1H), 2.33–2.17 (m, 1H), 2.04–1.88 (m, 1H), 1.71–1.58 (m, 1H), 1.47–1.37 (m, 1H), 1.24–1.15 (m, 2H), 1.11 (dd, J = 27.9, 6.7 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 173.54, 154.01, 153.6 (d, J = 246.1 Hz), 141.24 (d, J = 10.4 Hz), 128.39 (d, J = 13.3 Hz), 120.12 (q, J = 256.8 Hz), 113.57, 113.55, 105.65 (d, J = 23.6 Hz), 46.36, 43.48, 35.60, 32.77, 31.86, 31.66, 26.59, 17.17, 11.54. 19F NMR (CDCl3) δ −59.25 (d, J = 3.8 Hz), −127.19 (t, J = 7.5 Hz). MS (ESI) m/z = 406.2 (M + H+). Anal. (C18H23F4N3O3·0.95H2O): cald. C, 51.17; H, 5.94; N, 9.95. found. C, 51.11; H, 5.80; N, 9.80. [∝]D20= +10.4 (c = 0.506 g/100 mL EtOH).
EC5026 Metabolite Standard Synthesis
1-(3-Fluoro-4-(trifluoromethoxy)phenyl)-3-(1-(3-hydroxy-2-methylbutanoyl)piperidin-4-yl)urea (1, Most Abundant EC5026 Metabolite)
To a solution of the 1-(3-fluoro-4-(trifluoromethoxy)phenyl)-3-(piperidin-4-yl)urea 5 (100 mg, 0.31 mmol) and 3-hydroxy-2-methylbutanoic acid (40 mg, 0.34 mmol) in DMF (3 mL) were added PyBOP (208 mg, 0.40 mmol) followed by Et3N (66 μL, 0.47 mmol) at 0 °C. The reaction mixture was allowed to slowly warm to room temperature and stirred overnight. The solvent was removed in vacuo, and the residue was purified by column chromatography (3:7 hexanes-EtOAc containing 5% MeOH) to give the title compound, 111 mg (85%, purity = 93.75%) as a white solid. mp 53.9–63.5 °C. 1H NMR (600 MHz, DMSO-d6): δ 8.77 (d, J = 30.9 Hz, 1H), 7.67 (d, J = 13.3 Hz, 1H), 7.39 (t, J = 8.9 Hz, 1H), 7.11 (d, J = 8.9 Hz, 1H), 6.43–6.35 (m, 1H), 4.65–4.53 (m, 1H), 4.27–4.16 (m, 1H), 3.96–3.86 (m, 1H), 3.75–3.62 (m, 2H), 3.22–3.11 (m, 1H), 2.84–2.65 (m, 2H), 1.92–1.73 (m, 2H), 1.52–1.12 (m, 2H), 1.09–0.88 (m, 6H). MS (ESI) m/z: 420.1557 (M – H)−.
1-(3-Fluoro-4-(trifluoromethoxy)phenyl)-3-(1-(2-hydroxy-2-methylbutanoyl)piperidin-4-yl)urea (2, Minor EC5026 Metabolite)
To a solution of the 1-(3-fluoro-4-(trifluoromethoxy)phenyl)-3-(piperidin-4-yl)urea 5 (100 mg, 0.31 mmol) and 2-hydroxy-2-methylbutanoic acid (40 mg, 0.34 mmol) in DMF (3 mL) was added PyBOP (208 mg, 0.40 mmol) followed by Et3N (66 μL, 0.47 mmol) at 0 °C. The reaction mixture was allowed to slowly warm to room temperature and stirred overnight. The solvent was removed in vacuo ,and the residue was purified by column chromatography (3:7 Hexanes-EtOAc containing 5% MeOH) to give the title compound, 107 mg (82%, purity = 98.92) as a white solid. mp 188.2–189.9 °C. 1H NMR (600 MHz, DMSO-d6): δ 1H NMR (599 MHz, DMSO-d6) δ 8.75 (s, 1H), 7.67 (dd, J = 13.3, 2.5 Hz, 1H), 7.39 (t, J = 9.0 Hz, 1H), 7.11 (d, J = 8.9 Hz, 1H), 6.38 (d, J = 7.6 Hz, 1H), 5.22 (s, 1H), 4.75–4.13 (m, 2H), 3.75–3.66 (m, 1H), 3.25–3.18 (m, 1H), 2.82 (br s, 1H), 1.91–1.77 (m, 3H), 1.74–1.53 (m, 2H), 1.35–1.22 (m, 4H), 0.80 (t, J = 7.3 Hz, 3H). MS (ESI) m/z: 420.1566 (M – H)−.
Chronic Constriction Injury Model of Neuropathy
All animal experiments were performed based on protocols approved by the Animal Use and Care Committee of Antibody, Inc. Sprague–Dawley rats (male, 250–300 g) were purchased from Charles River Laboratories.
Pain Attenuation in a Chronic Constriction Injury Model of Neuropathy
Male SD rats underwent a chronic constriction injury surgery where 4 ligatures were loosely tied around the sciatic nerve. The rats were allowed to heal for 14 days and tested for allodynia in a von Frey assay. At 21 days post injury rats were treated via oral gavage with PEG 300 (vehicle), EC5026 or pregabalin as indicated and followed over a 6-h time course. The AUCs were calculated with the trapezoidal method for each animal and the average of the group reported ± SEM (n = 6–8/group). Kruskal–Wallis One Way Analysis of Variance on Ranks, H = 21.011 with three degrees of freedom with Dunn’s Method post hoc. EC5026 vs Peg300 (p ≤ 0.001), EC5026 vs pregabalin 30 (p = 0.039).
Blocking Withdrawal Pain in Morphine-Dependent Rats
Male SD rats were made morphine tolerant with twice daily treatment of 10 mg/kg s.c. morphine. After 10 days the morphine was withheld, and opioid withdrawal pain was measured with a von Frey assay 18 h after the last morphine dose. The opioid withdrawal baselines decreased 49% from pain-free naïve baseline scores. Rats were then dosed with vehicle or EC5026 (n = 4/group) via oral gavage and assessed for mechanical withdrawal thresholds at 30 min and 1 h post dose. Mann–Whitney Rank Sum Test, U Statistic = 10.0, T = 90.0 n(small) = 8, n(big) = 8, P(exact) = 0.021.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) SBIR Program R43/R44 ES025598, National Institute of Neurological Disorders and Stroke (NINDS) Blueprint Neurotherapeutics Network UG3/UH3NS094258, National Cancer Institute (NCI) R43CA233371, and National Institute on Drug Abuse (NIDA) Award Number UG3DA048767.
Glossary
Abbreviations Used
- AEPU
1-adamantanyl-3-{5-[2-(2-ethoxyethoxy)ethoxy]pentyl]}urea
- t-AUCB
trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- AUDA
12-(3-adamantan-1-yl-ureido)dodecanoic acid
- BCS
biopharmaceutics classification system
- sEH
soluble epoxide hydrolase
- DCC
dicyclohexyl carbodiimide
- DCU
dicyclohexyl urea
- DPN
diabetic peripheral neuropathy
- ECD
endothelial cell dysfunction
- EDHF
endothelium-derived hyperpolarizing factor
- EETs
cis-epoxyeicosatrienoic acids
- EpFA
epoxy-fatty acids
- IGRs
insect growth regulators
- sEH inhibitors
sEHI
- TMDD
target mediated drug disposition
- TPPU
1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea
- t-TUCB
trans-4-{4-[3-(4-trifluoromethoxy-phenyl)-ureido]-cyclohexyloxy}-benzoic acid
- UC
University of California.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01886.
This work was presented as Paper MEDI 217 in the “First-Time Disclosure of Clinical Candidates” at the American Chemical Society Meeting in San Diego, CA, 2019. The authors declare no competing financial interest other than obvious association with EicOsis Human Health and the University of California in some cases.
The authors declare the following competing financial interest(s): BDH, CBM, KW, AB, IC-P, GC, JY, WKS and SHH are employees of EicOsis LLC and KSSL is an employee of Synthia LLC.
Supplementary Material
References
- Kodani S. D.; Hammock B. D. The 2014 Bernard B. Brodie Award Lecture Epoxide Hydrolases: Drug metabolism to therapeutics for chronic pain. Drug Metab. Dispos. 2015, 43 (5), 788–802. 10.1124/dmd.115.063339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gill S. S.; Hammock B. D.; Yamamoto I.; Casida J. E.. Preliminary Chromatographic Studies on the Metabolites and Photodecomposition Products of the Juvenoid 1-(4′-ethylphenoxy-6,7-epoxy-3,7- dimethyl-2-octene. In Insect Juvenile Hormones: Chemistry and Action; Menn J. J., Beroza M., Eds.; Academic Press: New York, 1972; pp 177–189. [Google Scholar]; b Gill S. S.; Hammock B. D.; Casida J. E. Mammalian metabolism and environmental degradation of the juvenoid 1-(4’- ethylphenoxy)-3,7-dimethyl-6,7-epoxy-trans-2-octene and related compounds. J. Agric. Food Chem. 1974, 22 (3), 386–395. 10.1021/jf60193a058. [DOI] [PubMed] [Google Scholar]; c Hammock B. D.; Gill S. S.; Mumby S. M.; Ota K.. Comparison of Epoxide Hydrases in the Soluble and Microsomal Fractions of Mammalian Liver. In: Molecular Basis of Environmental Toxicity; Bhatnagar R. S., Eds.; Ann Arbor Science Publishers: Ann Arbor, MI, 1980; pp 229–272. [Google Scholar]
- Spector A. A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50, S52–56. 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C.; Hammock B. D. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 311–333. 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- Lee K. S. S.; Ng J. C.; Yang J.; Hwang S. H.; Morisseau C.; Wagner K.; Hammock B. D. Preparation and evaluation of soluble epoxide hydrolase inhibitors with improved physical properties and potencies for treating diabetic neuropathic pain. Bioorg. Med. Chem. 2020, 28 (22), 115735. 10.1016/j.bmc.2020.115735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinot F.; Grant D. F.; Beetham J. K.; Parker A. G.; Borhan B.; Landt S.; Jones A. D.; Hammock B. D. Molecular and biochemical evidence for the involvement of the Asp333-His523 pair in the catalytic mechanism of soluble epoxide hydrolase. J. Biol. Chem. 1995, 270 (14), 7968–7974. 10.1074/jbc.270.14.7968. [DOI] [PubMed] [Google Scholar]
- Morisseau C.; Goodrow M. H.; Dowdy D.; Zheng J.; Greene J. F.; Sanborn J. R.; Hammock B. D. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (16), 8849–8854. 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olearczyk J. J.; Field M. B.; Kim I.-H.; Morisseau C.; Hammock B. D.; Imig J. D. Substituted adamantyl-urea inhibitors of the soluble epoxide hydrolase dilate mesenteric resistance vessels. J. Pharmacol. Exp. Ther. 2006, 318 (3), 1307–1314. 10.1124/jpet.106.103556. [DOI] [PubMed] [Google Scholar]
- Liu J. Y.; Tsai H. J.; Morisseau C.; Lango J.; Hwang S. H.; Watanabe T.; Kim I. H.; Hammock B. D. In vitro and in vivo metabolism of N-adamantyl substituted urea-based soluble epoxide hydrolase inhibitors. Biochem. Pharmacol. 2015, 98 (4), 718–731. 10.1016/j.bcp.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig J. D.; Zhao X.; Capdevila J. H.; Morisseau C.; Hammock B. D. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 2002, 39 (2), 690–694. 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Schulz D.; Morisseau C.; Hammock B. D. High-throughput pharmacokinetic method: cassette dosing in mice associated with minuscule serial bleedings and LC/MS/MS analysis. Anal. Chim. Acta 2006, 559, 37–44. 10.1016/j.aca.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I.; Morisseau C.; Watanabe T.; Hammock B. D. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J. Med. Chem. 2004, 47, 2110–2122. 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- Shen H.; Hammock B. D. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55 (5), 1789–1808. 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Podolin P. L.; Bolognese B. J.; Foley J. F.; Long E. 3rd.; Peck B.; Umbrecht S.; Zhang X.; Zhu P.; Schwartz B.; Xie W.; Quinn C.; Qi H.; Sweitzer S.; Chen S.; Galop M.; Ding Y.; Belyanskaya S. L.; Israel D. I.; Morgan B. A.; Behm D. J.; Marino J. P. Jr.; Kurali E.; Barnette M. S.; Mayer R. J.; Booth-Genthe C. L.; Callahan J. F. In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediators 2013, 104–105, 25–31. 10.1016/j.prostaglandins.2013.02.001. [DOI] [PubMed] [Google Scholar]; b Lazaar A. L.; Yang L.; Boardley R. L.; Goyal N. S.; Robertson J.; Baldwin S. J.; Newby D. E.; Wilkinson I. B.; Tal-Singer R.; Mayer R. J.; cheriyan J. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. D.; Tsai H.-J.; Do Z. N.; Morisseau C.; Hammock B. D. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 2006, 16, 5212–5216. 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S. H.; Tsai H. J.; Liu J.-Y.; Morisseau C.; Hammock B. D. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem. 2007, 50 (16), 3825–3840. 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T. E.; Morisseau C.; Liu J.-Y.; Inceoglu B.; Jones P. D.; Sanborn J. R.; Hammock B. D. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure – activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem. 2010, 53 (19), 7067–7075. 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandan S.-K.; Webb H. K.; Chen D.; Wang Y.-X. J.; Aavula B. R.; Cases S.; Cheng Y.; Do Z. N.; Mehra U.; Tran V.; Vincelette J.; Waszczuk J.; White K.; Wong K. R.; Zhang L.-N.; Jones P. D.; Hammock B. D.; Patel D. V.; Whitcomb R.; MacIntyre D. E.; Sabry J.; Gless R. 1-(1-Acethyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg. Med. Chem. Lett. 2011, 21 (3), 983–988. 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S. S.; Liu J.-Y.; Wagner K. M.; Pakhomova S.; Dong H.; Morisseau C.; Fu S. H.; Yang J.; Wang P.; Ulu A.; Mate C. A.; Nguyen L. V.; Hwang S. H.; Edin M. L.; Mara A. A.; Wulff H.; Newcomer M. E.; Zeldin D. C.; Hammock B. D. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem. 2014, 57 (16), 7016–7030. 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer K. R.; Kubala L.; Newman J. W.; Kim I.-H.; Eiserich J. P.; Hammock B. D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (28), 9772–9777. 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K. M.; Gomes A.; McReynolds C. B.; Hammock B. D. Soluble epoxide hydrolase regulation of lipid mediators limits pain. Neurotherapeutics 2020, 17, 900–916. 10.1007/s13311-020-00916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S. K.; Rand A. A.; Wan D.; Yang J.; Inceoglu B.; Thomas M.; Morisseau C.; Yang G.-Y.; Hammock B. D. Pharmacological inhibition of soluble epoxide hydrolase or genetic deletion reduces diclofenac-induced gastric ulcers. Life Sci. 2017, 180, 114–122. 10.1016/j.lfs.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S. H.; Wecksler A. T.; Wagner K.; Hammock B. D. Rationally designed multitarget agents against inflammation and pain. Curr. Med. Chem. 2013, 20 (13), 1783–1799. 10.2174/0929867311320130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S. S.; Yang J.; Niu J.; Ng C. J.; Wagner K. M.; Dong H.; Kodani S. D.; Wan D.; Morisseau C.; Hammock B. D. Drug-target residence time affects in vivo target occupancy through multiple pathways. ACS Cent. Sci. 2019, 5, 1614–1624. 10.1021/acscentsci.9b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsume Y.; Mudie D. M.; Langguth P.; Amidon G. E.; Amidon G. L. The biopharmaceutics classification system: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci. 2014, 57, 152–163. 10.1016/j.ejps.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repka M. A.; Gerding T. G. Polyethylene Glycol Ointment for Apthous Ulcers. U.S. Patent 5,112,620, May 12, 1992.
- Wagner K. M.; McReynolds C. B.; Schmidt W. K.; Hammock B. D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. 10.1016/j.pharmthera.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez G. A.; Morisseau C.; Hammock B. D.; Christianson D. W. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry 2004, 43, 4716–4723. 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- Gomez G. A.; Morisseau C.; Hammock B. D.; Christianson D. W. Human soluble epoxide hydrolase: structural basis of inhibition by 4-(3-cyclohexylureido)-carboxylic acids. Protein Sci. 2006, 15, 58–64. 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S.; Henriksen N. M.; Ng C. J.; Yang J.; Jia W.; Morisseau C.; Andaya A.; Gilson M. K.; Hammock B. D. Probing the orientation of inhibitor and epoxy-eicosatrienoic acid binding in the active site of soluble epoxide hydrolase. Arch. Biochem. Biophys. 2017, 613, 1–11. 10.1016/j.abb.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmistrov V.; Morisseau C.; Karlov D.; Pitushkin D.; Vernigora A.; Rasskazova E.; Butov G. M.; Hammock B. D. Bioisosteric substitution of adamantane with bicyclic lipophilic groups improves water solubility of human soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2020, 30 (18), 127430. 10.1016/j.bmcl.2020.127430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S. S.; Morisseau C.; Yang J.; Wang P.; Hwang S. H.; Hammock B. D. Förster resonance energy transfer (FRET) competitive displacement assay for human soluble epoxide hydrolase. Anal. Biochem. 2013, 434 (2), 259–268. 10.1016/j.ab.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C.; Hammock B. D. Measurement of soluble epoxide hydrolase (sEH) activity. Curr. Protoc. Toxicol. 2007, 33, 4.23.1–4.23.18. 10.1002/0471140856.tx0423s33. [DOI] [PubMed] [Google Scholar]
- Inceoglu B.; Jinks S. L.; Ulu A.; Hegedus C. M.; Georgi K.; Schmelzer K. R.; Wagner K.; Jones P. D.; Morisseau C.; Hammock B. D. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (48), 18901–18906. 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A.; Nagata N.; AbouBechara D.; Chahed S.; Morisseau C.; Hammock B. D.; Haj F. G. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J. Biol. Chem. 2013, 288 (20), 14189–114199. 10.1074/jbc.M113.458414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamvimonvat N.; Ho C.-M.; Tsai H.-J.; Hammock B. D. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J. Cardiovasc. Pharmacol. 2007, 50, 225–237. 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- Xu D.; Li N.; He Y.; Timofeyev V.; Lu L.; Tsai H.-J.; Kim I.-H.; Tuteja D.; Mateo R. K. P.; Singapuri A.; Davis B.; Low R.; Hammock B. D.; Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18733–18738. 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Comerota M. M.; Wan D.; Chen F.; Propson N. E.; Hwang S. H.; Hammock B. D.; Zheng H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12 (573), eabb1206. 10.1126/scitranslmed.abb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Karmakar S.; Rashidian H.; Chan C.; Liu C.; Toth C. Investigating the role of neuropathic pain relief in decreasing gait variability in diabetes mellitus patients with neuropathic pain: a randomized, double-blind crossover trial. J. Neuroeng. Rehabil. 2014, 11, 125. 10.1186/1743-0003-11-125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kharasch E. D.; Clark J. D.; Kheterpal S. Perioperative Gabapentinoids: Deflating the Bubble. Anesthesiology 2020, 133 (2), 251–254. 10.1097/ALN.0000000000003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil P. R.; Wolfe J.; Said Q.; Thomas J.; Martin B. C. Opioid use in the management of diabetic peripheral neuropathy (DPN) in a large commercially insured population. Clin. J. Pain. 2015, 31 (5), 414–424. 10.1097/AJP.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K.; Yang J.; Inceoglu B.; Hammock B. D. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J. Pain 2014, 15 (9), 907–914. 10.1016/j.jpain.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J.; Gold M. S.; Backonja M. M. Combination drug therapy for chronic pain: a call for more clinical studies. J. Pain 2011, 12 (2), 157–166. 10.1016/j.jpain.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D.; Edin M. L.; Lee C. R.; Huang S.; Bielenberg D. R.; Butterfield C. E.; Barnes C. M.; Mammoto A.; Mammoto T.; Luria A.; Benny O.; Chaponis D. M.; Dudley A. C.; Greene E. R.; Vergilio J.-A.; Pietramaggiori G.; Scherer-Pietramaggiori S. S.; Short S. M.; Seth M.; Lih F. B.; Tomer K. B.; Yang J.; Schwendener R. A.; Hammock B. D.; Falck J. R.; Manthati V. L.; Ingber D. E.; Kaipainen A.; D’Amore P. A.; Kieran M. W.; Zeldin D. C. Epoxyeicosanoids stimulate mutiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest. 2012, 122 (1), 178–191. 10.1172/JCI58128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan D.; Yang J.; McReynolds C. B.; Barnych B.; Wagner K. M.; Morisseau C.; Hwang S. H.; Sun J.; Blocher R.; Hammock B. D. In vitro and in vivo metabolism of a potent inhibitor of soluble epoxide hydrolase, 1-(1-propionylpiperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea. Front. Pharmacol. 2019, 10, 464. 10.3389/fphar.2019.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schebb N. H.; Inceoglu B.; Ahn K. C.; Morisseau C.; Gee S. J.; Hammock B. D. Investigation of human exposure to triclocarban after showering and preliminary evaluation of its biological effects. Environ. Sci. Technol. 2011, 45 (7), 3109–3115. 10.1021/es103650m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D.; Gartung A.; Yang J.; Yang H.; Gilligan M. M.; Sulciner M. L.; Bhasin S. S.; Bielenberg D. R.; Chang J.; Schmidt B. A.; Piwowarski J.; Fishbein A.; Soler-Ferran D.; Sparks M. A.; Staffa S. J.; Sukhatme V.; Hammock B. D.; Kieran M. W.; Huang S.; Bhasin M.; Serhan C. N.; Sukhatme V. P. 2019. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J. Clin. Invest. 2019, 129 (7), 2964–2979. 10.1172/JCI127282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammock B. D.; Wang W.; Gilligan M. M.; Panigrahy D. Eicosanoids: the overlooked storm in COVID-19?. Am. J. Pathol. 2020, 190 (9), 1782–1788. 10.1016/j.ajpath.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Tan Y.; Chang L.; Hammock B. D.; Hashimoto K. Increased expression of soluble epoxide hydrolase in the brain and liver from patients with major psychiatric disorders: A role of brain - liver axis. J. Affective Disord. 2020, 270, 131–134. 10.1016/j.jad.2020.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes A.; Morisseau C.; Sole A.; Soares J. H.; Ulu A.; Dong H.; Hammock B. D. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet. Anaesth. Analg. 2013, 40 (4), 440–448. 10.1111/vaa.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes A.; Galuppo L.; Hood D.; Hwang S. H.; Morisseau C.; Hammock B. D. Soluble epoxide hydrolase activity and pharmacologic inhibition in horses with chronic severe laminitis. Equine Vet. J. 2017, 49 (3), 345–351. 10.1111/evj.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McReynolds C. B.; Hwang S. H.; Yang J.; Wan D.; Wagner K.; Morisseau C.; Li D.; Schmidt W. K.; Hammock B. D. Pharmaceutical effects of inhibiting the soluble epoxide hydrolase in canine osteoarthritis. Front. Pharmacol. 2019, 10, 533. 10.3389/fphar.2019.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Hammock B.; Lee K. S. S.; An G. Simultaneous target-mediated drug disposition (TMDD) model for two small-molecule compounds competing for their pharmacological target: soluble epoxide hydrolase. J. Pharmacol. Exp. Ther. 2020, 374 (1), 223–232. 10.1124/jpet.120.265330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.